

Abstract
The successes with chimeric antigen receptor (CAR) T cell therapy in early clinical trials involving patients with pre-B cell acute lymphoblastic leukaemia (ALL) or B cell lymphomas have revolutionized anticancer therapy, providing a potentially curative option for patients who are refractory to standard treatments. These trials resulted in rapid FDA approvals of anti-CD19 CAR T cell products for both ALL and certain types of B cell lymphoma — the first approved gene therapies in the USA. However, growing experience with these agents has revealed that remissions will be brief in a substantial number of patients owing to poor CAR T cell persistence and/or cancer cell resistance resulting from antigen loss or modulation. Furthermore, the initial experience with CAR T cells has highlighted challenges associated with manufacturing a patient-specific therapy. Understanding the limitations of CAR T cell therapy will be critical to realizing the full potential of this novel treatment approach. Herein, we discuss the factors that can preclude durable remissions following CAR T cell therapy, with a primary focus on the resistance mechanisms that underlie disease relapse. We also provide an overview of potential strategies to overcome these obstacles in an effort to more effectively incorporate this unique therapeutic strategy into standard treatment paradigms.
In April 2012, following successes first experienced with CD19-directed chimeric antigen receptor (CAR) T cell therapy in adults with follicular lymphoma or chronic lymphocytic leukaemia (CLL)1–3, the first child with acute lymphoblastic leukaemia (ALL) was infused with anti-CD19 CAR T cells4,5. Despite developing a life-threatening toxicity now referred to as cytokine-release syndrome (CRS), this child’s remarkable recovery and ongoing remission (now persisting for >6 years), alongside concurrent developments in other paediatric and adult patients, provided the first realization of the potential of CAR T cell therapy to induce durable remissions. Subsequently, unprecedented successes in early phase trials of anti-CD19 CAR T cell for the treatment of relapsed and/or refractory CD19-expressing B cell malignancies, with many patients achieving long-term remission and potentially cure6–10, have led to FDA approval of two distinct anti-CD19 CAR T cell products11–13 for the treatment of both B cell ALL and diffuse large B cell lymphoma and have revolutionized the field of anticancer immunotherapy. Furthermore, with >100 trials of CAR T cells ongoing globally14, comparable high rates of remission induction have now been demonstrated to be possible with CAR T cells targeting antigens other than CD19 (REF.15).
As more patients are treated and longer follow-up data are becoming available, we are realizing that approximately 30–50% of patients who achieve remission with anti-CD19 CAR T cells will have disease relapse, the majority within 1 year of treatment9,10. Relapses will not be unique to agents targeting CD19, as the initial clinical experience with other CAR targets, such as CD22, indicates that relapse will be a common and recurring challenge15. Furthermore, approximately 10–20% of patients fail to enter remission after receiving anti-CD19 CAR T cell therapy7–10. Loss or modulation of the target antigen15–17 and/or a lack of CAR T cell persistence18, as well as product manufacturing failures19,20 (FIG. 1), are among the more commonly cited impediments to effective CAR T cell therapy. Furthermore, similar successes with CAR T cells have not yet been achieved in diseases beyond B cell leukaemia and lymphoma. As antigen-directed CAR T cells become more widely used, understanding the limitations of CAR T cell therapy and overcoming these obstacles will be crucial to harnessing the full potential of this highly effective treatment modality.
Fig. 1 |. Limitations to durable remissions after CAR T cell therapy.
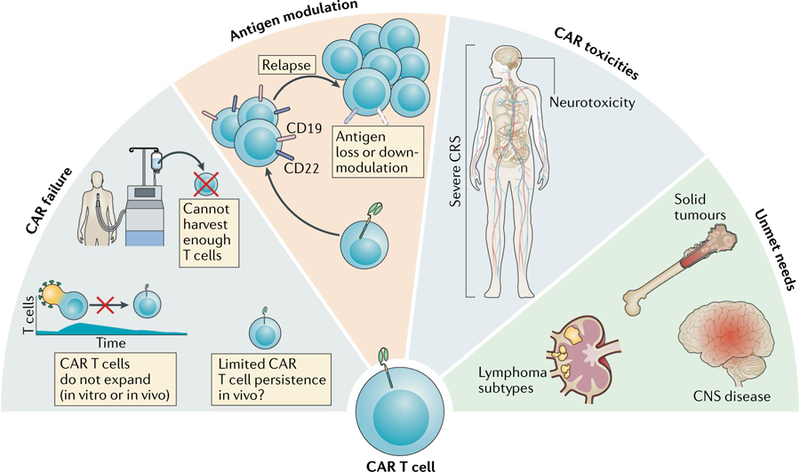
This figure summarizes several different limitations to achieving a durable remission with chimeric antigen receptor (CAR) T cell therapy. First, CAR T cell failures have several causes: for some patients, the CAR T cell product cannot be successfully manufactured or the generated CAR T cells do not expand sufficiently (either during manufacturing in vitro or after administration in vivo); in other patients, the problem of limited persistence in vivo is a potential mechanism underlying disease relapse. Second, antigen modulation — depicted by the loss or downregulation of CD19 and/or CD22 on malignant B cells — enables antigen escape as a mechanism of resistance to CAR T cell therapy, which can also be a problem in non-B cell malignancies, including solid tumours. Third, the characteristic toxicities of CAR T cell therapy — primarily severe cytokine-release syndrome (CRS) and/or neurotoxicity — can be fatal, thus abolishing the potential for therapeutic benefit in a small proportion of patients. Furthermore, data on the effect of therapeutic interventions for CRS on the durability of CAR remission remain unknown. Finally, unmet needs include some disease contexts that are a focus for ongoing research efforts to optimize the clinical utility of CAR T cell therapies. For example, although anti-CD19 CAR T cells can provide substantial benefit for adults with lymphoma, the complete remission rates are lower than those achieved in patients with leukaemia. Additionally, the outcomes of CAR T cell therapy in paediatric patients with lymphoma and in patients with central nervous system (CNS) involvement remain an area of ongoing investigation. Notably, such therapies currently have limited efficacy in patients with solid tumours, and approaches to optimize response are being explored.
In this Review, we summarize the rapidly evolving knowledge regarding the barriers to durable CAR T cell-induced remissions. With a primary focus on identifying mechanisms of emerging resistance to CAR T cell therapy associated with antigen escape, we also discuss antigen-positive relapses associated with poor CAR T cell persistence and address limitations associated with product manufacturing, nonresponsiveness or incomplete responses and toxicities. Furthermore, we will highlight areas of active research on potential strategies to overcome the limitations of CAR T cell therapies and novel indications for these treatments.
Barrier 1: failure to achieve remission
Potential benefit from CAR T cells first requires that a patient has access to the therapy and that a CAR T cell product is successfully manufactured, infused and effectively mediates a cytotoxic response, ideally resulting in complete remission. Remarkably, these prerequisites have proved to be feasible in most patients with ALL, although they still present potential barriers to effective CAR T cell therapy in some patients and, therefore, provide opportunities for improvement.
Product manufacturing.
Given the aggressive nature of most cancers for which treatment is indicated, the successful manufacture and infusion of CAR T cells in a timely manner remain barriers to the implementation of effective CAR T cell therapy. The first hurdle in this process is the collection of T cells to be used to generate a CAR-expressing product. Unfortunately, limited data are available on the actual percentage of patients with ALL for whom a sufficient number of T cells cannot be harvested, as patients are often excluded from consideration for CAR T cell therapy, typically as a result of pre-existing lymphopenia that can result from prior cytotoxic therapy. Even if T cells are collected successfully, patients might not receive therapy, for example, in the event of a product manufacturing failure. Notably, in one phase I study in which the baseline absolute lymphocyte counts (ALCs) of paediatric candidates for treatment with anti-CD19 CAR T cell therapy were analysed8, the median ALC was 1,228 cells/μl (range 168–4,488 cells/μl). This finding indicates that patient selection is highly skewed towards those with ALCs within the normal physiological range, which might result in the exclusion of patients who are heavily pretreated or receiving ongoing therapy. While groups at many centres are currently incorporating a minimum ALC or CD3+ cell count to proceed with apheresis, improvements in CAR T cell manufacturing will probably lower the limits for cell collection. Our experience at the National Cancer Institute (NCI) suggests that manufacturing of a CAR T cell product is feasible with a CD3+ cell count of ≥150 cells/μl. Specifically, an anti-CD22 CAR T cell product was successfully manufactured for 55 of 56 patients enrolled on a clinical trial performed at the NCI; the median CD3+ cell count in these patients was 567 cells/μl (range 145–2,144 cells/μl), and the median ALC was 775 cells/μl (range 230–4,620 cells/μl) (N.N.S. and T.J.F., unpublished observations). The likelihood of obtaining a sufficient number of T cells is certainly an important consideration, and additional data are needed in this area.
Characteristics of the apheresis product beyond cell quantity can also affect the ability to successfully manufacture a CAR T cell product or the quality of the manufactured CAR T cell product. Previous treatment with chemotherapy regimens, for example, those containing clofarabine or doxorubicin, has been implicated in the generation of quantitatively insufficient or poor-quality CAR T cell products7,21. Additionally, clinical data suggest that prior treatment with cyclophosphamide and cytarabine selectively reduces early lineage T cells that are associated with productive CAR T cell expansion22. Much of the data on CAR T cell manufacturing have been generated in patients with ALL or CLL; however, results published in the past 2 years indicate that manufacturing CAR T cell products from patients with high-risk solid tumours might be particularly challenging19,21. Reasons for this finding remain under investigation, but one potential explanation relates to the higher quantity of circulating myeloid-derived suppressor cells often found in patients with solid tumours than in those with haematological malignancies23,24. Implementation of CAR T cell manufacturing in patients with haematological malignancies beyond B cell leukaemias might also reveal further barriers to successful cell harvesting in certain patient populations and/or disease-specific contexts. Early collection of T cells in patients identified as having a high risk of relapse, or before treatment of those with relapsed disease, might improve the quality of the apheresis product and thus the resulting CAR T cells.
Other details of the CAR T cell manufacturing process are also likely to affect qualitative aspects of the resulting product and thus subsequent clinical responses. Current manufacturing protocols incorporate a T cell expansion step typically induced by activation through the T cell receptor (TCR)25,26. Both anti-CD3 and anti-CD28 bead-based expansion and anti-CD3 antibody expansion protocols are used in the generation of the FDA-approved anti-CD19 CAR T cell products27,28. The majority of ongoing trial protocols also use one of these approaches for CAR T cell expansion. To date, the superiority of one method of T cell expansion has not been established. Expansion protocols for CAR T cells also typically include the use of cytokines, such as IL-2, IL-7 and/or IL-15; again, the superiority of one method has not been established, but differential effects of these cytokines are likely to qualitatively affect the resultant CAR T cell product29.
Almost all CAR T cell products are generated using both CD4+ T cells and CD8+ T cells, and both of these cell populations probably contribute to therapeutic efficacy. The ratio of CD4+ to CD8+ T cells can vary substantially between patients and might also affect the CAR T cell product, although differences in efficacy between products with different ratios have not been established. On the basis of preclinical data demonstrating improved potency30, in some trials, separate CD4+ and CD8+ CAR T cell products have been generated for delivery to patients in a defined ratio8,31. Iterative modifications to the manufacturing process have the potential to improve the efficacy of CAR T cells, which will probably be of greater importance as CAR T cells are tested beyond B cell malignancies. Novel approaches to CAR T cell manufacturing, such as use of closed-system automated devices, incorporating the aforementioned foundations are just beginning to be implemented32,33; clinical trials using these approaches and further analysis of how these techniques will impact future CAR T cell therapy are warranted.
Even if a CAR T cell product can be manufactured successfully, the starting T cell phenotype has been demonstrated to be an important determinant of subsequent clinical activity. Selection of T cells with specific phenotypes, for example, central memory or stem cell-like memory T cells, before manufacturing22,34,35 or manipulation of the manufacturing conditions to skew CAR T cell production towards a particular T cell population might improve the likelihood of generating a successful product35. Preferential manufacturing approaches involving these unique T cell populations are under development. In a study of patients with CLL who received an anti-CD19 CAR T cell product36, responders were found to have a CAR T cell population that was enriched for expression of memory-related genes, compared with that of nonresponders, and had higher numbers of CD27+PD-1−CD8+ CAR T cells expressing high levels of the IL-6 receptor, thus leading to better tumour control; this cell phenotype was more predictive of a response than other disease and patient characteristics36.
Other cellular components in the apheresis product can affect CAR T cell manufacturing; this might be a particular challenge given patient heterogeneity and the presence of circulating blasts and nonmalignant cells, such as myeloid-derived suppressor cells, which can inhibit T cell growth37. A report of the incidental CAR transduction of B cells, which conferred resistance to subsequent CAR T cell therapy38, highlights the importance of having a purified starting product. Strategies to optimize the input apheresis product before CAR T cell manufacturing, generally through T cell selection and enrichment, have been developed by several groups19,37,39. Indeed, at the NCI, we have observed that, by further enhancing the purity of the starting T cell population and reducing the presence of additional inhibitory cells, the positive selection of CD4+ T cells and CD8+ T cells using a magnetic bead-based approach can result in successful manufacturing of CAR T cell from an apheresis product following failed manufacturing using a protocol with elutriation-based enrichment for lymphocytes. Furthermore, we have found that this manufacturing change can also increase the potency of the resulting CAR T cell product40. Guidelines for the development of commercial CAR cell products have been developed and are likely to evolve as our knowledge and experience grow41,42.
CAR construct design is another parameter that probably affects the characteristics of a CAR T cell product and subsequent in vivo behaviour of the modified cells, including their kinetics of expansion and duration of persistence. The majority of CAR T cell products being tested in clinical trials are second-generation agents, meaning that they contain both a TCR stimulatory domain (typically derived from the T cell surface glycoprotein CD3 ζ-chain (CD3ζ)) and a single co-stimulatory domain. The current FDA-approved products contain either a CD28 or a 4-1BB (also known as CD137) co-stimulatory domain. The effect of the co-stimulatory domain on response rates has not been systematically evaluated, although preclinical data43 and observations in patients44,45 indicate that this aspect of CAR design markedly affects the persistence of the cell product. Other details of CAR design, such as specific characteristics of the antigen-binding domain, the presence and structure of an extracellular hinge region and features of the transmembrane domain, might also affect CAR T cell attributes, but definitive data defining the effects of these design details have not yet been generated.
The current FDA-approved CAR T cell products and those used in the majority of clinical trials exploit retroviral vectors for insertion of the CAR gene construct into the genome of T cells46,47. Both gamma-retroviral vectors and HIV-derived lentiviral vectors have been successfully used for this purpose. Detailed descriptions of these vectors can be found elsewhere25,48,49 and are beyond the scope of this Review. Certain aspects of vector design are likely to affect the efficiency of gene transfer and, potentially, the activity of the resulting CAR T cell product, although these aspects have not been systematically evaluated. RNA transfection has also been used to deliver the CAR gene construct, resulting in transient CAR expression (because the CAR gene is not inserted into the genome of the T cells50). Such transient CAR-expressing T cell products have only short-lived benefits in preclinical models, with no durable responses observed51. Finally, non-viral vector-based introduction of the CAR gene into the T cell genome has been successfully achieved using a transposon–transposase system52,53, and clinical trials using such products have been reported, with the data demonstrating the safety and feasibility of this approach and providing preliminary evidence of CAR T cell activity54.
One important issue in the field of CAR T cell research is establishing reliable criteria to define the potency of CAR T cell products. At present, standard parameters that define T cell potency, such as markers of T cell exhaustion, have been disappointing in terms of predicting clinical efficacy55,56. Given the high response rates with CAR T cell therapy in patients with leukaemia and thus the small numbers of nonresponders, systematically evaluating and establishing parameters associated with a lack of responsiveness are difficult — particularly considering that the underlying causes are likely to be multifactorial and not fully attributable to product variables alone. In patients with CLL, however, among whom response rates have been substantially lower than those in patients with ALL or lymphoma, Fraietta et al.36 were able to identify favourable product characteristics, such as enrichment for IL-6–STAT3 signatures and an elevated frequency of CD29+CD45RO−CD8+ T cells before CAR T cell generation. Furthermore, pre-infusion anti-CD19 CAR T cell products comprising polyfunctional T cells subsets, defined on the basis of cytokine and chemokine expression profiles, have been associated with an improved response in patients with lymphoma compared with products without such polyfunctional activity57. Additional data will be required to establish desirable attributes of CAR T cells, but the optimal characteristics might differ depending on the CAR construct and the malignancy being targeted.
Generating a CAR T cell product using cells from healthy donors is an alternative strategy to circumventing issues of poor CAR T cell quality. Several groups have now tested donor-derived CAR-based treatment strategies both preclinically58 and in patients with post-transplant disease relapse using T cells harvested directly from their original allogeneic stem cell donors59–61. The results of these early studies demonstrate the feasibility of this approach, with only a low frequency of high-grade graft-versus-host disease (GVHD)59–61. Manufacturing CAR T cells from third-party donors might enable the development of universal, off-the-shelf products and is another method to overcome the problem of quantitatively insufficient or poor-quality CAR T cell products62,63. Generating such products will probably require additional genetic modification to limit CAR T cell rejection and/or GVHD but also offers opportunities to overcome resistance mechanisms, such as ‘fratricide’ reported with T cell antigen-targeted CAR T cell products64. Other novel strategies to enhance CAR-based therapeutic strategies and provide an alternative off-the-shelf product include CAR-engineered nature killer (NK) cells; indeed, preclinical and early phase clinical studies of such products are underway65–67. Additional potential benefits of incorporating NK cells into a CAR-based cell therapy include a more favourable adverse-effect profile than CAR T cell therapy alone, potentially owing to differential cytokine responses between CAR NK cells and conventional CAR T cells; however, further studies are needed to identify other features that determine the toxicity profiles of these two types of CAR-modified cell products68,69.
Infusion of CAR T cells.
Clearly, failure to infuse a CAR T cell product in a timely manner before patients develop progressive disease or disease-related complications can also preclude successful treatment. In the registration trial of the FDA-approved anti-CD19 CAR T cell product tisagenlecleucel in children and young adults with ALL10, 17 (18%) of 92 patients who had an apheresis product collected did not receive a CAR T cell infusion for reasons including tisagenlecleucel-related product issues in 7 patients (primarily from poor cell growth), death before CAR T cell infusion in 7 patients (attributable to disease progression in 4 patients and to infection-related complications in the others) and development of interval adverse events (fungal disease or GVHD) that rendered the patient ineligible to receive the CAR T cell infusion. In this study10, the median time to cell infusion was 45 days (range 30–105 days). Similarly, in a study performed at the Memorial Sloan Kettering Cancer Center9, 11 (14%) of 78 adult patients with ALL who underwent apheresis did not proceed with cell manufacturing (the majority sought alternative treatments), and 13 (19%) of the 67 patients for whom CAR T cell manufacturing was pursued did not undergo infusion owing in part to product failures, disease progression or complications rendering them ineligible to receive therapy. With 20–30% of candidates ultimately not being infused with CAR T cells, future strategies to shorten manufacturing times would be predicted to improve the likelihood that eligible patients will receive an infusion and thereby increase the number of patients who benefit from CAR T cell therapy. Commercialization of CAR T cell therapy and more readily accessible manufacturing programmes would hopefully decrease the delays in time to infusion.
CAR T cell activation and expansion.
Clinical studies have revealed that the dose of CAR T cells required for effective therapy is remarkably small, with the current dosing regimen of 0.2–5.0 × 106 transduced CAR T cells per kg or a total of 0.1–2.5 × 108 transduced CAR T cells per infusion, although activation and exponential expansion of the cells following infusion are essential6–10,70,71. As discussed, the quality and inherent T cell phenotype of the CAR T cell product can affect post-infusion CAR T cell behaviour. In addition, recipient-related factors are also important in CAR T cell expansion. For example, disease (and thus antigen) burden can positively influence the degree of cell expansion, which in turn might increase the risk and severity of CRS7,8. Lymphodepletion also seems to be important for CAR T cell expansion, with evidence indicating that certain chemotherapeutic agents, such as fludarabine, might be more effective in this regard31. However, fludarabine has been implicated as a potential contributor to CAR T cell-associated neurotoxicity, although the timing of classical fludarabine-associated neurotoxicity differs from that of CAR T cell-related toxicities, and the safety of lymphodepletion with this agent is generally supported72. Whether CAR T expansion can be achieved without the need for cytotoxic chemotherapy remains to be determined, and this requirement will probably vary between disease subtypes.
Access to CAR T cell therapy.
Despite the FDA approvals of CD19-directed CAR T cell products over the past 2 years11,12, access to these novel therapies remains limited. Nevertheless, ongoing efforts are improving the ability of patients to access CAR T cell therapy. With the FDA approval of two different anti-CD19 CAR constructs11,12, the most dramatic improvement in access reflects a very steady increase in the number of centres across the USA that are qualified to administer these products: at the time of the submission of this manuscript, 73 individual centres were listed as Kymriah Treatment Centers73 authorized to administer tisagenlecleucel, with a similar number of authorized axicabtagene ciloleucel treatment centres74. Thus, the ability of eligible patients to receive an FDA-approved CAR T cell product at their local cancer centre is increasing, thereby facilitating incorporation of these therapies into their individual treatment plan. Additionally, the international approvals of the same CAR T cell products in Europe75 and Canada76 further contribute to increasing access for patients in need and to reducing the regional disparities — although such limitations continue to exist77, at least partially owing to the fact that candidate treatment centres must have the capacity and capability to adhere to product manufacturing and administration protocols and safely manage the patient. Establishing standardized guidelines for CAR T cell therapy, determining which centres are most able to deliver this therapy and exploring the role for accreditation of sites are all subjects of ongoing research efforts aimed at improving access to this treatment modality.
Additionally, costs and insurance coverage are persistent barriers to expanding patient access to CAR T cell therapy78–80. Cost analyses and optimization of manufacturing strategies to reduce costs are needed as this therapy moves forward. In this regard, developing position statements supporting the utility of CAR T cell therapy as part of current treatment paradigms will probably facilitate a clearer path towards addressing issues of payment and insurance coverage. Many patients will continue to seek enrolment on clinical trials in order to circumvent limitations in access to FDA-approved constructs, but initial access to those FDA-approved agents would be more desirable than inclusion in a trial of an experimental agent — the benefits which are unlikely to have been established.
Additionally, eligibility criteria for receipt of the FDA-approved agents might be considered a limitation to patient access. For example, active central nervous system (CNS) involvement remains an exclusionary criterion among patients with B cell lymphoma; however, clinical trials to study the safety and efficacy of CAR T cell therapy in this population and others are essential, with the ultimate aim of extending the treatment indications to all populations that could potentially derive benefit.
Barrier 2: disease relapse
Disease relapse following anti-CD19 or anti-CD22 CAR T cell therapy can occur in up to 50% of patients with pre-B cell ALL by 12 months after infusion8–10,15 in two major patterns: early relapse of antigen-positive leukaemia or later relapse typically associated with antigen loss (FIG. 1). An increased understanding of the mechanisms underlying poor persistence of and/or resistance to CAR T cells and identifying patients with the highest likelihood of relapse will be crucial to optimizing CAR T cell therapy.
Antigen-positive relapse.
Early ALL relapse, typically within the first few months after successful induction of remission, is often associated with limited CAR T cell persistence and/or transient B cell aplasia, which suggests a loss of active CAR T cell-mediated surveillance of leukaemia6. Determinants of CAR T cell persistence remain to be fully determined but, in addition to inherent T cell quality (which might be patient-dependent and context-dependent81) and initial T cell phenotype (including the proportion of CD4+ versus CD8+ T cells82), include the co-stimulatory domain built into each unique CAR construct83, with preclinical reports indicating that CD28 co-stimulatory domain-containing CARs tend to persist less well than those containing a 4-1BB co-stimulatory domain45,84; clinical experience is consistent with these data6,7,85. In the aforementioned pivotal clinical trial of tisagenlecleucel10, 4-1BB-based CAR T cells persisted in the blood for a median duration of 168 days (range 20–617 days), generally with concurrent B cell aplasia in patients who remained in remission. By contrast, the median duration of CD28-based anti-CD19 CAR T cell persistence has been ~30 days, and these cells are rarely detected beyond 3 months7,86. The better persistence of the 4-1BB domain-containing CAR T cells might result, in part, from the reduced propensity for T cell exhaustion induced by tonic CAR signalling when co-stimulation is mediated by a 4-1BB versus a CD28 domain87. Efforts to calibrate the CAR activation potential in order to optimize durability of response and balance effector versus memory T cell expansion88, among other approaches89,90, are underway. Other co-stimulatory domains and multiple co-stimulatory domains have also been used in CAR constructs and will probably affect CAR T cell persistence. Additionally, further analysis of observations in patients with a notable expansion and/or persistence of the CAR T cells suggest a role for targeted genomic integration of the CAR construct to enhance persistence. For example, the anecdotal experience of clonal expansion in a patient with CLL revealed that TET2 disruption results in alterations in CAR T cell biology, leading to enhanced potency and a central memory phenotype91. Similarly, specific integration of the CAR gene into the TCRa constant (TRAC) locus of the T cell genome using CRISPR–Cas9 editing machineries results in better antitumour responses than those observed with conventionally transduced CAR T cells in preclinical models92. Advances in gene-editing technologies93 combined with enhanced understanding of determinants of CAR T cell potency and knowledge of determinants of therapeutic responsiveness will be crucial to the design of the next generation of CAR T cell technologies36,94. Thus, future directions to improve CAR persistence will rely heavily on understanding the optimal T cell biology in relation to CAR T cell functionality and subsequent optimization of CAR T cell design to promote persistence (when persistence is desired).
Strategies to improve persistence independently of CAR T cell design and manufacturing are being tested in the clinic; for example, administration of T cell-antigen-presenting cells (T-APCs) designed to activate anti-CD19 CAR T cells at regular intervals following remission induction in order to determine whether recurrent stimulation can reactivate and numerically expand the CAR T cells and prevent antigen-positive relapse ( NCT03186118). More broadly, the use of artificial antigen-presenting cells provides a potentially off-the-shelf approach for optimizing adoptive T cell immunotherapy by increasing the therapeutic efficacy and persistence of the infused T cells95–97. Shifting the CAR T cells towards a central memory or stem cell-like memory phenotype is another unique method of enhancing therapeutic responses and cell persistence35,36.
Combining CAR T cell therapy with immunecheckpoint inhibitors or other immunomodulatory therapies provides a synergetic approach to optimizing the rate, depth and durability of clinical responses98. Evidence for increased PD-1 expression in CAR T cells during the time from infusion to peak expansion has been demonstrated in clinical samples85, and preclinical data support a role of PD-1–PD-L1 blockade in improving the effectiveness of CAR T cell therapy99,100. Clinical testing of this strategy with FDA-approved immune-checkpoint inhibitors and CAR T cell products has provided anecdotal evidence of improved persistence101; future trials to test such approaches are under development.
The persistence of anti-CD19 CAR T cell has an important role in ongoing surveillance and seems to be important for durable remission in patients with ALL; however, whether cell persistence is absolutely necessary to maintain durable remissions achieved with all CAR T cell products remains unclear. To date, the longest follow-up data on CD28-based anti-CD19 CAR T cell therapies in patients with ALL have been reported by Park and colleagues9. In this study, CAR T cell persistence beyond induction of remission was rarely detected, yet the median event-free survival was 6.1 months overall and was 10.6 months in patients with a low disease burden (<5% bone marrow blasts)9. This duration of CAR T cell persistence is generally shorter than that of the 4-1BB-based constructs, although these findings nevertheless demonstrate a capacity for durable remission beyond the CAR T cell persistence, raising additional questions about the determinants of remission duration. Notably, the durability of remission in patients with B cell lymphoma has been relatively similar with both the 4-1BB-based and the CD28-based products71,102, thus suggesting that the relevance of CAR T cell persistence might vary by cancer type. Careful monitoring of outcomes following anti-CD19 CAR T cell therapy for B cell malignancies and following treatment with CAR T cells targeting other antigen and/or tumour types will be important to fully understand the impact of cell persistence on the durability of remissions. Regardless, the ability for CAR T cells to persist in the long term and thereby mediate ongoing surveillance against disease relapse is clearly a potential advantage of these agents over other targeted immunotherapies, such as bispecific or conjugated antibodies, which generally have a limited capacity to induce a long-term durable remission103–106.
Relapse with antigen-positive disease presents a potential opportunity for re-treatment with CAR T cells, although re-infusion strategies to treat antigen-positive relapse occurring with loss of CAR T cell persistence have unfortunately had limited success. Gardner et al.8 administered a second infusion of anti-CD19 CAR T cells to ten children and young adults with ALL. Eight of these patients had loss of CAR T cell persistence, only two of whom had CAR T cell expansion after re-infusion and only one of whom had a complete response8. The two remaining patients, who had detectable CAR T cells, were re-infused for persistent or relapsed CD19+ disease, but neither had a substantial CAR T cell re-expansion, B cell aplasia or anti-leukaemic effect8. Lee et al.7 described three patients who received a second infusion of CAR T cells for residual or recurrent CD19+ ALL at 2–5.5 months after the initial infusion, and none had an objective response. Maude et al.6 used a strategy with repeated re-infusions to counter early loss of CAR T cells and associated B cell recovery, which led to subsequent persistence of CAR T cells101. Turtle et al.31 reported on five patients with ALL who received an anti-CD19 CAR T cell re-infusion, none of whom had re-expansion, persistence or anti-leukaemia activity. Additionally, CAR-specific T cell responses were detected in all five patients, thus suggesting an immune-mediated rejection of CAR T cells upon repeat dosing31. The same group reported similarly poor outcomes following re-infusion of patients with B cell non-Hodgkin lymphoma, but use of an intensified lymphodepletion regimen containing fludarabine in addition to cyclophosphamide improved the re-infusion response, as well as improving initial CAR T cell expansion and persistence107. This concept of intensified lymphodepletion has similarly been used in re-infusion strategies for other CAR-based therapies, with improved clinical outcomes108. Alternative strategies to optimize the re-infusion approach include the use of a different CAR construct or even a CAR targeting a different antigen. The use of humanized CD19 CARs to overcome immune-mediated rejection of murine-derived anti-CD19 CAR constructs is one such strategy being tested in the clinic ( NCT02374333), with early data suggesting that this approach can induce complete remission109. Alternative targeting strategies (for example, with anti-CD22 CAR T cells) might also be effective in patients with disease relapse after prior CAR T cell therapy15. Thus, multiple-infusion strategies to treat or prevent disease relapse remain an active and important area of investigation.
Antigen loss or modulation as a mechanism of immune escape.
Target antigen modulation is one of the clearest mechanisms of disease relapse following successful remission induction using CAR T cells (TABLE 1) and has also been described with other targeted immunotherapeutic approaches, such as bispecific T cell-engager (BiTE) antibody constructs110–112 or monoclonal antibodies113. Established mechanisms leading to loss of CD19 expression include alternative splicing, which generates CD19 isoforms with disruption of the target epitope and/or reduced cell surface expression114,115, and interruption in the transport of CD19 to the cell surface116; other pathways leading to antigen loss are under active study. Complete antigen loss, however, might not be necessary for the development of resistance to initially effective CAR T cell therapy — even diminution of antigen expression can be sufficient. For example, with anti-CD22 CAR T cell therapy, a simple quantitative decrease in CD22 cell surface expression or antigen density in the leukaemic population was adequate to evade the CAR T cells, thus enabling leukaemic relapse despite ongoing CD22 positivity, with individual variation seen in the threshold antigen density that conferred relapse or resistance15. Notwithstanding, a minimum threshold of antigen expression is likely to be needed for functional and/or preserved CAR T cell activity — a concept that has been realized in preclinical models with targeting of both CD20 in B cell malignancies117 and ALK in neuroblastoma118, in which CAR T cell cytolytic activity and cytokine production were increased with higher levels of antigen expression. Along these lines, preclinical experience with an EGFR-targeted CAR T cell therapy suggests therapeutic potential that relies specifically on the differential antigen density of EGFR in tumours versus nonmalignant tissues to limit on-target, off-tumour toxicities while maintaining anticancer activity119.
Table 1 |.
Summary of antigen loss or modulation detected in published clinical trials of CAR T cell therapy
| Target antigen | Publication | Number of patients treated | Number of CRs (%) | Number of patients with antigen modulation (%) | Median time to antigen loss or modulation (months) | Comments |
|---|---|---|---|---|---|---|
| CD19 | Lee et al.7 | 21 | 14 (67) | 2 (14) | ~6 | 10 of 12 patients who were MRD-negative after CAR T cell therapy subsequently underwent HSCT |
| Maude et al.6 | 30 | 27 (90) | 4 (15) | ~3 | None | |
| Gardner et al.8 | 43 | 40 (93) | 7 (18) | ~3 | 11 of 40 subsequently underwent HSCT | |
| Park et al.9 | 53 | 44 (83) | 4 (9) | Unknown | None | |
| Maude et al.10 | 75 | 61 (81) | 15 (25) | Unknown | None | |
| CD22 | Fry et al.15 | 21 | 12 (57) | 7 (58) | ~3 | None |
CAR, chimeric antigen receptor; CRs, complete responses; HSCT, haematopoietic stem cell transplantation; MRD, minimal residual disease.
Antigen loss following effective CAR therapy has been best described in patients with ALL114,115 but is certainly not limited to this disease120. Findings of preclinical121 studies in solid tumour models and clinical studies in patients with glioblastoma122,123 have similarly implicated antigen modulation as a potential pitfall undermining the efficacy of CAR T cell therapy. In the design of future studies and novel CAR constructs, recognition of the role of antigen density in antitumour responses and identification of mechanisms leading to disrupted target expression will be needed in order to optimize CAR T cell responses.
The additive effect of prior targeted immunotherapies might further increase the complexity of immune evasion after CAR T cell therapy. For example, both the anti-CD19 BiTE blinatumomab and the anti-CD22 antibody–drug conjugate inotuzumab ozogamicin are FDA-approved therapies indicated for treatment of ALL, and loss of response with emergence of CD19– (REFS111,112) or CD22− (REF.113) escape variants has been reported in patients treated with these immunotherapies. Thus, such agents might render future therapy with CAR T cells targeting the same antigens less effective or decrease the durability of responses by increasing the risk of antigen-negative relapse. Notably, receipt of prior therapy with blinatumomab was an exclusion criterion of the pivotal registration trial of tisagenlecleucel in patients with ALL10. However, whether such avoidance of targeted immunotherapy before the administration of CAR T cells is necessary to achieve durable long-term responses remains to be determined.
Independent of treatment-related antigen loss or modulation, inherent tumour heterogeneity also has a role in predisposition to emergence of an antigen-negative clone. CD19 has been considered to be ubiquitously expressed on all pre-B cell ALL clones, with development of antigen-negative subclones upon CD19-targeted treatment; however, more-detailed analysis of pre-therapy CD19 expression is necessary, as rare patients have malignant cells with CD19 negativity or partial expression at diagnosis124. Indeed, we now have a greater appreciation that pre-existing CD19– subclones can be present at diagnosis115, with data from some studies indicating the possibility that the malignant B cell progenitors are CD19–, particularly in patients with BCR–ABL1 ALL125. CD22, although also expressed in a high percentage of pre-B cell ALL cells, has a well described heterogeneity in surface expression, particularly in infants with KMT2A (MLL)-rearranged ALL, in whom CD22– ALL cell subpopulations are more frequently detected124,126–128, leading to the emergence of CD22– or CD22dim populations following CD22-targeted therapy15,127.
Lineage switching is another mechanism for evading CAR T cells. This phenomenon was appreciated before the era of targeted therapies in the context of infant, KMT2A-rearranged leukaemia subtypes, which often present as mixed-lineage leukaemias, with patients having transformation to AML following ALL-specific therapy or vice versa129. In both the preclinical and the clinical setting, an analogous phenomenon with emergence of myeloid subtypes following CD19-directed immunotherapy has now been described in ALL with or without KMT2A rearrangement16,17,110,125. Similarly, targeting of FLT3 with CAR T cells in preclinical ALL models was found to induce a reversible B cell to T cell lineage switch (while effectively avoiding transformation to FLT3+ myeloid lineage leukaemias)130. Whether this resistance mechanism is active in non-leukaemic malignancies remains to be determined.
Despite the appreciation of antigen modulation as a mechanism of immune escape, we currently have a limited ability to predict which patients have a high risk of developing antigen-modulated relapsed disease, beyond those with identification of pre-existent antigen-negative subclones, those with established heterogeneity in antigen expression or those who have received prior immunotherapies targeting the same antigen. Minimal residual disease (MRD) monitoring strategies predicated on flow cytometry will be essential to identifying the existence of pretreatment subpopulations that might not be fully amenable to therapies targeting a single antigen, with a greater onus on actively screening for antigen-negative disease; thus, more complex flow cytometric gating methods and other improvements in the immunophenotypic characterization of single cells will be required, particularly when the disease burden is low. Tracking of leukaemic clones over time with adjunctive PCR and/or molecular assessments of VDJ immunoglobulin heavy chain (IgH) rearrangements might improve the prediction of disease relapse by identifying cell populations that are not fully eradicated by CAR T cell therapy or are not as easily identified using flow cytometry. Accordingly, in their phase I study of anti-CD19 CAR T cell therapy, Gardner et al.8 found that 27 (67.5%) of 40 patients who achieved an MRD-negative remission according to flow cytometry assessment had a malignant clone identified concurrently using next-generation sequencing. The majority of these patients (17 of 27; 65%) subsequently achieved a molecular complete remission by day 63, although several did not, suggesting the ongoing presence of leukaemic disease that might ultimately be the harbinger of future relapse.
Given the propensity of antigen modulation as a mechanism for evasion of effective immunotherapy, CAR constructs incorporating multi-antigen targeting are being developed to address inherent tumour heterogeneity and thus decrease the risk of leukaemic relapse. Preclinical data supporting the multi-targeted approaches include the use of tandem anti-CD19–CD20 CAR constructs131, combinatorial anti-CD19 and anti-CD123 (also known as IL-3Rα) strategies132 and CD19 and CD22 targeting using a single CAR construct targeting both antigens or two unique CARs targeting each antigen individually133, and several clinical trials of these strategies are underway (TABLE 2). Follow-up data on these protocols will help to determine whether dual-antigen-targeted approaches are adequate to prevent disease relapse or whether additional combinatorial strategies for targeting more than two antigens will be necessary to make CAR T cell therapy curative. In the development of combinatorial multi-antigen-targeted strategies, ensuring an effective response to each antigens is essential: preferential targeting of one antigen over another could lead to a predilection for a functional response to only a single antigen without obviating the problem of antigen-negative relapse. Developing functional multi-targeted constructs, however, is no easy task and is highly dependent on preclinical testing to identify biologically active constructs with equivalent capacity to target coincident antigens simultaneously, as has been nicely described in the work of Qin and colleagues133.
Table 2 |.
Active clinical trials of multi-antigen CAR T cells in the USA and UK
| Target antigens | Disease | Age group (years) | CAR construct signalling domains | Treatment centre | ClinicalTrials.gov reference number |
|---|---|---|---|---|---|
| CD19 and CD22 | ALL and NHL | 1–26 | CD3ζ–4-1BB (combinatorial approach with anti-CD19 CAR T cells, anti-CD22 CAR T cells and co-transduced anti-CD19 and anti-CD22 CAR T cells) | Seattle Children’s Hospital (Seattle, WA, USA) | NCT03330691 (PLAT-05) |
| ALL | 1–30 | CD3ζ–4-1BB | Lucile Packard Children’s Hospital, Stanford University (Palo Alto, CA, USA) | NCT03241940 | |
| ALL and DLBCL | ≥18 | CD3ζ–4-1BB | Stanford University (Palo Alto, CA, USA) | NCT03233854 | |
| ALL and NHL | 3–30 | CD3ζ –4-1BB | National Cancer Institute (Rockville, MD, USA) | NCT03448393 | |
| ALL | 1–24 | CD3ζ–OX40 (CD19) and CD3ζ–4-1BB (CD22) | Great Ormond Street Hospital (London, UK) | NCT03289455 | |
| CD19 and CD20 | NHL and CLL | 18–70 | CD3ζ–4-1BB | Medical College of Wisconsin (Milwaukee, WI, USA) | NCT03019055 |
ALL, acute lymphoblastic leukaemia; CAR, chimeric antigen receptor; CD3ζ, T cell surface glycoprotein CD3 ζ-chain; CLL, chronic lymphoblastic leukaemia; DLBCL, diffuse large B cell lymphoma; NHL, non-Hodgkin lymphoma.
The role of consolidative therapy after CAR T cell-induced remission.
Given the concern over antigenpositive or antigen-negative disease relapse, the need to consolidate CAR T cell-induced remissions needs to be considered. The first successful clinical applications of CAR T cell therapies have been in patients with haematological malignancies, for whom allogeneic haematopoietic stem cell transplantation (allo-HSCT) is a validated curative option; thus, consolidation treatment with allo-HSCT is a very relevant topic of discussion, particularly when no prior transplantation has been performed. In initial reports from Davila et al.86 and Lee et al.7,134, a high proportion of adults and children or young adults with ALL, respectively, who entered remission after treatment with CD28-based anti-CD19 CAR T cells proceeded to allo-HSCT, with overall improved outcomes. Specifically, in an analysis combining data from trials of anti-CD19 and anti-CD22 CAR T cell products performed at the NCI, 25 patients subsequently underwent allo-HSCT, and this was the first allo-HSCT for 19 patients134. Using a competing risks analysis (risk of relapse versus transplantation-related mortality), the 24-month cumulative incidence of post-allo-HSCT relapse in all 25 patients and in the 19 patients who underwent HSCT for the first time was 13.5% (95% CI 3.2–32.1%) and 11.3% (95% CI 1.7–31.1%), respectively134. Summers et al.135 reported a trend towards improved leukaemia-free survival in patients without a history of allo-HSCT who underwent consolidative allo-HSCT following anti-CD19, 4-1BB-based CAR T cell therapy compared with those who did not (P = 0.057)135. Specifically, among 64 evaluable patients, 17 had no prior history of allo-HSCT, 3 of whom chose not to proceed to allo-HSCT, and 2 (66.7%) of these 3 patients subsequently had disease relapse; among the 14 patients who proceeded to allo-HSCT, only 2 (14.2%) had disease relapse135. Park et al.9 reported that 17 (38.6%) of 44 patients with ALL proceeded to allo-HSCT after attaining complete remission with CD28-based anti-CD19 CAR T cell therapy; after a median follow-up duration of 29 months from CAR T cell administration, 12 (70.5%) of these 17 patients had died or had disease relapse after transplantation. Of the 26 patients who did not proceed to transplantation, 17 (65%) had relapsed or died9. Among 32 patients who obtained MRD-negative complete remission, those who proceeded to allo-HSCT had no difference in event-free survival or overall survival compared with those who did not (P = 0.64 and P = 0.89, respectively)9. Thus, the role of allo-HSCT following CAR T cell therapy needs to be better defined but is likely to be of greater importance with the use of shorter-acting CAR T cell products134, for example, CD28-based CAR T cells in patient with pre-B cell ALL. Regardless, a reduced reliance on consolidative therapy to achieve cure will be an important goal of efforts to optimize CAR T cells.
Barrier 3: CAR T cell-related toxicity
No article on CAR T cell therapy would be complete without a discussion of the related toxicities, particularly CRS; however, several in-depth reviews of the current state-of-the-art knowledge and management of CAR T cell-related toxicities have been published in this journal and elsewhere over the past few years136–142. Notably, efforts are underway to establish uniform multicentre grading scales and, in this regard, the original CRS grading scale proposed by Lee et al.141 has now been updated and published as the American Society for Blood and Marrow Transplant consensus guidelines143. Algorithms and treatment approaches to optimize the safety of CAR T cell therapies, including early intervention strategies144, and the incorporation of suicide genes and other genetic engineering strategies to reduce CAR T cell toxicity are under investigation145.
Of particular relevance to the discussion herein, severe or even fatal CRS and other toxicities are barriers to durable CAR T cell-induced remissions in some patients and, in this regard, in addition to established literature and guidelines in development, we offer two additional areas for consideration.
First, one must be cognizant of the fact that the literature on CRS management is predominantly based on the experience gained in trials of anti-CD19 CAR T cell therapy trials. As novel antigens are targeted, therefore, it will be important to recognize that not all cases of CRS will be the same and, as such, the appropriate intervention strategies might differ. For example, CRS observed after anti-CD22 CAR T cell therapy seemed to be less severe than that seen with anti-CD19 CAR T cells15, without the implementation of any early intervention strategies; however, novel toxicities, such as clinically relevant coagulopathy, occurred with CD22 targeting that required a different approach to treatment (N.N.F. and T.J.S., unpublished observations). When targeting solid tumours with CAR T cell-based strategies, the potential off-tumour, on-target toxicities might be less tolerable than the prolonged B cell aplasia associated with CAR T cells targeting B cell antigens, thus warranting close consideration.
Second, as the treatment of CRS and/or other toxicities becomes better established, evaluating the effects of such treatment, and particularly the effects of pre-emptive or prophylactic strategies to mitigate CRS, on anticancer activity will be equally important. Currently, the limited available data indicate that early intervention strategies can effectively reduce the severity of CRS without compromising peak CAR T cell expansion or functional persistence144. However, these data come from a single trial using a single CAR construct and, therefore, might not be broadly applicable. Furthermore, the effect of prolonged steroid use in those who develop severe CRS on CAR persistence needs to be further studied across various constructs. In addition, determining the ideal time to implement CRS intervention strategies while balancing the potential for an optimal anticancer response will be particularly challenging, compounded by the complexity of novel CAR constructs. Future studies are needed to establish the optimal strategies for intervention to negate toxicities and their impact on CAR persistence. A trial investigating the optimal timing of anti-IL-6 therapy with tocilizumab for the treatment of CRS associated with anti-CD19 CAR T cell therapy is underway ( NCT02906371).
Barrier 4: moving beyond leukaemia
Given the successes to date in heavily pretreated patients, earlier use of CAR T cells has considerable potential to change the treatment paradigm for children and adults with relapsed and/or refractory or high-risk B cell malignancies. Indeed, efforts are underway to understand how to best incorporate these therapies into upfront regimens in order to improve overall outcomes. However, substantial barriers exist when the use of CAR T cells is extended to cancers beyond ALL.
CAR T cell response, persistence and relapse in lymphoma.
With the FDA approval of axicabtagene ciloleucel and subsequently tisagenlecleucel, CAR T cell therapies provide a highly effective approach to the treatment of relapsed and/or refractory large B cell lymphoma; however, unique challenges exist in this setting, including lower remission rates (complete remission rates of 30% and 54% in the pivotal trials of axicabtagene ciloleucel71 and tisagenlecleucel102, respectively, versus 67–93% with anti-CD19 CAR T cells in patients with ALL; TABLE 1) and a limited understanding of the mechanisms of relapse. Antigen loss can be a mechanism of lymphoma relapse after CAR T cell therapy120,146 but seems to occur less often than in patients with ALL (TABLE 1). Notably, reports on the frequency of antigen loss in patients with B cell lymphoma are limited, probably owing to less frequent sampling of lymphomatous disease than of leukaemic disease, but it remains an important consideration for patients with lymphoma relapse after CAR T cell therapy. In addition, CAR T cell persistence might not be as necessary for a durable response in patients with lymphoma as it seems to be in those with ALL. With the seemingly shorter-acting CD28 domain-containing anti-CD19 CAR T cells, the longest follow-up data in patients with B cell lymphoma have been reported by Kochenderfer and colleagues147. In patients with diffuse large B cell lymphoma, specifically, these authors found that most patients had last measurable CAR T cell detection at <6 months after infusion, with 4 of 5 complete responses continuing (at 56, 51, 44 and 38 months) despite recovery of the nonmalignant B cell population147. Moreover, despite the FDA approvals for adults with large B cell lymphomas, the experience with CAR T cell therapies in paediatric lymphomas is scant, and responses have been limited, for reasons that are not fully understood.
Ongoing optimization of responses to CAR T cells in patients with lymphoma, using various aforementioned strategies that are being incorporated into the treatment of ALL, is underway. Testing of these approaches uniformly in paediatric patients with lymphoma, an area of unmet need (FIG. 1), remains a priority for future research.
CAR T cell strategies beyond B cell targeting.
CD19 is generally ubiquitously expressed on the majority of B cells, and targeting of both nonmalignant and malignant CD19+ cells is a therapeutic strategy with acceptable safety, whereas on-target, off-tumour toxicity might be more prohibitive in other cancers. With regard to AML, for example, broad targeting of myeloid lineage cells, particularly if sustained, is not acceptable nor can it be made more feasible using supportive interventions as simple as immunoglobulin replacement in patients with prolonged B cell aplasia. In such settings, use of a less persistent CAR T cell product, together with the administration of rescue stem cells from an allogeneic donor following an effective CAR T cell response, might be a reasonable strategy. A first-in-human study of myeloid cell targeting using anti-CD123, CD28 domain-containing CAR T cells revealed promising anti-leukaemia activity without prolonged myelosuppression148. Clinical testing of myeloid cell targeting with a CD28-based anti-CD33 CAR T cell product is planned149.
Going beyond haematological tumours, solid tumours present a new set of unique challenges to the development effective CAR T cell therapies, as reviewed elsewhere150–153. One key issue with solid tumours is that the inherent tumour heterogeneity is likely to be a substantial barrier to identifying an optimal target, and relatedly, antigen loss will probably be a key factor precluding curative remissions. Indeed, antigen density (as mentioned above regarding ALK in neuroblastoma cells118) and inherent tumour heterogeneity in antigen expression in solid tumours (for example, mesothelin154, HER2 (REF.155) or MUC1 (REF.156) expression in non-smallcell lung cancers) can limit the therapeutic potential of agents that target a single antigen and raise concerns over differing on-target, off-tumour toxicities. Among the CAR T cell-based strategies that are furthest in development, products targeting the disialoganglioside GD2 have demonstrated antitumour activity in patients with neuroblastoma, leading to complete remissions157, thus emphasizing the potential for CAR T cell therapy of solid tumours.
In addition, overcoming the immunosuppressive tumour microenvironment, which might render adoptively transferred T cells inactive, will be particularly relevant to effective CAR T cell therapy for solid tumours. Several approaches to overcome this issue are under evaluation, including optimization of the preparative regimen before CAR T cell infusion150,158. Armoured CAR T cells that constitutively secrete pro-inflammatory cytokines as a mechanism to overcome local immunosuppression are a novel concept. Similarly, incorporation of immune-checkpoint inhibition into CAR T cell-based strategies, by various combinatorial or built-in approaches, might improve responses98; the findings of several preclinical studies support the effectiveness of this approach, warranting further clinical translation98,152,159,160.
Targeting the CNS tumours demands careful consideration of optimal delivery mechanisms for CAR T cell therapy. A case report from 2016 described the use of CAR T cells targeting IL-13 receptor subunit α2 (IL-13Rα2) in a patient with glioblastoma, which incorporated multiple infusions over a 7-month period via two intracranial delivery routes (into the tumour resection cavity followed by infusions into the ventricular system)122. The patient had an impressive clinical response with complete regression of all intracranial and spinal tumours that lasted for 7.5 months122. Strategies to optimize IL-13Rα2-targeted strategies include designing a second-generation 4-1BB co-stimulatory domain-containing CAR product using enriched central memory T cells; in orthotopic glioblastoma models, this product was effective irrespective of corticosteroid treatment (a mainstay of glioblastoma therapy) and had higher efficacy with intraventricular infusions than intravenous delivery (and perhaps also than intratumour administration in the multifocal disease setting)161. Anti-GD2 CAR T cell therapy for histone H3 lysine 27-methylated (H3K27me) diffuse midline gliomas, a uniformly fatal type of paediatric CNS tumour, is another promising approach162. Anti-HER2, 4-1BB domain-containing CAR T cell-based targeting of medulloblastoma has also demonstrated efficacy in both mouse and non-human primate models, with good tolerance of intraventricular administration163. Most notably, HER2-specific CAR T cells have been tested in a phase I dose-escalation study involving patients with progressive glioblastoma, and preliminary results demonstrated the safety and feasibility of this approach and provided an early sign of activity by virtue of partial remission in one patient and disease stabilization in several others164. Similarly, EGFRvIII-directed CAR T cells were found to be safe and feasible in patients with glioblastoma but, as seen in patients with ALL, antigen loss and tumour heterogeneity were major determinants of immune escape123. Thus, optimizing CAR T cell approaches and testing novel delivery mechanisms for CAR T cells targeting CNS disease remains an active area of research.
Ultimately, many challenges must be overcome in the optimization of CAR T cell therapies for diseases beyond ALL, but much progress has been made in the past several years. Lessons learned from B cell-targeted strategies will provide a foundation for moving this field of research forward, but a cautious approach is needed to understand and attempt to mitigate novel CRS-related toxicities and off-tumour, on-target toxicities that might otherwise result in the premature abandonment of promising new CAR T cell-based strategies for treatment of solid tumours.
Conclusions
CAR T cell-based therapy is among the most promising anticancer therapies of all time. Many challenges remain, however, as we strive to make remissions induced by this therapy durable for all treated patients. CAR T cell manufacturing considerations — from patient selection to the characteristics of the infused product and the potential for subsequent cell expansion — reflect one particular aspect of efforts to improve outcomes, and advances in cell manufacturing will certainly make these products accessible for a greater number of patients. For those with response, confirming CAR T cell persistence and monitoring for antigen loss are necessary for relapse prediction and prevention strategies; however, antigen-negative disease relapse is currently difficult to treat, and even antigen-positive relapsed disease might not respond to CAR T cell re-infusion. Akin to multi-agent chemotherapy, multi-antigen-targeting strategies might address these mechanisms of relapse and, therefore, might be a pathway to more durable remissions. Optimizing this therapeutic approach in patients with non-B cell malignancies is the next frontier for research in this field of immunotherapy, but many obstacles need to be overcome, and close attention to the design of novel CAR constructs will be informative in identifying further barriers, such as those in solid tumours. In this fast-paced field of research, a shift in focus to overcome the obstacles identified (FIG. 1) will be imperative to make this promising therapy more broadly available, more effective and potentially curative for all those treated.
Key points.
Chimeric antigen receptor (CAR) T cell immunotherapy is a highly effective form of adoptive cell therapy, as demonstrated by the remission rates in patients with B cell acute lymphoblastic leukaemia or large B cell lymphoma, which have supported FDA approvals.
A complete understanding of the limitations of CAR T cell therapy will help to identify crucial areas requiring further research to improve patient outcomes.
Factors that can preclude durable remissions following CAR T cell therapy include CAR T cell manufacturing issues, limited CAR T cell expansion and/or persistence, various resistance mechanisms and toxicities.
Various intuitive strategies to overcome these obstacles are being investigated in order to optimize this unique therapeutic strategy and expand the indications for treatment.
Acknowledgements
The work of the authors is supported in part by the Intramural Research Program, the National Cancer Institute and the NIH Clinical Center.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Clinical Oncology thanks S. Grupp and other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's Disclaimer: DISCLAIMER
Publisher's Disclaimer: The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
References
- 1.Kalos M et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl Med 3, 95ra73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Porter DL, Levine BL, Kalos M, Bagg A & June CH Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med 365, 725–733 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kochenderfer JN et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116, 4099–4102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grupp SA et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med 368, 1509–1518 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenbaum L Tragedy, perseverance, and chance — the story of CAR-T therapy. N. Engl. J. Med 377, 1313–1315 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Maude SL et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee DW et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner RA et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 129, 3322–3331 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JH et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maude SL et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.US Food & Drug Administration. FDA approves CAR-T cell therapy to treat adults with certain types of large B cell lymphoma. FDA https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm581216.htm (2017).
- 12.US Food & Drug Administration. FDA approval brings first gene therapy to the United States. FDA https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm574058.htm (2017).
- 13.US Food & Drug Administration. FDA approves tisagenlecleucel for adults with relapsed or refractory large B cell lymphoma. FDA https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm606540.htm (2018).
- 14.Tang J, Hubbard-Lucey VM, Pearce L, O’Donnell-Tormey J & Shalabi A The global landscape of cancer cell therapy. Nat. Rev. Drug Discov 17, 465–466 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Fry TJ et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med 24, 20–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacoby E et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun 7, 12320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gardner R et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T cell therapy. Blood 127, 2406–2410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mueller KT et al. Cellular kinetics of CTL019 in relapsed/refractory B cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 130, 2317–2325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stroncek DF et al. Elutriated lymphocytes for manufacturing chimeric antigen receptor T cells. J. Transl Med 15, 59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ceppi F et al. Lymphocyte apheresis for chimeric antigen receptor T cell manufacturing in children and young adults with leukemia and neuroblastoma. Transfusion 58, 1414–1420 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Das RK, Storm J & Barrett DM T cell dysfunction in pediatric cancer patients at diagnosis and after chemotherapy can limit chimeric antigen receptor potential. Cancer Res 78 (Suppl), 1631 (2018). [Google Scholar]
- 22.Singh N, Perazzelli J, Grupp SA & Barrett DM Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci. Transl Med 8, 320ra3 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Zhang H et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood 122, 1105–1113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Veirman K et al. Myeloid-derived suppressor cells as therapeutic target in hematological malignancies. Front. Oncol 4, 349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levine BL, Miskin J, Wonnacott K & Keir C Global manufacturing of CAR T cell therapy. Mol. Ther. Methods Clin. Dev 4, 92–101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X & Riviere I Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol. Ther. Oncolyt 3, 16015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tumaini B et al. Simplified process for the production of anti-CD19-CAR-engineered T cells. Cytotherapy 15, 1406–1415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kochenderfer JN et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother 32, 689–702 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gargett T & Brown MP Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy 17, 487–495 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Sommermeyer D et al. Chimeric antigen receptor-modified T cells derived from defined CD8 + and CD4 + subsets confer superior antitumor reactivity in vivo. Leukemia 30, 492–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turtle CJ et al. CD19 CAR-T cells of defined CD4 + :CD8 + composition in adult B cell ALL patients. J. Clin. Invest 126, 2123–2138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang W, Jordan KR, Schulte B & Purev E Characterization of clinical grade CD19 chimeric antigen receptor T cells produced using automated CliniMACS Prodigy system. Drug Des. Devel Ther 12, 3343–3356 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu F et al. Closed-system manufacturing of CD19 and dual-targeted CD20/19 chimeric antigen receptor T cells using the CliniMACS Prodigy device at an academic medical center. Cytotherapy 20, 394–406 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Sabatino M et al. Generation of clinical-grade CD19-specific CAR-modified CD8 + memory stem cells for the treatment of human B cell malignancies. Blood 128, 519–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blaeschke F et al. Induction of a central memory and stem cell memory phenotype in functionally active CD4(+) and CD8(+) CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19(+) acute lymphoblastic leukemia. Cancer Immunol. Immunother 67, 1053–1066 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fraietta JA et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med 24, 563–571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stroncek DF et al. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy 18, 893–901 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruella M et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med 24, 1499–1503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fesnak A, Lin C, Siegel DL & Maus MV CAR-T cell therapies from the transfusion medicine perspective. Transfus. Med. Rev 30, 139–145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah NN et al. CD4/CD8 T-cell selection enhances CD22 CAR-T cell transduction and in-vivo CAR-T expansion: updated results on phase I anti-CD22 CAR dose expansion cohort. Blood 130, 809 (2017). [Google Scholar]
- 41.Vormittag P, Gunn R, Ghorashian S & Veraitch FS A guide to manufacturing CAR T cell therapies. Curr. Opin. Biotechnol 53, 164–181 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Perica K, Curran KJ, Brentjens RJ & Giralt SA Building a CAR garage: preparing for the delivery of commercial CAR T cell products at Memorial Sloan Kettering Cancer Center. Biol. Blood Marrow Transplant 24, 1135–1141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawalekar OU et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44, 380–390 (2016). [DOI] [PubMed] [Google Scholar]
- 44.June CH & Sadelain M Chimeric antigen receptor therapy. N. Engl. J. Med 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Stegen SJ, Hamieh M & Sadelain M The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov 14, 499–509 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cornetta K et al. Absence of replication-competent lentivirus in the clinic: analysis of infused T cell products. Mol. Ther 26, 280–288 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cornetta K et al. Screening clinical cell products for replication competent retrovirus: the National Gene Vector Biorepository experience. Mol. Ther. Methods Clin. Dev 10, 371–378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin DY et al. Paralleled comparison of vectors for the generation of CAR-T cells. Anticancer Drugs 27, 711–722 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Golumba-Nagy V, Kuehle J & Abken H Genetic modification of T cells with chimeric antigen receptors: a laboratory manual. Hum. Gene Ther. Methods 28, 302–309 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Riet T et al. Nonviral RNA transfection to transiently modify T cells with chimeric antigen receptors for adoptive therapy. Methods Mol. Biol 969, 187–201 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Panjwani MK et al. Feasibility and safety of RNA-transfected CD20-specific chimeric antigen receptor T cells in dogs with spontaneous B cell lymphoma. Mol. Ther 24, 1602–1614 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Monjezi R et al. Enhanced CAR T cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 31, 186–194 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Singh H, Huls H, Kebriaei P & Cooper LJ A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol. Rev 257, 181–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kebriaei P et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Invest 126, 3363–3376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Wolf C, van de Bovenkamp M & Hoefnagel M Regulatory perspective on in vitro potency assays for human T cells used in anti-tumor immunotherapy. Cytotherapy 20, 601–622 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Xu J, Melenhorst JJ & Fraietta JA Toward precision manufacturing of immunogene T cell therapies. Cytotherapy 20, 623–638 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Rossi J et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 132, 804–814 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghosh A et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat. Med 23, 242–249 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brudno JN et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J. Clin. Oncol 34, 1112–1121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Y et al. Donor-derived CD19-targeted T cell infusion induces minimal residual disease-negative remission in relapsed B cell acute lymphoblastic leukaemia with no response to donor lymphocyte infusions after haploidentical haematopoietic stem cell transplantation. Br. J. Haematol 179, 598–605 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Kochenderfer JN et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 122, 4129–4139 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Georgiadis C et al. Long terminal repeat CRISPR-CAR-coupled “universal” T cells mediate potent anti-leukemic effects. Mol. Ther 26, 1215–1227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Poirot L et al. Multiplex genome-edited T cell manufacturing platform for “off-the-shelf” adoptive T cell immunotherapies. Cancer Res 75, 3853–3864 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Cooper ML et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 32, 1970–1983 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Daher M & Rezvani K Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr. Opin. Immunol 51, 146–153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang X et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res 8, 1083–1089 (2018). [PMC free article] [PubMed] [Google Scholar]
- 67.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT03579927 (2019). [DOI] [PubMed]
- 68.Quintarelli C et al. CD19 redirected CAR NK cells are equally effective but less toxic than CAR T cells. Blood 132 (Suppl. 1), 3491 (2018). [Google Scholar]
- 69.Hofer E & Koehl U Natural killer cell-based cancer immunotherapies: from immune evasion to promising targeted cellular therapies. Front. Immunol 8, 745 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.US Food & Drug Administration. Package insert — Kymriah. FDA https://www.fda.gov/downloads/UCM573941.pdf (2018).
- 71.Neelapu SS et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lowe KL et al. Fludarabine and neurotoxicity in engineered T cell therapy. Gene Ther 25, 176–191 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Novartis. Kymriah treatment center locator. Kymriah https://www.us.kymriah.com/treatment-center-locator/ (2018).
- 74.Kite Pharma. Where can Yescarta be received? Yescarta https://www.yescarta.com/treatment-centers (2018).
- 75.European Medicines Agency. First two CAR-T cell medicines recommended for approval in the European Union. EMA https://www.ema.europa.eu/en/news/first-two-car-t-cell-medicines-recommended-approval-european-union (2018).
- 76.Novartis Pharmaceuticals Canada Inc. Novartis receives Health Canada approval of its CAR-T cell therapy, Kymriah™ (tisagenlecleucel)i. Newswire https://www.newswire.ca/news-releases/novartis-receives-health-canada-approval-of-its-car-t-cell-therapy-kymriah-tisagenlecleuceli-692581041.html (2018).
- 77.June CH, O’Connor RS, Kawalekar OU, Ghassemi S & Milone MC CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Shah GL, Majhail N, Khera N & Giralt S Value-based care in hematopoietic cell transplantation and cellular therapy: challenges and opportunities. Curr. Hematol. Malig. Rep 13, 125–134 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Caffrey M With approval of CAR T-cell therapy comes the next challenge: payer coverage. Am. J. Manag. Care https://www.ajmc.com/journals/evidence-based-oncology/2018/february-2018/with-approval-of-car-tcell-therapy-comes-the-next-challenge-payer-coverage (2018).
- 80.Bach PB National coverage analysis of CAR-T therapies — policy, evidence, and payment. N. Engl. J. Med 379, 1396–1398 (2018). [DOI] [PubMed] [Google Scholar]
- 81.Kotani H et al. Aged CAR T cells exhibit enhanced cytotoxicity and effector function but shorter persistence and less memory-like phenotypes. Blood 132, 2047 (2018). [Google Scholar]
- 82.Gardner R et al. Starting T cell and cell product phenotype are associated with durable remission of leukemia following CD19 CAR-T cell immunotherapy. Blood 132, 4022 (2018). [Google Scholar]
- 83.Fesnak AD, June CH & Levine BL Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 16, 566–581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao Z et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 28, 415–428 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kochenderfer JN et al. Chemotherapy-refractory diffuse large B cell lymphoma and indolent B cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol 33, 540–549 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Davila ML et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl Med 6, 224ra25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Long AH et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med 21, 581–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Feucht J et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med 25, 82–88 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sadelain M, Brentjens R & Riviere I The basic principles of chimeric antigen receptor design. Cancer Discov 3, 388–398 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maus MV & June CH Making better chimeric antigen receptors for adoptive T cell therapy. Clin. Cancer Res 22, 1875–1884 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fraietta JA et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558, 307–312 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eyquem J et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jung IY & Lee J Unleashing the therapeutic potential of CAR-T cell therapy using gene-editing technologies. Mol. Cells 41, 717–723 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maus MV Immunology: T cell tweaks to target tumours. Nature 543, 48–49 (2017). [DOI] [PubMed] [Google Scholar]
- 95.Hasan AN, Selvakumar A & O’Reilly RJ Artificial antigen presenting cells: an off the shelf approach for generation of desirable T-cell populations for broad application of adoptive immunotherapy. Adv. Genet. Eng 4, 130 (2015). [PMC free article] [PubMed] [Google Scholar]
- 96.Turtle CJ & Riddell SR Artificial antigen-presenting cells for use in adoptive immunotherapy. Cancer J 16, 374–381 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Butler MO & Hirano N Human cell-based artificial antigen-presenting cells for cancer immunotherapy. Immunol. Rev 257, 191–209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yoon DH, Osborn MJ, Tolar J & Kim CJ Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): combination or built-in CAR-T. Int. J. Mol. Sci 19, E340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cherkassky L et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest 126, 3130–3144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gargett T et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol. Ther 24, 1135–1149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li AM et al. Checkpoint inhibitors augment CD19-directed chimeric antigen receptor (CAR) T cell therapy in relapsed B-cell acute lymphoblastic leukemia. Blood 132 (Suppl. 1), 556 (2018). [Google Scholar]
- 102.Schuster SJ et al. Primary analysis of Juliet: a global, pivotal, phase 2 trial of CTL019 in adult patients with relapsed or refractory diffuse large B-cell lymphoma. Blood 130, 577 (2017). [Google Scholar]
- 103.Kantarjian HM et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med 375, 740–753 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gokbuget N et al. Blinatumomab for minimal residual disease in adults with B cell precursor acute lymphoblastic leukemia. Blood 131, 1522–1531 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kantarjian H et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med 376, 836–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Martinelli G et al. Complete hematologic and molecular response in adult patients with relapsed/refractory philadelphia chromosome-positive B-precursor acute lymphoblastic leukemia following treatment with blinatumomab: results from a phase II, single-arm, multicenter study. J. Clin. Oncol 35, 1795–1802 (2017). [DOI] [PubMed] [Google Scholar]
- 107.Turtle CJ et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8 + and CD4 + CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl Med 8, 355ra116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shalabi H et al. Intensification of lymphodepletion optimizes CAR re-treatment efficacy. Blood 130 (Suppl. 1), 3889 (2017). [Google Scholar]
- 109.Maude SL et al. Efficacy of humanized CD19-targeted chimeric antigen receptor (CAR)-modified T cells in children and young adults with relapsed/refractory acute lymphoblastic leukemia. Blood 128, 217 (2016).27207794 [Google Scholar]
- 110.Zoghbi A, Zur Stadt U, Winkler B, Muller I & Escherich G Lineage switch under blinatumomab treatment of relapsed common acute lymphoblastic leukemia without MLL rearrangement. Pediatr. Blood Cancer 64, e26594 (2017). [DOI] [PubMed] [Google Scholar]
- 111.Mejstrikova E et al. CD19-negative relapse of pediatric B cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J 7, 659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jabbour E et al. Outcome of patients with relapsed/refractory acute lymphoblastic leukemia after blinatumomab failure: no change in the level of CD19 expression. Am. J. Hematol 93, 371–374 (2018). [DOI] [PubMed] [Google Scholar]
- 113.Bhojwani D et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. Leukemia 10.1038/s41375-018-0265-z (2018). [DOI] [Google Scholar]
- 114.Sotillo E et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov 5, 1282–1295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fischer J et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J. Immunother 40, 187–195 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Braig F et al. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood 129, 100–104 (2017). [DOI] [PubMed] [Google Scholar]
- 117.Watanabe K et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8 + T cells. J. Immunol 194, 911–920 (2015). [DOI] [PubMed] [Google Scholar]
- 118.Walker AJ et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol. Ther 25, 2189–2201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Caruso HG et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res 75, 3505–3518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shalabi H et al. Sequential loss of tumor surface antigens following chimeric antigen receptor T cell therapies in diffuse large B cell lymphoma. Haematologica 103, e215–e218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Krenciute G et al. Transgenic expression of IL15 improves antiglioma activity of IL13Ralpha2-CAR T cells but results in antigen loss variants. Cancer Immunol. Res 5, 571–581 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Brown CE et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med 375, 2561–2569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.O’Rourke DM et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl Med 9, eaaa0984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Piccaluga PP et al. Surface antigens analysis reveals significant expression of candidate targets for immunotherapy in adult acute lymphoid leukemia. Leuk. Lymphoma 52, 325–327 (2011). [DOI] [PubMed] [Google Scholar]
- 125.Nagel I et al. Hematopoietic stem cell involvement in BCR-ABL1-positive ALL as a potential mechanism of resistance to blinatumomab therapy. Blood 130, 2027–2031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Raponi S et al. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: analysis of 552 cases. Leuk. Lymphoma 52, 1098–1107 (2011). [DOI] [PubMed] [Google Scholar]
- 127.Shah NN et al. Characterization of CD22 expression in acute lymphoblastic leukemia. Pediatr. Blood Cancer 62, 964–969 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chevallier P et al. Simultaneous study of five candidate target antigens (CD20, CD22, CD33, CD52, HER2) for antibody-based immunotherapy in B-ALL: a monocentric study of 44 cases. Leukemia 23, 806–807 (2009). [DOI] [PubMed] [Google Scholar]
- 129.Mitterbauer-Hohendanner G & Mannhalter C The biological and clinical significance of MLL abnormalities in haematological malignancies. Eur. J. Clin. Invest 34 (Suppl. 2), 12–24 (2004). [DOI] [PubMed] [Google Scholar]
- 130.Chien CD et al. FLT3 chimeric antigen receptor T cell therapy induces B to T cell lineage switch in infant acute lymphoblastic leukemia. Cancer Res 78 (Suppl), 1630 (2018). [Google Scholar]
- 131.Schneider D et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J. Immunother. Cancer 5, 42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ruella M et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Invest 126, 3814–3826 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Qin H et al. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol. Ther. Oncolyt 11, 127–137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shalabi H et al. Chimeric antigen receptor T-cell (CAR-T) therapy can render patients with ALL into PCR-negative remission and can be an effective bridge to transplant (HCT). Biol. Blood Marrow Transplant 24, S25–S26 (2018). [Google Scholar]
- 135.Summers C et al. Long term follow-up after SCRI-CAR19v1 reveals late recurrences as well as a survival advantage to consolidation with HCT after CAR T cell induced remission. Blood 132, 967 (2018). [Google Scholar]
- 136.Hay KA et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T cell therapy. Blood 130, 2295–2306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Neelapu SS et al. Chimeric antigen receptor T cell therapy — assessment and management of toxicities. Nat. Rev. Clin. Oncol 15, 47–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Teachey DT et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Cancer Discov 6, 664–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Porter D, Frey N, Wood PA, Weng Y & Grupp SA Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J. Hematol. Oncol 11, 35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gust J et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov 7, 1404–1419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lee DW et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124, 188–195 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mahadeo KM et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat. Rev. Clin. Oncol 16, 45–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lee DW et al. ASBMT Consensus Grading for cytokine release syndrome and neurological toxicity associated with immune effector cells. Biol. Blood Marrow Transplant 10.1016/j.bbmt.2018.12.758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gardner R et al. Decreased rates of severe CRS seen with early intervention strategies for CD19 CAR-T cell toxicity management. Blood 128, 586 (2016). [Google Scholar]
- 145.Perales MA, Kebriaei P, Kean LS & Sadelain M Reprint of: building a safer and faster CAR: seatbelts, airbags, and CRISPR. Biol. Blood Marrow Transplant 24, S15–S19 (2018). [DOI] [PubMed] [Google Scholar]
- 146.Yu H et al. Repeated loss of target surface antigen after immunotherapy in primary mediastinal large B cell lymphoma. Am. J. Hematol 92, E11–E13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kochenderfer JN et al. Long-duration complete remissions of diffuse large B cell lymphoma after anti-CD19 chimeric antigen receptor T cell therapy. Mol. Ther 25, 2245–2253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Budde L et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: a first-in-human clinical trial. Blood 130 (Suppl), 811 (2017). [Google Scholar]
- 149.Yang L et al. Preclinical efficacy of CD33 chimeric antigen receptor T cell immunotherapy in childhood acute myeloid leukemia. Pediatr. Blood Cancer 65 (Suppl.), O-100 (2018). [Google Scholar]
- 150.Schmidts A & Maus MV Making CAR T cells a solid option for solid tumors. Front. Immunol 9, 2593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Morgan MA & Schambach A Engineering CAR-T cells for improved function against solid tumors. Front. Immunol 9, 2493 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Watanabe K, Kuramitsu S, Posey AD Jr & June CH Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front. Immunol 9, 2486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.DeRenzo C, Krenciute G & Gottschalk S The landscape of CAR T cells beyond acute lymphoblastic leukemia for pediatric solid tumors. Am. Soc. Clin. Oncol. Educ. Book 38, 830–837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kachala SS et al. Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clin. Cancer Res 20, 1020–1028 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Heinmoller P et al. HER2 status in non-small cell lung cancer: results from patient screening for enrollment to a phase II study of herceptin. Clin. Cancer Res 9, 5238–5243 (2003). [PubMed] [Google Scholar]
- 156.Situ D et al. Expression and prognostic relevance of MUC1 in stage IB non-small cell lung cancer. Med. Oncol 28 (Suppl. 1), 596–604 (2011). [DOI] [PubMed] [Google Scholar]
- 157.Louis CU et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118, 6050–6056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Long AH et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol. Res 4, 869–880 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yeku OO, Purdon TJ, Koneru M, Spriggs D & Brentjens RJ Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep 7, 10541 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Avanzi MP et al. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep 23, 2130–2141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Brown CE et al. Optimization of IL13Ralpha2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol. Ther 26, 31–44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mount CW et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med 24, 572–579 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nellan A et al. Durable regression of medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 6, 30 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ahmed N et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol 3, 1094–1101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]