

Caught in the act, DNA reaction intermediates in bacterial genomes may illuminate disease-driving regions in human chromosomes.
Abstract
Chromosomal fragile sites are implicated in promoting genome instability, which drives cancers and neurological diseases. Yet, the causes and mechanisms of chromosome fragility remain speculative. Here, we identify three spontaneous fragile sites in the Escherichia coli genome and define their DNA damage and repair intermediates at high resolution. We find that all three sites, all in the region of replication termination, display recurrent four-way DNA or Holliday junctions (HJs) and recurrent DNA breaks. Homology-directed double-strand break repair generates the recurrent HJs at all of these sites; however, distinct mechanisms of DNA breakage are implicated: replication fork collapse at natural replication barriers and, unexpectedly, frequent shearing of unsegregated sister chromosomes at cell division. We propose that mechanisms such as both of these may occur ubiquitously, including in humans, and may constitute some of the earliest events that underlie somatic cell mosaicism, cancers, and other diseases of genome instability.
INTRODUCTION
Common fragile sites were found in human cells treated with replication-inhibiting drugs and were observed as chromosomal regions prone to gross-level cytologically visible gaps or breaks in chromatin and/or DNA (1). Genome rearrangements at human fragile sites underlie developmental disorders, cancers, and other diseases (2). The mechanisms of fragility are, therefore, central to understanding these pathologies. Replication inhibition, used to observe fragility in human cells, is presumed to increase numbers of events that occur similarly to spontaneous events (1); but, in human cells, spontaneous fragility is too infrequent to observe directly, making the assumption difficult to test. Many universal mechanisms of DNA damage and repair were found and characterized in bacteria (3), but the existence of analogous fragile sites in bacterial genomes and their potential utility for understanding human chromosome fragility have not been explored.
Holliday junctions (HJs) are X-shaped DNA junctions with four duplex arms (Fig. 1). HJs form during homology-directed repair (HR) of double-strand breaks (DSBs) (Fig. 1A, i) (4) and single-strand DNA (ssDNA) gaps (Fig. 1A, ii) (4, 5), and also when stalled replication forks are remodeled or “reversed” (Fig. 1A, iii) (6). In Escherichia coli, HR of DSBs is high fidelity but becomes error prone when cells are stressed (7). Thus, possible sites of recurrent HJs could be prone to genome rearrangements and other mutations. HJs can also form during genome-rearranging non-HR events such as microhomology-mediated break-induced replication (8, 9), which is thought to promote deletions associated with common fragile sites, and possibly expansion of simple repeated sequences nearby (2). Thus, because most types of DNA damage and repair implicated in eukaryotic fragile-site instability are likely to involve HJs, HJs might serve as molecular genomic markers for discovering fragile sites that were not known previously.
Fig. 1. Routes to HJ formation and the proteins that promote or nullify each.
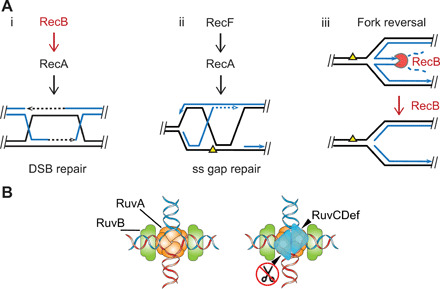
(A) HJ-generating processes and E. coli protein players: (i and ii) HR [reviewed in (4)] and (iii) RecBCD removal or prevention of reversed forks by its specific degradation of double-stranded DNA (dsDNA) ends, shown by Michel and colleagues (6, 15). Notched circle RecB indicates RecBCD nuclease, close parallel lines indicates base-paired DNA strands, and dashed lines indicates DNA repair synthesis. (B) Illustration of RuvCDef (blue triangles) binding to an HJ, taken from Xia et al. (5) (published under a CC BY-NC license, https://creativecommons.org/licenses/by-nc/4.0/).
Here, we identify sites of spontaneous, replication-associated HJs and DSBs in the E. coli genome at nucleotide resolution. We find that the chromosomal replication terminus region has prominent sites of recurrent HJs that, we show, are caused by DSB repair and, we find, have recurrent DSBs nearby. These fragile sites fall into two classes: those dependent on a replication barrier and those associated with the site and proteins of chromosome decatenation. We propose generalizable mechanisms by which either replication barriers or missteps in chromosome segregation underlie spontaneous chromosome fragility. Both may apply to fragile sites associated with human diseases.
RESULTS
HJ map reveals fragile sites
A molecular definition of fragile sites might be sites of recurrent DNA damage or breakage. We made a nucleotide-level map of spontaneous HJs in the E. coli genome using X-seq: chromatin immunoprecipitation and sequencing in cells that produce RuvCDefGFP (RDG) using RuvC antibody (Fig. 2) (5). RDG binds and traps HJs specifically (schematic in Fig. 1B) in living cells and as a purified protein (5, 10). We find that spontaneous X-seq signal is highest near the replication terminus region (Ter), where it forms three discrete peaks that span approximately 300 kb of the 4.6-Mb genome (Fig. 2, A, B, and E). The two largest peaks flank the dif site, at which sister chromosomes are decatenated (11, 12). Also, at dif, covalent dimers of the circular E. coli chromosome that are formed by HR are resolved by the XerCD site-specific endonuclease and resolvase. Both processes allow chromosome segregation (13).
Fig. 2. Spontaneous HJs near replication terminus and their formation by DSB repair.
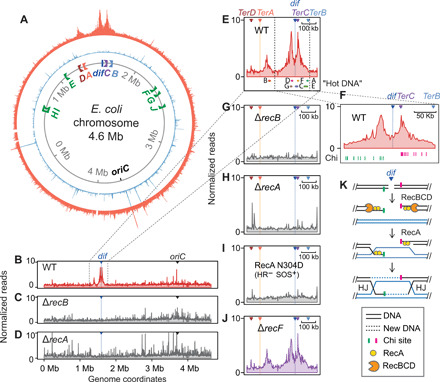
(A) Circular plot of normalized reads from X-seq shows three significant peaks of recurrent HJs. Replication terminus region at top, and origin, oriC, at the bottom; orange, X-seq with RuvC antibody; sky blue, chromatin immunoprecipitation and sequencing (ChIP-seq) with nonspecific immunoglobulin G2a (IgG2a). Italics capital letters A to H, locations and orientations of TerA-TerH sites. (B to D) RecB and RecA HR DSB-repair proteins are required for appearance of the three major X-seq peaks; whole-genome linear views of X-seq of indicated strains. WT, wild type. (E and F) Ter zoomed-in views at different scales; capital A to E, locations of HR hot DNA found previously (14). Green and pink hash marks in (F) represent Chi sites that curtail RecBCD nuclease activity of leftward- and rightward-moving RecBCD, respectively. (G to J) Ter zoomed-in views showing the dependence of HJ peaks on proteins of HR of DSBs. (K) Model: Exonucleolytic resection and RecA loading by RecBCD at a two-ended DSB near dif; HR will generate two HJs, one at each Chi cluster flanking dif.
The peak just to the left of dif (Fig. 2, A, E, and F) spans 9 to 91 kb to the left of dif with a summit 26 kb from dif. These coordinates and those below are the means of two independent experiments (repeats shown in fig. S1). The right-most peak (Fig. 2, A, E, and F) extends from 7 to 89 kb to the right of dif, with a summit 20 kb from dif. The farthest-left peak spans 28 to 85 kb to the right of TerA, with a summit 50 kb from TerA (orange line, Fig. 2E). The X-seq (HJ) peaks overlap seven of eight previously described "hot DNA" regions (Fig. 2E), which show elevated homologous recombination between plasmids and the chromosome (14).
Recurrent HJs from repair of DSBs, not reversed forks
HJs can form during HR of DSBs, HR of ssDNA gaps, or replication fork reversal, with each of these routes either dependent on or inhibited by specific proteins (Fig. 1A). The most definitive demonstration of reversed fork HJs is their destruction by RecBCD DSB end–dependent exonuclease (6, 15), which loads at, and degrades DNA specifically from, double-stranded DNA ends (Fig. 1A, iii). RecBCD removes or prevents reversed fork structures (Fig. 1A, iii) by its DSB nuclease activity so that reversed forks were detectable only in RecBCD null mutant cells previously (6). Reversed fork HJs, which were inferred previously by their RuvABC-dependent cleavage in cells, were too infrequent to observe in wild-type (RecBCD-proficient) cells (6). We find that, rather than accumulating in cells that lack RecB, the three terminal X-seq peaks are abolished in ∆recB cells. That is, they require functional RecB for their appearance (Fig. 2, C and G). These data show that the HJs in all three major peaks are not reversed forks and implicate DSB repair. The following data support HR repair of DSBs as the predominant source of HJs in all three peaks.
RecA and its orthologs are universally required HR proteins that catalyze DNA strand exchange in HR and also activate the SOS DNA damage response in bacteria (4). Biochemically, RecA also promotes fork reversal (16), also observed under conditions of RecA overproduction in living cells (5). RecA, however, is not required normally for fork reversal in living cells (6, 15). Thus, a requirement for RecA for HJ occurrence in cells implies that the HJs were formed either via HR or dependently on activation of the SOS response. We find that deletion of recA abolished all three major spontaneous X-seq peaks (Fig. 2, D and H, and replicates and negative controls in fig. S1A). A mutant RecA, RecA-N304D (17), that is capable of inducing SOS but defective for HR also abolished all three peaks of X-seq signal (Fig. 2I and fig. S1), demonstrating that RecA is required in its HR capacity.
HR of DSBs and ssDNA gaps use different proteins to load RecA onto a DNA strand for repair. The DSB-specific RecBCD multiprotein complex loads RecA at DSBs (Fig. 1A, i) (18), whereas RecF is required for loading RecA at ssDNA gaps (Fig. 1A, ii) (19). X-seq signal at all three spontaneous peaks required the RecB subunit of RecBCD and not RecF (Fig. 2, C, G, and J, and fig. S2). These data further support the conclusion that the HJs result from DSB repair by HR.
In addition, we find a notable colocalization of the X-seq peaks that flank dif with clusters of Chi sites (crossover hotspot instigator; Fig. 2F, hash marks). Chi sites are asymmetrical 8–base pair sequences that direct RecBCD to pause, reduce resection nuclease activity, and load RecA, which promotes strand exchange, HJ formation, and repair (18, 20). The X-seq peaks flanking dif colocalize with Chi clusters oriented so that they would impede RecBCD resection moving away from dif (toward the origin of replication) in each direction (Fig. 2, F and K). This correspondence is visible because the dif site itself resides in an unusually large, ~40 kb, Chi desert that is bounded by Chi clusters. The HJ peaks can, therefore, result from DSBs that occur throughout the large Chi desert, essentially all of which would be resected to the flanking Chi clusters at which strand-exchange HJs and DSB repair then occur (as per Fig. 2K). Thus, in addition to supporting DSB repair as the origin of the recurrent HJs, our data imply that spontaneous DSBs occur frequently and recurrently between the two Chi clusters in the Chi desert around the dif site (Fig. 2K).
Recurrent DSBs near terminal HJs
We sought and identified the predicted recurrent DSBs by modifying DNA-end sequencing or END-seq (21) for use in bacteria (Fig. 3). END-seq, used previously in mammalian cells, labels, purifies, and sequences DNA at DSB ends, mapping DSB ends to the nucleotide level (21). As illustrated in Fig. 3A, END-seq reads are strand specific and allow discrimination between one-ended and two-ended DSBs. They also reveal the orientations of DSB ends (Fig. 3A). We used END-seq to generate the first genome-wide maps of recurrent spontaneous DSBs in a bacterial genome (Fig. 3B and fig. S3). These are shown both in ∆recB cells, in which DSBs are not repaired (20), and in DSB repair-proficient wild-type cells (Fig. 3B).
Fig. 3. Recurrent genomic DNA breaks near X-seq HJ peaks at replication Ter and dif sites.
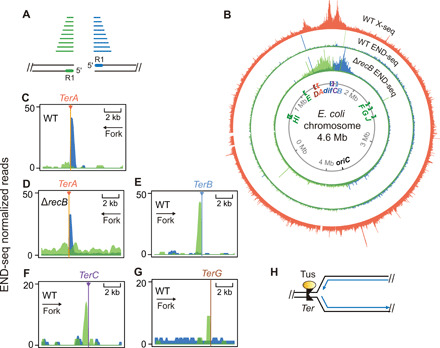
(A) Strand specificity of END-seq reads identifies DSB-end orientation. Green, DNA fragments with read 1 (5′ at DSB end) mapped to the bottom strand; blue, DNA fragments with read 1 mapped to the top strand. Green and blue lines, END-seq reads; black parallel lines, base-paired DNA strands. (B) Whole-genome maps of END-seq signal in the genomes of isogenic DSB repair-proficient (WT) and -deficient (∆recB) strains aligned with the X-seq map of recurrent genomic HJs (orange). Green and blue, DSB ends with the polarities shown in (A). Note strong one-ended DSB signal adjacent to and upstream of TerA that is not greater in repair-defective ∆recB cells, discussed in the main text. The largest localized DSB-end signal is seen flanking dif in DSB repair-defective ∆recB cells, near the two HJ peaks. Their absence in repair-proficient wild-type cells implies efficient repair. (C to G) END-seq signal indicating one-ended DSBs upstream of Ter sites A, B, C, and G, zoomed-in views. Persistent signal in repair-proficient wild-type cells (C) indicates poor or unsuccessful repair. (H) Diagram of Tus protein bound to unidirectional Ter site, which creates a replication barrier for forks traveling leftward. Blue lines, new DNA; arrow heads, 3′ ends.
We found the predicted large END-seq signals, representing recurrent DSBs, in the Ter region of the genome (Fig. 3). END-seq detected robust DSB signal surrounding the dif chromosome-decatenation site in repair-deficient ∆recB cells but not in wild-type E. coli (Fig. 3B), indicating that these DSB ends are repaired efficiently. The two peaks are of about equal size and have specific polarities, each end oriented “toward” dif (Fig. 3, A and B), suggesting that these may represent two-ended DSBs at or near dif (Fig. 3B). Their distribution “outward” from dif implies degradation by other nucleases in the absence of RecB, as reported previously (22–25).
In addition, we find one-ended END-seq peaks immediately adjacent to replication fork-arresting Ter sites, TerA and TerB, with smaller peaks at the TerC and TerG sites (Fig. 3, C to G). The TerA-adjacent one-ended DSBs (DSB ends) lie between TerA and its associated HJ peak (Figs. 3, A to D, and 4, A to F, top). Unlike the dif-proximal END-seq peaks, the Ter-proximal one-ended DSB peaks are present both in DSB repair–deficient ∆recB cells and in repair-proficient wild-type cells (Fig. 3, B to D, and fig. S3), indicating poor repair efficiency, as discussed below.
Fig. 4. Recurrent DSBs with HJs at a barrier; Tus and proliferation dependence.
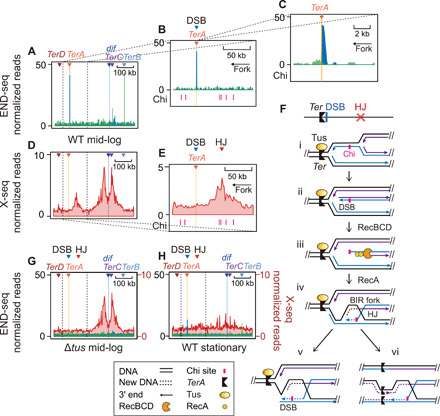
(A to C) END-seq data identify a “TerA-facing” DSB-end on the replication-arresting side of the TerA barrier; Ter zoomed-in views at different scales. Blue and green represent DSB-end polarities per Fig. 3A. (D and E) X-seq data show recurrent HJs next to the TerA barrier and its recurrent DSB peak (A); Ter zoomed-in views at scales aligned with genome locations in (A) and (B) above them, respectively. (F) Model for Tus/Ter-induced one-ended DSB formation (30) and subsequent HR that generates HJs upstream of the Tus/Ter barrier and its recurrent DSB ends. Black lines, template DNA strands; solid blue lines, nascent strands from the first round of replication; solid purple lines, nascent strands from a second round of replication; arrow heads, 3′ ends; dotted blue or purple lines, nascent strands during BIR repair. (G and H) Tus and proliferation dependence of the DSB and HJ peaks right of TerA, respectively (END-seq, blue and green; X-seq, red). Ter zoomed-in views.
Replication fork barriers as fragile sites
In E. coli, Tus protein binds DNA at 10 Ter sites to create unidirectional replication fork barriers (Fig. 3, B and H) (26). These Tus/Ter barriers facilitate the convergence of two replication forks within the terminus region and reduce the number of forks that pass dif and then travel in the wrong direction toward the origin (26), oriC (Fig. 3B). Similar replication fork barriers are found in yeast ribosomal DNA (rDNA) (27) and may occur generally in eukaryotic replication-termination zones (28). E. coli TerA and TerC are the first fork-blocking (nonpermissive) sites encountered by forks moving counterclockwise and clockwise past dif, respectively, and TerB is the second nonpermissive site for forks traveling clockwise (Fig. 3B).
We find that the DSB end and HJ peaks at TerA require replication fork arrest at TerA as follows. First, deletion of the tus gene, encoding the Tus replication barrier-binding protein (e.g., Fig. 3H), eliminated the END-seq signal at all Ter sites (Fig. 4G, blue, and fig. S3). Tus was also required for appearance of the X-seq peak next to TerA (Fig. 4G, red, and fig. S4). Thus, fragility near TerA is Tus dependent, supporting the hypothesis that replication fork arrest at Ter sites provokes the recurrent DSBs and HJs that occur there. Tus is not required for the X-seq peaks that flank dif (Fig. 4G), as discussed in the following section.
Further, cells that do not replicate DNA because they are in stationary phase show reduced END-seq signal at Ter sites, including TerA (Fig. 4H and fig. S4). X-seq signal is also reduced or eliminated at all three sites, supporting a requirement for replication for appearance of the recurrent HJs both at dif and TerA (Fig. 4H and fig. S4). These data support a role for DNA replication in formation of the spontaneous DSBs at Ter sites. The small, residual END-seq peaks that remain at TerA and TerB in stationary phase, relative to log-phase cultures, suggest that some DSB ends, potentially those arising in the last round of replication, are formed and not repaired.
We consider and then support a model for the mechanism of fragility at Ter replication barriers (Fig. 4F). In the model, (i) a replication fork stalls upstream of the Tus/Ter barrier; (ii) before the stalled fork is resolved, a second codirectional fork(s) arrives behind it and displaces the nascent leading strand, resulting in a DSB-end upstream of the barrier. (iii) RecBCD resects the DSB end to the nearest Chi site cluster and then loads RecA; and (iv) strand exchange leads to an HJ and allows establishment of a repair-generated replication fork, a process called break-induced replication or BIR (Fig. 4F, iv).
Note that the DSB end created (Fig. 4F, ii) cannot be repaired successfully unless two rightward-moving forks arrive from the left of the Ter site, the first fusing with the original fork stalled at Ter and the second fusing with the BIR fork (Fig. 4F, vi). Without the arrival of the converging forks, the BIR fork shown in Fig. 4F (iv) will collapse repeatedly as it reaches the Tus/Ter barrier, regenerating a DSB end each time in a futile cycle of incomplete repair (Fig. 4F, v). This failure to complete repair, which regenerates the DSB end repeatedly, can explain the persistent one-ended DSBs at Ter sites, which are visible in repair-proficient wild-type cells (Figs. 3, B to G, and 4, A to C). By contrast, the DSBs near dif (discussed in the following section) are repaired efficiently and so are visible only in repair-defective ∆recB cells (Figs. 3B and 5A). See Supplementary Text for discussion of the precision, i.e., the lack of erosion, of the Ter-proximal END-seq signals.
Fig. 5. Fragility near dif promoted by cell division, restricted by XerCD sister chromosome resolvase.
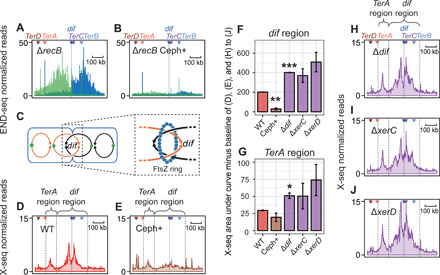
(A and B) Diffuse zone of END-seq–visualized DSBs in repair-deficient ∆recB cells and their inhibition by cell-division inhibitor cephalexin; Ter zoomed-in view. (C) Illustration of FtsZ ring at septation and catenated sister chromosomes following replication. (D and E) X-seq HJ peaks flanking dif are inhibited by cephalexin; Ter zoomed-in view. (F and G) Quantification of X-seq signal in (D), (E), and (H) to (J) in the dif and TerA region, respectively. N = 2 ± range; *P < 0.05, **P < 0.01, and ***P < 0.001, unpaired two-tailed t test. (H to J) Restriction of X-seq HJ signal to a narrow zone flanking dif is relaxed in mutants with reduced catenane/dimer resolution, which produce a wider zone. ∆dif cells lack the site of dimer resolution by XerCD nuclease/resolvase. ∆xerC and ∆xerD, partial-function mutants of essential XerCD resolution complex. Ter zoomed-in views.
We find that the X-seq peak near TerA overlaps with the first two properly oriented Chi sites encountered by RecBCD resecting a one-ended DSB rightward from TerA (Fig. 4E, Chi sites, pink hash marks). The distribution of Chi sites near TerB, conversely, is more uniform with no Chi clusters, which may explain why there is no single X-seq peak near TerB (fig. S5A).
As described above, our X-seq data are incompatible with models of HJ formation by fork reversal, followed by cleavage to make a DSB, because we found that HR proteins including RecBCD promote the TerA-proximal X-seq signal (Fig. 2, C, D, and G to I), whereas RecBCD removes reversed forks (Fig. 1A, iii) (6). We cannot rule out the possibility that Ter-proximal DSB ends form by cleavage of undetected reversed forks (RFs), but this is unlikely, given that X-seq detects RF HJs (5, 29), and our data demonstrate that the recurrent HJs are not RFs but rather result from repair of nearby recurrent DSBs (Figs. 1A and 2, C and H, as discussed above). Unlike DSBs at reversed forks (6), DSB-end formation at ectopic Ter sites did not require the HJ endonuclease RuvABC and did require additional rounds of replication (30), in support of the rereplication model in Fig. 4F. Moreover, two-dimensional (2D) gels of native Ter sites showed evidence of three-way stalled-fork structures but not four-way reversed fork HJs (31).
Together, all of these data support the model of chromosome fragility at Ter sites shown in Fig. 4F. The model is replication dependent, is Tus dependent, and features rereplication, DSB-end formation, and strand exchange, leading to incomplete repair in continuing futile cycles.
Chromosome segregation failure and fragility
Unlike the fragile site near TerA, the fragile-site HJ peaks flanking dif form independently of Tus (Fig. 4G), indicating that at least two independent mechanisms promote fragility in the terminus region of the genome. Further, the dif-associated DSBs are detected only in repair-defective (ΔrecB) cells and not in repair-proficient cells (Figs. 3B and 5A). Unlike X-seq signal, in which HJs are trapped and preserved indefinitely by RDG (5), END-seq signal reflects steady-state levels of DSB ends and is thus influenced by both DSB formation and repair rates. The observation that dif-proximal DSBs are detectable only in the absence of repair suggests their efficient repair by HR normally, a hypothesis supported by the prominent RecB- and RecA-dependent X-seq peaks in this region (Fig. 2, B to D and G to I).
The strand bias of the END-seq signal (Fig. 5A, green to the left, and blue to the right of dif, as defined in Fig. 3A) is most compatible with frequent formation of two-ended DSBs close to dif, followed by erosion of unrepaired DSB ends (Fig. 2K) (22–25). However, we cannot rule out the possibility that equal numbers of one-ended DSBs occur on each side of dif in different cells in the population.
Cell division promotes resolution of replicated sister chromosomes at dif (32, 33). We tested the possible role of cell division on DSB and HJ formation near dif using cephalexin, a drug that inhibits septation (Fig. 5). Cephalexin inactivates FtsI, a transpeptidase that licenses constriction of the tubulin-like FtsZ ring, which divides cells (Fig. 5C) (34). Cephalexin treatment blocked the appearance of dif-proximal DSBs (Fig. 5, A and B, and fig. S6) and HJs (Fig. 5, D to F, and fig. S7), without substantially reducing the formation of Ter site–associated DSBs and HJs (Fig. 5, B, E, and G). The data imply that cell division promotes DSBs near dif, perhaps by promoting breakage of unseparated sister chromosomes during segregation. In the growth conditions used here, cells have an average of four complete chromosomes (four termini) at the time of cell division (35), so most daughter cells will have an intact partner for HR of the broken chromosome and can form HJs if broken.
There are two main structures formed by sister chromosomes that, if not resolved, could lead to segregation failure and chromosome breakage: Catenanes (interlinked duplex DNA molecules) formed during each round of replication (Fig. 6A) (11, 33), and less frequent covalent chromosome dimers formed by HR-dependent crossing over between sister chromosomes (Fig. 6B) (32, 36). Typically, both types of attached chromosomes are resolved at dif (Fig. 6) (12, 13), either assisted or catalyzed by the dif-specific XerCD protein complex, respectively. For catenane resolution, XerCD binds topoisomerase (Topo) IV and brings it to dif (12) for Topo IV–mediated chromosome decatenation (Fig. 6A). For resolution of covalent dimers, XerCD, a dif site–specific recombinase, catalyzes strand exchange to form an HJ at dif that it then resolves (12, 37), separating the sister chromosomes (Fig. 6B).
Fig. 6. Interaction of XerCD recombinase with the FtsZ ring and dif localizes resolution of catenated sister chromosomes and covalent dimers to dif at cell division.
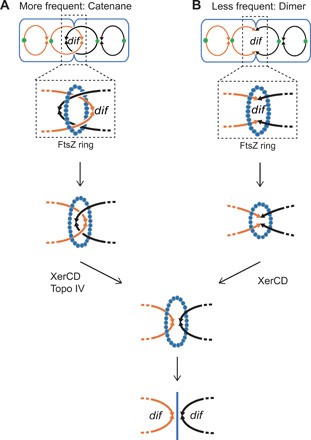
Single lines depict dsDNA. (A) Catenated sister chromosomes result frequently from DNA replication and are unlinked at cell division by topoisomerase (Topo) IV, a type II topoisomerase (11). Topo IV is brought to the dif site by interaction with XerCD (12), which interacts with the cell FtsZ ring, a tubulin-like polymer ring that constricts to divide cells (13). (B) Less frequently, chromosome dimers can result from crossing over during HR and are resolved by site-specific recombinase XerCD acting at the dif site, also linked with cell division.
In rapidly proliferating E. coli, these resolution mechanisms appear to fail frequently enough to produce the chromosome breakage and subsequent repair intermediates underlying fragility observed here as the major recurrent HJ and DSB signals (Figs. 3B and 5). The failed resolution and dif-associated reparable DSBs might result from partial reactions or failure of Topo IV during decatenation (Fig. 7A and Discussion) or from XerCD partial reactions or failure (Fig. 7B).
Fig. 7. Models: Mechanisms of fragility from failed resolution of sister chromosomes at segregation.
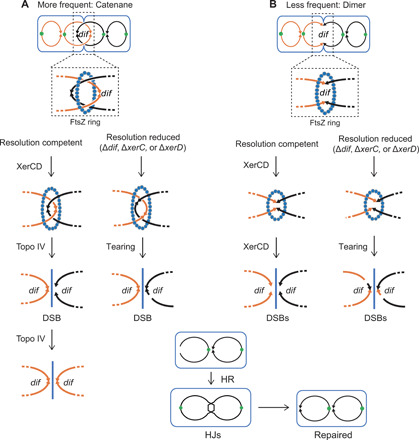
Single lines represent dsDNA. (A) Problems at sister chromosome decatenation. Left: Our data imply that some fraction of events in wild-type cells leads to breakage and highly efficient repair by HR at or close to dif (Fig. 5, A to F). The breakage could be half-reactions of type II topoisomerase Topo IV, which resolves catenated sister chromosomes at or near dif, facilitated by XerCD-dif interaction (11, 12), or might result from physical or other breakage before Topo IV has completed decatenation. Right: In resolution-reduced ∆dif, ∆xerC, or xerD mutants, the increase and broader zone of DSB-repair events around dif (Fig. 5, F, H to J) may result from chromosome tearing at segregation and, possibly, Topo IV half-reactions at sites near, but not confined to, dif. (B) Failed resolution of HR-generated covalent chromosome dimers. A single homologous crossover generates a dimer circle, which is usually resolved by site-specific recombinase XerCD at dif, in a process that is supposed not to generate DNA DSBs (Fig. 6) (13). Right: In resolution-compromised ∆dif, ∆xerC, or xerD mutants, breakage might occur (infrequently) by shearing of the infrequent unresolved chromosomes, causing breaks in a wider zone around the dif site.
We found that the deactivation of chromosome-dimer resolution by the deletion of dif or reduction of XerCD activity, by the deletion of xerC or xerD, increased X-seq signal around dif (Fig. 5, F and H to J, and figs. S8 and S9), supporting the hypothesis that unresolved chromosomes result in DSBs and repair HJs. Because the XerC and XerD activities are partially redundant, and the XerCD complex is required for viability, making the double mutant inviable, each single mutant has only partly impaired XerCD activity. This may explain why there are significantly more dif-proximal HJs in ∆dif cells (Fig. 5F), presumably breakage of more unresolved chromosomes, but the increase is not significant in ∆xerC or ∆xerD partial-function mutants (Fig 5F). In addition, the X-seq signal is broader in resolution mutants (xerC, xerD, or dif) (Fig. 5, H to J), indicating that XerCD binding of dif localizes any chromosome breakage from failed resolution of catenanes or dimers to a narrow region of the genome near dif (model, Fig. 7 and Discussion) (38, 39).
Fragility is common, and fragile sites interact
We estimated the frequency of genomes undergoing fragility by comparing spontaneous X-seq signal in the terminus region with X-seq signal at an enzymatically induced DSB (I-siteJ; Fig. 8A). The induced DSB is sustained by essentially all cells, shown by <1% survival in repair-deficient ΔrecB cells (Fig. 8A), and is repaired in at least 40% of the cells (Fig. 8A; DSB-survival data in wild type). As a first approximation, when we use a simplifying assumption of similar DNA copy number at the induced DSB and the terminus region, we estimate that TerA-associated HJs appear in a maximum of 9 to 12% of cells based on the size of the TerA-associated X-seq signal compared with the 49 or 43% of cells estimated to have repaired the engineered DSB (Fig. 8, A and B). The dif-associated HJs appear in maximally 13 to 14% under the simplifying assumption (Fig. 8B). However, proliferating cells have more DNA copies near the origin and fewer near the terminus. So, we can refine this estimate based on copy numbers between DNA near the I-site and the spontaneous loci, estimated from END-seq input libraries (fig. S3). These better estimates indicate that TerA-associated HJs occur in ~15 to 18% of cells and dif-associated HJs in ~21 to 23% of cells (Fig. 8B). However, some cells that form repair HJs near the I-Sce I DSB might die before repair is completed, so the frequency of TerA- and dif-associated HJs may be higher than estimated. Conversely because all of these estimates are based on X-seq with RuvCDef protein, which binds and remains bound to HJ DNA (5), and RuvCDef is produced for 3 to 4 hours and traps HJs blocking their resolution (5), these frequencies probably reflect accumulation of HJs during that time. Estimating roughly eight cell divisions and genome replications in 4 hours, the frequencies could be as low as 1 to 3% per genome replication. This is still unexpectedly frequent spontaneous fragility and repair due to normal cellular events in segregating sister chromosomes and is much higher than previous estimates of guillotined chromosomes (36). This discrepancy could be because division-induced DSBs are repaired efficiently (Fig. 3B; compare wild type with ΔrecB), whereas previous estimates of guillotined chromosomes were based on daughter cell filamentation (36), which results from unrepaired DSBs and their induction of SOS. Our data imply that most of the efficient repair events that X-seq detects do not result in an SOS response and daughter cell filamentation.
Fig. 8. Frequencies of recurrent spontaneous HJs in the replication terminus region.
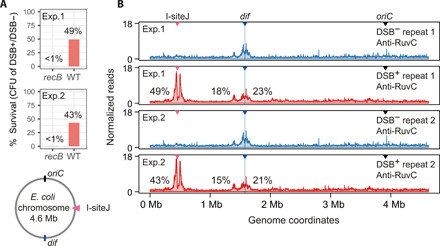
(A) Viable cells as CFU (colony-forming units) before and after the induction of an I–Sce I–induced DSB at I-siteJ. The percentage of cells that repair the DSB at I-siteJ is estimated by comparing the viability reduction of wild-type cells with that of repair-deficient recB cells. The roughly 40% of cells that survive when it is repair proficient (wild-type), but not when DSB repair deficient (recB), implies that about 40% of cells repaired DSBs that were not repaired in ∆recB, resulting in inviability. The diagram shows the E. coli chromosome (gray circle indicates duplex DNA). I-siteJ (pink triangle) is located about half way between oriC and dif. (B) X-seq of wild-type cells with (red)/without (blue) DSB induction by I–Sce I at I-siteJ, whole-genome views.
Although our data reveal two independent mechanisms of fragility in the terminus region, some interdependence between the two types of fragile sites is suggested. Chromosome-resolution mutants, which show increased HJs at dif (Fig. 5, F and H to J, and fig. S8), also show modestly increased X-seq signal near TerA (Fig. 5G and fig. S8). In addition, blocking cell division with cephalexin reduced END-seq and X-seq signal near dif (Fig. 5, B and D to F) and may cause slight reduction also at TerA (Fig. 5G and figs. S6 and S8). We propose a model in which most forks that stall and collapse at TerA begin at the chromosomal origin of replication, oriC (Fig. 9A), but a small number arise from BIR (repair replication) from dif-proximal chromosome cleavage (Fig. 9B). In this model, one-ended DSBs occur when BIR forks collapse at the Tus/Ter barrier (Fig. 9B, iv and v). The associated HJs could result from an unresolved HJ trailing the repair-replication bubble (Fig. 9A, bottom), the repair of a collapsed repair-induced fork (Fig. 9B, v) from dif repair or both.
Fig. 9. Models: Possible sources of replication forks that collapse upstream of TerA and their associated repair HJs.
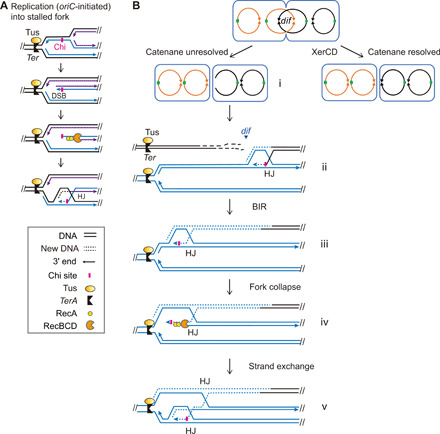
(A) Model: oriC-initiated replication leading to one-ended DSBs and HJs upstream of TerA. This is the major source. Black lines, template DNA strands; solid blue lines, nascent strands from the first round of replication; solid purple lines, nascent strands from a second round of replication; arrow heads, 3′ ends; dotted blue or purple lines, nascent strand during BIR repair. (B) Model: An alternative minor source of forks leading to DSBs and HJs upstream of TerA: BIR forks triggered by the repair of DSBs from unresolved catenanes at dif.
DISCUSSION
Technologies that capture specific DNA molecular-intermediate structures in living cells let us identify sites of spontaneous recurrent DNA breakage and repair, fragile sites, in the E. coli genome. We mapped them at high resolution with X-seq for HJs (5) and END-seq for DSBs (21). The bacterial fragile sites sustain DNA breakage and repair spontaneously and frequently, in 1% to more than 23% of the 4.6-Mb genomes (Fig. 8). Despite the much larger human genome, fragile sites in human cells are infrequent—observed, so far, only in cells treated with replication-inhibiting drugs—and are presumed to occur also spontaneously (2). Our identification of spontaneous fragile sites supports this hypothesis. The high sensitivity of RDG results from its trapping nature, which allows accumulation of HJs by preventing their further chemistry, both biochemically and in cells (5). Similar DNA structure-trapping proteins identify DSBs in human and bacterial cells (40), and DSBs plus another DNA damage structure(s) in bacteria (10, 41). HJ-trapping reagent(s) for human cells might improve detection of fragile sites and aid definition of their mechanisms of fragility. We are currently engineering HJ-trap(s) for human cells.
Repair HJs, not reversed forks
HJs can result from either reversed replication forks (Fig. 1A, iii) or HR (Fig. 1A, i and ii). Our data rule out reversed forks and demonstrate that both Ter- and dif-proximal HJs arise in DSB repair attempts because reversed forks are destroyed or prevented by RecBCD nuclease (6, 15) (illustrated in Fig. 1A, iii) and, before RDG (5), had been observed only in recB-null mutants (6, 15). By contrast, the Ter- and dif-proximal HJs require functional RecBCD for their appearance (Fig. 2, C and G, per Fig. 1A, i). The requirements for functional RecBCD and the RecA HR activity (Figs. 1A, i, and 2, C to I) identify the HJs, both at Ter barriers and dif sister chromosome resolution sites, as intermediates in HR DSB repair. Moreover, the TerA- and dif-proximal HJs align with Chi recombination hotspot sequences (Figs. 2F and 4E, respectively), which promote HJs as part of DSB repair (18, 20), illustrated in Figs. 2K and 4F (ii to iv). This and the recurrent DSBs near them (Figs. 3B and 4) also support the conclusion that the spontaneous HJs at the fragile sites are generated by HR of DSBs.
Failed repair of one-ended DSBs at a replication barrier
Fork stalling at replication barriers (e.g., Fig. 4F), either programmed or not, occurs in all organisms examined (42). We found poorly repaired or irreparable one-ended DSBs (single DSB ends) on the barrier sides of unidirectional Ter sites in the E. coli genome (Fig. 3), with recurrent HJs upstream of them (Fig. 4, A to F). The Ter-proximal DSBs and HJs result from replication fork arrest, as seen by their reduction or absence both in arrest-defective ∆tus mutants and nonreplicating cells (Fig. 4, G and H). These Ter-associated DSB ends resist repair, as seen by their visibility in repair-proficient wild-type cells (Fig. 3). By contrast, despite being much more numerous, reparable DSBs at dif are so well repaired that their END-seq peaks are visible only in repair-deficient ∆recB cells and not in wild-type cells (Fig. 4B).
A model that accounts for the poor repair and dependence on replication termination (Fig. 4, G and H) is shown in Figs. 4F and 9A. In the model, (i) replication forks stall upstream of the Tus/Ter barrier and, before they can be resolved by fusion with a converging fork, a subsequent codirectional fork arrives that (ii) displaces the nascent leading strand, which produces a DSB-end at the barrier (Fig. 4F, ii). (iii) RecBCD resects the DSB ends to Chi sites and then loads RecA, which (iv) generates HJs by strand exchange, initiating DSB-repair replication forks (Fig. 4F, iv) (43), a process called join-copy (43) or break-copy (44, 45) recombination, or BIR (Fig. 4F, iv). The BIR forks cannot repair the DSB ends unless they converge with an oncoming fork from the permissive side of the barrier (Fig. 4F, vi). Instead, BIR forks will most often be stopped by the barrier in continuous futile cycles of DSB-end regeneration and attempted repair (Fig. 4F, iii to v). The futile cycles create HJs without removal of DSB ends at the Ter site (Fig. 4F, v) such that DSBs are as numerous in repair-proficient wild-type cells as in repair-deficient ∆recB cells (for example, see Fig. 3B, TerA END-seq). Other models are possible.
Although previous studies suggested that the Ter region contains DNA damage and repair reaction intermediates, these studies could not define the specific intermediates or underlying mechanisms. DSBs and/or recombination hotspots were inferred from hot DNA (14), hotspots for Tn7 transposon insertion (46), RecB- and Chi site–dependent capture of small DNA fragments from the Ter region into engineered CRISPR arrays (47), and by division-induced loss of DNA in the Ter region in ΔrecB cells (48–50). These findings can be explained by our results and model (Fig. 9).
Supporting rereplication models, linear DNA fragments, detected by pulsed-field gel electrophoresis, arose from chromosomes of a strain with ectopic Ter sites flanking the replication origin (30). Their appearance required multiple rounds of replication (30). Under the growth conditions used here, multiple rounds of replication are initiated before completion of the previous rounds (35), making replication fork collisions of this type probable.
Parallels with eukaryotic chomosomes’ fragility and replication barriers
In yeast (51) and mammalian cells (52), E. coli Tus/Ter has been used as a model replication barrier. Fork stalling accompanied by the induction of HR was observed (51, 52), from which one-ended DSBs at the barrier were proposed (53). We have now observed one-ended DSBs upstream of replication barriers (Fig. 3), shown their dependence on Tus (replication-blocking) protein (Fig. 4G), and documented the HJs that result from their futile repair attempts (Figs. 3B; 4, D and E; and 8B).
In eukaryotes, programmed fork stalls occur within rDNA, at centromeres, and at other loci (e.g., yeast mating-type locus) (54). In human cells, one-ended DSBs, similar to those in Fig. 3, were observed upstream of the rDNA (55), a difficult region to replicate. These one-ended DSBs were proposed to arise by spontaneous breakage of stalled forks (55); alternatively, they might represent, presumably infrequent, rereplication as proposed here (Figs. 4F and 9A). Overreplication in eukaryotes is mostly prevented by the temporal separation of the licensing and firing steps of replication initiation into different cell cycle phases, G1 and S phase, respectively (56). But in cancers, many oncogenes dysregulate the cell cycle, potentially allowing overreplication (56). With human fragile sites, late replication and fork stalling are defining characteristics, including extending into G2 of the cell cycle (2), supporting involvement of replication barriers in their fragility. In noncancerous cells, BIR forks, which may result from any mechanism of DNA breakage, might generate second forks that could produce DSB ends at an initial stalled fork (Fig. 9B). The mechanisms uncovered here may occur and drive genome rearrangement and other mutagenesis during cancer formation and progression, more generally.
Sister chromosome segregation and fragility
In all organisms, DNA replication generates catenated sister chromosomes, which must be resolved before segregation into daughter cells (33) and which, we suggest, may provoke fragility, as seen here (Fig. 7A). In E. coli, decatenation requires cell division and occurs at dif, catalyzed by Topo IV (11), which is brought to dif by XerCD (13) (Fig. 6) (12). The largest spontaneous END-seq DSB signals and X-seq HJ signals in the genome flank the dif site (Figs. 3B and 4, A to D). Their appearance required replication/proliferation (Fig. 4H), not Tus/Ter (Fig. 4G), and was reduced by an inhibitor of cell division (Fig. 5, A to E) (39) implicating Topo IV and/or XerCD.
In Fig. 7A, we hypothesize that some of the chromosome-decatenation events go awry, leading to DSBs that require repair. Decatenation is carried out by type II topoisomerases (Fig. 7A, left) (11). Type II topoisomerases break both DNA strands, covalently attaching to each 5’ end; pass the unbroken duplex through the break; and then religate the DNA, detaching from the 5′ ends (Fig. 7A, left). We suggest that a small-fraction of decatenation events fail with Topo IV having broken but not religated the DNA, creating the reparable DSBs at dif, and the HJs that form during their repair (Figs. 3B and 7, bottom). Alternatively, complete failure of Topo IV would also lead to chromosome breakage by shearing of a chromosome as the sisters are segregated (Fig. 7A). In wild-type cells, the occasional breaks occur at dif (Figs. 5, A to E, and 7A, left), but in resolution mutants, e.g., ∆dif cells, there is more breakage and the repair HJs fall more broadly around dif (Figs. 5, F and H to J, and 7A, right), suggesting that segregation problems are worse and not dif-localized without the designated resolution mechanisms.
A similar mechanism seems likely to occur in human. The catenation link between sister chromatids in human cells underlies at least two classes of ultrafine bridges (UFBs) at common fragile sites: centromere-anchored UFBs (C-UFBs) (57) and rDNA-anchored UFBs (58). C-UFBs, the most common type, are found in all mitotic cells, including unstressed cells (57). Similarly to E. coli, the UFBs in eukaryotes are often resolved late and remain until the onset of anaphase (57). Unresolved UFBs at segregation could trigger DSBs and repair HJs, as here, and could activate the abscission checkpoint, which leads to cytokinesis failure and tetraploidization, posing a threat to genome integrity, and could drive cancer (59, 60). The bacterial model may help illuminate these events.
Possible support for the occurrence of a decatenation-related fragility mechanism in human is suggested to us by recent evidence that human TOPBP1 (DNA topoisomerase II–binding protein 1) is associated with suppression of formation of micronuclei (61): chromosome fragments released from chromosomes, a frequent anomaly in cancers. TOPBP1 interacts with human topoisomerase II beta, a type II topoisomerase, implicating a supportive role in breakage of DNA strands, as is done by E. coli Topo IV. The authors document recruitment of TOPBP1 to DSBs and suggest that TOPBP1 might tether broken chromosome ends so that they can be repaired after cell division. We suggest that, in addition, the TOPBP1 involvement with chromosome maintenance seems likely to be necessary particularly because its other client, topoisomerase II beta, actually makes those breaks during decatenation. Similarly to fragility at E. coli dif, decatenation might sometimes provoke fragility, DSBs, and their repair (HJs), which might, as we saw, be worse and more diffuse if the chromosome resolution machinery fails (Fig. 5, H to J).
Fragility and genome instability
Similar to the association of human fragile sites with hotspots for genomic rearrangements that drive genetic disorders, cancers, and other genetic diseases (2), the bacterial fragile sites described here are correlated with regions of both small mutations and rearrangements. Mutation accumulation (MA) studies in mismatch repair-deficient E. coli revealed a wave-like pattern of mutation frequencies across the E. coli genome with a local maximum spanning the terminus region (62, 63). This regional increase in mutation frequency was partially dependent on Tus and also constitutes the greatest mutation density in mismatch repair-proficient E. coli (63). A separate MA study implied that mobile element movement and other genome rearrangements cluster in the terminus region as well (P < 10−5, chi-square test) (64), as was shown for HR previously (65). Moreover, error-corrected sequencing of very rare variants in populations of E. coli revealed three major mutation hotspots in the terminus region, with the most prominent one located between dif and TerA (66), where we found fragile site DSBs and HJs. Together, these observations support the hypothesis that, similar to human chromosome fragility, bacterial DNA fragility provokes genome instability in E. coli. The mechanisms outlined here may underlie human chromosome fragility and the many important disease-driving events it instigates.
MATERIALS AND METHODS
Strains, media, and growth
Strains used in this study are summarized in table S1, and oligos are listed in table S2. E. coli K12 strains were grown in Luria Bertani Herskowitz (LBH)–rich medium (67). Other additives were used at the following concentrations: ampicillin (100 μg/ml), chloramphenicol (25 μg/ml), kanamycin (50 μg/ml), tetracycline (10 μg/ml), and sodium citrate (20 mM). P1 transductions were performed according to J. H. Miller (67). Genotypes were verified by antibiotic resistance, polymerase chain reaction (PCR), and, when relevant, ultraviolet sensitivity and sequencing.
Strains in each main figure
Strains used were as follows: SMR19425, SMR19407, SMR19406, SMR26434, and SMR19427 (Fig. 1); SMR19425, SMR6319, and SMR19460 (Fig. 2); SMR6319, SMR19425, SMR26432, and SMR26579 (Fig. 3); SMR19460, SMR19425, SMR26444, SMR26452, and SMR26454 (Fig. 4); and SMR22672 (Fig. 7). Strains used in extended data figures are listed in those figure legends.
X-seq library preparation and sequencing
Cultures were grown overnight shaking in LBH to saturation. For strains carrying PBAD I–Sce I and an I–Sce I cutsite, 0.1% glucose was added to reduce leaky expression of I–Sce I. The saturated cultures were diluted 500-fold into 80 ml of LBH with doxycycline (100 ng/ml) to induce RDG in 250-ml flasks and grown at 37°C, shaking at 225 rpm. After about 3 hours, cultures with an optical density at 600 nm (OD600) of 0.4 to 0.8 were used for later steps. For I–Sce I DSB induction, 0.005% arabinose was added, and cells were grown for another hour. For cephalexin treatment, cephalexin (10 μg/ml) was added to cultures with OD600 of about 0.2 and then incubated for 1.5 hours. For stationary-phase assays, overnight cultures (~15 hours) at OD600 > 4 were used. Cells were subjected to cross-linking, lysed, and sonicated as follows: 1% formaldehyde was added to cultures, and the cultures were incubated for 30 min at room temperature and then quenched by adding 0.5 M glycine. Cells were harvested by centrifugation and washed once with tris-buffered saline. Cells were lysed in lysis buffer (68) containing lysozyme (4 mg/ml). Sonication was performed using the Bioruptor Pico (Diagenode) for 30 cycles (30 s on, 30 s off) with 2 ml of lysate in 15-ml tubes containing sonication beads (Diagenode C01020031). The final DNA fragments were between 300 and 500 base pairs (bp). After sonication, lysates were centrifuged, and supernatants were collected and treated with ribonuclease A. The DNA concentration in lysates was measured and normalized to about 150 ng/μl. For each sample, two 1 ml aliquots of the same lysate were used and incubated separately with RuvC antibody (Santa Cruz Biotechnology, sc-53437) and nonspecific immunoglobulin G2a antibody (Santa Cruz Biotechnology, sc-3878) as a negative control. The protocol for immunoprecipitation and library preparation was modified as described (29): RuvC antibody was first incubated with Dynabeads protein A (Thermo Fisher Scientific, 10002D), and then the RuvC antibody–coated Dynabeads were incubated with cell lysates at room temperature for at least 1.5 hours. Blunting (New England Biolabs E0542L), A-tailing (New England Biolabs M0212L), and ligation (New England Biolabs E0542L) were performed while DNA fragments were still on Dynabeads, with multiple wash steps in between. Because the concentrations of immunoprecipitated DNA are low, samples were amplified briefly before size selection and, at the same time, barcoded using NEBNext Multiplex Oligos for Illumina (New England Biolabs E7335L, E7500L, E7700L, and E7730L). Two-sided size selection of adaptor-ligated DNA was performed on Agencourt AMPure XP Beads (Beckman Coulter A63881) at a ratio of 0.5 or 0.9. A second amplification was performed after size selection. Sequencing was performed on an Illumina MiSeq.
END-seq library preparation and sequencing
Cultures were grown overnight in LBH to saturation. Saturated cultures were diluted 500-fold into either 20 or 40 ml of fresh LBH in 250-ml flasks and grown at 37°C, shaking at 225 rpm. For cephalexin treatment, 40 ml of cultures was started and split in half when the OD600 reached about 0.2, with one half remaining untreated and the other treated with cephalexin (10 μg/ml). Both treated and untreated cultures were grown for an additional 1.5 hours, resulting in final OD600 readings of 0.6 to 1.0. All other mid-log cultures were harvested when OD600 reached 0.4 to 0.8, and stationary-phase cultures were harvested after 24 hours (OD600 > 4). Cells were harvested by centrifugation and washed twice with cold PBS and 50 mM EDTA.
Agarose plugs were prepared using the CHEF Bacterial Genomic DNA Plug Kit (Bio-Rad), as follows. Cell pellets were resuspended in cell suspension buffer, mixed with melted 2% CleanCut Agarose equilibrated to 50°C, and cast in 100-μl disposable molds. Plugs contained ~107 cells per plug with final agarose concentration of 0.75%. Plugs were chilled at 4°C until solid (30 min to 1 hour) and then expelled from molds. Plugs were treated with lysozyme and proteinase K according to the CHEF Bacterial Genomic DNA Plug Kit protocol. Two plugs per culture were used for END-seq library preparation.
Subsequent washes, ribonuclease A treatment, and enzymatic steps were performed as described for END-seq library preparation (21, 69), with the following minor adjustments. All enzymatic treatments and low volume washes were performed in 24-well plates instead of 1.5-ml tubes. After the ExoT blunting reaction, plugs were washed as described (69) and then stored overnight at 4°C. After ligation of the biotinylated END-seq adapter 1 and subsequent washes, plugs were transferred to 1.5-ml tubes and melted at 70°C for 10 min, followed by equilibration to 42°C for 10 min. Plugs were digested with 4 U of ß-agarase I (NEB) at 42°C for 1 hour. After drop dialysis and proteinase K treatment (Invitrogen), each DNA sample was brought to 100 μl with tris-EDTA (TE) and sheared with a Bioruptor Pico (Diagenode) for eight cycles of 10 s on and 90 s off at 4°C. Samples were vortexed and centrifuged midway through sonication to ensure optimal shearing. Following ethanol precipitation, DNA was quantified by NanoDrop and ~3 μg of DNA was used for downstream END-seq library construction. After streptavidin capture, blunting was performed using the Quick Blunting Kit (NEB) at 24°C for 30 min. A-tailing, ligation, hairpin digestion, PCR, and Agencourt AMPure XP bead (Beckman Coulter) clean up were performed as described (69). Libraries were amplified for an additional 5 to 10 PCR cycles using Illumina P5 and P7 primers and cleaned using 0.7× Agencourt AMPure XP beads (Beckman Coulter). Libraries were run on an 8% nondenaturing polyacrylamide gel, and DNA between 200 to 800 bp was cut from the gel. DNA was recovered by crushing gel slices, incubating crushed slices in gel extraction buffer [10 mM tris-HCl (pH 8.0), 0.3 M NaCl, and 1 mM EDTA] at 4°C overnight, and precipitation with isopropanol, as described (68).
A small fraction (~700 ng) of each END-seq sample was collected after sonication and dialysis and used to make input control libraries. Input libraries were constructed using the NEBNext Ultra II Kit (New England BioLabs) according to the manufacturers’ protocol.
Final END-seq and input libraries were quantified using the KAPA Library Quantification Kit for Illumina Platforms (KAPA Biosystems), pooled, and sequenced on an Illumina Nextseq550 (150-bp paired-end reads).
Analysis of sequencing data
Each sequencing run was checked for quality using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). When necessary, reads were trimmed by Trimmomatic (70) to remove sequencing adaptors and low-quality bases. Reads were then aligned by BWA-MEM (71) to the E. coli genome sequences of strains W3110 or MG1655 [National Center for Biotechnology Information Reference Sequence database accession: NC_007779.1 or NC_000913.3]. Reads that had multiple primary hits or low-mapping quality were discarded. Potential PCR duplicates were removed by Picard Tools MarkDuplicates (https://broadinstitute.github.io/picard/). Reads were counted in bins of 2 kb for X-seq and 100 bp for END-seq using the MOSAICS R package (version 2.18.0) (72). For each bin, the reads were then divided by the median read number of all bins. Therefore, most bins have normalized reads close to 1. Plots were generated in R.
Detecting peak boundaries in X-seq data
Because X-seq peaks are broad, as is expected from variable lengths of resection and the ability of HJs to branch migrate, conventional peak calling algorithms tend to break the broad peak into small peaks. Therefore, we use change point analyses to detect peak boundaries by detecting the change point in the mean for a minimum of two independent experiments and datasets. The cpt.mean function in the “changepoint” R package was used, with the PELT (Pruned Exact Linear Time) algorithm and Akaike’s information criterion as penalty (73). The input is normalized reads in 1-kb bins.
Quantification of peaks in X-seq data
The area under the curve of each peak is calculated, and the baseline was estimated by calculating the area under curve of random regions from the Ter half of the genome, excluding the Ter and dif region.
Acknowledgments
We thank C. Herman and D. Nelson for comments on the manuscript. Funding: This work was supported by an American Cancer Society postdoctoral fellowship (to D.M.F.), a gift from the WM Keck Foundation (to S.M.R.); U.S. NIH Director’s Pioneer Awards DP1-CA174424 and DP1-AG072751 (to S.M.R.); and grants R01-GM106373 (to P.J.H.), R35-GM122598 (to S.M.R.), and R01-CA250905 (to S.M.R.). Author contributions: S.M.R., Q.M., and D.M.F. conceived the study. Q.M. and D.M.F. performed experiments. J.L., J.X., J.P.P., Y.Z., R.B.N., J.P., H.L., and A.N. provided advice and/or assistance. Q.M., D.M.F., P.J.H., and S.M.R. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Raw sequencing data are available in the European Nucleotide Archive (ENA) under study accession number PRJEB39007. Bacterial strains are available by request (74, 75). Additional data related to this paper may be requested from the authors.
SUPPLEMENTAL MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabe2846/DC1
REFERENCES AND NOTES
- 1.Glover T. W., Berger C., Coyle J., Echo B., DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 67, 136–142 (1984). [DOI] [PubMed] [Google Scholar]
- 2.Glover T. W., Wilson T. E., Arlt M. F., Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 17, 489–501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wood R. D., Mitchell M., Sgouros J., Lindahl T., Human DNA repair genes. Science 291, 1284–1289 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Kuzminov A., Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 63, 751–813, table of contents (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xia J., Chen L. T., Mei Q., Ma C. H., Halliday J. A., Lin H. Y., Magnan D., Pribis J. P., Fitzgerald D. M., Hamilton H. M., Richters M., Nehring R. B., Shen X., Li L., Bates D., Hastings P. J., Herman C., Jayaram M., Rosenberg S. M., Holliday junction trap shows how cells use recombination and a junction-guardian role of RecQ helicase. Sci. Adv. 2, e1601605 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seigneur M., Bidnenko V., Ehrlich S. D., Michel B., RuvAB acts at arrested replication forks. Cell 95, 419–430 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Fitzgerald D. M., Hastings P. J., Rosenberg S. M., Stress-induced mutagenesis: Implications in cancer and drug resistance. Annu. Rev. Cancer Biol. 1, 119–140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slack A., Thornton P. C., Magner D. B., Rosenberg S. M., Hastings P. J., On the mechanism of gene amplification induced under stress in Escherichia coli. PLOS Genet. 2, e48 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hastings P. J., Ira G., Lupski J. R., A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLOS Genet. 5, e1000327 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia J., Mei Q., Rosenberg S. M., Tools to live by: Bacterial DNA structures illuminate cancer. Trends Genet. 35, 383–395 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X., Reyes-Lamothe R., Sherratt D. J., Modulation of Escherichia coli sister chromosome cohesion by topoisomerase IV. Genes Dev. 22, 2426–2433 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Sayyed H., Chat L. L., Lebailly E., Vickridge E., Pages C., Cornet F., Lagomarsino M. C., Espéli O., Mapping topoisomerase IV binding and activity sites on the E. coli genome. PLOS Genet. 12, e1006025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castillo F., Benmohamed A., Szatmari G., Xer site specific recombination: double and single recombinase systems. Front. Microbiol. 8, 453 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horiuchi T., Nishitani H., Kobayashi T., A new type of E. coli recombinational hotspot which requires for activity both DNA replication termination events and the Chi sequence. Adv. Biophys. 31, 133–147 (1995). [DOI] [PubMed] [Google Scholar]
- 15.Michel B., Ehrlich S. D., Uzest M., DNA double-strand breaks caused by replication arrest. EMBO J. 16, 430–438 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robu M. E., Inman R. B., Cox M. M., RecA protein promotes the regression of stalled replication forks in vitro. Proc. Natl. Acad. Sci. U.S.A. 98, 8211–8218 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adikesavan A. K., Katsonis P., Marciano D. C., Lua R., Herman C., Lichtarge O., Separation of recombination and SOS response in Escherichia coli RecA suggests LexA interaction sites. PLOS Genet. 7, e1002244 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Churchill J. J., Anderson D. G., Kowalczykowski S. C., The RecBC enzyme loads RecA protein onto ssDNA asymmetrically and independently of chi, resulting in constitutive recombination activation. Genes Dev. 13, 901–911 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morimatsu K., Kowalczykowski S. C., RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: A universal step of recombinational repair. Mol. Cell 11, 1337–1347 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Dillingham M. S., Kowalczykowski S. C., RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 72, 642–671 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Canela A., Sridharan S., Sciascia N., Tubbs A., Meltzer P., Sleckman B. P., Nussenzweig A., DNA breaks and end resection measured genome-wide by end sequencing. Mol. Cell 63, 898–911 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rinken R., Thomas B., Wackernagel W., Evidence that recBC-dependent degradation of duplex DNA in Escherichia coli recD mutants involves DNA unwinding. J. Bacteriol. 174, 5424–5429 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Razavy H., Szigety S. K., Rosenberg S. M., Evidence for both 3′ and 5′ single-strand DNA ends in intermediates in chi-stimulated recombination in vivo. Genetics 142, 333–339 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thoms B., Wackernagel W., Interaction of RecBCD enzyme with DNA at double-strand breaks produced in UV-irradiated Escherichia coli: Requirement for DNA end processing. J. Bacteriol. 180, 5639–5645 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thoms B., Borchers I., Wackernagel W., Effects of single-strand DNases ExoI, RecJ, ExoVII, and SbcCD on homologous recombination of recBCD+ strains of Escherichia coli and roles of SbcB15 and XonA2 ExoI mutant enzymes. J. Bacteriol. 190, 179–192 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill T. M., Arrest of bacterial DNA replication. Annu. Rev. Microbiol. 46, 603–633 (1992). [DOI] [PubMed] [Google Scholar]
- 27.Bastia D., Zaman S., Mechanism and physiological significance of programmed replication termination. Semin. Cell Dev. Biol. 30, 165–173 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fachinetti D., Bermejo R., Cocito A., Minardi S., Katou Y., Kanoh Y., Shirahige K., Azvolinsky A., Zakian V. A., Foiani M., Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol. Cell 39, 595–605 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xia J., Chiu L.-Y., Nehring R. B., Núñez M. A. B., Mei Q., Perez M., Zhai Y., Fitzgerald D. M., Pribis J. P., Wang Y., Hu C. W., Powell R. T., La Bonte S. A., Jalali A., Guzmán M. L. M., Lentzsch A. M., Szafran A. T., Joshi M. C., Richters M., Gibson J. L., Frisch R. L., Hastings P. J., Bates D., Queitsch C., Hilsenbeck S. G., Coarfa C., Hu J. C., Siegele D. A., Scott K. L., Liang H., Mancini M. A., Herman C., Miller K. M., Rosenberg S. M., Bacteria-to-human protein networks reveal origins of endogenous DNA damage. Cell 176, 127–143.e24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bidnenko V., Ehrlich S. D., Michel B., Replication fork collapse at replication terminator sequences. EMBO J. 21, 3898–3907 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duggin I. G., Bell S. D., Termination structures in the Escherichia coli chromosome replication fork trap. J. Mol. Biol. 387, 532–539 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Steiner W. W., Kuempel P. L., Cell division is required for resolution of dimer chromosomes at the dif locus of Escherichia coli. Mol. Microbiol. 27, 257–268 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Dewar J. M., Walter J. C., Mechanisms of DNA replication termination. Nat. Rev. Mol. Cell Biol. 18, 507–516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pogliano J., Pogliano K., Weiss D. S., Losick R., Beckwith J., Inactivation of FtsI inhibits constriction of the FtsZ cytokinetic ring and delays the assembly of FtsZ rings at potential division sites. Proc. Natl. Acad. Sci. U.S.A. 94, 559–564 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nielsen H. J., Youngren B., Hansen F. G., Austin S., Dynamics of Escherichia coli chromosome segregation during multifork replication. J. Bacteriol. 189, 8660–8666 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hendricks E. C., Szerlong H., Hill T., Kuempel P., Cell division, guillotining of dimer chromosomes and SOS induction in resolution mutants (dif, xerC and xerD) of Escherichia coli. Mol. Microbiol. 36, 973–981 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Aussel L., Barre F. X., Aroyo M., Stasiak A., Stasiak A. Z., Sherratt D., FtsK is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell 108, 195–205 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Corre J., Louarn J. M., Evidence from terminal recombination gradients that FtsK uses replichore polarity to control chromosome terminus positioning at division in Escherichia coli. J. Bacteriol. 184, 3801–3807 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kennedy S. P., Chevalier F., Barre F. X., Delayed activation of Xer recombination at dif by FtsK during septum assembly in Escherichia coli. Mol. Microbiol. 68, 1018–1028 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Shee C., Cox B. D., Gu F., Luengas E. M., Joshi M. C., Chiu L. Y., Magnan D., Halliday J. A., Frisch R. L., Gibson J. L., Nehring R. B., Do H. G., Hernandez M., Li L., Herman C., Hastings P. J., Bates D., Harris R. S., Miller K. M., Rosenberg S. M., Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. eLife 2, e01222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kotlajich M. V., Xia J., Zhai Y., Lin H. Y., Bradley C. C., Shen X., Mei Q., Wang A. Z., Lynn E. J., Shee C., Chen L. T., Li L., Miller K. M., Herman C., Hastings P. J., Rosenberg S. M., Fluorescent fusions of the N protein of phage Mu label DNA damage in living cells. DNA Repair 72, 86–92 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mirkin E. V., Mirkin S. M., Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 71, 13–35 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dannenberg R., Mosig G., Early intermediates in bacteriophage T4 DNA replication and recombination. J. Virol. 45, 813–831 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meselson M., Weigle J. J., Chromosome breakage accompanying genetic recombination in bacteriophage. Proc. Natl. Acad. Sci. U.S.A. 47, 857–868 (1961). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Motamedi M. R., Szigety S. K., Rosenberg S. M., Double-strand-break repair recombination in Escherichia coli: physical evidence for a DNA replication mechanism in vivo. Genes Dev. 13, 2889–2903 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peters J. E., Craig N. L., Tn7 transposes proximal to DNA double-strand breaks and into regions where chromosomal DNA replication terminates. Mol. Cell 6, 573–582 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Levy A., Goren M. G., Yosef I., Auster O., Manor M., Amitai G., Edgar R., Qimron U., Sorek R., CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature 520, 505–510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wendel B. M., Courcelle C. T., Courcelle J., Completion of DNA replication in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 111, 16454–16459 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sinha A. K., Durand A., Desfontaines J. M., Iurchenko I., Auger H., Leach D. R. F., Barre F. X., Michel B., Division-induced DNA double strand breaks in the chromosome terminus region of Escherichia coli lacking RecBCD DNA repair enzyme. PLOS Genet. 13, e1006895 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sinha A. K., Possoz C., Durand A., Desfontaines J. M., Barre F. X., Leach D. R. F., Michel B., Broken replication forks trigger heritable DNA breaks in the terminus of a circular chromosome. PLOS Genet. 14, e1007256 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larsen N. B., Liberti S. E., Vogel I., Jørgensen S. W., Hickson I. D., Mankouri H. W., Stalled replication forks generate a distinct mutational signature in yeast. Proc. Natl. Acad. Sci. U.S.A. 114, 9665–9670 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Willis N. A., Chandramouly G., Huang B., Kwok A., Follonier C., Deng C., Scully R., BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510, 556–559 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Willis N. A., Frock R. L., Menghi F., Duffey E. E., Panday A., Camacho V., Hasty E. P., Liu E. T., Alt F. W., Scully R., Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature 551, 590–595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gadaleta M. C., Noguchi E., Regulation of DNA replication through natural impediments in the eukaryotic genome. Genes 8, 98 (2017). [Google Scholar]
- 55.Tubbs A., Sridharan S., van Wietmarschen N., Maman Y., Callen E., Stanlie A., Wu W., Wu X., Day A., Wong N., Yin M., Canela A., Fu H., Redon C., Pruitt S. C., Jaszczyszyn Y., Aladjem M. I., Aplan P. D., Hyrien O., Nussenzweig A., Dual roles of poly(dA:dT) tracts in replication initiation and fork collapse. Cell 174, 1127–1142.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reusswig K. U., Pfander B., Control of eukaryotic DNA replication initiation-mechanisms to ensure smooth transitions. Genes 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y., Nielsen C. F., Yao Q., Hickson I. D., The origins and processing of ultra fine anaphase DNA bridges. Curr. Opin. Genet. Dev. 26, 1–5 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Nielsen C. F., Hickson I. D., PICH promotes mitotic chromosome segregation: Identification of a novel role in rDNA disjunction. Cell Cycle 15, 2704–2711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Steigemann P., Wurzenberger C., Schmitz M. H. A., Held M., Guizetti J., Maar S., Gerlich D. W., Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 136, 473–484 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Pampalona J., Frias C., Genesca A., Tusell L., Progressive telomere dysfunction causes cytokinesis failure and leads to the accumulation of polyploid cells. PLOS Genet. 8, e1002679 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zompit M. D. M., Mooser C., Adam S., Rossi S. E., Jeanrenaud A., Leimbacher P.-A., Fink D., Durocher D., Stucki M., The CIP2A-TOPBP1 complex safeguards chromosomal stability during mitosis. bioRxiv , (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Foster P. L., Hanson A. J., Lee H., Popodi E. M., Tang H., On the mutational topology of the bacterial genome. G3 3, 399–407 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Niccum B. A., Lee H., MohammedIsmail W., Tang H., Foster P. L., The symmetrical wave pattern of base-pair substitution rates across the Escherichia coli chromosome has multiple causes. mBio 10, e01226-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee H., Doak T. G., Popodi E., Foster P. L., Tang H., Insertion sequence-caused large-scale rearrangements in the genome of Escherichia coli. Nucleic Acids Res. 44, 7109–7119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Louarn J. M., Louarn J., Francois V., Patte J., Analysis and possible role of hyperrecombination in the termination region of the Escherichia coli chromosome. J. Bacteriol. 173, 5097–5104 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang X., Zhang X., Zhang X., Liao Y., Song L., Zhang Q., Li P., Tian J., Shao Y., AI-Dherasi A. M., Li Y., Liu R., Chen T., Deng X., Zhang Y., Lv D., Zhao J., Chen J., Li Z., Spatial vulnerabilities of the Escherichia coli genome to spontaneous mutations revealed with improved duplex sequencing. Genetics 210, 547–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.J. H. Miller, A Short Course in Bacterial Genetics – A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria (Cold Spring Harbor Laboratory Press, 1992). [Google Scholar]
- 68.Bonocora R. P., Wade J. T., ChIP-seq for genome-scale analysis of bacterial DNA-binding proteins. Methods Mol. Biol. 1276, 327–340 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Canela A., Maman Y., Jung S., Wong N., Callen E., Day A., Kieffer-Kwon K.-R., Pekowska A., Zhang H., Rao S. S. P., Huang S.-C., Mckinnon P. J., Aplan P. D., Pommier Y., Aiden E. L., Casellas R., Nussenzweig A., Genome organization drives chromosome fragility. Cell 170, 507–521.e18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.H. Li, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 (2013).
- 72.Kuan P. F., Chung D., Pan G., Thomson J. A., Stewart R., Keleş S., A statistical framework for the analysis of ChIP-seq data. J. Am. Stat. Assoc. 106, 891–903 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Killick R., Eckley I., Changepoint: An R package for changepoint analysis. J. Stat. Softw. 58, 3 (2014). [Google Scholar]
- 74.Datsenko K. A., Wanner B. L., One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cherepanov P. P., Wackernagel W., Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14 (1995). [DOI] [PubMed] [Google Scholar]
- 76.Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., Mori H., Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2, 2006.0008 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Magner D. B., Blankschien M. D., Lee J. A., Pennington J. M., Lupski J. R., Rosenberg S. M., RecQ promotes toxic recombination in cells lacking recombination intermediate-removal proteins. Mol. Cell 26, 273–286 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pennington J. M., Rosenberg S. M., Spontaneous DNA breakage in single living Escherichia coli cells. Nat. Genet. 39, 797–802 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabe2846/DC1