

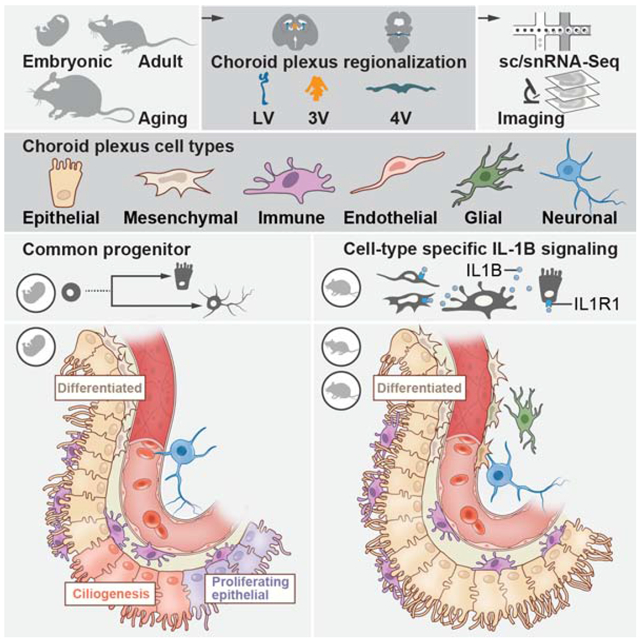
SUMMARY
The choroid plexus (ChP) in each brain ventricle produces cerebrospinal fluid (CSF) and forms the blood-CSF barrier. Here, we construct a single cell and spatial atlas of each ChP in the developing, adult, and aged mouse brain. We delineate diverse cell types, subtypes, cell states and expression programs in epithelial and mesenchymal cells across ages and ventricles. In the developing ChP, we predict a common progenitor pool for epithelial and neuronal cells, validated by lineage tracing. Epithelial and fibroblast cells showed regionalized expression by ventricle, starting at embryonic stages and persisting with age, with a dramatic transcriptional shift with maturation, and a smaller shift in each aged cell type. With aging, epithelial cells upregulated host defense programs and resident macrophages upregulated IL-1β signaling genes. Our atlas revealed cellular diversity, architecture and signaling across ventricles during development, maturation and aging of the ChP brain barrier.
Keywords: Choroid plexus, brain barrier, cerebrospinal fluid, single cell RNA-sequencing, single nucleus RNA-sequencing
Graphical Abstract
INTRODUCTION
The choroid plexus (ChP) is an essential brain barrier known primarily as a source of cerebrospinal fluid (CSF) and comprises an analogous set of four tissue structures located in each brain ventricle. The foundation of the ChP is an epithelial sheet that, together with a network of vasculature and diverse cell types, regulates CSF secretion and governs its composition by de novo protein synthesis and selective transcytosis of blood-borne factors (Damkier et al., 2013; Ghersi-Egea et al., 2018; Lun et al., 2015a; Saunders et al., 2018b). In addition, the ChP participates in regulating neurogenesis (Lehtinen et al., 2011; Silva-Vargas et al., 2016), homeostatic responses to arousal states (Mathew et al., 2016; Myung et al., 2018), inflammatory signals (Cui et al., 2020; Van Hove et al., 2019; Jordão et al., 2019a; Karimy et al., 2017; Reboldi et al., 2009; Schwartz and Baruch, 2014; Shechter et al., 2013), and atypical courses of neurodevelopment (Shannon et al., 2018; Tong et al., 2015) (reviewed in Fame and Lehtinen, 2020). Thus, the ChP could provide insights into disease pathology and provide potential therapeutic targets for a wide range of disorders.
Remarkably little is known about the molecular mechanisms governing these functions of the ChP, its cellular networks, and histology. Each ChP develops independently from distinct locations along the roofplate, where capillaries, mesenchymal and neural crest cells invaginate the neuroepithelium (Hunter and Dymecki, 2007; Lun et al., 2015a; Wilting and Christ, 1989). Bulk profiling of whole tissue (Liddelow et al., 2010; Lun et al., 2015b; Silva-Vargas et al., 2016) or sorted subpopulations (Lun et al., 2015b) indicates that each ChP retains a developmental blueprint of its origins along the body axis, with ventricle-specific transcriptomes and secretomes for the lateral and 4th ventricle ChPs (Lun et al., 2015a). The less-accessible third ventricle is the least explored, and boundaries of all ChPs with adjacent neural tissues remain poorly defined. Earlier efforts to address these deficiencies were limited by the lack of techniques to access, isolate, and comprehensively profile the ChP across brain ventricles and ages.
Here, we characterized the ChP across all brain ventricles in the developing, adult, and aging mouse brain, by single cell and single nucleus RNA-Seq (sc/snRNA-seq), and spatial mapping of specific RNAs and proteins. We profiled 15,620 cells and 83,040 nuclei, followed by multi-tiered validation with single molecule fluorescence in situ hybridization (smFISH), immunostaining, and transgenic lines. We used whole tissue imaging to map the identified cell types, sub-types and states to uncover specialized niches within ChPs and along transition areas between the ChP and adjacent brain parenchyma. Trajectory inference of developing ChP cells predicted a common progenitor pool for epithelial and neuronal cells, which we validated by fluorescent tagging and lineage tracing. While some expression signatures were invariant across ages and ventricles, such as the expression of the SARS-CoV-2 receptor ACE2 in mesenchymal and epithelial cells, others shifted with age or by ventricle, especially for epithelial and fibroblast cells. Ligand-receptor expression patterns across cell types predicted age- and ventricle- specific cell-cell interactions within the ChP, with a previously unappreciated role for mesenchymal cells (fibroblasts and pericytes) in the inferred cellular crosstalk, and an enhancement of IL-1β signaling from resident macrophage cells to endothelial and mesenchymal cells with age. Our molecular, cellular and spatial map of the ChP will be a resource for creating tools to access and control this essential brain-body barrier.
RESULTS
Molecular survey defines the cellular composition of the ChP across ventricles and ages
To chart a cell atlas of the developing, mature, and aging ChP, we profiled 15,620 single cells (scRNA-seq) and 83,040 single nuclei (snRNA-seq) from the lateral (LV), third (3V), and fourth (4V) brain ventricles of developing (embryonic day [E]16.5, n = 13), adult (4 months, n = 4) and aging (20 months, n = 9) mice (Figure 1A-C, F-H). We refined microdissection, cell dissociation and nucleus isolation techniques to obtain healthy single cells and nuclei from all ChPs (Figure 1A, STAR Methods). We conducted scRNA-seq for embryonic samples and snRNA-seq for all ages (Figure 1B, STAR Methods) (Gaublomme et al., 2019; Habib et al., 2017).
Figure 1. Single cell and single nucleus RNA-Seq of embryonic, adult, and aged ChPs.

(A) E16.5 brain and ChP from each ventricle in E16.5, adult, and aged brain. Arrows: anterior-posterior (A/P), dorso-ventral (D/V) and medial-lateral (M/L) axes. (B) Workflow. (C) Major cell types of E16.5 ChP. T-SNE of 15,620 single cell profiles (n = 9 mice, in pools of 3 animals per ventricle), colored by post hoc annotated cell type. (D) TUBB3-positive neurons (arrowheads) in E16.5 LV ChP. Inset a: High-magnification image. Schematic: dotted outline denotes region shown by immunostaining. Arrows: A/P and D/V axes. (E) Major cell type markers expressed in ChP. Top: H&E; bottom: Epithelial (AQP1), endothelial (PECAM1), mesenchymal (COL1A1) and immune cell (SPI1, arrowheads) immunostaining; Hoechst counterstain. (F) Major cell types of E16.5, adult, and aged ChP. UMAP of 83,040 single nucleus profiles (snRNA-seq, n=17 mice. 2-4 animals per replicate of LV, 3V and 4V ChP from E16.5, adult and aged mice), colored by post hoc annotated cell type. (G-H) UMAP as in F, colored by ventricle (in G) or age (in H). (I) Cell type marker genes by snRNA-seq. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected genes (rows) in each major cell population (columns). See also Figure S1.
Embryonic cells partitioned into clusters identifying epithelial, mesenchymal (mural and fibroblast), endothelial, immune, neuronal and glial cells in each ChP from all ventricles (by marker genes expression, Figure 1C, S1A,B,C, Table S1, STAR Methods). We determined the spatial positions of each major cell type within the ChP tissue from each ventricle by immunostaining of marker genes in whole explants and tissue slices of the developing brain (Figure 1D,E). Notably, actively cycling cell subsets were present within each of the six major cell classes in the developing ChP (Figure S1D) and were enriched along the base of the ChP proximal to the brain (Figure S1E).
SnRNA-seq of embryonic, adult, and aged ChP identified the same major cell classes in each ventricle (as in scRNA-seq of embryonic samples, Figure 1F), with conservation of overall cellular composition with age. While tissue maturation was accompanied by a distinct shift in gene expression between embryo and adult/aged ChP (Figure 1F-H, S1F,G), canonical cell-type markers were conserved across ages and ventricles (Figure 1I, Figure S1H,I).
Neuronal and glial cell populations in the ChP
We captured neuronal (Tubb3 expressing) and glia-like (Slc1a3/EAAT1 or GFAP expressing) cells in the ChP across all ages, validated by immunostaining (Figures 1C,D,I, S1J-L), in agreement with previous studies (Lindvall et al., 1978). The neuro-glial populations were proportionally low (8%/2.5% of cells/nuclei, Figure S1B,G), yet sub-clustering revealed substantial diversity within each population.
In embryos, subsets of glial cells expressed markers of neural progenitor/stem-like cells (e.g. Pax6, Nes, Sox2, Rspo2/3) or of oligodendrocyte precursor cells (OPCs, e.g. Olig1, Olig2) (Kriegstein and Alvarez-Buylla, 2009) (Figure 2A, S2A,B). Subsets of neurons expressed markers of intermediate progenitors (e.g. Eomes/Tbr2) or of immature neurons (e.g. Neurod1/2, Dlx1/2), and various neuropeptides (e.g. Vgf, Chgb) (Figure S2B).
Figure 2. Epithelial differentiation trajectory reveals progenitor and ciliogenesis programs of ChP epithelial cells during development.

(A) Embryonic neuronal and glia-like cell subsets. T-SNE of embryonic neuronal and glia-like single cell profiles (scRNA-seq, 955 cells). (B) Diffusion map embedding (components 1 and 3) of neuronal, glia-like and epithelial cell profiles (dots) from 3V ChP, colored by log2(TP10K+1) expression of marker genes of neurons (Tubb3), progenitors (Pax6, Rspo2), cycling (Mki67), ciliogenesis (Ccdc67), and mature epithelial (Krt18) cells. Right: Suggested differentiation trajectories. (C) Schematic of embryonic hindbrain; upper rhombic lip (URL), lower rhombic lip (LRL). Dotted box: area shown in panels (D-K). (D) Proliferating (KI67) progenitors (PAX6) in URL. (E) SmFISH markers of progenitors (Rspo2, Rspo3) and Wnt1. (F) SmFISH of Wnt1 and FoxJ1. (G) High magnification image of (F). (H) Confetti-labeling schematic. (I) E16.5 hindbrain region from Wnt1-CRE2 crossed with Confetti mouse shows distribution of Wnt1-derived cells. (J) Wnt1-derived cRFP or mCFP epithelial cells adjacent to URL. (K) Wnt1-derived cRFP and TUBB3 neurons in URL with DAPI. Inset: split cRFP (top) and TUBB3 (bottom). (L) Confetti labeled cells in 3V ChP. (M) Proliferating cells enriched at LV ChP base. Left: BrdU labeling; Middle: ISH of Ccdc67; Right: AQP1. (N) Left: Scanning EM of multi-ciliated epithelial cell. Right: Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of markers of ciliogenesis (columns) in ciliogenic (developing) and mature epithelial cells (rows). (O) Left: LV ChP immunostained for Ac-Tubulin and CCDC67/DEUP1. Middle and right: Ac-Tubulin with CCDC67 (middle) or SHISA8 (right). (P) Schematic: LV ChP maturation regions. (Q-R) Spatial mapping of maturation domains in 3V and 4V ChP marked by progenitors (Rspo2), ciliogenesis (Ccdc67), differentiated epithelial cells (Krt18) (Genepaint). Schematic: ChP regions with maturing epithelial cells. See also Figure S2.
Two neuro-glial subsets in the 3V expressed markers of developing pinealocytes (e.g. Crx, Krt19; Figure 2A, clusters 12 and 13, Figure S2B), which develop caudal to the 3V ChP. We leveraged the inclusion of adjacent brain tissue in our atlas to map the boundary between the 3V ChP and developing pineal gland using published ISH (Lein et al., 2007; Visel et al., 2004) of marker genes from our data (Figure S2C). The diversity of embryonic glia and neuronal populations observed in scRNA-seq was confirmed by snRNA-seq and expanded to adult and aged ChP (Figure S2D,E) (Dulken et al., 2017; Mizrak et al., 2019; Shah et al., 2018).
An inferred common progenitor pool for epithelial and neural cells validated by lineage tracing
The embryonic ChP scRNA-seq profiles of epithelial, glial, and neuronal cells were not discretely separable by low-dimensional embedding (Figure 1C, STAR Methods), suggesting a common progenitor pool for both epithelial and neuronal cells may exist in the ChP. We thus modeled the predicted continuum between glial, neuronal, and epithelial cells using a diffusion map (Haghverdi et al., 2016) of 3V ChP cells (Figure S1D). The analysis arranged neurons and mature epithelial cells at two opposite ends of a trajectory, with a progenitor glia-like population at the node between them (Figure 2B, further supported RNA velocity (La Manno et al., 2018) analysis, Figure S2F).
The predicted progenitors expressed proliferation (Mki67) and known markers (Pax6, Rspo2/3, Wnt5a/3a/1, Fabp7, clusters 7 and 8 in Figure 2A,B S2B; STAR Methods), previously reported for precursors of ependymal cells and cortical neurons (Kriegstein and Alvarez-Buylla, 2009), with Wnt1 having the most specific expression (Figure S2G). Wnt1 mutant mice have disrupted 3V and 4V ChP formation (Louvi and Wassef, 2000). Wnt1 contributes to lower rhombic lip (LRL)-derived 4V ChP (Awatramani et al., 2003; Hunter and Dymecki, 2007), but the upper rhombic lip (URL) Wnt1 progenitor pool contributes largely to cerebellar development (Wingate, 2001) and expression was noted in the ChP (Hagan and Zervas, 2012). Focusing on the URL, we validated that Wnt1+ cells localized near FoxJ1-expressing ChP epithelial cells (by smFISH and immunohistochemistry, Figures 2C-G).
We confirmed our hypothesis of a common progenitor pool by lineage tracing the progeny of Wnt1+ progenitors (by crossing Wnt1-CRE2 (Lewis et al., 2013) with R26R-Confetti mice (Livet et al., 2007; Snippert et al., 2010), Figure 2H,I. Wnt1-derived epithelial cells in the maturing 4V ChP extended away from the URL, and Wnt1-derived neurons were in close proximity to progenitors in the URL (Figure 2J,K). These data support our model of a common progenitor pool that can give rise to both cell types. Analysis of 3V ChP in Wnt1-CRE2::R26R-Confetti mice further revealed Wnt1-lineage contributions to 3V ChP epithelial cells (Figure 2L).
Differentiating epithelial cells had distinct spatial organization and ciliogenesis expression programs. The maturing ChP epithelial cells in each ventricle transiently expressed ciliogenesis genes including Ccdc67/Deup1 (Figures 2B, M-O), which drives centriole biogenesis, an essential step of multi-ciliation (Brooks and Wallingford, 2014). Shisa8, a reprogramming regulator (Schiebinger et al., 2019), was also expressed near the base of acetylated tubulin tufts in multi-ciliated epithelial cells (Figure 2N,O). We mapped the differentiating populations to distinct spatial boundaries within each ventricle (Figure 2P-R), with progenitor cells located near the ChP-brain boundary (by smFISH of Rspo1/2/3; Wnt2b/3a/5a; Axin2) (Figure S2H-K). Notably, the proportion of replicating epithelial cells was reduced from ~10% in embryonic ChP to ~1% in adult ChP, with an overall shift towards more mature cells compared to ciliogenic epithelial cells (p-value < 0.05, t-test on fraction of cells, Figure S2L). This maturation gradient of embryonic ChP epithelial cells matches prior models (Liddelow et al., 2010; Shuangshoti and Netsky, 1966) and provides a molecular handle for investigating distinct stages of epithelial cell development.
Epithelial cell programs are regionalized across brain ventricles in all ages
We next examined each of the major differentiated ChP cell populations – epithelial, mesenchymal, immune and endothelial. In each case, we first analyzed the embryonic cells (profiled by scRNA-seq) across the three ventricles, and then compared developing, adult, and aged mice (as profiled by snRNA-seq). Together these analyses identified shared and regionalized gene programs and distinguished those that are age-invariant and age-dependent.
In the embryo, epithelial cells formed the largest cell class in each ChP (by scRNA-seq, Figure S1B) and partitioned into subsets representing cycling, newly differentiated cells undergoing ciliogenesis (above, Figure 2N), and mature cells (Figure 3A). Mature epithelial cell profiles partitioned by ventricle (Figure 3A), matching differential gene expression between LV and 4V ChP by bulk RNA-seq (Lun et al., 2015b) (Figure S3A). To identify ventricle-specific expression programs of embryonic epithelial cells, we performed topic modeling (Bielecki et al., 2021; Blei et al., 2003; Dey et al., 2017; Pritchard et al., 2000) (STAR Methods), to learn expression programs (“topics”) and model cells as a mixture of topics. Briefly, a gene can belong to multiple topics with different weights, reflecting the gene’s role in each topic; and a topic’s weight for a given cell reflects the relative prominence of the expression program in that cell. We learned a topic model across all embryonic epithelial cells in the scRNA-seq dataset, and searched for topics that were differentially weighted in a subset of cells or enriched for genes of a specific biological process (Figure 3B, S3B,C STAR Methods, Table S2), such as ciliogenesis and immediate early genes (Figure S3B,C,D, Star Methods). Immediate early gene expression (Topic 19, Figure S3B,C) may reflect a response to tissue dissociation (van den Brink et al., 2017) as it was reduced in single nucleus profiles (Lacar et al., 2016) (Figure S3D).
Figure 3. Regionalized epithelial transcriptional programs across ventricles and ages.

(A) Distinct embryonic epithelial cells clusters by ventricle. T-SNE of embryonic epithelial cell profiles (dots, scRNA-seq), colored by ventricle. (B) For each ventricle associated topic, shown is a bar plot of topic scores for top ranked genes (left), and t-SNE of the single cell profiles (as in A) colored by topic’s weight per cell (right). (C-D) Regionalized genes in embryonic epithelial cells across ventricles. (C) Distribution of log2(TP10K+1) expression of each gene across ventricles. (D) Top rows: Regionalized mRNA expression from Genepaint. Bottom row: Ins2 smFISH. (E) Regionalized mRNA expression from Genepaint in 4V ChP with schematic. (F-G) T-SNE of single epithelial nucleus profiles colored by age (in F) or ventricle (in G). (H) Expression level scaled per gene (color scale) of transport and secretion genes, differential across ages (FDR<0.01) (see also (Xu et al., 2021) . (I-J) Heatmap of snRNA-seq data of differentially expressed genes across ventricles (FDR<0.01) that are age-invariant (I) or age-dependent (J). Scaled expression level per gene (columns). Left color bar: ventricle. Right color bar: Age. (K-L) Age-dependent genes (snRNA-seq, FDR<0.01) between embryonic and mature (adult and aged, in K) or between adult and aged (in L). Average expression per sample across ventricles, scaled per gene (rows). Right: Enriched pathways per age-signature (hypergeometric p-value). See Figure S3.
Several topics reflected cross-ventricle regionalized gene programs (Topics 3, 6, 11, 14, 16, and 23), having differential weights between cells from different ventricles (Figure 3B, S3C). We validated the ventricle-specific enrichment of key topic-associated genes using smFISH and in published data (Visel et al., 2004) (Figure 3C,D). For example, Ins2, encoding an insulin precursor and associated with Topic 16, was over-expressed in 3V epithelial cells in the embryo (in scRNA-seq data, Figure 3B-D). Regionalized differences identified by scRNA-seq data (Figure 3C) were recapitulated in the corresponding embryo snRNA-seq data (Figure S3E). For example, Ins2 was expressed in the 3V ChP, supporting the model that the ChP is an internal source of insulin for the brain (Mazucanti et al., 2019).
Other topics reflected spatial expression patterns within a ventricle. For example, epithelial cells in the ChP partitioned into two subpopulations by their weights of topics 6, 11 or 23 (Figure 3B,E, S3F). While these topics were highest in 4V, they also showed intra-ventricle variation in epithelial cells in other ventricles (Figure 3B). Mapping the expression of highly scoring genes for these topics in the 4V ChP in situ identified a rostro-caudal gradient of expression (Figure 3E), which may derive from the earliest stages of development, when roof plate progenitors originating from distinct rhombomeres give rise to hindbrain and 4V ChP (Hunter and Dymecki, 2007).
Comparing patterns across three age groups profiled by snRNA-seq, we found both age-invariant and age-dependent expression patterns, including regionalized patterns, with the main shift occurring from embryo to adult and then persisting into aging (Figure 3F,G). We found age-dependent expression of transporters and channels involved in ion and water movement across the epithelium, aligning with a key function of epithelial cells to produce CSF (Damkier et al., 2013; Fame and Lehtinen, 2020). For example, in adult ChP Slc12a2 (NKCC1), Kcnj13 (Kir7.1), and Slc4a5 (NBC4) were upregulated (see also (Xu et al., 2021)), while Aqp1 was modestly downregulated (Figure 3H, S3G). There were also age-dependent expression of genes encoding secreted proteins (Figure 3H, S3H, Table S3), including the “anti-aging” gene Klotho (Kuro-o et al., 1997; Zhu et al., 2018), which increased with age (Figure 3H).
Across all ages, differentially expressed genes were partitioned into three groups by age- and ventricle-dependent expression: First, age-invariant ventricle-dependent genes (e.g., Wls in 4V, Emx2 in LV, Figure 3I, Table S3); Second, age- and ventricle-dependent genes (e.g., Rspo3 in 3V, Figure 3J, Table S3), including a gene subset regionalized in adult and aged mice that were either not yet detected or not regionalized in the embryo at E16.5 (Figure S3I,J). Third, age-dependent and ventricle-invariant genes changed with age in all ventricles. These were enriched for specialized programs across ages: Embryonic epithelia upregulated genes for developmental processes (Figure 3K, S3K), while aged epithelia upregulated host-defense programs (Figure 3L, FDR q-value < 0.01, GOrilla (Eden et al., 2007, 2009;Baruch et al., 2014). Thus, epithelial cells likely adapt to fulfill age-dependent functions in the brain.
Mesenchymal cell programs are regionalized across brain ventricles
Mesenchymal cell profiles partitioned into fibroblasts and mural cells expressing pericyte and vascular smooth muscle cell (vSMC) markers (Figure 4A, S4A), consistent with cranial mesenchyme and neural crest contributions to the stromal space (Martik and Bronner, 2017; Wilting and Christ, 1989). Topic modeling of the embryonic cells recovered topics capturing mesenchymal cell types (e.g., pericytes (topic 3), proliferating cells (topics 12 and 16)), and ventricle-dependent topics within fibroblasts (e.g., LV (Topics 8 and 18), 3V (Topics 2 and 7) and 4V (Topic 5)) (Figure 4B, S4B,C, Table S4). We validated ventricle-dependent genes using published ISH data (Figure S4D, Visel et al., 2004). 3V ChP fibroblasts (Topic 7) shared expression signatures with meningeal fibroblasts (e.g., Apod, Slc1a3) and the ‘ceiling cell’ fibroblast subtype (e.g., Crym, Figure 4B, S4E; DeSisto et al., 2020). The ventricle-dependent topics revealed regionalized expression of genes encoding growth factors (Bmp4/7, Wnt4/2) and extracellular matrix proteins (Figure 4B, S4F). 4V ChP fibroblasts also expressed high levels of genes encoding signaling molecules with roles in hindbrain development (e.g., Hhip (Chuang and McMahon, 1999), Ptch1 (Huang et al., 2009), Rbp4 (Chang et al., 2016), and Wisp1 (Lun et al., 2015a, Figure 4B, S4D).
Figure 4. Regionalized mesenchymal transcriptional programs across ventricles and ages.

(A) Embryonic mesenchymal cells largely cluster by ventricle. T-SNE of embryonic mesenchymal cell profiles (dots) colored by ventricle. (B) Ventricle associated topics with a mesenchymal subtype: a bar plot of topic scores for top ranked genes (left), and t-SNE (as in A) colored by topic’s weight per cell (right) (topics associated with cycling cells in Figure S4B). (C-E) T-SNE of mesenchymal single nucleus profiles (dots), colored by cell type (C), age (D), and ventricle (E), and log2(TP10K+1) expression of pericyte markers Rgs5 and Des (F). (G-H) Heatmap of snRNA-seq data of differentially expressed mesenchymal genes across ventricles (FDR<0.01) that are age-invariant (G) or age-dependent (H). Scaled expression level per gene (columns). Left color bar: ventricle. Right color bar: Age. (I) Age-dependent genes (snRNA-seq, FDR<0.01) between embryonic and mature (adult and aged). Average expression per sample across ventricles, scaled per gene (rows). Right: Enriched pathways per age-signature (hypergeometric p-value). (J) SARS-CoV-2 cell entry and associated genes in embryonic scRNA-seq (left) and snRNA-seq (right). Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of markers (rows) across cell types (columns). (K) ACE2 expression in mesenchymal cells (COL1A1+) in 4V ChP. Coarse/fine dotted lines indicate apical/basal boundaries of epithelia. Arrowhead: ACE2+ cells. (L) ACE2 in adult epithelial cells (AQP1). Dotted line: base of epithelial cell. Arrowhead: stromal cell. Arrow: epithelial cell. (M) NRP1 expression in mesenchymal cells (COL1A1). See also Figure S4.
Comparing patterns across the three age groups using snRNA-seq data revealed all mesenchymal cell types – pericytes, fibroblasts, vSMCs, and meningeal fibroblasts (Figure 4C-F, S4G). In fibroblasts, we found both age-invariant and age-dependent patterns, including regionalized patterns (Figure 4G,H, Table S5). As in epithelial cells, age-dependent fibroblast genes typically changed postnatally and were maintained in aged ChP (Figure 4H). Among age-dependent ventricle-invariant genes, embryonic fibroblasts expressed higher levels of developmental and morphogenesis genes, whereas the adult and aged fibroblasts upregulated transporters, receptors, peptidase and proteolytic activity genes (Figure 4I, S4H,I).
ACE2, the SARS-CoV-2 receptor is expressed by mesenchymal cells in the ChP
Mesenchymal cells across all ages and ventricles expressed Ace2 (Angiotensin I converting enzyme 2; Figure 4J,S4J), the SARS-CoV-2 receptor, and other known and putative accessory components, including cathepsins (Ctsl and Ctsb (Muus et al., 2020)) and Neuropilin-1 (NRP1 (Cantuti-Castelvetri et al., 2020)) (Figure 4J, S4J). We validated ACE2 protein expression by mesenchymal cells in the stromal space (Figure 4K) and on some epithelial cells in embryonic, adult, and aged ChP (Figure 4K,L, S4K), similar to reports in human ChP (Yang et al., 2020). We also validated the expression of NRP1 protein, an effector of SARS-CoV-2 cell entry and infectivity, in mesenchymal cells (Cantuti-Castelvetri et al., 2020) (Figure 4M). These data highlight the potential of mesenchymal and epithelial ChP cells to facilitate viral particle entry into the CSF (Pellegrini et al., 2020b, 2020a; Wu et al., 2020) in all ventricles and ages and to contribute to neurological sequelae associated with SARS-CoV-2 infection (Ellul et al., 2020).
Homeostatic macrophage diversity within ChP across ages
In the embryonic ChP, scRNA-seq profiles spanned eight subsets of immune cells: B cells, lymphocytes, macrophages, basophils, mast cells, dendritic cells, monocytes and neutrophils (Figure 5A,B), each expressing specific cytokine, chemokine, and complement component genes (Figure S5A,B).
Figure 5. Immune cell diversity and macrophage niches within and across ChP.

(A) Embryonic immune cell subsets in the ChP. T-SNE of embryonic immune single cell profiles, colored by cluster. (B) Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected marker genes (columns) across immune cell subsets (rows). (C) Embryonic macrophage diversity. Diffusion map embedding of single cell macrophages, colored by log2(TP10K+1) expression of general macrophage marker (Cx3cr1) or of state specific genes (Lyve1, Spp1, Slc40a1, Spic). (D) Cx3cr1+/GFP mouse LV ChP immunostained with PECAM1 (red) and Hoechst (blue). Arrowhead: epiplexus cells. Dotted line: ChP apical surface. (E-F) Cx3cr1+/GFP LV ChP explant immunostained with PECAM1. (F) SLC40A1/FPN+ macrophages (arrowheads) along blood vessels. (G) Left: LYVE1+ epiplexus macrophages. Right: cross section at positions indicated by letters a, b and c in main panel. Arrows: stromal ChP macrophages. (H) UMAP of single immune nucleus profiles (dots), colored by immune cell type identity. (I) Volcano plot comparing embryonic to mature macrophages (snRNA-seq). (J) T-SNE of single immune nuclei (dots) colored by log2(TP10K+1) expression of genes marking macrophages (Cx3cr1), embryonically enriched gene (Mrc1/CD206) and adult enriched gene (Cd74). (K) Cx3cr1+/GFP 4V ChP immunostained with MRC1 in embryo (E16.5, left) and adult (right). (L) 4V ChP immunostained with CD74 and IBA1 (E16.5, left) and adult (right). (K-L) Arrowheads: stromal macrophages. Dotted line: epithelial apical boundary. See also Figure S5.
In macrophages (Cx3cr1+;Csf1R+), the largest class of immune cells in the ChP, we found heterogeneity within stromal and CSF-contacting epiplexus cells. Diffusion map embedding of scRNA-seq embryonic profiles showed graded gene expression patterns spanning three transcriptional states (Figure 5C, STAR Methods). Differentially expressed genes between states included the hyaluronan receptor Lyve1 (Jordão et al., 2019a; Lim et al., 2018); Spp1, a potential regulator of phagocytosis in the brain (Hammond et al., 2019); Slc40a1, an iron transporter; and Spic, which marks red pulp macrophages (Haldar et al., 2014) and bone marrow macrophages (Kohyama et al., 2009) that we confirmed by IHC (Figure 5C, S5C). A subset of Slc40a1+ macrophages expressed Spic and Clec4n (Figure S5D), corresponding to recently described Clec4n+ macrophages in postnatal ChP (Li et al., 2019). Macrophage states were associated with distinct spatial niches in the ChP, either basally under the epithelial cell monolayer or on the apical surface (e.g., epiplexus position, Figure 5D), by image analysis of Cx3cr1-GFP transgenic mouse. Cx3cr1-GFP-expressing cells tiled across the ChP (Figure 5E; Cui et al., 2020), with the subset expressing SLC40A1 located proximal to major vessels, suggesting roles in regulating iron homeostasis (Figure 5F). A subset of the Cx3cr1+ macrophages located on the apical ChP surface expressed LYVE1 (Figure 5G).
SnRNA-seq of embryonic, adult, and aged ChP across all ventricles identified macrophages, neutrophils, dendritic cells, plasma cells, and lymphoid (B- and T-) cells (Figure 5H, S5E,F). Macrophages were the most prevalent and were associated with blood vessels across all ages (Figure S5G; Shipley et al., 2020). The macrophages expressed markers of border associated macrophages (BAMs) (e.g., Apoe, Ms4a7, Ms4a6c) recently identified in periluminal regions, meninges, and ChP (Van Hove et al., 2019). In agreement with recent reports (Van Hove et al., 2019), we found diversity within the BAMs, which segregated into two subsets defined as major histocompatibility complex (MHC) class IIlow and MHC IIhigh. However, in contrast to Van Hove et al., our data revealed that this separation was age-dependent and potentially represented a maturation-dependent shift from MHC IIlow to MHC IIhigh (Figure 5I, S5I,J, Table S6). For example, CD206 (Mrc1, mannose receptor C-type I) expressed by embryonic macrophages was downregulated in adults, whereas CD74 (MHC class II associated protein) was upregulated in adult and aged macrophages vs. embryos (Figure 5J-L, Figure S5H). Similarly, we found a subset of macrophages expressing markers previously defined as subdural-specific BAMs (e.g., Lyve1, Ednrb, Colec12; Van Hove et al., 2019) that were enriched in embryonic ChP macrophages in our dataset (Figure S5H).
Arterio-venous zonation and blood brain barrier protein expression in ChP vasculature
The ChP serves as an important blood-CSF barrier. While the major vessels that feed each ChP are well described (Lun et al., 2015a), the identity, structure and development of vascular cell types within the ChP remain largely unknown. In the embryo, topic modeling of scRNA-seq data of vascular cells recovered expression programs associated with capillary (Topic 3), arterial (Topic 8 and 10) and blood-brain barrier (BBB)-associated (Topic 11) cells (Figure 6A, S6A,B, Table S7). We spatially mapped the developing arterio-venous zonation in the embryonic LV ChP in whole explants by combining the pan-endothelial marker PECAM1 with markers for arteries (Acta2+, Vwf+) and veins (Acta2−, Vwf+) (Vanlandewijck et al., 2018). Histological analyses revealed arteries developing obliquely across the ChP and veins running along the ventricular free margin of the tissue (Figure 6B). The arterio-venous zonation in the developing embryo was accompanied by enriched capillary marker expression (e.g. Esm1) along the ventricular margin of the ChP, suggesting zonated gene expression in vessels across the developing ChP (Figure 6C).
Figure 6. Vascular identity and BBB protein zonation within the ChP across ages.

(A) Embryonic endothelial cell transcriptional programs. For each topic, topic scores for top ranked genes (left) and t-SNE of single cell profiles colored by topic’s weight per cell (right). (B) LV ChP immunostained with PECAM1, ACTA2 and VWF, marking the arterial (ACTA2+) and venous (ACTA2-, VWF+) vessels. (C) Angiogenic zonation. Top: LV ChP immunostained with PECAM1 and ESM1 (green). Middle: Dotted line: LV ChP free margin. Double headed arrows: A/P and D/V axes. Bottom: ESM1, PECAM1 and Hoechst. (D) UMAP of single endothelial nucleus profiles (dots) colored by age (top), endothelial subtype score (bottom). (E) LV ChP immunostained as in (B) reveal arterio-venous organization in adult. Dotted line: region of interest as inset. (F) Aged LV ChP immunostained as in (B,E). Arrowheads: VWF accumulation in vessels. Dotted line: region of interest as inset. Inset: elevated VWF expression. (G) UMAP (as in D), colored by log2(TP10K+1) expression of genes Vwf and Selp. (H) Subset of aged vessels express VWF and SELP. (I) Distribution of Cldn5 expression across ages. (J) Embryonic LV ChP immunostained with CLDN5 and PECAM1. Dotted line: brain-ChP border. Bottom: Vessel at position ‘a’. (K) Adult LV ChP immunostained with CLDN5 and PECAM1. Arrowhead: CLDN5+ vessel at base of ChP. Dotted line: region enlarged on right. See also Figure S6.
Comparing single nucleus profiles across ages revealed that arterial, venous, and capillary identities were established across adult ages and ventricles (Figure 6D, S6C). In adult and aged LV ChP, IHC showed that arteries and veins were regularly segmented across the tissue (Figure 6E,F; Shipley et al., 2020) and connected by an expanded capillary-like network (Figure S6D) similar to human tissue (Hudson, 1960). Moreover, Plvap, which marks diaphragms of fenestrated endothelial cells (Herrnberger et al., 2012), scored highly in Topic 3 from scRNA-seq of embryonic cells (Figure 6A), and was also identified in adult and aged snRNA-seq profiles (Figure S6C). These findings match prior reports detailing the appearance of fenestrae during early embryonic rodent development (Figure S6E), which increase in number as the ChP matures (Keep and Jones, 1990). While we did not observe overt regionalization of endothelial cell programs, venous markers were enriched in the 3V ChP across all ages (Figure 6D, S6F,G), likely resulting from capture of the proximal vein of Galen (Mancini et al., 2015) (Figure S6H). The stereotyped vascular organization was retained in aged ChP (Figure 6F), where Vwf and Selp (P-selectin) expression were also detected (Figure 6G). IHC revealed VWF and SELP colocalized to enlarged luminal domains of vessels in aged mice (Figure 6H), suggesting age-associated vascular damage. Overall, these results identify molecular identities and vascular organization of ChPs across ventricles and ages that match vascular patterning in the human ChP (Hudson, 1960).
Notably, topic modeling of embryonic ChP cells also uncovered a previously unknown brain barrier transition zone embedded in the molecular and vascular map of the ChP, which we validated across ages. Specifically, non-capillary endothelial cells in the embryonic ChP expressed BBB markers (Cldn5 and Mfsd2a, Topic 11) (Figure 6A), even though the ChP is recognized to lack a classic BBB (Ben-Zvi et al., 2014; Saunders et al., 2018b). Cldn5 expression persisted in a subset of endothelial cells in snRNA-seq from all ages (Figure 6I, S6C), and CLDN5 protein was prominent in the major arteries bifurcating from the choroidal artery and extending into the ChP (Figure 6J, K, S6I).
Ligand-receptor analysis identifies mesenchymal cells as an interaction hub across ages and ventricles
Analysis of cognate ligand-receptor expression patterns across cell types, ventricles and ages (STAR Methods) predicted extensive cellular crosstalk involving all major ChP cell types (Figure 7). Briefly, for each ventricle and age, we identified putative interactions between each of the 6 major cell types by the expression of a ligand and its cognate receptor (through the CellPhoneDB (Efremova et al., 2020), STAR Methods). We estimated the significance of individual interactions (Efremova et al., 2020), and of the number and nature of observed interactions, between pairs of cell types by permutation tests (STAR Methods). We found significant individual interactions between each pair of cell types across all ventricles and ages, with specific cell type pairs having a significant enrichment in the number of interactions (Figure 7A, S7A,B, Table S8). We then focused all subsequent analyses only on significant interactions.
Figure 7. Mesenchymal, endothelial and immune cells contribute to cellular crosstalk in ChP.

(A) Number of statistically significant ligand-receptor pairs (LRPs, from CellPhoneDB (Efremova et al., 2020), STAR Methods) between pairs of ChP cell types, across ventricles in adult (top) and aged (bottom, embryo in Figure S7A). Significant pairs of cell types based on a randomization test marked as: *p≤0.05, **p≤0.01, ***p≤0.001. (B) Differential LRPs across ventricles or ages. Middle bar: cell types expressing the ligand (color coded). Right/left bars: cell types expressing the corresponding receptor. Edge width: Number of LRPs in each cell type pair that are specific to the specific ventricle (top) or age (bottom). Edge color: cell-type (middle bar). (C-E) Selected ventricle-dependent and age-dependent LRPs. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) in major cell population, across all ventricles (color coded) and ages (top: E16.5; middle: adult; bottom: aged). Dashed square: ventricle-dependent expression, solid line: age-dependent. Aging-dependent IL-1β-IL1R1 signaling (in E). (F) IL-1β immunostained macrophage in vessel like structures connecting ChP and brain parenchyma in aged brain. Inset: IL-1B macrophage. (G) Left: Whole mount aged hindbrain. Dotted lines outline 4V ChP. Right: IL1R1 immunostained endothelial cells (PECAM1) . Arrowheads: IL1R1+ blood vessels originating from the brain parenchyma and entering the ChP. See also Figure S7.
The number of significant ligand-receptor interactions between each pair of cell types changed across ventricles and ages (Figure 7A, Figure S7A,B). Overall, mesenchymal and epithelial cells had a high number of interactions (Figure 7A, Figure S7B). The majority of significantly interacting cell type pairs involved mesenchymal cells (FDR<0.05, 60%, 70% and 85% in embryonic, adult and aged, respectively), followed by epithelial cells (FDR<0.05, 45%, 55% and 42% in embryonic, adult and aged, respectively). For example, mesenchymal cells (fibroblasts and pericytes) expressed Pdgfra and Pdgfrb respectively, and epithelial and endothelial cells expressed their cognate ligands, Pdgfa and Pdgfb respectively (Figure S7C). Wnt5a, a regulator of 4V ChP and hindbrain development (Kaiser et al., 2019), was most robustly expressed by mesenchymal cells and its receptor Ror1 was expressed, amongst others, by epithelial cells, suggesting lifelong contributions to ChP function (Figure S7C). Other notable interactions included Vegfa in epithelial and mesenchymal cells and its receptor Flt1 on endothelial cells (Figure S7C).
Notably, in a more conservative, degree-preserving permutation test, the number of predicted interactions between most cell type pairs was not statistically significant (Figure S7D), indicating the dominance of a cell type in the crosstalk across all cells and not specific pairs. In addition, there were some differences between predicted significant interactions from scRNA-seq vs. snRNA-seq for embryo ChP, which may reflect differences in cell type recovery between methods (Figure S7A).
Comparing significant interactions across ventricles or ages revealed age- or ventricle-dependent interactions and overall differences in the number of interactions (Figure 7B-D). For example, Nov-Notch1 ligand receptor pair (LRP) expression in the 3V ChP and Dlk1-Notch LRP in 4V ChP predicted ventricle-specific signaling from mesenchymal cells to most other cell types (Figure 7C). In 3V and 4V ChP, Bmp5-Bmpr1b LRP expression predicted signaling from mesenchymal to neuro/glial and epithelial cells (Figure 7C). Among age-specific interactions, several LRPs were embryonically enriched. For example, Rspo2-Lgr4 LRP expression predicted signaling mainly from neuro/glial cells to epithelial and neuro/glial cells in the LV ChP. Lgr4 receptor is a key regulator of development and differentiation (Bell et al., 2008; Kinzel et al., 2014), consistent with our report of a common Rspo2+ progenitor for neurons and epithelial cells (Figure 2). Further, in the developing ChP, Efna5-Ephb2 LRP expression predicted signaling from epithelial cells to neuro/glial cells (Figure 7D), suggesting a role for Ephrin signaling in differentiation (similar to the gut; Kania and Klein, 2016). Finally, we predicted age-dependent signaling from mesenchymal, endothelial and epithelial cells expressing the insulin-like growth factor Igf2 to various cells expressing its receptors, Igflr and Igf2r, predominantly in embryonic ChP (Figure 7D, S7C), reinforcing the role of IGF signaling in development (Lehtinen et al., 2011). Among the ligand-receptor pairs expressed in adult that increased with age, Cxcl12-Ackr3 LRP expression predicted signaling from mesenchymal to endothelial and other cell types (Figure 7D).
Enhanced putative IL-1β signaling between macrophages, endothelial and mesenchymal cells in aging
Several interactions were predicted across all ventricles and ages, between immune cells and structural cells (epithelial, mesenchymal and endothelial), in line with the diverse functions assigned to immune cells in other barrier systems (Krausgruber et al., 2020). For example, Csf1, a signal for macrophage and monocyte differentiation was expressed by mesenchymal cells and endothelial cells, suggesting that myeloid cell signaling maturation for Csf1r expressing cells exists in the ChP throughout life (Figure S7C, Table S7).
Consistent with previous reports of overall induction of inflammatory signaling in the aged ChP (Baruch et al., 2014; Paré et al., 2018), IL-1β expression was induced in aged ChP macrophages and its receptor, IL1R1, was enriched in endothelial and mesenchymal cells (Figure 7E). We validated this observation, finding that IL-1β immunolabeled macrophages in the aged ChP localized to vessel-like structures bridging the ChP with the brain parenchyma (Figure 7F). Its cognate receptor, IL1R1, was expressed by endothelial, mesenchymal and epithelial cells, and IHC revealed enrichment along endothelial structures (Figure 7G). Overall, these interactions may contribute to age-dependent macrophage migration and infiltration of the ChP brain immune barrier.
DISCUSSION
The lack of a detailed cellular and molecular map for the ChP has been a major obstacle to elucidating its roles in instructing brain development, brain function, understanding its barrier properties, and unlocking its lifelong therapeutic potential. Here, we characterized ChP cell types across ventricles and ages, uncovered ventricle- and age-specific gene expression programs for epithelial and mesenchymal cells, described immune cell diversity, mapped developing and aging vascular structures, and uncovered age-dependent changes within ChP cellular signaling networks.
Our work builds on previous findings (Lun et al., 2015a) to demonstrate the ventricle-specific regionalization of epithelial and mesenchymal cells. Ventricle-specific identities were maintained in the adult and aged tissue, but additional genes became regionalized in the adult (and persisted into aging), either by activation of expression (for genes not expressed in the embryo) or by restriction of expression (for genes expressed across ventricles in the embryo). Gene programs and graded expression patterns, particularly across 4V ChP epithelial cells, may arise from the segmental development of the 4V ChP from distinct rhombomeres (Awatramani et al., 2003; Hunter and Dymecki, 2007).
The ChP is considered to be a primary producer of CSF and CSF-distributed factors (Ghersi-Egea et al., 2018; Lun et al., 2015a; Saunders et al., 2018b), and our cell atlas provides an opportunity to identify specifically which cells contribute. Essentially all major ChP cell types, and in particular ChP fibroblasts, also expressed many secreted factors found in CSF, including Igf2 (Lehtinen et al., 2011). Notably, mesenchymal cells were the cell type with the most predicted interactions with other ChP cell types. Future studies will determine if secreted factors of non-epithelial origin are restricted to targets within the ChP stromal space, or whether transport mechanisms allow them to contribute to CSF-based signaling in the brain.
Our profiles of ChP immune cell populations revealed spatial organization and age-dependent expression across ventricles and within the ChP. Most ChP macrophages shared signature genes recently described for adult ChP macrophages (Jordão et al., 2019a) and were Sall1 negative (Hammond et al., 2019). While we did not uncover a transcriptional signature purely for epiplexus cells, in validation experiments of Lyve1 expressing Cx3cr1-positive cells, we observed that some, but not all, epiplexus cells expressed LYVE1, demonstrating heterogeneity in immune cells positioned on the ChP surface. We noted age-dependent gene expression patterns in macrophages, such that genes upregulated during development, such as Cd206/Mrc1, were downregulated in adult and aged macrophages. Conversely, adult macrophages upregulated CD74, a chaperone of antigen presentation protein that was even further upregulated in the aged ChP. These maturation dependent changes agree with a recent report of BAMs (Van Hove et al., 2019). However, our data further clarify that this diversity is likely maturation-dependent, suggesting that the previously described diversity reflect instead a transition state between embryo and adult. Overall, we observed an upregulation of the inflammatory signals in the aged ChP in agreement with prior studies (Baruch et al., 2014) that was reflected in expression of Ifit1 and Il1b in aged epithelial cells. An improved understanding of ChP resident immune cells will facilitate future studies aimed at testing the protective and/or damaging consequences of ChP inflammation on brain function.
In summary, our study provides a comprehensive map of the molecular, cellular and spatial diversity of each ChP of the developing, adult, and aging mouse brain. Since the ChP-CSF system is implicated in a growing number of neurologic conditions, our dataset offers molecular insight that should accelerate future studies investigating the lifelong, active regulation of the ChP brain-body barrier.
Limitations of the study
To generate the atlas we overcame several challenges, while encountering new limitations. (1) To overcome the small tissue size, we isolated sufficient numbers of cells by pooling samples across donors. This approach was particularly salient for immune cells, which may nonetheless benefit from further enrichment for greater in-depth analysis. (2) We endeavored to isolate pure ChP by dissecting along anatomically defined tissue boundaries, which were at times limited (e.g., 3V ChP that has less defined borders). (3) We dissociated cells at different ages for whole cell sequencing. However, cellular health was particularly challenging for adult and aged tissues, ultimately leading us to favor differential sample processing (e.g., scRNA-seq for embryonic vs. frozen samples and snRNA-seq for adult/aged also applied to embryonic). This strategy subsequently introduced additional computational challenges in comparison of the two datasets. (4) Transitions between cell states and ages are continuous and required adjustments to our computational tools including the use of topic modeling. (5) We cannot rule out the possibility that we may miss some lower expressed genes of interest to ChP biology due to the limitations in depth of coverage.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maria K. Lehtinen, maria.lehtinen@childrens.harvard.edu.
Materials Availability
No new reagents generated.
Data and Code Availability
Raw and processed mouse sequencing data is available at https://portals.broadinstitute.org/single_cell and at the Gene Expression Omnbius (GEO) database GSE168704.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal models
All mouse work was performed in accordance with the Institutional Animal Care and Use Committees (IACUC) and relevant guidelines of Boston Children’s Hospital and Masaryk University.
Embryonic day (E) 16.5 embryos were obtained from time-pregnant CD-1 dams for embryonic scRNA-seq analyses. For snRNA-seq analyses, E16.5 embryos, adult (4 months) and aged (20 months) C57BL/6J males and females were used. All animals were housed under 12hr/12hr day night cycle with access to standard chow and water ad libitum.
CD1(ICR)– Charles River Laboratories Strain 022.
Cx3cr1-GFP (catalog #: 005582, The Jackson Laboratory)
C57BL/6 – Charles River Laboratories
C57BL/6J – (catalog #: 000664, The Jackson Laboratory)
Wnt1-Cre2 – (catalog #: 002137, The Jackson Laboratory)
R26R-Confetti – (catalog #: 013731, The Jackson Laboratory)
METHOD DETAILS
ChP tissue dissection
Whole ChP tissues from embryonic and adult were isolated as follows. LV ChP: Cerebral cortical hemispheres were separated. Following incisions into the cortical tissue at either end of the hippocampus, the hippocampus was rolled out using the flat surface of a microscalpel and the underlying LV ChP was separated from the hippocampus/fornix. 3V ChP: The dorsal midline of the midbrain was exposed and 3V ChP was isolated. Specific to adult brain, flushing the midline region with 1xHBSS helped visualize the ChP, which was identifiable by a blood vessel running along its rostro-caudal axis. The adult 3V ChP extends ventrally and rostrally, ultimately connecting to the ventral base of the LV ChP. 4V ChP: Hindbrain was separated from the brain. The cisterna magna was exposed by gently guiding away the developing or mature cerebellum to expose the 4V ChP.
FACS purification of healthy embryonic cells
LV, 3V and 4V ChP of three litters of embryonic E16.5 embryos were rapidly dissected and pooled together in dissecting medium (HBSS+0.6% Glucose+1xPen/Strep+Filtered medium 0.22 filter) (Corning, Cat:21-023-CV) + glucose (Sigma, Cat:7021-100G). Dissected ChPs were spun down at 300xG for 3 min. in a centrifuge at 4°C. Whole ChP was dissociated enzymatically by preparing fresh solution of collagenase II (Gibco, 17101-015) supplemented with 3 mM calcium (Sigma, C3881). Next, CP was tapped and flicked 30 times and incubated at 37°C incubator 3 consecutive times every 5 minutes over 15 minutes. The enzymatic solution with digested ChP tissue was then diluted using 1xHBSS, followed by pelleting down cells at 300xG for 3 mins at 4°C. TrypLE (Life Technologies, TrypLE, Catalog: 12604) was then added and samples were incubated for 5 min at 37°C followed by trituration using a micropipette. Samples were finally washed with ChP epithelial cell (CPEC) medium (DMEM+10%FBS+1xPen/strep) and resuspended in 1xHBSS + glucose + LIVE/DEAD staining kit (ThermoFisher, Catalog: L-3; 224). Live cells identified by green fluorescence (calcein-AM) (LifeTechnologies, Catalog L3224A) and lack of EtD-1 homodimer staining (L3224B) were selected and sorted by a MoFlo Astrios cell sorter (Beckman Coulter) into 500 μl filled into dissecting medium.
Single cell RNA-seq
Single cells were processed through the 10X Genomics Single Cell 3' platform using the Chromium Single Cell 3′ Library & Gel Bead Kit V1 and V2 kit (10X Genomics), as per the manufacturer’s protocol. Briefly, 7,000 cells were loaded on each channel and partitioned into Gel Beads in Emulsion in the Chromium instrument where cell lysis and barcoding occur. This step was followed by amplification, fragmentation, adaptor ligation, and index library PCR. Libraries were sequenced on an Illumina HiSeqX at a read length of 98 base pairs.
Single nucleus RNA-seq
Tissues were minced in 50 μl of CST lysis buffer [10 mM Tris, 146 mM NaCl, 1 mM CaCl2, 21 mM MgCl2, 0.1% CHAPS, 40 U/mL of RNAse inhibitor] in an eppendorf tube for 10 minutes. Volume in the tube was raised by addition of 1 mL CST lysis buffer and then transferred to a 15 mL conical tube on ice. The eppendorf tube was rinsed with 1 mL CST lysis buffer and the volume was added to the 15 mL conical tube. The nuclei suspension was diluted to 5 mL with ST buffer [10 mM Tris, 146 mM NaCl, 1 mM CaCl2, 21 mM MgCl2, 40 U/mL of RNAse inhibitor] and filtered through a 20 μm filter into a 15 mL conical tube prior to centrifugation at 500G for 5 minutes at 4°C in a swing bucket centrifuge. Nuclei pellets were then resuspended in ST [10 mM Tris; 146 mM NaCl; 1mM CaCl2; 21 mM MgCl2; 40U/mL of RNase Inhibitor] for a final volume of ~100 μl. Samples were normalized, pooled and filtered prior to loading 10X at a total of 10,000 nuclei per channel, which were run on the 10X Single Cell RNA-Seq Platform using the Chromium Single Cell 3’ Reagent Kits v3. Libraries were made following the manufacturer’s protocol. Briefly, single nuclei were partitioned into nanoliter scale Gel Bead-In-EMulsion (GEMs) in the Chromium controller instrument where cDNA share a common 10X barcode from the bead. Amplified cDNA was measured by Qubit HS DNA assay (Thermo Fisher Scientific: Q32851) and quality assessed by BioAnalyzer (Agilent: 5067-4626). This Whole Transcriptome Amplified (WTA) material was processed through 3’ V3 library construction, and resulting libraries were quantified again by Qubit and BioAnalzyer. Libraries from 4 channels were pooled and sequenced on 1 lane of Illumina HiSeqX by the Broad’s Genomics Platform, for a target coverage of around 1 million reads per channel.
Tissue fixation and processing
Dissected ChP were fixed in 4% paraformaldehyde (in 1X phosphate-buffered saline, pH 7.4). For microtome sectioning, embryonic brains were drop fixed in 4% paraformaldehyde, and incubated in the following series of solutions in 1X phosphate-buffered saline: 10% sucrose, 20% sucrose, 30% sucrose, 1:1 mixture of 30% sucrose and Optimal Cutting Temperature (OCT) compound (overnight) and finally in OCT compound alone (1 hour). Samples were then frozen in OCT compound.
Immunohistochemical analysis
Cryosections were permeabilized with 0.1% Triton X-100 in phosphate-buffered saline, incubated with primary antibodies overnight, and incubated with secondary antibodies for 2 hours. Sections were counterstained with Hoechst 33342 (Thermo Fisher Scientific) and mounted on slides using Fluoromount-G (SouthernBiotech, Birmingham, AL).
For Ki67 staining, an antigen retrieval step was included: A food steamer (catalog number 5712, Oster, Boca Raton, FL) was filled with water and preheated until the chamber temperature reached 100°C; sections were then immersed in boiling citric acid buffer (10 mmol/L sodium citrate, 0.05% Tween 20, pH 6) and placed in the steamer for 20 min. Sections were then cooled to room temperature.
Antibodies and probes
We used the following antibodies: Rabbit anti-5-HT (1:3000, S5545, Sigma), Mouse anti-AQP1 (1:100, sc-32737, Santa Cruz), Goat anti-ACE2 (1:250, AF3437, R&D Systems), Mouse anti-ACTA2-Cy3 (1:200, C6198, Sigma); Rabbit anti-CCDC67 (1:500, 24579-1-AP, Proteintech), Rat anti-CD74-488 (1:100, 151005, Biolegend), Rat anti-CD31/PECAM1 (1:200, 550274, BD Pharmingen), Goat anti-CD62P/P-Selectin (1:100, AF737, R&D Systems), Mouse anti-CLDN5-488 (1:400, 331588, Invitrogen), Rabbit anti-COL1A1 (1:250, AB34710, Abcam), Goat Anti-ESM1 (1:250, AF1999, R&D Systems), Chicken anti-GFP (1:250, ab13970, Abcam), Rabbit anti-GFAP (1: 250, Z0334, Dako), Hamster anti-IL1B (1:100, 503513, Biolegend), Goat anti-IL1R1 (1: 200, AF771, R&D Systems), Rabbit anti-IBA1 (1:250, 019-19741, Wako), Mouse anti-KI67 (1:50, 550609, BD Pharmingen), Rat anti-KI67 (1:250, 14-5698-80, ThermoFisher), Goat anti-LYVE1 (1:300, AF2125, R&D Systems), Rabbit anti-MRC1/CD206 (1:250, ab64693, Abcam), Goat anti-NRP1 (1:250, AF566, R&D Systems), Rabbit anti-PAX6 (1:500, ab2237, Millipore), Rat anti-SPI1/PU.1 (1:250, MAB7124-SP, Novus), Rabbit anti-SPIC (1:35, PA5-67537, ThermoFisher), Rabbit anti-SHISA8 (1:500, ab188621, Abcam), Rabbit anti-SLC40A1 (1:250, MTP11-A, Alpha Diagnostic International), Mouse anti-TUBB3 (1:100, 801202, Biolegend), Mouse anti-Acetylated Tubulin (1:250, T6793, Sigma), Rabbit anti-VWF (1:200, MA5-14029, ThermoFisher), Rat anti-WNT5A (1:250, MAB645, R&D Systems). Mouse anti-ZO1 (1:100, 33-9100, ThermoFisher).
We used the following RNAscope probes: Axin2 (mm-Axin2-C3, Ref: 400331-C3), FoxJ1 (mm-FoxJ1, Ref: 317091), Ins2 (mm-Ins2-C2, Ref: 310751-C2), Rspo1 (mm-Rspo1, Ref: 401991), Rspo2 (mm-Rspo2, Ref: 402001-C2), Rspo3 (mm-Rspo1, Ref: 402011-C3), Wnt1 (mm-Wnt1, Ref: 401091), Wnt2b (mm-Wnt2b-C2, Ref: 405031-C2), Wnt3a (mm-Wnt3a, Ref: 405041), Wnt5a (mm-Wnt5a-C3, Ref: 316791-C3),.
Whole explant and cortical section RNAscope in situ hybridization and immunohistochemistry
LV, 3V, and 4V ChP explants were dissected at E16.5 and fixed with 4% paraformaldehyde (PFA) for 10 min. at room temperature (RT) in a 9-well glass plate before beginning the manufacturer’s provided protocol for RNAscope Fluorescent Multiplex (ACD). Free-floating explants were incubated with Target Retrieval Reagent (ACD) in a steamer for 12 min. Subsequently, explants were washed 3x3 min. in double distilled water prior to incubation with Protease III Reagent (ACD) for 8 min. at 40°C, followed by another 3x3 min. wash cycle in double distilled water. Target Probes (ACD) were then hybridized and amplified according to the manufacturer’s specifications. Following hybridization, immunohistochemical staining was performed in a subset of explants and described above. For RNAscope and IHC of cortical sections used in confetti experiments, 14 μm cryosections were prepared. After isolation, embryos were immediately transferred and kept in fresh 4% PFA for 2 hours, washed briefly in ice cold PBS, incubated overnight in 30% sucrose solution, and frozen at −80°C. Transcripts were detected using the RNAscope 2.0 assay for fresh frozen tissue (Advanced Cell Diagnostics). Probes were designed and provided by Advanced Cell Diagnostics, Inc. Staining was performed using the RNAscope protocol according to the manufacturer’s instructions for fresh frozen sections processing (Wang et al., 2012, Kaiser et al., 2019). For immunostaining in confetti experiments, all sections underwent antigen retrieval by short boiling in the microwave followed by 15 min. incubation in 86°C pre-warmed water bath using antigen retrieval solution. Sections were washed in PBT (PBS with 0,5% Tween-20) and blocked in PBTA (PBS, 5% donkey serum, 0,3% Triton X-100, 1% BSA). Samples were incubated overnight at 4°C with primary antibodies diluted in PBTA. Following washes in PBT, samples were incubated with corresponding Alexa Fluor secondary antibodies for 1 hour at RT, followed by 5 min. incubation at RT with DAPI. Finally, samples were mounted and analyzed using Zeiss LSM 800 confocal microscope or Zeiss LSM 880 Airyscan for the analysis of the Confetti embryos. For the TUBB3 staining combined with in situ hybridization RNAscope analysis, the antigen retrieval step was excluded and the sections were washed using PBS followed by overnight incubation at 4°C with primary antibody in PBS containing 0.5% Triton X-100. Allen Brain Atlas (Lein et al., 2007) and Genepaint (Visel et al., 2004) were used to obtain in situ hybridization images of transcript localization within embryonic and adult brains.
Transmission electron microscopy
LV ChP was micro-dissected in HBSS (Thermo Fisher Scientific) and drop-fixed immediately in FGP fixative (5% Glutaraldehyde, 2.5% Paraformaldehyde and 0.06% picric acid in 0.2 M sodium cacodylate buffer, pH 7.4). After 2 hours at RT, the tissue was washed in 0.1M cacodylate buffer and postfixed with 1% Osmiumtetroxide (OsO4)/1.5% Potassiumferrocyanide (KFeCN6) for 1 hour, washed twice in water, washed once in 50mM Maleate buffer pH 5.15 (MB), incubated in 1% uranyl acetate in MB for 1 hour followed by one wash in MB, 2 washes in water, and subsequent dehydration in alcohol (10 min each; 50%, 70%, 90%, 2x10 min 100%). The samples were then put in propyleneoxide for 1 hour and infiltrated ON in a 1:1 mixture of propyleneoxide and TAAB Epon (TAAB Laboratories Equipment Ltd, https://taab.co.uk). The following day, the samples were embedded in TAAB Epon and polymerized at 60°C for 48 hours. Ultrathin sections (about 80 nm) were cut on a Reichert Ultracut-S microtome, picked up on to copper grids stained with lead citrate. Images were acquired with a JEOL 1200EX transmission electron microscope, and recorded with an AMT 2k CCD camera (Biological Electron Microscopy Facility, Harvard Medical School).
Scanning electron microscopy
LV ChP was micro-dissected in HBSS (Thermo Fisher Scientific) and drop-fixed immediately in FGP fixative (5% Glutaraldehyde, 2.5% Paraformaldehyde and 0.06% picric acid in 0.2 M sodium cacodylate buffer, pH 7.4). Post-fixation was performed in 1% osmium tetroxide in 0.1M sodium cacodylate buffer for 1 hour. This step was followed by rinsing in 0.1M sodium cacodylate buffer 3 times 15 min. each. The samples were dehydrated in a graded series of ethanol to 100% and dried using a Tousimis PVT-3D critical point dryer (Tousimis Research Corp., Rockville, MD). The dried samples were then attached to sample mounts using double sided conductive carbon adhesive tabs and sputter coated with 5 nm of platinum with a Cressington 208HR sputter coater (Cressington Scientific Instruments, Walford, UK). SEM images were acquired on a Hitachi S-4800 FE-SEM (Hitachi Ltd., Tokyo, Japan).
Hematoxylin and Eosin (H&E) staining
Paraffin-embedded brains were sectioned to a thickness of 5 μm. The sections were deparaffinized in xylene and then rehydrated via successive incubations in 100% ethanol, 95% ethanol, and water. Sections were incubated in Gill 3 hematoxylin (Sigma Aldrich, St. Louis, MO) for 2 min., followed by a 5-second incubation in 0.5% ammonia water to increase the contrast of the hematoxylin stain. Next, sections were rinsed in water and incubated in 1% alcoholic eosin for 3 min. Finally, sections were dehydrated via successive incubations in 95% ethanol and 100% ethanol, and mounted using Permount (Thermo Fisher Scientific).
Regionalized proliferation in the LV ChP
Tissue was stained with KI67 and Hoechst and imaged using laser scanning confocal microscopy with tiling function (LSM 710, Zeiss). The tissue images were divided into three equal regions along the dorso-ventral: (1) brain-proximal, (2) -medial, and (3) -distal regions. Nuclei were identified by Hoechst staining and proliferating cells were identified by overlapped KI67 staining. Percent proliferating cells were counted in each region. A total of four independent explants were quantified. Multiple comparisons ANOVA was employed using statistical software package Graphpad PRISM. Data are represented as mean ± SEM. P < 0.05 was considered significant.
Pre-processing of droplet-based scRNA-seq
De-multiplexing, alignment to the mm10 transcriptome and unique molecular identifier (UMI)-collapsing were performed using the CellRanger toolkit from 10X Genomics (version 1.1.0 for the first batch of embryonic scRNA-seq, which used V1 chemistry, and version 2.0. for experiments in batch two and batch three, which used 10x Genomics V2 chemistry). For each cell, we quantified the number of genes for which at least one read was mapped, and then excluded all cells with fewer than 500 detected genes. Since the total number of UMI and genes detected varies across cell types (Figure S1A), we further excluded epithelial cells with fewer than 10,000 total UMI and mesenchymal cells with fewer than 6,000 total UMIs. Genes that were detected in less than 5 cells were excluded. Expression values Ei,j for gene i in cell j were calculated by dividing UMI counts for gene i by the sum of the UMI counts in cell j, to normalize for differences in coverage, and then multiplying by 10,000 to create TPM-like values (TP10K), and finally computing log2(TP10K + 1). Batch correction was performed for each cell type separately using ComBat as implemented in the R package sva (Leek et al., 2012), using the default parametric adjustment mode. The output was a corrected expression matrix, which was used as an input to further analysis.
Pre-processing of droplet-based snRNA-seq data
De-multiplexing, alignment to the mm10 transcriptome with retained intronic regions (mm10_premrna_v1.2.0) and UMI-collapsing were performed using the CellRanger toolkit (version 3.1.0, V3 chemistry) from 10X Genomics. To address ambient (background) RNA and other confounders we applied CellBender (Fleming et al., 2019) to each sample separately with expected number of nuclei ranging between ~4,500 and ~8,000, depending on the sample, 15,000 total droplets included for the analysis and 200 epochs. For each nucleus, we quantified the number of genes for which at least one read was mapped, and then excluded all nuclei with fewer than 500 detected genes, or 200 detected genes for immune cell nuclei. Furthermore, we excluded nuclei that had more than 10% of total UMI’s mapped to mitochondrial genes. Genes that were detected in less than 10 nuclei were excluded. Expression values Ei,j for gene i in nucleus j were calculated by dividing UMI counts for gene i by the sum of the UMI counts in nucleus j, to normalize for differences in coverage, and then multiplying by 10,000 to create TPM-like values (TP10K), and finally computing log2(TP10K + 1).
Identifying variable genes
Selection of variable genes was performed by fitting a logistic regression model to the cellular detection fraction (often referred to as α), using the total number of UMIs per cell/nucleus as a predictor as in (Montoro et al., 2018). Outliers from this curve are genes that are expressed in a lower fraction of cells/nuclei than would be expected given the total number of UMIs mapping to that gene, that is, likely cell-type or state-specific genes. We used a threshold of deviance between <−0.15 and <−0.3. For the embryonic scRNA-seq data, if there were enough cells from all three batches, variable genes were calculated within each batch, and only the intersection of variable genes in each batch were taken for downstream analysis. This was the case when analyzing all cells (Figure 1C), and epithelial and mesenchymal cells. Batch was ignored for computing variable genes for immune, endothelial and neuro-/glia-like cells. Batch was ignored for all snRNA-Seq data. We restricted the expression matrix to this subset of variable genes and values were centered and scaled and capped at a z-score of 10.
Dimensionality reduction using PCA and visualization using t-SNE or UMAP
We restricted the expression matrix to the subsets of variable genes and high-quality cells/nuclei noted above, and then centered and scaled values before inputting them into principal component analysis (PCA), implemented using ‘RunPCA’ in Seurat which runs the irlba function. For the snRNA-seq data, which included mixed sexes, we removed Y-chromosome and X-inactivation genes (Xist, Tsix) from the variable genes prior to running PCA. The cell embeddings were either the singular vectors themselves or the singular vectors multiplied with the singular values depending on the cell type. After PCA, significant principal components were identified using the elbow-method, when looking at the distribution of singular values. Scores from only those significant principal components were used as the input to further analysis. For visualization purposes, the dimensionality of the datasets was further reduced to 2D embeddings using t-SNE (for scRNA-seq) (Van Der Maaten and Hinton, 2008) or UMAP (Becht et al., 2019) (for snRNA-seq) on the significant PCs using, respectively, the RunTSNE() or RunUMAP() functions of the Seurat package in R.
Clustering and removing doublets in scRNA-seq data
To cluster single cells by their expression, we used an unsupervised clustering approach, based on the Infomap graph-clustering algorithm (Girvan and Newman, 2002). In brief, we constructed a k-nearest-neighbor graph on the data using, for each pair of cells, the Euclidean distance between the scores of significant principal components to identify k nearest neighbors. The parameter k was chosen to be consistent with the size of the dataset. Specifically, k was set to 40 for neuronal/glia-like cells and to 20 for immune cells, for which subclusters of macrophages were post-hoc merged together based on expression of canonical markers. The nearest-neighbor graph was computed using the function nng from the R package cccd. The k-nearest-neighbor graph was then used as the input to Infomap (Girvan and Newman, 2002), implemented using the infomap.community function from the igraph R package. For major cell types (Figure 1C), clusters were post-hoc merged to six major cell populations using canonical markers for all cell types detected.
Doublets were clearly identifiable as forming their own clusters with dual expression of marker genes of distinct subsets (e.g., epithelial genes and endothelial genes) and were removed from further analysis and any visualizations.
Removing doublets in snRNA-seq data
Nucleus doublets were identified using the R package DoubletFinder (version DoubletFinder_2.0.2, (McGinnis et al., 2019). Briefly, artificial doublets are generated from the data, and for each real data point the fraction of artificial doublets of the nearest neighbors in a latent space is computed. If this fraction is above a certain cutoff, the data point is considered to be a doublet. Doublets were calculated for each sample separately after initial data filtering by genes detected and mitochondrial gene expression as per instructions. For each sample, an optimal pK (defines the PC neighborhood size) was chosen by computing a parameter sweep (paramSweep_v3 function) and a pK was chosen that maximized the BCmvn (mean-variance-normalized bimodality coefficient). Doublets were then calculated using the function doubletFinder_v3() with parameters; PCs=1:30, pN=0.25, nEXP=15% of cells in sample and removed from downstream analysis.
Scoring cells using signature gene sets
In order to score cells using a gene sets, such as cell cycle (Figure 2A) or arterial / venous gene expression (Figure S5A), we calculate the following gene signature score: Mean program expression was calculated by averaging over the genes in each program of the centered and scaled gene expression table and transforming to a z-score over 1,000 randomly selected gene sets with matched mean-variance patterns. First, genes were grouped into 10 bins based on their mean expression, and into 10 (separate) bins based on their variance of expression across all cells. Given a list of genes (e.g. genes in a program), a cell-specific signature score was computed for each cell as follows: First, 1,000 random gene lists were generated, where each instance of a random gene-list was generated by sampling (with replacement) for each gene in the gene-list a gene from the equivalent mean and variance bin it was placed in. Then, the sum of centered and scaled gene expression in the given cell was computed for all 1000 random gene-lists generated and the z-score of the original gene-list for the generated 1,000 sample distribution is returned, as in (Singer et al., 2016). For cycling cells, cells with a z-score above 2 were classified as cycling cells.
Topic modeling
We performed topic modeling using Latent Dirichlet Allocation (LDA). LDA was computed on epithelial, mesenchymal and endothelial cells separately in the embryo, and on epithelial cells in adult. Specifically, we used the FitGoM() function from the CountClust R package (Dey et al., 2017) to fit LDA topic models to the UMI counts (Bielecki et al., 2021). To improve topic signals, genes expressed in more than 98% of cells or less than 2% of cells were removed from the count matrix prior to fitting the topic models (analogous to the removal of highly abundant words or extremely rare words in document analysis), except for endothelial cells. The number of topics to fit (K) and the tolerance value are required to run FitGoM() function. Thus, for each cell type and developmental status, we fit a range of K and tolerance values and picked values that gave us robust topics and where we mostly saturated the number of informative topics found. For the embryo data, this was achieved with a tolerance of 0.1, and a K of 16 for endothelial cells, K of 26 for epithelial and mesenchymal cells, and K of 14 and tolerance of 0.01 for adult epithelial nuclei. The top genes to highlight for each topic were selected using the ExtractTopFeatures() function. Topics were annotated post-hoc. Notably, in the analysis of epithelial cells, one topic was enriched for immediate early genes (Topic 19, Figure S3B,C) and may reflect a response to tissue dissociation (van den Brink et al., 2017) because it was reduced in single nucleus profiles (Lacar et al., 2016) (Figure S3D).
Defining cell-type or cluster signatures
Differential expression between cell types in scRNA-Seq of embryo (Figure 1) and between clusters of immune cells and neuro-/glia-like cells in scRNA-Seq was performed using MAST (Finak et al., 2015), which fits a hurdle model to the expression of each gene, consisting of logistic regression for the zero component (i.e. whether the gene is expressed) and linear regression for the continuous component (i.e. the expression level). The regression model includes terms to capture the effects of the cell subset or cluster, while controlling for cell complexity (i.e. the total number of UMIs (nUMI)). Specifically, we used the regression formula, Yi ~ X + N, where Yi is the standardized log2(TP10K+1) expression vector for gene i across all cells, X is a factor variable reflecting cell subset or cluster membership, N is the scaled nUMI in each cell. In all cases, the discrete and continuous coefficients of the model were retrieved and p-values were calculated using the likelihood ratio test in MAST. Q-values were estimated using the Benjamini-Hochberg correction. In order to identify cell-type specific or cluster specific markers, an FDR cutoff for the hurdle model was chosen using the elbow-method and a small mastfc cutoff to exclude genes with very small effect sizes. If a gene passed the FDR and the mastfc cutoff in only one cluster / cell type, it was considered to be specific.
Cell type classification of scRNA-seq dataset using snRNA-seq dataset of embryo
To compare embryo scRNA-seq dataset to embryo snRNA-seq, a random forest classifier was used from the R package ‘randomForest’. First, common variable genes were computed between the two datasets by taking the intersection of the variable genes in each dataset. Then, the classifier was trained on the scaled expression matrix of the common variable genes in the embryo snRNA-seq dataset. The out-of-bag error was 1.7%. The classifier then ran on the scaled expression matrix of the common variable genes in the embryo scRNA-seq dataset. We then compared the predicted cell type assignment of the random forest classifier to our own cell type assignment shown in Figure 1C.
Diffusion map
For diffusion analysis of embryo epithelial and neuronal cells, we selected cells from only 3V ChP, where most neuronal and glial cells were sampled from, and only from replicate 3, because of strong batch effects between replicates. Furthermore, the epithelial cell clusters scoring highly for IEG genes (Figure S2D) and oligodendrocyte precursor cells were also excluded from this analysis. Of the remaining cells, variable genes were computed as described above. Diffusion components were calculated on a gene expression matrix limited to variable genes within the selected cells (not correcting for batch). Diffusion components were calculated using the DiffusionMap function from the destiny package in R (Angerer et al., 2016) with a k of 30 and a local sigma.
For macrophages, cells of replicate 3 were excluded for this analysis because they had very high expression of mitochondrial genes. Diffusion components were calculated as above on a gene expression matrix limited to variable genes within the macrophages (not correcting for batch) with a k of 20 and a local sigma.
RNA Velocity analysis of epithelial-neuronal trajectory
To study the inferred trajectory of epithelial-neuronal differentiation (Figure 2), RNA velocity analysis was performed, which compares reads mapped to exonic versus intronic regions to assess the future transcriptional state of the cell. We ran the function gene.relative.velocity.estimates() of the velocyto package in R on the cells used in the trajectory analysis in Figure 2 and variable genes as described above. We used the 1-correlation of the first 16 diffusion components as a cell distance measure, and deltaT=1, kCells=25, fit.quantile=0.02. The RNA velocity vectors were visualized on the trajectory embedding from the diffusion analysis.
Pseudo-bulk analysis of epithelial and fibroblast cells to test for significantly differentially expressed genes between age and ventricle
To conservatively identify high confidence genes that vary with age and/or ventricle, we performed pseudo-bulk analysis for epithelial cells and fibroblast (excluding meningeal cells, pericytes and vSMCs). To that end, all UMIs were summed within each sample of a specific age and ventricle. Because the samples consisted of pooled mice from both sexes, the fraction of male genes expression in each sample was normalized by the sum of all UMIs from y-chromosome genes (by introducing a covariate was termed tUMI.ygens). Then, we performed differential expression analysis using DESeq2 package in R, where the data was the total UMIs as described above per sample. The expression modeling was ‘~ tUMI.ygenes + Stage + Ventricle + Stage:Ventricle, where Stage denotes pre- or post-natal (including both adult and aged samples). A separate DESeq2 analysis was performed for testing for differentially expressed genes between Adult and Aged, with a likelihood-ratio test against a reduced model of ‘~tUMI.ygenes + Ventricle’ to identify age-effects (adjusted p-value cutoff used by elbow method was 10−5 for epithelial cells and fibroblast) or ‘~tUMI.ygenes + Stage’ to identify regionalized genes (adjusted p-value cutoff used by elbow method; Epithelial = 10−15, Fibroblast = 10−3) and required the absolute log2FoldChange to be >1 to filter out genes with significant but small effect size. For visualization of the significantly differentially expressed genes, normalized values of these pseudo-bulk samples were scaled, and capped at an absolute z-score of 2.5.
Cell-cell interactions
To quantify cell-cell interactions between major cell types across age, we used CellPhoneDB (Efremova et al., 2020) on all cells of embryo, adult and aged ChP separately. In short, this method computes the average expression of a ligand-receptor pair for each pair of cell type provided as a meta file. It then computes an empirical p-value to the specificity of this interaction by permuting cell types identity 1,000 times and comparing the real average expression against this null distribution. As CellPhoneDB is developed for human ligand-cognate receptor pairs, gene names were converted to their human homolog, removing genes for which no one-to-one human homolog could be associated.
Significant cell-type pair ligand-receptor interactions were used as edges for a directed cell-cell interaction graph. For pairs lacking clear ligand and receptor notations, we could not infer the directionality of the interaction by the annotation, and thus the directionality of the edge was determined by the CellPhoneDB annotation (given that the statistical significance assigned to each pair of cells has a clear direction). Next, to calculate the statistical significance of the strength of estimated interaction between pairs of cell types (in a specific age and ventricle), we performed two types of permutation tests – in each test we perform 10,000 permutations and calculate an empirical p-value for the number of interactions found for a pair of cell types. The tests were: (1) maintaining the total number of edges and randomizing the ligand- and receptor-cell type assignments; and (2) maintaining the total number of edges per each cell type (the degree of the nodes) and permutating the cell type source and destination of the edge (the ligand-receptor pair).
Differential interaction graphs, over different ages or ventricles were constructed based on the unique edges of each dataset. We define a differential edge, as a ligand-receptor pair that were found to be both statistically significant expressed in the respective pair of cell types under one condition (age and ventricle) but not in another condition compared to it. A similar permutation strategy was used to compute an empirical p-value for the differential interaction graphs.
To visualize interaction graphs, we used Sankey plots, using the Sankey Network function of the networkD3 package, showing the number of edges between ligand expressing- and receptor expressing cell types. We combined two Sankey plots to show the differential graphs between two conditions.
Supplementary Material
Figure S1, related to Figure 1. Cellular markers, identity and organization within the ChP. (A) Embryonic cell quality of scRNA-seq. Distribution of number of transcripts (log10(nUMI), left) genes (log10(nGene), right) across cell types, grouped and colored by batch. (B) Proportions of cell types captured from each ChP by embryonic scRNA-seq. Fractions (y axis) of each major cell type identified (x axis) per ventricle and batch (color). (C) Expression of cell type markers by scRNA-seq of embryonic ChP. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) across marker genes (columns) for each cell type cluster (rows). (D) Proliferating cells across cell types in the ChP. Left: t-SNE (as in Figure 1C) of cell profiles (dots), with cells classified as cycling colored black (STAR Methods). Right: Mean fraction of cycling cells (y axis) in each cell type (x axis), and in each batch replicate (dots). Line: standard deviation (S.D.). (E) A proliferating cell niche in the ChP. Left: Proliferation marker KI67 staining (magenta) of whole explant of LV ChP. Dotted brackets mark regions of interest that are proximal, medial and distal relative to the ventral margin of the ChP near the brain. Double headed arrows mark Anterior (A)/Posterior (P) and Dorsal (D)/Ventral (V) axes. Scale bar = 500 μm. Right: Percent of proliferating cells (KI67+) out of all cells in proximal, medial and distal regions of the LV ChP (n=4, Scale bar = 500 μm; one-way ANOVA, ** p<0.01. Error bars indicate S.E.M.). (F) snRNA-seq nuclei library quality. Distribution of number of genes (nGene, left) and transcripts (nUMI, right) across cell types, grouped and colored by age. (G) Proportions of cells captured from each ChP and age by snRNA-seq. Fractions (y axis) of each major cell type identified (x axis) per ventricle across ages (color). (H) Single nuclei batch across cell clusters. UMAP embedding (as in Figure 1F) of single nucleus profiles colored by batch, showing uniform distribution of batches across clusters. (I) A random forest classifier assigns cell types across scRNA-seq and snRNA-seq datasets in embryo ChP. Fraction of cells (color and circle size) per cell type in embryo scRNA-seq (rows) that were mapped to embryo snRNA-seq cell types (columns) (STAR Methods). Columns add up to 1. (J) Resident neurons in embryonic LV ChP. Top: Representative neuron co-stained with anti-serotonin (5-HT) (red) and anti-TUBB3 (green) and nuclei counterstained with Hoechst in embryonic ChP (Scale bar = 10 μm). Bottom: Split channel showing anti-serotonin staining alone. (K) Resident neurons in adult and aged 4V ChP. Representative image of neuronal cell bodies stained with anti-TUBB3 (green) and nuclei (Hoechst) in adult (top) and aged (bottom) ChP. Scale bar = 10 μm. (L) Resident glial cells in adult and aged LV ChP. Representative image of glial cells stained with anti-GFAP (red) antibodies in LV ChP in adult (left) and aged (right). Scale bar = 20 μm.
Figure S2, related to Figure 2. Neuronal and glial cell types across ages and ventricles. (A) Glial and neuronal cell markers. T-SNE of neuronal and glia-like cell profiles from scRNA-seq in embryonic ChP (dots, as in Figure 2A), colored by expression (log2(TP10K+1)) of Tubb3 (neuronal marker, top) and Slc1a3 (glial marker, bottom). (B) Neuronal/glia-like subset markers and neuropeptides. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected genes (columns) in neuronal and glia-like subsets (rows). (C) Organization of the developing 3V ChP. In situ hybridization images (Allen Brain Atlas and Genepaint) of marker genes for epithelial cells (Ttr), progenitor cells (Rspo3) and developing pineal gland clusters (Krt19, Crx), localizing the progenitors adjacent to the pineal gland and choroid plexus epithelial cells. Rightmost panel: Schematic of the 3V structure. (D) Neuro-glial subclusters. UMAP of neuronal and glia-like nucleus profiles across age groups from snRNA-seq from the three ventricles, colored by age (left) and cluster membership (right). (E) Neuro-glial marker gene expression across snRNA-seq clusters. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected genes (columns) across clusters (rows). (F) RNA Velocity analysis of inferred differentiation trajectory of neuronal and epithelial cells from a predicted common progenitor pool. Diffusion map embedding of cells (dots) from embryonic 3V ChP (scRNA-seq) (as in Figure 2B). Arrows mark the direction of cell transition as inferred by RNA velocity algorithm (La Manno et al., 2018). (G) Wnt1 restricted expression in the progenitor pool. Diffusion map embedding of cells as in F colored by log2(TP10K+1) expression level of Wnt1. (H-K) Distribution of progenitor domains near the root and base of the LV ChP (dashed lines). Tissue sections of embryonic LV ChP labelled with progenitor markers (arrowheads): immunostained for PAX6 (H), or RNA smFISH for R-spondin genes (Rspo1, Rspo2, and Rspo3, I, Inset: Cells expressing all three markers, Scale bar = 20 μm), Wnt genes (Wnt2b, Wnt3a and Wnt5a, J), and WNT-mediated signaling (Axin2, Wnt5a, K). Scale bar = 50 μm for (H-K). (L) Reduced proportion of ciliogenic and cycling epithelial cells in adult and aged ChP compared to embryos. Fraction of ciliogenic (left) and cycling (right) epithelial cells out of the total epithelial cells, in embryo, adult and aged ChP. P-value of T-test on the proportion of cycling or ciliogenic cells comparing between every pair of age groups, *p<0.05.
Figure S3, related to Figure 3. Regionalized transcriptional programs in epithelial cell populations. (A) Regionalized expression programs of epithelial cells in embryonic ChP from scRNA-seq agrees with bulk RNA-seq. Differentially expressed genes between LV and 4V in bulk RNAseq (Lun et al., 2015a) (Left: up-regulated in 4V ChP. Right: up-regulated in LV ChP) averaged across single cells in 4V ChP (x axis) and LV ChP (y axis). Red line indicates x=y. All genes show a coordinated difference between the datasets. (B) Topic modeling of embryonic epithelial cells captures distinct transcriptional programs. Embryonic epithelial transcriptional programs associated with cell cycle (Topics 4 and 9), ciliogenesis (Topic 24) and immediate early gene expression (Topic 19). For each topic, a bar plot of topic scores is shown for top ranked genes (left), and t-SNE of the cell profiles (as in Figure 3A) colored by topic’s weight per cell (right). (C) Ventricle associated transcriptional programs in embryonic epithelial cells. Empirical cumulative distribution (y axis) of topic’s weight (x axis), grouped and colored by ventricle. (D) Immediate early gene (IEG) expression across all cells is higher in scRNA-seq compared to snRNA-seq data. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected genes (columns) in our scRNA-seq and snRNA-seq data (rows). (E) snRNA-seq analysis recapitulates regionalized gene expression as compared to scRNA-seq. Distribution of expression level (y axis) of regionalization marker genes across ventricles (colors) in embryonic epithelial cells. (F) Rostral and caudal gene expression pattern in embryonic 4V ChP epithelium shown in situ. Top: ISH images from Genepaint showing regionalization of Shh and Serpinb1a (Scale bar = 100 μm). Bottom: T-SNE of epithelial cell profiles (dots), colored by log2(TP10K+1) expression levels of genes shown in Figure 3E. (G-H) Maturation-dependent shifts in transporter (G) and secreted factor (H) expression. Heatmap of gene expression from snRNA-seq across ages (left color bar) and ventricles (right color bar). Scaled across all nuclei (columns) by z-score. Examples of genes marked on the bottom. (I) Age- and ventricle-dependent transcriptional programs in epithelial cells by snRNA-seq. Distribution of expression level (y axis) of regionalized genes (Tns4 and Slit2) and age-dependent genes (Gpc3 and Prlr) across ages (top row x axis) and ventricles (colors). (J-K) Validations of ventricle- and age- dependent expression by ISH images from Allen Brain Atlas (Lein et al., 2007). Regionalization and relative expression (color) of Tns4 (Top, J) and Slit2 (Bottom, J) genes in adult LV, 3V and 4V ChP. Relative expression in embryo and adult of Gpc3 (Top, K) and Prlr (Bottom, K) in LV ChP. Scale bar = 100 μm.
Figure S4, related to Figure 4. Regionalized transcriptional programs in mesenchymal cell populations. (A) Embryonic mesenchymal cells partition into fibroblast and mural cells (pericytes and smooth muscle cells). T-SNE of mesenchymal cell profiles (dots, as in Figure 4A), colored by log2(TP10K+1) expression level of marker genes for: pericytes (Des), fibroblasts (Pdgfra) and smooth muscle actin positive cells (Acta2). (B) Topic modeling of embryonic mesenchymal cells captures distinct transcriptional programs. Embryonic mesenchymal transcriptional programs associated with: cell cycle (Topics 12,16) and LV and 3V fibroblasts shared program (Topic 8). For each topic, a bar plot is shown of topic scores for top ranked genes (left), and t-SNE of the cell profiles (as in Figure 4B) colored by topic’s weight per cell (right). (C) Ventricle associated transcriptional programs in mesenchymal cells. Empirical cumulative distribution (y axis) of topic’s weight (x axis), grouped and colored by ventricle. (D) Regionalized embryonic mesenchymal transcriptional programs. In situ hybridization (Genepaint) in sagittal sections of ChP for genes with ventricle specific enrichment in fibroblasts of the LV (Lox), 3V (Adamts2, Fbln1), 4V (Hhip), pericyte marker (Rgs5) and 3V/meningeal fibroblast markers (Fbln1). Scale bar for LV = 50 μm; 3V and 4V = 10 μm. (E) In situ hybridization from Genepaint and Allen Brain Atlas of sagittal sections of embryonic 3V ChP showing expression of fibroblast markers (Apod, Crym, Slc6a13) shared by meninges and 3V ChP stroma. Bottom right: Composite model identifying organization of 3V/meningeal fibroblasts in the developing brain. Scale bar = 100 μm. (F) Regionalized expression of extracellular matrix (ECM) protein in mesenchymal cells. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of ECM genes within top 50 features (rows) across ventricles (columns). (G) Mesenchymal marker expression across mesenchymal subtype clusters. Dot plots as in F of marker genes (columns) across mesenchymal sub-types (rows). (H-I) Regionalized expression of receptors (H) and transporters (I). Heatmap of gene expression from snRNA-seq across ages (right color bar) and ventricles (left color bar). Scaled across all nuclei (columns) by z-score. (J) SARS-CoV-2 cell entry receptor Ace2 gene expression in mesenchymal cells (including pericytes) across ages. UMAP of mesenchymal single nuclei profiles (snRNA-seq as in Figure 4C), colored by log2(TP10K+1) expression level of Ace2. (K) Distribution of ACE2 expression in fibroblast cells across age. Co-staining of ChP tissue sections in embryo (left), adult (middle) and aged (right) with anti-ACE2 (green) and stromal cell marker anti-COL1A1 (red) in 4V ChP. Arrowheads denote ACE2 and COL1A1 positive staining cells. Horizontal arrows denote ACE2 positive COL1A1 negative cells. Scale bar = 20 μm.
Figure S5, related to Figure 5. Cytokine and chemokine expression across ChP immune cells. (A-B) Gene expression of cytokines and chemokines across embryonic immune cell subsets. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of cytokines (A, columns) and chemokines (B, columns) in immune cell subsets (rows) from scRNA-seq in embryonic ChP across all ventricles. (C) SPIC expressing macrophages in embryonic ChP stromal space. LV ChP explants from Cx3cr1+/GFP mice stained with anti-SPIC (magenta, white arrowhead). Scale bar = 20μm. (D) Correlated gene expression in Spic expressing embryonic macrophages. Partial correlation (normalized for nUMI, y axis) between gene expression level and diffusion component 2 score (DC2, from diffusion map in Figure 5C) per cell. Marking gene names of highest correlated (e.g. Spic, Hmox1, Fth1) and anticorrelated genes with DC2. (E) Age and ventricle specific enrichment of immune cell populations. UMAP of snRNA-seq immune cell profiles (dots, as in Figure 5H) color coded by age (left) and ventricle (right). (F) Lymphoid populations. Left: UMAP of snRNA-seq immune cell profiles (dots, as in Figure 5H) color coded by marking T/B cell populations (blue). Right: UMAP of the T/B population colored by log2(TP10K+1) expression level of marker genes identifying T and B cell sub-populations. (G) Macrophage tiling patterns retained in adult and aged ChP. Co-staining of LV ChP explants for anti-PECAM1 (endothelial marker) and anti-IBA1 (macrophage marker) in adult (left two panels) and aged (right two panels). Scale bar = 10 μm. (H) Differentially expressed BAM genes (Van Hove et al., 2019) are age-dependent in the ChP. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected genes (columns) in embryonic, adult and aged macrophage cells (rows). (I) Aging associated shifts in gene expression. Bar plots for top enriched pathways (hypergeometric test −log10(FDR), x axis) upregulated (left) or down-regulated (right) in aged macrophage cells. (J) Aged stromal macrophages express CD74 protein. Tissue sections of aged 4 V ChP co-stained for anti-CD74 and anti-IBAl (macrophage marker). Arrowheads denote CD74 and IBA1 positive staining cells. Scale bar = 10 μm.
Figure S6, related to Figure 6. Molecular identities of ChP endothelial cells. (A) Arterial and venous transcriptional programs in embryonic endothelial cells (scRNA-seq data). T-SNE (as in Figure 6A) of endothelial cell profiles (dots), colored by signature scores for arterial (left) and venous (right) gene sets (gene signatures taken from (Vanlandewijck et al., 2018)). (B) Ventricle associated transcriptional programs in embryonic endothelial cells. Empirical cumulative distribution (y axis) of topic’s weight (x axis), grouped and colored by ventricle (scRNA-seq data). (C) Blood brain barrier markers in endothelial cells across age. UMAP of endothelial nucleus profiles from snRNA-seq (as in Figure 6D), colored by log2(TP10K+1) expression level of BBB marker (Cldn5), fenestrated vessel marker (Plvap), capillary marker (Esm1) and venous marker (Nr2f2). (D) Adult ChP capillaries express ESM1 protein. LV ChP explant co-stained with endothelial marker (anti-PECAM1, green) and capillary marker (anti-ESM1, magenta). Scale bar = 20 μm. (E) Fenestrae in embryonic and adult ChP blood vessels. Transmission electron microscopy image of developing fenestrae in the embryonic vessels (top, black arrow heads), and of the well-defined fenestrae in the adult endothelia (bottom, black arrow heads). Scale bar = 50 nm. (F) Venous signature enrichment in 3V ChP across all ages. Violin plot shows distribution of vein signature (defined as in A) by z-score in embryo, adult and aged across ventricles (colors). (G) Diversity of endothelial cells across ventricles. UMAP of snRNA-seq of endothelial cell profiles across ages (as in C), colored by ventricle. (H) Vein of Galen organization adjacent to 3V ChP. In situ hybridization for endothelial marker Flt1 and venous marker Nr2f2 (Allen brain Atlas) shows location of intracerebral vessel proximal to the 3V ChP (Scale bar = 100 μm) that is a likely candidate of a regionalized endothelial cell population. (I) BBB organization in the aged ChP. Whole explant co-staining for anti-CLDN5 (green) and anti-PECAM1 (red), showing CLDN5-positive staining endothelial cells continuous with brain vessels (arrow) that vascularize the ChP. Scale bar = 500 μm.
Figure S7, related to Figure 7. Mesenchymal, endothelial and immune cells contribute to cellular cross-talk in ChP. (A) Directional ligand-receptor interaction networks across cell types. Number of statistically significant (color bar, as estimated by CellPhoneDB, Star Methods) ligand (rows) receptor (columns) between pairs of ChP cell types, across ventricles (LV,3V,4V, from left to right), in embryonic nuclei (top) and embryonic cells (bottom, adult and aged nuclei in Figure 7A). Including ligand-receptor pairs marked as significantly expressed by CellPhoneDB (Efremova et al., 2020). Significant pairs of cell types based on a randomization test marked as: *p≤0.05, **p≤0.01, ***p≤0.001. (B) Number of significant interactions varies by cell type, ventricle and age. Bar plot of the percent of ligand-receptor pairs involving each cell type out of the total significant pairs, for each age (color) and ventricle (rows). (C) Examples of ligand-receptor pairs involving mesenchymal cells. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected ligands and receptors (columns) in each major cell population (rows), across all ventricles (color coded) and ages (top: embryonic; middle: adult; bottom: aged). (D) Directional ligand-receptor interaction networks across cell types. Number of significant ligand-receptors per pair of cell types for adult and aged ChP, across ventricles (as in Figure 7A). Significant pairs of cell types marked by asterisk, from a permutation test, constrained to preserve the total number of interactions per cell type, *p≤0.05, **p≤0.01.
Table S1, related to Figure 1, Figure 2 and Figure 5. Markers for cell types (Figure 1C), neuronal and glia-like clusters (Figure 2A) and immune clusters (Figure 5A).
Table S2, related to Figure 3B. Top 50 genes in each topic revealed by topic modeling on epithelial cells of developing ChPs.
Table S3, related to Figure 3I,J, Figure S3G,H. Epithelial DEG Supplemental table
Table S4, related to Figure 4B. Top 50 genes in each topic revealed by topic modeling on mesenchymal cells of developing ChPs.
Table S7, related to Figure 6A. Top 50 genes in each topic revealed by topic modeling on endothelial cells of developing ChPs.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-BrdU | Abcam | #ab6326; RRID: AB_305426 |
| Goat polyclonal anti-mouse ACE2 | R&D Systems | #AF3437; RRID: AB_2223140 |
| Mouse monoclonal anti-ACTA2-Cy3 | Sigma-Aldrich | #C6198, RRID: AB_476856 |
| Mouse monoclonal anti-AQP1 | Santa Cruz | #32737; RRID: AB_626692 |
| Rabbit polyclonal anti-CCDC67 | Proteintech | #24579-1-AP |
| Goat polyclonal anti-CD62P/P-Selectin | R&D Systems | #AF737; RRID: AB_2285644 |
| Rat monoclonal anti-CD74-488 | Biolegend | #151005; RRID: AB_2750325 |
| Mouse monoclonal anti-CLDN5-488 | ThermoFisher | #352588; RRID: AB_2532189 |
| Rabbit polyclonal anti-COL1A1 | Abcam | #ab34710, RRID: AB_731684 |
| Goat polyclonal Anti-ESM1 | R&D Systems | #AF1999, RRID: AB_2101810 |
| Chicken polyclonal anti-GFP | Abcam | #ab13970; RRID: AB_300798 |
| Rabbit polyclonal anti-GFAP | Dako | #Z0334; RRID: AB_10013382 |
| Rabbit polyclonal anti-IBA1 | Wako | #019-19741, RRID: AB_839504 |
| Hamster monoclonal anti-IL1B | Biolegend | #503513, RRID: AB_2814399 |
| Goat polyclonal anti-IL1R1 | R&D Systems | #AF771, RRID: AB_355587 |
| Mouse monoclonal anti-KI67 | BD PharMingen | #550609; RRID: AB_393778 |
| Rat monoclonal anti-KI67 | ThermoFisher | #14-5698-80; RRID: AB_10853185 |
| Goat polyclonal anti-LYVE1 | R&D Systems | #AF2125, RRID: AB_2297188 |
| Rabbit polyclonal anti-MRC1/CD206 | Abcam | #ab64693, RRID: AB_1523910 |
| Goat polyclonal anti-NRP1 | R&D Systems | #AF566; RRID: AB_355445 |
| Rabbit polyclonal anti-PAX6 | Millipore | #ab2237, RRID: AB_1587367 |
| Rat monoclonal anti-PECAM1/CD31 | BD PharMingen | #550274, RRID: AB_393571 |
| Rabbit polyclonal anti-SHISA8 | Abcam | #ab188621 |
| Rat monoclonal anti-SPI1/PU.1 | Novus Bio | #MAB7124-SP |
| Rabbit polyclonal anti-SPIC | ThermoFisher | #PA5-67537, RRID: AB_2692238 |
| Rabbit anti-SLC40A1 | Alpha Diagnostic International | #MTP11-A, RRID: AB_1619475 |
| Mouse monoclonal anti-TUBB3 | Biolegend | #801202, RRID: AB_10063408 |
| Mouse monoclonal anti-Acetylated Tubulin | Sigma-Aldrich | T6793, RRID: AB_477585 |
| Rabbit polyclonal anti-VWF | ThermoFisher | #MA5-14029; RRID: AB_305689 |
| Rat monoclonal anti-WNT5A | R&D Systems | #MAB645; RRID: AB_10571221 |
| Mouse monoclonal anti-ZO1 | ThermoFisher | #33-9100; RRID: AB_2533147 |
| Rabbit polyclonal anti-5HT | Sigma-Aldrich | #S5545; RRID: AB_477522 |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase II | GIBCO | 17101-015 |
| TrypLE | Life Technologies | 12604 |
| LIVE/DEAD staining kit | ThermoFisher | L-3224 |
| 10% Tween-20 (used for TST) | VWR Teknova | T0710 |
| NP-40 Surfact-Amps Detergent Solution (used for NST) | ThermoFisher | 28324 |
| CHAPS, Molecular Biology Grade, (used for CST) | EMD Millipore | 220201-1GM |
| Nuclei EZ Prep Nuclei Isolation Kit | Sigma | NUC101-1KT |
| Critical commercial assays | ||
| 10X Genomics Single Cell 3′ Gel Bead Kit V1 | 10X Genomics | PN-120231 |
| 10X Genomics Single Cell 3′ Gel Bead Kit V2 | 10X Genomics | PN-120237 |
| 10X Genomics Single Cell 3′ Gel Bead Kit V3 | 10X Genomics | PN-1000075 |
| RNAscope Fluorescent Multiplex | ACD | #320851 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE168704 |
| Experimental models: Organisms/strains | ||
| Mouse: CD-1 (ICR) | Charles River Laboratories | Strain 022 |
| Mouse: C57BL/6J | Jackson Laboratory | 000664 |
| Mouse: C57BL/6 | Charles River Laboratories | N/A |
| Mouse: B6.129P2(Cg)-Cx3cr1tm1Litt/J | Jackson Laboratory | 005582 |
| Mouse: 129S4.Cg-E2f1Tg(Wnt1-cre)2Sor/J | Jackson Laboratory | 002137 |
| Mouse: Gt(ROSA) 26Sortm1(CAG-Brainbow2.1)Cle/J | Jackson Laboratory | 013731 |
| Oligonucleotides | ||
| mm-Axin2-C3 | ACD | 400331-C3 |
| mm-FoxJ1 | ACD | 317091 |
| mm-Ins2-C2 | ACD | 310751-C2 |
| mm-Rspo1 | ACD | 401991 |
| mm-Rspo2-C2 | ACD | 402001-C2 |
| mm-Rspo3-C3 | ACD | 402011-C3 |
| mm-Wnt1 | ACD | 401091 |
| mm-Wnt2b-C2 | ACD | 405031-C2 |
| mm-Wnt3a | ACD | 405041 |
| mm-Wnt5a-C3 | ACD | 316791-C3 |
| Software and algorithms | ||
| Cellranger | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome | N/A |
| CellPhoneDB | Efremova et al., 2020 | N/A |
| Seurat | https://github.com/satijalab/seurat | N/A |
| R | https://www.r-project.org/ | N/A |
| Other | ||
| Noyes Spring Scissors – Tungsten Carbide/Straight | Fine Science Tools | #15514-12 |
| Dounce Tissue Grinder Set | Sigma | #D8938-1SET |
ACKNOWLEDGEMENTS
We thank members of the Lehtinen, Regev, Andermann, Fleming, Hla, and Stevens labs, and L. Tsai for reagents and helpful discussions; C. Smillie, M. Shannon, H. Zucker, X. Adiconis, and M. Kumar, M. Ericsson and HMS EM Facility, W. Fowle and Northeastern EM Facility, the Flow Cytometry facility at the Broad Institute for assistance and advice; A. Hupalowska and L. Gaffney for illustrations. This work was supported by: Glenn/AFAR Postdoctoral Fellowship, Reagan Sloane Shanley Research Internship (N.D.), William Randolph Hearst Fellowship (N.D. and J.C.); NIH T32 HL110852 (J.C.); NSF Graduate Research Fellowship (F.B.S.); HHMI fellow of the Helen Hay Whitney Foundation (N.H.); Neuron Fund for Support of Science (23/2016) and Czech Science Foundation (GA17-16680S)(V.B.); the core facility CELLIM supported by the Czech-BioImaging large RI project (LM2018129 funded by MEYS CR); RVO 68378050 from the Czech Academy of Sciences and the LM2018126l CZ.1.05/2.1.00/19.0395, CZ.02.1.01/0.0/0.0/16_013/0001789 grants by MEYS and ERDF, OP RDI to Czech Centre for Phenogenomics; Israel Science Foundation (ISF) research grant no. 1709/19, Myers Foundation, Goren-Khazzam (N.H.); Boston Children’s Hospital-Broad Institute Collaboration Grant, Pediatric Hydrocephalus Foundation, NIH R01 NS088566 (M.K.L.); the New York Stem Cell Foundation (M.K.L. and F.Z.); BCH IDDRC 1U54HD090255; and the Klarman Cell Observatory. F.Z. and A.R. are Investigators of the Howard Hughes Medical Institute. F.Z. and M.K.L. are New York Stem Cell Foundation – Robertson Investigators.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Aviv Regev is a co-founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas, and was an SAB member of ThermoFisher Scientific, Syros Pharmaceuticals, Neogene Therapeutics and Asimov until July 31, 2020. From August 1, 2020, A.R. is an employee of Genentech. From August 16, 2020, R.H.H. is an employee of Immunai. A.R., R.H.H., N.H., M.K.L., N.D., and A.J. are co-inventors on a provisional patent application filed by the Broad Institute relating to this manuscript.
REFERENCES
- Angerer P, Haghverdi L, Büttner M, Theis FJ, Marr C, and Buettner F (2016). Destiny: Diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243. [DOI] [PubMed] [Google Scholar]
- Awatramani R, Soriano P, Rodriguez C, Mai JJ, and Dymecki SM (2003). Cryptic boundaries in roof plate and choroid plexus identified by intersectional gene activation. Nat. Genet 35, 70–75. [DOI] [PubMed] [Google Scholar]
- Awatramanil R, Soriano P, Rodriguez C, Mai JJ, and Dymecki SM (2003). Cryptic boundaries in roof plate and choroid plexus identified by intersectional gene activation. Nat. Genet 35, 70–75. [DOI] [PubMed] [Google Scholar]
- Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, et al. (2014). Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science (80-. ). 346, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–47. [DOI] [PubMed] [Google Scholar]
- Bell SM, Schreiner CM, Wert SE, Mucenski ML, Scott WJ, and Whitsett JA (2008). R-spondin 2 is required for normal laryngeal-tracheal, lung and limb morphogenesis. Development 135, 1049–1058. [DOI] [PubMed] [Google Scholar]
- Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, and Gu C (2014). Mfsd2a is critical for the formation and function of the blood–brain barrier. Nature 509, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielecki P, Riesenfeld SJ, Hutter JC, Torlai Triglia E, Kowalczyk MS, Ricardo-Gonzalez RR, Lian M, Amezcua Vesely MC, Kroehling L, Xu H, et al. (2021). Skin-resident innate lymphoid cells converge on a pathogenic effector state. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blei D, Ng A, and Jordan M (2003). Latent Dirichlet Allocation. [Google Scholar]
- van den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson-Maduro J, Baron CS, Robin C, and van Oudenaarden A (2017). Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936. [DOI] [PubMed] [Google Scholar]
- Brooks ER, and Wallingford JB (2014). Multiciliated Cells. Curr. Biol 24, R973–R982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, van der Meer F, Kallio K, Kaya T, Anastasina M, et al. (2020). Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science (80-. ). 370, eabd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JT, Lehtinen MK, and Sive H (2016). Zebrafish cerebrospinal fluid mediates cell survival through a retinoid signaling pathway. Dev. Neurobiol 76, 75–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang P-T, and McMahon AP (1999). Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 397, 617–621. [DOI] [PubMed] [Google Scholar]
- Cui J, Shipley FB, Shannon ML, Alturkistani O, Dani N, Webb MD, Sugden AU, Andermann ML, and Lehtinen MK (2020). Inflammation of the Embryonic Choroid Plexus Barrier following Maternal Immune Activation. Dev. Cell 55(5), 617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damkier HH, Brown PD, and Praetorius J (2013). Cerebrospinal Fluid Secretion by the Choroid Plexus. Physiol. Rev 93, 1847–1892. [DOI] [PubMed] [Google Scholar]
- DeSisto J, O’Rourke R, Jones HE, Pawlikowski B, Malek AD, Bonney S, Guimiot F, Jones KL, and Siegenthaler JA (2020). Single-Cell Transcriptomic Analyses of the Developing Meninges Reveal Meningeal Fibroblast Diversity and Function. Dev. Cell 54, 43–59.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey KK, Hsiao CJ, and Stephens M (2017). Visualizing the structure of RNA-seq expression data using grade of membership models. PLOS Genet. 13, e1006599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulken BW, Leeman DS, Boutet SC, Hebestreit K, and Brunet A (2017). Single-Cell Transcriptomic Analysis Defines Heterogeneity and Transcriptional Dynamics in the Adult Neural Stem Cell Lineage. Cell Rep. 18, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Lipson D, Yogev S, and Yakhini Z (2007). Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol 3, 0508–0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc 15, 1484–1506. [DOI] [PubMed] [Google Scholar]
- Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, Kneen R, Defres S, Sejvar J, and Solomon T (2020). Neurological associations of COVID-19. Lancet Neurol. 19(9):767–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fame RM, and Lehtinen MK (2020). Emergence and Developmental Roles of the Cerebrospinal Fluid System. Dev. Cell 52, 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SJ, Marioni JC, and Babadi M (2019). CellBender remove-background: A deep generative model for unsupervised removal of background noise from scRNA-seq datasets. BioRxiv 791699. [Google Scholar]
- Gaublomme JT, Li B, McCabe C, Knecht A, Yang Y, Drokhlyansky E, Van Wittenberghe N, Waldman J, Dionne D, Nguyen L, et al. (2019). Nuclei multiplexing with barcoded antibodies for single-nucleus genomics. Nat. Commun 10(1), 2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghersi-Egea J-F, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, and Engelhardt B (2018). Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol. 135, 337–361. [DOI] [PubMed] [Google Scholar]
- Girvan M, and Newman MEJ (2002). Community structure in social and biological networks. Proc. Natl. Acad. Sci. U. S. A 99, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, et al. (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan N, and Zervas M (2012). Wnt1 expression temporally allocates upper rhombic lip progenitors and defines their terminal cell fate in the cerebellum. Mol. Cell. Neurosci 49, 217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Büttner M, Wolf FA, Buettner F, and Theis FJ (2016). Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848. [DOI] [PubMed] [Google Scholar]
- Haldar M, Kohyama M, So AY-L, KC W, Wu X, Briseño CG, Satpathy AT, Kretzer NM, Arase H, Rajasekaran NS, et al. (2014). Heme-Mediated SPI-C Induction Promotes Monocyte Differentiation into Iron-Recycling Macrophages. Cell 156, 1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. (2019). Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50, 253–271.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrnberger L, Seitz R, Kuespert S, Bösl MR, Fuchshofer R, and Tamm ER (2012). Lack of endothelial diaphragms in fenestrae and caveolae of mutant Plvap-deficient mice. Histochem. Cell Biol 138, 709–724. [DOI] [PubMed] [Google Scholar]
- Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Rita Pombo Antunes A, De Prijck S, Vandamme N, De Schepper S, Van Isterdael G, Scott CL, et al. (2019). A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci 22(6):1021–1035. [DOI] [PubMed] [Google Scholar]
- Huang X, Ketova T, Fleming JT, Wang H, Dey SK, Litingtung Y, and Chiang C (2009). Sonic hedgehog signaling regulates a novel epithelial progenitor domain of the hindbrain choroid plexus. Development 136, 2535–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson AJ (1960). The development of the vascular pattern of the choroid plexus of the lateral ventricles. J. Comp. Neurol 115, 171–186. [DOI] [PubMed] [Google Scholar]
- Hunter NL, and Dymecki SM (2007). Molecularly and temporally separable lineages form the hindbrain roof plate and contribute differentially to the choroid plexus. Development 134, 3449–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordão MJC, Sankowski R, Brendecke SM, Sagar, Locatelli G, Tai Y-H, Tay TL, Schramm E, Armbruster S, Hagemeyer N, et al. (2019a). Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363, eaat7554. [DOI] [PubMed] [Google Scholar]
- Kaiser K, Gyllborg D, Procházka J, Salašová A, Kompaníková P, Molina FL, Laguna-Goya R, Radaszkiewicz T, Harnoš J, Procházková M, et al. (2019). WNT5A is transported via lipoprotein particles in the cerebrospinal fluid to regulate hindbrain morphogenesis. Nat. Commun 10(1):1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kania A, and Klein R (2016). Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol 17, 240–256. [DOI] [PubMed] [Google Scholar]
- Karimy JK, Zhang J, Kurland DB, Theriault BC, Duran D, Stokum JA, Furey CG, Zhou X, Mansuri MS, Montejo J, et al. (2017). Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat. Med 23, 997–1003. [DOI] [PubMed] [Google Scholar]
- Keep RF, and Jones HC (1990). A morphometric study on the development of the lateral ventricle choroid plexus, choroid plexus capillaries and ventricular ependyma in the rat. Dev. Brain Res 56, 47–53. [DOI] [PubMed] [Google Scholar]
- Kinzel B, Pikiolek M, Orsini V, Sprunger J, Isken A, Zietzling S, Desplanches M, Dubost V, Breustedt D, Valdez R, et al. (2014). Functional roles of Lgr4 and Lgr5 in embryonic gut, kidney and skin development in mice. Dev. Biol 390, 181–190. [DOI] [PubMed] [Google Scholar]
- Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, Frazier WA, Murphy TL, and Murphy KM (2009). Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 457, 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausgruber T, Fortelny N, Fife-Gernedl V, Senekowitsch M, Schuster LC, Lercher A, Nemc A, Schmidl C, Rendeiro AF, Bergthaler A, et al. (2020). Structural cells are key regulators of organ-specific immune responses. Nature 583, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, and Alvarez-Buylla A (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci 32, 149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, et al. (1997). Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51. [DOI] [PubMed] [Google Scholar]
- Lacar B, Linker SB, Jaeger BN, Krishnaswami S, Barron J, Kelder M, Parylak S, Paquola A, Venepally P, Novotny M, et al. (2016). Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun 7:11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, and Storey JD (2012). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, et al. (2011). The Cerebrospinal Fluid Provides a Proliferative Niche for Neural Progenitor Cells. Neuron 69, 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Lewis AE, Vasudevan HN, O’Neill AK, Soriano P, and Bush JO (2013). The widely used Wnt1-Cre transgene causes developmental phenotypes by ectopic activation of Wnt signaling. Dev. Biol 379, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE, et al. (2019). Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 101, 207–223.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Dziegielewska KM, VandeBerg JL, and Saunders NR (2010). Development of the lateral ventricular choroid plexus in a marsupial, Monodelphis domestica. Cerebrospinal Fluid Res. 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HY, Lim SY, Tan CK, Thiam CH, Goh CC, Carbajo D, Chew SHS, See P, Chakarov S, Wang XN, et al. (2018). Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity 49, 326–341.e7. [DOI] [PubMed] [Google Scholar]
- Lindvall M, Edvinsson L, and Owman C (1978). Sympathetic nervous control of cerebrospinal fluid production from the choroid plexus. Science 201, 176–178. [DOI] [PubMed] [Google Scholar]
- Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, Sanes JR, and Lichtman JW (2007). Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 450, 56–62. [DOI] [PubMed] [Google Scholar]
- Louvi A, and Wassef M (2000). Ectopic Engrailed 1 expression in the dorsal midline causes cell death, abnormal differentiation of circumventricular organs and errors in axonal pathfinding. Development 127, 4061–4071. [DOI] [PubMed] [Google Scholar]
- Lun MP, Monuki ES, and Lehtinen MK (2015a). Development and functions of the choroid plexus–cerebrospinal fluid system. Nat. Rev. Neurosci. 16, 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun MP, Johnson MB, Broadbelt KG, Watanabe M, Kang YJ, Chau KF, Springel MW, Malesz A, Sousa AM, Pletikos M, et al. (2015b). Spatially heterogeneous choroid plexus transcriptomes encode positional identity and contribute to regional CSF production. J Neurosci 35, 4903–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Maaten L, and Hinton G (2008). Visualizing Data using t-SNE. [Google Scholar]
- Mancini M, Greco A, Tedeschi E, Palma G, Ragucci M, Bruzzone MG, Coda ARD, Torino E, Scotti A, Zucca I, et al. (2015). Head and neck veins of the mouse. A magnetic resonance, micro computed tomography and high frequency color Doppler ultrasound study. PLoS One 10(6):e0129912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martik ML, and Bronner ME (2017). Regulatory Logic Underlying Diversification of the Neural Crest. Trends Genet. 33, 715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew RS, Mullan H, Blusztajn JK, and Lehtinen MK (2016). Comment on "Multiple repressive mechanisms in the hippocampus during memory formation". Science 353, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazucanti CH, Liu QR, Lang D, Huang N, O’Connell JF, Camandola S, and Egan JM (2019). Release of insulin produced by the choroids plexis is regulated by serotonergic signaling. JCI Insight 4(23):e131682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrak D, Levitin HM, Delgado AC, Crotet V, Yuan J, Chaker Z, Silva-Vargas V, Sims PA, and Doetsch F (2019). Single-Cell Analysis of Regional Differences in Adult V-SVZ Neural Stem Cell Lineages. Cell Rep. 26, 394–406.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, et al. (2018). A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muus C, Luecken M, Eraslan G, Waghray A, Heimberg G, Sikkema L, Kobayashi Y, Vaishnav ED, Subramanian A, Smilie C, et al. (2020). Integrated analyses of single-cell atlases reveal age, gender, and smoking status associations with cell type-specific expression of mediators of SARS-CoV-2 viral entry and highlights inflammatory programs in putative target cells. BioRxiv 2020.04.19.049254. [Google Scholar]
- Myung J, Schmal C, Hong S, Tsukizawa Y, Rose P, Zhang Y, Holtzman MJ, De Schutter E, Herzel H, Bordyugov G, et al. (2018). The choroid plexus is an important circadian clock component. Nat. Commun 9, 1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paré A, Mailhot B, Lévesque SA, Juzwik C, Doss PMIA, Lécuyer MA, Prat A, Rangachari M, Fournier A, and Lacroix S (2018). IL-1β enables CNS access to CCR2hi monocytes and the generation of pathogenic cells through GM-CSF released by CNS endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 115, E1194–E1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L, Albecka A, Mallery DL, Kellner MJ, Paul D, Carter AP, James LC, and Lancaster MA (2020a). SARS-CoV-2 Infects the Brain Choroid Plexus and Disrupts the Blood-CSF Barrier in Human Brain Organoids. Cell Stem Cell. 27(6):951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L, Bonfio C, Chadwick J, Begum F, Skehel M, and Lancaster MA (2020b). Human CNS barrier-forming organoids with cerebrospinal fluid production. Science. 369(6500):eaaz5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, and Donnelly P (2000). Inference of Population Structure Using Multilocus Genotype Data. Genetics 155(2):945–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, and Sallusto F (2009). C-C chemokine receptor 6–regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol 10, 514–523. [DOI] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018a). Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders NR, Dziegielewska KM, Møllgård K, and Habgood MD (2018b). Physiology and molecular biology of barrier mechanisms in the fetal and neonatal brain. J. Physiol 596, 5723–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebinger G, Shu J, Tabaka M, Cleary B, Subramanian V, Solomon A, Gould J, Liu S, Lin S, Berube P, et al. (2019). Optimal-Transport Analysis of Single-Cell Gene Expression Identifies Developmental Trajectories in Reprogramming. Cell 176, 928–943.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, and Baruch K (2014). The resolution of neuroinflammation in neurodegeneration: Leukocyte recruitment via the choroid plexus. EMBO J. 33, 7–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PT, Stratton JA, Stykel MG, Abbasi S, Sharma S, Mayr KA, Koblinger K, Whelan PJ, and Biernaskie J (2018). Single-Cell Transcriptomics and Fate Mapping of Ependymal Cells Reveals an Absence of Neural Stem Cell Function. Cell 173, 1045–1057.e9. [DOI] [PubMed] [Google Scholar]
- Shannon ML, Fame RM, Chau KF, Dani N, Calicchio ML, Géléoc GS, Lidov HGW, Alexandrescu S, and Lehtinen MK (2018). Mice Expressing Myc in Neural Precursors Develop Choroid Plexus and Ciliary Body Tumors. Am. J. Pathol 188, 1334–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, Kim K-W, Klein E, Kalchenko V, Bendel P, et al. (2013). Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 38, 555–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley FB, Dani N, Xu H, Deister C, Cui J, Head JP, Sadegh C, Fame RM, Shannon ML, Flores VI, et al. (2020). Tracking Calcium Dynamics and Immune Surveillance at the Choroid Plexus Blood-Cerebrospinal Fluid Interface. Neuron. 108(4):623–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuangshoti S, and Netsky MG (1966). Histogenesis of choroid plexus in man. Am. J. Anat 118, 283–315. [DOI] [PubMed] [Google Scholar]
- Silva-Vargas V, Maldonado-Soto ARR, Mizrak D, Codega P, and Doetsch F (2016). Age-Dependent Niche Signals from the Choroid Plexus Regulate Adult Neural Stem Cells. Cell Stem Cell 19, 643–652. [DOI] [PubMed] [Google Scholar]
- Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, et al. (2016). A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 166, 1500–1511.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, et al. (2010). Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143, 134–144. [DOI] [PubMed] [Google Scholar]
- Tong Y, Merino D, Nimmervoll B, Gupta K, Wang YD, Finkelstein D, Dalton J, Ellison DW, Ma X, Zhang J, et al. (2015). Cross-Species Genomics Identifies TAF12, NFYC, and RAD54L as Choroid Plexus Carcinoma Oncogenes. Cancer Cell 27, 712–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. [DOI] [PubMed] [Google Scholar]
- Visel A, Thaller C, and Eichele G (2004). GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 32, D552–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang L-C, Bui S, Nielson A, Wu X, Vo H-T, Ma X-J, and Luo Y (2012). RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn 14, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilting J, and Christ B (1989). An experimental and ultrastructural study on the development of the avian choroid plexus. Cell Tissue Res. 255, 487–494. [DOI] [PubMed] [Google Scholar]
- Wingate RJT (2001). The rhombic lip and early cerebellar development. Curr. Opin. Neurobiol 11, 82–88. [DOI] [PubMed] [Google Scholar]
- Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, Liu C, and Yang C (2020). Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain. Behav. Immun 87, 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Fame RM, Sadegh C, Sutin J, Naranjo C, Della Syau, Cui J, Shipley FB, Vernon A, Gao F, et al. (2021). Choroid plexus NKCC1 mediates cerebrospinal fluid clearance during mouse early postnatal development. Nat. Commun 12(1):447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AC, Kern F, Losada PM, Maat CA, Schmartz G, Fehlmann T, Schaum N, Lee DP, Calcuttawala K, Vest RT, et al. (2020). Broad transcriptional dysregulation of brain and choroid plexus cell types with COVID-19. BioRxiv 2020.10.22.349415. [Google Scholar]
- Zhu L, Stein LR, Kim D, Ho K, Yu G-Q, Zhan L, Larsson TE, and Mucke L (2018). Klotho controls the brain-immune system interface in the choroid plexus. Proc. Natl. Acad. Sci. U. S. A 115, E11388–E11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, related to Figure 1. Cellular markers, identity and organization within the ChP. (A) Embryonic cell quality of scRNA-seq. Distribution of number of transcripts (log10(nUMI), left) genes (log10(nGene), right) across cell types, grouped and colored by batch. (B) Proportions of cell types captured from each ChP by embryonic scRNA-seq. Fractions (y axis) of each major cell type identified (x axis) per ventricle and batch (color). (C) Expression of cell type markers by scRNA-seq of embryonic ChP. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) across marker genes (columns) for each cell type cluster (rows). (D) Proliferating cells across cell types in the ChP. Left: t-SNE (as in Figure 1C) of cell profiles (dots), with cells classified as cycling colored black (STAR Methods). Right: Mean fraction of cycling cells (y axis) in each cell type (x axis), and in each batch replicate (dots). Line: standard deviation (S.D.). (E) A proliferating cell niche in the ChP. Left: Proliferation marker KI67 staining (magenta) of whole explant of LV ChP. Dotted brackets mark regions of interest that are proximal, medial and distal relative to the ventral margin of the ChP near the brain. Double headed arrows mark Anterior (A)/Posterior (P) and Dorsal (D)/Ventral (V) axes. Scale bar = 500 μm. Right: Percent of proliferating cells (KI67+) out of all cells in proximal, medial and distal regions of the LV ChP (n=4, Scale bar = 500 μm; one-way ANOVA, ** p<0.01. Error bars indicate S.E.M.). (F) snRNA-seq nuclei library quality. Distribution of number of genes (nGene, left) and transcripts (nUMI, right) across cell types, grouped and colored by age. (G) Proportions of cells captured from each ChP and age by snRNA-seq. Fractions (y axis) of each major cell type identified (x axis) per ventricle across ages (color). (H) Single nuclei batch across cell clusters. UMAP embedding (as in Figure 1F) of single nucleus profiles colored by batch, showing uniform distribution of batches across clusters. (I) A random forest classifier assigns cell types across scRNA-seq and snRNA-seq datasets in embryo ChP. Fraction of cells (color and circle size) per cell type in embryo scRNA-seq (rows) that were mapped to embryo snRNA-seq cell types (columns) (STAR Methods). Columns add up to 1. (J) Resident neurons in embryonic LV ChP. Top: Representative neuron co-stained with anti-serotonin (5-HT) (red) and anti-TUBB3 (green) and nuclei counterstained with Hoechst in embryonic ChP (Scale bar = 10 μm). Bottom: Split channel showing anti-serotonin staining alone. (K) Resident neurons in adult and aged 4V ChP. Representative image of neuronal cell bodies stained with anti-TUBB3 (green) and nuclei (Hoechst) in adult (top) and aged (bottom) ChP. Scale bar = 10 μm. (L) Resident glial cells in adult and aged LV ChP. Representative image of glial cells stained with anti-GFAP (red) antibodies in LV ChP in adult (left) and aged (right). Scale bar = 20 μm.
Figure S2, related to Figure 2. Neuronal and glial cell types across ages and ventricles. (A) Glial and neuronal cell markers. T-SNE of neuronal and glia-like cell profiles from scRNA-seq in embryonic ChP (dots, as in Figure 2A), colored by expression (log2(TP10K+1)) of Tubb3 (neuronal marker, top) and Slc1a3 (glial marker, bottom). (B) Neuronal/glia-like subset markers and neuropeptides. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected genes (columns) in neuronal and glia-like subsets (rows). (C) Organization of the developing 3V ChP. In situ hybridization images (Allen Brain Atlas and Genepaint) of marker genes for epithelial cells (Ttr), progenitor cells (Rspo3) and developing pineal gland clusters (Krt19, Crx), localizing the progenitors adjacent to the pineal gland and choroid plexus epithelial cells. Rightmost panel: Schematic of the 3V structure. (D) Neuro-glial subclusters. UMAP of neuronal and glia-like nucleus profiles across age groups from snRNA-seq from the three ventricles, colored by age (left) and cluster membership (right). (E) Neuro-glial marker gene expression across snRNA-seq clusters. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected genes (columns) across clusters (rows). (F) RNA Velocity analysis of inferred differentiation trajectory of neuronal and epithelial cells from a predicted common progenitor pool. Diffusion map embedding of cells (dots) from embryonic 3V ChP (scRNA-seq) (as in Figure 2B). Arrows mark the direction of cell transition as inferred by RNA velocity algorithm (La Manno et al., 2018). (G) Wnt1 restricted expression in the progenitor pool. Diffusion map embedding of cells as in F colored by log2(TP10K+1) expression level of Wnt1. (H-K) Distribution of progenitor domains near the root and base of the LV ChP (dashed lines). Tissue sections of embryonic LV ChP labelled with progenitor markers (arrowheads): immunostained for PAX6 (H), or RNA smFISH for R-spondin genes (Rspo1, Rspo2, and Rspo3, I, Inset: Cells expressing all three markers, Scale bar = 20 μm), Wnt genes (Wnt2b, Wnt3a and Wnt5a, J), and WNT-mediated signaling (Axin2, Wnt5a, K). Scale bar = 50 μm for (H-K). (L) Reduced proportion of ciliogenic and cycling epithelial cells in adult and aged ChP compared to embryos. Fraction of ciliogenic (left) and cycling (right) epithelial cells out of the total epithelial cells, in embryo, adult and aged ChP. P-value of T-test on the proportion of cycling or ciliogenic cells comparing between every pair of age groups, *p<0.05.
Figure S3, related to Figure 3. Regionalized transcriptional programs in epithelial cell populations. (A) Regionalized expression programs of epithelial cells in embryonic ChP from scRNA-seq agrees with bulk RNA-seq. Differentially expressed genes between LV and 4V in bulk RNAseq (Lun et al., 2015a) (Left: up-regulated in 4V ChP. Right: up-regulated in LV ChP) averaged across single cells in 4V ChP (x axis) and LV ChP (y axis). Red line indicates x=y. All genes show a coordinated difference between the datasets. (B) Topic modeling of embryonic epithelial cells captures distinct transcriptional programs. Embryonic epithelial transcriptional programs associated with cell cycle (Topics 4 and 9), ciliogenesis (Topic 24) and immediate early gene expression (Topic 19). For each topic, a bar plot of topic scores is shown for top ranked genes (left), and t-SNE of the cell profiles (as in Figure 3A) colored by topic’s weight per cell (right). (C) Ventricle associated transcriptional programs in embryonic epithelial cells. Empirical cumulative distribution (y axis) of topic’s weight (x axis), grouped and colored by ventricle. (D) Immediate early gene (IEG) expression across all cells is higher in scRNA-seq compared to snRNA-seq data. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected genes (columns) in our scRNA-seq and snRNA-seq data (rows). (E) snRNA-seq analysis recapitulates regionalized gene expression as compared to scRNA-seq. Distribution of expression level (y axis) of regionalization marker genes across ventricles (colors) in embryonic epithelial cells. (F) Rostral and caudal gene expression pattern in embryonic 4V ChP epithelium shown in situ. Top: ISH images from Genepaint showing regionalization of Shh and Serpinb1a (Scale bar = 100 μm). Bottom: T-SNE of epithelial cell profiles (dots), colored by log2(TP10K+1) expression levels of genes shown in Figure 3E. (G-H) Maturation-dependent shifts in transporter (G) and secreted factor (H) expression. Heatmap of gene expression from snRNA-seq across ages (left color bar) and ventricles (right color bar). Scaled across all nuclei (columns) by z-score. Examples of genes marked on the bottom. (I) Age- and ventricle-dependent transcriptional programs in epithelial cells by snRNA-seq. Distribution of expression level (y axis) of regionalized genes (Tns4 and Slit2) and age-dependent genes (Gpc3 and Prlr) across ages (top row x axis) and ventricles (colors). (J-K) Validations of ventricle- and age- dependent expression by ISH images from Allen Brain Atlas (Lein et al., 2007). Regionalization and relative expression (color) of Tns4 (Top, J) and Slit2 (Bottom, J) genes in adult LV, 3V and 4V ChP. Relative expression in embryo and adult of Gpc3 (Top, K) and Prlr (Bottom, K) in LV ChP. Scale bar = 100 μm.
Figure S4, related to Figure 4. Regionalized transcriptional programs in mesenchymal cell populations. (A) Embryonic mesenchymal cells partition into fibroblast and mural cells (pericytes and smooth muscle cells). T-SNE of mesenchymal cell profiles (dots, as in Figure 4A), colored by log2(TP10K+1) expression level of marker genes for: pericytes (Des), fibroblasts (Pdgfra) and smooth muscle actin positive cells (Acta2). (B) Topic modeling of embryonic mesenchymal cells captures distinct transcriptional programs. Embryonic mesenchymal transcriptional programs associated with: cell cycle (Topics 12,16) and LV and 3V fibroblasts shared program (Topic 8). For each topic, a bar plot is shown of topic scores for top ranked genes (left), and t-SNE of the cell profiles (as in Figure 4B) colored by topic’s weight per cell (right). (C) Ventricle associated transcriptional programs in mesenchymal cells. Empirical cumulative distribution (y axis) of topic’s weight (x axis), grouped and colored by ventricle. (D) Regionalized embryonic mesenchymal transcriptional programs. In situ hybridization (Genepaint) in sagittal sections of ChP for genes with ventricle specific enrichment in fibroblasts of the LV (Lox), 3V (Adamts2, Fbln1), 4V (Hhip), pericyte marker (Rgs5) and 3V/meningeal fibroblast markers (Fbln1). Scale bar for LV = 50 μm; 3V and 4V = 10 μm. (E) In situ hybridization from Genepaint and Allen Brain Atlas of sagittal sections of embryonic 3V ChP showing expression of fibroblast markers (Apod, Crym, Slc6a13) shared by meninges and 3V ChP stroma. Bottom right: Composite model identifying organization of 3V/meningeal fibroblasts in the developing brain. Scale bar = 100 μm. (F) Regionalized expression of extracellular matrix (ECM) protein in mesenchymal cells. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of ECM genes within top 50 features (rows) across ventricles (columns). (G) Mesenchymal marker expression across mesenchymal subtype clusters. Dot plots as in F of marker genes (columns) across mesenchymal sub-types (rows). (H-I) Regionalized expression of receptors (H) and transporters (I). Heatmap of gene expression from snRNA-seq across ages (right color bar) and ventricles (left color bar). Scaled across all nuclei (columns) by z-score. (J) SARS-CoV-2 cell entry receptor Ace2 gene expression in mesenchymal cells (including pericytes) across ages. UMAP of mesenchymal single nuclei profiles (snRNA-seq as in Figure 4C), colored by log2(TP10K+1) expression level of Ace2. (K) Distribution of ACE2 expression in fibroblast cells across age. Co-staining of ChP tissue sections in embryo (left), adult (middle) and aged (right) with anti-ACE2 (green) and stromal cell marker anti-COL1A1 (red) in 4V ChP. Arrowheads denote ACE2 and COL1A1 positive staining cells. Horizontal arrows denote ACE2 positive COL1A1 negative cells. Scale bar = 20 μm.
Figure S5, related to Figure 5. Cytokine and chemokine expression across ChP immune cells. (A-B) Gene expression of cytokines and chemokines across embryonic immune cell subsets. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of cytokines (A, columns) and chemokines (B, columns) in immune cell subsets (rows) from scRNA-seq in embryonic ChP across all ventricles. (C) SPIC expressing macrophages in embryonic ChP stromal space. LV ChP explants from Cx3cr1+/GFP mice stained with anti-SPIC (magenta, white arrowhead). Scale bar = 20μm. (D) Correlated gene expression in Spic expressing embryonic macrophages. Partial correlation (normalized for nUMI, y axis) between gene expression level and diffusion component 2 score (DC2, from diffusion map in Figure 5C) per cell. Marking gene names of highest correlated (e.g. Spic, Hmox1, Fth1) and anticorrelated genes with DC2. (E) Age and ventricle specific enrichment of immune cell populations. UMAP of snRNA-seq immune cell profiles (dots, as in Figure 5H) color coded by age (left) and ventricle (right). (F) Lymphoid populations. Left: UMAP of snRNA-seq immune cell profiles (dots, as in Figure 5H) color coded by marking T/B cell populations (blue). Right: UMAP of the T/B population colored by log2(TP10K+1) expression level of marker genes identifying T and B cell sub-populations. (G) Macrophage tiling patterns retained in adult and aged ChP. Co-staining of LV ChP explants for anti-PECAM1 (endothelial marker) and anti-IBA1 (macrophage marker) in adult (left two panels) and aged (right two panels). Scale bar = 10 μm. (H) Differentially expressed BAM genes (Van Hove et al., 2019) are age-dependent in the ChP. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected genes (columns) in embryonic, adult and aged macrophage cells (rows). (I) Aging associated shifts in gene expression. Bar plots for top enriched pathways (hypergeometric test −log10(FDR), x axis) upregulated (left) or down-regulated (right) in aged macrophage cells. (J) Aged stromal macrophages express CD74 protein. Tissue sections of aged 4 V ChP co-stained for anti-CD74 and anti-IBAl (macrophage marker). Arrowheads denote CD74 and IBA1 positive staining cells. Scale bar = 10 μm.
Figure S6, related to Figure 6. Molecular identities of ChP endothelial cells. (A) Arterial and venous transcriptional programs in embryonic endothelial cells (scRNA-seq data). T-SNE (as in Figure 6A) of endothelial cell profiles (dots), colored by signature scores for arterial (left) and venous (right) gene sets (gene signatures taken from (Vanlandewijck et al., 2018)). (B) Ventricle associated transcriptional programs in embryonic endothelial cells. Empirical cumulative distribution (y axis) of topic’s weight (x axis), grouped and colored by ventricle (scRNA-seq data). (C) Blood brain barrier markers in endothelial cells across age. UMAP of endothelial nucleus profiles from snRNA-seq (as in Figure 6D), colored by log2(TP10K+1) expression level of BBB marker (Cldn5), fenestrated vessel marker (Plvap), capillary marker (Esm1) and venous marker (Nr2f2). (D) Adult ChP capillaries express ESM1 protein. LV ChP explant co-stained with endothelial marker (anti-PECAM1, green) and capillary marker (anti-ESM1, magenta). Scale bar = 20 μm. (E) Fenestrae in embryonic and adult ChP blood vessels. Transmission electron microscopy image of developing fenestrae in the embryonic vessels (top, black arrow heads), and of the well-defined fenestrae in the adult endothelia (bottom, black arrow heads). Scale bar = 50 nm. (F) Venous signature enrichment in 3V ChP across all ages. Violin plot shows distribution of vein signature (defined as in A) by z-score in embryo, adult and aged across ventricles (colors). (G) Diversity of endothelial cells across ventricles. UMAP of snRNA-seq of endothelial cell profiles across ages (as in C), colored by ventricle. (H) Vein of Galen organization adjacent to 3V ChP. In situ hybridization for endothelial marker Flt1 and venous marker Nr2f2 (Allen brain Atlas) shows location of intracerebral vessel proximal to the 3V ChP (Scale bar = 100 μm) that is a likely candidate of a regionalized endothelial cell population. (I) BBB organization in the aged ChP. Whole explant co-staining for anti-CLDN5 (green) and anti-PECAM1 (red), showing CLDN5-positive staining endothelial cells continuous with brain vessels (arrow) that vascularize the ChP. Scale bar = 500 μm.
Figure S7, related to Figure 7. Mesenchymal, endothelial and immune cells contribute to cellular cross-talk in ChP. (A) Directional ligand-receptor interaction networks across cell types. Number of statistically significant (color bar, as estimated by CellPhoneDB, Star Methods) ligand (rows) receptor (columns) between pairs of ChP cell types, across ventricles (LV,3V,4V, from left to right), in embryonic nuclei (top) and embryonic cells (bottom, adult and aged nuclei in Figure 7A). Including ligand-receptor pairs marked as significantly expressed by CellPhoneDB (Efremova et al., 2020). Significant pairs of cell types based on a randomization test marked as: *p≤0.05, **p≤0.01, ***p≤0.001. (B) Number of significant interactions varies by cell type, ventricle and age. Bar plot of the percent of ligand-receptor pairs involving each cell type out of the total significant pairs, for each age (color) and ventricle (rows). (C) Examples of ligand-receptor pairs involving mesenchymal cells. Median expression level in expressing cells (color) and proportion of expressing cells (circle size) of selected ligands and receptors (columns) in each major cell population (rows), across all ventricles (color coded) and ages (top: embryonic; middle: adult; bottom: aged). (D) Directional ligand-receptor interaction networks across cell types. Number of significant ligand-receptors per pair of cell types for adult and aged ChP, across ventricles (as in Figure 7A). Significant pairs of cell types marked by asterisk, from a permutation test, constrained to preserve the total number of interactions per cell type, *p≤0.05, **p≤0.01.
Table S1, related to Figure 1, Figure 2 and Figure 5. Markers for cell types (Figure 1C), neuronal and glia-like clusters (Figure 2A) and immune clusters (Figure 5A).
Table S2, related to Figure 3B. Top 50 genes in each topic revealed by topic modeling on epithelial cells of developing ChPs.
Table S3, related to Figure 3I,J, Figure S3G,H. Epithelial DEG Supplemental table
Table S4, related to Figure 4B. Top 50 genes in each topic revealed by topic modeling on mesenchymal cells of developing ChPs.
Table S7, related to Figure 6A. Top 50 genes in each topic revealed by topic modeling on endothelial cells of developing ChPs.
Data Availability Statement
Raw and processed mouse sequencing data is available at https://portals.broadinstitute.org/single_cell and at the Gene Expression Omnbius (GEO) database GSE168704.