

The functional interaction landscape of HSV-1 infection uncovers a link between immune sensing and the DNA damage response.
Abstract
Dynamically shifting protein-protein interactions (PPIs) regulate cellular responses to viruses and the resulting immune signaling. Here, we use thermal proximity coaggregation (TPCA) mass spectrometry to characterize the on-off behavior of PPIs during infection with herpes simplex virus 1 (HSV-1), a virus with an ancient history of coevolution with hosts. Advancing the TPCA analysis to infer associations de novo, we build a time-resolved portrait of thousands of host-host, virus-host, and virus-virus PPIs. We demonstrate that, early in infection, the DNA sensor IFI16 recruits the active DNA damage response kinase, DNA-dependent protein kinase (DNA-PK), to incoming viral DNA at the nuclear periphery. We establish IFI16 T149 as a substrate of DNA-PK upon viral infection or DNA damage. This phosphorylation promotes IFI16-driven cytokine responses. Together, we characterize the global dynamics of PPIs during HSV-1 infection, uncovering the co-regulation of IFI16 and DNA-PK functions as a missing link in immunity to herpesvirus infection.
INTRODUCTION
Dynamic cascades of virus–host protein interactions underlie the ability of viruses to replicate within host cells, as well as the communication between infected cells and their microenvironment. This is well portrayed for viruses that share an ancient history of coevolution with their hosts, such as members of the family Herpesviridae (1), where an expansive range of interactions reflect adaptations of cellular processes for the benefit of the virus or the host. The large genomes of herpesviruses, which frequently encode more than 70 viral proteins, drive the formation of hundreds of spatially and temporally tuned protein-protein interactions (PPIs) that regulate each stage of the replication cycle—viral entry, trafficking, replication, assembly, and egress events (Fig. 1A) (2). Dynamic protein interactions also allow the host cell to initiate defense mechanisms such as apoptosis, autophagy, and immune signaling.
Fig. 1. Leveraging thermal proteome profiling to characterize protein interactions at a global scale during HSV-1 infection.
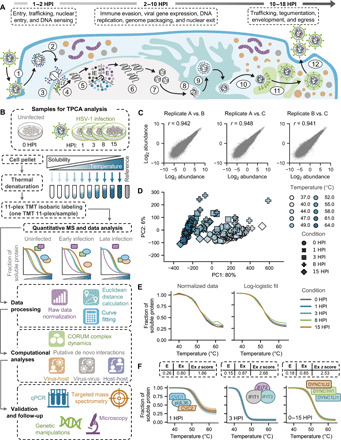
(A) Schematic representation of the herpes simplex virus 1 (HSV-1) life cycle from cellular entry; expression of the immediate early (IE), early (E), and late (L) genes; genome replication; and to virus assembly and egress over a time period of ~18 hours. (B) Workflow for thermal proteome profiling (TPP) sample preparation and subsequent data processing and computational analysis of uninfected and HSV-1–infected cells. (C) Overall consistency of protein signal detected across three biological replicates (A, B, and C), where the r value represents the pairwise Pearson correlation between replicates across all conditions (see also fig. S1C). (D) Principal components analysis (PCA) plot of all normalized protein abundances demonstrating the separation of different tandem mass tagging (TMT) channels (e.g., temperatures) and HSV-1 conditions (e.g., infection time points). x and y axes denote the percent of the data variance explained by principal components (PC) 1 and 2, respectively (see also fig. S1D). (E) Mean normalized (left) and log-logistic fitted (right) protein solubility curves for each condition across the temperature gradient. (F) Overlapping smoothed protein coaggregation curves for several expected interactions (i.e., known interactions) represented in the dataset. Solid lines and shaded regions represent the mean and SD, respectively, for a given protein across conditions and biological replicates. Average Euclidean distance (E), Ex, and Ex z score values for each set of proteins are also reported.
At early stages of infection with nuclear-replicating herpesviruses, after virus entry into host cells, the viral capsid traffics through the cytoplasm and then docks at the nuclear pore. As the viral genome is ejected into the nucleus (Fig. 1A, steps 3 and 4), there is a crucial moment before the initiation of viral transcription when the viral DNA (vDNA) can be detected by host DNA sensors to trigger cytokine expression and alert nearby cells to mount defense responses. To control herpesviruses, which establish lifelong latent infections in their hosts, this innate DNA sensing event is needed for the later establishment of an adaptive immune response that inhibits virus reactivation. One such nuclear DNA sensor is the interferon (IFN)–inducible protein 16 (IFI16), which moves to the nuclear periphery and forms oligomeric assemblies upon binding to vDNA (3–6). This host protein–vDNA interaction serves as a platform and launching point of antiviral signaling cascades that stimulate cytokine expression and suppress herpesvirus transcription (7–10). Although the capacity of IFI16 to elevate cytokine expression is now recognized during a number of viral infections, many aspects of this host defense response remain unknown. It is not yet understood what regulatory mechanisms drive the localization of IFI16 to the nuclear periphery and whether dynamic protein interactions are needed for downstream immune signaling and communication from the nucleus to other subcellular compartments. Several studies show that the nuclear IFI16 can communicate with the endoplasmic reticulum adaptor protein stimulator of interferon genes (STING), activating the TBK1-IRF3 (TANK-binding kinase 1 and Interferon regulatory factor 3) axis, although STING-independent mechanisms have also been proposed. Furthermore, as IFI16 must be constitutively expressed to function as an antiviral sensor, the dynamic remodeling of its interactome after detection of vDNA is likely a central regulatory mechanism that prevents overstimulation and autoimmunity.
It is also known that, in response to IFI16-mediated host defense mechanisms, herpesviruses have acquired strategies to inhibit IFI16 functions. Herpes simplex virus type 1 (HSV-1) induces IFI16 proteolytic degradation through expression of the virally encoded E3-ubiquitin ligase, ICP0 (11), and the human cytomegalovirus (HCMV) tegument protein pUL83 interacts with IFI16 to block its aggregation (12). Other instances where viral proteins intersect with host responses to evade innate immunity are evident throughout the replication cycles of herpesviruses and can occur simultaneously within different cellular compartments. For example, early during HSV-1 infection, the tegument protein pUL41 inhibits host transcription and translation to block immune responses that are triggered during capsid trafficking (13). The HSV-1 pUL37 and HCMV pUL31 proteins, among others, interact directly with the cytosolic DNA cyclic GMP-AMP synthase (cGAS) to inhibit its immune functions (14–16).
These intertwined relationships between anti- and proviral mechanisms underscore the need to understand the global dynamics of protein associations as they occur throughout infected cells. Immunoaffinity purification (IP) mass spectrometry (MS) and proximity labeling approaches ascorbic acid peroxidase (APEX) or biotin identification (BioID) have been proven valuable for uncovering virus–host protein interactions (2, 17). However, these methods are not designed to simultaneously track numerous protein complexes in order to provide a global view of the interactome or to capture the dynamic behavior of proteins cell-wide. For example, while providing an in-depth view of specific bait-prey protein interactions, IP-MS and other affinity-based or proximity-based methods cannot be easily scaled to resolve the subsequent interactions of a prey protein after it has dissociated from the bait. Yet, these dynamic and cascading protein interactions are at the core of the activation of cytokine expression by immune sensors.
Here, we used the thermal proximity coaggregation (TPCA) method to globally define the dynamics of host-host, virus-host, and virus-virus protein associations during HSV-1 infection, uncovering interactions that regulate IFI16-mediated antiviral responses. TPCA, which builds on the principle of thermal proteome profiling by MS (TPP-MS), provides an avenue to interrogate protein interactions throughout an entire cell system (18, 19). This method leverages the stabilizing nature of PPIs during exposure to a gradient of increasing temperatures, which causes interacting proteins to exhibit similar aggregation profiles, as quantified by MS. Upon stepwise heat denaturation, isobaric labeling and MS are used for measuring protein abundances, allowing melting curves to be simultaneously generated for thousands of proteins. The presence or absence of a protein interaction can be inferred by assessing melting curve similarity (or proximity) for different proteins. To date, TPCA has been used to identify drug targets, characterize PPI dynamics during cell cycle progression, and investigate the impact of a PTM on thermal stabilities (18–22). Demonstrating its applicability to viral infection studies, our laboratory has previously used TPCA to monitor the temporal assembly and disassembly of protein complexes during HCMV infection (23). HCMV infection occurs over a relatively long period of time (120 hours) and with relatively well-defined, synchronous stages of infection. In contrast, HSV-1 infection takes place in less than 24 hours, with temporal overlap in key events in the virus replication cycle, which brings additional challenges to deciphering how PPIs are regulated on a system-wide scale during infection. In the present study, we construct a system-level portrait of protein interactions as they fluctuate throughout the HSV-1 replication cycle. We characterize the regulation of hundreds of functional cellular protein complexes, as well as expand the TPCA method to infer PPIs de novo. With a focus on uncovering interactions driving immune signaling responses, we temporally resolve protein interactions with IFI16. This analysis uncovers that, already by 1 hour postinfection (HPI), IFI16 associates with a master regulatory kinase of the DNA damage response (DDR), DNA-PK. We interrogate the function of this temporal association by integrating microscopy, virology, phosphoproteomics, and mutagenesis assays. We show that IFI16 is necessary for the recruitment of active DNA-PK at sites of viral genome deposition within the nuclear periphery. Furthermore, IFI16 and DNA-PK have coordinated functions in driving host antiviral responses. We establish IFI16 as a substrate of DNA-PK and discover the IFI16 T149 phosphorylation site as a regulatory hub for immune response. Together, this study provides unprecedented insights into the PPIs that drive HSV-1 replication, the cellular response to herpesvirus infections, and the strategies that these viruses may use to suppress antiviral responses.
RESULTS
Leveraging TPCA to temporally resolve the global dynamics of protein interactions during HSV-1 infection
With the goal of globally characterizing the dynamic assembly and disassembly of protein complexes during HSV-1 infection, we performed TPCA throughout the viral replication cycle (Fig. 1A). Samples were collected at 0, 1, 3, 8, and 15 HPI in primary human foreskin fibroblast (HFF), a gold-standard model system for studying primary HSV-1 infection (Fig. 1B). These time points span stages of HSV-1 replication, including innate immune sensing and viral immediate early (IE) protein expression (1 to 3 HPI), vDNA replication (3 to 8 HPI), and assembly and egress of newly formed virus particles (8 to 15 HPI). Samples were divided into ten fractions, whereafter each fraction was exposed to increasing temperatures (37° to 64°C in 3°C increments), which was shown previously to be sufficient to produce log-logistic protein denaturation curves that are needed to assess protein interactions via TPCA (19, 23). The soluble proteins were then quantified by tandem mass tagging (TMT)–MS. To augment protein detection across samples, we also included a pooled reference channel of uninfected and 18 HPI samples. Of the ~6400 proteins that were detected, ~5200 proteins were quantifiable across all temperatures, time points, and three biological replicates with at least two peptides (fig. S1A and table S1). Among these were 50 to 60 viral proteins, representing ~75% of viral genes (fig. S1B). High reproducibility was observed between biological replicates (Fig. 1C and fig. S1C). Principal components analysis (PCA) showed that the overall variability was primarily driven by temperature changes, rather than variability between replicates and time points of infection (Fig. 1D and fig. S1D), suggesting that HSV-1 infection induces targeted rather than global changes to protein complex dynamics.
To process data for coaggregation analysis, protein abundance values were normalized across temperatures to generate standardized solubility curves that are fit by a three-parameter log-logistic function (Fig. 1E). To quantify the extent of overlap between protein solubility curves, we calculated the Euclidean distance between all pairwise combinations of proteins in a given sample (fig. S1E). We also calculated Ex distances to constrain Euclidean distances between zero and one and better fit a normal distribution (fig. S1F). From these transformed Ex values, we then determined standardized Ex z scores, which represent how a given Ex value compares to the rest of the population (fig. S1G). Possible protein associations were suggested by low Euclidean distance values between thermal curves, which correspond to high Ex and Ex z scores.
Confirming the ability of TPCA to capture PPIs, a number of known protein complexes were evident at different infection time points (Fig. 1F). This included the capsid-associated viral proteins CVC1 (capsid vertex component 1), CVC2, and pUL36, which exhibited overlapping solubility curves as early as 1 HPI, likely representing incoming viral particles. We also observed an extensive overlap between the cellular IFN-inducible proteins IFIT1, IFIT2, and IFIT3 at 3 HPI, a time point when host cells initiate a range of antiviral responses. The assembly of dynein complex subunits was observed in both uninfected and infected states, likely because of the roles of these proteins in trafficking of virus particles (24). Together, the assessment of the data quality and reproducibility and the observed known interactions support the ability of TPCA to map dynamic protein complex assemblies throughout the relatively rapid HSV-1 infection cycle.
Convergent and divergent regulation of protein complex dynamics during herpesvirus infections
A strength of the TPCA method is the capacity to monitor the dynamics of functionally annotated protein complexes, which can reveal signature complex activities that may affect infection. Therefore, we measured the association of known functional protein complexes from the CORUM (Comprehensive Resource of Mammalian Protein Complexes) database (25) to determine whether they are represented in our dataset and how they are regulated during HSV-1 infection. Within our dataset, we found 1187 CORUM complexes with >40% of subunits present for complexes with ≥3 members or 100% for two-member complexes (table S2). A range of different complex sizes (member numbers) was represented in the dataset (fig. S2A). Of these complexes, more than 200 were observed as being significantly associated (z score ≥ 1.5) at any given time point. The complexes that passed our significance threshold did not show a bias for complex size when compared to the overall population distribution (fig. S2A). Many of these complexes displayed dynamic behavior during infection (Fig. 2A). The observed changes included time point–dependent associations and dissociations or gradual changes in complex membership. Hierarchical clustering of these dynamic complexes revealed functional clusters linked to cell cycle control, DNA and RNA regulation and metabolic processes, and translation (Fig. 2A).
Fig. 2. CORUM complex dynamics during HSV-1 infection and a comparative analysis with HCMV infection.

(A) Hierarchical clustering of CORUM complex dynamics during HSV-1 infection. Any complex passing filtering criteria with an average Ex z score ≥ 1.5 across subunits is represented, and both Ex z scores and changes in complex abundance (relative to mock) are plotted. The table below the heatmaps shows the names of selected complexes denoted in the above heatmaps. (B) Dynamic interaction properties of NFKB1-STAT3 (top), Condensin I (middle), and ESCRT II (bottom) complexes during infection with HSV-1. For each complex, all subunits are represented, and each data point represents the Ex z score for a given pair of subunits. Gray lines connect data points for a given pair of subunits across infection time. (C) Comparison of CORUM complex dynamics during HSV-1 (this study) versus HCMV (23) infection. Heatmaps reflect the average change in complex Ex z scores (relative to the previous time point) and abundances (relative to mock) for each complex across both infections for early (HSV-1, 3 HPI; HCMV, 24 HPI) and late (HSV-1, 15 HPI; HCMV, 96 HPI) infection time points. Only complexes meeting our filtering and significance criteria for both viruses are shown.
The nature and temporality of the complex dynamics that we characterized were consistent within the context of HSV-1 replication. For example, by 1 to 3 HPI, we saw robust association of the NFKB1 (Nuclear Factor Kappa B Subunit 1)–signal transducer and activator of transcription 3 (STAT3) complex, which is involved in immune signaling (26) and implicated in viral gene expression (Fig. 2B) (27). We also observed an association of Condensin I complex subunits at 3 HPI, followed by their dissociation at 8 HPI when the viral genome replicates. Given that Condensin I is active after cellular DNA replication to block reamplification of the genome (28), this temporal dissociation by HSV-1 may be a strategy allowing for multiple rounds of vDNA synthesis. As another example, we found that the ESCRT (endosomal sorting complex required for transport) II complex assembles late during infection, when it may play a role in virus egress. The importance of ESCRT complexes, particularly ESCRT III, during herpesvirus infection is well established (29). However, whether ESCRT II plays a role in this process and how this may manifest remain unclear.
Next, to determine whether these complex regulatory events are unique to HSV-1 infection or more broadly conserved between several herpesviruses, we compared this temporally resolved CORUM complex analysis with our previous TPCA study during HCMV infection (23). Hierarchical clustering of changes in protein complex assembly and subunit abundance was performed to compare early and late stages of HSV-1 and HCMV infections (Fig. 2C). An example of a difference between these viruses is our finding that HSV-1 increased protein abundances and tighter associations within 40S and 60S ribosomal subunits (Fig. 2C). This may reflect an increased demand for viral protein expression that is required by the α-herpesvirus cycle, which must accomplish many of the same tasks and the same amount of virus production in a shorter period of time. Conversely, ribosomal subunit complex associations diminished during early HCMV infection, likely reflecting the host response, and increased late during infection when the viruses induces high expression of the eIF4 (eukaryotic initiation factor 4) proteins to increase translation (30). We additionally observed several protein complexes that were similarly regulated by both viral infections. For example, the NFKB1-STAT3 complex that we observed to associate early during HSV-1 infection also assembled upon HCMV infection. The CyclinD3-CDK4-CDK6-P21 complex that regulates S phase entry (31) was disassembled during the early stages of both infections, which may reflect the replication of herpesviruses in G1 phase. Last, the CCC complex that we previously reported to be assembled late during HCMV infection to promote viral production (23) was also induced during the late stage of HSV-1 infection. Together, this analysis of CORUM complex assemblies highlights the idea that TPCA can provide a functional and cell-wide snapshot that can contextualize cellular processes disrupted by viral infection or induced for antiviral responses.
Investigating the functional landscape of thermal profiling proteomics data
Although the TPCA method has only recently been used to interrogate protein interactions on a broad scale, one limitation that has become evident is that some protein aggregation curves are not well fit by the log-logistic function. To better understand the functional landscape of protein associations that can be probed by TPCA, we calculated the mean square error (MSE) for protein solubility curves compared to their fitted, log-logistic representations (Fig. 3A and table S3). As expected, most proteins were well modeled by the log-logistic function, as shown by small MSE values (<0.01) on average across all conditions. A small proportion (~2%) of proteins had relatively large MSE values (>0.2) and did not fit a log-logistic pattern (Fig. 3B). Overrepresentation analysis of this subset showed enrichment for proteins associated with cellular membranes and secretory vesicles (fig. S2B). This is perhaps expected since membranous proteins are difficult to isolate using weak lysis conditions that help maintain PPIs. The future implementation of different lysis buffer conditions that provide access to distinct subcellular compartments while retaining protein associations may help to interrogate interactions with proteins that are not well captured via TPCA. However, it was reassuring to observe that, of the proteins well represented by the TPCA data, there was a good representation across all major organelles and a wide variety of cellular processes (Fig. 3C and fig. S2, C and D). Represented in the data were nearly 500 different pathways from Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome, as well as more than 400 different protein domains and 1500 functional ontology pathways (fig. S2C). This underscores the broad scope of biological processes and pathways that can be monitored simultaneously by this method.
Fig. 3. Investigating the functional landscape of thermal profiling data and developing an approach to enrich for putative de novo interactions.
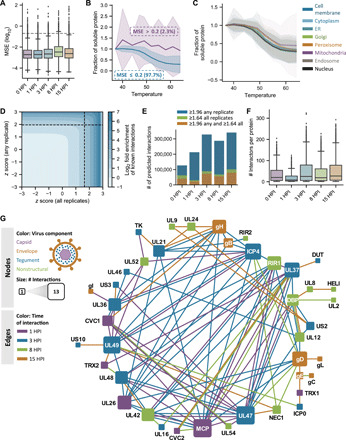
(A) Box-and-whisker plots representing the MSE (i.e., prediction error) for fitted log-logistic protein curves versus normalized solubility values. Boxes span the interquartile range (IQR), while whiskers extend to 1.5*IQR, and outliers outside of this range are plotted in black. The gray dashed line represents an MSE cutoff of 0.2 used for subsequent analyses. (B) Coaggregation curves for proteins with MSE values above and below 0.2. Solid lines and shaded regions represent the mean and SD, respectively, for each category across proteins, conditions, and replicates. (C) Coaggregation curves for proteins localized to different subcellular compartments. Solid lines and shaded regions represent the mean and SD, respectively, for proteins annotated to reside within a given organelle across conditions and replicates. (D) Contour plot showing known interaction enrichment (as represented in CORUM and STRING databases) at increasing Ex z score values when the Ex z scores for a given combination of proteins reach a specific cutoff for all (x axis) or any (y axis) replicate(s). The black dashed lines represent the significance cutoff used to define putative predicted interactions in the rest of this study. (E) Number of putative de novo interactions obtained by applying different Ex z score cutoffs. (F) Box-and-whisker plots showing the distribution of predicted number of interactions per protein at a given time point upon thresholding z scores at the specified cutoff described in the manuscript. (G) Temporal interactions between viral proteins across infection time. Edges represent pairwise Ex z scores between proteins that pass de novo interaction significance thresholding.
De novo modeling of virus-virus and virus-host interactions throughout HSV-1 infection
To further advance the TPCA analysis toward a system-level tool for interrogating protein-protein associations, we sought to complement our analysis of known protein complexes by also extracting putative de novo interactions from the dataset. We first asked to what extent different Ex z score cutoffs enrich for known interactions represented in the CORUM and STRING databases. By examining the relative enrichment for known interactions when the z score for a set of proteins passes a specific cutoff across any and/or all replicates, we observed that increasing Ex z scores highly enriches for known interactions. This enrichment increased as z scores approached 2.0. To therefore establish a set of criteria to predict de novo interactions, we used an iterative testing of z score cutoffs to determine their capacity to enrich for known interactions (Fig. 3D). This led to a first cutoff requiring a z score ≥ 1.96 (top 5% of putative interactions) in at least one replicate, which selected for high-confidence putative interactions. An additional cutoff was set, requiring the Ex z score for a protein pair to be ≥1.64 (top 10% of putative interactions) across all replicates for a given time point, which selected for likely consistent interactions. Our application of this dual threshold helped filter out putative interactions with a spuriously high z score in a single replicate (Fig. 3E). Using these parameters for predicting de novo interactions, we observed that any given protein forms 0 to 300 putative interactions that, on average, yielded Ex z scores exceeding 2.0 (Fig. 3F and table S4).
Among the resulting putative de novo interactions, we first interrogated the temporal dynamics of viral protein associations. The observed virus-virus interactions matched the progression of the HSV-1 replication cycle (fig. S3). For example, hubs of interactions were observed for the viral capsid and tegument, as illustrated by the tegument proteins pUL36, pUL37, and pUL49. Temporal regulation was also evident for interactions between components of the viral replicase complex [UL30, DBP (Major DNA-binding protein), and UL2], which occurred at 8 HPI when viral genome amplification is robust (fig. S3). We further visualized the temporality of virus-virus interactions by constructing a network of TPCA-inferred associations (Fig. 3G). Consistent with its role as a viral transcription factor that orchestrates events early in infection, ICP4 was a hub for viral PPIs at 3 HPI. Alternatively, the viral glycoprotein gD, which is necessary for envelopment, served as a focal point for PPIs at 8 and 15 HPI when mature virions are produced.
In addition to virus-virus protein interactions, our analysis also pointed to the formation of numerous temporally regulated virus-host PPIs (Fig. 4A). The number of interactions per protein was monitored in conjunction with the temporal abundance of the viral proteins (Fig. 4A). In agreement with the cascade of viral gene expression, an increasing number of virus-host associations was observed with the progression through the replication cycle (Fig. 4B). From the ~1500 TPCA-derived virus-host PPIs, we saw representation of a wide range of biological processes that are known to be affected during viral infection, including intracellular transport, metabolism, and mRNA splicing (Fig. 4C). Viral proteins that drove these virus-host interaction hubs were also evident, but these seem to be different from the hubs driving virus-virus PPIs (Fig. 4A). The temporality of these interactions again corresponded with key events in viral infection. For example, UL42, a component of the viral replicase, gained interactions at 3 HPI, a time point when vDNA replication begins. We also noticed more specific viral protein interactions with CORUM complexes, such as the dynamic association at 8 HPI between the viral exonuclease, UL12, and the KU complex that is important for DNA repair (Fig. 4D). UL12 is required for DNA repair events that produce vDNA that can be packaged and has been shown to interact with cellular DNA repair proteins, including the KU complex (32). In addition, UL48, a viral protein that regulates assembly and egress, associated with the ESCRT II complex at 8 HPI, highlighting the need for future investigation of this complex in the context of infection. Another cluster of interactions was observed for the viral dUTPase (deoxyuridine 5′-triphosphate nucleotidohydrolase) protein, DUT (or UL50), which changed between 3 and 8 HPI. Specifically, we noticed that DUT interacted dynamically with the NFKB1-STAT3 complex by 8 HPI (Fig. 4D) and also gained interactions with nuclear factor κB (NF-κB)–linked proteins, including IFI16 and IKBKB (Inhibitor of NF-kB kinase subunit beta) (Fig. 4E). Although no such interaction has been reported for HSV-1, DUT regulates inflammatory responses and triggers NF-κB activation during Epstein-Barr virus infections, respectively (33). Although NF-κB is well known to mediate immune responses, its inhibition decreases herpesvirus replication (27), indicating that this transcription factor also has a proviral role. Overall, we found that virus–host protein interactions were poised to regulate critical cellular defense processes, such as the regulation of IFI16 and NF-κB.
Fig. 4. Virus–host protein interaction dynamics throughout HSV-1 infection.
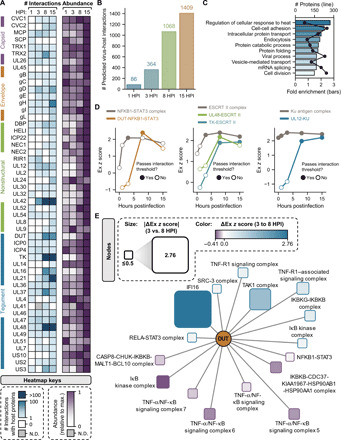
(A) Heatmap depicting the number of predicted virus-host interactions for each detected viral protein and relative viral protein abundances throughout infection (scaled to their maximum value across all conditions). (B) Number of predicted virus–host protein interactions at each time point following infection. (C) Bar plot showing terms that are overrepresented in the subset of host proteins that are predicted to interact with viral components. (D) Ex z scores for NFKB1-STAT3 complex interactions across infection time with itself and with the viral protein DUT. Solid data points represent Ex z scores surpassing the de novo interaction threshold. Ex z scores for ESCRT II complex interactions across infection time with itself and with viral proteins UL48 and TK. Ex z scores for Ku antigen complex interactions across infection time with itself and with the viral protein UL12. (E) Interaction network of DUT interactions with immune-related complexes and the antiviral DNA sensor IFI16 at 8 HPI relative to 3 HPI. N.D., not detected.
IFI16 interacts with DNA-PK at sites of vDNA deposition at the nuclear periphery by 1 HPI
To better understand the host antiviral response, we next characterized the temporal interactions of the DNA sensor IFI16 (Fig. 5A). Although known to be critical for the innate response to herpesvirus infections (3, 4, 6), the dynamic processes that allow IFI16 to localize and bind to vDNA and then trigger cytokine expression remain unknown. From our TPCA analysis, we predicted 154 proteins that putatively interact with IFI16 at any time point of infection or in uninfected cells (Fig. 5A). Several clusters of interactions were linked to translation, splicing, DNA repair, IFN signaling, and the NF-κB response. This fits well with the known IFI16 functions to suppress viral transcription and stimulate cytokine expression (5, 10). We overlaid our TPCA-inferred interactions with prior knowledge from IP-MS studies (Fig. 5B and table S5) (5, 10, 34, 35), which allowed us to understand the temporality of known interactions, as well as uncover previously unrecognized interactions. This comparison showed that ~40% of TPCA-predicted interactions were identified by IP-MS analysis with high specificity. For example, TPCA analysis revealed that the known IFI16 interaction with STAT1, STAT2, and STAT3 proteins is temporally regulated, and the association with STAT1 and STAT3 appears to be mutually exclusive. Of the interactions that were previously unreported, many proteins have STRING associations with known IFI16 interactors (Fig. 5A). This observation highlights an advantage of TPCA to identify PPIs that may be stabilized through nucleic acid intermediates or that associate only under native cellular conditions. By considering these dynamic IFI16 associations in conjunction with changes in protein abundances (Fig. 5C), additional insights into the regulation of these interactions were obtained. For example, IFI16 coaggregated with the NF-κB activator, IKBKB, at 3 HPI, a time point that coincides with a sharp decrease in IKBKB abundance and an increase in NFKB1 levels (Fig. 5C). By the next time point of infection (8 HPI), IFI16 gained an association with NFKB1 and STAT3, which may induce an inflammatory response during infection.
Fig. 5. IFI16 interacts with the DNA-PK complex at vDNA in the nuclear periphery.
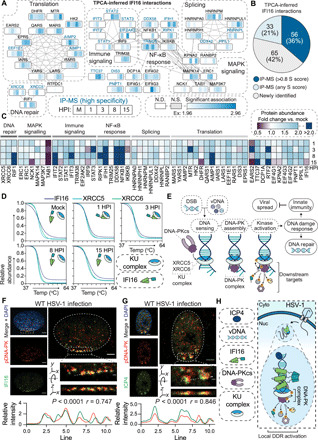
(A) Protein interaction network of TPCA-derived IFI16 PPIs. Gray lines indicate STRING interactions, and proteins are clustered by annotation (STRING and Reactome). Heatmaps above the nodes represent significant Ex z scores for IFI16 interactions with that node at 0, 1, 3, 8, and 15 HPI time points. Proteins previously identified to associate with IFI16 by IP-MS are labeled in blue. N.S., not significant. (B) Pie chart showing the proportion of overlap with IFI16 IP-MS studies for TPCA-inferred interactions. (C) Heatmap of protein abundances over the HSV-1 infection time course relative to the uninfected control. (D) Smoothed protein aggregation curves for IFI16 (dark blue), XRCC5 (teal), and XRCC6 (green) at each time point of infection. (E) Model for the DNA-PK DDR to DSBs and vDNA. (F and G) DNA-PKcs pS2056 (red) staining for immunofluorescent microscopy (IFA) at 100× after wild-type (WT) HSV-1 infection (3 HPI) with staining (green) for either (F) IFI16 or (G) ICP4. Scale bars, 5 or 1 μm in the inset (dashed box). The dashed line in the inset represents line scans for spatial quantification of pDNA-PK versus IFI16 or pDNA-PK versus ICP4 intensities for 30 nuclei across biological replicates (n = 3). Representative images are shown. Pearson’s correlation coefficient (PCC) between pDNA-PK versus IFI16 or ICP4 is represented for the displayed line scan. (H) Model representing IFI16 association with kinase activated DNA-PK holoenzyme with ICP4-labeled vDNA in the nuclear periphery.
We were particularly interested in proteins that gained an interaction with IFI16 rapidly after viral infection, as these may contribute to DNA sensing or the regulation of the antiviral response (5, 36). One such interaction that satisfied these criteria was with XRCC5 (X-ray repair cross complementing 5) and XRCC6, which we predicted to associate with IFI16 by 1 HPI and then disassemble after 8 HPI (Fig. 5D), which coincides with a dynamic association between IFI16 and DUT and the KU complex with UL12 discussed above (Fig. 4, D and E). XRCC5 and XRCC6 form a heterodimer (KU complex) that is essential for the recognition of double-stranded DNA breaks (DSBs) in the nucleus and vDNA in the cytosol (37). After binding to DNA, the KU complex recruits the kinase subunit of the DNA-PK holoenzyme, DNA-PK catalytic subunit (DNA-PKcs), to activate the DDR and stimulate cytokine secretion (Fig. 5E) (37).
IFI16 coaggregated with the KU complex at 1 to 3 HPI, a time point that coincides with IFI16 binding to incoming vDNA at the nuclear periphery (5) Therefore, we next assessed the subnuclear localization of this interaction event using microscopy. To specifically visualize the active DNA-PK kinase, we stained for autophosphorylation at S2056 (pDNA-PK) (Fig. 5F) (38). By costaining for IFI16 and pDNA-PK at 3 HPI, we observed their robust colocalization at the nuclear periphery. Furthermore, upon staining for ICP4, a viral transcription factor that marks the vDNA, we found that pDNA-PK localizes to the viral genome at the nuclear periphery (Fig. 5G). Together, our findings demonstrate that IFI16 forms an early and rapid interaction with the active DNA-PK holoenzyme at sites of viral genome deposition into the nucleus (Fig. 5H).
Recruitment of active DNA-PK to incoming vDNA for antiviral response is IFI16 dependent
To coordinate an antiviral response, IFI16 serves as a platform to recruit antiviral cofactors to the nuclear periphery upon infection (10). Given our finding of an IFI16–DNA-PK interaction, we asked whether IFI16 facilitates DNA-PK activation at viral genomes. We used recombinantly purified CRISPR-Cas9 to generate IFI16 knockout (KO) HFF cells and measured DNA-PK activation at the nuclear periphery after infection (Fig. 6, A and B, and fig. S4A). In the presence of IFI16 (control cells), pDNA-PK was enriched at ICP4-labeled transcription compartments. However, in the absence of IFI16 (ΔIFI16), DNA-PK activation displayed reduced correlation with ICP4 staining, while remaining evident throughout the nucleoplasm (Fig. 6, A and B). These results indicate that IFI16 is required for DNA-PK enrichment at vDNA.
Fig. 6. DNA-PK initiates an antiviral DDR that inhibits HSV-1 replication.
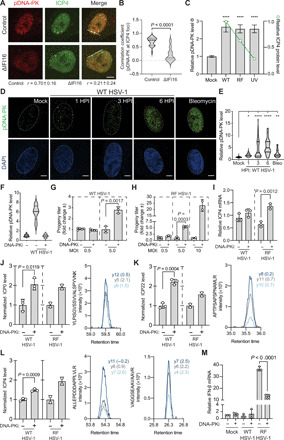
(A and B) WT HSV-1–infected (3 HPI) control and ΔIFI16 HFF cells were stained for DNA-PK activation (pDNA-PK) and ICP4 expression. Colocalization (PCC) was measured at the line (20 nuclei per n; n = 2). (C) ICP4 (green line) and pDNA-PK (gray bars) levels during WT, RF, or ultraviolet (UV)–inactivated WT HSV-1 infection (1 HPI) by IFA. Shown is mean ± 95% confidence interval for 50 nuclei per n (n = 3). (D and E) Time course of DNA-PK activation (green) during WT HSV-1 infection by IFA. Scale bars, 10 μm at 60× magnification. Fifty nuclei per n (n = 4). (F) DNA-PK inhibition (DNA-PKi; NU7441 at 2 μM) efficiency was assessed by pDNA-PK staining in the dimethyl sulfoxide (DMSO) control (−) compared to DNA-PKi (+) (n = 2). (G) WT [multiplicity of infection (MOI) 0.5: n = 5; MOI 5: n = 3] or (H) RF (MOI 0.5: n = 3; MOI 5: n = 3; MOI 10: n = 2) HSV-1 titers (24 HPI) after DNA-PKi or DMSO treatment. Replicates were normalized by the replicate average. (I) ICP4 mRNA levels were quantified by qPCR (ΔΔCt against GAPDH) at 6 HPI. (J to L) (Left) Targeted MS analysis (PRM) of the HSV-1 genes (J) ICP0, (K) ICP22, and (L) ICP4 during WT (n = 3) or RF HSV-1 (n = 2) infection. (Right) The most intense peptide transitions with mass error [parts per million (ppm)]. (M) IFN-β mRNA levels as in (I). Significance was determined by analysis of variance (ANOVA) for (C) and (E) and Student’s t test for all others. Bar plots are mean ± SD and all replicates are biological. See also fig. S4. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
It is known that a mechanism through which HSV-1 suppresses host antiviral responses is by using its viral E3-ubiquitin ligase, ICP0, to induce the proteolytic degradation of IFI16, DNA-PK, and promyelocytic leukemia protein (PML), among others (39). As an ICP0 substrate, DNA-PK is thought to be inactivated during viral infection (39); however, our data show that, early in infection, DNA-PK kinase activity is present and localized to viral replication compartments. Therefore, we next sought to characterize the virus trigger and temporality of DNA-PK activation during infection. We infected HFF cells with wild-type (WT) HSV-1, an HSV-1 strain that harbors an inactivating mutation in the ring finger domain of the ICP0 viral E3-ubiquitin ligase (strain denoted as HSV-1 RF), or infection with ultraviolet (UV)–inactivated WT HSV-1, and assessed DNA-PK activation by microscopy (Fig. 6C). Compared to mock (uninfected) cells, all infection conditions led to similar levels of DNA-PK stimulation, suggesting that kinase activation is independent of downstream events of viral infection. Further, these findings suggest that although ICP0 eventually targets DNA-PK for proteolytic degradation, its activity does not substantially reduce DNA-PK activity at this early stage of infection (1 HPI). We next characterized the duration of the DNA-PK response by staining for pDNA-PK at 1, 3, and 6 HPI with WT HSV-1 (Fig. 6, D and E). We observed discrete punctate pDNA-PK staining at 1 HPI, which progressed to a robust staining throughout the nucleus by 6 HPI, resembling bleomycin treatment, a positive control for DNA-PK activation that causes DSBs. These findings show that DNA-PK mounts a rapid and robust kinase response, which lasts throughout the first 6 hours of HSV-1 infection, despite being later targeted by ICP0 for degradation. Therefore, IFI16 and DNA-PK may have the opportunity to coordinate antiviral functions early in infection.
To evaluate whether DNA-PK contributes to host antiviral response to HSV-1, we inhibited its activity and assessed the impact of virus production. As DNA-PKcs KO cell lines have delayed growth kinetics (40), we chemically inhibited DNA-PK kinase activity with NU7441 (DNA-PKi) (38), which we found sufficient for preventing DNA-PK activation by infection (Fig. 6F and fig. S5, D and H). Next, we infected HFF cells with WT or RF HSV-1 and measured viral titers at 24 HPI, representing a full round of viral amplification. DNA-PKi enhanced WT HSV-1 production by ~3-fold (Fig. 6G and fig. S4B) and RF HSV-1 titers by ~6-fold (Fig. 5H and fig. S4C) compared to the vehicle control. These findings demonstrate that DNA-PK kinase activation is antiviral and that ICP0 activity at least partially inhibits this DNA-PK function.
We were surprised to see that the antiviral activity of DNA-PK correlates positively with higher multiplicities of infection (MOIs) (Fig. 6, G and H). A recent study showed that DNA-PK can play a proviral role during WT HSV-1 infection, which specifically becomes evident when infecting cells at a low MOI (MOI 0.01) (41). Given that the KU complex is essential for DNA repair by the nonhomologous end joining (NHEJ) pathway, it is thought that DNA-PK may mediate antiviral DNA repair that inhibits proper viral replication (42). This is evidenced by the observation that HSV-1 replicates more efficiently in XRCC6-deficient (43) and DNA-PK–deficient cells (44). Contradictorily, studies that have silenced XRCC4 and ligase IV, proteins that facilitate end joining in the NHEJ pathway (45), showed that these proteins are necessary for high levels of viral amplification (46). To clarify how DNA-PK suppressed progeny titers at higher MOIs, we next asked whether, similar to IFI16, DNA-PK suppresses viral transcription. We inhibited DNA-PK and then measured the expression of viral transcripts using reverse transcription quantitative polymerase chain reaction (RT-qPCR). At an early time point (3 HPI), the mRNA levels of representative viral immediate early (IE) (ICP4) and early (ICP8) genes were increased by DNA-PK inhibition during both WT and RF HSV-1 infections (fig. S4, D and E). By 6 HPI, ICP4 expression was no longer enhanced by DNA-PKi during WT HSV-1 infection, while retaining sensitivity to the inhibitor during RF HSV-1 infection (Fig. 6I). Given that we found that activated DNA-PK levels were still high at 6 HPI (Fig. 6, D and E), it is possible that DNA-PK associates with another cofactor that is targeted for degradation by ICP0 in order to suppress viral transcription. Last, we asked whether the increase in IE gene transcript levels upon DNA-PKi is also reflected at the viral protein levels. We designed a targeted MS approach [based on parallel reaction monitoring (PRM)] for the detection of signature peptides of the IE proteins ICP0, ICP4, and ICP22. We observed that DNA-PKi results in increased viral protein abundances at early time points of infection (Fig. 6, J to L, and table S6), which we further confirmed by microscopy (fig. S4F).
In addition to suppressing viral transcription, IFI16 is also known to promote cytokine expression; therefore, we next asked whether DNA-PK activates innate immunity to HSV-1. As above, we infected HFF cells with WT or RF HSV-1, inhibited DNA-PK, and harvested RNA at 6 HPI, a common time point for assessing cytokine production in response to HSV-1 (11). As previously reported (34), RF HSV-1 infection induced IFN-β expression, while WT HSV-1 mostly counteracted the cytokine response (Fig. 6M). DNA-PKi reduced IFN-β mRNA levels by ~3-fold compared to the vehicle-treated control, establishing that DNA-PK contributes to robust IFN signaling during HSV-1 infection. These observations were supported by measurement of additional inflammatory cytokines at 3 HPI (fig. S4, G to I). Our findings are consistent with earlier reports that DNA-PK activates interleukin-6 (IL-6) expression in response to viral infection, IRF3 responses to transfected DNA, and STING-independent immune responses (37, 47, 48). Taking all these observations into consideration, we have found that IFI16 is necessary for the localization of active DNA-PK to viral genomes at the nuclear periphery and that DNA-PK mounts an antiviral response that, similar to IFI16, suppresses viral mRNA and protein levels and virus titers, while stimulating antiviral cytokines.
DNA-PK phosphorylates IFI16 at T149 to stimulate cytokine expression
Given our findings that IFI16 is required for DNA-PK activation at the nuclear periphery and that the antiviral responses mounted by DNA-PK are phenotypically similar to IFI16 functions, we next investigated the co-regulation of these antiviral factors. Specifically, we asked whether IFI16 is a substrate for phosphorylation by DNA-PK. To explore this possibility, we induced DNA-PK activation by either RF HSV-1 infection or an optimized bleomycin treatment (fig. S5), in combination with DNA-PK inhibition (Fig. 7A). We then performed phosphopeptide enrichment, followed by MS analysis, to search for IFI16 peptides that may be phosphorylated in a DNA-PK–dependent manner. These analyses revealed nine IFI16 phosphosites that were identified with high confidence (Fig. 7B, fig. S6, and table S6), of which four sites were previously reported (S95, S106, S153, and S575) (4, 49). One of the five phosphorylation sites that were not previously known (S110, S123, T149, S248, and S776) displayed dynamic levels that indicated its regulation by active DNA-PK. IFI16 T149 phosphorylation was substantially induced following DNA damage and RF HSV-1 infection but was decreased upon DNA-PK inhibition (Fig. 7, B and C, and fig. S6). Although phosphorylation may occur at either T149 or S145 (blue mark), the ptmRS prediction algorithm (50) assigned the modification to T149 with >98.9% confidence. In addition, the T149 residue is positioned within a canonical motif (QTQ) for DNA-PK kinase activity.
Fig. 7. IFI16 and DNA-PK coordinate an intrinsic immune response to HSV-1 infection.
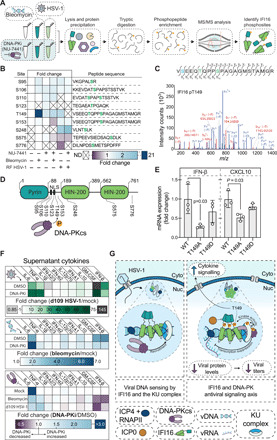
(A) Schematic for phosphopeptide enrichment following viral infection or DNA damage, using DNA-PKi to identify DNA-PK–dependent IFI16 modifications. (B) Heatmap for phosphopeptide abundances of IFI16 phosphosites. Abundances were normalized per peptide to the untreated control if it was detected in that condition, or otherwise to the bleomycin control (n = 3 to 5 biological replicates). (C) (Top) Amino acid sequence for the doubly phosphorylated peptide containing T149 detected in (B). Highlighted fragment ions detected in the tandem MS (MS/MS) assignment below are indicated in the fragment map. Green highlighted residues represent high-confidence assignment of the phosphosite by the ptmRS algorithm in Proteome Discoverer 2.4. Blue represents a potential phosphosite localization with low confidence. (Bottom) MS/MS of the above amino acid sequence with fragment ions mapped to the observed ions from Proteome Discoverer 2.4. Blue and red highlighted ions are y- and b-ions, respectively. (D) Schematic of IFI16 phosphosites relative to their location on IFI16. (E) WT, phospho-null (T149A), or phosphomimetic (T149D) IFI16 was expressed in ΔIFI16 HFF cells and then IFN-β and CXCL10 levels (mean ± SD) were measured by RT-qPCR at 6 HPI with RF HSV-1. (F) Secreted cytokine protein levels were measured after DNA damage (bleomycin; 24 HPT) and HSV-1 infection (d109; MOI 15; 24 HPT) in DMSO- or DNA-PKi–treated HFF cells (n = 3 biological replicates). (G) Model of IFI16 and DNA-PK interaction during the antiviral response to HSV-1. See also figs. S5 and S6.
To determine whether this DNA-PK–mediated phosphorylation affects IFI16 antiviral functions, we performed mutagenesis analyses. T149 was converted to either a phospho-null (alanine) or a phosphomimetic (aspartic acid) residue. As the T149 resides in the region that contains the IFI16 multipartite nuclear localization signal (Fig. 7D) (4), we assessed whether the mutagenized IFI16 constructs remain localized in the nucleus. Microscopy analyses following the expression of the IFI16 mutants in HFF cells showed their localization to the nucleus, thereby indicating that the mutation of the T149 residue does not affect cellular localization (fig. S5E). We then expressed WT, T149A, or T149D IFI16 in IFI16 knockout HFF cells. To determine whether IFI16 T149 phosphorylation affects cytokine expression during infection, IFN-β and CXCL10 (C-X-C Motif Chemokine Ligand 10) mRNA levels were measured by RT-qPCR at 6 HPI with RF HSV-1 (Fig. 7E). Decreased IFN-β and CXCL10 mRNA levels were observed in T149A cells when compared to cells expressing WT IFI16. This defect in cytokine expression was partially rescued in the T149D-expressing cells.
Given that IFI16 T149 phosphorylation was induced by DNA-PK activation after both viral infection and DNA damage (Fig. 7B) and that this phosphorylation was required for efficient expression of IFN-β mRNA (Fig. 7E), we next asked whether DNA-PK affects the levels of secreted cytokines during the inflammatory response. First, we infected DNA-PKi or dimethyl sulfoxide (DMSO)–treated HFF cells with a mutant HSV-1 (d109) that has mutations in the IE gene promoters to block the temporal cascade of viral gene expression (51), thereby allowing investigation of active immune signaling upon infection. We used flow cytometry to measure the levels of 10 secreted cytokines [IFN-β, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-10, IFN-λ1, IL-12p70, IFN-α2, IFN-λ2/3, IL-1β, tumor necrosis factor–α (TNF-α), and IL-6] in the supernatant (Fig. 7F and fig. S5, F to J). HFF cells responded to viral infection by substantially up-regulating IFN-β, GM-CSF, TNF-α, and IL-6. As we observed at the mRNA level, DNA-PK activation was required for robust IFN-β secretion after HSV-1 infection (Fig. 7F and fig. S6F). Similarly, DNA-PK inhibition also substantially decreased GM-CSF secretion, which is an important component of the immune response to HSV-1 (52). Reciprocally, IL-6 and TNF-α were further increased by DNA-PK inhibition, suggesting that either an alternative antiviral response is initiated when DNA-PK is attenuated or DNA-PK suppresses the full activation of these cytokines. Given the strong, DNA-PK–dependent up-regulation of IFI16 phospho-T149 after DNA damage, we also assessed the role of DNA-PK in cytokine secretion following bleomycin treatment. As expected, DNA damage led to induction of the innate immune response but did not induce inflammatory signaling as robustly as viral infection (Fig. 7F). We found a correlation between the DNA-PK–driven cytokine responses following DNA damage and viral infection, with higher IFN-β and GM-CSF expression linked to DNA-PK activation, and increased TNF-α and IL-6 expression following DNA-PK inhibition (fig. S5, F to K). Together, these findings reveal a conserved immune signaling axis that is driven by DNA-PK and IFI16 to coordinate cytokine secretion and inflammatory responses after both viral infection and DNA damage.
Together, our results establish IFI16 as a substrate for DNA-PK phosphorylation, uncovering T149 as the IFI16-specific residue regulated by the kinase and demonstrating the function of this site in cytokine expression during HSV-1 infection (Fig. 7F). Given that IFI16 T149 phosphorylation was also elevated after DNA damage, it is possible that this site also plays a role in DSB-linked immune responses, whereby this DNA-PK–mediated phosphorylation could link IFI16 to the DDR.
DISCUSSION
Virus modulation of the host microenvironment begins at the moment of its cellular attachment and is evident at every stage of infection thereafter, including for immune evasion, metabolic reprogramming to accommodate viral genome synthesis, and alteration of trafficking pathways for egress (2, 16, 17). The ancient evolutionary history of Herpesviridae, coupled with their large coding capacity, has produced a family of viruses that all exert precise control over the infected host cell, but each with a distinct modality of replication. For example, despite encountering the same challenges and host defenses faced by all herpesviruses, HSV-1 and other α-herpesviruses have comparatively much faster replication cycles, which underscores the rapidity of the virus-host interactions that must be formed to drive HSV-1 replication. To understand the strategies used by HSV-1 to avoid antiviral responses and repurpose host factors to complete its infectious cycle, as well as how these strategies differ from the more slowly replicating β-herpesvirus HCMV, we performed TPCA analysis and characterized dynamic protein interactions throughout the HSV-1 replication cycle. This analysis now stands as a detailed portrait of protein interactions that assemble and disassemble during HSV-1 infection. We have further expanded the TPCA approach to infer PPIs de novo in order to interrogate host-host, virus-host, and virus-virus PPIs at a system scale.
The time-resolved TPCA analysis of global interactions during HSV-1 infection, in conjunction with integrative omic approaches, microscopy, and molecular virology, uncovered an antiviral signaling axis, uniting IFI16 and the master DDR kinase, DNA-PK. IFI16 coaggregated with DNA-PK immediately upon infection, which we showed to correspond to their association at sites of viral genome deposition at the nuclear periphery. IFI16 promoted DNA-PK activation at the viral genome. In addition, DNA-PK kinase activity suppressed viral gene expression, protein levels, and virion production. Given our finding that DNA-PK was further needed to activate cytokine signaling through phosphorylation of IFI16 at the T149 residue, our findings show that IFI16 and DNA-PK form an antiviral signaling axis that governs innate immunity to HSV-1 infection.
Placing these findings into a broader context, the intersection between IFI16 and the DDR is of particular interest. Others have shown that IFI16 can regulate P53 transcriptional activity (53) and interact with BRCA1 to enhance apoptosis and cell cycle arrest following DNA damage (54). A recent investigation found that ATM (ataxia telangiectasia mutated) (55), another DDR master kinase, can mediate STING and NF-κB activation in response to DNA damage through IFI16 and P53, leading to downstream IFN-β production. It is not yet understood whether this signaling pathway can contribute to viral innate immunity, as ATM is robustly activated by nearly all studied nuclear-replicating viruses and is largely proviral (56). Although future investigations will be needed to understand whether IFI16 is linked to inflammation following DNA-PK activation due to DNA damage, our phosphopeptide enrichment showed a substantial increase in IFI16 T149 phosphorylation levels after DSB induction with bleomycin. Such investigations may shed light onto cellular strategies that control IFI16-linked autoimmunity (57). Conversely, DNA-PK is essential for the expansion of the adaptive immune repertoire by V(D)J recombination (58), and mice and humans with mutations in DNA-PK are severely immunocompromised. Therefore, DNA-PK activation of IFI16 phosphorylation may link DNA repair in V(D)J recombination to proliferative cytokine signaling. In addition, IFI16 is commonly mutated in cancer cells, whereas DNA-PK is required in highly proliferative cells (59). It remains to be seen whether mutation of IFI16 during oncogenic transformation can act as an escape strategy to avoid increased immune signaling via IFI16 T149 phosphorylation by DNA-PK activation that is linked to higher replication stress in cancer cells. Further investigations will determine how IFI16 T149 phosphorylation can change its biophysical properties to regulate its function and how this may be linked to antiviral response, inflammation, and lymphocyte development and differentiation.
MATERIALS AND METHODS
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, I.M.C. (icristea@princeton.edu).
Experimental model and subject details
Primary cultures, cell lines, and transfections
WT primary HFF cells [American Type Culture Collection (ATCC) no. SCRC-1041] were used as the model for HSV-1 primary infection and were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-Products, 100-106), 1% penicillin, and 1% streptomycin. HFF cells were maintained between passages 14 and 18 for the studies herein. U-2 OS cells (ATCC HTB-96, female) were maintained as with HFF cells and were used for plaque assays to determine HSV-1 titers.
IFI16 CRISPR-Cas9 KO cell lines were generated using passage 8 HFF cells via the TrueCut system by Invitrogen with matched scramble controls. Briefly, cells were seeded the evening before transfection at 0.9 × 105 cells/ml. IFI16 sequence-specific TrueGuide single-guide RNA (sgRNA) (Invitrogen #A35533) targeting 5′-GACCAGCCCTATCAAGAAAG-3′ and scrambled negative-control sgRNA (Invitrogen #A35526) were combined in equal molar ratios with TrueCut Cas9 Protein V2 (Invitrogen #A36499) in Opti-MEM (Thermo Fisher Scientific #31985062) and 2:1 CRISPRMAX transfection reagent (Thermo Fisher Scientific #CMAX0001). Transfection and editing efficiency were >90% for all constructs.
For plasmid transfections, HFF cells were seeded at 1 × 105 cells/ml the evening before transfection with 3 μg of DNA and a 1:3 ratio with an X-tremeGENE transfection reagent (MilliporeSigma #6366244001) in Opti-MEM. Cells were allowed to recover for 24 hours before infection.
Virus strains and infections
WT HSV-1 17+ strain, a gift from B. Sodeik (Hannover Medical School, Hannover, Germany), was propagated as previously described (34), aliquoted, snap-frozen, and stored at −80°C. Briefly, WT HSV-1 was produced by electroporating U-2 OS cells with pBAC-HSV-1 that was purified from Escherichia coli strain GS1783 to generate a P0 stock. The ICP0-RF HSV-1 mutant was a gift from B. Roizman (University of Chicago, Chicago, IL, USA) and S. Silverstein (Columbia University, New York, NY, USA). The d109 HSV-1 and complementing Vero F06 cells were gifts from N. DeLuca of University of Pittsburgh (Pittsburgh, PA, USA). Working stocks were generated from the P0 stock by infecting U-2 OS or Vero F06 cells at a low MOI [0.001 plaque-forming units (PFU) per cell]. Both culture supernatant and cells were buffered with MNT buffer [200 mM MES, 30 mM tris-HCl, and 100 mM NaCl (pH 7.4)]. Supernatants were subjected to ultracentrifugation [20,000 rpm, 2 hours, 4°C with SW28 swinging bucket rotor (Beckman Coulter)] over a 10% Ficoll cushion to concentrate virus. Cell-associated virus was collected by sonication and combined with concentrated cell-free virus. Virus stock titers were determined by plaque assay on U-2 OS or Vero F06 monolayers.
For all experiments herein, infection was conducted at an MOI of 5 PFU per cell for both WT and ICP0 RF HSV-1 unless otherwise noted in the figure legend. Infections were conducted in DMEM supplemented with 2% FBS with intermittent rocking at 37°C and 5% CO2. Inoculum was removed after 1 hour, and media were replaced with growth media as above. For the purposes of our study, 0 HPI is considered to be 60 min after addition of the inoculum to account for the adsorption of the virus. HSV-1 titers were calculated by plaque assay. Briefly, cells were lysed by freezing, and both cell-associated and cell-free viruses were collected, briefly sonicated, and serially diluted. Infections were as above, but after 1 hour, the inoculum was replaced with 1% METHOCEL (w/v) in DMEM with 10% FBS. After ~72 hours, when plaques were evident, the cells were incubated with crystal violet [1% crystal violet (w/v) and 50% methanol (v/v)] for 15 min at room temperature before rinsing and plaque quantification. For UV-inactivated virus, a sample of WT HSV-1 was thawed and split into two fractions. One fraction was used as a control and the other was exposed to UV radiation in a UVC 500 Ultraviolet Crosslinker (Amersham Biosciences) at 60 mJ/cm2 for 2 min. UV-inactivated and control viruses were then used immediately for infections at equal, high MOIs to ensure equal infection of cells. ICP4 staining was used to verify virus inactivation (Fig. 6C).
Inhibitors and constructs
DNA-PK kinase inhibition was performed by addition of 2 μM NU-7441 (Selleckchem #S2638) in DMSO. Equivalent concentrations of DMSO were used as a vehicle control. Inhibitors were added directly after exchanging viral inoculum with growth media, as indicated, or in tandem with the addition of bleomycin (10 mg/ml in PBS; ApexBio #A8331) when inducing DNA damage. The concentration of NU-7441 and the concentration, duration, and recovery kinetics of bleomycin exposure were all validated before the study (fig. S5, A to H).
Point mutations were introduced into green fluorescent protein (GFP)–tagged IFI16 (pEGFP-N1-IFI16) (35) by PCR, using the KOD Hot Start DNA Polymerase Kit (MilliporeSigma #71086).PCRs for single amino acid mutations were run for 35 cycles of 30 s at 95°C and 25 s at 65°C, followed by 5 min 50 s at 70°C. The resulting mutant plasmids were verified by DNA sequencing. Primers for mutagenesis were as follows: IFI16 T149A, 5′-GTAAGGTGTCCGAGGAACAGGCTCAGCC-3′ (forward) and 5′-CCTGCAGGAGAGGGAGGCTGAGCCTGTT-3′ (reverse); IFI16 T149D, 5′-GTAAGGTGTCCGAGGAACAGGACCAGCC-3′ (forward) and 5′-CCTGCAGGAGAGGGAGGCTGGTCCTGTT-3′ (reverse).
Sample preparation for liquid chromatography–tandem mass spectrometry
Detailed sample preparation methods for TMT TPP-MS, PRM, and phosphopeptide enrichment are described in the Supplementary Materials.
Peptide liquid chromatography–tandem mass spectrometry analysis
TPCA TMT liquid chromatography–tandem mass spectrometry analysis
Samples were analyzed on a Q-Exactive HF mass spectrometer equipped with an EASY-Spray ion source (Thermo Fisher Scientific). Peptides were resolved for nanoscale liquid chromatography–MS (nLC-MS) analysis with a Dionex UltiMate 3000 nRSLC (Thermo Fisher Scientific) equipped with an EASY-Spray C18 column (2 μm particle size, 75 μm diameter, 500 mm length; Thermo Fisher Scientific, ES903). Peptides were separated with a 90-min gradient of 0.1% formic acid in LC-MS–grade water (solvent A) and 0.1% formic acid in 97% LC-MS–grade acetonitrile and 2.9% LC-MS–grade water (solvent B). Peptides were resolved with a linear gradient of 6 to 18% solvent B for 60 min, followed by a linear gradient of 18 to 29% solvent B for 30 min, at a flow rate of 250 nl/min. MS and tandem MS (MS/MS) spectra were automatically obtained in a full MS/data-dependent MS2 method. The MS1 method operated with a full scan range of 350 to 1800 mass/charge ratio (m/z) with a resolution of 120,000 and a target AGC (automatic gain control) of 3 × 106 with a maximum injection time (MIT) of 30 ms, and spectra were recorded in profile. For the data-dependent MS2 scans, higher-energy collision-induced dissociation (HCD) was performed on the top 20 most intense precursor ions, with a 25-s dynamic exclusion duration. MS2 isolation windows were set to 1.2 m/z, with a target AGC of 1 × 105 and a MIT of 72 ms, a fixed first mass of 100 m/z, a resolution of 45,000, and a normalized collision energy (NCE) of 34, and centroided spectral data were recorded. These settings were used for both TMT test mix samples and the fractionated TMT samples, except that the MS2 isolation windows for the fractionated TMT samples were set to 0.8 m/z.
PRM LC-MS/MS analysis
Samples were analyzed on a Q-Exactive HF mass spectrometer equipped with an EASY-Spray ion source (Thermo Fisher Scientific). Peptides were resolved for nLC-MS analysis with a Dionex UltiMate 3000 nRSLC (Thermo Fisher Scientific) equipped with an EASY-Spray C18 column (2 μm particle size, 75 μm diameter, 250 mm length; Thermo Fisher Scientific, ES902). Peptides were separated with 60-min gradient using solvent A and solvent B (2 to 22% solvent B over 45 min, 22 to 38% solvent B over 15 min, both at a flow rate of 250 nl/min). One full duty cycle of the instrument consisted of a single MS-SIM MS1 scan followed by 30 PRM scans. For the full scan MS1, the instrument was set to 400 to 2000 m/z full scan range with a 15,000 resolution, 15 ms MIT, and 3 × 106 AGC target. For the PRM scans, the instrument was set to 30,000 resolution, 60 ms MIT, 1 × 105 AGC target, 0.8 m/z isolation window, NCE of 27, and 125 m/z fixed first mass. Spectrum data for both the MS1 and PRM scans were recorded in profile.
Phosphopeptide LC-MS/MS analysis
Samples were analyzed on a Q-Exactive HF mass spectrometer equipped with a Nanospray Flex Ion Source (Thermo Fisher Scientific). Peptides were resolved for nLC-MS analysis with a Dionex UltiMate 3000 nRSLC (Thermo Fisher Scientific) equipped with an in-house packed 50-cm column (360 μm outer diameter, 75 μm inner diameter; Thermo Fisher Scientific) packed with ReproSil-Pur C18 material (120 Å pore size, 1.9 μm particle size; ESI Source Solutions) equipped with a stainless steel emitter (Thermo Fisher Scientific). Peptides were resolved with a linear 150-min gradient using solvent A and solvent B (3 to 35% B over 150 min at a 250 nl/min flow rate). MS and MS/MS spectra were automatically obtained in a full MS/data-dependent MS2 method. The MS1 method operated with a full scan range of 350 to 1800 m/z with a resolution of 120,000 and a target AGC of 3 × 106 with a MIT of 30 ms, and spectra were recorded in profile. For the data-dependent MS2 scans, HCD was performed on the top 10 most intense precursor ions, with a 30-s dynamic exclusion duration. MS2 isolation windows were set to 1.6 m/z, with a target AGC of 1 × 105 and a MIT of 150 ms, a fixed first mass of 100 m/z, a resolution of 30,000, and an NCE of 28, and centroided spectral data were recorded.
Peptide identification and quantification
TPCA TMT data
All TPCA TMT MS/MS spectra were compared with protein sequences from a combined human and herpesvirus HSV-1 UniProt-SwissProt database downloaded in November 2018 and December 2018, respectively, combined with common contaminants, using the SEQUEST algorithm in Proteome Discoverer v2.3 (Thermo Fisher Scientific). A spectral recalibration node was used for offline recalibration of the mass accuracy. The database was constrained to fully tryptic peptides, with a maximum missed cleavage of 2, and a static cysteine carbamidomethylation modification and dynamic TMT labeling on the peptide N terminus and lysine for the test mix searches, or static TMT labeling at these sites for the fractionated samples for quantitative analysis. Additional dynamic modifications included methionine oxidation, asparagine deamination, and protein N-terminal methionine loss and acetylation. Precursor mass tolerance was set to 4 parts per million (ppm), and fragment ion mass tolerance was set to 0.02 Da. The false discovery rate (FDR) of the matched spectra was determined using Percolator (FDR of 1%) using a reversed sequence database search. For the reporter ion quantifier node, the integration tolerance was set to 10 ppm, and the integration method was set to the most confident centroid. In the consensus workflow reporter ions quantifier node, the co-isolation threshold and average reporter S/N threshold was set to 30 and 8, respectively.
Phosphopeptide data
All phosphopeptide MS/MS spectra were compared with protein sequences from the same human UniProt-SwissProt database used for the TPP-MS TMT data, combined with the same common contaminants database; however, data were searched using the SEQUEST algorithm in Proteome Discoverer v2.4 (Thermo Fisher Scientific). The phosphopeptide searches were performed similarly to the TMT searches, except for the following changes: No static TMT modifications were included, but dynamic phosphorylation modifications on serine, threonine, or tyrosine were included; an IMP-PtmRS node was used with default settings to determine phosphosite localization; the reporter ion quantifier node was not used, but, rather, the Minora feature detector node with default settings was used to detect chromatographic peaks and features for label-free MS1 quantification; and the reporter ion quantifier node was not used in the consensus workflow, but, rather, the feature mapper node was used to perform chromatographic retention time alignment with a 15-min maximum retention time window, a 6 ppm mass tolerance, and a minimum S/N threshold of 4. For quantitation, in instances where multiple peptides for a single phosphosite were detected (i.e., EVDATpSPAPSTSSTVK and KEVDATpSPAPSTSSTVK), abundance values were summed across identical phosphosites. When different peptides due to differences in methionine oxidation states were detected (i.e., VSEEQTQPPpSPAGAGMSTAMGR and VSEEQTQPPpSPAGAGoxMSTAoxMGR), the peptide with the most reproducible detection across the dataset was used for quantitation (table S6). The resulting abundances were analyzed in GraphPad Prism, and two-tailed Student’s t test was performed to determine statistically significant differences.
PRM data analysis
RAW files containing targeted PRM LC-MS/MS spectra were imported into Skyline to extract product ion chromatograms and calculate peak areas. The PRM method monitored three HSV-1 IE proteins (ICP0, ICP4, and ICP22), with four proteotypic peptides per protein selected based on ion intensity, peak area reproducibility, and LC reproducibility from other datasets produced within the laboratory. From this initial analysis, a single peptide was monitored for ICP0, and two peptides from ICP4 and ICP22 were selected for reproducibility within this dataset. A single peptide from MYH9 (myosin heavy chain 9) was also monitored for data normalization. Peptide detection was first verified with at least six transitions specific and unique to each peptide, and quantitative analysis was performed with the top three most intense transition ions with the highest mass accuracy per peptide. The peak areas for each of the three transitions for each peptide were summed, and the resulting summed peak areas were either scaled to the average replicate peak area (ICP0, ICP4, and ICP22) or scaled to the average peak area across all replicates (MYH9). Scaled ICP0, ICP4, and ICP22 peak areas were then normalized to the scaled MHY9 peak area (table S6). The resulting normalized scaled peak area values were analyzed in GraphPad Prism, and two-tailed Student’s t test was performed to determine statistically significant differences.
Processing of TPP-MS data
In this analysis, we identified 6451 proteins assembled with a single unique peptide per protein at a 1% FDR. To increase the confidence of these protein identifications, we identified 5260 proteins when we required a 1% FDR and at least two unique peptides per protein. As expected with these data, when we required reporter ion quantification values across all TMT plexes, we obtained highly quantifiable data for 2301 proteins. Because of the decreasing abundances of proteins at higher melting temperatures, we did not require the presence of the reporter ion in all TMT channels within each 11-plex but imputed these abundances during the log-logistics transformation of the data (see Supplementary Text under the “Processing of TPP-MS Data” section). Consequently, we used the list of 5260 proteins identified at a 1% FDR and with at least two unique peptides per protein for the remaining data analysis. For a detailed description of TPCA data normalization, missing value imputation, and log-logistic parameter fitting, consult Supplementary Materials and Methods.
Principal components analysis
PCA was performed using the Python Scikit-learn package decomposition.PCA function fitted against normalized solubility values for each condition and replicate represented in C, where missing values were imputed as described above.
Euclidian distance calculation
To quantify the similarity between protein-normalized solubility curves (C), we calculated the pairwise Euclidean distance between all possible combinations of proteins detected in a given sample, where the distance between two proteins, p and q in the sample corresponding to condition i and replicate j across y temperatures, is calculated as . This calculation was performed using the Python SciPy package spatial.distance.cdist function with default parameters to yield pairwise Euclidean distances across all possible combinations of proteins detected in each sample. To transform values such that they better fit a normal distribution, we calculated the derivative of Euclidean distance Exijpq = 1/(1 + Dijpq).
De novo interaction prediction via Ex z score calculation
To predict de novo interactions, we first standardized the transformed Euclidean distance values by calculating their z scores: , where and σ are the mean and SD, respectively, of Exijpq along the p and q axes. We then asked to what extent increasing z scores enriched for known interactions represented in the CORUM and STRING databases. We assumed that each pairwise combination of subunits within a given CORUM complex represents a predicted binary interaction, and we additionally queried the STRING database for interactions between proteins in the dataset that have a STRING score ≥ 0.4. For each condition, we then calculated ni = (ni, CORUM + ni, STRING)/∣zi, j, p, q∣, where ni, CORUM and ni, STRING represent the sum of known interactions from CORUM and STRING, respectively, in zi, j, p, q along the j axis, and ∣zi, j, p, q∣ corresponds to the number of entries in zi, j, p, q along the j axis. For a given set of z score cutoffs u and v, we then calculated the enrichment ratio where mi is the subset of ni ≥ u across all replicates and ni ≥ v in any replicate for a given condition. We then plotted φu, v across linearly distributed combinations of u and v within the interval [−3, 3] to inform our final cutoffs for de novo interaction prediction.
Bioinformatic analysis
Overrepresentation analyses
Overrepresentation analyses represented in Fig. 3 were performed via the Database for Annotation, Visualization, and Integrated Discovery using a curated list of proteins that are expressed in human fibroblast cells as a background gene set (table S4). For analyses in fig. S2B and Fig. 3C, GOTERM_CC_ALL and GOTERM_BP_DIRECT gene ontology databases, respectively, were selected as the enrichment query population. Terms with Bonferroni-adjusted P values ≤ 0.05 were considered significant and subsequently reported.
Hierarchical clustering
Hierarchical clustering of CORUM complex Ex z scores and abundance was performed using the Python Seaborn package clustermap function, which leverages the SciPy cluster.hierarchy.linkage function to calculate the Ward linkage between each complex.
Immunofluorescence microscopy and analysis
HFF cells were seeded onto autoclaved 15-mm glass coverslips (VWR) at 1.5 × 105 cells/ml and infected or treated as indicated. Samples were fixed in 4% paraformaldehyde for 15 min at room temperature and washed three times with 1× PBS (Sigma-Aldrich D8537-6X1Ld) with 0.2% Tween 20 (PBST). Cells were permeabilized in 0.1% Triton X-100 in PBST for 15 min at room temperature, washed twice with PBST, and then blocked in 2.5% human serum and 2% bovine serum albumin in PBST for 60 min. Samples were incubated for 1 hour with the primary antibodies diluted into block: ICP4 (Mouse, 1:500; Abcam, Ab6514) to mark viral transcription compartments, DNA-PK pS2056 (Rabbit, 1:500; Abcam, Ab18192) to measure DNA-PK kinase activity, IFI16 (Mouse, 1:250; Sigma-Aldrich, WH0003428M3), or ICP0 (Mouse, 1:2000; Santa Cruz Biotechnology, SC-53070). After primary staining, cells were washed three times in PBST and then incubated with 4′,6-diamidino-2-phenylindole (DAPI) (1:1000; Thermo Fisher Scientific) and appropriate secondary antibody diluted 1:2000 in block for 1 hour: goat anti-Ms immunoglobulin G (IgG) highly cross-adsorbed Alexa Fluor 488 (1:2000; Thermo Fisher Scientific, A-11001) or goat anti-Rb IgG cross-adsorbed Alexa Fluor 568 (1:2000; Thermo Fisher Scientific, A11011). Coverslips were mounted using ProLong Diamond and were imaged in the Princeton Confocal Imaging Core using an inverted fluorescence confocal microscope (Nikon Ti-E) equipped with a Yokogawa spinning disc (CSU-21) and digital CMOS camera (Hamamatsu ORCA-Flash TuCam) using a Nikon 100× Plan Apo objective with a 100× magnification (maximum projections centered around the nuclear center) or 60× magnification (single slice) as Nikon 60× Plan Apo objective as indicated. Image analysis was performed using ImageJ. Average fluorescence intensity within a nucleus was quantified following definition of the nucleus as an ROI defined by DAPI staining and background subtraction in the channel to be quantified using a rolling ball radius of 150 to 200.
RNA isolation and quantitative RT-PCR
A detailed protocol is provided in Supplementary Materials and Methods. Briefly, RNA extraction was by the RNeasy Mini Kit (Qiagen) from 1.75 × 105 HFF; then, cDNA was prepared using the RETROscript Reverse Transcription Kit (Life Technologies) and the SuperScript IV First-Strand Synthesis Kit (Thermo Fisher Scientific) as per the manufacturer’s instructions. Gene-specific primers and the SYBR green PCR master mix (Life Technologies) were used to quantify cDNA by qPCR on the ViiA 7 Real-Time PCR Systems (Applied Biosystems). Relative mRNA quantities were determined using the ∆∆CT method with glyceraldehyde phosphate dehydrogenase (GAPDH) as an internal control [as previously described (10)]. Following quantification, data were normalized by the average within each replicate.
Supernatant cytokine measurement
A detailed protocol is provided in Supplementary Materials and Methods. Briefly, supernatant-associated cytokines were measured via the BioLegend Human Antiviral Response Kit (catalog no. 740390) according to the manufacturer’s protocol on an LSR II flow cytometer (Princeton Flow Cytometry Core).
Quantification and statistical analysis
Data processing and large-scale analyses were performed using Python 2.7 in conjunction with Pandas, NumPy, SciPy, Scikit-learn, Seaborn, and Requests libraries. Cytoscape was used to generate interaction networks. Microscopy images were analyzed in ImageJ (2.1.0). Statistical analysis was performed using GraphPad Prism 9. Significance was determined by two-tailed Student’s t test (n = 3 biological replicates) unless otherwise stated. Where applicable, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Figures were constructed in either Adobe Illustrator or Microsoft PowerPoint.
Acknowledgments
Funding: We are grateful for funding provided by NIH NIGMS (GM114141), the Edward Mallinckrodt Foundation, and the Princeton Catalysis Initiative to I.M.C.; NSF Graduate Research Fellowship (DGE-1656466) to M.A.K.; China Scholarship Council (CSC) scholarship (201506210052) to B.S.; and the NIH training grant from NIGMS (T32GM007388). We also thank J. Federspiel for the PRM design, G. Laevsky (Princeton Microscopy Facility), the Princeton Genomics Core, and Princeton Flow Cytometry Facility and staff for help. Author contributions: J.L.J., M.A.K., J.E.H., and I.M.C. conceptualized the study. J.L.J., M.A.K., J.E.H., B.S., D.L., B.P., and I.M.C. contributed to investigation and methodology. Data curation, analysis, visualization, and validation were carried out by J.L.J., M.A.K., J.E.H., and I.M.C. The manuscript was written, reviewed, and edited by J.L.J., M.A.K., J.E.H., and I.M.C. Funding was primarily acquired by I.M.C., with M.A.K. and B.S. supported by fellowships. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The mass spectrometry proteomics TPCA data have been deposited to the ProteomeXchange Consortium via the PRIDE (60) partner repository with the dataset identifier PXD023167.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabg6680/DC1
REFERENCES AND NOTES
- 1.Whitley R. J., Roizman B., Herpes simplex virus infections. Lancet 357, 1513–1518 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Greco T. M., Diner B. A., Cristea I. M., The impact of mass spectrometry-based proteomics on fundamental discoveries in virology. Annu. Rev. Virol. 1, 581–604 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unterholzner L., Keating S. E., Baran M., Horan K. A., Jensen S. B., Sharma S., Sirois C. M., Jin T., Latz E., Xiao T. S., Fitzgerald K. A., Paludan S. R., Bowie A. G., IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li T., Diner B. A., Chen J., Cristea I. M., Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. U.S.A. 109, 10558–10563 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diner B. A., Lum K. K., Toettcher J. E., Cristea I. M., Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. MBio 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kerur N., Veettil M. V., Sharma-Walia N., Bottero V., Sadagopan S., Otageri P., Chandran B., IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrone S. R., Wang T., Constantoulakis L. M., Hooy R. M., Delannoy M. J., Sohn J., Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc. Natl. Acad. Sci. U.S.A. 111, E62–E71 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merkl P. E., Knipe D. M., Role for a filamentous nuclear assembly of IFI16, DNA, and host factors in restriction of herpesviral infection. MBio 10, e02621 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy A., Ghosh A., Kumar B., Chandran B., IFI16, a nuclear innate immune DNA sensor, mediates epigenetic silencing of herpesvirus genomes by its association with H3K9 methyltransferases SUV39H1 and GLP. eLife 8, e49500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lum K. K., Howard T. R., Pan C., Cristea I. M., Charge-mediated pyrin oligomerization nucleates antiviral IFI16 sensing of herpesvirus DNA. MBio 10, e01428-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orzalli M. H., DeLuca N. A., Knipe D. M., Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U.S.A. 109, E3008–E3017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li T., Chen J., Cristea I. M., Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14, 591–599 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzutani T., Nagamine M., Shibaki T., Ogasawara M., Yoshida I., Daikoku T., Nishiyama Y., Azuma M., The role of the UL41 gene of herpes simplex virus type 1 in evasion of non-specific host defence mechanisms during primary infection. J. Gen. Virol. 81, 1763–1771 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Zhang J., Zhao J., Xu S., Li J., He S., Zeng Y., Xie L., Xie N., Liu T., Lee K., Seo G. J., Chen L., Stabell A. C., Xia Z., Sawyer S. L., Jung J., Huang C., Feng P., Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe 24, 234–248.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang Z. F., Zou H. M., Liao B. W., Zhang H. Y., Yang Y., Fu Y. Z., Wang S. Y., Luo M. H., Wang Y. Y., Human cytomegalovirus protein UL31 inhibits DNA sensing of cGAS to mediate immune evasion. Cell Host Microbe 24, 69–80.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Howard T. R., Cristea I. M., Interrogating host antiviral environments driven by nuclear DNA sensing: A multiomic perspective. Biomolecules 10, 1591 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillen J., Nita-Lazar A., Experimental analysis of viral-host interactions. Front. Physiol. 10, 425 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Savitski M. M., Reinhard F. B., Franken H., Werner T., Savitski M. F., Eberhard D., Martinez Molina D., Jafari R., Dovega R. B., Klaeger S., Kuster B., Nordlund P., Bantscheff M., Drewes G., Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Tan C. S. H., Go K. D., Bisteau X., Dai L., Yong C. H., Prabhu N., Ozturk M. B., Lim Y. T., Sreekumar L., Lengqvist J., Tergaonkar V., Kaldis P., Sobota R. M., Nordlund P., Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science 359, 1170–1177 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Dai L., Zhao T., Bisteau X., Sun W., Prabhu N., Lim Y. T., Sobota R. M., Kaldis P., Nordlund P., Modulation of protein-interaction states through the cell cycle. Cell 173, 1481–1494.e13 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Huang J. X., Lee G., Cavanaugh K. E., Chang J. W., Gardel M. L., Moellering R. E., High throughput discovery of functional protein modifications by Hotspot Thermal Profiling. Nat. Methods 16, 894–901 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Määttä T. A., Rettel M., Sridharan S., Helm D., Kurzawa N., Stein F., Savitski M. M., Aggregation and disaggregation features of the human proteome. Mol. Syst. Biol. 16, e9500 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hashimoto Y., Sheng X., Murray-Nerger L. A., Cristea I. M., Temporal dynamics of protein complex formation and dissociation during human cytomegalovirus infection. Nat. Commun. 11, 806 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye G. J., Vaughan K. T., Vallee R. B., Roizman B., The herpes simplex virus 1 U(L)34 protein interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. J. Virol. 74, 1355–1363 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruepp A., Waegele B., Lechner M., Brauner B., Dunger-Kaltenbach I., Fobo G., Frishman G., Montrone C., Mewes H. W., CORUM: The comprehensive resource of mammalian protein complexes—2009. Nucleic Acids Res. 38, D497–D501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan Y., Mao R., Yang J., NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 4, 176–185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amici C., Rossi A., Costanzo A., Ciafrè S., Marinari B., Balsamo M., Levrero M., Santoro M. G., Herpes simplex virus disrupts NF-κB regulation by blocking its recruitment on the IκBα promoter and directing the factor on viral genes. J. Biol. Chem. 281, 7110–7117 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Johzuka K., Terasawa M., Ogawa H., Ogawa T., Horiuchi T., Condensin loaded onto the replication fork barrier site in the rRNA gene repeats during S phase in a FOB1-dependent fashion to prevent contraction of a long repetitive array in Saccharomyces cerevisiae. Mol. Cell. Biol. 26, 2226–2236 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arii J., Watanabe M., Maeda F., Tokai-Nishizumi N., Chihara T., Miura M., Maruzuru Y., Koyanagi N., Kato A., Kawaguchi Y., ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat. Commun. 9, 3379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walsh D., Perez C., Notary J., Mohr I., Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J. Virol. 79, 8057–8064 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Topacio B. R., Zatulovskiy E., Cristea S., Xie S., Tambo C. S., Rubin S. M., Sage J., Kõivomägi M., Skotheim J. M., Cyclin D-Cdk4,6 drives cell-cycle progression via the retinoblastoma protein’s C-terminal helix. Mol. Cell 74, 758–770.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grady L. M., Szczepaniak R., Murelli R. P., Masaoka T., Le Grice S. F. J., Wright D. L., Weller S. K., The exonuclease activity of herpes simplex virus 1 UL12 is required for production of viral DNA that can be packaged to produce infectious virus. J. Virol. 91, e01380-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ariza M.-E., Glaser R., Kaumaya P. T., Jones C., Williams M. V., The EBV-encoded dUTPase activates NF-κB through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 182, 851–859 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Diner B. A., Lum K. K., Javitt A., Cristea I. M., Interactions of the antiviral factor interferon gamma-inducible protein 16 (IFI16) mediate immune signaling and herpes simplex virus-1 immunosuppression. Mol. Cell. Proteomics 14, 2341–2356 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diner B. A., Li T., Greco T. M., Crow M. S., Fuesler J. A., Wang J., Cristea I. M., The functional interactome of PYHIN immune regulators reveals IFIX is a sensor of viral DNA. Mol. Syst. Biol. 11, 787 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson K. E., Bottero V., Flaherty S., Dutta S., Singh V. V., Chandran B., IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLOS Pathog. 10, e1004503 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferguson B. J., Mansur D. S., Peters N. E., Ren H., Smith G. L., DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 1, e00047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yanai M., Makino H., Ping B., Takeda K., Tanaka N., Sakamoto T., Yamaguchi K., Kodani M., Yamasaki A., Igishi T., Shimizu E., DNA-PK inhibition by NU7441 enhances chemosensitivity to topoisomerase inhibitor in non-small cell lung carcinoma cells by blocking DNA damage repair. Yonago Acta Med. 60, 9–15 (2017). [PMC free article] [PubMed] [Google Scholar]
- 39.Orzalli M. H., Broekema N. M., Knipe D. M., Relative contributions of herpes simplex virus 1 ICP0 and vhs to loss of cellular IFI16 vary in different human cell types. J. Virol. 90, 8351–8359 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruis B. L., Fattah K. R., Hendrickson E. A., The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol. Cell. Biol. 28, 6182–6195 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun X., Liu T., Zhao J., Xia H., Xie J., Guo Y., Zhong L., Li M., Yang Q., Peng C., Rouvet I., Belot A., Shu H. B., Feng P., Zhang J., DNA-PK deficiency potentiates cGAS-mediated antiviral innate immunity. Nat. Commun. 11, 6182 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith S., Weller S. K., HSV-I and the cellular DNA damage response. Future Virol. 10, 383–397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor T. J., Knipe D. M., Proteomics of herpes simplex virus replication compartments: Association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 78, 5856–5866 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parkinson J., Lees-Miller S. P., Everett R. D., Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 73, 650–657 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costantini S., Woodbine L., Andreoli L., Jeggo P. A., Vindigni A., Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair 6, 712–722 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Muylaert I., Elias P., Knockdown of DNA ligase IV/XRCC4 by RNA interference inhibits herpes simplex virus type I DNA replication. J. Biol. Chem. 282, 10865–10872 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Karpova A. Y., Trost M., Murray J. M., Cantley L. C., Howley P. M., Interferon regulatory factor-3 is an in vivo target of DNA-PK. Proc. Natl. Acad. Sci. U.S.A. 99, 2818–2823 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burleigh K., Maltbaek J. H., Cambier S., Green R., Gale M., James R. C., Stetson D. B., Human DNA-PK activates a STING-independent DNA sensing pathway. Sci. Immunol. 5, eaba4219 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M., Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Taus T., Köcher T., Pichler P., Paschke C., Schmidt A., Henrich C., Mechtler K., Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Samaniego L. A., Neiderhiser L., DeLuca N. A., Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72, 3307–3320 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsuboi K., Kimura T., Sugiura K., Hashimoto I., Nishikawa M., Uyama M., Fujisawa J. I., Granulocyte-macrophage colony-stimulating factor expressed in T cells mediates immunity against herpes simplex virus type 1 encephalitis. J. Infect. Dis. 178, 16–26 (1998). [DOI] [PubMed] [Google Scholar]
- 53.Johnstone R. W., Wei W., Greenway A., Trapani J. A., Functional interaction between p53 and the interferon-inducible nucleoprotein IFI 16. Oncogene 19, 6033–6042 (2000). [DOI] [PubMed] [Google Scholar]
- 54.Dutta D., Dutta S., Veettil M. V., Roy A., Ansari M. A., Iqbal J., Chikoti L., Kumar B., Johnson K. E., Chandran B., BRCA1 regulates IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon-β responses. PLOS Pathog. 11, e1005030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Dunphy G., Flannery S. M., Almine J. F., Connolly D. J., Paulus C., Jønsson K. L., Jakobsen M. R., Nevels M. M., Bowie A. G., Unterholzner L., Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after Nuclear DNA damage. Mol. Cell 71, 745–760.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spriggs C. C., Laimins L. A., Human papillomavirus and the DNA damage response: Exploiting host repair pathways for viral replication. Viruses 9, 232 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mondini M., Vidali M., Airò P., De Andrea M., Riboldi P., Meroni P. L., Gariglio M., Landolfo S., Role of the interferon-inducible gene IFI16 in the etiopathogenesis of systemic autoimmune disorders. Ann. N. Y. Acad. Sci. 1110, 47–56 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Kienker L. J., Shin E. K., Meek K., Both V(D)J recombination and radioresistance require DNA-PK kinase activity, though minimal levels suffice for V(D)J recombination. Nucleic Acids Res. 28, 2752–2761 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choubey D., Deka R., Ho S.-m., Interferon-inducible IFI16 protein in human cancers and autoimmune diseases. Front. Biosci. 13, 598–608 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., Tiwary S., Cox J., Audain E., Walzer M., Jarnuczak A. F., Ternent T., Brazma A., Vizcaíno J. A., The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabg6680/DC1