

Abstract
Medulloblastoma (MB) is the most common malignant brain tumor in childhood. According to the World Health Organization (WHO) classification of central nervous system (CNS) tumors, this embryonal tumor is divided into a wingless (WNT)-activated, Sonic hedgehog (SHH)-activated, and non-WNT/non-SHH entity. The latter is poorly defined but frequently carries mutations in Brahma-related gene 1 (BRG1) or amplifications of MYCN. Here, we investigated whether a combination of a Brg1 knockout and an overexpression of MYCN in cerebellar granule neuron precursors or multipotent neural stem cells is sufficient to drive brain tumor formation in mice. To this end, we generated Math1-creERT2::Brg1fl/fl::lslMYCN and hGFAP-cre::Brg1fl/fl::lslMYCN mice, respectively. We did not observe brain tumor formation in any of these models. hGFAP-cre::Brg1fl/fl::lslMYCN mice revealed severe CNS abnormalities with short survival, similar to the situation with a sole loss of Brg1, as we previously described. Investigation of Math1-creERT2::Brg1fl/fl::lslMYCN mice with a tamoxifen induction at postnatal day 3 revealed a regular survival but significant increase in cerebellar granule neuron precursor proliferation, followed by a delayed inward migration of these cells. This is in stark contrast to the hypoplastic cerebellum that we previously observed after embryonic deletion of Brg1 in Math1 positive cerebellar granule neurons. Our results indicate a time-specific function of Brg1 in cerebellar granule neuron precursors. Yet, the exact temporal and spatial origin of non-WNT/non-SHH MB remains unclear.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12311-020-01219-2.
Keywords: Medulloblastoma, BRG1, MYCN, Mouse, Migration deficit
Introduction
Medulloblastoma (MB) is the most common malignant brain tumor in children [1]. This embryonal tumor arises in the posterior fossa and can be divided into at least three molecular subgroups: wingless (WNT)-activated MB, Sonic hedgehog (SHH)-activated MB, and non-WNT/non-SHH MB [2]. WNT and SHH MBs are characterized by activating mutations in the respective pathways, but driver mutations in non-WNT/non-SHH, which can be further segregated into Group 3 and Group 4 MBs, are less well understood [3]. A number of large-scale sequencing studies revealed that missense mutations in the epigenetic modifier Brahma-related gene 1 (BRG1, SMARCA4, BAF190A) are found in 4.3–8.8% of all MBs, but especially in WNT and Group 3 MBs [4–6]. In the study performed by Jones et al., BRG1 was the most common mutated gene in Group 3 MB. BRG1 encodes one of the two mutually exclusive ATPase subunits of the SWItch/sucrose nonfermenting (SWI/SNF) chromatin remodeling complex, which regulates gene expression by increasing nucleosome mobility [7–9]. It has been described as a tumor suppressor in different cancer entities, such as rhabdoid tumors, small cell carcinoma of the ovary, hypercalcemic type (SCCOHT), and lung cancer [10–14]. Still, a Brg1 knockout in various different neural stem cell (NSC) populations in mice was not sufficient to drive tumor development but caused hypoplasia of diverse brain regions [15–17]. Amplifications of the proto-oncogene MYCN have been identified in several cancer entities, including tumors of the central nervous system (CNS) such as high-grade gliomas, spinal ependymoma and MB [18–21]. In MB, they are found in SHH, Group 3 and Group 4 MBs and are associated with a poor outcome [4–6, 19, 22]. MYCN is a transcription factor of the MYC family and is important for cell growth, apoptosis, tumor cell metabolism and normal cerebellar development [21, 23, 24]. Swartling et al. demonstrated that overexpression of MYCN in Glutamate transporter 1 (Glt1) positive cells gives rise to murine MB [25]. However, these tumors occurred rather late in development and accumulated sporadic p53 mutations in addition to the MYCN alteration [26]. Furthermore, due to the broad expression of the promoter and the late tumor detection, the exact anatomical origin of these tumors remains obscure. The same group recently reported that ectopic expression of MYCN in human induced pluripotent stem cell-derived neuroepithelial stem (NES) cells increases proliferation in vitro. Orthotopic transplantation of these NES cells into mice results in tumor formation resembling human SHH MB, again without precise information about the tumor origin [27]. In hGFAP expressing cells, MYCN amplification did not result in MB formation, possibly due to the lack of a second hit [28]. In this study, we were interested whether Brg1 knockout and MYCN overexpression might have synergistic effects in the development of tumors in the posterior fossa. Therefore, we generated Math1-creERT2::Brg1fl/fl::lslMYCNfl/fl (Mert::Bfl/fl::Nfl/fl) and hGFAP-cre::Brg1fl/fl::lslMYCNfl/fl (hG::Bfl/fl::Nfl/fl) mice to investigate the combination in postnatal granule neuron precursor cells (GNPCs) and multipotent NSCs, respectively [29, 30]. Mice carrying the inducible creERT2 transgene under the control of the Math1 promoter received tamoxifen at postnatal day 3 (P3) to induce the cre recombinase. In hG::Bfl/fl::Nfl/fl mice, the cre recombinase was constitutively active from embryonal day 13.5 (E13.5) onwards [30]. We observed that the loss of Brg1 in combination with an additional overexpression of MYCN in Math1 expressing cells resulted in changes regarding cell survival parameters in the external granular layer (EGL) of the cerebellum. Furthermore, GNPCs showed delayed inward migration. However, the adult cerebellum appeared normal with regular organization into inner granular layer (IGL), Purkinje cell layer (PCL) and molecular layer (ML) and without any signs of tumor development. Amplification of MYCN in addition to the loss of Brg1 in hGFAP expressing cells did not rescue the phenotype caused by the deprivation of Brg1 alone. As we published earlier, Brg1 deficiency in hGFAP positive cells causes a hypoplastic cerebellum [17]. Likewise, hG::Bfl/fl::Nfl/wt, and hG::Bfl/fl::Nfl/fl mice presented with an underdeveloped cerebellum without organization into lobuli and with absence of the characteristic cerebellar layering. They died after about 2 weeks without any signs of tumor formation. Hence, we conclude that MYCN amplification and Brg1 deficiency disturb cerebellar development but are not sufficient to drive brain tumors originating from hGFAP or Math1 positive precursor cells.
Material and Methods
Mice
hGFAP-cre (JAX #004600) and Math1-creERT2 (JAX #7684) mice were purchased from The Jackson Laboratory [30, 31]. Brg1fl/fl and lox-STOP-lox-MYCNfl/fl (lslMYCNfl/fl) mice have previously been generated and described [32, 33]. These mice were crossed to generate Math1-creERT2::Brg1fl/wt (Mert::Bfl/wt), Math1-creERT2::Brg1fl/fl (Mert::Bfl/fl), Math1-creERT2::Brg1fl/fl::lslMYCNfl/wt (Mert::Bfl/fl::Nfl/wt), Math1-creERT2::Brg1fl/fl::lslMYCNfl/fl (Mert::Bfl/fl::Nfl/fl), hGFAP-cre::Brg1fl/wt::lslMYCNfl/wt (hG::Bfl/wt::Nfl/wt), hGFAP-cre::Brg1fl/wt::lslMYCNfl/fl (hG::Bfl/wt::Nfl/fl), hGFAP-cre::Brg1fl/fl::lslMYCNfl/wt (hG::Bflfl::Nfl/wt), and hGFAP-cre::Brg1fl/fl::lslMYCNfl/fl (hG::Bflfl::Nfl/fl) mice. They were kept on a C57Bl/6J background. Mice carrying the Math1-creERT2 knock-in and respective controls were treated with a single dose of 0.6 mg tamoxifen (Sigma-Aldrich) dissolved in corn oil at P3 via intraperitoneally (i.p.) injections. To label proliferating cells in vivo, 5-bromo-2-deoxyuridine (BrdU, Sigma-Aldrich) was injected i.p. (25 μg/g bodyweight) 2 h prior to sacrifice. Genotyping of genomic DNA from mouse ear biopsies or tail tips was performed by PCR. Mice were kept on a 12 h dark/light cycle; water and food were available ad libitum. Animals of both sexes were used for the experiments. All experimental procedures were approved by the Government of Hamburg, Germany (113/16).
Immunohistochemistry
For hematoxylin and eosin (H&E) and immunohistochemistry (IHC) stains, brain tissue was fixed in 4% paraformaldehyde/PBS for at least 12 h. The tissue was dehydrated, embedded in paraffin, and sectioned at 4 μm according to standard protocols. All IHC stains were performed on a Ventana System (Roche) using standard protocols. The following antibodies were used: mouse anti-BrdU (Invitrogen, clone MoBU-1, #B35128, 1:100), rabbit anti-Brg1 (Abcam, ab110641, 1:200), rabbit anti-cleaved Caspase 3 (cl. Casp3; Asp175; Cell Signaling Technology, #9664, 1:100), rabbit anti-MycN (Cell Signaling, #51705, 1:100), mouse anti-Pax6 (DSHB, 1:50), mouse anti-phospho-Histone H3 (pHH3, Cell Signaling Technology, #9706, 1:200), rabbit anti-S100 (DAKO, Z0311, 1:100).
Image Quantifications and Statistical Analysis
In order to quantify the proportion of cells positive for a specific marker, we used IHC stained sagittal sections and counted their numbers in relation to the total number of cells per field using the ImageJ software (Wayne Rasband, National Institute of Health, USA). For marker expression in the EGL, we quantified the cells in the EGL between lobuli V and VI. To analyze the proportion of Pax6 positive cells in the ML, we counted the cells in lobulus VI. Marker expression was quantified for at least three animals per genotype by a blinded investigator. All statistical analyses were performed using Prism Software Version 7 (GraphPad Software, Inc., San Diego, USA). For quantifications of marker expression in the EGL and the ML, a Tukey’s multiple comparisons test and for survival analyses, a log-rank Mantel-Cox test was performed.
Results
A Postnatal Loss of Brg1 With or Without Additional Amplification of MYCN in Math1 Expressing Cells Causes an Increase in Proliferation
A prenatal loss of Brg1 in Math1 positive cells results in cerebellar hypoplasia [16]. Therefore, we chose the tamoxifen-inducible creERT2 recombinase to generate mice with a postnatal Brg1 loss either alone or with additional overexpression of the human MYCN in Math1 expressing cells. We injected tamoxifen at P3 to activate cre-mediated recombination. In a first approach, we were interested in the short-term effects and sacrificed the animals 5 days after tamoxifen injections at P8. At this time, proliferation of GNPCs peaks during normal murine cerebellar development [34, 35]. H&E stains revealed no morphological alterations at P8 as animals of all genotypes had normal cerebella with regular lobuli and distinct layers (Fig. 1a, b, h, i, o, p, v, w, ac, ad). We further investigated the extent of Brg1 loss and MYCN overexpression by IHC. In controls and in Mert::Bfl/wt animals, all cells were positive for Brg1 and MYCN was expressed in the outer EGL (Fig. 1c, d, j, k). The EGL of the remaining three genotypes consisted of Brg1 negative cells in the outer EGL and Brg1 positive cells in the inner EGL (Fig. 1q, x, ae). This result was expected, since we induced Brg1 loss by tamoxifen treatment not before P3. The number of MYCN expressing cells was significantly increased in the presence of the lslMYCN transgenes (71% and 89% in Mert::Bfl/fl::Nfl/wt and Mert::Bfl/fl::Nfl/fl, respectively) compared to controls (33%), Mert::Bfl/wt (31%) and Mert::Bfl/fl (38%) mice (Fig. 1y, af, aj).
Fig. 1.

Mert::Bfl/fl::Nfl/wt and Mert::Bfl/fl::Nfl/fl mice display significant alterations in the expression of markers for apoptosis and cell proliferation in the external granule layer. Representative sagittal H&E sections of mouse cerebella are shown at P8 (a, b, h, i, o, p, v, w, ac, ad). There are no morphological alterations in the EGL in any of the mutants compared to controls. Brg1 knockout and MYCN amplification are confirmed by IHC (c, d, j, k, q, r, x, y, ae, af). Apoptosis induction is indicated by cl. Casp3 stainings (e, l, s, z, ag). Proliferating cells are identified by BrdU and pHH3 stainings (f, g, m, n, t, u, aa, ab, ah, ai). Quantifications of MYCN, cl. Casp3, BrdU and pHH3 positive cells are shown in aj, ak, al and am, respectively. The control group includes Bfl/fl, Bfl/wt, Bfl/fl::Nfl/wt and Bfl/fl::Nfl/fl mice (n = 7). The mutant groups are Mert::Bfl/wt (n = 3), Mert::Bfl/fl (n = 7), Mert::Bfl/fl::Nfl/wt (n = 5), and Mert::Bfl/fl::Nfl/fl (n = 7). The scale bar in A corresponds to 400 μm and is representative for h, o, v and ac. The scale bar in B corresponds to 50 μm and is representative for all other panels. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s., not significant
Next, we investigated the influence of Brg1 loss and MYCN overexpression on cell survival by examining apoptosis and proliferation marker expression. We stained for cl. Casp3 as an indicator for apoptosis induction (Fig. 1e, l, s, z, ag, ak). Quantification of cl. Casp3 stainings revealed no significant differences in the number of apoptotic cells in the EGL of control mice (0.5%) and mice with both a hetero- (0.47%) and a homozygous loss of Brg1 (0.76%). However, in Mert::Bfl/fl::Nfl/fl mice, but not in Mert::Bfl/fl::Nfl/wt mice, the additional overexpression of MYCN caused a significant increase in apoptosis in the EGL as 2.24% and 1.36% of cells were cl. Casp3 positive, respectively (Fig. 1ak). In order to investigate proliferation, we pulsed the animals 2 h prior to sacrifice with BrdU, a thymidine analog that is incorporated into newly synthesized DNA during the S phase of the cell cycle. We stained for BrdU and additionally for pHH3 to examine two independent proliferation markers (Fig. 1f, g, m, n, t, u, aa, ab, ah, ai). There were no significant differences in the number of BrdU positive cells in the EGL of control (27.0%), Mert::Bfl/wt (23.6%) and Mert::Bfl/fl mice (32.7%), even though there was a trend towards more proliferating cells in the latter (Fig. 1al). However, in case of a simultaneous MYCN amplification, there were significantly more BrdU positive cells in the EGL. In Mert::Bfl/fl::Nfl/wt mice, 40.2% of cells and in Mert::Bfl/fl::Nfl/fl mice, 39.5% of cells had incorporated BrdU during S phase (Fig. 1al). Likewise, the percentage of pHH3 expressing cells was insignificantly different in case of a Brg1 knockout without MYCN amplification (Fig. 1am). In the EGL of controls, Mert::Bfl/wt mice and Mert::Bfl/fl mice, 7.86%, 9.42%, and 10.8% were pHH3 positive, respectively. Again, these results indicate that a Brg1 deficiency caused a trend towards more proliferating cells. Amplification of MYCN caused a significant rise in the number of pHH3 expressing cells in the EGL of Mert::Bfl/fl::Nfl/fl mice (14.5%), but not of Mert::Bfl/fl::Nfl/wt mice (11.2%).
Taken together, the combination of a Brg1 deficiency with MYCN overexpression significantly alters the expression of cell survival parameters in the EGL at P8.
Brg1 Deficiency and MYCN Amplification Decreases Inward Migration of Granule Neurons
In mice, cerebellar cortical development is finalized at an age of 3 weeks resulting in three distinct layers: ML, PCL and IGL [36]. In order to analyze how the postnatal knockout of Brg1 with or without simultaneous overexpression of MYCN influenced the ontogenesis of the cerebellum, we sacrificed the mice at P21 and investigated the morphology by H&E stains (Fig. 2a, b, e, f, i, j, m, n, q, r). In Mert::Bfl/fl mice, it appeared as if more cells were present in the ML compared to control and Mert::Bfl/wt mice. This effect seemed to be even more pronounced in Mert::Bfl/fl::Nfl/wt mice and Mert::Bfl/fl::Nfl/fl mice. To validate, whether the supernumerary cells in the ML were Brg1 deficient, we stained for Brg1 (Fig. 2c, g, k, o, s). In all mice not carrying a homozygously floxed Brg1 allele, Brg1 was expressed in all cells. In all three mutants, Brg1 negative cells were not only observed in the IGL, but also in the ML. This suggested that Brg1 loss resulted in decelerated migration of GNPCs from the EGL to the IGL. In order to verify that these cells were GNPCs, we stained for Pax6 and quantified the proportion of Pax6 expressing cells in the ML (Fig. 2d, h, l, p, t, y). In both, the ML of controls and of Mert::Bfl/wt mice, 24.6% and 25.4% of cells were Pax6 positive. In Mert::Bfl/fl, Mert::Bfl/fl::Nfl/wt, and Mert::Bfl/fl::Nfl/fl mice, there were significantly more GNPCs in the ML, as indicated by Pax6 positivity (55.8%, 66.2%, and 78.7%, respectively).
Fig. 2.

Mert::Bfl/fl::Nfl/wt and Mert::Bfl/fl::Nfl/fl mice display increased numbers of Pax6 positive cells in the molecular layer. Representative sagittal H&E sections of whole cerebella are shown at P21 and at adult age (a, e, i, m, q and u). Brg1 knockout (c, g, k, o, s, w) is examined by IHC. Sagittal H&E (b, f, j, n, r, v) and Pax6 (d, h, l, p, t, x) stainings of the EGL and IGL display an aggregation of Brg1 deficient granule cells. Quantification of Pax6 expressing cells in the ML of animals at P21 are shown in Y. The control group includes Bfl/fl::Nfl/wt and Bfl/fl::Nfl/fl mice (n = 4). The mutant groups (Mert::Bfl/wt, Mert::Bfl/fl, Mert::Bfl/fl::Nfl/wt and Mert::Bfl/fl::Nfl/fl) include 3 animals each. The scale bar in A corresponds to 400 μm and is representative for e, i, m, q, and u. The scale bar in B corresponds to 50 μm and is representative for all other panels. **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s., not significant
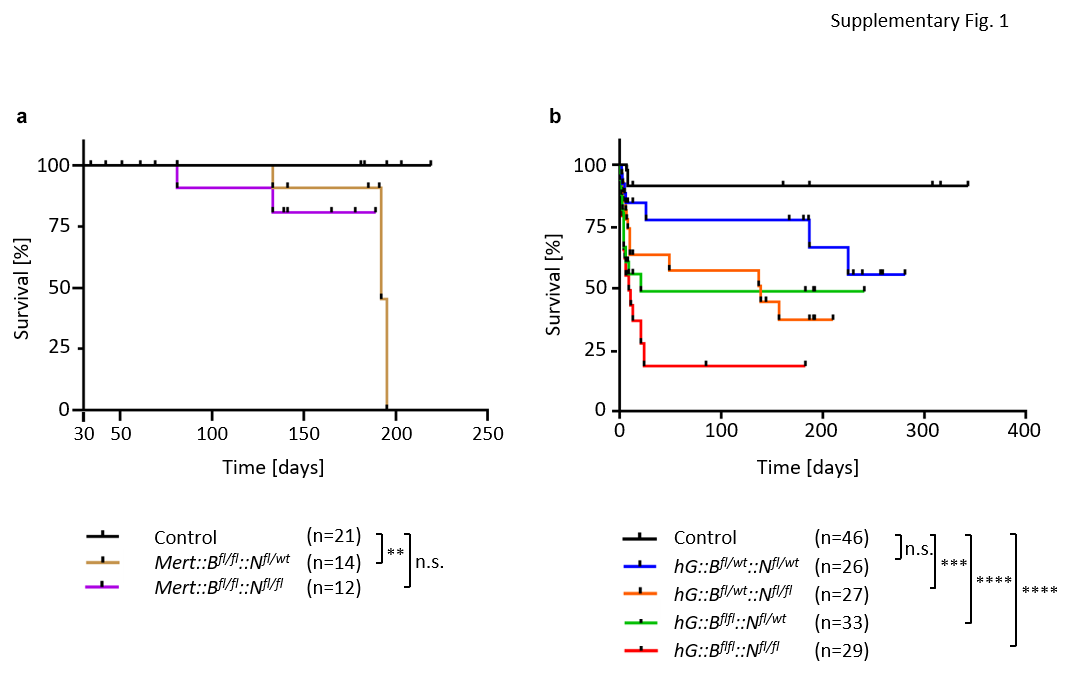
In order to validate how the changes in apoptosis and proliferation of GNPCs at P8 and the alterations in the GNPCs’ migratory behavior affected the morphology of the adult cerebellum, we examined the brains of 6-month-old mice. Similar to Mert::Bfl/wt, Mert::Bfl/fl, and Mert::Bfl/fl::Nfl/wt mice (data not shown), H&E analysis of cerebella from Mert::Bfl/fl::Nfl/fl mice revealed that the overall morphology appeared normal (Fig. 2u–x). Brg1 negative cells were found in both, the ML and the IGL, and only a very small fraction of Pax6 expressing cells was present in the ML, similar to the expression profile of control P21 cerebella. We therefore conclude that GNPC inward migration is delayed after loss of Brg1 and overexpression of MYCN but is completed at the age of 6 months. In order to exclude tumor growth in a small proportion of such animals, we observed a total of 14 Mert::Bfl/fl::Nfl/wt and 12 Mert::Bfl/fl::Nfl/fl animals over a period of 6 months (Supplementary Fig. 1a). Three and two animals of these cohorts died, respectively, without any preceding symptoms. Therefore, we were not able to analyze their brains or bodies. All other animals underwent full autopsy after 6 months, but we did not detect any tumor growth or other abnormalities. Although we cannot report on the autopsy of 5 out of 25 animals, we assume that a simultaneous loss of Brg1 and overexpression of MYCN in Math1 positive GNPCs is not sufficient to drive tumor growth.
Amplification of MYCN in Multipotent NSCs Does Not Rescue the Effects Caused by the Loss of Brg1
In order to examine the combination of Brg1 deficiency and MYCN amplification in a spatially and temporally different setting, we generated hG::B::N mice. In contrast to the induced cre recombinase activity in postnatal GNCPs in Mert::B::N animals, hGFAP drives the constitutive expression of the cre recombinase in multipotent NSCs from E13.5 onwards. Consequently, Brg1 loss and MYCN overexpression can be observed in almost all cerebellar cells, except for Purkinje cells and choroid plexus epithelium [30]. We previously reported that multipotent NSCs marked by hGFAP expression depend on the proper expression of Brg1 to give rise to all major brain regions [17]. For instance, the cerebella of hGFAP-cre::Brg1fl/fl mice are hypoplastic without foliation or lamination. In the present study, we investigated, whether an additional MYCN amplification was able to rescue this phenotype or would even give rise to brain tumors. For this purpose, we generated hG::Bfl/wt::Nfl/wt, hG::Bfl/wt::Nfl/fl, hG::Bflfl::Nfl/wt and hG::Bflfl::Nfl/fl mice. In case of a heterozygous Brg1 deficiency, the cerebella at P8 appeared normal in H&E stains compared to controls (Fig. 3a, b, f, g, k, l). As expected, Brg1 was detectable in all cells of these mice (Fig. 3c, h, m), whereas the number of MYCN positive cells seemed increased in the EGL of hG::Bfl/wt::Nfl/wt and hG::Bfl/wt::Nfl/fl mice (Fig. 3d, i, n)In contrast, the cerebella of hG::Bflfl. ::Nfl/wt and hG::Bflfl::Nfl/fl mice were severely underdeveloped and resembled those of hGFAP-cre:.Brg1fl/fl mice. Neither lobules nor distinct layers were detectable in H&E stains (Fig. 3p, q, u, v). Like in hGFAP-cre::Brg1fl/fl mice, Brg1 expression was lost in a fraction of cells distributed across the entire cerebellum of hG::Bflfl::Nfl/wt and hG::Bflfl::Nfl/fl mice (Fig. 3r, w). As the hGFAP promoter is expressed in the majority of cerebellar cells or their ancestors [30], Brg1 deficiency was likely to be present in most cell types. However, only a small proportion of cells were positive for MYCN (Fig. 3s, x). This indicates either that the recombination of two floxed transgenes was not efficient or that especially those cells with enhanced MYCN expression were depleted from the developing brain until P8.
Fig. 3.

hG::Bfl/fl::Nfl/wt and hG::Bfl/fl::Nfl/fl mice display a hypoplastic cerebellum. Representative sagittal H&E sections of mouse cerebella at P8 indicate that compared to controls (a, b) a heterozygous Brg1 knockout has no morphological consequences (f, g, k, l), whereas a homozygous Brg1 loss leads to a hypoplastic cerebellum (p, q, u, v). Panels c, h, m, r and w show Brg1 stains and d, i, n, s, and x MYCN stains. S100 stains are presented in panels e, j, o, t, y. The control group includes Bfl/fl::Nfl/wt and Bfl/fl::Nfl/fl mice (n = 5). The mutant groups include hG::Bfl/wt::Nfl/wt (n = 4), hG::Bfl/wt::Nfl/fl (n = 3), hG::Bfl/fl::Nfl/wt (n = 4), and hG::Bfl/fl::Nfl/fl (n = 4). The scale bar in A corresponds to 400 μm in a, f, k, p, and u and the scale bar in B corresponds to 50 μm and is representative for all other panels
Next, we examined, whether the absence of distinct layers in hG::Bflfl::Nfl/wt and hG::Bflfl::Nfl/fl cerebella was caused by impaired neuronal migration due to altered Bergmann glia cell morphology. Proper neuronal migration is highly dependent on Bergmann glia cells [37], and hGFAP-mediated cre expression is present in these cells [30]. We employed S100 as a Bergmann glia marker [38] that stains the entire cells including their radial processes. In controls, hG::Bfl/wt::Nfl/wt and hG::Bfl/wt::Nfl/fl mice, Bergmann glia cells were located in the PCL and their processes extended to the pial surface (Fig. 3e, j, o). In hG::Bflfl::Nfl/wt and hG::Bflfl::Nfl/fl cerebella, there were almost no S100 positive cells detectable and those that were present did not have any radial processes (Fig. 3t, y).
Finally, we investigated the mice for the development of disease-related symptoms for half a year (Supplementary Fig. 1b). Fifteen out of 33 animals with a homozygous loss of Brg1 had to be sacrificed in the first 2 weeks of life, indicating that the overexpression of MYCN did not compensate for the loss of Brg1. In the few animals that survived into adulthood, we did not observe any Brg1 deficient cells in their brains (data not shown). This suggests that in these mice, the simultaneous recombination of the two floxed transgenes (Brg1 and lslMYCN) was not efficient, causing long-term survival. Of note, none of these mice developed a brain tumor. However, some hG::Bfl/wt::Nfl/wt, hG::Bfl/wt::Nfl/fl and hG::Bfl/fl::Nfl/wt mice died due to neuroendocrine tumors of the pancreas and the pituitary gland as already reported for hGFAP-cre::lslMYCN mice (data not shown) [28]. Hence, the development of these tumors was likely caused by the amplification of MYCN but not by the Brg1 deficiency.
In summary, we did not observe any cooperative effects of Brg1 loss and MYCN amplification in these mice, as hG::Bfl/wt::Nfl/wt and hG::Bfl/wt::Nfl/fl mice developed similar phenotypes as hGFAP-cre::lslMYCN mice. Likewise, hG::Bflfl::Nfl/wt and hG::Bflfl::Nfl/fl mice developed morphological alterations in the brain, which resembled the abnormalities observed in hGFAP-cre:.Brg1fl/fl mice.
Discussion
Mutations in BRG1 and amplifications of MYCN have frequently been reported in MBs, especially those of the non-WNT/non-SHH subgroup. This subgroup comprises two thirds of all MBs, but the molecular mechanisms driving oncogenesis are insufficiently understood [4–6, 19, 39]. Therefore, the aim of this study was to elucidate, whether the combination of Brg1 loss and MYCN overexpression in postnatal GNPCs or multipotent NSCs in mice could model the development of human non-WNT/non-SHH MB. To this end, we used Mert::B::N and hG::B::N mice, and examined histological alterations and survival of these animals.
First, we studied the short-term effects of a postnatal Brg1 loss and MYCN amplification on cell survival parameters of the CGNPs in the EGL of Mert::B::N mice. At P8, MYCN overexpression in a Brg1 deprived EGL caused a significant increase in both, proliferating and apoptotic cells. Normal cerebellar development is a highly orchestrated sequence of proliferation, differentiation, migration and programmed cell death. The latter is important to eliminate supernumerary cells from the developing brain (reviewed by [40]). Hence, the increase in cl. Casp3 expressing cells indicating an increase of apoptotic cells in the EGL might just be a compensatory mechanism for the enhancement of proliferation caused by Brg1 loss and MYCN overexpression. MYCN is known to be essential for SHH-mediated CGNP proliferation and is amplified and upregulated in human and murine MB, respectively [21, 24, 41, 42]. Previous results already suggested that the enhancement in proliferation caused by MYCN amplification alone does not result in MB formation from CGNPs [28]. Even though the postnatal loss of Brg1 alone appeared to show a trend towards enhanced proliferation, the combination of MYCN overexpression and Brg1 loss did not synergize and did still not result in tumor formation. Nonetheless, we cannot completely preclude that knockdown of Brg1 and overexpression of MYCN might not be fully parallelized events as it might take longer/shorter to accumulate MYCN protein than to lose the Brg1 protein.
The finding of a slightly increased (although not significant) EGL proliferation in Mert::Bfl/fl mice after application of tamoxifen at P3 is still noteworthy, since it contradicts previous results from (non-inducible) Math1-cre::Brg1fl/fl mice, in which the ATPase is lost at around E10.5. The latter suffer from a severely hypoplastic cerebellum caused by decreased proliferation of GNPCs [16]. Similarly, Zhan et al. reported that loss of Brg1 at P0 in Nestin-creERT2::Brg1fl/fl mice resulted in smaller cerebella [43]. Together, in combination with these published data, our here presented results highlight the time-specific role of Brg1 in the developing cerebellum, a phenomenon that has also been described for other major players in cerebellar development [44, 45].
In addition to the changes in proliferation and apoptosis at P8, we recognized that the induced Brg1 deficiency in combination with MYCN amplification at P3 resulted in the accumulation of cells in the ML at P21. During murine cerebellar development, CGNPs in the EGL first stop proliferating, then start to differentiate and finally migrate towards the IGL, where they start to form synaptic connections [46]. Quantification of Pax6 positive cells revealed that Brg1 loss by itself or in combination with MYCN amplification caused a significant increase in the proportion of granule cells in the ML. This indicates that the migratory behavior of granule neurons was impaired in Mert::Bfl/fl, Mert::Bfl/fl::Nfl/wt and Mert::Bfl/fl::Nfl/fl mice. Of note, many of the cells in the ML were negative for Brg1 in IHC, supporting the hypothesis that the genetic alterations in our mutant mice were directly responsible for the observed phenotype. Likewise, Brg1 deprivation in cerebellar explants from hGFAP-cre::Brg1fl/fl mice lead to decreased migration of late-migrating cells in vitro [17]. Additionally, in hG::Bflfl::Nfl/wt and hG::Bflfl::Nfl/fl mice, the number and morphology of Bergmann glia was severely diminished, indicating that the observed lack of layering in these mice, might be partially explained by disturbed migration as well. Our results suggest that the addition of MYCN overexpression enhanced the migration deficit caused by the Brg1 deficiency. MYCN is essential for normal cerebellar development as indicated by different mouse models examining MYCN knockout [23, 47]. However, to our knowledge the need for balanced MYCN levels for granule cell migration has not been described before. In the adult mice, i.e. 6 months or older, there were no misplaced Pax6 positive cells left in the ML, indicating that granule cell migration was delayed but not permanently inhibited in our model.
A fraction of hG::B::N mice developed neuroendocrine tumors of the pancreas and the pituitary gland as previously published for hGFAP-cre::lslMYCN mice [28]. Consequently, in these mice, the amplification of MYCN determined the phenotype, whereas the heterozygous Brg1 loss (or the incomplete recombination in mice with a homozygous Brg1 loss) had no obvious impact. Vice versa, hG::Bflfl::Nfl/wt and hG::Bflfl::Nfl/fl mice presented with symptoms resembling those of hGFAP-cre::Brg1fl/fl mice, including early postnatal death, hydrocephalus and hypoplasia of the cerebrum and cerebellum [17]. In these mice, the effect of the homozygous Brg1 deficiency was dominant and the additional overexpression of MYCN did not seem to have an impact. However, as we did not detect MYCN protein expression by IHC in the majority of cerebellar cells at P8, we cannot rule out that the recombination efficiency of the loxP sites of the lslMYCN transgene was decreased in case of the additional presence of a homozygously floxed Brg1 gene. Another more likely explanation for the lack of MYCN expressing cells might be that most of the cells hit by the loss of Brg1 and MYCN overexpression have not at all developed or have died by P8. Most importantly, Brg1 deficiency in combination with overexpression of MYCN in hGFAP positive NSCs does not result in brain tumor formation.
Conclusion
In this study, we demonstrate that the combination of a Brg1 deficiency with MYCN amplification in mice is not sufficient to drive tumor formation, neither in postnatal Math1 expressing cells nor in hGFAP positive multipotent NSCs. Consequently, either these cells do not represent cells of origin for non-WNT/non-SHH MBs or the combination of Brg1 loss and MYCN amplification does not provide the genetic basis for tumorigenesis. Still, the animals presented with minor developmental disturbances. Furthermore, in Math1 positive cells, the combination of the two genetic events seems to have additive effects regarding survival and migration of granule neurons. In contrast, the homozygous knockout of Brg1 defined the phenotype in hG::Bflfl::Nfl/wt and hG::Bflfl::Nfl/fl mice, whereas the addition of MYCN amplification had no obvious additional effect.
Supplementary Information
{kind=link}
Kaplan-Meier curves of Mert::Bfl/fl::Nfl/wt, Mert::Bfl/fl::Nfl/fl, hG::Bfl/wt::Nfl/wt, hG::Bfl/wt::Nfl/fl, hG::Bflfl::Nfl/wt and hG::Bflfl::Nfl/fl mice. Kaplan-Meier curves from P30 onwards of Mert::Bfl/fl::Nfl/wt mice (brown) and Mert::Bfl/fl::Nfl/fl (purple) shows the decreased survival compared to respective controls (black) in A. However, none of the animals developed a tumor. The survival curves of hG::Bfl/wt::Nfl/wt (blue), hG::Bfl/wt::Nfl/fl (orange), hG::Bflfl::Nfl/wt (green) and hG::Bflfl::Nfl/fl mice (red) show diminished survival compared to respective controls (black) and are depicted in B. Still, none of the animals developed a brain tumor. **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s., not significant. (PNG 51.4 kb).
{kind=link}
Acknowledgments
We thank Dr. J. Schulte (Charité Berlin, Germany) for providing lslMYCNfl/fl mice. We further thank Anne Reichstein, Jacqueline Kolanski, Vanessa Thaden, and Kristin Hartmann for expert technical assistance. Finally, we acknowledge the support of the Small animal models core facility of the Heinrich Pette Institute.
Author Contribution
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Dörthe Holdhof and Ji Hoon On. The first draft of the manuscript was written by Dörthe Holdhof. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work received funding from the Deutsche Krebshilfe, and U.S. was additionally supported by the Fördergemeinschaft Kinderkrebszentrum Hamburg. JHO was supported by a Juli Harnack fellowship for medical students from the Fördergemeinschaft Kinderkrebszentrum Hamburg.
Compliance with Ethical Standards
Conflict of Interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dörthe Holdhof and Ji Hoon On contributed equally to this work.
References
- 1.Ostrom QT, de Blank PM, Kruchko C, Petersen CM, Liao P, Finlay JL, Stearns DS, Wolff JE, Wolinsky Y, Letterio JJ, Barnholtz-Sloan JS. Alex’s Lemonade Stand Foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro-oncology. 2015;16(Suppl 10):x1–x36. doi: 10.1093/neuonc/nou327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louis D, Ohgaki H, Wiestler O, Cavenee W. WHO classification of tumours of the central nervous system. Lyon: International Agency for Research on Cancer; 2016. [Google Scholar]
- 3.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, Greulich H, Lawrence MS, Lennon NJ, McKenna A, Meldrim J, Ramos AH, Ross MG, Russ C, Shefler E, Sivachenko A, Sogoloff B, Stojanov P, Tamayo P, Mesirov JP, Amani V, Teider N, Sengupta S, Francois JP, Northcott PA, Taylor MD, Yu F, Crabtree GR, Kautzman AG, Gabriel SB, Getz G, Jäger N, Jones DTW, Lichter P, Pfister SM, Roberts TM, Meyerson M, Pomeroy SL, Cho YJ. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, Chalhoub N, Baker SJ, Huether R, Kriwacki R, Curley N, Thiruvenkatam R, Wang J, Wu G, Rusch M, Hong X, Becksfort J, Gupta P, Ma J, Easton J, Vadodaria B, Onar-Thomas A, Lin T, Li S, Pounds S, Paugh S, Zhao D, Kawauchi D, Roussel MF, Finkelstein D, Ellison DW, Lau CC, Bouffet E, Hassall T, Gururangan S, Cohn R, Fulton RS, Fulton LL, Dooling DJ, Ochoa K, Gajjar A, Mardis ER, Wilson RK, Downing JR, Zhang J, Gilbertson RJ. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv. 2015;1:e1500447. doi: 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alfert A, Moreno N, Kerl K. The BAF complex in development and disease. Epigenetics Chromatin. 2019;12:19. doi: 10.1186/s13072-019-0264-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, Giangaspero F, Giannini C, Judkins AR, Frühwald MC, Obser T, Schneppenheim R, Siebert R, Paulus W. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol. 2011;35:933–935. doi: 10.1097/PAS.0b013e3182196a39. [DOI] [PubMed] [Google Scholar]
- 11.Hasselblatt M, Nagel I, Oyen F, Bartelheim K, Russell RB, Schüller U, Junckerstorff R, Rosenblum M, Alassiri AH, Rossi S, Schmid I, Gottardo NG, Toledano H, Viscardi E, Balbin M, Witkowski L, Lu Q, Betts MJ, Foulkes WD, Siebert R, Frühwald MC, Schneppenheim R. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 2014;128:453–456. doi: 10.1007/s00401-014-1323-x. [DOI] [PubMed] [Google Scholar]
- 12.Schneppenheim R, Frühwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U, Kreuz M, Leuschner I, Subero JIM, Obser T, Oyen F, Vater I, Siebert R. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet. 2010;86:279–284. doi: 10.1016/j.ajhg.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lang JD, Hendricks WPD. Identification of driver mutations in rare cancers: the role of SMARCA4 in small cell carcinoma of the ovary, hypercalcemic type (SCCOHT) Methods Mol Biol. 2018;1706:367–379. doi: 10.1007/978-1-4939-7471-9_20. [DOI] [PubMed] [Google Scholar]
- 14.Reisman DN, Sciarrotta J, Wang W, Funkhouser WK, Weissman BE. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res. 2003;63:560–566. [PubMed] [Google Scholar]
- 15.Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–215. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moreno N, Schmidt C, Ahlfeld J, Pöschl J, Dittmar S, Pfister SM, et al. Loss of Smarc proteins impairs cerebellar development. J Neurosci. 2014;34:13486–13491. doi: 10.1523/jneurosci.2560-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holdhof D, Schoof M, Hellwig M, Holdhof NH, Niesen J, Schüller U. hGFAP-positive stem cells depend on Brg1 for proper formation of cerebral and cerebellar structures. Cereb Cortex. 2020;30:1382–1392. doi: 10.1093/cercor/bhz173. [DOI] [PubMed] [Google Scholar]
- 18.Ghasemi DR, Sill M, Okonechnikov K, Korshunov A, Yip S, Schutz PW, Scheie D, Kruse A, Harter PN, Kastelan M, Wagner M, Hartmann C, Benzel J, Maass KK, Khasraw M, Sträter R, Thomas C, Paulus W, Kratz CP, Witt H, Kawauchi D, Herold-Mende C, Sahm F, Brandner S, Kool M, Jones DTW, von Deimling A, Pfister SM, Reuss DE, Pajtler KW. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol. 2019;138:1075–1089. doi: 10.1007/s00401-019-02056-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, Gröbner S, Segura-Wang M, Zichner T, Rudneva VA, Warnatz HJ, Sidiropoulos N, Phillips AH, Schumacher S, Kleinheinz K, Waszak SM, Erkek S, Jones DTW, Worst BC, Kool M, Zapatka M, Jäger N, Chavez L, Hutter B, Bieg M, Paramasivam N, Heinold M, Gu Z, Ishaque N, Jäger-Schmidt C, Imbusch CD, Jugold A, Hübschmann D, Risch T, Amstislavskiy V, Gonzalez FGR, Weber UD, Wolf S, Robinson GW, Zhou X, Wu G, Finkelstein D, Liu Y, Cavalli FMG, Luu B, Ramaswamy V, Wu X, Koster J, Ryzhova M, Cho YJ, Pomeroy SL, Herold-Mende C, Schuhmann M, Ebinger M, Liau LM, Mora J, McLendon RE, Jabado N, Kumabe T, Chuah E, Ma Y, Moore RA, Mungall AJ, Mungall KL, Thiessen N, Tse K, Wong T, Jones SJM, Witt O, Milde T, von Deimling A, Capper D, Korshunov A, Yaspo ML, Kriwacki R, Gajjar A, Zhang J, Beroukhim R, Fraenkel E, Korbel JO, Brors B, Schlesner M, Eils R, Marra MA, Pfister SM, Taylor MD, Lichter P. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–317. doi: 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, Sill M, Buchhalter I, Northcott PA, Leis I, Ryzhova M, Koelsche C, Pfaff E, Allen SJ, Balasubramanian G, Worst BC, Pajtler KW, Brabetz S, Johann PD, Sahm F, Reimand J, Mackay A, Carvalho DM, Remke M, Phillips JJ, Perry A, Cowdrey C, Drissi R, Fouladi M, Giangaspero F, Łastowska M, Grajkowska W, Scheurlen W, Pietsch T, Hagel C, Gojo J, Lötsch D, Berger W, Slavc I, Haberler C, Jouvet A, Holm S, Hofer S, Prinz M, Keohane C, Fried I, Mawrin C, Scheie D, Mobley BC, Schniederjan MJ, Santi M, Buccoliero AM, Dahiya S, Kramm CM, von Bueren AO, von Hoff K, Rutkowski S, Herold-Mende C, Frühwald MC, Milde T, Hasselblatt M, Wesseling P, Rößler J, Schüller U, Ebinger M, Schittenhelm J, Frank S, Grobholz R, Vajtai I, Hans V, Schneppenheim R, Zitterbart K, Collins VP, Aronica E, Varlet P, Puget S, Dufour C, Grill J, Figarella-Branger D, Wolter M, Schuhmann MU, Shalaby T, Grotzer M, van Meter T, Monoranu CM, Felsberg J, Reifenberger G, Snuderl M, Forrester LA, Koster J, Versteeg R, Volckmann R, van Sluis P, Wolf S, Mikkelsen T, Gajjar A, Aldape K, Moore AS, Taylor MD, Jones C, Jabado N, Karajannis MA, Eils R, Schlesner M, Lichter P, von Deimling A, Pfister SM, Ellison DW, Korshunov A, Kool M. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell. 2016;164:1060–1072. doi: 10.1016/j.cell.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruiz-Perez MV, Henley AB, Arsenian-Henriksson M. The MYCN protein in health and disease. Genes (Basel). 2017;8. 10.3390/genes8040113. [DOI] [PMC free article] [PubMed]
- 22.Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J, Wittmann A, Devens F, Gerber NU, Joos S, Kulozik A, Reifenberger G, Rutkowski S, Wiestler OD, Radlwimmer B, Scheurlen W, Lichter P, Korshunov A. Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol. 2009;27:1627–1636. doi: 10.1200/jco.2008.17.9432. [DOI] [PubMed] [Google Scholar]
- 23.Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–2712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by Sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- 25.Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD, Lau J, Masic S, Nguyen K, Yakovenko S, Zhe XN, Flynn Gilmer HC, Collins R, Nagaoka M, Phillips JJ, Jenkins RB, Tihan T, Vandenberg SR, James CD, Tanaka K, Taylor MD, Weiss WA, Chesler L. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010;24:1059–1072. doi: 10.1101/gad.1907510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, Boult JKR, Williamson D, Ahmad Z, Hallsworth A, Ryan SL, Poon E, Robinson SP, Ruddle R, Raynaud FI, Howell L, Kwok C, Joshi A, Nicholson SL, Crosier S, Ellison DW, Wharton SB, Robson K, Michalski A, Hargrave D, Jacques TS, Pizer B, Bailey S, Swartling FJ, Weiss WA, Chesler L, Clifford SC. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell. 2015;27:72–84. doi: 10.1016/j.ccell.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang M, Tailor J, Zhen Q, Gillmor AH, Miller ML, Weishaupt H, et al. Engineering genetic predisposition in human neuroepithelial stem cells recapitulates medulloblastoma tumorigenesis. Cell Stem Cell. 2019;25:433–46.e7. doi: 10.1016/j.stem.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fielitz K, Althoff K, De Preter K, Nonnekens J, Ohli J, Elges S, et al. Characterization of pancreatic glucagon-producing tumors and pituitary gland tumors in transgenic mice overexpressing MYCN in hGFAP-positive cells. Oncotarget. 2016;7:74415–74426. doi: 10.18632/oncotarget.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Machold R, Fishell G. Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron. 2005;48:17–24. doi: 10.1016/j.neuron.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 30.Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 31.Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Indra AK, Dupe V, Bornert JM, Messaddeq N, Yaniv M, Mark M, et al. Temporally controlled targeted somatic mutagenesis in embryonic surface ectoderm and fetal epidermal keratinocytes unveils two distinct developmental functions of BRG1 in limb morphogenesis and skin barrier formation. Development. 2005;132:4533–4544. doi: 10.1242/dev.02019. [DOI] [PubMed] [Google Scholar]
- 33.Althoff K, Beckers A, Bell E, Nortmeyer M, Thor T, Sprussel A, et al. A Cre-conditional MYCN-driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene. 2015;34:3357–3368. doi: 10.1038/onc.2014.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujita S, Shimada M, Nakamura T. H3-thymidine autoradiographic studies on the cell proliferation and differentiation in the external and the internal granular layers of the mouse cerebellum. J Comp Neurol. 1966;128:191–208. doi: 10.1002/cne.901280206. [DOI] [PubMed] [Google Scholar]
- 35.Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, McMahon AP. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol. 2004;270:393–410. doi: 10.1016/j.ydbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 36.Sillitoe RV, Joyner AL. Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Annu Rev Cell Dev Biol. 2007;23:549–577. doi: 10.1146/annurev.cellbio.23.090506.123237. [DOI] [PubMed] [Google Scholar]
- 37.Rakic P. Neuron-glia relationship during granule cell migration in developing cerebellar cortex. A Golgi and electronmicroscopic study in Macacus rhesus. J Comp Neurol. 1971;141:283–312. doi: 10.1002/cne.901410303. [DOI] [PubMed] [Google Scholar]
- 38.Landry CF, Ivy GO, Dunn RJ, Marks A, Brown IR. Expression of the gene encoding the beta-subunit of S-100 protein in the developing rat brain analyzed by in situ hybridization. Brain Res Mol Brain Res. 1989;6:251–262. doi: 10.1016/0169-328x(89)90071-5. [DOI] [PubMed] [Google Scholar]
- 39.Menyhárt O, Giangaspero F, Győrffy B. Molecular markers and potential therapeutic targets in non-WNT/non-SHH (group 3 and group 4) medulloblastomas. J Hematol Oncol. 2019;12:29. doi: 10.1186/s13045-019-0712-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lossi L, Castagna C, Merighi A. Caspase-3 mediated cell death in the normal development of the mammalian cerebellum. Int J Mol Sci. 2018;19. 10.3390/ijms19123999. [DOI] [PMC free article] [PubMed]
- 41.Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, Wickramasinghe R, Scott MP, Wechsler-Reya RJ. Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci U S A. 2003;100:7331–7336. doi: 10.1073/pnas.0832317100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatton BA, Knoepfler PS, Kenney AM, Rowitch DH, de Alborán IM, Olson JM, Eisenman RN. N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 2006;66:8655–8661. doi: 10.1158/0008-5472.Can-06-1621. [DOI] [PubMed] [Google Scholar]
- 43.Zhan X, Shi X, Zhang Z, Chen Y, Wu JI. Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc Natl Acad Sci U S A. 2011;108:12758–12763. doi: 10.1073/pnas.1018510108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hellwig M, Lauffer MC, Bockmayr M, Spohn M, Merk DJ, Harrison L, Ahlfeld J, Kitowski A, Neumann JE, Ohli J, Holdhof D, Niesen J, Schoof M, Kool M, Kraus C, Zweier C, Holmberg D, Schüller U. TCF4 (E2-2) harbors tumor suppressive functions in SHH medulloblastoma. Acta Neuropathol. 2019;137:657–673. doi: 10.1007/s00401-019-01982-5. [DOI] [PubMed] [Google Scholar]
- 45.Merk DJ, Ohli J, Merk ND, Thatikonda V, Morrissy S, Schoof M, et al. Opposing effects of CREBBP mutations govern the phenotype of Rubinstein-Taybi syndrome and adult SHH medulloblastoma. Dev Cell. 2018;44:709–24.e6. doi: 10.1016/j.devcel.2018.02.012. [DOI] [PubMed] [Google Scholar]
- 46.Hatten ME, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- 47.Sun R, Zhao K, Shen R, Cai L, Yang X, Kuang Y, et al. Inducible and reversible regulation of endogenous gene in mouse. Nucleic Acids Res. 2012;40:e166. doi: 10.1093/nar/gks738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Kaplan-Meier curves of Mert::Bfl/fl::Nfl/wt, Mert::Bfl/fl::Nfl/fl, hG::Bfl/wt::Nfl/wt, hG::Bfl/wt::Nfl/fl, hG::Bflfl::Nfl/wt and hG::Bflfl::Nfl/fl mice. Kaplan-Meier curves from P30 onwards of Mert::Bfl/fl::Nfl/wt mice (brown) and Mert::Bfl/fl::Nfl/fl (purple) shows the decreased survival compared to respective controls (black) in A. However, none of the animals developed a tumor. The survival curves of hG::Bfl/wt::Nfl/wt (blue), hG::Bfl/wt::Nfl/fl (orange), hG::Bflfl::Nfl/wt (green) and hG::Bflfl::Nfl/fl mice (red) show diminished survival compared to respective controls (black) and are depicted in B. Still, none of the animals developed a brain tumor. **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s., not significant. (PNG 51.4 kb).