

CONSPECTUS:
Continued, rapid development of antimicrobial resistance has become worldwide health crisis and a burden on the global economy. Decisive and comprehensive action is required to slow down the spread of antibiotic resistance, including increased investment in antibiotic discovery, sustainable policies that provide returns on investment for newly launched antibiotics, and public education to reduce the over usage of antibiotics, especially in livestock and agriculture. Without significant changes in the current antibiotic pipeline, we are in danger of entering a post-antibiotic era.
In this account, we summarize our recent efforts to develop next-generation streptogramin and lankacidin antibiotics that overcome bacterial resistance by means of modular chemical synthesis. First, we describe our highly modular, scalable route to four natural group A streptogramins antibiotics in 6-8 steps from seven simple chemical building blocks. We next describe the application of this route to the synthesis of a novel library of streptogramin antibiotics informed by in vitro and in vivo biological evaluation and high-resolution cryo-electron microscopy. One lead compound showed excellent inhibitory activity in vitro and in vivo against a longstanding streptogramin-resistance mechanism, virginiamycin acetyltransferase. Our results demonstrate that the combination of rational design and modular chemical synthesis can revitalize classes of antibiotics that are limited by naturally arising resistance mechanisms.
Second, we recount our modular approaches toward lankacidin antibiotics. Lankacidins are a group of polyketide natural products with activity against several strains of Gram-positive bacteria, but have not been deployed as therapeutics due to their chemical instability. We describe a route to several diastereomers of 2,18-seco-lankacidinol B in a linear sequence of ≤ 8 steps from simple building blocks, resulting in a revision of the C4 stereochemistry. We next detail our modular synthesis of several diastereoisomers of iso-lankacidinol that resulted in the structural reassignment of this natural product. These structural revisions raise interesting questions about the biosynthetic origin of lankacidins, all of which possessed uniform stereochemistry prior to these finding. Finally, we summarize the ability of several iso- and seco-lankacidins to inhibit the growth of bacteria and to inhibit translation in vitro, providing important insights into structure-function relationships for the class.
Graphical Abstract

1. Introduction
The introduction of penicillin is regarded as one of the greatest discoveries in the history of human medicine. This serendipitous discovery, along with isolation of streptomycin in 1943, paved the way to the golden era of antibiotics, in which the majority of classes of clinically used antibiotics were unearthed by investigating the metabolites of specialized bacteria and fungi.5 However, over the past 60 years, continued use and overuse of antibiotics in humans and livestock has contributed to the emergence of several resistant bacterial pathogens including multidrug-resistant strains that are impervious to most or all clinically available antibiotics.
According to CDC’s 2019 Antibiotic Resistance Threats report, more than 2.8 million antibiotic-resistant infections occur in the US each year, resulting in more than 35,000 deaths.6 Worldwide, according to the 2014 “The Review on Antimicrobial Resistance (AMR)”, drug-resistant superbugs could claim 10 million lives and cost the global economy a cumulative US$100 trillion each year by 2050.7 Combined with a dramatic reduction in antibiotic research in global pharmaceutical industry, if we fail to act on a global scale immediately, our society is in danger of entering a post-antibiotic era, in which common infections or minor injuries could once again carry a major risk of death. Our group is interested in combating bacterial resistance by revisiting abandoned or understudied classes of antibiotics discovered in the golden era, such as streptogramin and lankacidin antibiotics (Figure 1A), and optimizing them through modular chemical synthesis. Herein we present a full account of these endeavors, comprising modular chemical synthesis of group A streptogramin antibiotics and lankacidin antibiotics, evaluation of in vitro and in vivo activity, and elucidation of molecular mechanisms of action.
Figure 1.

Representative streptogramin and lankacidin antibiotics. A. Selected natural streptogramins and lankacidins. B. Semisynthetic streptogramins.
2. Platform for the discovery of streptogramin antibiotics that overcome resistance
2.1. Modular chemical synthesis of group A streptogramin antibiotics
Streptogramin antibiotics are produced by several species of Streptomyces and comprise two structurally distinct subgroups: group A (23-membered macrocyclic polyketide/nonribosomal peptide hybrids, virginiamycin M1 (1), virginiamycin M2 (2), madumyicin I (3) and madumycin II (5)) and group B (19-membered macrocyclic depsipeptides,virginiamycin S1 (4) and pristinamycin IIA (6)).8 Streptogramins have been used as livestock feeds for decades,9 were not approved by the FDA until the introduction of Synercid® (a 30:70 (w/w) combination of quinupristin (10) and dalfopristin (9)) in 1999 which were derived from pristinamycin IA (6) and virginiamycin M1 (1) semisynthetically.10 However, due to its IV-only formulation and narrow spectrum of activity, Synercid is only reserved for hospitalized patients with multidrug-resistant skin and skin-structure infections or with bacteremia caused by vancomycin-resistant Enterococcus faecium.10 Recently, an orally bioavailable combination of semisynthetic streptogramins flopristin (11) and linopristin (12) known as NXL-103, underwent a successful phase II clinical trial in 2010, but has not progressed further in the clinic yet.11 The sophisticated architecture and therapeutic potential of group A streptogramins, the major components of combination therapies in the class, have attracted considerable interest from the synthetic community. Before our research, Schlessinger’s group12a, Uguen group12b and Panek’s group12c,d reported the total syntheses of virginiamycin M2 (2). Syntheses of madumycin II (5)13 and of closely related streptogramins14 have also been disclosed. Fully synthetic routes to madumycin I (3) and virginiamycin M1 (1) had not been developed prior to our work.
Our retrosynthetic analysis of madumycin I (3) is summarized in Scheme 1A. Based on analysis of the common structural features of group A streptogramin antibiotics, we envisioned that madumycin I (3) could be disconnected into left half 13 and right half 14, which have comparable structural complexity, by means of amide bond formation and intramolecular transition metal coupling. Each of the two halves could be derived from 3-4 readily available and simple building blocks.
Scheme 1.

A. Retrosynthetic analysis of madumycin I (3). B. Total synthesis of madumycin I (3) and madumycin II (5).
Our first modular synthetic route to madumycin I (3) from seven simple building blocks is described in Scheme 1B.1,15 The construction of left half 13 commenced with formation of α,β-unsaturated ester 17 from silyl dienol ether 18 and isobutyraldehyde (19) in 94% yield and 87% ee, by means of oxazaborolidine-catalyzed asymmetric vinylogous Mukaiyama aldol reaction.16 Trimethylaluminum-mediated amidation between methyl ester 17 and propargyl amine (15), followed by hydrostannylation of the resulting terminal alkyne under stoichiometric stannylcupration conditions, produced vinyl tin 25 in 90% yield over 2 steps. Esterification of 25 with Fmoc-D-Ala-OH (16) in the presence of DCC and catalytic DMAP, followed by dilution of the reaction mixture with Et2NH to remove the Fmoc group, delivered the left half 13 in 88% yield in a single operation. Overall, the synthesis of left half 13 proceeded in 4 steps and 74% yield from building blocks 15, 16, 18 and 19, and three out of five chiral centers were introduced. For right half 29, aldol condensation between (E)-3-bromobut-2-enal 22 and acetyl thiazolidinethione 23 acted as the first step, providing 26 in 64% yield as a single diastereomer.17 In order to avoid retro-aldol reaction in the following manipulations, 26 was protected with TBSOTf in 92% yield. Nucleophilic substitution of the chiral auxiliary with the dianion of oxazole 28 successfully delivered the right half 29 in 70% yield. It is worth noting that the trimethylsilyl group at the C5 position of the oxazole ring is indispensable to prevent deprotonation at the oxazole ring.18 The route to the right half 29 proceeds in 3 steps and 42% overall yield from 22, 23, and 28. The macrocycle precursor 30 was delivered in 82% yield by coupling the left half and the right half with HATU. We anticipated challenges with the intramolecular Stille macrocyclization to form the 23-membered macrocycle. Upon optimizing the intramolecular Stille coupling on a related substrate,1 we found that sterically hindered phosphine ligands like BrettPhos, which contains two methoxy groups which share the same aryl with phosphine, provided the cyclized product in 39% yield. Further investigation of BrettPhos-motif type phosphine ligands led us to JackiePhos, which contains two electron-withdrawing P-bound 3,5-(bis)trifluoromethylphenyl groups and was originally designed to facilitate challenging transmetalation.19 Thus, intramolecular Stille macrocyclization of precursor 30 could be carried out with 20% catalyst loading at 50 °C to provide protected macrocycle in 64% yield. Finally, removal of the two silyl groups with tetrabutylammonium fluoride buffered with imidazole hydrochloride provided madumycin I (3) with an overall yield of 38% from 18 and 19 or 21% from 22 and 23. Stereoselective reduction of β-hydroxy ketone of madumycin I (3) using NaBH4 as a hydride source and Et2BOMe as complexing reagent20 gave access to madumycin II (5) in 72% yield as a single diastereomer, representing the first reported interconversion of these two natural products.
To demonstrate the modularity of our route, we sought to apply it to the synthesis of virginiamycin M1 (1) and virginiamycin M2 (2) (Scheme 2). By replacing Fmoc-D-Ala-OH with Fmoc-D-Pro-OH and following the exact sequence to madumycin I (3), a total synthesis of virginiamycin M2 (2) was achieved in 31% yield from 18 and 19 (7 steps) or 18% overall yield from 22 and 23 (6 steps). However, access to virginiamycin M1 (1) proved to be more challenging due to its special 2,3-dehydroproline function. Amine 31 was efficiently and selectively oxidized to imine 32 by iodosylbenzene.21 Owing to the lower nucleophilicity of imine function of 32 than amine function of 31, it was difficult to form the amide bond under universal peptide-forming reagents. The macrocycle precursor was finally reached by coupling imine 32 with a newly formed acid chloride of the right half 29, which was synthesized from 29 using Ghosez’s reagent22 in situ, in 65% yield on multigram scale. The resulting precursor could be transferred to virginiamycin M1 through Stille macrocyclization, which required increased catalyst loading (30%) and higher temperature (80 °C), and removal of two silyl groups with tetrabutylammonium fluoride and imidazole hydrochloride. Overall, the route to virginiamycin M1 proceeded in 15% yield from 18 and 19 (8 steps) or 9% overall yield from 22 and 23 (6 steps), which also represented the first fully synthetic access to virginiamycin M1 (1).
Scheme 2.

Modular chemical synthesis of virginiamycin M1 and virginiamycin M2.
Although it was high yielding and modular, our first-generation route suffered from several limitations including acid lability of several intermediates, reliance on toxic organotin reagents, generation of organotin byproducts, and a moderate-yielding Stille macrocyclization. We developed a shorter, higher-yielding route to virginiamycin M2 (2) featuring a ring-closing metathesis that proceeded in 6 steps from building blocks 18 and 19 or 23 and 36 in 24-43% overall yield and that overcame the limitations associated with organotin compounds (Scheme 3).15,23 In addition to removing tin-associated liabilities, this route also features a shorter route (3 steps) to the left half (35) by virtue of the removal of the hydrostannylation step. Macrocyclization was achieved with ring-closing metathesis on an unprotected macrocycle precursor 38, and was higher yielding than the Stille macrocyclization. The choice of alkene stereochemistry was critical to the success of the macrocyclization, with the (Z,E)-diene on the right half providing the highest yield of macrocycle. Extensive details of both catalyst screens and substrate modifications that led to the optimized route can be found in the original publication.23 This represents the shortest and most efficient route to virginiamycin M2 reported to date.
Scheme 3.

Second-generation route to virginiamycin M2
In short, we achieved a modular, scalable and fully synthetic route to access four streptogramin A antibiotics and an improved tin-free route to virginiamycin M2. Every natural antibiotic was assembled from seven simple building blocks that contribute every non-hydrogen atom in the final products. The high degree of convergency, robustness and scalability of the route certainly facilitates access to next-generation group A streptogramins with the aims of improving their pharmacological properties, expanding their spectra of activity, and increasing their potency against multidrug-resistant strains of pathogenic bacteria.
2.2. A modular platform for discovering new streptogramin antibiotics that overcome resistance
Next, in order to overcome multidrug resistance and naturally occurred Vat-resistance, we chose virginiamycin M2 (2) as our parent scaffold for the synthesis of analogs. We made some technical improvements (Scheme 4) that increase yield through both the left-half sequence (31% to 40%) and the right-half sequence (18% to 28%), and simplified the purification of several intermediates compared to the previously reported route.1,2
Scheme 4.

A practical improvement of our first-generation route to virginiamycin M2 (2).
Through a fruitful collaboration with the Fraser group at UCSF we were able to obtain a 2.5–Å cryo-EM structure of virginiamycin M2 (2) bound to the large subunit of the E. coli ribosome (Figure 2).2 This was the first structural data for 2 available in the literature and the highest resolution of any streptogramin–ribosome complex. We used the structure to inform systematic modification of group A streptogramins by exchanging building blocks. By virtue of the modular, convergent route (~7 steps longest linear sequence) with substantial scalability (each “half” was pooled in decagram quantities), this enabled rapid access to 15 unnatural group A streptogramin antibiotics that had not previously been explored with semisynthetic approaches (Table 1). We were able to make at least one change per building block, demonstrating the functional group tolerance of the approach.
Figure 2.

2.5-Å cryo-EM structure of virginiamycin M2 (2) bound to the large subunit of the E. coli ribosome. Coulomb potential density is contoured at 4.0σ. Colors of the atoms in 2 correspond to building blocks used in the fully synthetic route.
Table 1.
Fully synthetic group A streptogramin analogs generated by systematic exchange of building blocks using a modular, convergent approach.
 |
One drawback of diversification by building block exchange is that each analog requires up to seven steps to procure (sometimes more, if the building block itself is not commercially available). In order to rapidly increase the diversity of our library, we introduced an alcohol functional group to the C3 sidechain that could serve as a handle for late-stage diversification (Scheme 5). Thus, C3-functionalized intermediates 69 and 70, which differ in their C29 stereochemistry, each require only one building block exchange, but can be used to generate large pools of analogs in the last few steps of the synthesis. For example, the C29R intermediate 69 was fluorinated to generate 71 (Scheme 5A) by treatment with diethylaminosulfur trifluoride (DAST). Three secondary amine analogs 72-74 (Scheme 5B) were reached by means of oxidation/reductive amination of 70. As a testament to the rapid diversification possible with this approach, 69 and 70 were carried forward to 34 aryl carbamates (Scheme 5C). Furthermore, inspired by the discovery of flopristin (11) by transferring 16-ketone to 16-fluorine, we installed a fluorine at C16 by a 4-step sequence (Scheme 5D), providing the clinical candidate flopristin (11, and several fluorinated analogs (79-81). Overall, our platform efficiently provided 62 new group A streptogramin analogs, four natural products, and the first fully synthetic sample of the clinical candidate flopristin (11).
Scheme 5.

Fully synthetic group A streptogramin analogs by late-stage diversification.
With these structure-diversified streptogramin A analogs in hand, we evaluated their inhibitory activities against a panel of 20 pathogens (10 Gram-positive and 10 Gram-negative bacterial strains), including three strains with well-studied mechanisms of streptogramin resistance, including VatA expressing Staphylococcus aureus, Cfr-mediated resistant S. aureus, and ABC-F in Enterococcus faecalis (selected data shown in Figure 3). We also measured in vitro ribosomal translation inhibitory activities for selected analogues (at 10 μM) using an in vitro translation (IVT) assay, as indicated by the blue bars on the right of the table. A smaller bar indicates less translation, and thus a more potent inhibitor. While many of our analogs lacked significant activity in both assays, we were delighted to find that some modifications increased activity in some strains. For non-fluorinated analogs, modifications of C3 (teal building blocks) and C4 (blue building blocks) resulted in improved IVT values and improved MICs in most strains, including a group A streptogramin-resistant strain of S. aureus expressing virginiamycin acetyltransferase A (VatA). C16-fluorination resulted in a further improvement in activity, as can be seen in the activities of 79-81. It is interesting to note that C3-modified, C16-fluoro analog 79 showed better activity in the VatA streptogramin-resistant strain of S. aureus than in the WT strain. Thiazole-containing C16-fluoro analogs 59 and 80 displayed comparable activity to their oxazole-containing counterparts 2 and 11. Encouragingly, C4-allyl, C16-fluoro analog 81 exhibited up to a 128-fold improvement in MIC comparing to the parent compound virginiamycin M2 (2). This analog even had moderate activity (32 μg/mL) against ABC-F-expressing E. faecalis24 and Gram-negative E. coli (16 μg/ml), strains which are highly resistant to streptogramins. These results show that engineering group A streptogramins using a fully synthetic approach can lead to improved activities in both streptogramin-susceptible and streptogramin-resistant strains.
Figure 3.

Inhibitory activities of selected fully synthetic group A streptogramins.
We next evaluated the inhibitory activity of analogs 79 and 81 in the presence of the group B streptogramin virginiamycin S1 (4). Although one might expect improved activity of an individual component to result in improved activity of the combination, this is not always the case. For example, if the C4 extension of 81 were to clash with 4, they would be very unlikely to show cooperative inhibition of the ribosome. Much to our delight, we found that the combinations of 79+4 and 81+4 were extremely potent in most bacterial strains tested. In many cases, the combinations fully inhibited bacterial growth even at the lowest concentration tested (0.06 μg/mL). These results show that combinations of engineered group A streptogramins with natural group B streptogramins can be extremely potent inhibitors of bacterial growth.
Furthermore, 81 showed more potent activity than flopristin (11) against many multidrug-resistant clinical isolates of S. aureus with resistance to group A and group B streptogramins (data not shown).2 Finally, in a mouse thigh model of infection using streptogramin-resistant S. aureus CIP 111304 (Figure 4), treatment with compound 81 was more than 10-fold effective than flopristin (11) at decreasing bacterial load (Figure 4).2 It is especially notable that compound 81 demonstrates notable potency in this demanding model of infection, even without combining with a group B streptogramin partner.
Figure 4.

Mouse thigh model of infection with a streptogramin-resistant strain of S. aureus (CIP 111304). Mice were treated with compound or vehicle 2 h post-infection, and bacterial load was quantified after 24 h. Adapted with permission from ref. 2. Copyright 2020 Nature Publishing Group.
Overall, we built a platform for discovering next-generation streptogramin A antibiotics via modular chemical synthesis. Compound 81 showed more potent activity than clinical candidate flopristin (11) against streptogramin-resistant bacterial pathogens both in vitro and in vivo. We anticipate further medicinal chemistry efforts to yield a safe and efficacious lead compound.
3. Modular approaches to lankacidin antibiotics
Lankacidins are a class of polyketide/nonribosomal peptide natural products isolated from the soil bacterium Streptomyces rochei. Their most common structural feature is a β-keto-δ-lactone core that is often buried within a 17-membered macrocycle.25 The structures of lankacidins C (82) and A (83) were assigned in part by X-ray crystallographic analysis of p-bromophenylsulfonylhydrazone derivatives.26 In 2018, two new family members of lankacidins group which lack the C2-C18 bond (seco-lankacidinols 84 and 85) were isolated by Wang and co-workers (Figure 5).27 In the past two years, errors were identified in the assigned structures of both seco-lankacidinols and revised structures were determined by chemical synthesis. Hong and co-workers reassigned 2,18-seco-lankacidionol A (84) to contain a 4-oxazolidinone motif (90).28 Our group has revised the C4 stereochemistry of 2,18-seco-lankacidinol B (85) to the S configuration (7), which differs from all other family members.3 More recently, we found the C2 and C5 stereocenters of iso-lankacidinol (86) were incorrect and reassigned the structure to iso-lankacidinol (8).4
Figure 5.

Members of the lankacidin class of natural products.
Lankacidins exhibit potent activity against several strains of Gram-positive bacteria (including resistant strains of staphylococci), mycobacteria, and some Gram-negative bateria.29 Lankacidin C (82) and lankacidinol (88) are inhibitors of protein synthesis with similar activity to macrolides such as erythromycin, but different resistance profiles, making them promising for development.29e Additionally, lankacidin C (82)30 and 2,18-seco-lankacidinol A (84) and B (85)26 exhibit moderate antitumor activities in vitro. Lankacidin C 14-butyrate showed antitumor effects by oral administration in mice.29d
Semisynthesis of lankacidins offers a potential pathway towards a derivative worthy of clinical development. However, semisynthetic manipulations have primarily been restricted to modification of the alcohol functional groups due to the instability of the β-keto-δ-lactone core (Scheme 6).31 Early chemical degradation experiments showed that lankacidinol (88) was reactive under either mildly acidic conditions, resulting in opening of the macrocycle via cleavage of the C2─C18 bond, or basic conditions, breaking the β-keto-δ-lactone via decarboxylation. Fully synthetic routes that incorporate this functionality at a late stage offer a potential solution to the challenged posed by the chemical instability of the class.
Scheme 6.

Selected transformations between lankacidin family members
The transformation between lankacidinol (88) and iso-lankacidinol (86) was achieved in very low yield (<5%) in refluxing pyridine. It was proposed that this induced a retro-Michael-Michael cascade that resulted in epimerization of the C5 stereochemistry.26 We were suspicious of the β-keto-acid intermediate, which would be especially prone to decarboxylation (leading to lancacyclinol (87)). We hypothesized that a retro-Mannich-Mannich cascade that results in inversion of the C2 stereochemistry would be more likely, and we proposed 8 as a structural reassignment.
A combination of non-ribosomal peptide synthetase (NRPS) and modular-iterative mixed polyketide synthetase (PKS) for the lankacidin biosynthetic pathway was proposed (Figure 6).32 The core of the lankacidin molecule is constructed from acyl-CoA and amino acid building blocks, which operate in a standard assembly-line fashion. The full-length chain is released by means of a thioesterase (TE) that generatres the β-keto-δ-lactone. LkcE then catalyzes the post-assembly of macrocyclization via oxidation (N-acyliminium)/Mannich reaction. This could explain the existence of iso-lankacidinol in nature, which would result from imperfect stereochemical control of the ring-forming Mannich reaction.
Figure 6.

Proposed biosynthetic pathway of lankacidinol.
The various biological activities observed within the lankacidin group make this family an attractive target for both chemical and biological research communities. While lankacidin-class natural products have received much interest from the total synthesis chemists for decades, only the groups of Kende, Williams, Hong, and Seiple have developed fully synthetic routes to natural products within the class (Scheme 7). Any synthetic approach to the lankacidins has to address the question of stereochemical control, particularly at the quaternary center C2. In 1993, the first total synthesis of (–)-lankacidin C was accomplished by Kende and coworkers through a convergent, enantioselective sequence starting from D-arabinose and L-aspartic acid in 34 steps (longest linear sequence).33 In 2000, Williams and coworkers reported a synthesis of lankacyclinol (87) featuring a Horner–Wadsworth–Emmons macrocyclization.34 In 2017, Hong and colleagues reported an elegant biomimetic approach to finish the total synthesis of lankacidinol in 8 steps.35 Three years later, the same group revised the structure of 2,18-seco-lankacidinol A (90) on the basis of a biosynthetic proposal.28 During the past three decades, Thomas’ group made great contributions toward lankacidin group, including a phenylnitrone approach to access a partially substituted core structure36a and a β-lactam approach to access a fully substituted (but unstable) core structure.36b
Scheme 7.
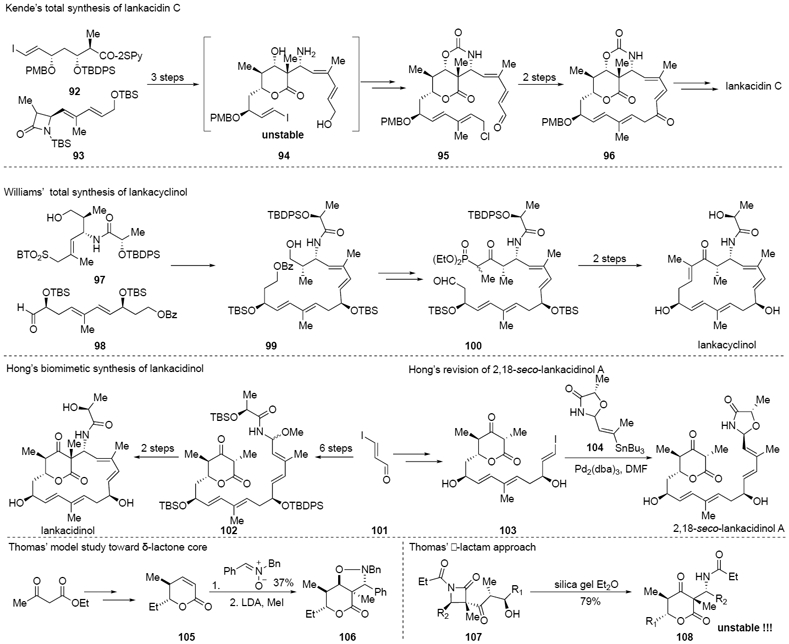
Previous syntheses of lankacidin family members and derivatives thereof.
3.1. Synthesis and Structural Reassignment of 2,18-seco-Lankacidinol B
2,18-seco-Lankacidinol B (7), the first acyclic member of the lankacidin class, was our first target due to the simplicity of the skeleton, which could pave the way toward syntheses of other family members. The synthesis commenced with epoxide opening and the resulting alcohol was quenched by TBSCl to generate protected diol 111 in 78% yield in one pot. A sequence of oxidative deprotection, Mitsunobu reaction with benzo[d]thiazole-2-thiol, and thioether oxidation delivered sulfone 112 in 77% yield over 2 steps. NaHMDS-promoted Julia–Kociensky olefination between sulfone 112 and commercially available aldehyde 113 provided dienoate 114 in 80% yield as a single stereoisomer. DIBAL-H reduction followed by palladium-catalyzed hydrostannylation gave right half 36 in 84% yield. The route to 115 requires 6 steps from simple building blocks epoxide 109 and lithium acetylide 110 and proceeds in 40% overall yield in multi-gram scales (Scheme 8A).
Scheme 8.

Synthesis and structural reassignment of 2,18-seco-lankacidinol B
We next began our synthesis of the the δ-lactone motif. As shown in Scheme 8B, aldehyde 117 could be easily prepared from an aldol reaction between aldehyde 101 and thiazolidinethione 23, followed by TBS protection and DIBAL-H reduction in 3 steps. By leveraging the Evans aldol reaction, a two-step, one-pot procedure was developed for formation of the δ-lactone motif based on from chemistry developed by Hong and coworkers in their elegant synthesis of lankacidinol and lankacyclinol.35 Evans aldol reaction between aldehyde 117 with chiral β-keto imide 118 followed by NaOMe-promoted lactonization to remove the chiral auxiliary resulted in formation of the desired δ-lactone 119 in one pot. This protocol enabled access to the left half in decagram scale. Following a similar sequence, we could access the other three isomers 121, 123 and 124 with high efficiency. The left half 121 was converted into its ferrocene carboxylate 122 which crystallized from diethyl ether. X-ray crystallographic analysis confirmed its relative and absolute stereochemistry.
With both right half 115 and the four left-half diastereomers in hand, various conditions were tested to facilitate Stille cross-coupling.37 We found that the use of Ph2PO2NBu4 as a tin scavenger and DMSO as solvent was required to reach the final products (73% to 78% yield).38 Desilylation with hydrogen fluoride/pyridine provided the C4 and C5 stereoisomers of 2,18-seco-lankacidinol B (Scheme 8C). After comparison of the 1H- and 13C-NMR spectra of the original report and the four synthetic 2,18-seco-lankacidinol B isomers, we found the originally assigned 4(R),5(R)-2,18-seco-lankacidinol B (85) was incorrect. Fortunately, the spectra 4(S),5(R)-2,18-seco-lankacidinol B (7) matched the reported spectra. This structural reassignment makes 2,18-seco-lankacidinol B (7) stereochemically unique among reported lankacidin-class natural products, all of which have been assigned the opposite stereochemistry at C4.
3.2. Synthesis and structural reassignment of iso-lankacidinol
While the synthesis of 2,18-seco-lankacidinol B (7) laid the groundwork for concise synthetic access to the lankacidin family, the challenge to construct the labile β-keto-δ-lactone motif for more complex family members remained. Our investigations in this direction first lead us to construct the lankacidin C (82) by closing the macrocycle with a Tsuji–Trost reaction based on our 2,18-seco-lankacidinol B (7) approach and late stage nitrene insertion at C18 position (Scheme 9A).39 Attempted ring closure on acetate, allylic alcohol, carbonate or phosphonate intermediate 127 with a variety of conditions failed to produce measurable amounts of macrocycle 128. We were able to successfully affect ring closure on a derivative with an extra methyl group at C4 (resulting in a quaternary center), supporting the hypothesis that the β-keto-δ-lactone motif decomposes via β-elimination.4 We next screened a Mannich approach for macrocycle formation by means of photochemical imine formation (Scheme 9B).40 Azide 129, generated with a Mitsunobu reaction, was transformed into the imine in the presence of a ruthenium catalyst and light, followed by in situ acylation delivered N-acylimine 130 in one pot. Unfortunately, all attempts to cyclize this intermediate failed, also likely due to the instability of the β-keto-δ-lactone.
Scheme 9.

Failed strategies for macrocycle formation and retrosynthetic plan for iso-lankacidinol
To circumvent these obstacles, we devised an alternative route to access other lankacidin family members. We noted that lankacidinol 88 and iso-lankacidinol 86 shared the same carbon skeleton except for the C5 stereochemistry (although we had reason to doubt this, as mentioned above). We aimed to develop a modular approach to complete both family members, incorporating a late-stage β-keto-δ-lactone formation to circumvent structural instability of intermediates. Retrosynthetically, we envisioned lankacidinol 88 or iso-lankacidinol 86 arising from three fragments by means of the sequential esterification, Julia–Kociensky olefination, and Stille coupling (Scheme 9C). It is worth mentioning that the top fragment could be derived from aspartic acid, which is a low cost, high abundance starting material with built-in absolute stereochemistry. For the left fragment, we planned to borrow from our 2,18-seco-lankacidinol B (7) synthesis. By simply varying the stereochemistry on the left fragment, we could access either lankacidinol (88) or the reported structure for iso-lankacidinol (86). The three fragments could be accessed from eight simple building blocks (23, 101, 109, 110, and 132-135).
We began our synthesis of iso-lankacidinol 86 and lankacidinol 88 by synthesizing the bottom fragment through a palladium-catalyzed hydrostannylation of primary alcohol 111, followed by DMP oxidation in 58% yield over two steps. TiCl4 mediated syn-selective aldol reaction between 117 and 132 provided secondary alcohol 137 (left fragment) in 93% yield and >20:1 daistereomeric ratio, with stereochemistry at C5 that matched the reported structure of iso-lankacidinol (86).41 Synthesis of the top fragment (140) started with a Wittig reaction between the aldehyde 13342 and ylide 134 to provide β-lactam 137 in 68% yield.43 Reduction of the aldehyde followed by Mitsunobu reaction with benzo[d]thiazole-2-thiol provided allylic thioether 139 in 85% yield over two steps. Silyl deprotection and lactam opening was achieved under acidic conditions. The solution was basified 12 M NaOH and then treated with acid chloride 135 furnished amide 140 (top fragment). Stereochemistry of each of the fragments (136, 137, and 140) is either known from chiral pool starting materials or predicable through established reactions, reducing the ambiguity in structural assignment of the final product.
We next sought to develop a reliable sequence for the assembly of the three fragments. Ester 141 was formed between left fragment 137 and top fragment 140 with EDCI in the presence of DMAP in 60% yield. Molybdenum promoted thioether oxidation, Julia–Kociensky olefination with aldehyde 136, and Stille macrocyclization in the presence of Pd2(dba)3 provided macrocycle 142 in 62% over three steps. Importantly, the sequence to assemble all three fragments and close the macrocyclic ring is highly convergent, requiring only four steps, and the sensitive β-keto-δ-lactone has still not been assembled at this stage. LiHMDS provided the desired Dieckmann cyclization product 143, albeit in low yield (5%).44 C2 Methylation followed by global deprotection with hydrofluoric acid/pyridine provided lankacidinol 144 in 92% yield over two steps as a single diastereomer (Scheme 10). The 1H- and 13C-NMR spectra of 144 did not match the reported spectra iso-lankacidinol 86, although without conclusive evidence for the C2 stereochemistry, we could not rule out at this stage if we had synthesized the C2 epimer. We next turned our attention to the other stereocenters around the δ-lactone ring to find the source of potential error in the original assignment.
Scheme 10.

Synthesis of the reported structure of iso-lankacidinol.
We hypothesized that the stereochemistry of C4 and C5 of iso-lankacidinol might match that of 2,18-seco-lankacidinol B (7) and set out to synthesize the (4S,5R) stereoisomer. Evans aldol reaction between imide (S)-132 and aldehyde 117 could quickly access the 4(S),5(R)-syn-aldol product in high yield for the left fragment. Macrocycle 145 was prepared by means of a four-step sequence similar to the one in Scheme 10. Treatment with sodium hydride affected the Dieckmann cyclization, and methylation was achieved in the same pot overall 30% yield. Global desilylation gave 2(R),4(S),5(R)-iso-lankacidinol 146 in 95% yield. Unfortunately, the 1H- and 13C-NMR spectra of 146 did not match the reported spectra of the natural product. X-ray structure of compound 146 revealed that the stereochemistry of C2 methyl group, installed in the penultimate step of our synthesis, was inverted compared to other family members. Comparison of the 1H- and 13C-NMR spectra to the natural sample led us to suspect that the “inverted” C2 methyl stereochemistry was actually the correct orientation, and that the stereochemistry at C4 and C5 might match the rest of the family members. With this in mind, we modified our synthesis to access the C4,C5-anti stereochemistry using an Oppholzer sultam approach.45 Aldolization of aldehyde 117 with the silyl enol ether 147 followed by methanolysis provided methyl ester 148 in 85% yield over two steps. Esterification, oxidation, Julia–Kociensky olefination, and Stille coupling furnished macrocycle 150 in 44% yield over four steps. LiHMDS-promoted Dieckmann cyclization proceeded in 35% yield, and C2-methylation was highly stereoselective (>20:1). Desilylation provided the final compound 8 in 95% yield, the structure of which was confirmed by X-ray crystallography (Scheme 11B). The 1H- and 13C-NMR spectra of the compound 8 perfectly matched the reported spectra of iso-lankacidinol, which confirmed the iso-lankacidinol is the C2 isomer of the lankacidinol (88).
Scheme 11.

Reassignment of iso-lankacidinol by means of modular chemical synthesis.
3.3. Antimicrobial activity of seco- and iso-lankacidinols and biosynthetic considerations
We next evaluated the antimicrobial activity of our fully synthetic seco-lankacidinols and iso-lankacidinols. Many lankacidin antibiotics exhibit antimicrobial activity by inhibiting the bacterial ribosome, and for well-known members of the class such as lankacidin C (82), activities against several strains of bacteria and the binding mode to the ribosome have been reported.30 We measured the activity of 2,18-seco-lankacidinols 85 (originally reported structure), 7 (reassigned structure) and its derivatives 125 and 126 against a panel of 10 Gram-positive and Gram-negative bacteria (Figure 7a). None of these compounds exhibited substantial activity against any of the common pathogens in our panel. This suggests that the macrocyclic ring, the C18 pyruvamide, and C2 stereochemsitry are important for substantial antibacterial activity within the lankacidin class.
Figure 7.

(a) Antimicrobial activity of seco-lankacidinols; (b) Inhibition of translation by iso-lankacidinols.
To rule out cellular accumulation as a roadblock for activity, we next measured the ability of iso-lankacidinols to inhibit the ribosome by means of an IVT assay. We evaluated inhibition the E. coli ribosome for three iso-lankacidinols at a single concentration of 10 μM (Figure 7b). The diastereomeric iso-lankacidinols (144 and 146) did not inhibit translation at this concentration compared to DMSO control. The natural product iso-lankacidinol (8) showed small but measurable inhibition of translation. These results suggest that the stereochemistry within and around the β-keto-δ-lactone core of lankacidinols is important for translation inhibitory activity in cyclic members of the lankacidin class.
4. Conclusion and perspective
Owing to the urgency of the antimicrobial resistance (AMR) crisis, many strategies should be pursued, such as discovering new therapeutics, improving existing ones, and revisiting or repurposing old drugs. Over the past 5 years, research in our laboratory has focused on building new molecules from simple chemical building blocks much like “legos” to revisit and revitalize antibiotics that haven’t reached their full potential. Through this approach, we have improved antibiotics known as streptogramins which was limited by clinical resistance. Additionally, we have explored structural assignments and structure–activity relationships of lankacidin antibiotics. We hope that these efforts will contribute to the continual effort to outmaneuver resistant bacteria, and we also hope this strategy will be able to encourage scientists to continue chemistry to access complex molecules with promising biological profiles.
Funding Sources
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (R35GM128656). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. L.C. is grateful for funding from the National Science Foundation of China (22001124).
Biography
Lingchao Cai received his B.S. and M.S. from Nankai University in China. He got his Ph. D from the University of California, Los Angeles in 2017 under the direction of Professor Ohyun Kwon and his research is focusing on the total synthesis of indole alkaloid. After graduation, he moved to the University of California, San Francisco for his postdoctoral research in Professor Ian Seiple’s lab working on total syntheses and structure modification of Lankacidin antibiotics natural products. Since July 2020, he began his independent career at Nanjing Forestry University in China and his research group works on methodology development for derivatization of fine chemicals from biomass and total synthesis of limonoid natural products.
Ian B. Seiple received his B.S. from the University of California at Berkeley under the tutelage of Prof. Dirk Trauner, and conducted his doctoral research laboratory of Prof. Phil S. Baran at The Scripps Research Institute as an NSF Predoctoral Fellow. During his graduate studies he synthesized anti-tumor and antibiotic marine alkaloids. He was an NIH postdoctoral fellow with Andy Myers at Harvard University where he developed fully synthetic routes to macrolide antibiotics. He started at University of California, San Francisco in 2015, where his lab focuses on the strategic application of chemical synthesis to address prevailing challenges in biology and medicine.
Qi Li obtain his B. Sc. and M. Sc.in Medicinal Chemistry at Shenyang Pharmaceutical University under the guidance of Prof. Ping Gong in 2004 and 2009. After receiving his Ph. D. at Institute of Chemistry, CAS, he moved to Tsinghua Unversity as a postdoctoral researcher. He joined the Seiple Group at University of California, San Francisco in 2016. Dr. Li began his independent career in late 2020 at Shanghai Institute of Materia Medica, CAS at where he is currently a professor. His research focuses on natural products-based drug discovery combating antimicrobial resistance.
REFERENCES
- (1).Li Q; Seiple IB Modular, Scalable Synthesis of Group A Streptogramin Antibiotics. J. Am. Chem. Soc 2017, 139, 13304–13307.This publication outlines a modular, scalable synthesis of four natural group A streptogramin antibiotics from seven simple building blocks.
- (2).Li Q; Pellegrino J; Lee DJ; Tran AT; Chaires HA; Wang R; Park JE; Ji K; Chow D; Zhang N; Brilot AF; Biel JT; van Zundert G; Borrelli K; Shinabarger D; Wolfe C; Murray B; Jacobson MP; Mühle E; Cheaneau O; Fraser JS; Seiple IB Synthetic group A streptogramin antibiotics that overcome Vat resistance. Nature 2020, 586, 145–150.By combining modular chemical synthesis with rational drug design and cryo-electron microscopy, we developed a platform to discover next-generation group A streptogramins antibiotics. One lead compound showed very potent activity against streptogramin-resistant S. aureus both in vitro and in vivo.
- (3).Yao Y; Cai L; Seiple IB Synthesis, structural reassignment, and antibacterial evaluation of 2,18-seco-lankacidinol B Angew. Chem. Int. Ed 2018, 57, 13551–13554.We developed a fully synthetic route to 2,18-seco-lankacidinol B, an acyclic member of the lankacidin class, in a linear sequence of ≤8 steps from simple precursors. This resulted in a reassignment of the stereochemistry at the C4 position of the natural product.
- (4).Cai L; Yao Y; Yeon SK; Seiple IB Modular approaches to lankacidin antibiotics. J. Am. Chem. Soc 2020, 142, 15116–15126.This article outlines the reassignment of two natural products of the lankacidin class and evaluation of the ability of several iso- and seco-lankacidins to inhibit the bacterial growth and protein synthesis. This work grants insight into the rich chemical complexity of this class of antibiotics and provides an avenue for further structural derivatization.
- (5).Brown ED ; Wright GD Antibaterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [DOI] [PubMed] [Google Scholar]
- (6).Centers for Disease Control and Prevention, Office of Infectious Disease Antibiotic resistance threats in the United States, November., 2019. Available at (https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf).
- (7).Review on Antimicrobial Resistance: Tackling drug-resistant infections globally: final report and recommendations. May 2016. Available at: https://amr-review.org/sites/default/files/160525_Final%20paper_with%20cover.pdf).
- (8).(a) Charney J; Fisher WP; Curran C; Machlowitz RA; Tytell AA Streptogramin, a new antibiotic. Antibiot. Chemother 1953, 3, 1283. [PubMed] [Google Scholar]; (b) Vazquez D In Antibiotics; Gottlieb D, Shaw PD, Eds.; Springer: New York, 1967; Vol. 1, pp 387–403. [Google Scholar]
- (9).Yates JD; Schaible PJ Virginiamycin as an antibiotic for poultry feeds. Nature 1962, 194, 183–184. [DOI] [PubMed] [Google Scholar]
- (10).(a) Delgado G Jr.; Neuhauser MM; Bearden DT; Danziger LH Quinupristin-dalfopristin: an overview. Pharmacotherapy 2000, 20, 1469–1485. [DOI] [PubMed] [Google Scholar]; (b) Pavan B Curr. Opin. Invest. Drugs 2000, 1, 173–180. [PubMed] [Google Scholar]; (c) Bonfiglio G; Furneri PM Novel streptogramin antibiotics. Expert Opin. Invest. Drugs 2001, 10, 185–198. [DOI] [PubMed] [Google Scholar]; (d) Allington DR; Rivey MP Quinupristin/dalfopristin: a therapeutic review. Clin. Ther 2001, 23, 24–44. [DOI] [PubMed] [Google Scholar]
- (11).(a) Politano AD; Sawyer RG NXL-103, a combination of flopristin and linopristin, for the potential treatment of bacterial infections including community-acquired pneumonia and MRSA. Curr. Opin. Invest. Drugs 2010, 11, 225–236. [PMC free article] [PubMed] [Google Scholar]; (b) Pankuch GA; Lin G; Clark C; Appelbaum PC Time-kill activity of the streptogramin NXL-103 against Gram-positive and -negative bacteria. Antimicrob. Agents Chemother 2011, 55, 1787–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Schlessinger RH; Li Y-J Total Synthesis of (–)-Virginiamycin M2 Using Second-Generation Vinylogous Urethane Chemistry. J. Am. Chem. Soc 1996, 118, 3301–3302. [Google Scholar]; (b) Breuilles P; Uguen D Total synthesis of pristinamycin IIB. Tetrahedron Lett. 1998, 39, 3149–3152. [Google Scholar]; (c) Wu J; Panek JS Total synthesis of (–)-virginiamycin M2. Angew. Chem., Int. Ed 2010, 49, 6165–6168. [DOI] [PubMed] [Google Scholar]; (d) Wu J; Panek JS Total synthesis of (–)-virginiamycin M2: application of crotylsilanes accessed by enantioselective Rh(II) or Cu(I) promoted carbenoid Si–H insertion. J. Org. Chem 2011, 76, 9900–9918. [DOI] [PubMed] [Google Scholar]
- (13).(a) Tavares F; Lawson JP; Meyers AI Total synthesis of streptogramin antibiotic, madumycin II. J. Am. Chem. Soc 1996, 118, 3303–3304. [Google Scholar]; (b) Ghosh AK; Liu W A convergent, enatioselective total synthesis of streptogramin antibiotic (–)-madumycin II. J. Org. Chem 1997, 62, 7908–7909. [DOI] [PubMed] [Google Scholar]
- (14).(a) Entwistle DA; Jordan SI; Montgomery J; Pattenden G Total synthesis of the virginiamycin antibiotic 14,15-anhudropristinamycin IIB. J. Chem. Soc., Perkin Trans 1 1996, 1315–1317. [Google Scholar]; (b) Entwistle DA; Jordan SI; Montgomery J; Pattenden G Total synthesis of oxazole-based virginiamycin antibiotic: 14,15-anhydropristinamycin IIB. Synthesis 1998, 603–612. [Google Scholar]; (c) Dvorak CA; Schmitz WD; Poon DJ; Pryde DC; Lawson JP; Amos RA; Meyers AI The synthesis of streptogramin antibiotic: (–)-griseoviridin and it’s C-8 epimer. Angew. Chem., Int. Ed 2000, 39, 1664–1666. [DOI] [PubMed] [Google Scholar]
- (15).Li Q; Seiple IB Synlett 2020, 31, DOI: 10.1055/a-1293-9555. [DOI] [Google Scholar]
- (16).Simsek S; Kalesse M Enantioselective synthesis of polyketide segments through vinylogous Mukaiyama aldol reactions. Tetrahedron Lett. 2009, 50, 3485–3488. [Google Scholar]
- (17).González A; Aiguadé J; Urpí F; Vilarrasa J Tetrahedron Lett. 1996, 37, 8949–8952. [Google Scholar]
- (18).Wood RD; Ganem B A simple solution to the oxazole problem of virginiamycin M. Tetrahedron Lett. 1983, 24, 4391–4392. [Google Scholar]
- (19).Hicks JD; Hyde AM; Cuezva AM; Buchwald SL Pd-catalyzed N-arylation of secondary acyclic amides: catalyst development, scope, and computational study. J. Am. Chem. Soc 2009, 131, 16720–16734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chen K-M; Hardtmann GE; Prasad K; Repič, O.; Shapiro, M. J. 1,3-syn-Diastereoselective reduction of β-hydroxy ketones utilizing alkoxydialkylboranes. Tetrahedron Lett. 1987, 28, 155–158. [Google Scholar]
- (21).Ochiai M; Inenaga M; Nagao Y; Moriarty RM; Vaid RK; Duncan MP Oxidative decarboxylation of cyclic amino acids and dehydrogenation of cyclic secondary amines with iodosobenzene. Tetrahedron Lett. 1988, 29, 6917–6920. [Google Scholar]
- (22).Devos A; Remion J; Frisque-Hesbain A-M; Colens A; Ghosez L Synyhesis of acyl halides under very mild conditions. J. Chem. Soc., Chem. Commun 1979, 1180–1181. [Google Scholar]
- (23).Li Q; Seiple IB A concise route to virginiamycin M2. Tetrahedron 2019, 75, 3309–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) Sharkey LKR; O’Neill AJ Antibiotic resistance ABC-F proteins: bringing target protection into the limelight. ACS Infect. Dis 2018, 4, 239–246. [DOI] [PubMed] [Google Scholar]; (b) Sharkey LKR, Edwards TA & O’Neill AJ ABC-F proteins mediate antibiotic resistance through ribosomal protection. MBio. 2016, 7, e01975–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).(a) Gäumann E; Hütter R; Keller-Schierlein W; Neipp L; Prelog V; Zähner H Stoffwechselprodukte von Actnomyceten. 21. Mitteilung. Lankamycin und Lankacidin. Helv. Chim. Acta 1960, 43, 601–606. [Google Scholar]; (b) Harada S; Higashide E; Fugono T; Kishi T Isolation and Structures of T-2636 Antibiotics. Tetrahedron Lett. 1969, 10, 2239–2244. [DOI] [PubMed] [Google Scholar]
- (26).(a) Kamiya K; Harada S; Wada Y; Nishikawa M; Kishi T X-Ray Analysis of An Antibiotic, T-2636 A (Bundlin B). Tetrahedron Lett. 1969, 27, 2245–2258. [DOI] [PubMed] [Google Scholar]; (b) Uramoto M; Otake N; Ogawa Y; Yonehara H The Structures of Bundlin A (Lankacidin) and Bundlin B. Tetrahedron Lett. 1969, 27, 2249. [DOI] [PubMed] [Google Scholar]; (c) Harada S Studies on Lankacidin-Group (T-2636) Antibiotics. VI. Chemical Structures of Lankacidin-Group Antibiotics. II. Chem. Pharm. Bull 1975, 23, 2201–2210. [DOI] [PubMed] [Google Scholar]; (d) Suzuki T; Mochizuki S; Yamamoto S; Arakawa K; Kinashi H Regulation of Lankamycin Biosynthesis in Streptomyces rochei by Two SARP Genes, srrY and srrZ. Bioscience, Biotechnology, and Biochemistry 2010, 74, 819–827. [DOI] [PubMed] [Google Scholar]
- (27).Lu C; Li J; Qi H; Zhang H; Zhang J; Xiang W; Wang J; Wang X Two New Lankacidin-Related Metabolites from Streptomyces sp. HS-NF-1178. J. Antibiot 2018, 71, 397–401. [DOI] [PubMed] [Google Scholar]
- (28).Zheng K; Hong R Postulated Biogenesis-Guided Total Synthesis and Structural Revision of 2,18-seco-Lankacidinol A. Org. Lett 2020, 22, 3785–3788. [DOI] [PubMed] [Google Scholar]
- (29).(a) Higashide E; Fugono T; Hatano K; Shibata M T-2636 Antibiotics. I. Taxonomy of Strptomyces Rochei Var Volubilis Var Nov. and Production of the Antibiotics and an Esterase. J. Antibiot 1971, 24, 1–12. [PubMed] [Google Scholar]; (b) Fugono T; Harada S; Higashide E; Kishi T T-2636 Antibiotics. III. A new component, T-2636F. J. Antibot 1971, 24, 23–28. [DOI] [PubMed] [Google Scholar]; (c) Tsuchiya K; Yamazaki T; Takeuchi Y; Oishi T Studies on T-2636 Antibiotics. IV. In Vitro and in Vivo Antibacterial Activity of T-2636 Antibiotics. J. Antibiot 1971, 24, 29–41. [DOI] [PubMed] [Google Scholar]; (d) Harada S; Yamazaki T; Hatano K; Tsuchiya K; Kishi T Studies on Lankacidin-Group (T-2636) Antibiotics. J. Antibiot 1973, 26, 647–657. [DOI] [PubMed] [Google Scholar]; (e) Hayashi T; Suenaga I; Narukawa N; Yamazaki T In Vitro and In Vivo Activities of Sedecamycin Against Treponema Hyodysenteriae. Antimicrob. Agents Chemother 1988, 32, 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).(a) Ootsu K; Matsumoto T Effects of Lankacidin Group (T2636) Antibiotics on the Tumor Growth and Immune Response Against Sheep Erythrocytes in Mice. Gann 1973, 64, 481–492. [PubMed] [Google Scholar]; (b) Ootsu K; Matsumoto T; Harada S; Kishi T Antitumor and Immunosuppressive Activities of Lankacidin-Group Antibiotics: Structure-Activity Relationships. Cancer Chemother. Rep 1975, 59, 919–928. [PubMed] [Google Scholar]; (c) Auerbach T; Mermershtain I; Davidovich C; Bashan A; Belousoff M; Wekselman I; Zimmerman E; Xiong L; Klepacki D; Arakawa K; Kinashi H; Mankin AS; Yonath A The Structure of Ribosome-Lankacidin Complex Reveals Ribosomal Sites for Synergistic Antibiotics. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 1983–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Belousoff MJ; Shapira T; Bashan A; Zimmerman E; Rozenberg H; Arakawa K; Kinashi H; Yonath A Crystal Structure of the Synergistic Antibiotic Pair, Lankamycin and Lankacidin, in Complex with the Large Ribosomal Subunit. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 2717–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).McFarland JW; Pirie DK; Retsema JA; English AR Side Chain Modifications in Lankacidin Group Antibiotics. Antimicrob. Agents Chemother 1984, 25, 226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dorival J; Risser F; Jacob C; Collin S; Dräger G; Paris C; Chagot B; Kirschning A; Gruez A; Weissman KJ Insights into a Dual Function Amide Oxidase/Macrocyclase from Lankacidin Biosynthesis. Nat. Commun 2018, 9, 3998–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kende AS; Koch K; Dorey G; Kaldor I; Liu K Enantioselective Total Synthesis of Lankacidin C. J. Am. Chem. Soc 1993, 115, 9842–9843. [Google Scholar]; (b) Kende AS; Liu K; Kaldor I; Dorey G; Koch K Total Synthesis of the Macrolide Antitumor Antibiotic Lankacidin C. J. Am. Chem. Soc 1995, 117, 8258–8270. [Google Scholar]
- (34).(a) Williams DR; Cortez GS; Bogen SL; Rojas CM Total Synthesis of Lankacyclinol. Angew. Chem., Int. Ed 2000, 39, 4612–4616. [DOI] [PubMed] [Google Scholar]; (b) Williams DR; Rojas CM; Bogen SL Studies of Acyl Nitrene Insertions. A Stereocontrolled Route toward Lankacidin Antibiotics. J. Org. Chem 1999, 64, 736–746. [DOI] [PubMed] [Google Scholar]
- (35).Zheng K; Shen D; Hong R Biomimetic Synthesis of Lankacidin Antibiotics. J. Am. Chem. Soc 2017, 139, 12939–12942. [DOI] [PubMed] [Google Scholar]
- (36).(a) Stereoselective Addition of Benzonitrile Oxide and N-Benzyl-C-phenylnitrone to (5RS, 6SR)-5,6-Dihydro-6-Ethyl-5-Methylpyran-2(2H)-one. Crystal Structure of (1RS, 4RS, 5RS, 6RS, 9SR)-8-Benzyl-1,5-Dimethyl-4-Ethyl-9-Phenyl-3,7-Dioxa-8-azabicyclo[4. 3. 0]nonan-2-one. J. Chem. Soc., Chem. Commun 1985, 2763–2767. [Google Scholar]; (b) Roe JM; Thomas EJ Development of a Synthesis of Lankacidins: Synthesis of the C(14) –C(6) Fragment and Introduction of the C(10) –C(13) diene. J. Chem. Soc., Perkin Trans 1 1995, 359–368. [Google Scholar]
- (37).Cordovilla C; Bartolomé C; Martínez-Ilarduya JM; Espinet P The Stille Reaction, 38 Years Later. ACS Catal. 2015, 5, 3040–3053 [Google Scholar]
- (38).(a) Srogl J; Alled GD; Liebeskind LS; Sulfonium Salts: Participants par Excellence in Metal-Catalyzed Carbon–Carbon bond-forming reactions. J. Am. Chem. Soc 1997, 119, 12376–12377. [Google Scholar]; (b) Fürstner A; Ackerstaff J; Formal Total Synthesis of Haouamine A. Chem. Commun 2008, 2870–2872. [DOI] [PubMed] [Google Scholar]
- (39).(a) Negishi E; Matsushita H; Chatterjee S; John RA Selective Carbon-Carbon Bond Formation via Transition Metal Catalysis. 29. A Highly Regio- and Stereospecific Palladium-Catalyzed Allylation of Enolates Derived from Ketones. J. Org. Chem 1982, 47, 3188–3190. [Google Scholar]; (b) Trost BM; Self CR On the Palladium-Catalyzed Alkylation of Silyl-Substituted Allyl Acetates with Enolates. J. Org. Chem 1984, 49, 468–473. [Google Scholar]; (c) Trost BM; Van Vranken DL Asymmetric Transition Metal-Catalyzed Allylic Alkylations. Chem. Rev 1996, 96, 395–422. [DOI] [PubMed] [Google Scholar]; (d) Sha S-C; Zhang J; Carroll PJ; Walsh PJ Raising the pKa Limit of “Soft” Nucleophiles in Palladium-Catalyzed Allylic Substitutions: Application of Diarylmethane Pronucleophiles. J. Am. Chem. Soc 2013, 135, 17602–17609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).(a) Lee JH; Gupta S; Jeong W; Rhee YH; Park J Characterization and Utility of N-Unsubstituted Imines Synthesized from Alkyl Azides by Ruthenium Catalysis. Angew. Chem. Int. Ed 2012, 51, 10851–10855. [DOI] [PubMed] [Google Scholar]; (b) Kwon Y; Rhee YH; Park J Chemoselective, Isomerization-Free Synthesis of N-Acylketimines from N-H Imines. Kwon Y; Rhee YH; Park, J. Adv. Synth. Catal 2017, 359, 1503–1507. [Google Scholar]
- (41).Crimmins MT; She J An Improved Procedure for Asymmetric Aldol Additions with N-Acyl Oxazolidinones, Oxazolidinethiones and Thiazolidinethiones. Synlett 2004, 8, 1371–1374. [Google Scholar]
- (42).Cundy DJ; Donohue AC; McCarthy TD An Asymmetric Synthesis of ADDA and ADDA-Glycine Dipeptide Using the β-Lactam Synthon Method. J. Chem. Soc., Perkin Trans 1 1999, 559–567. [Google Scholar]
- (43).Schlessinger RH; Poss MA; Richardson S; Lin P The Vinylogation of Aldehydes: an Improved Method for the Preparation of Alpha Formylethylidenetriphenylphosphorane, and an Improved Alpha Silyl Imine Reagent of Propionaldehyde. Tetrahedron Lett. 1985, 26, 2391–2394. [Google Scholar]
- (44).Takada H; Yamada T; Hirose T; Ishihara T; Nakashima T; Takahashi Y; Omura S; Sunazuka T Total Synthesis and Determination of the Absolute Configuration of Naturally Occurring Mangromicin A, with Potent Antitrypanosomal Activity. Org. Lett 2017, 19, 230–233. [DOI] [PubMed] [Google Scholar]
- (45).Heravi MM; Zadsirjan V Recent Advances in the Application of the Oppolzer Camphorsultam As a Chiral Auxiliary. Tetrahedron: Asymmetry 2014, 25, 1061–1090. [Google Scholar]
- (46).Zheng K; Xie C; Hong R Concise Synthesis and Revision of Proposed Biogenesis of Helicascolides. Tetrahedron Lett. 2017, 58, 4459–4464. [Google Scholar]