

Abstract
Ivosidenib is a potent, targeted, orally active, small‐molecule inhibitor of mutant isocitrate dehydrogenase 1 (IDH1) that has been approved in the United States for the treatment of adults with newly diagnosed acute myeloid leukemia (AML) who are greater than or equal to 75 years of age or ineligible for intensive chemotherapy, and those with relapsed or refractory AML, with a susceptible IDH1 mutation. Ivosidenib is an inducer of the CYP2B6, CYP2C8, CYP2C9, and CYP3A4 and an inhibitor of P‐glycoprotein (P‐gp), organic anion transporting polypeptide‐1B1/1B3 (OATP1B1/1B3), and organic anion transporter‐3 (OAT3) in vitro. A physiologically‐based pharmacokinetic (PK) model was developed to predict drug‐drug interactions (DDIs) of ivosidenib in patients with AML. The in vivo CYP3A4 induction effect of ivosidenib was quantified using 4β‐hydroxycholesterol and was subsequently verified with the PK data from an ivosidenib and venetoclax combination study. The verified model was prospectively applied to assess the effect of multiple doses of ivosidenib on a sensitive CYP3A4 substrate, midazolam. The simulated midazolam geometric mean area under the curve (AUC) and maximum plasma concentration (Cmax) ratios were 0.18 and 0.27, respectively, suggesting ivosidenib is a strong inducer. The model was also used to predict the DDIs of ivosidenib with CYP2B6, CYP2C8, CYP2C9, P‐gp, OATP1B1/1B3, and OAT3 substrates. The AUC ratios following multiple doses of ivosidenib and a single dose of CYP2B6 (bupropion), CYP2C8 (repaglinide), CYP2C9 (warfarin), P‐gp (digoxin), OATP1B1/1B3 (rosuvastatin), and OAT3 (methotrexate) substrates were 0.90, 0.52, 0.84, 1.01, 1.02, and 1.27, respectively. Finally, in accordance with regulatory guidelines, the Simcyp modeling platform was qualified to predict CYP3A4 induction using known inducers and sensitive substrates.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The physiologically‐based pharmacokinetic (PBPK) model was developed previously to assess the drug interaction potential of ivosidenib as a victim.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study aimed to determine the drug interaction potential of ivosidenib as a perpetrator in patients with cancer (acute myeloid leukemia) at clinically relevant exposures.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study illustrates the utility of the PBPK modeling to assess drug‐drug interactions (DDIs) due to induction of CYPs (CYP2B6, CYP2C8, CYP2C9, and CYP3A4) and inhibition of transporters (OATP1B1/1B3 and OAT‐3) by ivosidenib and inform drug labeling. This study also highlights the qualification of Simcyp software in predicting DDIs owing to CYP3A4 induction.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The concepts utilized in this study can be applied to prospectively predict DDIs in patients with cancer using in vitro and healthy participant clinical data.
INTRODUCTION
Isocitrate dehydrogenase 1 and 2 (IDH1/2) are key enzymes involved in cellular metabolism, and mutated forms of these enzymes in cancer produce an oncometabolite, D‐2‐hydroxyglutarate (2‐HG), thought to play a role in the formation and progression of many cancers. 1 IDH1 mutations were reported in ~ 70% of conventional chondrosarcomas, greater than 80% of lower‐grade and secondary glioblastomas, ~ 20% of intrahepatic cholangiocarcinomas, and 6–16% of acute myeloid leukemias (AMLs). 2 Ivosidenib (AG‐120) targets the mutant IDH1 enzyme and prevents accumulation of 2‐HG. 3 Ivosidenib is approved in the United States as a single agent for the treatment of adults with newly diagnosed AML who are greater than or equal to 75 years of age or ineligible for intensive chemotherapy, and those with relapsed or refractory AML, with a susceptible IDH1 mutation. 4
Physiologically‐based pharmacokinetic (PBPK) modeling approaches are emerging as an alternative to clinical studies to quantitatively predict pharmacokinetic (PK)‐based drug interactions. PBPK modeling integrates physicochemical properties of a new chemical entity and key in vitro data with species‐specific parameters to predict drug‐drug interactions (DDIs). 5 PBPK modeling provides benefits by adopting mechanistic approaches, including simultaneous inhibition and induction effects, fraction metabolized in the liver (fm) and gut (fg), and changing the concentrations of substrate and perpetrator, as well as modification of enzyme(s)/transporters. In addition, the PBPK approach allows the evaluation of various doses and dosing schedules to predict the DDI potential for untested scenarios. With appropriate verification and qualification, modeling approaches are being used effectively to support drug labeling. 6 Although cytochrome P450 (CYP) inhibition‐based PBPK modeling approaches are well‐established, examples of induction‐based DDI predictions by PBPK modeling are limited.
Ivosidenib is primarily metabolized by CYP3A4 and is an inducer of multiple CYPs, including CYP3A4, CYP2B6, and CYP2Cs. Ivosidenib is also an inhibitor of P‐glycoprotein (P‐gp), organic anion transporting polypeptide‐1B1/1B3 (OATP1B1/1B3), and organic anion transporter‐3 (OAT3). The apparent clearance (CL/F) in patients with AML was approximately threefold lower than in healthy participants (HPs) after a single oral dose of ivosidenib, and was attributed to a food and dose‐dependent effect on bioavailability and disease or age effect on the systemic clearance (CL) of the drug. 7 , 8 , 9 , 10 In addition, ivosidenib also exhibits autoinduction following multiple‐dose administration to patients with AML. Hence, it is very important to build and verify a model that can capture DDIs at steady‐state. Owing to the difference in CL/F between HPs and patients with AML and lack of multiple dose PKs in HPs, a separate model was developed to accurately capture and verify multiple dose PKs of ivosidenib in the patient population. 10 Development of the model, predictive performance of PKs, and drug interactions as a victim were presented in another publication. 10
The aim of the current the study was to (i) predict the effect of multiple doses of ivosidenib on venetoclax (sensitive CYP3A4 substrate) and verify the model using results from a clinical study; (ii) simulate the magnitude of DDIs between ivosidenib and midazolam (a sensitive CYP3A4 substrate); (iii) evaluate the effect of ivosidenib on the PK of the CYP2B6, CYP2C8, CYP2C9, P‐gp, OATP1B1/1B3, and OAT3 substrates; and (iv) qualify the Simcyp platform for the assessment of CYP3A4 induction predictability using sensitive CYP3A4 substrates and inducers. This article provides a comprehensive summary of simulations of ivosidenib drug interactions as a perpetrator in patients with cancer, and the predictive performance of the Simcyp platform for CYP3A4 induction.
METHODS
In vitro studies
Hepatocyte cultures (3 donors; Xenotech LLC) were treated with ivosidenib (0.05, 0.1, 1, 5, 10, 25, 50, or 90 μM) or positive controls (omeprazole [CYP1A2], phenobarbital [CYP2B6], and rifampicin [CYP3A4, CYP2C8, and CYP2C9]), or a negative control (flumazenil) once daily for 3 consecutive days to assess CYP induction. After 72 h, cell lysates were prepared. Total RNA was isolated and single‐stranded cDNA was prepared from the RNA. 11 Quantitative real‐time reverse transcription polymerase chain reaction (RT‐PCR) was performed on cDNA isolated from the RNA; the relative quantity of the target cDNA versus control cDNA (glyceraldehyde 3‐phosphate dehydrogenase [GAPDH]) was determined by the ΔΔCt method 12 and half‐maximal effective concentration (EC50) and maximum effect (E max) were calculated.
A P‐gp inhibition assay was performed using Caco‐2 cells cultured on 24‐well transwell plates (0.3 to 0.4 × 106 cells). Incubation medium containing solvent control, digoxin (10 µM), control inhibitor (valspodar [1 µM] or verapamil [60 µM], or six concentrations of ivosidenib (0.5, 1, 5, 10, 50, and 100 µM) was added to the donor chamber. Samples were collected (donor: 0 and 120 min; receiver: 120 min) and analyzed. The inhibition of bidirectional permeability (A→B or B→A) of digoxin by ivosidenib was assessed and the half‐maximal inhibitory concentration (IC50) was calculated.
For the transporter inhibition assessment, 11 human embryonic kidney (HEK293) cells (transfected with vectors containing human transporter cDNA) were pre‐incubated with medium containing ivosidenib (0.1, 0.3, 1, 3, 10, 30, and 100 µM for OATP1B1/1B3; 0.1, 0.3, 1, 3, 10, 30, and 65 µM for OAT1/OAT3/OCT2) or positive control (OATPs: rifampicin; OATs: probenecid; and OCT2: quinidine), or solvent control (0.3 ml) for 15 min. The medium was replaced with an incubation medium containing ivosidenib, positive control, or solvent control, and the probe substrates ([3H]‐estradiol‐17β‐glucuronide [OATPs], [3H]‐p‐aminohippurate [OAT1], [3H]‐estrone‐3‐sulfate [OAT‐3], and [14C]‐metformin [OCT2]) and incubated for 2 min for all transporters except OAT1 (1 min). The accumulation of probe substrates was assessed using radioactivity measurements and IC50 values were calculated.
Ivosidenib model development and verification
Full details on the development of the PBPK model have been discussed elsewhere. 10 Briefly, two ivosidenib drug models, one in HPs and one in patients with AML, were developed using Simcyp version V15.1 (Simcyp Ltd., a Certara Company; Figure 1). The predictive performance of the drug patient model was verified with single‐ and multiple‐dose (100 [b.i.d.], 300, 500, 800, 1200 mg once daily [q.d.]) PK of ivosidenib. The autoinduction of CYP3A4 under steady‐state conditions in patients was supported by the in vivo effect of ivosidenib on the hepatic CYP3A4 marker4β‐OHC and its established relationship with CYP3A4 induction. The fm (0.865) was also supported by the multiple‐dose data (the time dependency in ivosidenib CL/F) in patients at different dose levels. PBPK simulations were performed at recommended daily oral dose (500 mg) of ivosidenib. The predicted area under the curve (AUC) and maximum plasma concentration (Cmax) values were within 0.78‐fold and 1.11‐fold of the observations, upon single and multiple doses of 500 mg ivosidenib. The developed model was deemed robust for the subsequent model application at this dose level.
FIGURE 1.
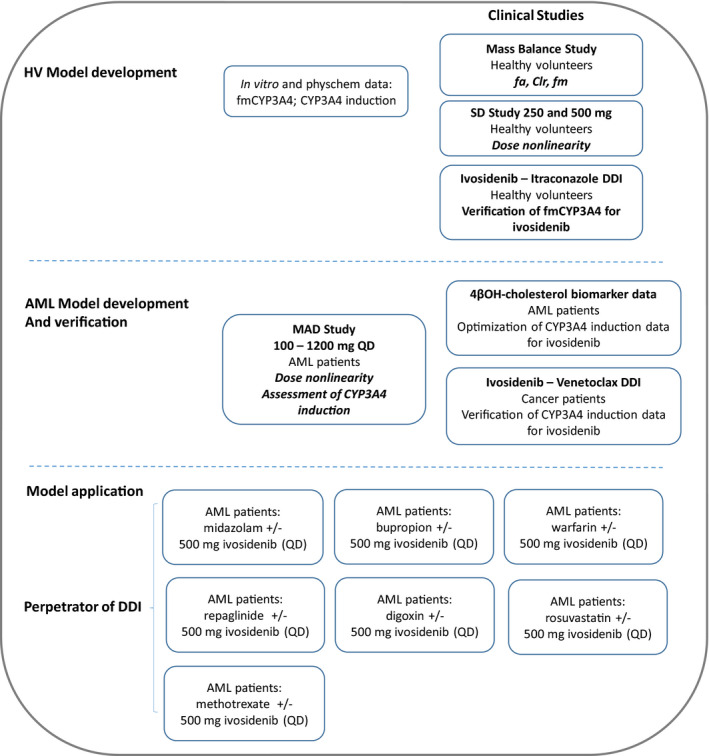
Schematic representation of ivosidenib physiologically‐based pharmacokinetic model. AML, acute myeloid leukemia; DDI, drug‐drug interaction; HV, healthy volunteer; MAD, multiple ascending dose
A sensitivity analysis was performed on the induction parameters observed in vitro in relation to the observed accumulation ratio. For DDI predictions (ivosidenib as a perpetrator), the default Sim‐in vivo (SV)‐bupropion, SV‐repaglinide, Sim‐S‐Warfarin, Sim‐midazolam, SV‐digoxin, and SV‐rosuvastatin models (within Simcyp version 15.1) were used without any modification. A methotrexate PBPK model was developed as an OAT3/ multidrug resistance‐associated protein (MRP) substrate and the contribution of OAT3 to methotrexate disposition was verified against the observed DDI data, with probenecid as a perpetrator.
Venetoclax model development
The venetoclax PBPK model was built on the basis of published data 13 , 14 (Table S1). Simulations were performed with default Simcyp parameter values and have been described previously. 15 Venetoclax PK exposure in healthy subjects was similar to that in patients with cancer, based on a cross‐study comparison of PK and population PK analyses. 16 The input parameters for intrinsic clearance (CLint) were back‐calculated from the observed mean CL/F (14.9 L/h) using the well‐stirred liver model (Equation 1).
| (1) |
CLpo: oral clearance; B:P: blood to plasma ratio; fub: fraction unbound drug in blood; QH: blood flows in the hepatic vein; fg: fraction escaping gut metabolism; CLR: renal clearance; and fa: fraction absorbed.
Venetoclax exhibited a biphasic PK profile. The minimal PBPK model with single adjusting compartment was applied to all simulations of the plasma concentration‐time profiles for venetoclax. The value of volume of distribution at steady state (Vss; =0.85 L/kg) was predicted by method 2 17 with a global scalar of 3. The estimated mean Vss/F from clinical data obtained from patients was 3.67 L/kg. 16 This corresponds to a Vss range of 0.36 to 1.78 L/Kg after correction for F (0.1 to 0.48 assumed). The model was applied to all simulations without any modification using patients receiving multiple oral doses of 400 mg venetoclax once daily in the absence and presence of ivosidenib dosing (500 mg q.d.) for 14 days.
Simcyp platform qualification
The prediction accuracy of CYP3A4‐mediated DDIs by the Simcyp Simulator (version 15.1) was assessed using CYP3A4 substrates (midazolam, nifedipine, triazolam, and alfentanil). These substrates were selected based on their high fmCYP3A4 value (fmCYP3A4 ≥ 90%), as well as the availability of data to verify the fmCYP3A4. The fg of the substrates ranged from 0.52 to 0.71. The strong and moderate inducers of CYP3A4 (rifampicin, carbamazepine, phenobarbital, efavirenz, and rifabutin) were used in this analysis. In Simcyp version 15.1, the in vivo maximum fold induction expressed as a fold over vehicle control; maximum fold induction (Indmax) for rifampicin was set to 16 on the basis of DDI predictions involving 10 intravenous (i.v.) and 19 oral studies (mainly midazolam) of rifampicin. 18 The Indmax values for all other CYP3A4 inducers included were calibrated against the rifampicin in vivo Indmax of 16. Values of the intrinsic turnover of hepatic and gut CYP3A4 (kdeg) used in the simulations were 0.019 h−1 and 0.03 h−1, respectively. 19
The University of Washington Database was used to identify clinical studies in which DDIs involving greater than 20% decreases in the exposure of CYP3A4 substrates in the presence of the moderate and strong inducers mentioned above were identified. 18 The most representative rifampicin DDI studies were included in the current analysis. Twenty studies were identified for inclusion in the analysis: 15 involved rifampicin, 1 involved carbamazepine, 2 involved efavirenz, 1 involved phenobarbital, and 1 involved rifabutin.
The AUC and Cmax ratios were calculated with observed (or predicted) values with or without inducers in a clinical study (or PBPK model). The PBPK model predicted AUC and Cmax ratios were compared with observed mean AUC or Cmax ratios from each clinical study. Several success criteria (1.25‐fold or 2‐fold range) have been used in the literature 20 to assess model accuracy for DDI predictions. The acceptance criteria proposed by Guest et al. is a more sensitive measure of concordance in reflecting absolute changes in AUC, especially when these are small. 21 Equations 2 and 3 were used to calculate the mean‐fold error (MFE) and the root‐mean square error (RMSE), which were used to assess the bias and precision of the predictions, respectively. In the presence of an enzyme inducer, the AUC ratios were less than 1; for the calculation of MFE and RMSE, the reciprocal of this ratio has been used to yield ratios greater than 1 in the presence of an enzyme inducer.
| (2) |
| (3) |
RESULTS
In vitro studies
Ivosidenib showed no significant induction of CYP1A2 mRNA and increased CYP2B6 mRNA levels, with mean Emax and EC50 2.85 ± 1.53 and 2.93 ± 0.65 μM, respectively. Ivosidenib also caused an increase in mean CYP3A4 (Emax: 78.2 ± 113; EC50: 12.5 ± 9.2 μM), CYP2C8 (Emax: 3.79 ± 1.28; EC50: 8.1 ± 3.3 μM), and CYP2C9 (Emax: 3.39 ± 0.63; EC50: 3.23 ± 1.04 μM) mRNA levels. Under the conditions of this study, treatment of cultured human hepatocytes with prototypical inducers, omeprazole (CYP1A2), phenobarbital (CYP2B6), and rifampicin (CYP3A4) caused an increase in mRNA levels of 104 ± 97‐fold, 13 ± 4.7‐fold, and 58.2 ± 64‐fold, respectively. Because the positive control rifampicin failed to induce CYP2C19 mRNA levels in all three hepatocyte cultures, the effect of ivosidenib on CYP2C19 mRNA levels was inconclusive and this phenomenon is known in the literature. 22
Ivosidenib inhibited human P‐gp–mediated transport of digoxin in a concentration‐dependent manner, resulting in an IC50 value of 19.6 μM. Under the conditions examined, ivosidenib appeared to be an inhibitor of OATP1B1‐, OATP1B3‐, and OAT3‐mediated uptake of the probe substrate, resulting in IC50 values of 9.56, 22.8, and 0.322 µM, respectively. In the presence of ivosidenib (up to 65 µM), less than 50% inhibition of organic anion transporter‐1 (OAT1)‐ and organic cation transporter‐2 (OCT2)‐mediated uptake of the probe substrate (IC50 >65 µM) was observed.
Verification of the model and simulation of the effect of ivosidenib on the PK of CYP3A4 substrates owing to induction
CYP3A4 induction parameters were optimized using 4β‐OHC data. 4β‐OHC increased by up to 371% (i.e., nearly 4‐fold) after 28 days of treatment with 100 mg b.i.d. and 100–1200 mg q.d. ivosidenib, 10 , 23 which is comparable with the strong CYP3A4 inducer rifampicin. 24
In the current study, the venetoclax PBPK model was developed to provide additional verification to the model. The predicted mean AUC and Cmax values were 1.04 to 1.11‐fold and 0.78 to 1.0‐fold of the observed values, respectively after 50 mg, 100 mg single dose and 400 mg steady‐state (Table S2). Venetoclax is a CYP3A4 substrate; the clinical effects of the strong CYP3A inhibitor ketoconazole on venetoclax PK have been evaluated in patients with non‐Hodgkin’s lymphoma. 13 The results demonstrated that co‐administration of 400 mg ketoconazole q.d. with a single 50 mg venetoclax dose resulted in a 2.3‐ and 6.4‐fold increase in the venetoclax Cmax and AUC, respectively . Similarly, multiple doses of rifampicin (600 mg q.d.) decreased venetoclax Cmax and AUC by ~ 70% and 80%, respectively. The predicted and observed AUC ratio on co‐administration of ketoconazole and rifampicin were 0.95 and 0.95, respectively (Table S3). Based on the above data, the model was considered verified. The verified model was applied to assess the CYP3A4‐mediated DDIs following multiple doses of venetoclax (400 mg q.d.) as a victim owing to concurrent ivosidenib treatment (500 mg q.d.) in patients with cancer. The predicted AUC and Cmax ratios at steady‐state of venetoclax were 0.27 and 0.40, versus the observed ratios of 0.36 and 0.42, respectively (Table 1 and Figure 2), after a 500 mg q.d. dose of ivosidenib for 14 days.
TABLE 1.
Geometric mean plasma Cmax and AUC (predicted) values and corresponding ratios after single and multiple oral doses of enzyme/transporter substrates in the absence (control) and presence of multiple doses of ivosidenib
| Substrates of enzyme/transporter | Enzyme/ transporter | Cmax (ng/ml) | AUC (ng.hr/ml) | Cmax ratio a | AUC ratio a |
|---|---|---|---|---|---|
| Bupropion | 2B6 | 188.1 | 1357.6 | 0.92 | 0.90 |
| Bupropion + ivosidenib | 173.6 | 1218.1 | |||
| Repaglinide | 2C8 | 4.3 | 10.1 | 0.61 | 0.52 |
| Repaglinide + ivosidenib | 2.6 | 5.2 | |||
| Warfarin | 2C9 | 1033 | 41465 | 0.99 | 0.84 |
| Warfarin + ivosidenib | 1019 | 34856 | |||
| Midazolam | 3A4 | 20.8 | 69.5 | 0.27 | 0.18 |
| Midazolam + ivosidenib | 5.7 | 12.5 | |||
| Venetoclax | 3A4 | 2265 | 37,173 | 0.40 (0.42 b ) | 0.27 (0.36 b ) |
| Venetoclax + ivosidenib | 900 | 9855 | |||
| Digoxin | P‐gp | 2.4 | 33.3 | 1.02 | 1.01 |
| Digoxin + ivosidenib | 2.4 | 33.7 | |||
| Rosuvastatin | OATP1B1 | 9.6 | 111.0 | 1.03 | 1.02 |
| Rosuvastatin + ivosidenib | 9.9 | 113.7 | |||
| Methotrexate | OAT3 | 90,830 | 63,540 | 1.00 | 1.27 |
| Methotrexate + ivosidenib | 90,897 | 80,965 |
Abbreviations: AUC, area under the curve; Cmax, maximum plasma concentration.
AUC and Cmax ratios were calculated by comparing the exposure of a substrate treated with and without ivosidenib.
Observed values.
FIGURE 2.
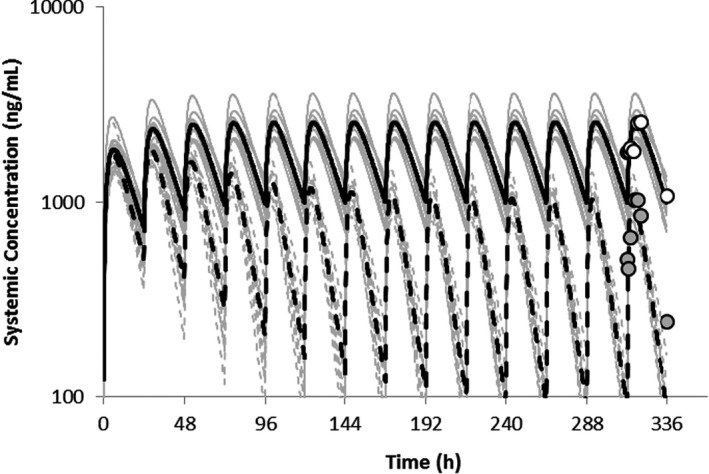
Simulated mean plasma concentration‐time profiles of venetoclax following the administration of multiple doses of 400 mg q.d. in the absence of and on co‐administration with ivosidenib (500 mg q.d.) for 14 days. The gray lines represent the outcomes of simulated individual trials (10 × 7) and the solid black line is the mean data for the simulated population (n = 7)
Following multiple doses of ivosidenib (500 mg q.d.), the CYP3A4‐mediated CLint of midazolam increased by 3.1‐fold (Figure 3a). The interaction of ivosidenib and the sensitive CYP3A4 substrate were simulated with subjects receiving ivosidenib for 15 days and a single oral dose of 5 mg midazolam on day 15 (Figure 3b ). The predicted midazolam geometric mean AUC and Cmax ratios were 0.18 (95% confidence interval [CI]: 0.16–0.20) and 0.27 (95% CI: 0.26–0.29), respectively (Table 1). The significant decrease in midazolam exposure (AUC and Cmax) following co‐administration with multiple doses of ivosidenib was indicative of a strong induction effect of ivosidenib on CYP3A substrates.
FIGURE 3.
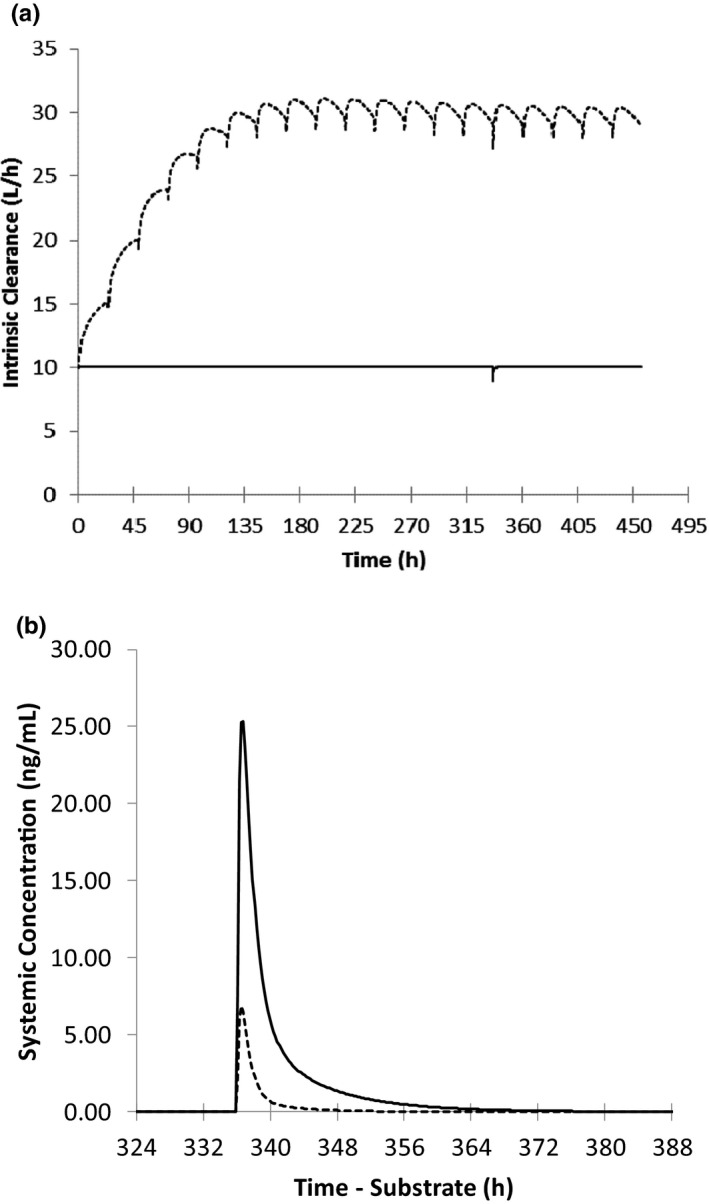
Simulated mean (a) gut intrinsic clearance profile for the formation of 1‐hydroxymidazolam after a single oral dose of 5 mg midazolam in the presence (dashed line) and absence (solid line) of ivosidenib and (b) midazolam after a single oral dose of 5 mg in the presence (dashed line) and absence (solid line) of multiple daily doses of ivosidenib (500 mg q.d. for 19 days)
Simulation of the effect of ivosidenib on CYP2B6, CYP2C8, and CYP2C9 substrates owing to induction
The effect of co‐administration of ivosidenib with CYP2B6, CYP2C8, and CYP2C9 substrates bupropion, repaglinide, and warfarin, respectively, were simulated using the patient PBPK model (Table 1). The predicted bupropion geometric mean AUC and Cmax ratios were 0.90 and 0.92, respectively, after a single oral dose of 150 mg bupropion (on day 15) in the presence and absence of ivosidenib 500 mg (for 19 days; Table 1). Simulated concentration‐time profile of bupropion (single dose) with and without co‐administration of ivosidenib (multiple doses) is shown in Figure 4a. A sensitivity analysis revealed that lowering half‐maximal induction (IndC50) by 10‐fold (Figure 4b) resulted in a predicted bupropion area under the curve ratio (AUCR) of 0.52. The predicted AUC and Cmax ratios of the CYP2C8 substrate repaglinide (0.25 mg orally) following co‐administration with ivosidenib were 0.52 and 0.61, respectively. In addition to CYP2C8 (fm = 61.3%), CYP3A4 (fm = 38.6%) also significantly contributed to the metabolism of repaglinide; the induction effect on repaglinide could be mainly driven by CYP3A4‐mediated induction. To support this, a separate repaglinide DDI simulation was conducted in which the induction of CYP2C8 alone was considered: the simulated AUC and Cmax ratio of repaglinide was 0.91 and 0.95, respectively. Repaglinide also subjects to OATP1B1‐mediated hepatic uptake. However, the inhibitory effect on OATP1B1 is expected to be small as the simulated AUC ratios following multiple doses of ivosidenib (500 mg q.d.) and a single dose of rosuvastatin was 1.02, using the in vitro Ki for OATP1B1. Co‐administration of ivosidenib with the CYP2C9 substrate warfarin (10 mg orally) predicted AUC and Cmax ratios of 0.84 and 0.99, respectively (Table 1). A sensitivity analysis showed that lowering IndC50 by 10‐fold resulted in a predicted warfarin AUCR of 0.42.
FIGURE 4.
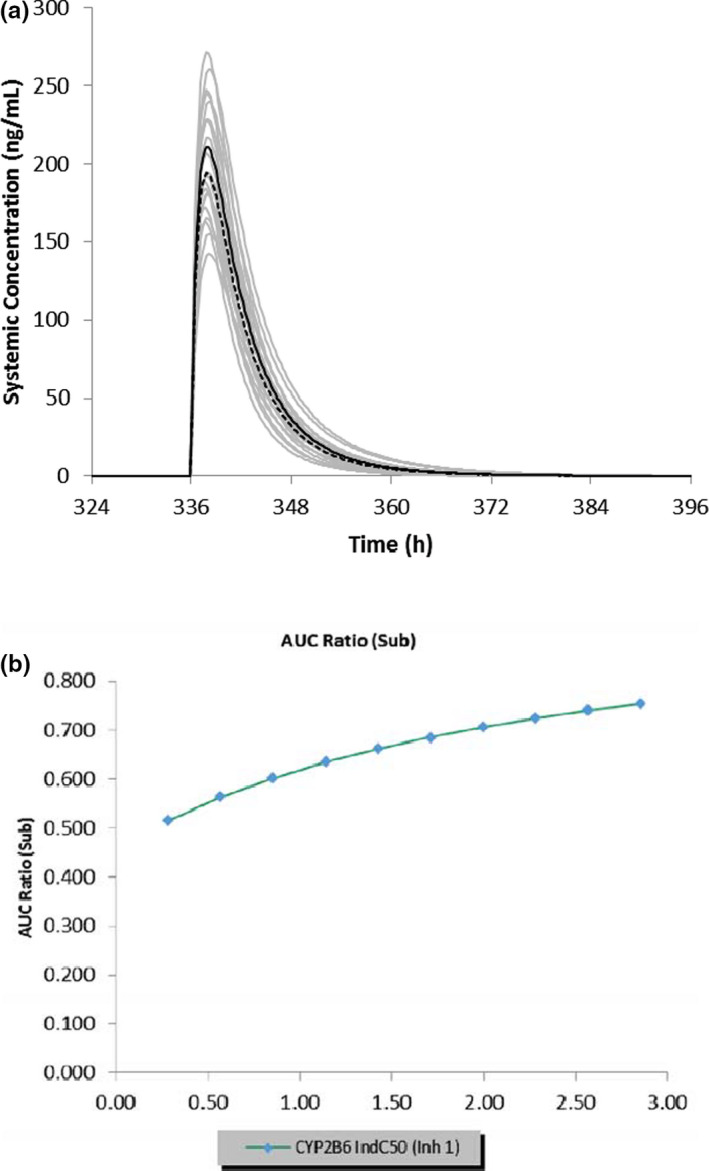
(a) Simulated mean plasma concentration‐time profiles of bupropion after a single oral dose of 150 mg in the presence (dashed line) and absence (solid line) of multiple daily doses of ivosidenib (500 mg q.d. for 19 days). The gray lines represent the outcomes of simulated individual trials (10 × 10) and the solid/dashed black line is the mean data for the simulated population (n = 100). (b) Sensitivity analysis of CYP2B6 IndC50 for ivosidenib in the range of 0.285‐2.85 µM on the predicted bupropion area under the curve (AUC) ratio
Simulation of the effect of ivosidenib on the PK of P‐gp, OATP‐1B1/1B3, and OAT‐3 substrates owing to inhibition
The DDIs following multiple doses (19 days) of 500 mg ivosidenib and a single oral dose (day 15) of 0.5 mg digoxin (P‐gp substrate), 20 mg rosuvastatin (OATP1B1/1B3 substrate), and a 200 mg/m2 i.v. dose of methotrexate (OAT3 substrate) were simulated using a patient (AML) model. Simulated AUC and Cmax ratios of digoxin and rosuvastatin (20 mg) ranged from 1.01 to 1.03 (Table 1). Simulated concentration‐time profile of rosuvastatin (single dose) with and without co‐administration of ivosidenib (multiple doses) is shown in Figure 5a. Sensitivity analysis of both P‐gp and OATPs inhibition constant (Ki) was performed at 1200 mg, the highest dose tested. A sensitivity analysis of P‐gp Ki showed that digoxin AUCR is insensitive to P‐gp Ki greater than 5 µM; when P‐gp Ki was lowered by 15‐fold, the predicted digoxin AUCR was 1.12. Similarly, a sensitivity analysis of OATP1B1/1B3 by a 10‐fold lowering of Ki predicted (Figure 5b) that rosuvastatin AUCR was 1.36. A mechanistic kidney model for methotrexate was developed and verified against clinical PK data arising from i.v. 15–200 mg/m2 and oral 7.5–15 mg doses of methotrexate in patient populations (Table S4). The OAT3‐mediated uptake into the kidney cells and MRP2/4‐mediated efflux into the urine were incorporated into the model using in vitro uptake data and fitted relative expression factor (REF) values of 12 and 20 for OAT3 and MRPs, respectively. The REF of 12 for OAT3 was optimized to recover to plasma PK profiles for methotrexate, and the REF of 20 for MRP was optimized to recover to urine PK profiles for methotrexate. To verify the contribution of OAT3 to methotrexate disposition, the methotrexate model was applied to evaluate probenecid DDI. The predicted methotrexate AUCRs of 1.64‐ to 1.68‐fold, using the lowest reported probenecid OAT3 Ki (=0.76 µM), were comparable with the reported AUCR of 1.57‐fold. The predicted and observed total CL and CLR ratio in the presence and absence of probenecid treatment ranged from 0.9 to 1.04 (Table S5). 25 Based on a sensitivity analysis, the lowest reported probenecid OAT3 Ki (=0.76 µM) was used for DDI simulation. A verified methotrexate model was used to predict ivosidenib DDIs owing to OAT‐3 inhibition. The predicted AUC and Cmax ratios were 1.27 and 1.0, respectively, after a single i.v. administration of methotrexate and multiple doses of ivosidenib in patients with AML. Sensitivity analysis with 10‐fold lower OAT3 Ki, model predicted an increase of exposure of methotrexate by 60% suggesting a weak interaction of ivosidenib with OAT3 substrates. 26
FIGURE 5.
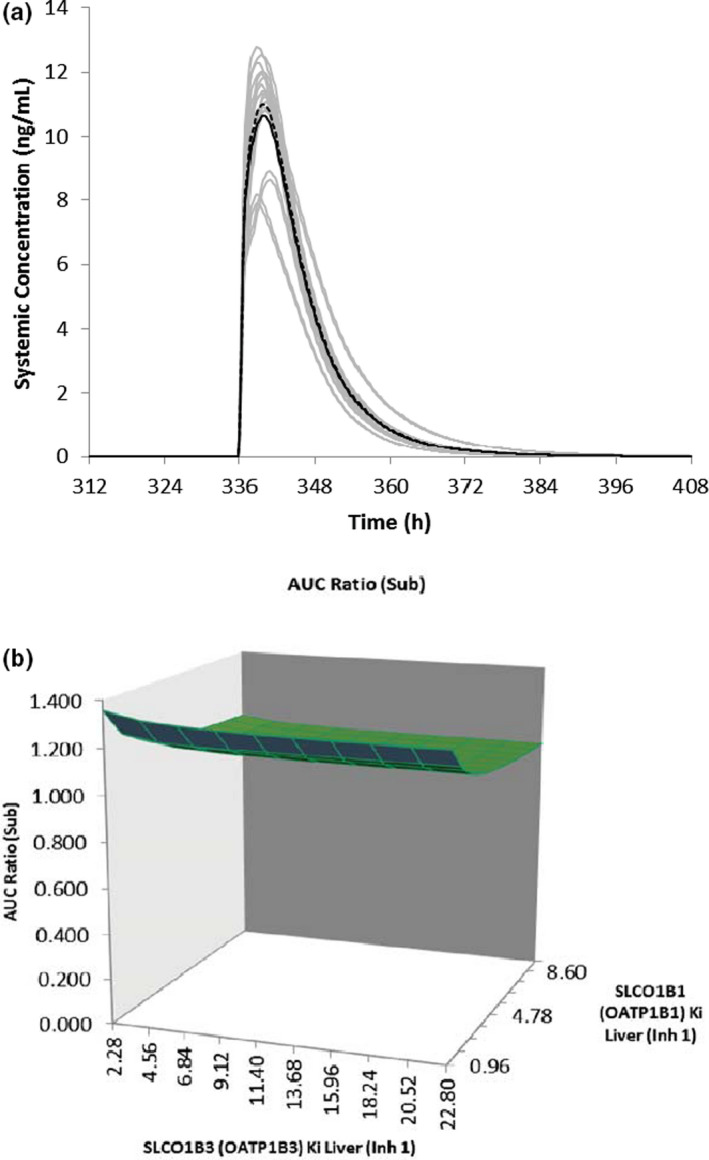
(a) Simulated mean plasma concentration‐time profiles of rosuvastatin after a single oral dose of 20 mg in the presence (dashed line) and absence (solid line) of multiple daily doses of ivosidenib (500 mg q.d. for 19 days). The gray lines represent the outcomes of simulated individual trials (10 × 10) and the solid/dashed black line is the mean data for the simulated population (n = 100). (b) Sensitivity analysis of ivosidenib OATP1B1/1B3 Ki on the predicted rosuvastatin area under the curve ratio (AUCR)
Simcyp platform qualification for predicting CYP3A4 induction
To assess the prediction accuracy of the PBPK modeling platform (Simcyp Simulator version 15.1) with respect to the induction of CYP3A4‐mediated DDIs, 20 clinical studies were identified for this analysis. The predicted versus observed AUC and Cmax ratios for the 20 DDI simulations are shown in Figure 6 (Tables S6 and S7). In 100% of the cases, the predicted mean AUC ratios were within 2‐fold of the observed data or criteria described by Guest et al. 21 In 63% of the cases, the predicted AUCRs were within 1.25‐fold of the observed data. The MFE (bias) and RMSE (precision) were estimated to be 1.23 and 3.73, respectively, for the predicted data relative to the observed 1/AUCRs. In 94% of the cases, the predicted mean Cmax ratios were within 2‐fold of the observed data. In 50% of the cases, the predicted Cmax ratios were within 1.25‐fold of the observed data. In 75% of the cases, the predicted Cmax ratios were within the criteria described by Guest et al. The MFE (bias) and RMSE (precision) were estimated to be 1.31 and 2.77, respectively, for the predicted ratios relative to observed 1/Cmax ratios.
FIGURE 6.
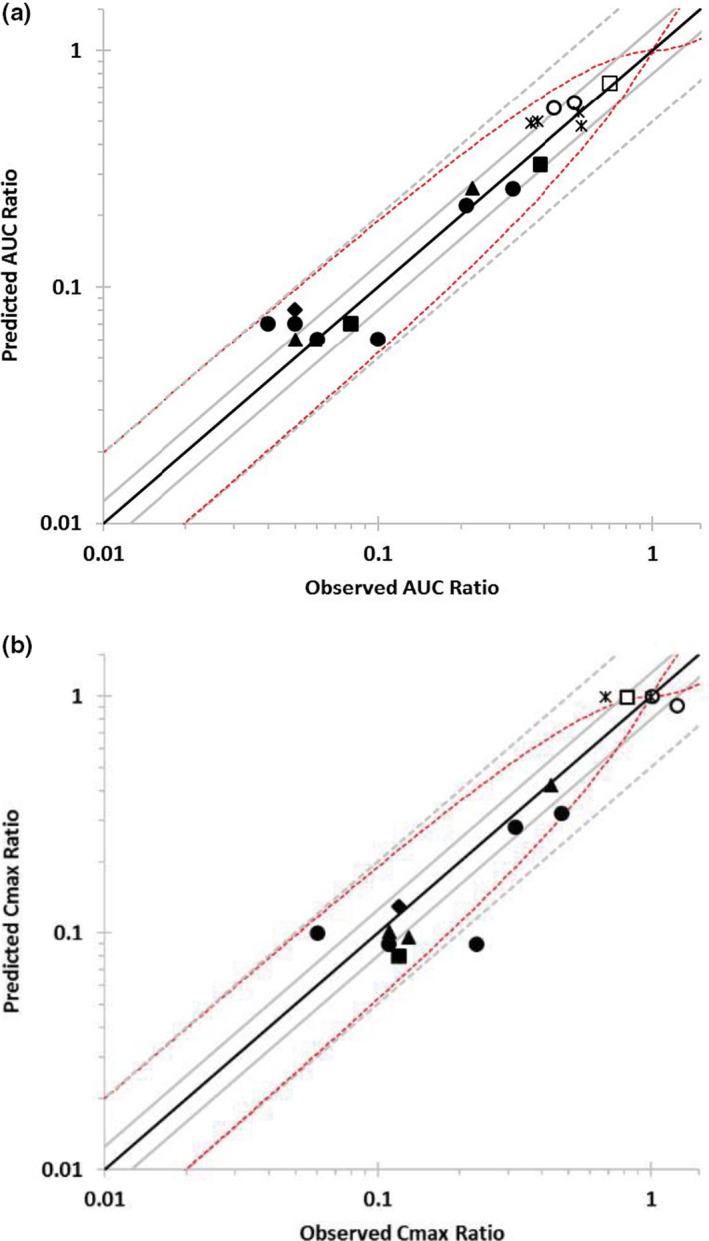
Predicted versus observed (a) area under the curve (AUC) (b) maximum plasma concentration (Cmax) ratios for CYP3A4 substrates. Simulations were conducted as 10 trials of the number of patients described in the clinical study, with study‐matched dosage regimen, age range, and proportion of female patients. Data points are shown for midazolam i.v. (○), midazolam p.o. (●), nifedipine i.v. (□), nifedipine p.o. (■), triazolam p.o. (♦), alfentanil i.v. (*), and alfentanil p.o. (▲). Lines represent unity (solid black), predicted/observed fold error between 0.8 and 1.25 (solid gray) or within 2‐fold (dashed gray), according to the criteria described by Guest et al. (2011) (dashed red)
DISCUSSION
The patient PBPK model of ivosidenib was developed and verified using single‐ and multiple‐dose PK in patients with AML. 10 The verified model was used to determine CYP and transporter‐mediated drug interactions, and to support DDI labeling. Ivosidenib is primarily metabolized by CYP3A4 and is an auto‐inducer.
Ivosidenib exhibited lower CL/F in patients with AML compared with HPs after a single oral dose. The reduced CL/F in patients with AML was attributable to food‐ and dose‐dependent effects on bioavailability and potential effects of disease or age on the systemic CL of ivosidenib. 7 , 8 , 9 , 10 , 23 In addition, high levels of circulating cytokines, including IL‐6, were observed in patients with AML. 27 , 28 Literature evidence shows that increased cytokines suppress CYP3A4 expression and metabolic activity, which could also be a cause of lower clearance of ivosidenib in patients with AML after a single dose. 29 Because multiple dose studies in patients present ethical (particularly induction) and safety challenges, extensive PBPK modeling was used to predict the drug interactions of ivosidenib as a perpetrator. The fmCYP3A4 used in the patient model was assumed to be the same as in HPs after a single dose and to increase under multiple‐dose conditions owing to autoinduction. The autoinduction of CYP3A4 under steady‐state conditions in patients was supported by the in vivo effect of ivosidenib on the hepatic CYP3A4 marker 4β‐OHC and its established relationship with the effects of the sensitive CYP3A4 substrate midazolam. 24 A study conducted with 600 mg rifampicin (strong CYP3A4 inducer) once daily for 14 days and single‐dose midazolam (sensitive CYP3A4 substrate) gave rise to a 220% increase in 4β‐OHC plasma concentration. 30 Midazolam AUC was reduced by 92% and 51% following multiple oral and i.v. doses of rifampicin, respectively. This relationship was also used to support the PBPK prediction of the CYP3A4 induction effect of midostaurin and its metabolites at steady‐state. 31 Similarly, in the current study, the CYP3A4 induction effect of ivosidenib was supported by the relationship between 4β‐OHC and oral midazolam AUC. 24 , 30 Mean 4β‐OHC levels were increased by ~ 199% following treatment with 500 mg ivosidenib, corresponding to a midazolam AUCR of 0.15, suggesting a strong induction effect of ivosidenib on CYP3A4 substrates. Venetoclax is an oral targeted BCL2 inhibitor and a sensitive CYP3A4 substrate. 32 , 33 Recently, a phase Ib/II study was conducted to evaluate the safety, tolerability, PK profiles, and efficacy of combined ivosidenib and venetoclax in patients with myelodysplastic syndromes or AML with IDH1 mutations. 34 The verified ivosidenib model successfully predicted the observed DDIs at steady‐state (Table 1). The simulated midazolam results showed that ivosidenib is a potent inducer of CYP3A4 and can be classified as a strong inducer as per regulatory guidance. 35 Hence, no clinical study was conducted and the PBPK results informed drug labeling.
Apart from CYP3A induction, ivosidenib also induces CYP2B6, CYP2C8, and CYP2C9. Although the magnitude of DDI owing to CYP2B6 induction tends to be underpredicted by the Simcyp platform, the predicted versus observed values seem to correlate well at lower magnitudes of CYP2B6 DDI. 36 Repaglinide is a substrate of CYP3A4, CYP2C8, and the hepatic uptake transporter OATP1B1, and hence DDI potential could be complex following co‐administration with ivosidenib. A simulated 50% reduction in repaglinide AUC could also be because of the contribution of CYP3A4 induction. A negligible DDI was observed with warfarin, a CYP2C9 substrate, following repeated administration of ivosidenib. These predictions are consistent with the available knowledge, indicating that the effects of a PXR‐activating inducer on CYP2C9 will be less marked than the effects on CYP3A4. 37 , 38 Thus, PBPK simulations suggest that clinically significant interactions following co‐administration of ivosidenib with CYP2B6, CYP2C8, and CYP2C9 substrates are minimal.
The use of PBPK models to predict enzyme‐mediated drug interactions is more established than for transporter‐mediated drug interactions. Competing passive and active processes, along with lack of a good understanding of transporter biology, make it challenging to predict the quantitative contribution of transporters to drug disposition. There is a lot of uncertainty associated with the determination of IC50 values in vitro for transporters and the values are often overpredicted. 39 , 40 However, PBPK models are being used to delineate transporter and transporter‐enzyme interplay and predict in vivo outcomes using in vitro parameters. In the current study, an independent evaluation of the in vitro P‐gp inhibition data (IC50 or Ki) of four P‐gp inhibitors, including clarithromycin, nifedipine, ritonavir, and verapamil, indicated that lower Ki values were required to recover the interaction in all four cases. The extent of this decrease ranged from 4‐ to 26‐fold (average 14.7‐fold) when comparing the lowest Ki value of each compound (unpublished data). These optimizations may reflect the fact that for some P‐gp substrates or inhibitors, the “intracellular” concentrations are substantially higher than the measured concentration in the medium. Thus, a sensitivity analysis of ivosidenib Ki showed that digoxin AUCR is insensitive to P‐gp at Ki greater than 5 µM; when P‐gp Ki was lowered by 15‐fold, the predicted digoxin AUCR was 1.12, suggesting minimal impact of ivosidenib on P‐gp inhibition compared with the model compounds used in this study. Rosuvastatin is an index probe substrate that has been used to regularly assess OATP‐mediated DDI studies. Multiple transporters, including OATP1B1, OATP1B3, and sodium‐taurocholate co‐transporting polypeptide (NTCP; and/or OATP2B1), are involved in hepatic uptake of rosuvastatin. The Simcyp platform was successfully used to predict OATP‐mediated DDIs. 41 In the current study, the relative contributions of OATP1B1/1B3 and NTCP transporters to the uptake of rosuvastatin were 65% and 35%, respectively. 41 A sensitivity analysis showed that when both the OATP1B1 and OATP1B3 Ki values were lowered by 10‐fold (Figure 5b), the predicted rosuvastatin AUCR was 1.36. To assess the predictability of DDIs owing to OAT3 inhibition, a separate model was built, with methotrexate as a substrate and probenecid as a perpetrator. This model was verified with the observed data (Table S5) and used to predict ivosidenib DDI as a perpetrator. 25 The predicted renal DDI owing to inhibition of OAT3 by ivosidenib is marginal. In addition, the literature suggests that the maximum DDI predicted with OAT3 substrates and a strong inhibitor, probenecid, is approximately twofold, supporting the predicted ivosidenib DDI owing to OAT3 inhbition. 42 In a sensitivity analysis with worst case scenario, ivosidenib showed a minimal DDI effect owing to inhibition of P‐pg, OATP1B1/1B3, and OAT3. Although limited, there are a number of cases that have successfully used PBPK models to predict transporter‐mediated drug interactions. 43 The PBPK model presented in this article was considered adequate for prediction of DDIs of ivosidenib with CYP3A4, CYP2B6, CYP2C8, CYP2C9, OATP1B1/OATP1B3, and OAT3 substrates by the US Food and Drug Administration (FDA). 26 , 44 The capability of the model to predict DDI owing to ivosidenib as a P‐gp inhibitor was considered inadequate by the agency due to lack of mechanism‐based oral absorption model. 26 , 44 Although modeling results showed that the inhibition of OAT3 and P‐gp by ivosidenib will have minimal clinical relevance at 500 mg dose level, based on the agency’s feedback, the language for these two transporters was derived for ivosidenib label.
PBPK modeling platforms, such as Simcyp, are being extensively used to predict DDIs, and are qualified for various DDI prediction applications and regulatory decision making. 45 , 46 , 47 The ability of PBPK modeling to predict the effect of strong and moderate CYP3A inducers on drugs that are eliminated through CYP3A4‐catalyzed metabolism has been established by Wagner et al. 20 However, because CYP3A substrates were selected from new drug applications, actual CYP3A4 substrates were not disclosed in this study. The current analysis was performed with known substrates and inducers. Simcyp performed well (AUC: 100%; Cmax: 75%) in predicting CYP3A induction‐mediated DDIs within the criteria described by Guest et al. (Figure 6). 21 The results from this study demonstrated the suitability of the SimCYP platform to predict CYP3A induction‐mediated DDIs. This is the first report of the qualification of the Simcyp platform for predicting CYP3A induction.
In summary, PBPK modeling was used extensively to predict multiple DDIs of ivosidenib as a perpetrator. In this modeling and simulation exercise, along with in vitro data, autoinduction, 4β‐OHC, PK at various doses, and results from a combination (venetoclax plus ivosidenib) study, were used to build, verify, and assess the prediction accuracy of the PBPK model. This multifactorial approach allowed to us to predicted DDIs of ivosidenib with enough confidence to inform labeling.
CONFLICT OF INTEREST
C.P. is a current employee and stockholder of Agios. J.B. and H.Y. are former employees of Agios Pharmaceuticals, A.K. is an employee of Simcyp UK Ltd. The authors declared no other conflict of interest.
AUTHOR CONTRIBUTIONS
J.B. and C.P. wrote the manuscript. J.B., A.K., H.Y., and C.P. designed the research. J.B., A.K., and C.P. performed the research and analyzed the data.
Supporting information
Table S1‐S7
ACKNOWLEDGEMENTS
The authors would like to thank Bin Fan, Kha Le, and Luke Utley for technical support or scientific insight into this work. We thank Miranda Dixon, Excel Medical Affairs, Horsham, UK for Editorial assistance.
Funding information
This study was funded by Agios Pharmaceuticals, Inc.
REFERENCES
- 1. Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;8:5562‐5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pelosi E, Castelli G, Testa U. Isocitrate dehydrogenase mutations in human cancers: physiopathologic mechanisms and therapeutic targeting. J Explor Res Pharmacol. 2016;1:20‐34. [Google Scholar]
- 3. Popovici‐Muller J, Lemieux RM, Artin E, et al. Discovery of AG‐120 (Ivosidenib): a first‐in‐class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett. 2018;9:300‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ivosidenib label. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211192s001lbl.pdf. Accessed December 29, 2020.
- 5. Zhuang X, Lu C. PBPK modeling and simulation in drug research and development. Acta Pharm Sin B. 2016;6:430‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shebley M, Sandhu P, Emami Riedmaier A, et al. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin Pharmacol Ther. 2018;104:88‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mangoni AA, Jackson SHD. Age‐related changes in pharmacokinetics and pharmacodynamics: basic principles and practical applications. Br J Clin Pharmacol. 2004;57:6‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheeti S, Budha NR, Rajan S, Dresser MJ, Jin JY. A physiologically based pharmacokinetic (PBPK) approach to evaluate pharmacokinetics in patients with cancer. Biopharm Drug Dispos. 2013;34:141‐154. [DOI] [PubMed] [Google Scholar]
- 9. Hurria A, Lichtman SM. Clinical pharmacology of cancer therapies in older adults. Br J Canc. 2008;98:517‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prakash C, Fan B, Ke A, Le K, Yang H. Physiologically based pharmacokinetic modeling and simulation to predict drug‐drug interactions of ivosidenib with CYP3A perpetrator in patients with acute myeloid leukemia. Cancer Chemother Pharmacol. 2020;86:619‐632. [DOI] [PubMed] [Google Scholar]
- 11. Chen N, Cui D, Wang Q, et al. In vitro drug–drug interactions of budesonide: inhibition and induction of transporters and cytochrome P450 enzymes. Xenobiotica. 2018;48:637‐646. [DOI] [PubMed] [Google Scholar]
- 12. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402‐408. [DOI] [PubMed] [Google Scholar]
- 13. Freise KJ, Shebley M, Salem AH. Quantitative prediction of the effect of CYP3A inhibitors and inducers on venetoclax pharmacokinetics using a physiologically based pharmacokinetic model. J Clin Pharmacol. 2017;57:796‐804. [DOI] [PubMed] [Google Scholar]
- 14. Emami Riedmaier A, Lindley DJ, Hall JA, et al. Mechanistic physiologically based pharmacokinetic modeling of the dissolution and food effect of a biopharmaceutics classification system IV compound ‐ the venetoclax story. J Pharm Sci. 2018;107:495‐502. [DOI] [PubMed] [Google Scholar]
- 15. Howgate EM, Rowland‐Yeo K, Proctor NJ, Tucker GT, Rostami‐Hodjegan A. Prediction of in vivo drug clearance from in vitro data: impact of interindividual variability. Xenobiotica. 2006;36:473‐497. [DOI] [PubMed] [Google Scholar]
- 16. US Food and Drug Administration , CDER . Venetoclax “Clinical pharmacology & Biopharmaceutics review”. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208573Orig1s000ClinPharmR.pdf Accessed October 17, 2020.
- 17. Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95:1238‐1257. [DOI] [PubMed] [Google Scholar]
- 18. Almond LM, Mukadam S, Gardner I, et al. Prediction of drug‐drug interactions arising from CYP3A induction using a physiologically based dynamic model. Drug Metab Dispos. 2016;44:821‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rowland‐Yeo K, Walsky RL, Jamei M, Rostami‐Hodjegan A, Tucker GT. Prediction of time‐dependent CYP3A4 drug–drug interactions by physiologically based pharmacokinetic modelling: Impact of inactivation parameters and enzyme turnover. Eur J Pharm Sci. 2011;43:160‐173. [DOI] [PubMed] [Google Scholar]
- 20. Wagner C, Pan Y, Hsu V, Sinha V, Zhao P. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: an analysis of PBPK submissions to the US FDA. Clin Pharmacokinet. 2016;55:475‐483. [DOI] [PubMed] [Google Scholar]
- 21. Guest EJ, Aarons L, Houston JB, Rostami‐Hodjegan A, Galetin A. Critique of the two‐fold measure of prediction success for ratios: application for the assessment of drug‐drug interactions. Drug Metab Dispos. 2011;39:170‐173. [DOI] [PubMed] [Google Scholar]
- 22. Hariparsad N, Ramsden D, Palamanda J, et al. Considerations from the IQ Induction Working Group in response to drug‐drug interaction guidance from regulatory agencies: focus on downregulation, CYP2C induction, and CYP2B6 positive control. Drug Metab Dispos. 2017;45:1049‐1059. [DOI] [PubMed] [Google Scholar]
- 23. Fan B, Dai D, DiNardo CD, et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib in patients with advanced hematologic malignancies with an IDH1 mutation. Cancer Chemother Pharmacol. 2020;85:959‐968. [DOI] [PubMed] [Google Scholar]
- 24. Leil TA, Kasichayanula S, Boulton DW, LaCreta F. Evaluation of 4β‐hydroxycholesterol as a clinical biomarker of CYP3A4 drug interactions using a Bayesian mechanism‐based pharmacometric model. CPT Pharmacometrics Syst Pharmacol. 2014;3:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aherne GW, Marks V, Mould GP, Piall E, White WF. The interaction between methotrexate and probenecid in man. Br J Pharmacol. 1978;63:369P. [PMC free article] [PubMed] [Google Scholar]
- 26. US FDA Drug Approval Package: Tibsovo (ivosidenib) www.211192Orig1s000MultidisciplineR.pdf (fda.gov). Accessed December 21, 2020.
- 27. Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer. 2005;104:788‐793. [DOI] [PubMed] [Google Scholar]
- 28. Tsimberidou AM, Estey E, Wen S, et al. The prognostic significance of cytokine levels in newly diagnosed acute myeloid leukemia and high‐risk myelodysplastic syndromes. Cancer. 2008;113:1605‐1613. [DOI] [PubMed] [Google Scholar]
- 29. Evers R, Dallas S, Dickmann LJ, et al. Critical review of preclinical approaches to investigate cytochrome P450‐mediated therapeutic protein drug‐drug interactions and recommendations for best practices: a white paper. Drug Metab Dispos. 2013;41:1598‐1609. [DOI] [PubMed] [Google Scholar]
- 30. Kasichayanula S, Boulton DW, Luo W‐L, et al. Validation of 4β‐hydroxycholesterol and evaluation of other endogenous biomarkers for the assessment of CYP3A activity in healthy subjects. Br J Clin Pharmacol. 2014;78:1122‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gu H, Dutreix C, Rebello S, et al. Simultaneous physiologically based pharmacokinetic (PBPK) modeling of parent and active metabolites to investigate complex CYP3A4 drug‐drug interaction potential: a case example of midostaurin. Drug Metab Dispos. 2018;46:109‐121. [DOI] [PubMed] [Google Scholar]
- 32. University of Washington DDI database. https://didb.druginteractionsolutions.org/uploads/documents/v‐637304113661269648/DIDB%20List%20of%20CYP3A%20Sensitive%20Substrates%20July%202020.pdf. Accessed October 17, 2020.
- 33. Richard‐Carpentier G, DiNardo CD. Venetoclax for the treatment of newly diagnosed acute myeloid leukemia in patients who are ineligible for intensive chemotherapy. Ther Adv Hematol. 2019;10:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dinardo C, Takahashi K, Kadia T, et al. A phase 1b/2 clinical study of targeted IDH1 inhibition with ivosidenib, in combination with the BCL2 inhibitor venetoclax, for IDH1‐mutated myeloid malignancies. HemaSphere. 2019;3:97. [Google Scholar]
- 35. US Food and Drug Administration Guidance . In Vitro Drug Interaction Studies — Cytochrome P450 Enzyme‐ and Transporter‐Mediated Drug Interactions Guidance for Industry (2020). https://www.fda.gov/media/134582/download. Accessed October 17, 2020.
- 36. Fahmi OA, Shebley M, Palamanda J, et al. Evaluation of CYP2B6 induction and prediction of clinical drug–drug interactions: considerations from the IQ consortium induction working group—an industry perspective. Drug Metab Dispos. 2016;44:1720‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lutz JD, Kirby BJ, Wang LU, et al. Cytochrome P450 3A induction predicts P‐glycoprotein induction; part 2: prediction of decreased substrate exposure after rifabutin or carbamazepine. Clin Pharmacol Ther. 2018;104:1191‐1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lutz JD, Kirby BJ, Wang LU, et al. Cytochrome P450 3A induction predicts P‐glycoprotein induction; part 1: establishing induction relationships using ascending dose rifampin. Clin Pharmacol Ther. 2018;104:1182‐1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Varma MVS, Lai Y, Feng B, et al. Physiologically based modeling of pravastatin transporter‐mediated hepatobiliary disposition and drug‐drug interactions. Pharm Res. 2012;29:2860‐2873. [DOI] [PubMed] [Google Scholar]
- 40. Li R, Barton HA, Varma MV. Prediction of pharmacokinetics and drug‐drug interactions when hepatic transporters are involved. Clin Pharmacokinet. 2014;53:659‐678. [DOI] [PubMed] [Google Scholar]
- 41. Jamei M, Bajot F, Neuhoff S, et al. A mechanistic framework for in vitro‐in vivo extrapolation of liver membrane transporters: prediction of drug‐drug interaction between rosuvastatin and cyclosporine. Clin Pharmacokinet. 2014;53:73‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ivanyuk A, Livio F, Biollaz J, Buclin T. Renal drug transporters and drug interactions. Clin Pharmacokinet. 2017;56:825‐892. [DOI] [PubMed] [Google Scholar]
- 43. Taskar KS, Pilla Reddy V, Burt H, et al. Physiologically‐based pharmacokinetic models for evaluating membrane transporter mediated drug–drug interactions: current capabilities, case studies, future opportunities, and recommendations. Clin Pharmacol Ther. 2020;107:1082‐1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang X, Yang Y, Grimstein M, et al. Application of PBPK modeling and simulation for regulatory decision making and its impact on us prescribing information: an update on the 2018–2019 submissions to the US FDA’s Office of Clinical Pharmacology. J Clin Pharmacol. 2020;60:S160‐S178. [DOI] [PubMed] [Google Scholar]
- 45. Wagner C, Pan Y, Hsu V, et al. Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin Pharmacokinet. 2015;54:117‐127. [DOI] [PubMed] [Google Scholar]
- 46. Fahmi OA, Hurst S, Plowchalk D, et al. Comparison of different algorithms for predicting clinical drug‐drug interactions, based on the use of CYP3A4 in vitro data: predictions of compounds as precipitants of interaction. Drug Metab Dispos. 2009;37:1658‐1666. [DOI] [PubMed] [Google Scholar]
- 47. Luzon E, Blake K, Cole S, Nordmark A, Versantvoort C, Berglund EG. Physiologically based pharmacokinetic modeling in regulatory decision‐making at the European Medicines Agency. Clin Pharm Ther. 2017;102:98‐105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S7