

Abstract
Background:
Exaggerated Toll-like receptor (TLR) signaling and intestinal dysbiosis are key contributors to necrotizing enterocolitis (NEC). Lactobacillus rhamnosus GG (LGG) decreases NEC in preterm infants, but underlying mechanisms of protection remain poorly understood. We hypothesized that LGG alleviates dysbiosis and upregulates TLR inhibitors to protect against TLR-mediated gut injury.
Methods:
Effects of LGG (low- and high-dose) on intestinal pro-inflammatory TLR signaling and injury in neonatal mice subjected to formula-feeding (FF) and NEC were determined. 16S sequencing of stool and expression of anti-TLR mediators Single Immunoglobulin Interleukin-1 Related Receptor (SIGIRR) and A20 were analyzed.
Results:
FF induced mild intestinal injury with increased expression of interleukin-1 beta (IL-1β) and KC (mouse homolog of IL-8) compared to controls. LGG decreased IL-1β and KC in association with attenuated TLR signaling and increased SIGIRR and A20 expression in a dose-dependent manner. Low- and high-dose LGG had varying effects on gut microbiome despite both doses providing gut protection. Subsequent experiments of LGG on NEC revealed that pro-inflammatory TLR signaling and intestinal injury were also decreased, and SIGIRR and A20 expression increased, in a dose-dependent manner with LGG pre-treatment.
Conclusion:
LGG protects against intestinal TLR-mediated injury by upregulating TLR inhibitors without major changes in gut microbiome composition.
INTRODUCTION
Necrotizing enterocolitis (NEC) is a devastating disease of preterm infants characterized by rapid and uncontrolled inflammation of the intestinal tract. It is estimated that about 5% to 10% of infants born less than 32 weeks gestation at birth develop NEC. About 25% to 40% of infants who develop NEC will die because of the disease, while infants who survive remain at risk for serious long-term morbidities including intestinal failure and neurodevelopmental delays (1). Pathologic activation of the Toll-Like Receptor (TLR) family of innate immune receptors has been identified as the key signaling pathway through which uncontrolled inflammation occurs in NEC (2, 3). Intestinal microbiome studies of preterm infants with and without NEC suggest that this excessive TLR activity is triggered by abnormal gut bacterial colonization (4–6). Dysbiotic patterns found to precede the development of NEC include lower gut microbial diversity, increased abundance of γ-Proteobacteria, and decreased abundance of Firmicutes (5, 6).
Based on the strong link between gut dysbiosis and NEC, clinical trials of probiotic supplementation in preterm infants have been conducted, and evidence is accumulating regarding its benefit in reducing NEC (7, 8). While it was commonly believed that the protection conferred by probiotics was related to changing gut microbiota signatures and mitigating dysbiosis, recent studies have begun to challenge this hypothesis (9, 10). Zmora et al. (11) demonstrated that minimal changes in gut bacterial composition occurred in probiotic-treated adult mice compared to placebo-treated mice or to their own pre-probiotic baseline. Nevertheless, despite the modest changes in gut microbiome, probiotic consumption did lead to significant transcriptional changes in the ileum, with a majority of genes related to the intestinal innate immune system.
Prior genetic studies from our laboratory have identified Single-Immunoglobulin Interleukin-1 Related Receptor (SIGIRR), an immune gene that normally functions as a negative regulator of TLR signaling, as a potential candidate gene for NEC susceptibility. We found that loss of SIGIRR worsens NEC-like injury in mice, suggesting that SIGIRR is important in maintaining intestinal TLR homeostasis and protection against NEC (12, 13). The impact of probiotics in modulating the balance between inhibitors and mediators of TLR4 signaling in the neonatal intestine is poorly understood. In this study, we sought to determine whether the probiotic Lactobacillus rhamnosus GG (LGG), which is the most commonly used probiotic in the United States for the prevention of NEC (14), protected against NEC-like intestinal injury in mice by effecting changes in microbiota or by modulating gene expression of TLR4 signaling mediators. We hypothesized that LGG will confer protection from excessive TLR activation by inducing expression of SIGIRR and other TLR inhibitors and by alleviating microbial dysbiosis in the neonatal gut.
METHODS:
A full description of the materials and methods is available as supplementary material at the Pediatric Research website (Supplemental Text - Methods, online).
RESULTS
LGG treatment attenuates formula-induced inflammation and TLR signaling concurrent with increased SIGIRR and A20 expression in a dose-dependent fashion
To determine the effects of formula feeding and probiotics on intestinal inflammation, we first compared cytokine gene expression between dam-fed and formula-fed C57BL6 neonatal mice. P5 pups administered formula for 3 days exhibited upregulation of inflammatory cytokines IL-1β, KC and inflammatory marker ICAM1 in the gut compared to dam-fed controls (Fig 1A). Increasing doses of LGG were then administered to formula-fed mice to evaluate the effects of LGG on formula-induced inflammation and we found that LGG treatment decreased expression of inflammatory cytokines in a dose-dependent manner (Fig 1A).
Fig 1. Lactobacillus rhamnosus GG (LGG) dose-dependently increases SIGIRR and decreases TLR-mediated ileal inflammation induced by formula.

(A) mRNA expression of ICAM1, IL-1β, and KC increased in formula-fed pups (F) and decreased dose-dependently with LGG treatment in FP and FPP. (n≥5 per group; ***p<0.001; Comparisons: BM vs. BMP, BM vs. F, F vs. FP, F vs. FPP). (B) SIGIRR and A20 mRNA expression increased dose-dependently with LGG treatment accompanied by decrease in TLR4 and IRAK-1 mRNA expression. (n≥5 per group; ***p<0.001, *p<0.05, §p=0.06; Comparisons: BM vs. BMP, BM vs. F, F vs. FP, F vs. FPP). (C-D) Western blot showing increased protein expression of phosphorylated IKKβ, p65, IL-1β, and ICAM1 indicating pro-inflammatory canonical TLR4 signaling is activated with formula feeding. LGG treatment increased protein expression of SIGIRR in association with reduction in protein levels of phosphorylated IKKβ, p65, IL-1β, and ICAM1. (n≥5 per group; ***p<0.001, *p<0.05; Comparisons: BM vs. BMP, BM vs. F, F vs. FP, F vs. FPP).
To investigate the mechanisms by which LGG ameliorates formula-induced inflammation, we evaluated the balance between pro-inflammatory and anti-inflammatory TLR pathway genes in the intestinal tract. We found that compared to BM controls, pups fed with formula had increased RNA expression of TLR signaling mediators TLR4 and IRAK1 (TLR signaling mediators). In contrast, RNA expression of TLR inhibitor SIGIRR was induced with LGG in breast fed pups (BM vs. BMP, p<0.05), and showed a trend towards increase in formula-fed pups treated with high-dose LGG (F vs. FPP; p=0.067, Fig 1B). LGG treatment also significantly induced the TLR inhibitor A20 in both the breast milk and formula-fed groups (Fig 1B). Induction of TLR inhibitors SIGIRR and A20 in formula-fed mice correlated with a reduction in TLR mediators TLR4 and IRAK1, both of which occurred in a dose-dependent manner with LGG treatment (Fig 1B).
To further explore these findings, we investigated whether canonical TLR signaling, which drives intestinal inflammation, was also suppressed with LGG administration (Fig 1 C–D). Protein expression of phosphorylated IKKβ, phosphorylated p65, ICAM1, and IL-1β were increased in formula-fed mice compared to BM controls, while formula-fed pups treated with LGG exhibited decreased levels of TLR signaling and inflammation (Fig 1 C–D). This decreased expression of phosphorylated IKKβ, phosphorylated p65, and pro-inflammatory cytokines correlated in a dose-dependent manner with the induced expression of SIGIRR and A20 from LGG treatment (Fig 1 C–D). Together, these data suggest that LGG ameliorates formula-induced TLR-dependent inflammation by inducing expression of TLR inhibitors in the gut.
Intestinal apoptosis and injury from formula feeding is decreased with LGG treatment
We next investigated the effect of formula and LGG on intestinal apoptosis by evaluating protein levels of Cleaved Caspase 3 (CC3) (Fig 2 A–B). Formula-fed pups had increased CC3 levels in the intestinal tract compared to BM controls, while LGG treatment decreased CC3 levels of formula-fed pups to levels similar to BM controls, signifying a protective effect of LGG against formula-induced enterocyte apoptosis. The impact of formula and LGG on intestinal injury was also assessed by using a validated intestinal injury scoring tool (Fig 2 C–D). Histologic evaluation of the terminal ileum showed mild increase in intestinal injury scores of pups fed with formula compared to controls. Intestinal injury scores of formula-fed pups were unchanged with low-dose LGG treatment, but decreased when high-dose LGG was administered.
Fig 2. Formula causes apoptosis and mild injury to immature intestine that is ameliorated by treatment with Lactobacillus rhamnosus GG (LGG).
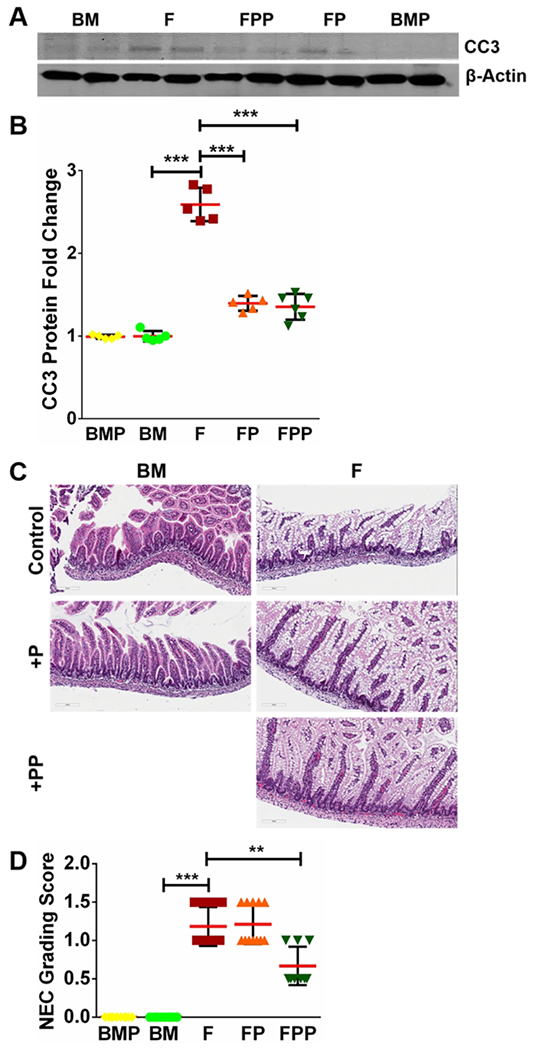
(A-B) Quantification of CC3 protein using densitometry demonstrated formula-induced increase in CC3 levels which were attenuated with LGG treatment in FP and FPP groups. (n≥5 per group; ***p<0.001). (C-D) Examination of terminal ileum of formula-fed pups shows damage to intestinal epithelial cells compared to controls which was partially decreased with LGG treatment. Quantification of intestinal injury by a validated scoring tool shows decrease in injury score following high-dose LGG (FPP), but not following low-dose LGG (FP). (n≥6 per group; ***p<0.001, **p<0.01).
Taxonomic changes in the bacterial constituents of colon fecal microbiome following LGG treatment
To determine if mouse pups fed breast milk versus those receiving formula had differences in microbial community profiles, and whether LGG treatment influenced gut microbiota, we performed MiSeq v3 PE-300 bp sequencing of 72plex V3/V4 16S amplicon library pool. The sequencing provided average of ~400,000 reads per sample. After quality filtering and removal of chimeras and non-bacterial sequences, barcoded 16S rRNA amplicons generated a total of 60,000 sequences in the colonic fecal samples. Prior to estimating microbial diversity, phylum level analyses revealed that among the 12 phyla, four phyla, Firmicutes, Bacteroidetes, Proteobacteria and Verrucomicrobia, were dominant in all five groups (BM, BMP, F, FP & FPP; Fig 3A). In the formula group receiving higher doses of LGG (FPP), Bacteroidetes phyla was more abundant compared to any other group (Fig 3A and Supplemental Fig S2A, online).
Fig 3. Changes in gut bacterial composition and diversity in response to LGG.
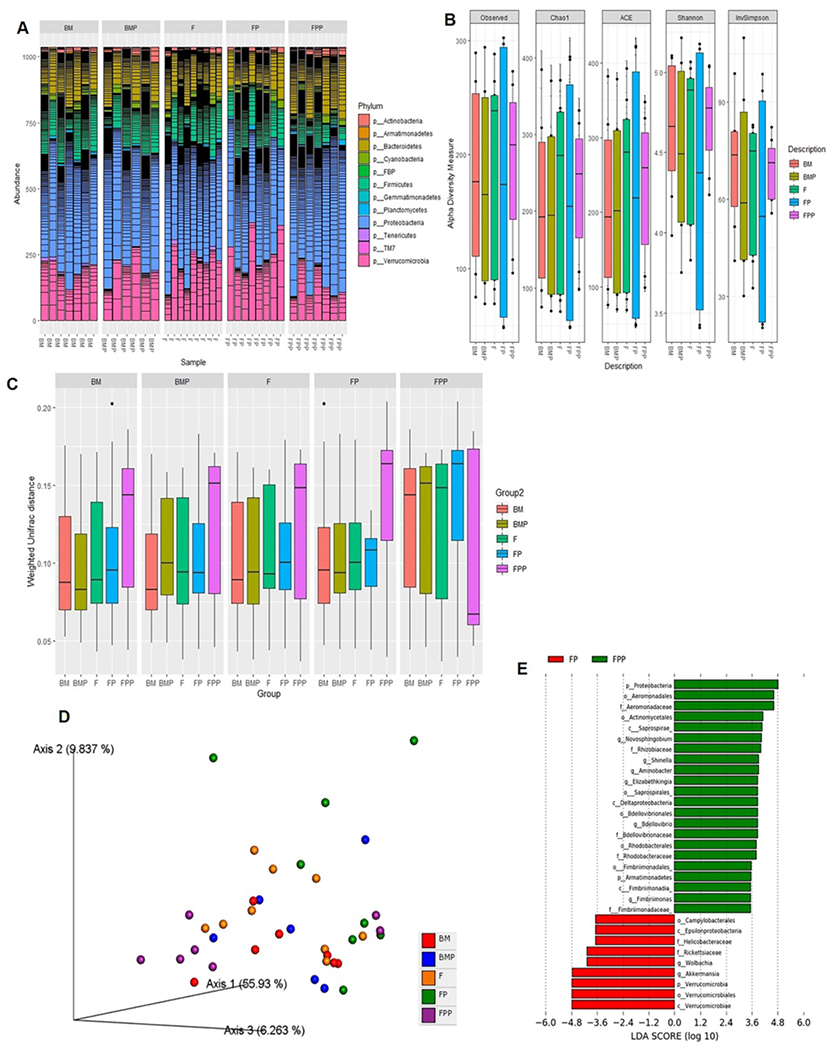
(A) Estimation of taxonomic abundance showing compositional shifts at the phyla level. (B) Box and Whisker plots showing fecal microbial diversity based on observed OTUs, Chao1 index, abundance-based coverage estimators (ACE), Shannon and InvSimpson indices in the five groups of mice. The horizontal line in the middle of each box represents the median, while the top and bottom borders of the box represent the 75th and 25th percentiles, respectively. P values for Shannon indices: BM (n=7) vs. BMP (n=6): <0.4795; FP (n=8) vs. FPP (n=7): <0.008151. P values for Chao1 indices: BM (n=7) vs. BMP (n=6): <0.125; FP (n=8) vs. FPP (n=7): <0.008151. P values for ACE indices: BM (n=7) vs. BMP (n=6): <0.668; FP (n=8) vs. FPP (n=7): not significant. (C) Box plot of inter-group β-distance amongst the five groups (pairwise Anosim analysis in the five groups of mice (BM, BMP, F, FP, FPP): p values for BM vs. BMP F, FP and FPP: 0.671, .556, .824 and 0.311; p values for BMP vs. F, FP, FPP: 0.413,0.301,0.580; p values for F vs. FP and FPP: 0.649, 0.461; p values for FP vs. FPP: 0.157). (D) Principal Coordinates Analysis (PCoA) based on weighted UniFrac distances between bacterial communities. P values: (FP vs FPP <0.05). (E) Histogram of the LDA scores (log 10) computed for features that were differentially abundant in FPP vs. FP group.
To evaluate the species richness, evenness and diversity of the microbial communities, the observed OTUs, Chao1 index, abundance-based coverage estimators (ACE), Shannon and Simpson indices were calculated for samples within each group. The observed OTUs, Chao1 and Shannon indices in the FP group were significantly more variable compared to any other group. We saw similar trend with ACE and InvSimpson indices respectively (Fig 3B). These indices in the FPP group interestingly were higher but not as variable than those in any other group (Fig 3B). This suggests that while both low- and high-dose LGG conferred protection against formula-induced gut injury, low-dose LGG promoted more variability while high-dose LGG showed reduced variability but higher microbial diversity.
Next, we estimated β-diversity after samples were rarefied to the lowest library size (4600 reads/sample). Weighted UniFrac distance metric was used to examine differences in community structure. As depicted in Fig 3C, FPP group consistently showed higher β-diversity compared to any other group. This was confirmed by both ANOSIM and PERMANOVA (P < 0.05) that indicated significant differences in the microbial community structure specially between FP and FPP groups. Principal Coordinates Analysis (PCoA) based on 16S rRNA community profiles further demonstrated that in contrast to the more uniform pattern of clustering in either BM or BMP, FP and FPP groups compared to F, were clustered in separate coordinates indicating discrete colonization types (Fig 3D). Lastly, linear discriminant analysis (LDA) was performed to determine differentially abundant bacterial taxa between the groups. A total of 22 bacterial groups were differentially expressed (LDA score > 3.0) in the FPP group. Proteobacteria were overrepresented, while Verrucomicrobia and others including Akkermansia, were underrepresented in the FP group (Fig 3E). Cladogram, plotted from LEfSe analysis, further demonstrated significant differences in the FP vs. FPP group (Supplemental Fig. S2B, online).
LGG pre-treatment increases SIGIRR and A20 expression and attenuates canonical TLR signaling and inflammatory cytokine expression in mice with NEC
Our experiments with formula-induced gut injury indicated that both low-dose and high-dose LGG increased SIGIRR and A20 expression and decreased pro-inflammatory TLR signaling in the neonatal intestinal tract. As TLR4 is central to NEC pathogenesis, we next investigated whether low-dose LGG pre-treatment confer similar protective effects in mice with experimental NEC. We posited that LGG pre-treatment also increases intestinal SIGIRR and A20 expression and ameliorates intestinal injury from NEC. To mimic how probiotics are used clinically in preterm infants, we pre-treated mice with LGG for 3 days prior to NEC induction with formula, hypoxia, and LPS. As expected, increased expression of inflammatory cytokines KC, IL-1β and inflammation marker ICAM1 was observed in mice following NEC induction compared to controls. (Fig 4A) When mice were pre-treated with LGG for 3 days prior to NEC induction, expression of TLR4 was attenuated in association with increased SIGIRR and A20 expression (Fig 4B), suggesting that LGG pre-treatment programs resistance against pro-inflammatory TLR signaling by inducing negative regulators of TLR.
Fig 4. Low-dose LGG pre-treatment induces SIGIRR and ameliorates TLR-mediated inflammation in mice with experimental NEC.
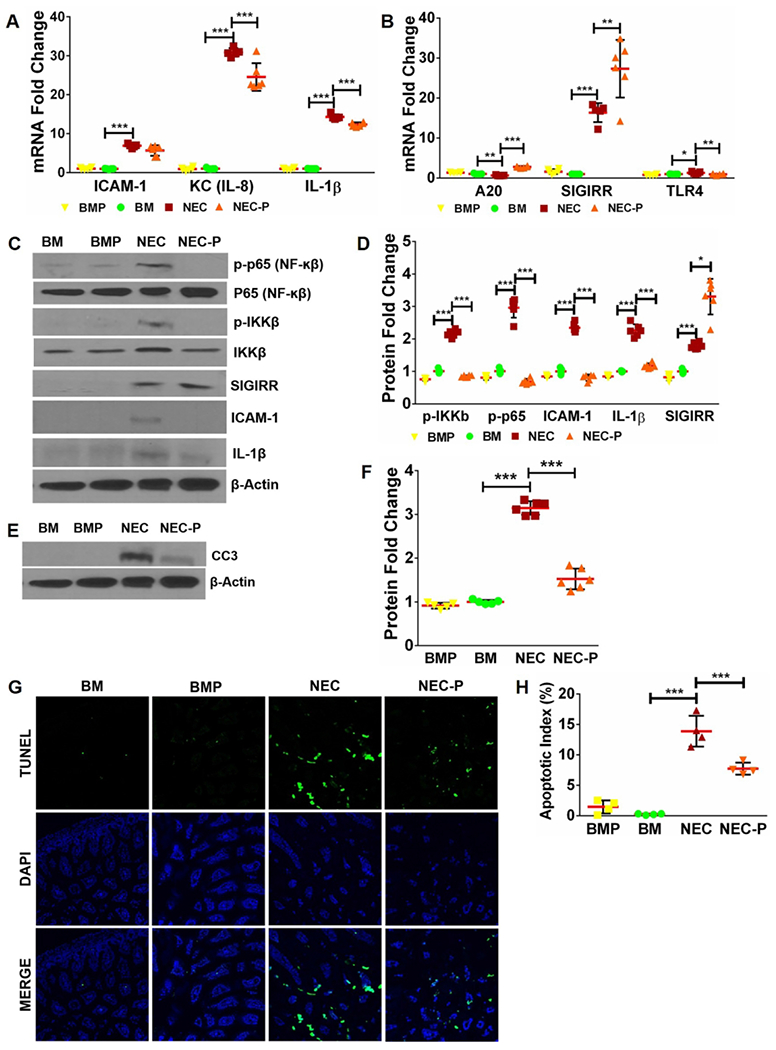
(A) Gene expression of inflammatory cytokines ICAM1, KC, and IL-1β increased with NEC and decreased in NEC-P group pre-treated with LGG. (n≥5 per group; ***p<0.001). (B) Pups pre-treated with LGG exhibit higher SIGIRR and A20 expression compared to untreated mice with NEC. (n≥5 per group. ***p<0.001, **p<0.01, *p<0.05). (C) Western blot analysis of canonical TLR4 signaling mediators and downstream inflammatory effectors demonstrated increased levels of phosphorylated p65, phosphorylated IKKβ, ICAM1, and IL-1β with NEC induction, all of which were attenuated in mice pre-treated with LGG. Increased SIGIRR protein expression in mice pre-treated with LGG (NEC-P) was also evident. (D) Graphical representation of densitometric quantification of protein expression showing decreased canonical TLR4 inflammatory signaling in mice pre-treated with LGG prior to NEC induction. (n≥5 per group. ***p<0.001, *p<0.05). (E) CC3 protein expression of BM and NEC mice at baseline and following LGG treatment was examined by western blotting of ileal homogenates. (F) Quantification of CC3 protein using densitometry demonstrated increased CC3 levels with NEC that were attenuated with LGG treatment. (n≥5 per group. ***p<0.001). (G) Representative ileal sections from BM, BMP, NEC, and NEC-P pups show reduction in apoptotic cells (TUNEL, green stained) with LGG pre-treatment. (H) Quantification of apoptotic index (apoptotic cells/total cells) in intestinal sections demonstrate statistically significant decrease in apoptotic index in NEC-P mice compared to NEC. (n≥4 per group; ***p<0.001, **p<0.01).
To further investigate TLR pathway signaling during NEC after LGG pre-treatment, we assessed activation of TLR4 signaling mediators, SIGIRR, and downstream TLR4-mediated inflammatory cytokines (Fig 4 C–D). Phosphorylated p65, phosphorylated IKKβ, IL-1β, and ICAM1 were increased with NEC compared to controls, indicating robust activation of pro-inflammatory TLR pathway activation. Pre-treatment with LGG prior to NEC induction resulted in higher levels of SIGIRR protein and decreased levels of TLR signaling and inflammation (Fig 4 C–D). We also assessed CC3 protein levels and TUNEL staining to assess apoptosis and found that intestinal apoptosis was increased in NEC and decreased in pups pre-treated with LGG (Fig 4 E–H).
Bacterial invasion and intestinal injury are decreased with low-dose LGG pre-treatment
Direct invasion of intestinal mucosa by bacteria is an important contributor in NEC pathogenesis, and improved barrier function has been proposed as a mechanism by which probiotics protect against NEC (17, 18). We therefore investigated the effect of low-dose LGG pre-treatment on bacterial community proximity and invasiveness into the colonic epithelium using the universal bacterial probe, EUB338. As seen in Fig 5A, there is clear separation of bacteria from intestinal mucosa in dam-fed mice indicating an intact mucosal barrier function; while mice with NEC exhibited invasion of bacteria into the mucosa. In contrast, mice pre-treated with LGG prior to NEC induction had decreased invasion of bacteria into the mucosa (Fig 5A). We also performed analysis with standard H&E staining to assess intestinal injury in NEC pups pre-treated with LGG. Compared to untreated pups with NEC, we found that intestinal injury scores were decreased in pups who received LGG prior to experimental NEC induction. (Fig 5 B–C)
Fig 5. Pre-treatment with low-dose LGG protects against bacterial invasion of intestinal mucosa and ameliorates intestinal injury from experimental NEC.

(A) BM control mice demonstrate separation of bacteria (stained red) from intestinal mucosa (stained blue with DAPI), while mice with experimental NEC exhibit invasion of bacteria into the mucosa. Bacterial invasion was ameliorated when mice were pre-treated with low-dose LGG prior to NEC induction. (n≥5 per group). (B) Histologic examination of terminal ileum shows moderate damage to intestinal epithelial cells with destruction of villi architecture in experimental NEC. Pups pre-treated with LGG prior to NEC induction show less damage to intestinal epithelial cells with preservation of villi. (C) Intestinal injury scores decreased significantly in pups pre-treated with LGG. (n≥ 5 per group, representative samples shown; ***p<0.001, *p<0.05).
Pre-treatment with high-dose LGG also increases SIGIRR and confers protection against intestinal injury from experimental NEC
As our prior data indicated that high-dose LGG exhibited more substantial protective effects with formula-induced injury, we investigated the effects of high-dose LGG pre-treatment on subsequent experimental NEC. Overall, we found consistent findings of decreased inflammation, decreased TLR signaling activity, and increased SIGIRR expression at both RNA and protein level in mice pre-treated with high-dose LGG. (Fig 6 A–D) Pre-treatment with high-dose LGG also conferred protection from gut injury with decreased apoptosis (Fig 6 E–G) and lower intestinal injury (Fig 6 H–I) than low-dose LGG.
Fig 6. Pre-treatment with high-dose LGG (P5-7) conferred similar protection from TLR4-mediated inflammation, apoptosis, and injury caused by experimental NEC induction (P8-10).
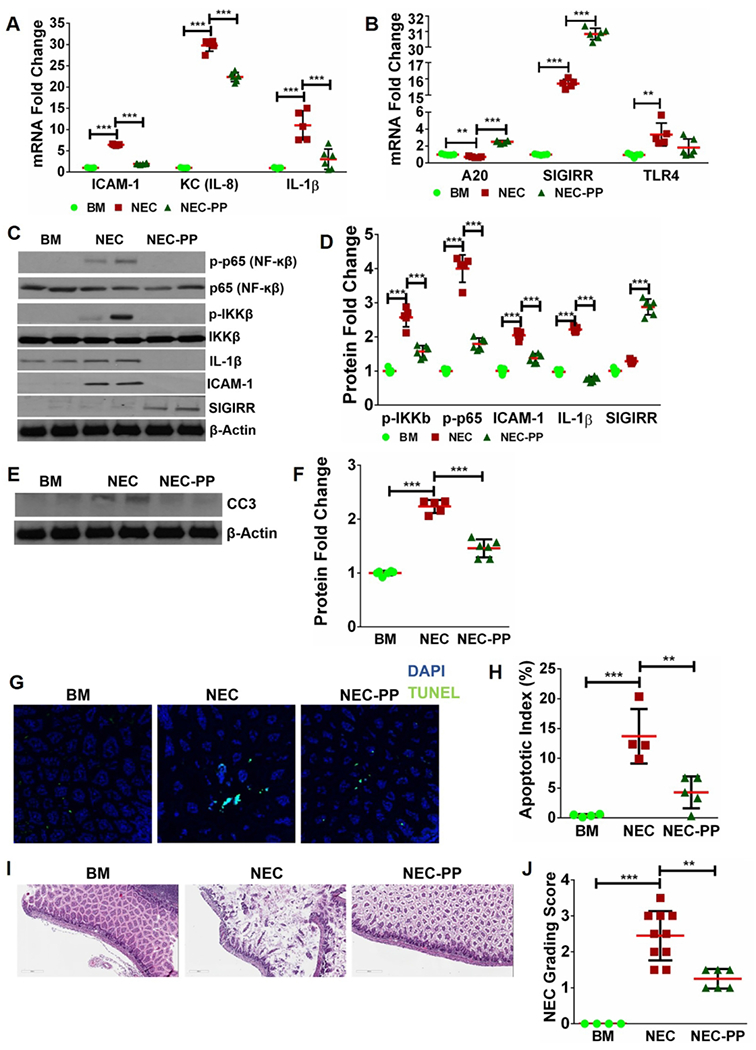
(A-B) mRNA levels of pro-inflammatory and anti-inflammatory mediators in the gut showing amelioration of TLR4-mediated inflammation in conjunction with increased levels of SIGIRR and A20 induced by high-dose LGG (NEC-PP). (n≥5/group. ***p<0.001, **p<0.01). (C-D) Western blot analysis showed reduction in canonical TLR4 signaling mediators (phosphorylated p65, phosphorylated IKKβ) and downstream inflammatory markers (IL-1β, ICAM1) with increased SIGIRR levels in NEC pups pre-treated with high-dose LGG (NEC-PP). (n≥5/group. ***p<0.001, **p<0.01). (E-F) Protein levels of CC3 were increased with NEC and decreased with high-dose LGG pre-treatment (NEC-PP). (G, H) TUNEL staining demonstrated increased intestinal apoptosis with NEC induction with subsequent reduction in pups pre-treated with high-dose LGG (NEC-PP). (n≥4/group. ***p<0.001, **p<0.01). (I) Representative sections of terminal ileum showed decreased injury with less mucosal edema and villi damage in pups pre-treated with high-dose LGG (NEC-PP) compared to untreated NEC pups. (J) Graph of intestinal injury scores showing that injury is decreased in pups pre-treated with high-dose LGG (NEC-PP) compared to untreated NEC pups. (n≥5/group. ***p<0.001, **p<0.01).
DISCUSSION
Evidence is accumulating regarding the benefit of prophylactic probiotics in reducing the risk for NEC, yet the mechanisms by which probiotics confer protection remain poorly understood (19). In this study, we showed that concurrent LGG supplementation in mice subjected to formula-feeding decreases intestinal TLR signaling, inflammation, and injury. Pre-treatment with probiotics prior to experimental NEC induction – a strategy which mimics how probiotics are used in preterm infants – also conferred similar protection against TLR-mediated inflammation, apoptosis, and injury in the gut. These protective effects occurred in a dose-dependent manner with increasing intestinal SIGIRR and A20 expression as higher dose of LGG was used, despite variable changes in gut microbial diversity and richness. Additional protection against bacterial mucosal invasion in NEC was also provided by LGG pre-treatment. Together, our findings suggest that altering intestinal gene expression to favor an anti-inflammatory milieu, as well as enhanced mucosal barrier protection against bacteria, are primary mechanisms by which probiotics afford protection against NEC.
Nanthakumar et al. (20) studied ileal tissue resected from infants with surgical NEC and found that mRNA expression of TLR inhibitors SIGIRR and A20 were decreased in relation to increased levels of TLR4 signaling, suggesting that imbalances between TLR4 and its negative regulators contribute to the exaggerated inflammatory TLR activation of NEC. Among the TLR inhibitors identified in Nanthakumar’s study include SIGIRR and A20. SIGIRR is a well-known negative regulator of TLR signaling in the gut (21, 22). The importance of SIGIRR in human NEC was demonstrated in genetic studies that identified loss-of-function mutations of SIGIRR were enriched in preterm infants with NEC compared to controls (12). Moreover, functional studies using neonatal SIGIRR knock-out mice confirmed that a pro-inflammatory phenotype of intestinal TLR hyper-responsiveness was present when SIGIRR is dysfunctional (13). A20 is an ubiquitin-editing enzyme that limits NF-κB activation by targeting several proximal mediators of TLR signaling for ubiquination and subsequent degradation (23, 24). Mice deficient in A20 develop severe intestinal inflammation early in life, while mice with dendritic cell –specific A20 deletion demonstrate increased mortality following exposure to LPS compared to controls (25, 26). In our study, administration of LGG to neonatal mice induced gene expression of SIGIRR and A20 in the gut and was associated with amelioration of TLR mediated-intestinal injury from formula-feeding and experimental NEC. The use of higher-dose LGG further induced SIGIRR and A20 levels and enhanced protection from gut injury. Together, our results suggest that probiotics could potentially restore imbalance between TLR activators and inhibitors in the gut and protect against NEC.
A few other studies have investigated the effect of probiotics on the balance between TLR signaling and negative regulators of TLR. Ganguli et al. (27) studied immature intestinal xenografts and primary enterocyte cultures exposed to probiotic-conditioned media (PCM) from Lactobacillus acidophilus and Bifidobacterium infantis then treated with LPS to induce TLR4-mediated inflammation. They found that PCM attenuated LPS-induced inflammation in relation to increased mRNA levels of SIGIRR and Toll-interacting protein (TOLLIP), another TLR inhibitor in the gut. Important limitations of their study include the use of an ex vivo model which may or may not accurately represent the complex physiology that occurs in vivo, and the lack of comprehensive analysis of canonical TLR signaling beyond gene expression studies. Wu et al. (28) used a rodent model of NEC to explore the protective effect of probiotics on NEC and found that Bifidobacterium adolescentis inhibited TLR4 expression and alleviated ileal injury in relation to increased SIGIRR and TOLLIP expression in the intestine. Although a well-established in vivo model of NEC was employed, their study used a probiotic strain that has not been tested in clinical studies and also did not examine changes in gut microbiota.
In our study, we evaluated the protective effects of LGG – a widely-used probiotic against NEC in preterm infants – in neonatal mice subjected to formula-feeding and experimental NEC. We first performed formula-feeding experiments to determine whether formula alone induces injury to the gut and if so, whether LGG treatment confers protection against formula-induced injury. We then tested the effects of LGG on the full NEC model with formula feeding, hypoxia, and LPS. Neonatal mice were pre-treated with LGG for 3 days prior to NEC induction to mimic how probiotics are clinically used in preterm infants. We used low-dose and high-dose LGG to determine dose-dependent effects. We also performed a comprehensive analysis of the effects of LGG on canonical TLR signaling, microbiome composition with 16S sequencing, and bacterial invasion with EUB338 staining. In both models, we demonstrated that LGG attenuated TLR-mediated inflammation and apoptosis as well as overall injury gut injury in relation with increased expression of negative TLR regulators. Taken in conjunction with prior studies, our data suggest that modulation of TLR inhibitors to maintain intestinal TLR homeostasis is a primary mechanism by which probiotics confer protection in NEC.
Favorable alteration of intestinal microbiome has been proposed as another mechanism by which probiotics protect against NEC (29, 30). In our study, inconsistent changes in alpha diversity by LGG supplementation were demonstrated, with low-dose LGG showing higher variability in the FP group while high-dose LGG exhibited higher microbial diversity in the FPP group, respectively. Similarly, high-dose LGG increased beta-diversity based on weighted Unifrac distance metric; and PCoA visualization and LDA demonstrated significant differences in microbial communities of FPP pups treated with LGG compared to FP. Although the biological explanation for varying effects of LGG supplementation on the microbiome is unknown, our results indicate that both low-dose and high-dose LGG remained capable of inducing SIGIRR and A20 expression and decreasing TLR-mediated gut injury. Our findings are similar to that of Underwood et al. (31) who showed protective benefits of the probiotic Bifidobacterium infantis in a rat model of experimental NEC despite inconsistent changes in the gut microbiome. How probiotics confer intestinal protection despite heterogenous changes in gut microbiome may be explained by its effects on expression of several important immune genes in the gut. This was elegantly shown by Zmora et al. (11) who found minimal changes in gut microbial composition in adult mice and humans but observed marked differences in mRNA expression of mostly immune-related genes in the intestinal mucosa following probiotic supplementation. Our results in neonatal mice are thus consistent with this study and suggest that protective effects of probiotics may not be necessarily mediated by changes in microbial composition but by transcriptional-level changes in the intestinal mucosa. The exact mechanisms by which probiotics modulate intestinal gene expression and subsequent host function needs further elucidation but may likely be mediated by epigenetic regulation of TLR pathway mediators and inhibitors (32, 33).
In our study, supplementation with high-dose LGG resulted in increased abundance of Bacteroidetes in the gut. These results were similar to another study in which supplementation with Lactobacillus rhamnosus increased microbial abundance of Bacteroidetes and improved lipid metabolism of hyperlipidemic rats (34). As reduction in the relative abundance of Bacteroidetes has been identified as a distinct microbial signature that precedes NEC in preterm infants (6), the restoration of Bacteroidetes abundance by LGG supplementation may be a potential strategy for NEC prevention. The exact mechanism by which LGG, which belongs to the phylum Firmicutes, increases microbial abundance of Bacteroidetes remains unclear. Recent evidence suggests that complex interaction of several factors including genetics, diet, antibiotics, existing gut microbiota, and microbiota-derived metabolites contribute to gut microbiome modulation (35, 36).
Another potential mechanism by which probiotics prevent NEC is by enhancing intestinal epithelial barrier function. Under physiologic conditions, the intestinal epithelium is separated from gut bacteria by an epithelial barrier consisting mainly of a dense mucus layer (37). Disruption of this mucosal barrier with subsequent bacterial invasion and TLR pathway activation has been postulated as a central mechanism underlying NEC pathogenesis. Using the bacterial probe EUB338, our study demonstrated a decrease in bacterial invasion in NEC pups pre-treated with LGG, suggesting that probiotic pre-treatment confers protection against NEC-like injury in part by enhancing the intestinal mucosal barrier.
Our study has a number of strengths. We employed two in vivo models of gut injury that relate to the clinical condition in premature infants – a formula-feeding model and an experimental NEC model – to study the effects of LGG supplementation on neonatal intestinal injury. Several protective mechanisms of probiotics were investigated including immune modulation by negative regulators of TLR, alteration of microbial composition, and maintenance of mucosal barrier. The use of colonic stool directly obtained at time of death to assess the microbiome was another strength of the study. A limitation is the absence of microbiome data on the ileum, which is the site where most NEC injury occur. We chose to focus on colonic stool in this study as bacteria are most concentrated in the colon. Future studies will be needed to assess the role of microbiome in other portions of the neonatal gut in NEC. Our study was also limited by its investigation of a single probiotic strain which does not allow generalization of our results to other probiotic strains. Several clinical studies have demonstrated that LGG used alone or in combination with other probiotic strains or prebiotics protects against NEC in preterm infants (38, 39). In one observational study (40) LGG supplementation was unexpectedly associated with increased rates of NEC; however, their results were limited by its single-center observational study design, selection bias with regards to which infants receive probiotics, and uncertainty whether practice changes during the study period could have affected NEC rates. Future mechanistic studies using similar combinations of different probiotic strains with or without prebiotics are also warranted.
The enthusiasm for probiotics use in preterm neonates stems from evidence suggesting that they protect against NEC and late onset sepsis, putatively by alleviating gut dysbiosis. Our data suggest that concurrent probiotic use with formula feeding or pre-treatment strategies reflecting the clinical situation, both suppress TLR pathway activation and intestinal injury. However, protective effects of LGG were related to upregulation of the TLR pathway inhibitors, SIGIRR and A20, but not significant changes in the gut microbiota. Interestingly, we also noted less bacterial invasion through preservation of the intestinal barrier. While deeper metagenomics approaches and assessment of small intestinal microbiome may reveal subtle changes in gut microbiota with probiotics use in the developing gut, our results are consistent with an emerging body of literature indicating changes in intestinal gene expression that tamponade excessive TLR signaling may be the predominant mechanism of action with probiotics. These considerations are directly relevant to the choice of probiotic use in preterm neonates as efforts to standardize preparations for clinical use are accelerated.
Supplementary Material
ACKNOWLEDGMENT
We would like to thank Dr. Tamas Jilling and Dr. Namasivayam Ambalavanan from University of Alabama at Birmingham for providing training and support in establishing the mouse model of NEC used in this study.
Statement of financial support: This study was supported by institutional funds at Children’s Mercy Hospital (VS), and by the Paul Henson Award (AC). VS, WY, HM were partly supported by 1R01 DK117296 [VS]
Footnotes
Disclosure statement: The authors have no potential or perceived conflicts of interest to disclose.
Category of Study: Basic Science
REFERENCES
- 1.Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med. 364:255–264 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jilling T, et al. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol. 177:3273–3282 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leaphart CL, et al. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol. 179:4808–4820 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Mai V, et al. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS One 6:e20647 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrow AL, et al. Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants. Microbiome 1:13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pammi M, et al. Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: a systematic review and meta-analysis. Microbiome 5:31 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.AlFaleh K, Anabrees J. Probiotics for prevention of necrotizing enterocolitis in preterm infants. Cochrane Database Syst Rev. 10;(4):CD005496 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Underwood MA. Probiotics and the prevention of necrotizing enterocolitis. J Pediatr Surg. 54:405–412 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Suez J, Zmora N, Segal E, Elinav E. The pros, cons, and many unknowns of probiotics. Nat Med. 25:716–729 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Litvak Y, Baumler AJ. The founder hypothesis: A basis for microbiota resistance, diversity in taxa carriage, and colonization resistance against pathogens. PLoS Pathog. 15:e1007563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zmora N, et al. Personalized Gut Mucosal Colonization Resistance to Empiric Probiotics Is Associated with Unique Host and Microbiome Features. Cell 174:1388–1405 e1321 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Sampath V, et al. SIGIRR genetic variants in premature infants with necrotizing enterocolitis. Pediatrics 135:e1530–1534 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fawley J, et al. Single-Immunoglobulin Interleukin-1-Related Receptor regulates vulnerability to TLR4-mediated necrotizing enterocolitis in a mouse model. Pediatr Res. 83:164–174 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Viswanathan S, Lau C, Akbari H, Hoyen C, Walsh MC. Survey and evidence based review of probiotics used in very low birth weight preterm infants within the United States. J Perinatol. 36:1106–1111 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansson ME, et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 105:15064–15069 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hackam DJ, Good M, Sodhi CP. Mechanisms of gut barrier failure in the pathogenesis of necrotizing enterocolitis: Toll-like receptors throw the switch. Semin Pediatr Surg. 22:76–82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergmann KR, et al. Bifidobacteria stabilize claudins at tight junctions and prevent intestinal barrier dysfunction in mouse necrotizing enterocolitis. Am J Pathol. 182:1595–1606 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin CR, Walker WA. Probiotics: role in pathophysiology and prevention in necrotizing enterocolitis. Semin Perinatol. 32:127–137 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Nanthakumar N, et al. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: an immature innate immune response. PLoS One 6:e17776 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin J, Qian Y, Yao J, Grace C, Li X. SIGIRR inhibits interleukin-1 receptor- and toll-like receptor 4-mediated signaling through different mechanisms. J Biol Chem. 280:25233–25241 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Wald D, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 4:920–927 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430:694–699 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Heyninck K, Beyaert R. A20 inhibits NF-kappaB activation by dual ubiquitin-editing functions. Trends Biochem Sci. 30:1–4 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Xuan NT, et al. A20 expression in dendritic cells protects mice from LPS-induced mortality. Eur J Immunol. 45:818–828 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Ouyang Y, Guner Y, Ford HR, Grishin AV. Ubiquitin-editing enzyme A20 promotes tolerance to lipopolysaccharide in enterocytes. J Immunol. 183:1384–1392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganguli K, et al. Probiotics prevent necrotizing enterocolitis by modulating enterocyte genes that regulate innate immune-mediated inflammation. Am J Physiol Gastrointest Liver Physiol. 304:G132–141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu W, et al. Bifidobacterium adolescentis protects against necrotizing enterocolitis and upregulates TOLLIP and SIGIRR in premature neonatal rats. BMC Pediatr. 17:1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Igarashi M, et al. Alteration in the gastric microbiota and its restoration by probiotics in patients with functional dyspepsia. BMJ Open Gastroenterol. 4:e000144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koga Y, Ohtsu T, Kimura K, Asami Y. Probiotic L. gasseri strain (LG21) for the upper gastrointestinal tract acting through improvement of indigenous microbiota. BMJ Open Gastroenterol. 6:e000314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Underwood MA, et al. Bifidobacterium longum subsp. infantis in experimental necrotizing enterocolitis: alterations in inflammation, innate immune response, and the microbiota. Pediatr Res. 76:326–333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghadimi D, Helwig U, Schrezenmeir J, Heller KJ, de Vrese M. Epigenetic imprinting by commensal probiotics inhibits the IL-23/IL-17 axis in an in vitro model of the intestinal mucosal immune system. J Leukoc Biol. 92:895–911 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Vahamiko S, et al. The impact of probiotic supplementation during pregnancy on DNA methylation of obesity-related genes in mothers and their children. Eur J Nutr. 58:367–377 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Chen D, et al. Effect of Lactobacillus rhamnosus hsryfm 1301 on the Gut Microbiota and Lipid Metabolism in Rats Fed a High-Fat Diet. J Microbiol Biotechnol. 25:687–695 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Li Z, et al. Effects of Metabolites Derived From Gut Microbiota and Hosts on Pathogens. Front Cell Infect Microbiol. 8:314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasan N, Yang H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ. 7:e7502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohland CL, Macnaughton WK. Probiotic bacteria and intestinal epithelial barrier function. Am J Physiol Gastrointest Liver Physiol. 298:G807–819 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Meyer MP, Alexander T. Reduction in necrotizing enterocolitis and improved outcomes in preterm infants following routine supplementation with Lactobacillus GG in combination with bovine lactoferrin. J Neonatal Perinatal Med. 10:249–255 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Fernandez-Carrocera LA, et al. Double-blind, randomised clinical assay to evaluate the efficacy of probiotics in preterm newborns weighing less than 1500 g in the prevention of necrotising enterocolitis. Arch Dis Child Fetal Neonatal Ed. 98:F5–9 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Kane AF, Bhatia AD, Denning PW, Shane AL, Patel RM. Routine Supplementation of Lactobacillus rhamnosus GG and Risk of Necrotizing Enterocolitis in Very Low Birth Weight Infants. J Pediatr. 195:73–79 e72 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.