

Abstract
Merck KGaA observed slight differences in the dissolution of Concor® (bisoprolol) batches over the years. The purpose of this work was to assess the impact of in vitro dissolution on the simulated pharmacokinetics of bisoprolol using in vitro–in vivo relationship established with available in vitro dissolution and corresponding plasma concentrations‐time data for several bisoprolol batches. A mechanistic absorption model/physiologically based pharmacokinetics model linked with a biopharmaceutics tool such as dissolution testing, namely, physiologically based biopharmaceutics modeling (PBBM), can be valuable in determining a dissolution “safe space.” A PBBM for bisoprolol was built using in vitro, in silico, and clinical data. We evaluated potential influences of variability in dissolution of bisoprolol batches on its clinical performance through PBBM and virtual bioequivalence (BE) trials. We demonstrated that in vitro dissolution was not critical for the clinical performance of bisoprolol over a wide range of tested values. Based on virtual BE trials, safe space expansion was explored using hypothetical dissolution data. A formulation with in vitro dissolution reaching 70% dissolved in 15 min and 79.5% in 30 min was shown to be BE to classical fast dissolution of bisoprolol (>85% within 15 min), as point estimates and 90% confidence intervals of the maximum plasma concentration and area under the concentration‐time curve were within the BE limits (0.8–1.25).
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The in vitro dissolution testing plays an important role in assuring the quality and performance of a drug product by specifying acceptance criteria according to current guidance. However, this can be a conservative approach.
WHAT QUESTION DID THIS STUDY ADDRESS?
The present work illustrates the utility of physiologically based biopharmaceutics modeling to address the question of whether meeting the acceptance criterion for in vitro dissolution is crucial for the clinical performance of bisoprolol, a highly soluble and rapidly dissolving drug product.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The mechanistic modeling and virtual bioequivalence approach allowed researchers to determine safe space by defining the range of hypothetical in vitro dissolution profiles that should not affect in vivo product performance.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
For a well‐characterized BCS class I drug with a wide therapeutic window such as bisoprolol, a thorough mechanistic‐based approach can support biowaiver by challenging the bioequivalence requirement of rapid dissolution for immediate release oral solid dosage form.
INTRODUCTION
Mechanistic absorption/physiologically based pharmacokinetic (PBPK) modeling has been well established and used for drug development by industry and accepted by regulatory agencies. 1 This approach has been used to address questions that arise within the context of supporting drug product quality. Regulatory authorities encourage modeling and simulation approaches to support drug product changes and demonstrate bioequivalence (BE), allowing the pharmaceutical industry to save time and resources by avoiding unnecessary clinical trials. 1 Mechanistic absorption and PBPK modeling in conjunction with in vitro biopharmaceutics tools such as in vitro dissolution testing, namely, physiologically based biopharmaceutics modeling (PBBM), can serve as a powerful aid to establish a dissolution “safe space” and widen dissolution criteria. 2
Bisoprolol is a β‐1 selective adrenoceptor blocking agent used in the treatment of hypertension and angina pectoris. Orally administered bisoprolol was almost completely absorbed. 3 Bisoprolol is subjected to a low first‐pass effect (<10%); hence, absolute oral bioavailability is high at ~90%. 3 , 4 It is equally cleared by metabolic and renal routes. 3 , 4 It has a long elimination half‐life (~10–11 h), ideal for a once‐a‐day dosing regimen. 4 Bisoprolol exhibits linear pharmacokinetics (PK) in the dose range of 2.5–100 mg. 4 The therapeutic dose range is 1.25–20 mg once daily. 5
Bisoprolol is an immediate release (IR) oral tablet product containing bisoprolol in the form of bisoprolol fumarate and manufactured by Merck KGaA, Darmstadt, Germany. It is a rapidly disintegrating tablet with more than 85% dissolved within 15 min. Bisoprolol is very soluble in water and is a highly permeable drug substance with low PK variability. Therefore, it is considered to be a class I compound of the Biopharmaceutical Classification System (BCS). 6 The BCS biowaiver approach allows the approval of drug product changes that are not expected to impact in vivo behavior. Thus, major manufacturing and process changes implemented for bisoprolol could be waived through in vitro dissolution data under the BCS framework if comparable in vitro dissolution profiles were presented. However, the impact of dissolution profiles that are not comparable for bisoprolol tablets on the in vivo performance of bisoprolol has never been studied methodically. Specifically, given that the dissolution criterion was not met (e.g., the drug products were not rapidly dissolving), BCS could not be applied.
As part of the internal quality system, Merck tests dissolution on a limited number of batches using the approved quality control method. Over the years, slight differences in dissolution of bisoprolol batches have been observed. Although some batches showed slightly faster dissolution (hereafter referred to as “fast dissolution”), other batches showed slightly slower dissolution (hereafter referred to as “slow dissolution”).
We present a case study where PBBM simulations were conducted to assess potential influences of dissolution variability of bisoprolol batches on the PK of bisoprolol, a well‐studied and established drug. The first objective was to build and verify a PBBM model for bisoprolol using its physicochemical and biopharmaceutical properties that would explain observed mean Cp‐time profiles following single and/or multiple dose oral administrations in healthy humans. The second objective was to establish an in vitro–in vivo relationship (IVIVR) by comparing in vitro dissolution data to in vivo release data (obtained by deconvolving the in vivo dissolution profile from the average observed Cp‐time profile) of several batches of bisoprolol. The IVIVR was deemed validated if the absolute percent prediction error (%PE), calculated by comparing observed values with the simulated maximum plasma concentration (Cmax) and area under the concentration‐time curve (AUC) (obtained using the observed in vitro dissolution as input), met the criteria for individual predictability per in vitro–in vivo correlation (IVIVC) guidance. The third objective was to assess virtual BE for the slow and fast dissolution batches. Bioequivalence was concluded according to current European Medicines Agency 7 and US Food and Drug Administration (FDA) 8 guideline requirements (point estimate and 90% confidence interval [CI] of the geometric mean ratio [GMR] of test and reference within the range 0.8–1.25). The fourth objective was to determine the dissolution range (i.e., safe space) with no impact on systemic exposure (i.e., Cmax and AUC) to define the minimum dissolution level of a bisoprolol batch that would satisfy the BE criteria (0.8–1.25) if compared with a batch used in a clinical PK study.
The present model integrated physicochemical and absorption, distribution, metabolism and excretion properties of the compound with physiology in the PBPK modeling platform. The baseline model was developed using intravenous, oral solution, and IR tablet literature data. 3 , 9 , 10 , 11 , 12 , 13 The model was further verified with several different PK data sets obtained from Merck's clinical studies. Once the confidence in the predictive performance of the model was established, the verified model was extended to examine the impact of variability in dissolution of bisoprolol batches on the PK of the product to establish a dissolution “safe space.”
METHODS
Software
Model development and verification were conducted using GastroPlus® version 9.6 (Simulations Plus Inc., Lancaster, CA). Several features of GastroPlus® were used to achieve study objectives. The ADMET Predictor® module (version 9.0) was used to obtain in silico estimates of key physicochemical and biopharmaceutical properties from the structure. The in silico values were used to parameterize the model where experimentally determined values were not available or to provide an objective alternative to experimental data. The Advanced Compartmental Absorption and Transit model 14 was used to describe the in vivo dissolution and intestinal absorption of bisoprolol after oral administration. Physiologies, including age and body weight, matched tissue volumes, and blood perfusion rates were generated by the Population Estimates for Age‐Related Physiology module. 14 The PBPKPlus™ module was used to simulate systemic distribution and elimination of bisoprolol. The Population Simulator mode was used to run crossover virtual BE trials.
Physicochemical and biopharmaceutical parameters
The physicochemical and biopharmaceutical properties for bisoprolol were defined using a combination of in silico estimates, measured in vitro data obtained from the literature and/or Merck, and fitted values. The values of bisoprolol parameters are listed in Table 1.
TABLE 1.
Key physicochemical and biopharmaceutical parameters for bisoprolol used in GastroPlus® simulations
| Parameter | Value | Reference |
|---|---|---|
| Log of the octanol‐water partition coefficient (LogP) | 2.15 | 29 |
| Acid dissociation constant (pKa) | Base: 9.57 | 29 |
| Reference solubility, mg/mL | 2.24 @ pH 10.7 | 30 |
| Human effective jejunal permeability (P eff), cm/s | 3.28 × 10−4 | 17 |
| Drug particle density, g/mL | 1.20 | GastroPlus® default value |
| Particle size, µm | 25 | GastroPlus® default value |
| Diffusion coefficient, cm2/s | 0.66 × 10−5 | ADMET Predictor® (Simulations Plus, Inc.) |
| Blood:plasma concentration ratio (Rbp) | 1.10 | Optimized |
| Fraction unbound in plasma (fup) | 70.0% | 31 |
| Adjusted fup a | 69.7% | GastroPlus® algorithm |
Adjusted fup was calculated from experimental fup and logD @ pH = 7.4 using the default. GastroPlus® equation 14 was used in the simulation and tissue:plasma partition coefficient calculation in place of fup.
In vitro dissolution data
The analytical method employed to test dissolution of the bisoprolol product used a paddle (European Pharmacopoeia apparatus 2) at a rotation speed of 100 rpm in different media at 37.0°C (±0.5°C): simulated gastric fluid without enzymes and dissolution mediums pH 1.2, pH 4.5, and pH 6.8. Dissolution data of bisoprolol batches used in clinical trials are listed in Table S1.
Clinical data
Clinical studies used for model development and verification/validation are summarized in Table S2. Studies used healthy subjects with intravenous and oral doses ranging from 5–40 mg. Published data were used for model development, 3 , 9 , 10 , 11 , 12 , 13 and data from clinical studies (ALO‐P8‐481, ESO‐P0‐180, EMR200006‐001, CAEP 43.001.15) were used for model verification/validation.
Bisoprolol PBPK model in healthy volunteers
The workflow for the PBBM modeling and simulation is shown in the supplementary materials (Figure S1).
Systemic disposition of bisoprolol was simulated with a full PBPK model. Individual tissues were modeled as perfusion limited, and drug tissue:plasma partition coefficients (Kps) were estimated using the default Lukacova method from drug (Log of the octanol‐water partition coefficient, acid dissociation constant [pKa], fraction unbound in plasma, and R bp [blood to plasma concentration ratio]) and system‐specific (tissue composition) properties. 14 , 15 Intravenous plasma data from the literature 3 were used to calibrate volume of distribution at steady state (V ss). Due to lack of information, R bp was fitted (1.1) to account for the effect of lysosomal trapping on V ss following intravenous administration.
The baseline model was developed using default built‐in fasted physiology in human. 14 The GastroPlus® default Johnson model 16 was used to model drug dissolution in individual intestinal compartments. Default values were used for particle size (25.0 µm), shape factor (1), and density (1.20 gm/ml). For IVIVR and virtual BE trials, single Weibull parameters fitted to in vitro dissolution data were used as in vivo dissolution input.
Intestinal absorption of bisoprolol was modeled as passive diffusion (transcellular and paracellular). The human effective jejunal permeability of 3.28 × 10−4 cm/s was obtained by converting the experimental Caco‐2 value of 2 × 10−5 cm/s (apical to basolateral) from literature. 17
To explain the observed delay in the time of Cmax (Tmax), lysosomal trapping in enterocytes was implemented. To this end, fraction unbound in enterocytes (fuent) was fitted to 5% (default value = 100%) using available observed Cp‐time profiles after oral administration. The likelihood of lysosomal trapping was verified using MembranePlusTM (Simulations Plus, Inc.) software. Gut first‐pass effect was not included in the model because it is negligible. 4 In vivo PK study showed that 50% drug undergoes metabolism while the remaining bisoprolol is excreted unchanged in urine. 4 The total systemic clearance included metabolic clearance in liver and renal excretion. In vitro clearance 18 was not predictive of in vivo clearance and therefore liver clearance was scaled to match in vivo Cp‐time profiles. Renal excretion was modeled as fraction of kidney blood flow method, where the fraction of 0.12 was fitted to match urinary excretion of unchanged bisoprolol (~50%) as reported in literature. 3
In vitro–in vivo relationship (IVIVR)
Merck's in vitro dissolution data measured in 0.005 N HCL, 0.1 N HCL/pH 1.2, pH 4.5, and 6.8 media were similar and available for all batches used in clinical studies (Table S1).
To build the IVIVR, deconvolved in vivo dissolution profiles were obtained using observed average Cp‐time profiles of each clinical study. These in vivo dissolution profiles were visually compared with all in vitro dissolution data of corresponding batches. The relationship was established based on the in vitro/in vivo data that met the %PE criterion for validation. %PE was calculated by comparing the observed values with simulated Cmax and AUC by using in vitro dissolution profiles measured at lower and higher pH values (at 1.2/0.1 N HCL and 6.8 media) only. Several in vitro dissolution profiles were similar irrespective of pH, and therefore profiles measured only at lower and higher pH values were chosen. Single Weibull parameters fitted to in vitro dissolution data measured in 0.1 N HCL/pH 1.2 and pH 6.8 were provided as input.
In the absence of an observed Cp‐time profile for batch 231975, which had the slowest observed dissolution, the safe space IVIVR was established by predicting the Cp‐time profile of this batch using an in vitro dissolution profile measured at pH 6.8 as input and comparing it to thr reference formulation (PK data from clinical batch 5080504 used in study ALO‐P8‐481).
Virtual populations
To incorporate intersubject variability, the Population Simulator mode was used, which generates virtual subjects by random sampling of selected variables such as physiological, PK parameters, and formulation. Each variable is defined as means and lower and upper limits along with coefficients of variation and distributions (normal, log‐normal, or uniform). 14
Individual subject data (N = 27) from study ALO‐P8‐481 were used to generate virtual populations with matching mean demographics (White males, age = 33.0 years, body weight = 75.0 kg, body mass index = 24.3 kg/m2) for virtual BE evaluations. Default percent coefficient of variation (CV%) was used for all parameters except stomach and intestinal transit time, muscle Kp, and fuent to capture variability in the data.
Establishment of dissolution safe space via virtual BE
Clinical batch 5080504 (study ALO‐P8‐481) was selected as the reference formulation because it is representative of the observed mean dissolution profiles for which plasma concentrations (Cp)‐time profiles were available. In vitro dissolution data of the slow dissolution batch (231975 @ pH 6.8) were first used to assess BE by comparing them to the reference formulation. Single Weibull parameters were then fitted to in vitro dissolution data of the slow dissolution batch and used as in vivo dissolution input for the crossover BE simulations.
To determine the dissolution range that allows new batches to continue to meet BE criteria using the bisoprolol formulation as a reference, several hypothetical dissolution profiles were explored in crossover virtual trials to assess their BE with the reference formulation. The hypothetical dissolution profiles were created by lowering the percentage of bisoprolol dissolved per single timepoint including the final timepoint at 45 min. The overall shape of the dissolution profile is maintained to fit to common dissolution profiles of the drug product. This way an expanded safe space was established by extrapolation using the hypothetical dissolution profiles beyond the knowledge space (namely, the observed in vitro and corresponding in vivo data available) allowing for the determination of the slowest dissolution that could fulfill the BE criteria.
RESULTS
The PBPK model was able to reproduce the Cp‐time profile following intravenous administration 3 (Figure 1a,b). The calculated mean V ss was 219 L/kg, which was in agreement with the literature‐reported value of 226 ± 37.0 L/kg (mean ± SD). 3 To account for differences in clearance between studies, bisoprolol in vitro intrinsic clearance 18 was scaled 6‐fold to 13‐fold to match the observed plasma data, which resulted in liver clearance values ranging from 3.16 to 9.69 L/h following a single administration. This range agrees with the literature‐reported values of liver clearance (3.10–13.10 L/h 3 ). Total systemic clearance (including metabolic liver clearance and renal excretion) of analyzed PK data from the literature and clinical studies was estimated to be in the range of 12.4–18.4 L/h. This is in agreement with the literature‐reported range of total systemic clearance (9.00–22.2 L/h 3 ).
FIGURE 1.
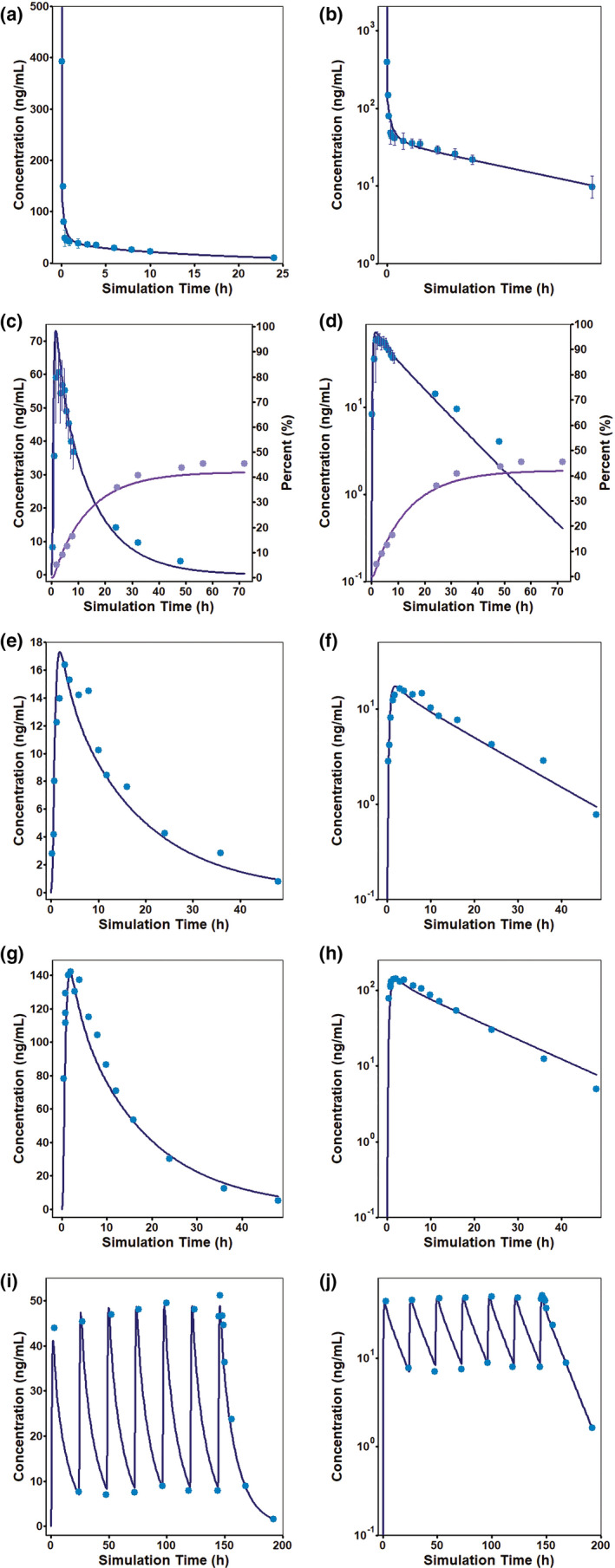
Literature mean observed (symbols) and simulated (lines) bisoprolol plasma concentrations in the fasted state following (a,b) 10 mg intravenous bolus dose, 3 (c,d) a single oral dose of 20 mg solution, 3 (e,f) a single oral dose of a 5 mg immediate release (IR) tablet, 11 (g,h) a single oral dose of a 40 mg (4 × 10) IR tablet, 11 and (i,j) once‐a‐day oral doses of a 10 mg IR tablet for 7 days. 12 Concentrations are shown on linear scales (a,c,e,g,i) as well as on log scales (b,d,f,h,j). Error bars represent the percent coefficient of variation. (c,d) Literature 3 mean observed (symbols) and simulated (lines) bisoprolol urine concentrations. Cumulative amount excreted in urine (purple) are shown as percent of the administered dose (y axis on the right)
Default fasted state stomach transit time (0.25 h) and fitted fuent (5%) were used to model all available Cp‐time profiles after oral dosing from the literature 3 , 9 , 10 , 11 , 12 , 13 and Merck. To verify lysosomal trapping, simulated lysosomal concentration was compared for two pH conditions in MembranePlus. At a lysosomal pH = 4.0, the simulated lysosome concentration was approximately two orders of magnitude higher than the cytoplasm concentration. With the lysosomal pH = 6.5, the accumulation in the lysosomal compartment was reduced to only fivefold above the cytoplasm concentration (data not shown). The simulations suggested lysosomal trapping for bisoprolol. The model estimated complete absorption following instant dissolution after oral administration, which is in agreement with the literature finding that bisoprolol is completely absorbed. 3 It is subject to a very low hepatic first‐pass effect (~10% of dose), and the absolute oral bioavailability of bisoprolol is reported to be 90% after IR solution administration. 3 , 4 The model estimated absolute oral bioavailability of 89%, which is in agreement with the reported value. The reported urinary excretion profile of unchanged bisoprolol after 20 mg IR solution dosing (47.8% ± 10.5%) 3 was accurately captured by the proposed model (estimated urinary excretion ~42%) (Figure 1c,d).
The model (Table 1) accurately explains the observed oral Cp‐time profiles obtained from the literature (Figure 1e–j) and clinical studies conducted by Merck (Figure 2a–h). Simulated bisoprolol oral Cp‐time profiles were within the BE limit of 0.8–1.25 of the clinically observed mean data. Observed and simulated PK parameters (Cmax and area under the concentration‐time curve from time zero to time t) from all studies are summarized in Table 2.
FIGURE 2.
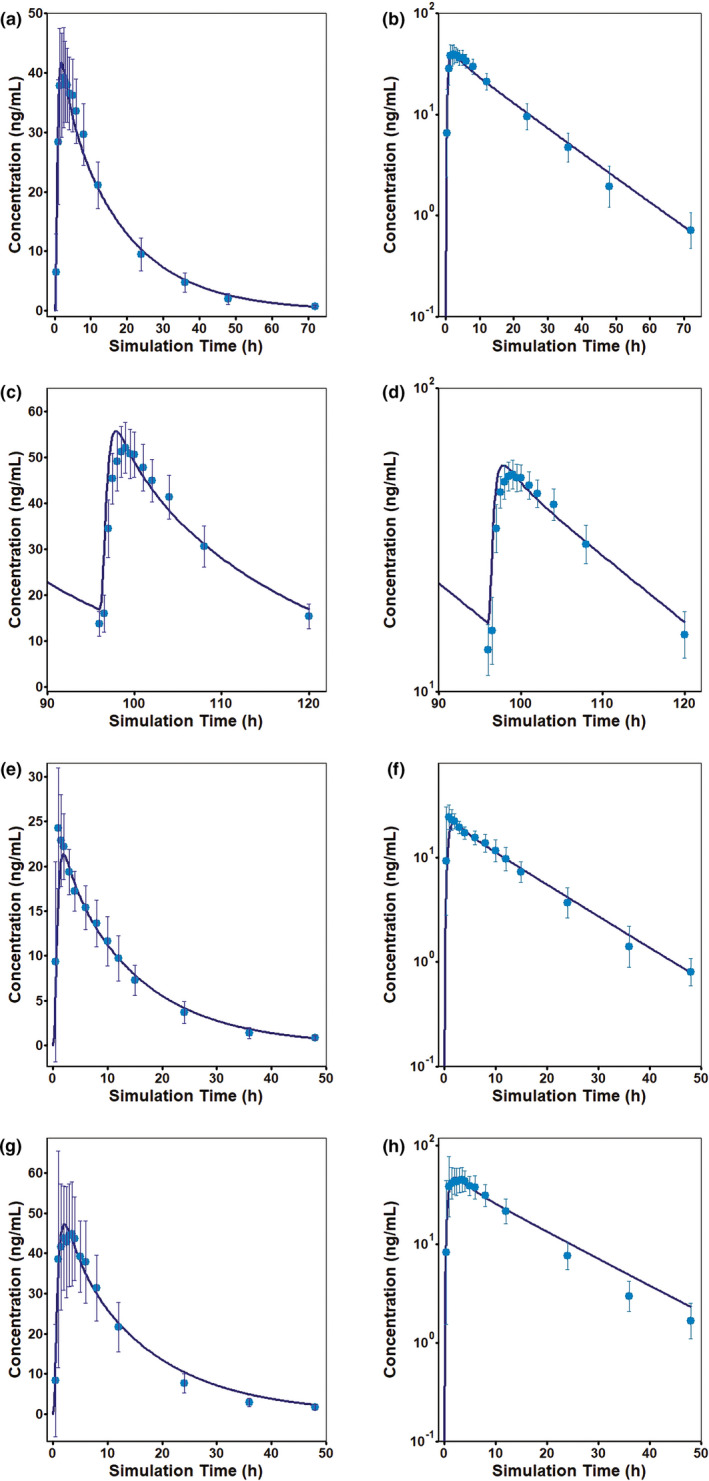
Merck clinical studies of the mean observed (symbols) and simulated (lines) bisoprolol plasma concentrations in the fasted state following (a,b) a single oral dose of a 10 mg immediate release (IR) tablet (study ALO‐P8‐481), (c,d) once‐a‐day doses of a 10 mg IR tablet for 5 days (study ESO‐P0‐180), (e,f) a single oral dose of a 10 mg IR tablet (study EMR200006‐001), and (g,h) a single oral dose of a 10 mg IR tablet (study CAEP 43.001.15). Concentrations are shown on linear scales (a,c,e,g,i) as well as on log scales (b,d,f,h,j). Error bars represent the percent coefficient of variation
TABLE 2.
Simulated versus observed pharmacokinetic parameters of bisoprolol in healthy human subjects
| Dose (mg) | Mean Cmax (ng/mL) | Mean AUC0−t (ng·h/mL) a | Cmax | AUC0−t | Reference | ||
|---|---|---|---|---|---|---|---|
| Observed mean | Simulated mean | Observed mean | Simulated mean | Simulated/observed | Simulated/observed | ||
| 10 | 597.90 | 570.80 | 0.95 | 3 | |||
| 20 | 60.50 | 72.09 | 990.60 | 935.40 | 1.19 | 0.94 | 3 |
| 5 | 16.38 | 17.30 | 293.70 | 260.50 | 1.06 | 0.89 | 11 |
| 10 | 34.60 | 35.36 | 565.80 | 532.30 | 1.02 | 0.94 | 11 |
| 20 | 72.80 | 70.72 | 1099.60 | 1064.60 | 0.97 | 0.97 | 11 |
| 40 | 142.00 | 141.50 | 2238.30 | 2129.10 | 1.00 | 0.95 | 11 |
| 10 | 39.60 | 38.67 | 565.60 | 576.10 | 0.98 | 1.02 | 9 |
| 10 | 51.10 | 48.79 | 778.85 | 650.95 | 0.95 | 0.84 | 12 |
| 10 | 49.30 | 48.84 | 635.10 | 590.87 | 0.99 | 0.93 | 13 |
| 5 | 19.00 | 17.80 | 256.80 | 244.20 | 0.94 | 0.95 | 10 |
| 10 | 39.16 | 41.71 | 697.60 | 694.40 | 1.07 | 1.00 | ALO‐P8‐481 |
| 10 | 52.00 | 55.68 | 765.80 | 779.26 | 1.07 | 1.02 | ESO‐P0‐180 |
| 5 | 24.30 | 21.35 | 297.50 | 300.80 | 0.88 | 1.01 | EMR200006‐001 |
| 10 | 44.89 | 47.24 | 662.00 | 713.90 | 1.05 | 1.08 | CAEP 43.001.15 |
Abbreviation: AUC0–t, area under the concentration‐time curve from time zero to time t; Cmax, maximum plasma concentration.
Last measured plasma concentration timepoint.
Deconvolved in vivo dissolution profiles obtained using the observed average Cp‐time profile of each clinical study were comparable (more than 80% dissolved in 15 min) with the in vitro dissolution data of corresponding batches measured in all different dissolution media (Figure S2).
For IVIVR validation, %PE for simulated Cp‐time profiles based on in vitro dissolution profiles as in vivo input were ≤15% (within the internal validation criteria per IVIVC guidance) as shown in Table S3. These results suggested that in vitro dissolution (measured in 0.005 N HCL or 0.1 N HCL/pH 1.2 and 6.8 media) was predictive of in vivo performance. In addition, comparison of simulated PK with in vivo dissolution calculated based on particle size by the Johnson model and in vitro dissolution data represented by the Weibull function provided equivalent results.
The variability in the observed PK data from study ALO‐P8‐481 was not fully captured with default CV% for all model parameters (Figure 3a,b). Parameter sensitivity analysis (Figure S3) and modeling of individual representative subjects were performed to identify key parameters that could affect PK of bisoprolol. These simulations suggested that gastrointestinal transit times, V ss, and fuent were likely to contribute the most to observed PK variability. Variability in observed PK data from ALO‐P8‐481 was captured reasonably well after modifying CV% for the following parameters: stomach transit time (default = 20 CV%; modified = 50 CV%), intestinal transit time (default = 20 CV%; modified = 50 CV%), muscle Kp (default = 10 CV%; modified = 50 CV%), and fuent (default = 10 CV%; modified = 20 CV%). Simulation results (Figure 3c,d) were bioequivalent to the observed data because point estimates and 90% CIs of the GMR (simulated/observed) of all end points (Cmax and AUC) were within BE limits (data not shown).
FIGURE 3.
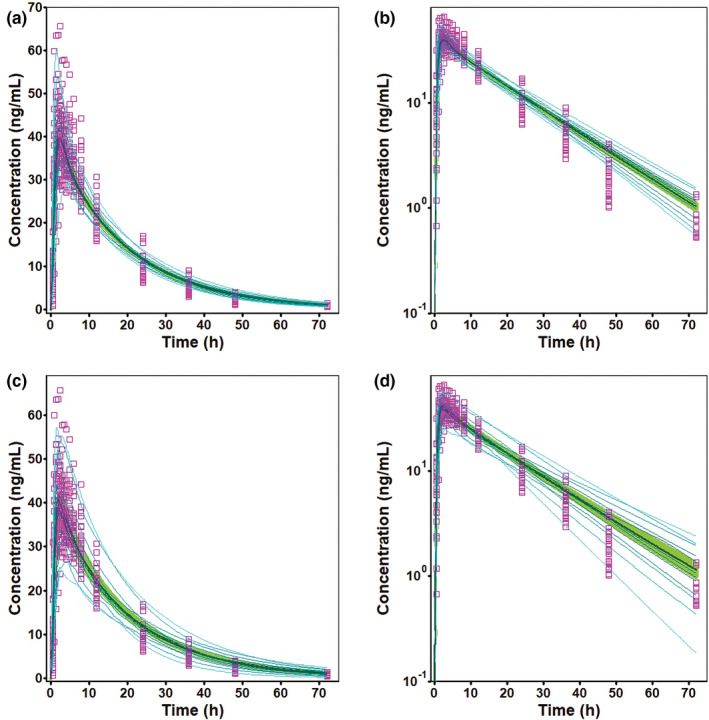
Observed (symbols) (study ALO‐P8‐481) and simulated (lines) bisoprolol plasma concentrations after a single oral dose of a 10 mg immediate release (IR) tablet in the fasted state with default (a,b) and modified (c,d) percent coefficient of variation in population simulation. The pink square symbols are the observed plasma concentration data in 27 subjects, and the solid line corresponds to the mean simulated plasma concentrations; 90% CI is displayed as a green band, and the light blue lines represent the probability contours. Concentrations are shown on linear scales (a,c) as well as on log scales (b,d)
Simulated systemic exposure from 10 virtual trials for the batch with slow dissolution (231975 @ pH 6.8) was within the BE limits of the observed PK data from the clinical batch used in ALO‐P8‐481 (Table S4). Similarly, the crossover virtual trial demonstrated that the point estimates and 90% CIs of the GMR of the simulated PK of the fast‐dissolving batch (229619 @ pH 6.8) were within the BE limits of 0.8–1.25 of the observed PK parameters of the clinical batch used in ALO‐P8‐481 (Table 3).
TABLE 3.
Point estimates and 90% CIs for pharmacokinetics of batch 229619 @ pH 6.8 (fast‐dissolving batch) and hypothetical dissolution profiles from a single crossover virtual trial
| Dissolution profiles | Cmax (ng/ml) | AUC0−t (ng·h/ml) a | AUC0−inf (ng·h/ml) | |||
|---|---|---|---|---|---|---|
| Point estimates b | 90% CI | Point estimates b | 90% CI | Point estimates a | 90% CI | |
|
Batch 229619 @ pH 6.8 (fast) |
92.36 | 87.04–98.73 | 107.70 | 100.72–113.11 | 106.80 | 99.75–112.13 |
| Hypothetical example 1 | 87.05 | 82.11–92.29 | 108.70 | 102.74–114.97 | 107.40 | 101.50–113.59 |
| Hypothetical example 2 | 85.86 | 80.69–91.82 | 108.40 | 101.42–113.89 | 107.00 | 99.98–112.42 |
| Hypothetical example 3 | 84.10 | 78.96–89.95 | 107.30 | 100.29–112.61 | 105.80 | 98.78–111.07 |
Abbreviations: AUC0–inf, area under the concentration‐time curve from time zero to infinity; AUC0–t, area under the concentration‐time curve from time zero to time t; CI, confidence interval; Cmax, maximum plasma concentration.
t = 72 h.
Calculated (geometric mean test/reference × 100).
To expand the safe space via virtual BE trials, several hypothetical dissolution profiles were explored (Figure 4). These profiles had a mean dissolution of 72% (example 1), 70% (example 2), and 69% (example 3) at 15 min and 81% (example 1), 79.5% (example 2), and 78% (example 3) at 30 min. The hypothetical profile with 70% and 79.5% dissolved at 15 and 30 min, respectively (example 2), was the slowest dissolution profile that was within BE criteria, and the profile with 69% dissolved at 15 min and 78% dissolved at 30 min (example 3) was marginally outside of the BE limits (Table 3). These simulations suggest that provided the shapes of the profiles are comparable, minimum 70% dissolution in 15 min and 79.5% at 30 min is sufficient for new batches to be bioequivalent with the current formulation.
FIGURE 4.
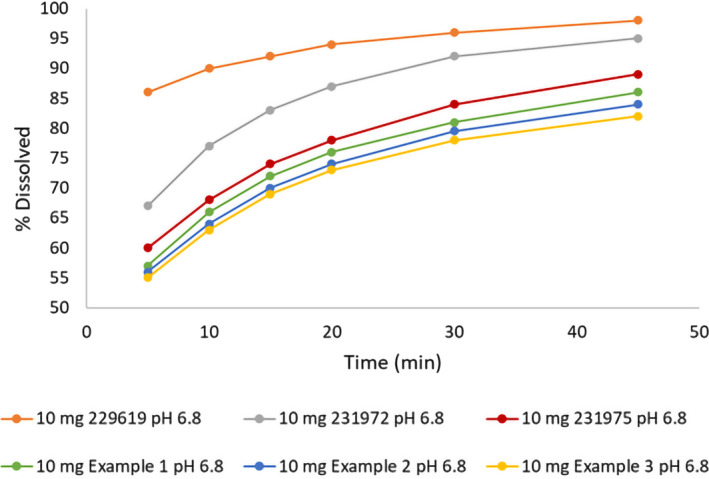
Experimental (the three fastest) and hypothetical examples (the three slowest) of the dissolution profiles for a 10 mg immediate release tablet
DISCUSSION
IR drug products composed of BCS class I compounds, which rapidly dissolve in mild conditions, are expected to have fast oral absorption resulting in short Tmax values. However, bisoprolol, a highly soluble and permeable compound, has a Tmax of 2.70 ± 1.60 h (mean ± SD) following an oral solution administration. 4 This phenomenon can be explained by lysosomal sequestration 19 , 20 in the enterocytes’ lysosomes—acidic organelles (pH 4.5), which serve as reservoirs where lipophilic amines with pKa > 6 can easily diffuse and get positively charged and subsequently sequestered, resulting in a slower entry into the portal vein and effectively longer Tmax. Kazmi et al. 19 showed evidence that propranolol, very similar structurally and property‐wise to bisoprolol, undergoes lysosomal trapping and also has a Tmax similar to bisoprolol (~2 h). Also, structurally alike labetalol was marked as possible to undergo lysosomal trapping. 19 The effects of lysosomal trapping in the enterocytes were simulated by reducing fuent to 5% (default = 100%). The reduction of fuent effectively reduces the rate of mass transfer from inside of the enterocytes across the basolateral membrane into the portal vein. The model with fitted fuent allowed reproduction of the observed Tmax. Other possible reasons for delayed Tmax, including possible involvement of transporters in absorption, were also explored. The model with P‐glycoprotein efflux transporter at the apical side in gut produced later Tmax but resulted in ~72% bioavailability (the reported bioavailability for bisoprolol is >90% 4 ). There are published bisoprolol PBPK models where the longer Tmax was accounted for by their authors with prolonged stomach‐emptying time 21 or fitted—lower than the value obtained from the in vitro measurement—permeability. 22 We strongly believe that the model with lysosomal trapping is the most probable when considering bisoprolol's physicochemical properties.
Once the predictive performance was verified, a PBBM‐based IVIVR was established to assess the impact of the in vitro dissolution data of different bisoprolol batches on simulated PK. Clinical bisoprolol batches used in several PK studies conducted by Merck had fast dissolution and hence very narrow safe space was determined based on knowledge space. In the absence of a measured Cp‐time profile for the slow dissolution batch (231975), an extrapolated safe space was created using the observed data (ALO‐P8‐481’s study) to (1) expand the safe space and (2) determine the clinical impact of batches with slower dissolution profiles.
Recently, the PBBM approach has been widely used to justify BE waivers. In this regard, the mechanistic absorption model (MAM) approach was used to assess the impact of in vitro dissolution on in vivo performance of Zurampic® (lesinurad) tablets and was further used to explore dissolution space using a bioinequivalent in vivo batch and theoretical dissolution profile. 23 The model proposed dissolution specifications of Q = 80% in 30 min for drug product batches to be bioequivalent with the clinical reference batch. Modeling results were submitted to the US FDA and resulted in the acceptance of the proposed specifications for dissolution and particle size. Likewise, Kesisoglou et al. 24 studied the impact of dissolution rate differences on the bioavailability of losartan tablets. In this case study, similar in vivo performance of slow and fast tablets of losartan compared with the target formulation was demonstrated using MAM by incorporating the available in vitro dissolution data. 24 A recent research article published by the US FDA demonstrated the utility of the PBPK absorption modeling approach to set clinically relevant dissolution “extrapolated” safe space for oseltamivir oral dosage forms with theoretical dissolution profiles as inputs using a virtual BE simulation in different age groups. 25 Similarly, virtual BE assessment through a robust PBBM model approach for several drugs have been discussed in the literature. 26 , 27 The literature has shown irrelevance of in vitro dissolution for dextromethorphan, which is subjected to lysosomal trapping. 28
The Cp‐time profiles from batch 231975 with the slowest measured dissolution were not available. From the available PK data sets (N = 4), study ALO‐P8‐481’s Cp‐time profiles with the slowest dissolution batch (5080504) were chosen as a reference to create a representative virtual population. The same virtual population was used to run crossover virtual BE trials using in vitro data as dissolution input of the batch (231975 @ pH 6.8) with the slowest dissolution. The simulated PK for this batch was within the BE limits of observed PK data from the clinical batch used in study ALO‐P8‐481. The dissolution profile of this batch (5080504) falls between the available dissolution profiles of fast‐dissolving and slow‐dissolving batches. The comparison of in vitro dissolution profiles of different batches is shown in Figure S4.
The validated PBBM model, which was used to run several crossover virtual BE trials, demonstrated that differences in dissolution of fast‐dissolving and slow‐dissolving batches did not impact the PK of bisoprolol.
Wider safe space (extrapolated) was explored based on virtual BE trials using dissolution profiles outside (lower bound) of the knowledge space. The results of the virtual BE trials for the hypothetical dissolution profiles showed that the PK of bisoprolol was not affected by differences in dissolution, which are likely to be expected in bisoprolol batches. The lower bounds of the dissolution safe space established via virtual BE trials using the observed clinical batch 5080504 as a reference were 70% dissolution in 15 min and 79.5% in 30 min. It should be noted that if BE analysis is performed between batch 231975 as a reference (with the slowest measured dissolution closely meeting the dissolution criterion per the US FDA 2018 guidance for highly soluble compounds: Q = 80% dissolved in 30 min) and hypothetical profiles (e.g., with slower dissolution compared with batch 231975), the resulting safe space “dimensions” could be much wider (e.g., one could justify wider dissolution acceptance criterion for this drug product). Given bisoprolol's highly soluble and permeable profile, the use of extrapolated dissolution safe space to define the edge of failure is deemed low risk, especially considering that the proposed lower dissolution boundary corresponds to a dissolution performance of 80% at 30 min.
The present study illustrates that the PBBM modeling and simulation approach provides an opportunity to build enhanced drug product understanding and support flexibility in regulatory assessment (e.g., reducing the need for clinical studies). The study showed that there was no impact of in vitro drug release on in vivo performance over a wide‐defined range of bisoprolol dissolution. This work demonstrated the possibility of developing an approach for requesting a biowaiver for a bisoprolol drug product that exhibits varied release properties. This analysis suggests that extrapolation outside the knowledge space may be extended to other BCS class I compounds with well‐defined dissolutions (e.g., rapid dissolution) and well‐characterized absorption properties (e.g., no change in excipients) because the risk for lack of in vivo performance similarity is very low.
CONFLICT OF INTEREST
J.S.M. and G.F. are employees of Simulations Plus, Inc., developer GastroPlus®. M.B., P.K., and S.A.P. are employed by Merck KGaA, producer Concor®.
AUTHOR CONTRIBUTIONS
J.S.M. wrote the manuscript. J.S.M., G.F., M.B., P.K., and S.A.P. designed the research. J.S.M. performed the research. J.S.M., G.F., M.B., P.K., and S.A.P. analyzed the data.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Table S1
Table S2
Table S3
Table S4
ACKNOWLEDGMENTS
The authors thank Sandra Suarez‐Sharp for her support and scientific discussions. The authors are grateful to Lisa Behr and Victor Aguilar for their help with figure preparation.
Funding information
The research that was part of this publication was funded by Merck KGaA. Simulations Plus, Inc. has covered the expenses of employee time spent on writing this publication.
REFERENCES
- 1. US Food and Drug Administration . Impact story: modeling tools could modernize generic drug development. https://www.fda.gov/drugs/regulatory‐science‐action/impact‐story‐modeling‐tools‐could‐modernize‐generic‐drug‐development. Accessed July 19, 2020.
- 2. Pepin XJH, Parrott N, Dressman J, et al. Current state and future expectations of translational modeling strategies to support drug product development, manufacturing changes and controls: a workshop summary report. J Pharm Sci. 2020;110:555‐566. [DOI] [PubMed] [Google Scholar]
- 3. Leopold G, Pabst J, Ungethum W, Buhring KU. Basic pharmacokinetics of bisoprolol, a new highly beta 1‐selective adrenoceptor antagonist. J Clin Pharmacol. 1986;26:616‐621. [DOI] [PubMed] [Google Scholar]
- 4. Leopold G. Balanced pharmacokinetics and metabolism of bisoprolol. J Cardiovasc Pharmacol. 1986;8(suppl 11):S16‐S20. [DOI] [PubMed] [Google Scholar]
- 5. Bethge H, Leopold G, Wagner G. Bisoprolol in angina pectoris. Cardiovasc Drug Rev. 1991;9:110‐122. [Google Scholar]
- 6. Ramirez E, Laosa O, Guerra P, et al. Acceptability and characteristics of 124 human bioequivalence studies with active substances classified according to the Biopharmaceutic Classification System. Br J Clin Pharmacol. 2010;70:694‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. European Medicines Agency . Guideline on the investigation of bioequivalence. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐investigation‐bioequivalence‐rev1_en.pdf. Accessed June 6, 2020. [DOI] [PubMed]
- 8. US Food and Drug Administration . Bioavailability studies submitted in NDAS or INDS—general considerations guidance for industry. https://www.fda.gov/media/121311/download. Accessed February 9, 2021.
- 9. Buś‐Kwaśnik K, Rudzki PJ, Ksycińska H, et al. Bioequivalence study of 2.5 mg film‐coated bisoprolol tablets in healthy volunteers. Kardiol Pol. 2017;75:48‐54. [DOI] [PubMed] [Google Scholar]
- 10. Ding L, Zhou X, Guo X, et al. LC‐ESI‐MS method for the determination of bisoprolol in human plasma. J Pharm Biomed Anal. 2007;44:520‐525. [DOI] [PubMed] [Google Scholar]
- 11. Dutta A, Lanc R, Begg E, et al. Dose proportionality of bisoprolol enantiomers in humans after oral administration of the racemate. J Clin Pharmacol. 1994;34:829‐836. [DOI] [PubMed] [Google Scholar]
- 12. Kirch W, Rose I, Demers HG, Leopold G, Pabst J, Ohnhaus EE. Pharmacokinetics of bisoprolol during repeated oral administration to healthy volunteers and patients with kidney or liver disease. Clin Pharmacokinet. 1987;13:110‐117. [DOI] [PubMed] [Google Scholar]
- 13. Kirch W, Rose I, Klingmann I, Pabst J, Ohnhaus EE. Interaction of bisoprolol with cimetidine and rifampicin. Eur J Clin Pharmacol. 1986;31:59‐62. [DOI] [PubMed] [Google Scholar]
- 14. Simulations Plus, Inc . GastroPlus® User Manual for Version 9.7. Lancaster, CA: Simulations Plus, Inc; 2019. [Google Scholar]
- 15. Lukacova V, Parrott N, Lave T, Fraczkiewicz G, Bolger M, Woltosz W. General approach to calculation of tissue:plasma partition coefficients for physiologically based pharmacokinetic (PBPK) modeling. Paper presented at: American Association of Pharmaceutical Scientists Annual Meeting; November 16‐20, 2008; Atlanta, GA.
- 16. Lu AT, Frisella ME, Johnson KC. Dissolution modeling: factors affecting the dissolution rates of polydisperse powders. Pharm Res. 1993;10:1308‐1314. [DOI] [PubMed] [Google Scholar]
- 17. Charoo NA, Shamsher AAA, Lian LY, et al. Biowaiver monograph for immediate‐release solid oral dosage forms: bisoprolol fumarate. J Pharm Sci. 2014;103:378‐391. [DOI] [PubMed] [Google Scholar]
- 18. Horikiri Y, Suzuki T, Mizobe M. Stereoselective metabolism of bisoprolol enantiomers in dogs and humans. Life Sci. 1998;63:1097‐1108. [DOI] [PubMed] [Google Scholar]
- 19. Kazmi F, Hensley T, Pope C, et al. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N‐4 cells). Drug Metab Dispos. 2013;41:897‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nadanaciva S, Lu S, Gebhard DF, Jessen BA, Pennie WD, Will Y. A high content screening assay for identifying lysosomotropic compounds. Toxicol In Vitro. 2011;25:715‐723. [DOI] [PubMed] [Google Scholar]
- 21. Li G‐F, Wang K, Chen R, Zhao H‐R, Yang J, Zheng Q‐S. Simulation of the pharmacokinetics of bisoprolol in healthy adults and patients with impaired renal function using whole‐body physiologically based pharmacokinetic modeling. Acta Pharmacol Sin. 2012;33:1359‐1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hansmann S, Darwich A, Margolskee A, Aarons L, Dressman J. Forecasting oral absorption across biopharmaceutics classification system classes with physiologically based pharmacokinetic models. J Pharm Pharmacol. 2016;68:1501‐1515. [DOI] [PubMed] [Google Scholar]
- 23. Pepin XJH, Flanagan TR, Holt DJ, Eidelman A, Treacy D, Rowlings CE. Justification of drug product dissolution rate and drug substance particle size specifications based on absorption PBPK modeling for lesinurad immediate release tablets. Mol Pharm. 2016;13:3256‐3269. [DOI] [PubMed] [Google Scholar]
- 24. Kesisoglou F, Mitra A. Application of absorption modeling in rational design of drug product under quality‐by‐design paradigm. AAPS J. 2015;17:1224‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miao L, Mousa YM, Zhao L, Raines K, Seo P, Wu F. Using a physiologically based pharmacokinetic absorption model to establish dissolution bioequivalence safe space for oseltamivir in adult and pediatric populations. AAPS J. 2020;22:107. [DOI] [PubMed] [Google Scholar]
- 26. Li X, Yang Y, Zhang YU, et al. Justification of biowaiver and dissolution rate specifications for piroxicam immediate release products based on physiologically based pharmacokinetic modeling: an in‐depth analysis. Molec Pharmaceut. 2019;16:3780‐3790. [DOI] [PubMed] [Google Scholar]
- 27. Zhang S, Fang M, Zhang Q, Li X, Zhang T. Evaluating the bioequivalence of metronidazole tablets and analyzing the effect of in vitro dissolution on in vivo absorption based on PBPK modeling. Drug Dev Ind Pharm 2019;45:1646‐1653. [DOI] [PubMed] [Google Scholar]
- 28. Bolger MB, Macwan JS, Sarfraz M, Almukainzi M, Lobenberg R. The irrelevance of in vitro dissolution in setting product specifications for drugs like dextromethorphan that are subject to lysosomal trapping. J Pharm Sci. 2019;108:268‐278. [DOI] [PubMed] [Google Scholar]
- 29. Cheymol G, Poirier JM, Carrupt PA, et al. Pharmacokinetics of beta‐adrenoceptor blockers in obese and normal volunteers. Br J Clin Pharmacol. 1997;43:563‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meylan WM, Howard PH, Boethling RS. Improved method for estimating water solubility from octanol/water partition coefficient. Environ Toxicol Chem. 1996;15:100‐106. [Google Scholar]
- 31. Bühring KU, Sailer H, Faro H‐P, Leopold G, Pabst J, Garbe A. Pharmacokinetics and metabolism of bisoprolol‐14C in three animal species and in humans. J Cardiovasc Pharmacol. 1986;8(Suppl 11):S21‐S28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Table S1
Table S2
Table S3
Table S4