

Alpha-dystroglycanopathy (α-DGP) is a subtype of congenital muscular dystrophies (CMDs) with autosomal recessive inheritance. Its main pathogenesis is the defect of post-translational O-glycosylation in α-dystroglycan (α-DG). α-DGP presents a wide clinical spectrum that ranges from the most severe CMDs such as Walker-Warburg syndrome (WWS), muscle-eye-brain disease (MEB), and Fukuyama congenital muscular dystrophy to the mildest limb-girdle muscular dystrophy. The B3GALNT2 (NM 152490) is one of the pathogenic genes of α-DGP. It encodes an enzyme that produces a unique carbohydrate structure, nitroacetylgalactosamine-β-1,3-nitroacetylglucosamine (GalNAc-β-1–3GlcNAc), which is essential for O-glycosylation of α-DG.[1] Most patients with B3GALNT2 mutations in previous studies presented with the most severe WWS or less severe MEB[2] and those with WWS generally had a very short lifespan. They universally exhibited severe muscle weakness, ocular anomalies, including congenital cataract, glaucoma, severe myopia, and optic nerve atrophy. Their brain imaging showed cobblestone lissencephaly or polymicrogyria, cerebellum cysts, and dysplasia.
To date, there are rare studies on Chinese patients with B3GALNT2 gene mutations. Here, we described two Chinese patients who carried compound heterozygous mutations of B3GALNT2. They presented with MEB-like phenotypes, which were very similar to MEB but milder than it. The research protocol was reviewed and approved by the Ethics Committee of Peking University First Hospital (No. 2015[916]). Written informed consent for the research and publication of medical data was obtained from the patients and their parents.
Patient 1 was a 2-year and 11-month-old girl who was the second child of healthy and non-consanguineous parents without a family history of neuromuscular disorders. She was born at full-term without perinatal complications. She had a poor interaction with people at 10 months of age and could not stand independently until 2 years old. Physical examination at 13 months of age showed a head circumference of 44.5 cm (<P25), proximal muscle weakness, and decreased tendon reflexes without joint contractures. At the present age, she could walk independently for a few minutes but had a wide-base gait and could not understand instructions or say any complicated words. Her neuropsychological evaluation at 10 months of age revealed that the developmental quotient (DQ) was 66, which meant mild to moderate mental retardation. The serum creatine kinase (CK) level at 10 months of age, auditory test, and fundus examination at 1 year of age were all normal. The brain magnetic resonance imaging (MRI) at 11 months of age demonstrated polymicrogyria, increased T2 signal in the white matter of cerebrum and cerebellum, mildly enlarged ventricles, cerebellum cysts, and dysplastic cerebellum and brainstem [Figure 1A].
Figure 1.
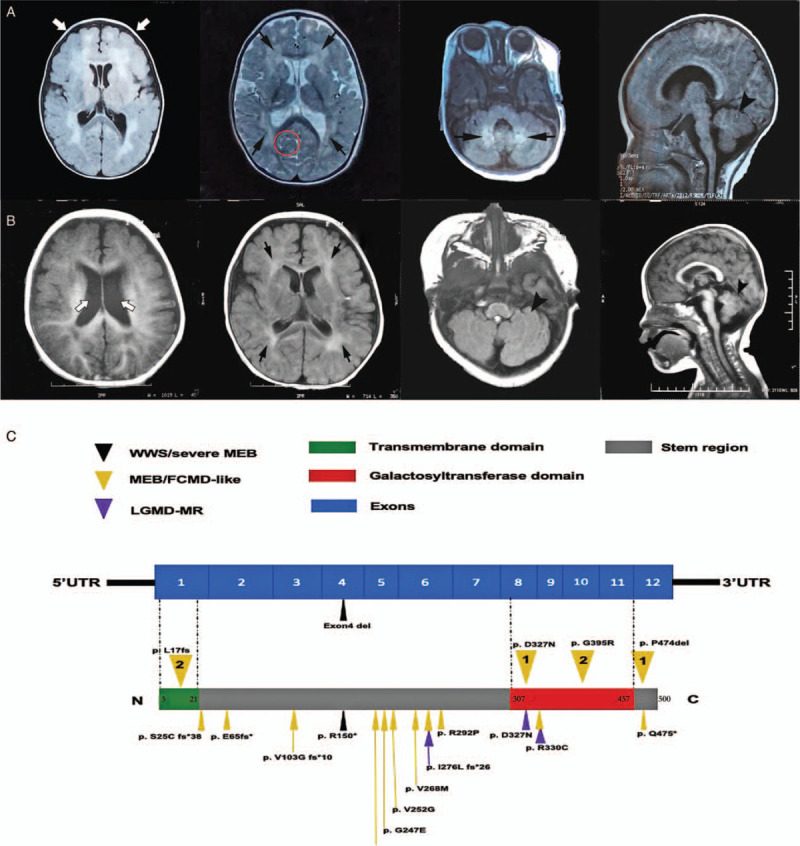
(A) The brain MRI of patient 1 at 11 months of age showed polymicrogyria (white arrows), abnormal signal intensity in the white matter of bilateral cerebrum and cerebellum (black arrows), cerebellum cysts (red circle), and dysplastic cerebellum and brainstem (arrowhead). (B) The brain MRI of patient 2 at 7 months of age showed mildly enlarged ventricles (white arrows), increased signals in the white matter (black arrows), brainstem, and cerebellum dysplasia (arrowheads). (C) Mutational sites of B3GALNT2 and the corresponding phenotypes. The transmembrane domain of B3GALNT2 is encoded by exon 1. The catalytic domain is encoded by exon 8–11. The numbers in the yellow inverted triangles represent our patients: 1, Patient 1; 2, Patient 2. The triangles represent mutations reported previously. MRI: Magnetic resonance imaging.
Patient 2 was a 1-year and 9-month-old girl who was the first child of non-consanguineous and healthy parents. She was born at full-term without perinatal complications. She could say one- or two-syllable words at the age of 8 months, stand with assistance at the age of 1 year, and walk with support at the age of 15 months but without any progress since then. She had no ocular or auditory impairment. Psychological evaluation at 15 months of age showed the DQ was 57, which meant moderate to severe mental retardation. Physical examination showed a 45 cm (<P25) head circumference at 17 months of age. She had decreased muscle strength and no joint contractures. Her knee reflexes could be elicited bilaterally. The serum CK level at 17 months of age was 565 U/L (normal value <170 U/L). Her brain MRI at 7 months of age showed slightly increased T2 signals in the white matter of the cerebrum, brainstem, cerebellum dysplasia, and mildly enlarged ventricles [Figure 1B].
The whole exon sequencing was performed on both patients, and candidate variants were validated by Sanger sequencing in the families. If the next-generation sequencing data revealed different number of sequence reads between patients and control samples, a copy number variant (CNV) was confirmed by the quantitative polymerase chain reaction. Both patients were found to carry variants of B3GALNT2 and their parents were carriers. Patient 1 had compound heterozygous variants of c.979G>A (p.D327N) and c.1421_c.1423delCTC (p.P474del), which had been reported previously[3–5] and no CNVs [Supplementary Figure 1A]. Patient 2 harbored novel compound heterozygous variants of c.48dupG (p.L17fs) and c.1183G>A (p.G395R) [Supplementary Figure 1B]. Furthermore, she had a CNV which was a duplication of 550-kilo base pairs on chromosome 12: 94075229-94625165. According to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines, these two novel variants were predicted to be pathogenic and likely pathogenic, respectively. The CNV was not recorded in the chromosome CNVs polymorphism database of the normal population, and there was no smaller pathogenic duplication reports than this region in the DatabasE of genomiC varIation and Phenotype in Humans using Ensemble Resources database (DECIPHER). Based on the earlier evidence, its pathogenicity was considered to be uncertain.
These two patients manifested early onset of muscle weakness, psychomotor, and language retardation. Together with microcephaly, brain structural abnormalities but without eye anomalies, we considered them to have MEB-like phenotypes as their presentations were relatively mild.
The B3GALNT2 gene at chromosome 1q42.3 contains 12 exons and encodes approximately 500 amino acids. It comprises one transmembrane domain, one stem region, and a galactosyltransferase catalytic domain. The catalytic domain is located between amino acids 307 and 457.[1]
Five Iranian siblings who carried homozygous mutations of p.D327N manifested much milder than our patient 1. They only had psychomotor and language retardation and non-specific cerebral white matter change. They had no brain structural deformity or ocular anomalies except that they all had epilepsy. Strikingly, their muscle involvement was much milder than those reported previously.[4] The manifestations of one Swedish patient with mutations of p.D327N and p.E65fs∗ were very similar to our patient 1.[5] It seems that p.D327N affects protein function slightly despite it is located in the catalytic domain. The mutation p.P474del combined with p.S25Cfs∗38 had been reported to cause severe MEB.[3] Although p.P474del is outside the catalytic domain, it is a conservative site in many species. Thus, it is probably the mutation p.P474del that contributes to the severity of clinical manifestations in patient 1 rather than p.D327N. The severities of clinical manifestations may depend on the degree of impairment in the protein function caused by the mutation rather than the region in which the mutation is located. The missense mutation p.G395R and the frameshift mutation p.L17fs of patient 2 are novel and located in the catalytic and the transmembrane domain, respectively. Both sites are conservative, and the frameshift mutation may produce truncated protein. We summarized all the reported mutations and their corresponding phenotypes in Figure 1C.
In conclusion, these two Chinese patients with B3GALNT2 gene mutations expand the mutational and phenotypic spectrum of α-DGP. The relationship between these mutations and their effects on enzyme activities had not been elucidated. Further studies are necessary to investigate the pathogenicity of these mutations and their molecular mechanisms.
Acknowledgements
The authors deeply appreciate the families who participated in this study.
Funding
This study was supported by the Development Program of China (No. 2016YFC0901505), National Natural Science Foundation of China (No. 81571220), and Beijing Key Laboratory of Molecular Diagnosis and Study on Pediatric Genetic Diseases (No. BZ0317).
Conflicts of interest
None.
Supplementary Material
Footnotes
How to cite this article: Chen XY, Song DY, Fan YB, Tan DD, Chang XZ, Xiao JX, Toda T, Xiong H. Novel mutations in B3GALNT2 gene causing α-dystroglycanopathy in Chinese patients. Chin Med J 2020;134:1483–1485. doi: 10.1097/CM9.0000000000001283
Supplemental digital content is available for this article.
References
- 1.Hiruma T, Togayachi A, Okamura K, Sato T, Kikuchi N, Kwon YD, et al. A novel human beta1,3-N-acetylgalactosaminyltransferase that synthesizes a unique carbohydrate structure, GalNAcbeta1-3GlcNAc. J Biol Chem 2004; 279:14087–14095. doi: 10.1074/jbc.M310614200. [DOI] [PubMed] [Google Scholar]
- 2.Sframeli M, Sarkozy A, Bertoli M, Astrea G, Hudson J, Scoto M, et al. Congenital muscular dystrophies in the UK population: clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul Disord 2017; 27:793–803. doi: 10.1016/j.nmd.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Alby C, Malan V, Boutaud L, Marangoni MA, Bessieres B, Bonniere M, et al. Clinical, genetic and neuropathological findings in a series of 138 fetuses with a corpus callosum malformation. Birth Defects Res A Clin Mol Teratol 2016; 106:36–46. doi: 10.1002/bdra.23472. [DOI] [PubMed] [Google Scholar]
- 4.Maroofian R, Riemersma M, Jae LT, Zhianabed N, Willemsen MH, Wissink-Lindhout WM, et al. B3GALNT2 mutations associated with non-syndromic autosomal recessive intellectual disability reveal a lack of genotype-phenotype associations in the muscular dystrophy-dystroglycanopathies. Genome Med 2017; 9:118.doi: 10.1186/s13073-017-0505-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hedberg C, Oldfors A, Darin N. B3GALNT2 is a gene associated with congenital muscular dystrophy with brain malformations. Eur J Hum Genet 2014; 22:707–710. doi: 10.1038/ejhg.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.