

Abstract
Both sepsis and treatment of cancer with chemotherapy are known to cause neurologic dysfunction. The primary defects seen in both groups of patients are neuropathy and encephalopathy; the underlying mechanisms are poorly understood. Analysis of preclinical models of these disparate conditions reveal similar defects in ion channel function contributing to peripheral neuropathy. The defects in ion channel function extend to the central nervous system where lower motoneurons are affected. In motoneurons the defect involves ion channels responsible for subthreshold currents that convert steady depolarization into repetitive firing. The inability to correctly translate depolarization into steady, repetitive firing has profound effects on motor function, and could be an important contributor to weakness and fatigue experienced by both groups of patients. The possibility that disruption of function, either instead of, or in addition to neurodegeneration, may underlie weakness and fatigue leads to a novel approach to therapy. Activation of serotonin (5HT) receptors in a rat model of sepsis restores the normal balance of subthreshold currents and normal motoneuron firing. If an imbalance of subthreshold currents also occurs in other central nervous system neurons, it could contribute to encephalopathy. We hypothesize that pharmacologically restoring the proper balance of subthreshold currents might provide effective therapy for both neuropathy and encephalopathy in patients recovering from sepsis or treatment with chemotherapy.
Keywords: motoneuron, motor neuron, excitability, action potential, sepsis, ICUAW, oxaliplatin, chemotherapy, subthreshold current, neuropathy
Introduction
At first glance, patients critically ill with sepsis and patients receiving chemotherapy for treatment of cancer have little in common. Sepsis, the “life-threatening organ dysfunction caused by a dysregulated host response to infection”(Singer and others 2016) is caused by acute infection. Patients receiving chemotherapy for cancer suffer from complications of treatment. There is no a priori reason to suspect similar mechanisms at play in the two conditions. However, both groups share difficulties with neurologic deficits during recovery (Fig. 1). The primary neurological deficit complicating recovery in both groups is neuropathy. As defined by the National Institutes of Health Common Terminology Criteria for Adverse Events (Version 5.0, 2017), peripheral sensory/motor neuropathy is “A disorder characterized by damage or dysfunction of the peripheral sensory/motor nerves” (https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf). While this definition does not infer mechanism, it is often assumed in reviews of neuropathy following sepsis and chemotherapy that the mechanism of dysfunction is degeneration of peripheral nerve axons (Dorsey and others 2019; Friedrich and others 2015; Miltenburg and Boogerd 2014). In this review we consider evidence suggesting that defective repetitive firing mechanisms in motoneurons contribute significantly to neurologic symptoms experienced by patients in these two different disorders.
Figure 1.
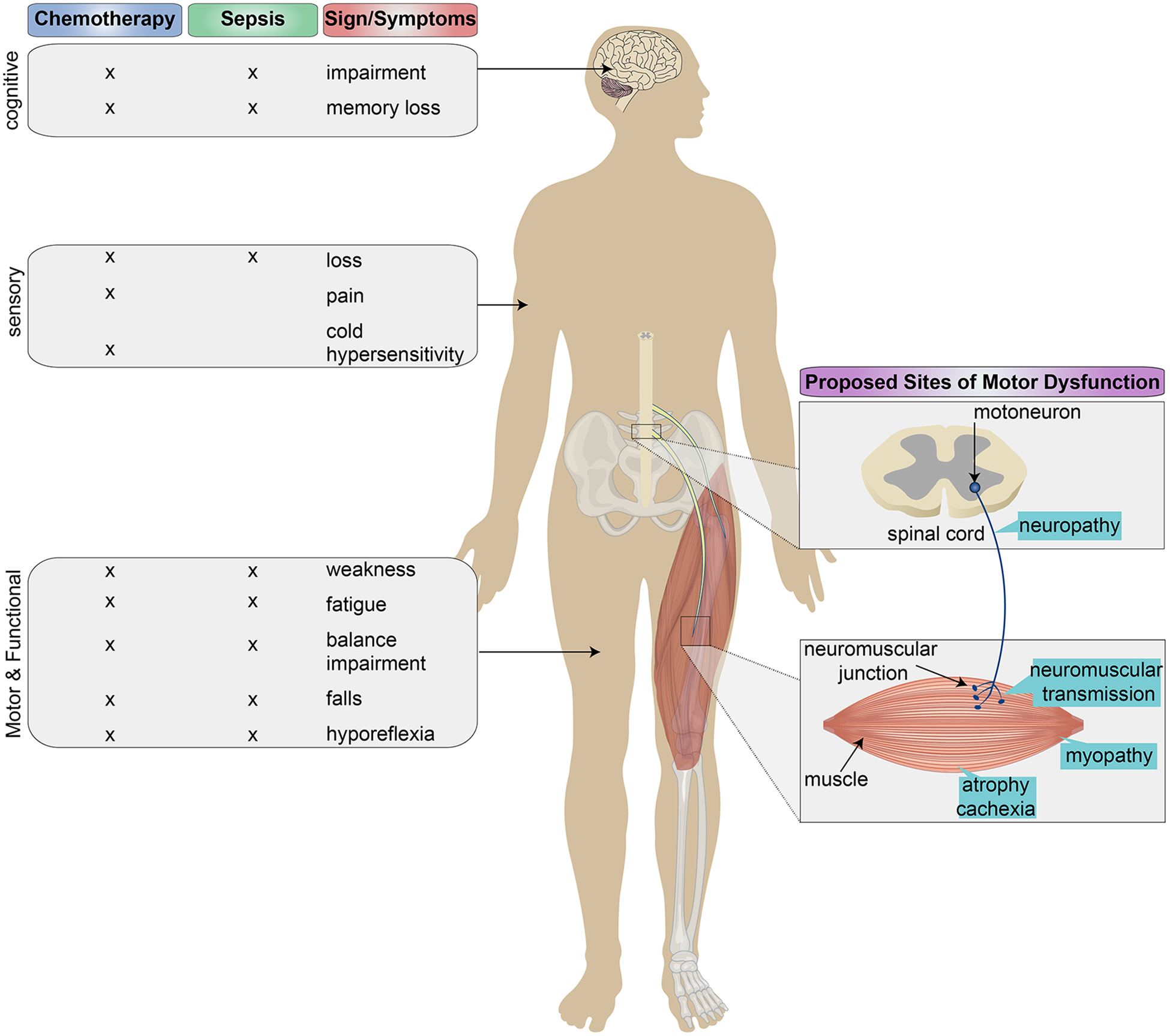
Neurological manifestations of sepsis-induced intensive care unit–acquired weakness (ICUAW) and chemotherapy induced neurotoxicity (CIN) with proposed loci of motor dysfunction. Neurological manifestations of CIN and ICUAW are diverse and can be classified by four major dimensions, including cognitive, sensory, autonomic (not shown), and motor/functional. Among the dimensions, motor and functional presentations such as muscle weakness, fatigue, and other behavioral impairments demonstrate the greatest overlap between ICUAW and CIN. Multiple sites of motor dysfunction, for example neuropathy, neuromuscular junction insufficiency, etc. have been proposed to explain both clinical presentations, yet little attention has been paid to the central nervous system component of the motor unit, that is, motoneurons.
Clinical Presentations of Neurologic Complications Following Sepsis and Treatment With Oxaliplatin
ICU-Acquired Weakness
One of the primary complications of sepsis is intensive care unit acquired weakness (ICUAW), which is defined as, “a syndrome of generalized limb weakness that develops while the patient is critically ill and for which there is no alternative explanation other than the critical illness itself” (Fan and others 2014). ICUAW affects approximately one third of critically ill patients and is associated with increased hospital length of stay, mortality, long-term functional disability and reduced quality of life (Batt and others 2019; Herridge and others 2003; Latronico and Bolton 2011).
The diagnosis of ICUAW is often made when the patient begins to recover from the acute illness. As sedation is discontinued and encephalopathy begins to resolve, difficulty weaning from the ventilator is often the first indication that there is a problem (Friedrich and others 2015; Latronico and Bolton 2011). The diagnosis is made by physical examination of volitional strength using tests such as the Medical Research Council sum score or a handgrip dynamometer. The weakness is flaccid, symmetric, and involves both limb and respiratory muscles (Batt and others 2019; Friedrich and others 2015; Latronico and Bolton 2011). In addition to weakness some patients have sensory loss; however, sensory examination in patients recovering from critical illness is often complicated by encephalopathy (Friedrich and others 2015; Latronico and Bolton 2011). Loss of reflexes is inconsistent, with areflexia in some patients and preserved reflexes in others (Friedrich and others 2015; Latronico and Bolton 2011).
Chemotherapy-Induced Neurotoxicity
Platinum-based chemotherapies such as oxaliplatin, are central pillars of contemporary cancer treatment (André and others 2009; de Gramont and others 2000; Fu and others 2006) used in approximately 50% of patients (Galanski and others 2005; Johnstone and others 2014), which extend lifespans by up to 80% (Galanski and others 2005; Johnstone and others 2014; Montagnani and others 2011). Despite impressive efficacy, nearly all patients treated with oxaliplatin and other agents are afflicted by severe off-target, neurotoxic side effects (Bennett and others 2012). This constellation of dose-limiting side effects, collectively referred to as chemotherapy-induced neurotoxicity (CIN), diminishes quality of life and limits functional capacity (Alcindor and Beauger 2011; Argyriou and others 2008; Argyriou and others 2014; André and others 2004; Avan and others 2015; Beijers and others 2014; Cavaletti and others 1995; Mols and others 2013; Quasthoff and Hartung 2002; Tofthagen and others 2013).
Signs and symptoms of oxaliplatin-induced neurotoxicity, hereafter identified as OIN, are divided into two distinct phases differentiated by their temporal proximity to treatment. Up to 95% of patients experience acute OIN immediately after and for days following infusion. Acute OIN is characterized by pain, muscle spasms, and cold hypersensitivity and is associated with sensory and motor nerve hyperexcitability (Krishnan and others 2006). Signs and symptoms frequently worsen after each treatment cycle in a dose-dependent manner. In a significant proportion of patients (50%−70%, Zajączkowska and others 2019; 26%−46%, Beijers and others 2014); 84%, Briani and others 2014), OIN can progress after discontinuation of treatment, leaving many patients with chronic OIN (cOIN) that does not improve for months or even years (Aaronson and others 2014; Dietrich and others 2006; Marshall and others 2017; Park and others 2011; Park and others 2013; Seretny and others 2014). Chronic OIN is characterized by a complicated clinical presentation, including but not limited to, ataxia, weakness, reduced sensation, and reduction or loss of deep tendon reflexes (Marshall and others 2017; Saif and others 2010). Sensorimotor and proprioceptive-like disorders are classically attributed to length-dependent axon degeneration (Argyriou and others 2014; Avan and others 2015; Burakgazi and others 2011).
Peripheral Neuropathy: A Shared Mechanism Underlying Neurologic Dysfunction Following Both Sepsis and Treatment With Oxaliplatin
Diagnosis of peripheral neuropathy is considered in patients with generalized weakness in the ICUAW or sensorimotor disorders following chemotherapy. Although underlying causes cannot be assigned based on clinical presentation, the mechanism is generally thought to be peripheral nerve degeneration (Cavaletti and Marmiroli 2015; Dorsey and others 2019; Friedrich and others 2015; Miltenburg and Boogerd 2014). Evidence for axon degeneration comes from histological studies of peripheral nerves sampled in biopsy or autopsy (Latronico and others 1996; Zochodne and others 1987). Histological examination of epidermal skin biopsies demonstrates degeneration expressed as decreased density of sensory nerve terminals in OIN (North and others 2019). Degeneration of axons is also inferred from conventional electrophysiological studies of nerve conduction, which show decreases in the amplitudes of electrically evoked compound action potentials (CAPs) produced by stimulating either mixed nerves to generate compound muscle action potentials (CMAPs) or sensory nerves to produce compound sensory action potentials (SNAPs or CSAPs) (Griffith and others 2014; Latronico and others 1996; Velasco and others 2014; Zochodne and others 1987). Instances in which it is found that abnormal nerve conduction findings correlate with symptom severity support a role for nerve degeneration in OIN (Griffith and others 2014).
There are discrepancies, however, in the correspondence between nerve conduction findings and symptoms in both ICUAW and OIN. In ICUAW, the decline in SNAPs is not always correlated with pathologic abnormality (Latronico and others 1996). In some cases, reductions in SNAPs and CMAPs can recover within days, that is, faster than axons can possibly regenerate (Novak and others 2009). For chemotherapy induced neuropathy, “conventional electrophysiology often does not mirror the patient’s symptoms” (Staff and others 2017), and in some patients reporting sensorimotor symptoms with cOIN, neither SNAPs nor epidermal nerve density measures give evidence of axon degeneration (Burakgazi and others 2011). Furthermore, some signs of cOIN, notably reduction or loss of deep tendon reflexes, do not correlate with the severity of other symptoms (Binda and others 2015) suggesting that not all signs and symptoms share the same underlying mechanism. Collectively, these observations suggest that a complete explanation of both ICUAW and cOIN requires additional factors.
Neuropathy by definition (see Introduction section), is a disorder that encompasses not only structural degeneration but also functional impairment of peripheral nerves. Although measurement of functional impairments in human peripheral nerves is limited by technical difficulties, one methodological approach provides direct evidence of motor and sensory nerve dysfunction. Excitability testing of peripheral nerves tracks action potential threshold during the different phases of depolarization and repolarization of sensory or motor CAPs. Changes within a suite of threshold measures provide assessment of various functional abnormalities in peripheral nerves and are used to support inferences about underlying axon biophysics and properties of voltage-gated ion channels (Kiernan and others 2020). When applied in patients with ICUAW, excitability testing demonstrates threshold changes consistent with hypoexcitabillity of nerves coincident with muscle weakness (Z’Graggen and others 2006). Excitability testing also reveals functional changes in sensory and motor nerves in patients with cOIN. Changes in measures of CAP recovery threshold are prominent soon and for months after cessation of oxaliplatin therapy (Krishnan and others 2006; Park and others 2009a). Correlations between changes in these excitability parameters and the severity of patient symptoms scored by clinical grading scales suggest dysfunction of ion channels may be a significant contributor to the peripheral neuropathy in OIN (Park and others 2009a; Park and others 2009b). Ion channel dysfunction is also consistent with observations of pathologic spontaneous firing recorded from human peripheral nerves and dorsal root ganglia (Hill and others 2010; Lehky and others 2004; North and others 2019; Wilson and others 2002).
Deficits in the Peripheral Nervous System Are Insufficient to Explain ICUAW and OIN
Several observations point to central nervous system (CNS) participation in mediating behavioral disorders in ICUAW and OIN. Neural networks in the CNS are likely to promulgate behavioral disorders simply by integrating faulty sensory information received from neurons undergoing neuropathy. Cognitive disabilities, confusion, and memory loss demonstrate that the CNS can also be the origin of behavioral disorders in both groups of patients (Dietrich 2010; Mazeraud and others 2020; Pandharipande and others 2013; Simó and others 2013). Further inferential evidence derives from demonstration that infusion of lipopolysaccharide, a pathogen stimulating inflammatory responses in sepsis, produces acute weakness in human subjects before the onset of degeneration or dysfunction in peripheral nerve, neuromuscular junction, or skeletal muscle (McNicol and others 2010). Direct evidence obtained in both ICUAW and cOIN show physical alterations in the brain, as well as changes in brain activity (Kaiser and others 2014; Mazeraud and others 2020). These observations strongly suggest that CNS dysfunction in ICUAW and cOIN combines with neuropathy in producing behavioral disorders, but CNS contributions remain underexplored.
While neuronal dysfunction in the CNS remains virtually unknown in ICUAW and cOIN, the hallmark signs of sensorimotor disability, fatigue, and weakness suggest a possible role for motoneurons. Motoneurons are essential to regulating skeletal muscle force and are participants in the pathophysiology of a variety of disorders. They are also propitious targets for studying biophysical impairments responsible in vivo for altering neuronal excitability. These advantages led us to explore impaired excitability in motoneurons as a potentially shared mechanism between the two pathologies using the rodent models described below.
Rodent Models of Sepsis and cOIN
Currently and for the foreseeable future, animal experimentation is indispensable for obtaining mechanistic understanding about neural dysfunction in disorders such as those occurring in sepsis and OIN. In vitro studies of neural tissue from rodents have identified numerous mechanisms related to changes in neuronal excitability, for example, channelopathies induced by sepsis and antineoplastic agents (Boyette-Davis and others 2015; Haeseler and others 2008; Housley and others 2020a; Rossignol and others 2007; Sittl and others 2012). In vivo studies of the effects of sepsis and cOIN are fewer, but they allow examination of neurons without disturbing their structural integrity. Here we describe two experimental models providing in vivo access to the central nervous system that enabled the biophysical studies described below.
While there are several experimental animal models of ICUAW and cOIN, we will focus on the rat model of each disease that was used to study motoneuron excitability in the studies described below. Both sepsis and oxaliplatin in rats are effective for inducing sensorimotor disorders similar to those observed in patients and for exposing myriad cellular effects, including changes in neural excitability (Bullinger and others 2011; Nardelli and others 2013; Novak and others 2009; Vincent and others 2016). Before describing key features of these models, however, it is important to address the fact that preclinical findings have not yet translated into successful clinical trials. This disappointment has several plausible explanations, including the straightforward possibility that the canonical causes have not yet been found or identified. Additional challenges to translation include deficiencies in experimental design and implementation of both clinical and preclinical studies, as well as misinterpretation of preclinical findings and consideration of species differences (Dejager and others 2011; Dyson and Singer 2009; Housley and others 2020a; Pitts and Simpson 2010). Clinical conditions introduce a wide range of confounding variables, for example, preexisting conditions, medications, lifestyle, genomic heterogeneity, disease process, and so on in various permutations that have unknown effects. All these factors require consideration for their capacity to confound preclinical findings in ways that complicate translation.
The Cecal Ligation and Puncture Model of Sepsis
Cecal ligation and puncture in adult rodents is an acknowledged gold standard for clinically relevant models of sepsis in human subjects (Alverdy and others 2020; Dejager and others 2011). This model of sepsis recapitulates the trauma that can occur following abdominal injury or surgery in patients. Sepsis in the surgical cecalligation rat model is validated by the occurrence of bacteremia and acute inflammation which are the hallmarks of sepsis in patients (Alverdy and others 2020; Dejager and others 2011). Within a day of surgery, the model reproduces the rapid onset of muscular weakness seen in humans treated with bacterial lipopolysaccharide, a significant contributor to the pathogenesis in some cases of septic shock (McNicol and others 2010; Nardelli and others 2013). In the model, as in patients treated with lipopolysaccharide, acute weakness occurs independently of dysfunction in peripheral nerve, neuromuscular junction, or skeletal muscle (McNicol and others 2010; Nardelli and others 2016), pointing to contributions made in the central nervous system control of muscle contraction. At later time points, the cecal ligation and puncture model reproduces the reversible (over days to weeks) drop in SNAPs seen in a subset of patients with ICUAW (Nardelli and others 2013; Novak and others 2009). This drop in SNAPs is accompanied by hypoexcitability of axons (Novak and others 2009). Both the rapid development of reversible motor deficits due to dysfunction of the CNS and the hypoexcitability of axons suggest ion channel dysfunction may be an important contributor to neuropathology in sepsis.
The Rat Model of cOIN
Repeated delivery of oxaliplatin is commonly used to generate an OIN model that reproduces many important features depending on the time, varying dosing parameters and route of administration (Cavaletti and others 2001). Behavioral signs including a constellation of acute and chronic symptoms including but not limited to temperature hypersensitivity, allodynia, and movement disorders (Cavaletti and Marmiroli 2010; Pasetto and others 2006). At the systems level, reduced weight gain, altered inflammatory and oxidative signaling, impaired memory, and changes in neurophysiological measures, for example, reduction in CAPs mirror those observed clinically. Degeneration of peripheral nerves and dorsal root ganglia give direct evidence of neuropathy. OIN models also recapitulate factors relevant to pathogenesis, including the biodistribution of oxaliplatin throughout various organ systems and nervous system structures, for example, nerves, dorsal root ganglia, brain, and cerebrospinal fluid.
In order to initiate in vivo study of cellular deficits responsible for sensorimotor disorders in the coasting period when symptoms intensify after cancer treatment, we studied a rat model of cOIN. Weeks after human-scaled treatment, the model recapitulates clumsiness, ataxia, and other dysfunctions similar to those described clinically as proprioceptive in origin. Specifically, rats fail to successfully perform skilled sensorimotor tasks, demonstrating errors in accurate and precision fore- and hindlimb placement (Housley and others 2020a; Vincent and others 2016). These deficits are consistent with those ascribed to neuropathy in patients (Marshall and others 2017; Pasetto and others 2006). In our model, neuropathy manifests as deficits in sensory encoding of naturalistic limb movements rather than degeneration of axons (Bullinger and others 2011; Housley and others 2020a; Vincent and others 2016). The deficits in sensory encoding together with altered axonal excitability found in patients (Heide and others 2018; Krishnan and others 2006; Park and others 2009a; Park and others 2009b) suggest dysfunction of ion channels may be an important contributor to sensory dysfunction in cOIN.
Inability of Motoneurons to Convert Depolarization Into Steady, Repetitive Firing Following Sepsis and Oxaliplatin
One of the most accessible CNS neurons for physiologic study is the spinal alpha motoneurons. These motoneurons, which are the central focus of mechanistic considerations below, span both peripheral and central nervous systems. The soma, dendrites and proximal axon of each motoneuron reside centrally within the spinal cord or brainstem, while the axon projects peripherally to synapse with skeletal muscle fibers comprising motor units. Centrally, motoneurons integrate synaptic input together with their intrinsic excitability to encode the action potential trains which are then conducted by the peripheral axon to regulate skeletal muscle contraction. As such, motoneurons embody sites and functions susceptible to damage not only in the periphery, but also within the central nervous system where pathophysiological mechanisms are accessible for examination in vivo in preclinical studies described below.
Motoneurons are the ultimate encoders of neural instructions for moving our bodies through space. They receive all information relevant to activating skeletal muscle and for translating that activity into movement. Much is known about the biophysics of this process (Heckman and Enoka 2012). Synaptic current converging on motoneurons from many sources is variably amplified by intrinsic currents to yield membrane depolarization that is encoded into temporally sequenced repetitive firing of action potentials or spikes (Heckman and Enoka 2012). The resultant encoded spike trains sent to muscle control both the magnitude and temporal features of muscle contraction. Despite its crucial role at the ultimate step in neural control of muscle force generation, neuronal encoding by motoneurons, the so-named final common pathway in movement control has gone underappreciated for its potential to cause muscle weakness, fatigue and movement discoordination in ICUAW and cOIN.
Motoneuron Firing Defects Induced by Sepsis
In healthy animals, motoneurons typically fire orderly trains of action potentials. These trains are conveniently observable as EMG signals generated in skeletal muscles by a motoneuron’s composite motor unit as it engages in diverse movement tasks performed by conscious animals, including humans. Motor units, and motoneurons by direct inference, sustain regular firing that slowly adapts over several seconds or longer during steady muscle contraction; they modulate firing rate in direct relation to the force of graded muscle contractions; and they fire with similar patterns and rates in replicate muscle contractions (Heckman and Enoka 2012). These firing profiles express rules employed by the motoneuron as it first integrates and then, through the central encoding process, translates intrinsic currents into spike trains. Encoding rules can be determined in current clamp experiments that examine the relation between experimentally controlled current injected by micropipette into a motoneuron and features of the evoked repetitive firing. This experimental approach bypasses synaptic current to isolate the contribution of a motoneuron’s intrinsic excitability to the encoding process. Ample characterization of spike train encoding under these conditions establishes that motoneurons reproducibly encode steady current into continuous and slowly adapting repetitive firing that varies rate with current amplitude (Fig. 2). These spiking behaviors measured from motoneurons in anesthetized animals approximate those recorded in freely moving healthy animals, including humans (Heckman and Enoka 2012).
Figure 2.
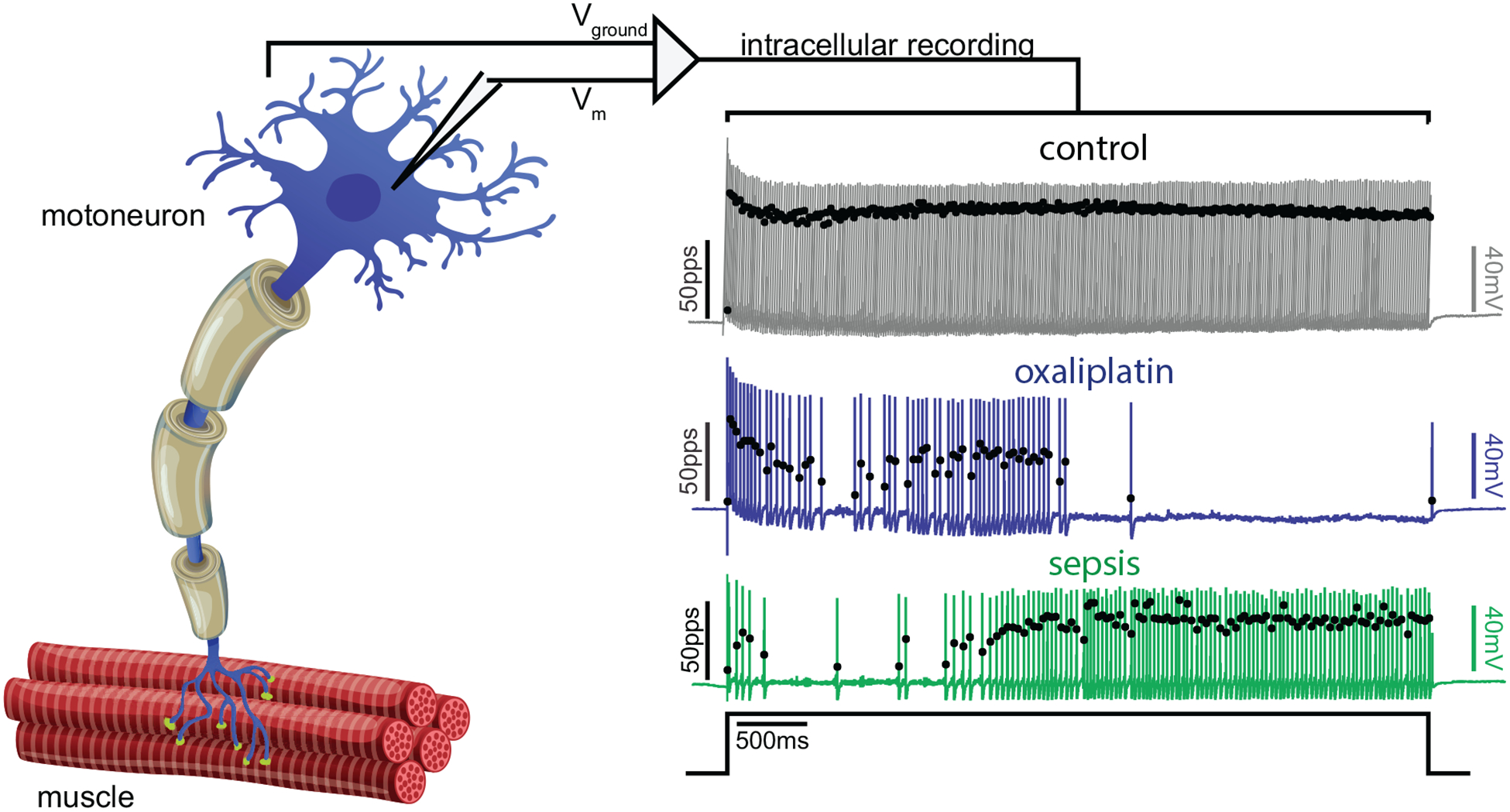
Disrupted repetitive firing of motoneurons in both sepsis and chronic oxaliplatin-induced neurotoxicity (cOIN). On the left is a cartoon depiction of a single motor unit. Placement of a recording electrode into the motoneuron cell body allows for recordings of the motor unit firing patterns shown on the right. The records show representative instantaneous firing rates (filled black circles: pulses per second [pps]) superimposed on action potentials (colored lines: mV) recorded intracellularly from motoneurons from a control rat (top: gray), a rat 5 weeks after clinically relevant chemotherapy treatment with oxaliplatin (middle: blue) and from a rat 3 days after induction of sepsis by cecal ligation and puncture (bottom: green) in response to matched depolarizing current injection (bottom trace: nA). Note reduced number of action potentials, reduced firing rate and increased variation in firing rate following sepsis and oxaliplatin. For this and all subsequent figures cOIN traces are blue and sepsis traces are green.
In the only report to our knowledge of motor-unit activity in ICUAW patients, we found motor units firing slowly and irregularly during maximal voluntary contractions (Nardelli and others 2013). This behavior expresses a functional deficit in motor units as opposed to structural degeneration, especially having been observed in proximal muscles that are not typically involved in neuropathy caused by sepsis (Nardelli and others 2013). Recognizing functional origins opens novel possibilities for explaining weakness in ICUAW. Aberrant motor unit firing might arise from defects in the synaptic input received by motoneurons or in the way the motoneurons decode that input or both. Distinguishing these sources of motor unit misbehavior is needed to achieve the mechanistic understanding required for developing effective treatment. Limitations in studying human subjects brought us to pursue this line of investigation using the rodent model of sepsis detailed above. Experimental access to motoneurons in this model permitted us to isolate the encoding properties of motoneurons under conditions in which synaptic input is substituted with current injected into the motoneuron from an extrinsic source under experimental control. Motoneurons in rats with sepsis fire at slow and irregular rates similar to those observed in patients with ICUAW (Fig. 2). In addition, motoneuron firing rate in this sepsis model is abnormally less sensitive to variations in current intensity (Nardelli and others 2013; Nardelli and others 2016). These firing aberrations under experimental control of input current are attributable to defects in spike-train encoding by motoneurons.
Motoneuron Firing Defects in cOIN
Patients with OIN also exhibit abnormal motor unit activity, but in the direction of increased firing. In the acute phase of OIN, during the several weeks of repeated administration of platinum-based compounds, motor units fire sporadically in high-frequency bursts when muscles are either at rest or actively contracting (Webster and others 2005; Wilson and others 2002). This deviant behavior seems a reasonable explanation for acute muscle spasms and cramps that fade near the time of chemotherapy treatment completion (Lehky and others 2004). At the end of chemotherapy during the coasting period, patients exhibit phenotypically different symptoms that define chronic OIN, including physical fatigue and sensorimotor disability. Data on motor unit firing are absent to our knowledge during this chronic phase, but we wondered whether sensorimotor disability might be accompanied by abnormally slow and irregular motoneuron firing rates in our cOIN model as it is in the sepsis model. Current-clamp experiments reveal encoding defects remarkably similar to those reported following sepsis (Fig. 2). These findings suggest that platinum-based chemotherapy induces chronic defects in motoneuronal encoding, and they lead to a plausible explanation for weakness/fatigue in cOIN as described below.
Motoneuron firing relies not only on intrinsic excitability but also on synaptic current from sensory and other neurons carrying the motor commands for movement. Aberrant motoneuron firing in behaving animals might also derive, therefore, from defects in strength or timing of current from synaptic input. Although synaptic input to motoneurons has not been measured following either sepsis or oxaliplatin, abnormality seems likely to arise from two already identified sources of dysfunction of the peripheral nervous system. The first is degeneration that reduces the number of somatosensory neurons transmitting current to motoneurons. The second source is modification of synaptic input transmitted by proprioceptive encoding that is blunted, delayed, and abbreviated (Bullinger and others 2011; Housley and others 2020a; Vincent and others 2016). Whether by structural or functional neuropathy, disruption of synaptic current, unless otherwise compensated, would compound the abnormality in motoneuron firing caused by defective motoneuron encoding.
Impact of Defective Encoding on Muscle Force Production and Fatigue
In healthy adult vertebrates, the central nervous system regulates the force of skeletal muscle contraction by activating motoneurons and their corresponding motor units in different numbers, rates, and patterns (Heckman and Enoka 2012). Among these force regulators, varying the rate of repetitive firing (rate modulation) achieves the greatest modulation of total force produced by single muscles. Losing control of motor unit rate modulation in ICUAW and cOIN poses two significant problems for regulating muscle force production (Fig. 3). First, dampened rate modulation results in weakness from lower than intended force production, leaving recruitment of additional motor units as the only remaining option for increasing muscle force. However, if the additional motor units recruited also lack rate modulation, then normal force may never be achieved. Some compensation might be achieved by recruiting many more motor units than normally required to perform a task, but it is likely to increase fatigue because the usual orderly recruitment of motor units following the size principle will engage units progressively more susceptible to fatigue. Second, the larger than normal fluctuations in firing rate produces instability in muscle force, presenting a significant challenge to achieving steady movement and posture. The only way of partially compensating for this instability is to activate more motor units, again making the subject vulnerable to fatigue. In these ways, defective repetitive firing in motoneurons alone has the capacity to result in the weakness, unsteadiness, and fatigue observed in ICUAW and cOIN just as it is proposed to do in the paretic limbs of patients following stroke (Hu and others 2016; McManus and others 2017).
Figure 3.
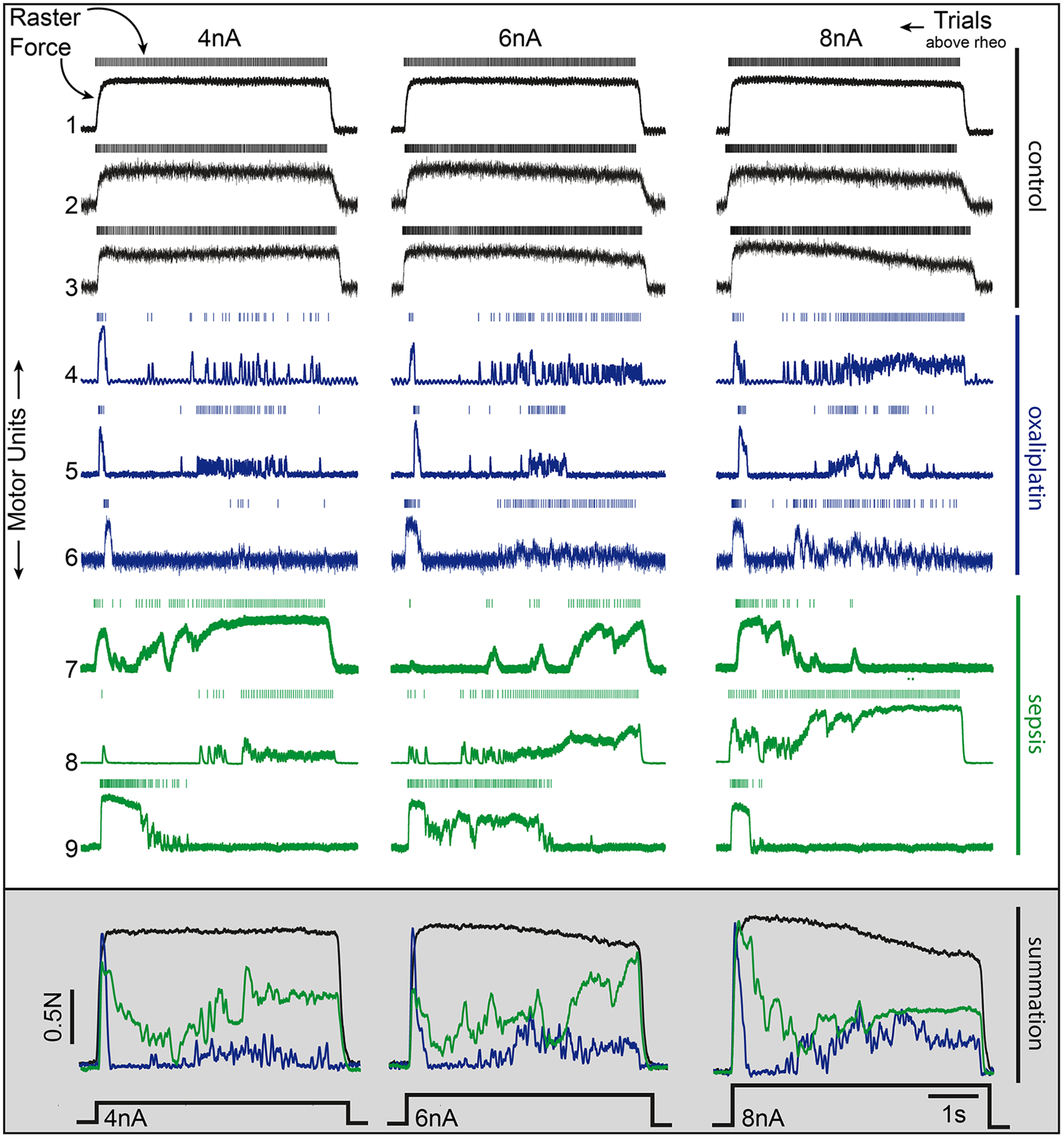
The functional consequence of impaired rate modulation on motor function. Motor unit recordings of motoneuron action potentials (raster: top) and isometric force generated by the motor unit (the muscle fibers innervated by the individual motoneurons being stimulated, lower trace), from three independent, representative motoneurons (rows) in each experimental group in response to 4, 6, and 8 nA of current injected above rheobase (the minimal current necessary to trigger firing). Firing rates and resultant force were disrupted at all current strengths in chemotherapy and septic motoneurons. The traces at the bottom demonstrate the effect of summing the force generated by the three motor units shown in each group (simulated recruitment of three motor units) across each injected current strength. Summing multiple motor units (in this simulated recruitment analysis) does improve resultant muscle force yet fails to restore the smooth, predictable motor unit output observed in control. Note that increasing motoneuron depolarization provides little to no improvement in motor unit force output, indicating that rate-modulation cannot compensate for the defect in force production.
While motoneuron firing defects necessarily impact movement, sepsis- and cancer-induced nerve and muscle dysfunction are also important contributors that have been extensively reviewed (Batt and others 2019; Fearon and others 2011; Friedrich and others 2015; Latronico and Bolton 2011; Porporato 2016; Schefold and others 2010; Tisdale 2002). Both sepsis and cancer trigger cachexia that primarily targets skeletal muscle (Friedrich and others 2015; Porporato 2016; Puthucheary and others 2013). Cachexia is associated with severe muscle wasting, reduced physical function, reduced tolerance to anticancer therapy, and reduced survival in a variety of clinical populations (Batt and others 2019; Fearon and others 2011; Herridge and others 2003). Another contributor to weakness in ICUAW is myosin loss, which causes reduction in specific force (Friedrich and others 2015; Latronico and Bolton 2011). There is a similar reduction in specific force in preclinical studies of cancer, although it is not known whether myosin loss contributes (Roberts and others 2013; VanderVeen and others 2017). In both ICUAW and cancer, there are alterations in muscle Ca handling and release (Batt and others 2019; Friedrich and others 2015; Ochala and Larsson 2008). Oxaliplatin may exacerbate the effects of cancer on muscle as it causes atrophy and similar changes in gene expression (Feather and others 2018; Sakai and others 2014). Furthermore, oxaliplatin as well as sepsis are shown to impair transmission at the neuromuscular junction (Lehky and others 2004; Webster and others 2005). These peripheral defects would further worsen weakness, fatigue and movement disorders arising from defective motoneuron firing behavior.
Biophysical Mechanisms Underlying Defective Motoneuron Firing Following Sepsis and Oxaliplatin
No Change in Motoneuron Passive Electrical Properties and Action Potentials
Several properties of motoneurons are unaffected in rat models of ICUAW and cCOIN (Housley and others 2020b; Nardelli and others 2016). No changes emerge in passive electrical properties of motoneurons assessed from resting membrane potential or input conductance (Housley and others 2020b; Nardelli and others 2013; Nardelli and others 2016). Neither are changes expressed in action potentials. Single evoked action potentials remain normal as assessed by rheobase current, that is, threshold current, and from action potential amplitude and rates of change in voltage.
Action potentials generated during repetitive firing were also indistinguishable from normal (Fig. 4). The primary current responsible for triggering action potentials is the voltage-gated Na current. Normally, inactivation of the Na current responsible for repetitive firing of action potentials occurs at depolarized voltages such that less current is available to fire the next action potential. With reduction in Na current, there is reduction in the rate of rise of the action potential in association with attenuated firing rates (Bean 2007; Brownstone and others 2010; Miles and others 2005). If build-up of inactivation of Na channels during repetitive firing is responsible for stuttering in firing, then the rate of action potential rise should be slowest prior to pauses in firing and greatest following resumption of firing, indicating that firing resumed due to recovery of Na current from inactivation. These observations were not made in either sepsis or cOIN (Housley and others 2020b; Nardelli and others 2013). Rates of action potential rise were lowest following resumption of firing and increased during the first few action potentials. These data argue strongly against inactivation of Na channels as a cause of stuttering in firing. High-threshold, voltage-gated K channels also regulate the rate of repolarization during the descending phase of the action potential. Reduction in these K current causes widening of action potentials (Bean 2007). However, action potential half width was normal following both sepsis and treatment with oxaliplatin (Housley and others 2020b; Nardelli and others 2013) Another measure of K current is the rate of action potential repolarization, which was normal in both sepsis and cOIN. Collectively, the apparent normalcy of action potentials leads to the conclusion that suprathreshold currents are insufficient to explain abnormal motoneuron firing behavior.
Figure 4.
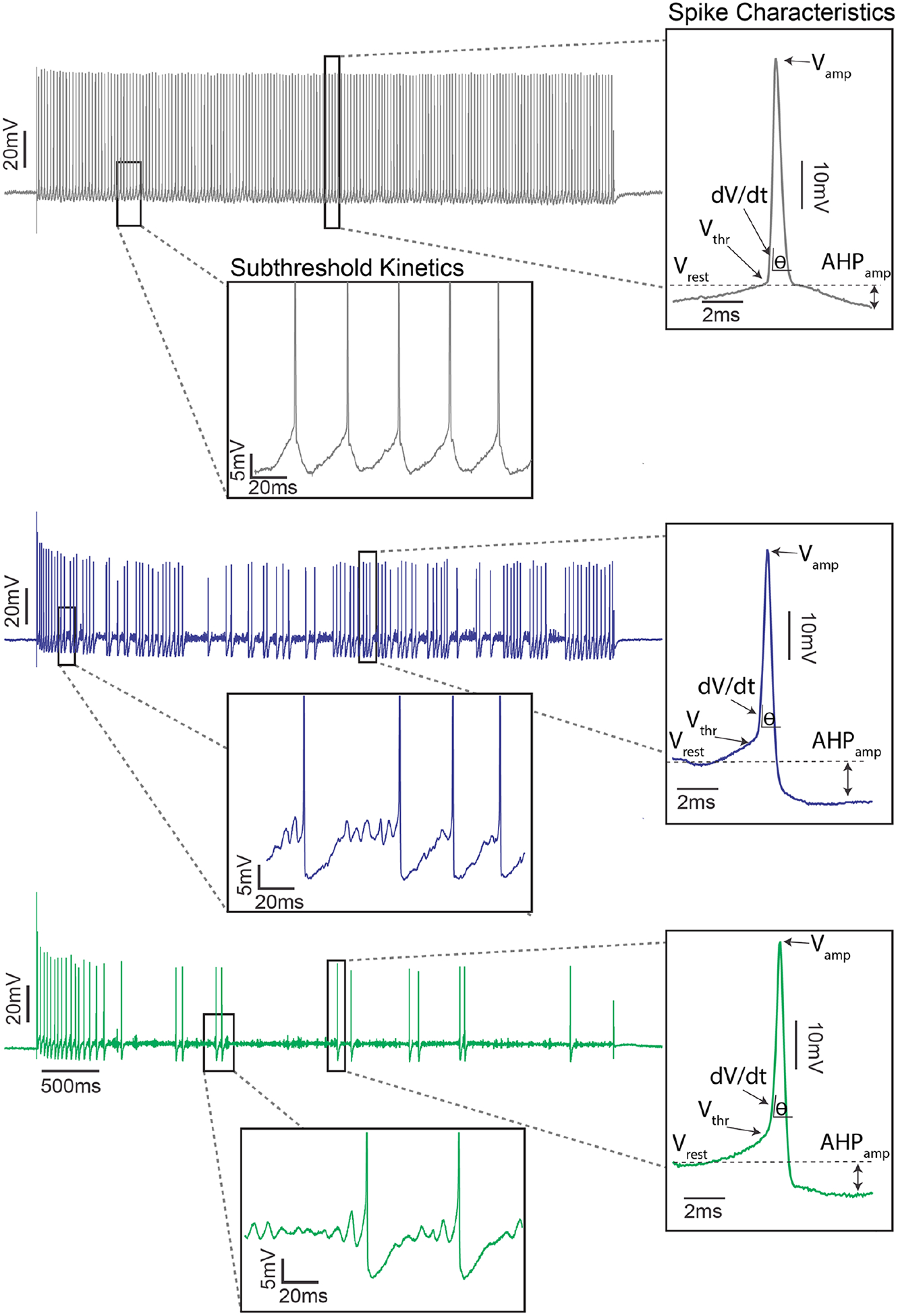
Subthreshold membrane potential oscillations with unaltered action potentials in motoneurons following both sepsis and chemotherapy. Representative intracellular records of membrane potential for control (top: gray), chemotherapy (middle: blue), and septic (bottom: green) motoneurons in response to matched depolarizing current injection. Subthreshold oscillations in membrane potential following sepsis and oxaliplatin are highlighted in left inset with expanded axes. The oscillations precede the generation of action potentials and are absent in control. Right inset shows temporally expanded view of single action potentials (spikes) during repetitive firing. Measured properties included (Vrest: resting membrane potential [mV]; Vthr: voltage threshold for action potential generation [mV]; Vamp: action potential amplitude [mV]; AHPamp: after hyperpolarization amplitude [mV]) and derived (dV/dt: first derivative of action potential voltage [mV/ms]). None of these action potential characteristics were significantly different between septic, oxaliplatin treated, and control motoneurons.
An Imbalance in Subthreshold Currents Underlies Inability to Sustain Regular Firing
Close examination of the membrane potential between spikes offered a clue to the mechanism responsible for abnormal repetitive firing. In healthy motoneurons, there is a smooth depolarization to action potential threshold during repetitive firing (Fig. 4), except over a very narrow range of just suprathreshold current injection (Jensen and others 2018; Manuel and others 2009). The rate of depolarization is similar for each spike such that firing rate is stable. In sepsis or cOIN, this was not the case. There were oscillations in the membrane potential associated with irregular rates of depolarization prior to each spike (Fig. 4) (Housley and others 2020b; Nardelli and others 2017). During pauses in firing, the oscillations in membrane potential persisted until a depolarizing oscillation was large and fast enough to reach action potential threshold. These data suggested a defect in subthreshold currents, which activate and shut off at membrane potentials near the resting membrane potential of motoneurons.
Oscillations in the membrane potential have been observed in motoneurons when excitability is low (Iglesias and others 2011; Sciamanna and Wilson 2011). The oscillations were suggested to result from an imbalance in the ratio of depolarizing (inward) to hyperpolarizing (outward) voltage gated currents first activated in the subthreshold range. When the ratio of depolarizing to hyperpolarizing subthreshold currents is high, there is rapid depolarization to action potential threshold such that there is steady repetitive firing (Nardelli and others 2017). When the ratio is low, there are oscillations in membrane potential and stuttering of firing (Golomb and others 2007; Gutfreund and others 1995; Klink and Alonso 1993).
The most likely depolarizing and hyperpolarizing conductances contributing to high-frequency oscillations and irregular firing are slowly inactivating Na currents, also known as Na persistent inward currents (NaP) and subthreshold potassium current (Ksthr most likely Kv1; Bean 2007). Both NaP and Ksthr activate below threshold and have rapid kinetics (Chatelier and others 2010; Johnston and others 2010). Low NaP/Ksthr ratios are expected to lead to irregular firing whereas high ratios are thought to underlie more regular discharge. This possibility was explored using computer simulation. When the ratio of NaP/Ksthr was high, simulation produced rapid depolarization toward action potential threshold and a steep increase in firing rate with increased current injection (Housley and others 2020b; Nardelli and others 2017). When the ratio was low there were subthreshold oscillations in membrane potential and inconsistent repetitive firing at low rates that mimicked the oscillations and inconsistent repetitive firing following both sepsis and cOIN. What mattered was the ratio of NaP to Ksthr rather than the absolute amount of either current. This suggests that either reduction in NaP or an increase in Ksthr lead to the same defect in repetitive firing.
To directly test the capacity for reduced ratios of subthreshold depolarizing to hyperpolarizing currents to cause irregular firing, we used dynamic clamp in vivo to experimentally manipulate current ratios in motoneurons. Dynamic clamp injects motoneurons with inward or outward currents modeled on the basis of their voltage dependence and kinetics. By changing the sign of a current, it can be added or subtracted. Reduced NaP or increased Ksthr current in healthy motoneurons reproduced the irregular firing seen in sepsis or cOIN (Fig. 5). Conversely, when NaP was increased or Ksthr was reduced, firing of septic motoneurons was normalized (Nardelli and others 2017). These data demonstrate that reduction in the ratio of depolarizing to hyperpolarizing subthreshold currents was sufficient to explain the defect in firing of motoneurons.
Figure 5.
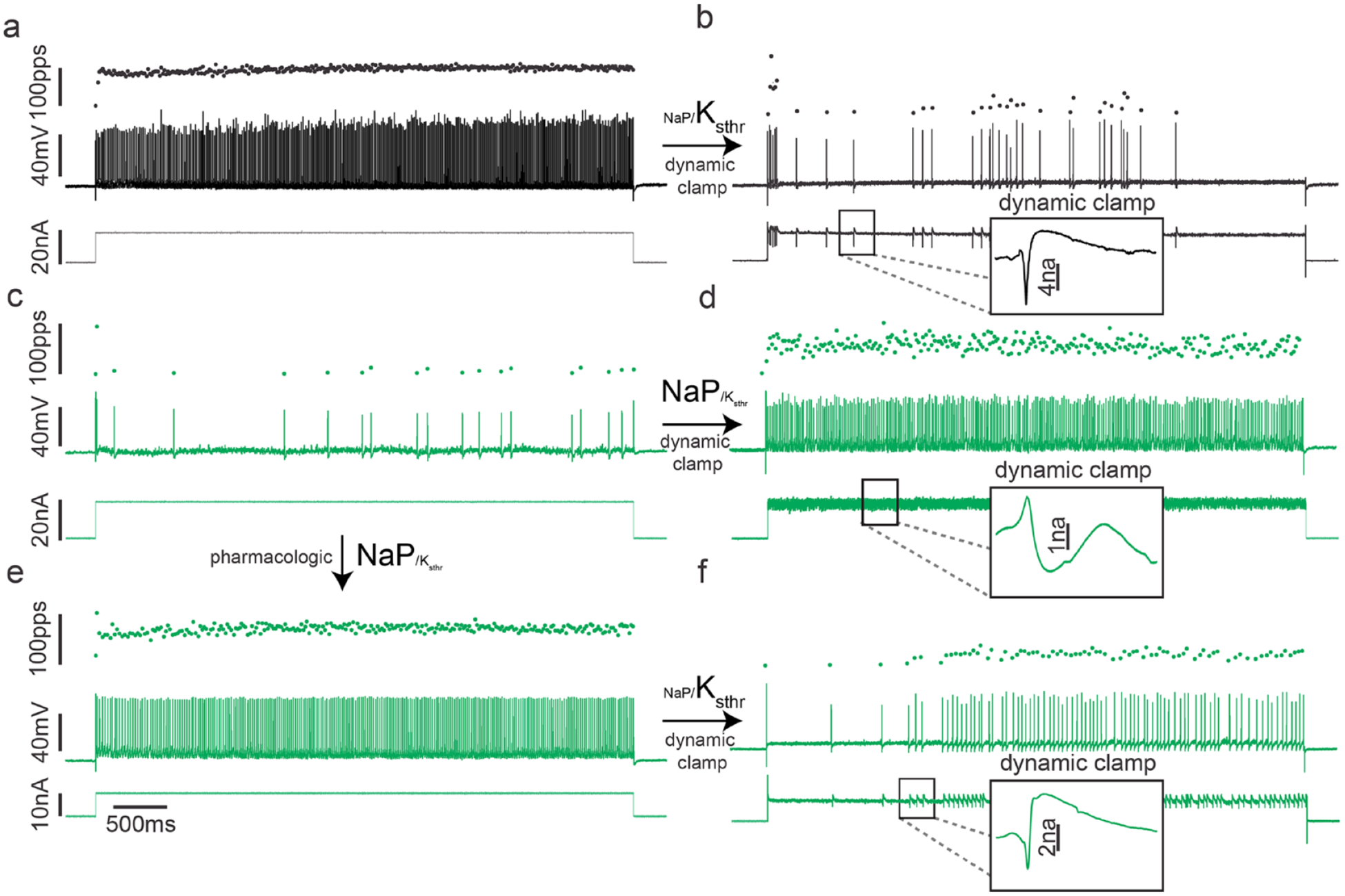
Stable repetitive firing depends on the proper balance of Na persistent inward current (NaP) and subthreshold potassium current (Ksthr). Each record (A-F) shows representative instantaneous firing rates (filled black circles: pulses per second [pps], top trace), and action potentials (lines: mV, middle trace) and current injection (nA, lower trace) during a 5-second in vivo intracellular recording from a rat spinal cord motoneuron. (a) A record from a healthy motoneuron. (b) Lowering the persistent inward current NaP/Ksthr ratio in the motoneuron shown in A reproduces impaired repetitive firing observed for septic and chemotherapy motoneurons. Note that with dynamic clamp, current injection is varied, rather than continuous (insets in b, d, and f). (c) A record from a motoneuron in a rat that had been septic for 2 days. The motoneuron fires erratically and cannot sustain repetitive firing throughout the 5-second current injection. (d) Increasing the NaP/Ksthr ratio via dynamic clamp in the motoneuron shown in C normalized repetitive firing. (e) A further record from the same motoneuron 20 minutes after pharmacologic treatment to increase the NaP/Ksthr ratio (3 mg/kg lorcaserin). Motoneuron firing is normalized. (f) Firing of the same motoneuron after lowering the NaP/Ksthr ratio with dynamic clamp, which reversed the effect of lorcaserin. When dynamic clamp is turned off, the effect of lorcaserin treatment is again evident, as firing is rapid and steady throughout the current injection (data not shown). Arrows indicate the path from original motoneuron recording (a and c) through pharmacologic and dynamic clamp tests of impaired repetitive firing’s dependency on NaP/Ksthr ratio.
Activation of Serotonin Receptors as Potential Therapy
Activation of monoaminergic receptors, for example, serotonin receptors (5HT2) increases persistent inward currents such as NaP and increases motor neuron excitability (Bennett and others 2001; Harvey and others 2006a; Harvey and others 2006b; Lee and Heckman 1999; Murray and others 2010; Murray and others 2011; Perrier 2013). Exploiting this avenue to increase NaP afforded a means of normalizing the reduced NaP/Ksthr ratio in motoneurons observed in septic rats. We administered the 5HT receptor agonists quipazine and lorcaserin to septic rats and found both improved motoneuron repetitive firing and muscle force generation (Fig. 5) (Nardelli and others 2017). 5HT agonists have many effects on the nervous system such that the normalization of firing could be due to indirect effects. To examine this possibility, we used dynamic clamp to reduce the NaP/Ksthr ratio following treatment with lorcaserin. After reduction of the NaP/Ksthr ratio, lorcaserin was no longer effective in normalizing firing (Fig. 5). These data suggest that lorcaserin restores the balance of subthreshold currents in motoneurons. While we have not yet tested the efficacy of lorcaserin on motoneuron firing in cOIN, the similarity in the defects suggests that activation of 5HT receptors will be similarly beneficial. Taken together, our data strongly suggest that a decrease in the ratio of depolarizing to hyperpolarizing subthreshold currents is responsible for defective repetitive firing in motoneurons in sepsis and possibly also in cOIN.
The Biophysical Determinants of Repetitive Firing
The demonstration that pharmacologic and dynamic clamp manipulations of subthreshold currents in motoneurons can bestow or remove the ability to fire repetitively may apply to other neuron types. It has been suggested that in sensory neurons when slowly activating depolarizing subthreshold currents predominate, neurons fire repetitively (integrators) and when slowly activating hyperpolarizing currents predominate, neurons can only fire a few spikes (differentiators) (Ratté and others 2015). The ratio of subthreshold voltage-gated excitatory to inhibitory conductances is the key determinant of the ability to repetitively fire (Fig. 6). When the ratio of NaP and Ksthr is normal, the capacity to sustain steady repetitive firing, that is, excitability is normal. When the ratio is high, the neuron is hyperexcitable and when the ratio is low it is hypoexcitable. The ratio rather than the absolute value of excitatory and inhibitory conductances is the determinant of excitability. This conceptual framework suggests that what might seem like subtle changes in the absolute amount of subthreshold currents (which are often small), could have profound function impact on neuronal excitability and thus circuit behavior.
Figure 6.

Model of biophysical determinants of impaired motoneuron repetitive firing. Two-dimensional conductance space is constructed from parameters utilized in dynamic clamp experimental study of motoneurons from control and septic rats. Individual neurons are indicated by small outlined, color-coded circles projected onto the conductance space. Two-dimensional ellipsoids enclosing 95% of experimental group data were computed with least-squares elliptical fitting. The magnitude and direction of dynamic clamp manipulations (perturbations) of Na persistent inward current (NaP) and subthreshold potassium current (Ksthr) are indicated along the horizontal and vertical axes respectively. The origin was defined as the mean dynamic clamp manipulation (nS) to NaP (94.2 nS) and Ksthr (−44.7 nS) to restore normal repetitive firing of septic motoneurons (gray neurons). A linear transformation of these two values was applied to all neurons. This provided a simple reorientation of the parameter space, centering normal firing at the origin and allowed easy comparisons with restored firing while preserving the interneuron comparisons. There is a relatively narrow operating range around the origin affording stable repetitive firing (total range of 180 nS for NaP and 120 nS for Ksthr). The plot is subdivided into four quadrants with varying ratios of NaP/Ksthr: <1 (top-left), ~1 (top-right), >1 (bottom right), and ~1 (bottom left). An additional diagonal dotted line is drawn from bottom left to top right indicating the iso-NaP/Ksthr where the balance of currents is normal. Wild type motoneurons in which the ratio of NaP/Ksthr was reduced to mimic sepsis (green) are clustered along a line orthogonal to the iso-NaP/Ksthr and exclusively located in the low NaP/Ksthr quadrant. After pharmacologic restoration of repetitive firing in septic motoneuron, the same dynamic clamp parameters are required to reinduce unstable firing (blue). Note that nearly all of the blue two-dimensional ellipsoid’s area is enclosed in the green ellipsoid, indicating the dependency of unstable repetitive firing on low NaP/Ksthr. Arrows indicate the phase transitions between the three experimental tests indicating ability to induce, correct and reintroduce corrupt repetitive firing. Note that all nS comparisons indicate relative changes induced by dynamic clamp and are not representative of absolute conductances required of healthy motoneurons.
Could Imbalanced Subthreshold Currents Also Contribute to Encephalopathy Following Sepsis and Chemotherapy?
Sensorimotor dysfunction is not the only neurologic complication shared by ICUAW and cOIN. In both settings there is significant dysfunction of the cortex (encephalopathy). In sepsis the disorder has variously been termed septic encephalopathy, sepsis-associated brain dysfunction or sepsis-associated delirium. Following chemotherapy the dysfunction is termed “chemobrain.”
Septic encephalopathy is the most frequent sepsis-associated organ dysfunction, often occurring before any other organs are involved (Bolton and others 1993). In prospective studies of sepsis and critical illness, encephalopathy was found to occur in approximately 70% of patients (Pandharipande and others 2013; Young and others 1990). The severity of encephalopathy ranges from delirium to coma. Development of septic encephalopathy is associated with increased mortality (Eidelman and others 1996), and many patients are left with long-term cognitive deficits (Pandharipande and others 2013; Semmler and others 2013). A number of theories have been proposed, including neuroinflammation, excitotoxicity and impaired perfusion (Mazeraud and others 2020; Robba and others 2018), but there is currently no clear consensus as to which of these mechanisms predominate.
A substantial number of cancer patients receiving chemotherapy report cognitive disturbances that include problems with memory and confusion, which can be accompanied by mood disorders and fatigue (Dantzer and others 2012; Winocur and others 2018). In patients with cancer it is difficult to confidently ascribe changes in cognition to chemotherapy rather than the underlying illness or the associated stress. However, treatment of healthy rodents with chemotherapy has yielded strong evidence of effects on cognition, suggesting treatment with chemotherapy itself is an important contributor to the cognitive decline (Winocur and others 2018). Similar to septic encephalopathy, a number of contributors are suspected, but there is little consensus (Dantzer and others 2012; Winocur and others 2018).
Could an imbalance of subthreshold currents contribute to encephalopathy following sepsis and chemotherapy? Subthreshold membrane potential oscillations have been reported in rodent cortical neurons (Klink and Alonso 1993; Yoshida and others 2011). Complete block of Na channels not only eliminated firing of action potentials, but also eliminated the subthreshold oscillations, suggesting involvement of NaP (Klink and Alonso 1993). If sepsis and chemotherapy alter the ratio of inward to outward subthreshold currents in cortical neurons as they do in central encoding processes of motoneurons, then similar imbalances and resultant firing defects may contribute to encephalopathy.
Conclusions
Detailed in vivo studies of physiologic function of rat lower motoneurons following sepsis and weeks following chemotherapy with oxaliplatin revealed a similar defect in excitability specific to repetitive firing. The discovery of a shared defect specific to the subthreshold currents responsible for repetitive firing raises the possibility of a shared mechanism underlying motor dysfunction in what have previously been considered to be distinct syndromes. Further work is needed to determine whether imbalanced subthreshold currents contribute to disordered excitability in other diseases affecting motoneurons such as amyotrophic lateral sclerosis, spinal muscular atrophy, and spinal cord injury. Restoration of the normal balance between depolarizing and hyperpolarizing subthreshold currents holds the potential for development of novel therapy for patients with weakness and fatigue following sepsis and chemotherapy. In addition, the theoretical framework for understanding the essential and unique role of subthreshold currents in repetitive firing developed here is applicable to all situations in which steady depolarization is converted into repetitive firing of electrically active cells.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported National Insitutes of Health (NIH) grants AR074985 (M.M.R.) and CA221363 (T.C.C).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Aaronson NK, Mattioli V, Minton O, Weis J, Johansen C, Dalton SO, and others. 2014. Beyond treatment—psychosocial and behavioural issues in cancer survivorship research and practice. Eur J Cancer Suppl 12:54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcindor T, Beauger N. 2011. Oxaliplatin: a review in the era of molecularly targeted therapy. Curr Oncol 18:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverdy JC, Keskey R, Thewissen R. 2020. Can the cecal ligation and puncture model be repurposed to better inform therapy in human sepsis? Infect Immun 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, and others. 2004. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer N Engl J Med 350:2343–51. [DOI] [PubMed] [Google Scholar]
- André T, Boni C, Navarro M, Tabernero J, Hickish T, Topham C, and others. 2009. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol 27:3109–16. [DOI] [PubMed] [Google Scholar]
- Argyriou AA, Kyritsis AP, Makatsoris T, Kalofonos HP. 2014. Chemotherapy-induced peripheral neuropathy in adults: a comprehensive update of the literature. Cancer Manage Res 6:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyriou AA, Polychronopoulos P, Iconomou G, Chroni E, Kalofonos HP. 2008. A review on oxaliplatin-induced peripheral nerve damage. Cancer Treat Rev 34:368–77. [DOI] [PubMed] [Google Scholar]
- Avan A, Postma TK, Ceresa C, Avan A, Cavaletti G, Giovannetti E, and others. 2015. Platinum-induced neurotoxicity and preventive strategies: past, present, and future. Oncologist 20:411–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batt J, Herridge MS, Dos Santos CC. 2019. From skeletal muscle weakness to functional outcomes following critical illness: a translational biology perspective. Thorax 74:1091–8. [DOI] [PubMed] [Google Scholar]
- Bean BP. 2007. The action potential in mammalian central neurons. Nat Rev Neurosci 8:451. [DOI] [PubMed] [Google Scholar]
- Beijers AJM, Mols F, Vreugdenhil G. 2014. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 22:1999–2007. [DOI] [PubMed] [Google Scholar]
- Bennett BK, Park SB, Lin CS-Y, Friedlander ML, Kiernan MC, Goldstein D. 2012. Impact of oxaliplatin-induced neuropathy: a patient perspective. Support Care Cancer 20: 2959–67. [DOI] [PubMed] [Google Scholar]
- Bennett DJ, Li Y, Siu M. 2001. Plateau potentials in sacrocaudal motoneurons of chronic spinal rats, recorded in vitro. J Neurophysiol 86:1955–71. [DOI] [PubMed] [Google Scholar]
- Binda D, Cavaletti G, Cornblath DR, Merkies ISJ, and, and CI-PeriNomS Study Group. 2015. Rasch-Transformed Total Neuropathy Score clinical version (RT-TNSc(©)) in patients with chemotherapy-induced peripheral neuropathy. J Peripher Nerv Syst 20:328–32. [DOI] [PubMed] [Google Scholar]
- Bolton CF, Bryan GY, Zochodne DW. 1993. The neurological complications of sepsis. Ann Neurol. 33:94–100. [DOI] [PubMed] [Google Scholar]
- Boyette-Davis JA, Walters ET, Dougherty PM. 2015. Mechanisms involved in the development of chemotherapy-induced neuropathy. Pain 5:285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briani C, Argyriou AA, Izquierdo C, Velasco R, Campagnolo M, Alberti P, and others. 2014. Long-term course of oxaliplatin-induced polyneuropathy: a prospective 2-year follow-up study. J Peripheral Nerv Syst 19:299–306. [DOI] [PubMed] [Google Scholar]
- Brownstone RM, Krawitz S, Jordan LM. 2010. Reversal of the late phase of spike frequency adaptation in cat spinal motoneurons during fictive locomotion. J Neurophysiol 105:1045–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullinger KL, Nardelli P, Wang Q, Rich MM, Cope TC. 2011. Oxaliplatin neurotoxicity of sensory transduction in rat proprioceptors. J Neurophysiol 106:704–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burakgazi AZ, Messersmith W, Vaidya D, Hauer P, Hoke A, Polydefkis M. 2011. Longitudinal assessment of oxaliplatin-induced neuropathy. Neurology 77:980–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaletti G, Tredici G, Petruccioli MG, Donde E, Tredici P, Marmiroli P, and others. 2001. Effects of different schedules of oxaliplatin treatment on the peripheral nervous system of the rat. Eur J Cancer 37:2457–63. [DOI] [PubMed] [Google Scholar]
- Cavaletti G, Bogliun G, Marzorati L, Zincone A, Marzola M, Colombo N, and others. 1995. Peripheral neurotoxicity of taxol in patients previously treated with cisplatin. Cancer 75:1141–50. [DOI] [PubMed] [Google Scholar]
- Cavaletti G, Marmiroli P. 2010. Chemotherapy-induced peripheral neurotoxicity. Nat Rev Neurol 6:657–66. [DOI] [PubMed] [Google Scholar]
- Cavaletti G, Marmiroli P. 2015. Chemotherapy-induced peripheral neurotoxicity. Curr Opin Neurol 28:500–7. [DOI] [PubMed] [Google Scholar]
- Chatelier A, Zhao J, Bois P, Chahine M. 2010. Biophysical characterisation of the persistent sodium current of the Na v 1.6 neuronal sodium channel: a single-channel analysis. Pflügers Archiv–Eur J Physiol 460:77–86. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Meagher MW, Cleeland CS. 2012. Translational approaches to treatment-induced symptoms in cancer patients. Nat Rev Clin Oncol 9:414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J, and others. 2000. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol 18:2938–47. [DOI] [PubMed] [Google Scholar]
- Dejager L, Pinheiro I, Dejonckheere E, Libert C. 2011. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol 19:198–208. [DOI] [PubMed] [Google Scholar]
- Dietrich J 2010. Chemotherapy associated central nervous system damage. Adv Exp Med Biol 678:77–85. [DOI] [PubMed] [Google Scholar]
- Dietrich J, Han R, Yang Y, Mayer-Pröschel M, Noble M. 2006. CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J Biol 5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsey SG, Kleckner IR, Barton D, Mustian K, O’Mara A, St Germain D, and others. 2019. The National Cancer Institute Clinical Trials Planning Meeting for prevention and treatment of chemotherapy-induced peripheral neuropathy. J Natl Cancer Inst 111:531–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson A, Singer M. 2009. Animal models of sepsis: why does preclinical efficacy fail to translate to the clinical setting? Crit Care Med 37(1 suppl):S30–7. [DOI] [PubMed] [Google Scholar]
- Eidelman LA, Putterman D, Putterman C, Sprung CL. 1996. The spectrum of septic encephalopathy: definitions, etiologies, and mortalities. JAMA 275:470–3. [PubMed] [Google Scholar]
- Fan E, Cheek F, Chlan L, Gosselink R, Hart N, Herridge MS, and others. 2014. An official American Thoracic Society Clinical Practice guideline: the diagnosis of intensive care unit–acquired weakness in adults. Am J Respir Crit Care Med 190:1437–46. [DOI] [PubMed] [Google Scholar]
- Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, and others. 2011. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 12:489–95. [DOI] [PubMed] [Google Scholar]
- Feather CE, Lees JG, Makker PGS, Goldstein D, Kwok JB, Moalem-Taylor G, and others. 2018. Oxaliplatin induces muscle loss and muscle-specific molecular changes in mice. Muscle Nerve 57:650–8. [DOI] [PubMed] [Google Scholar]
- Friedrich O, Reid MB, Van den Berghe G, Vanhorebeek I, Hermans G, Rich MM, and others. 2015. The sick and the weak: neuropathies/myopathies in the critically ill. Physiol Rev 95:1025–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Kavanagh JJ, Hu W, Bast RC. 2006. Clinical application of oxaliplatin in epithelial ovarian cancer. Int J Gynecol Cancer 16:1717–32. [DOI] [PubMed] [Google Scholar]
- Galanski M, Jakupec MA, Keppler BK. 2005. Update of the preclinical situation of anticancer platinum complexes: novel design strategies and innovative analytical approaches. Curr Med Chem 12:2075–94. [DOI] [PubMed] [Google Scholar]
- Golomb D, Donner K, Shacham L, Shlosberg D, Amitai Y, Hansel D. 2007. Mechanisms of firing patterns in fast-spiking cortical interneurons. PLoS Comput Biol 3:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith KA, Dorsey SG, Renn CL, Zhu S, Johantgen ME, Cornblath DR, and others. 2014. Correspondence between neurophysiological and clinical measurements of chemotherapy-induced peripheral neuropathy: secondary analysis of data from the CI-PeriNomS study. J Peripheral Nerv Syst 19:127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutfreund Y, Yarom Y, Segev I. 1995. Subthreshold oscillations and resonant frequency in guinea-pig cortical neurons: physiology and modelling. J Physiol 483:621–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeseler G, Foadi N, Wiegand E, Ahrens J, Krampfl K, Dengler R, and others. 2008. Endotoxin reduces availability of voltage-gated human skeletal muscle sodium channels at depolarized membrane potentials. Crit Care Med 36:1239–47. [DOI] [PubMed] [Google Scholar]
- Harvey PJ, Li X, Li Y, Bennett DJ. 2006a. 5-HT 2 receptor activation facilitates a persistent sodium current and repetitive firing in spinal motoneurons of rats with and without chronic spinal cord injury. J Neurophysiol 96:1158–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey PJ, Li Y, Li X, Bennett DJ. 2006b. Persistent sodium currents and repetitive firing in motoneurons of the sacrocaudal spinal cord of adult rats. J Neurophysiol 96:1141–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman CJ, Enoka RM. 2012. Motor unit. Compr Physiol 2:2629–82. [DOI] [PubMed] [Google Scholar]
- Heide R, Bostock H, Ventzel L, Grafe P, Bergmans J, Fuglsang-Frederiksen A, and others. 2018. Axonal excitability changes and acute symptoms of oxaliplatin treatment: in vivo evidence for slowed sodium channel inactivation. Clin Neurophysiol 129:694–706. [DOI] [PubMed] [Google Scholar]
- Herridge MS, Cheung AM, Tansey CM, Matte-Martyn A, Diaz-Granados N, Al-Saidi F, and others. 2003. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 348:683–93. [DOI] [PubMed] [Google Scholar]
- Hill A, Bergin P, Hanning F, Thompson P, Findlay M, Damianovich D, and others. 2010. Detecting acute neurotoxicity during platinum chemotherapy by neurophysiological assessment of motor nerve hyperexcitability. BMC Cancer 10:451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housley SN, Nardelli P, Carrasco D, Rotterman TM, Pfahl E, Matyunina LV, and others. 2020a. Cancer exacerbates chemotherapy-induced sensory neuropathy. Cancer Res 80:2940–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housley SN, Nardelli P, Powers RK, Rich MM, Cope TC. 2020b. Chronic defects in intraspinal mechanisms of spike encoding by spinal motoneurons following chemotherapy. Exp Neurol 331:113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Suresh AK, Rymer WZ, Suresh NL. 2016. Altered motor unit discharge patterns in paretic muscles of stroke survivors assessed using surface electromyography. J Neural Eng 13:046025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias C, Meunier C, Manuel M, Timofeeva Y, Delestrée N, Zytnicki D. 2011. Mixed mode oscillations in mouse spinal motoneurons arise from a low excitability state. J Neurosci 31:5829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen DB, Stecina K, Wienecke J, Hedegaard A, Sukiasyan N, Hultborn HR, and others. 2018. The subprimary range of firing is present in both cat and mouse spinal motoneurons and its relationship to force development is similar for the two species. J Neurosci 38:9741–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J, Forsythe ID, Kopp-Scheinpflug C. 2010. Symposium review. Going native: voltage-gated potassium channels controlling neuronal excitability. J Physiol 588:3187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone TC, Park GY, Lippard SJ. 2014. Understanding and improving platinum anticancer drugs–phenanthriplatin. Anticancer Res 34:471–6. [PMC free article] [PubMed] [Google Scholar]
- Kaiser J, Bledowski C, Dietrich J. 2014. Neural correlates of chemotherapy-related cognitive impairment. Cortex 54:33–50. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Bostock H, Park SB, Kaji R, Krarup C, Krishnan AV, and others. 2020. Measurement of axonal excitability: consensus guidelines. Clin Neurophysiol 131:308–23. [DOI] [PubMed] [Google Scholar]
- Klink R, Alonso A. 1993. Ionic mechanisms for the subthreshold oscillations and differential electroresponsiveness of medial entorhinal cortex layer II neurons. J Neurophysiol 70:144–57. [DOI] [PubMed] [Google Scholar]
- Krishnan AV, Goldstein D, Friedlander M, Kiernan MC. 2006. Oxaliplatin and axonal Na+ channel function in vivo. Clin Cancer Res. 12:4481–4. [DOI] [PubMed] [Google Scholar]
- Latronico N, Bolton CF. 2011. Critical illness polyneuropathy and myopathy: a major cause of muscle weakness and paralysis. Lancet Neurol 10:931–41. [DOI] [PubMed] [Google Scholar]
- Latronico N, Recupero D, Candiani A, Guarneri B, De Maria G, Antonini L, and others. 1996. Critical illness myopathy and neuropathy. Lancet 347:1579–82. [DOI] [PubMed] [Google Scholar]
- Lee RH, Heckman CJ. 1999. Enhancement of bistability in spinal motoneurons in vivo by the noradrenergic α1 agonist methoxamine. J Neurophysiol 81:2164–74. [DOI] [PubMed] [Google Scholar]
- Lehky TJ, Leonard GD, Wilson RH, Grem JL, Floeter MK. 2004. Oxaliplatin-induced neurotoxicity: acute hyperexcitability and chronic neuropathy. Muscle Nerve 29:387–92. [DOI] [PubMed] [Google Scholar]
- Manuel M, Iglesias C, Donnet M, Leroy F, Heckman CJ, Zytnicki D. 2009. Fast kinetics, high-frequency oscillations, and subprimary firing range in adult mouse spinal motoneurons. J Neurosci 29:11246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall TF, Zipp GP, Battaglia F, Moss R, Bryan S. 2017. Chemotherapy-induced-peripheral neuropathy, gait and fall risk in older adults following cancer treatment. J Cancer Res Pract 4:134–8. [Google Scholar]
- Mazeraud A, Righy C, Bouchereau E, Benghanem S, Bozza FA, Sharshar T. 2020. Septic-associated encephalopathy: a comprehensive review. Neurotherapeutics 17:392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus L, Hu X, Rymer WZ, Suresh NL, Lowery MM. 2017. Motor unit activity during fatiguing isometric muscle contraction in hemispheric stroke survivors. Front Hum Neurosci 11:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicol FJ, Hoyland JA, Cooper RG, Carlson GL. 2010. Skeletal muscle contractile properties and proinflammatory cytokine gene expression in human endotoxaemia. Br J Surg 97:434–42. [DOI] [PubMed] [Google Scholar]
- Miles GB, Dai Y, Brownstone RM. 2005. Mechanisms underlying the early phase of spike frequency adaptation in mouse spinal motoneurones. J Physiol 566:519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miltenburg NC, Boogerd W. 2014. Chemotherapy-induced neuropathy: a comprehensive survey. Cancer Treat Rev 40:872–82. [DOI] [PubMed] [Google Scholar]
- Mols F, Beijers T, Lemmens V, van den Hurk CJ, Vreugdenhil G, van de Poll-Franse LV. 2013. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 31:2699–707. [DOI] [PubMed] [Google Scholar]
- Montagnani F, Turrisi G, Marinozzi C, Aliberti C, Fiorentini G. 2011. Effectiveness and safety of oxaliplatin compared to cisplatin for advanced, unresectable gastric cancer: a systematic review and meta-analysis. Gastric Cancer 14:50–5. [DOI] [PubMed] [Google Scholar]
- Murray KC, Nakae A, Stephens MJ, Rank M, D’amico J, Harvey PJ, and others. 2010. Recovery of motoneuron and locomotor function after spinal cord injury depends on constitutive activity in 5-HT 2C receptors. Nat Med 16:694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KC, Stephens MJ, Ballou EW, Heckman CJ, Bennett DJ. 2011. Motoneuron excitability and muscle spasms are regulated by 5-HT2B and 5-HT2C receptor activity. J Neurophysiol 105:731–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardelli P, Khan J, Powers R, Cope TC, Rich MM. 2013. Reduced motoneuron excitability in a rat model of sepsis. J Neurophysiol 109:1775–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardelli P, Powers R, Cope TC, Rich MM. 2017. Increasing motor neuron excitability to treat weakness in sepsis. Ann Neurol 82:961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardelli P, Vincent JA, Powers R, Cope TC, Rich MM. 2016. Reduced motor neuron excitability is an important contributor to weakness in a rat model of sepsis. Exp Neurol 282:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RY, Li Y, Ray P, Rhines LD, Tatsui CE, Rao G, and others. 2019. Electrophysiological and transcriptomic correlates of neuropathic pain in human dorsal root ganglion neurons. Brain 142:1215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak KR, Nardelli P, Cope TC, Filatov G, Glass JD, Khan J, and others. 2009. Inactivation of sodium channels underlies reversible neuropathy during critical illness in rats. J Clin Invest 119:1150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochala J, Larsson L. 2008. Effects of a preferential myosin loss on Ca2+ activation of force generation in single human skeletal muscle fibres. Exp Physiol 93:486–95. [DOI] [PubMed] [Google Scholar]
- Pandharipande PP, Girard TD, Jackson JC, Morandi A, Thompson JL, Pun BT, and others. 2013. Long-term cognitive impairment after critical illness. N Engl J Med 369:1306–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SB, Goldstein D, Krishnan AV, Lin CS-Y, Friedlander ML, Cassidy J, and others. 2013. Chemotherapy-induced peripheral neurotoxicity: a critical analysis. CA: A Cancer J Clin 63:419–37. [DOI] [PubMed] [Google Scholar]
- Park SB, Goldstein D, Lin CS-Y, Krishnan AV, Friedlander ML, Kiernan MC. 2009a. Acute abnormalities of sensory nerve function associated with oxaliplatin-induced neurotoxicity. J Clin Oncol 27:1243–9. [DOI] [PubMed] [Google Scholar]
- Park SB, Lin CS-Y, Krishnan AV, Goldstein D, Friedlander ML, Kiernan MC. 2009b. Oxaliplatin-induced neurotoxicity: changes in axonal excitability precede development of neuropathy. Brain 132:2712–23. [DOI] [PubMed] [Google Scholar]
- Park SB, Lin CS-Y, Krishnan AV, Goldstein D, Friedlander ML, Kiernan MC. 2011. Long-term neuropathy after oxaliplatin treatment: challenging the dictum of reversibility. Oncologist 16:708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasetto LM, D’Andrea MR, Rossi E, Monfardini S. 2006. Oxaliplatin-related neurotoxicity: how and why? Crit Rev Oncol/Hematol 59:159–68. [DOI] [PubMed] [Google Scholar]
- Perrier J-F. 2013. Modulation of motoneuron activity by serotonin. Curr Pharm Des 19:4371–84. [DOI] [PubMed] [Google Scholar]
- Pitts LR, Simpson SQ. 2010. From mice to men: systematic reviews of animal data could make sepsis trials safer and more productive. Crit Care Med 38:2420–2. [DOI] [PubMed] [Google Scholar]
- Porporato PE. 2016. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 5:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthucheary ZA, Rawal J, McPhail M, Connolly B, Ratnayake G, Chan P, and others. 2013. Acute skeletal muscle wasting in critical illness. JAMA 310:1591–600. [DOI] [PubMed] [Google Scholar]
- Quasthoff S, Hartung HP. 2002. Chemotherapy-induced peripheral neuropathy. J Neurol 249:9–17. [DOI] [PubMed] [Google Scholar]
- Ratté S, Lankarany M, Rho Y-A, Patterson A, Prescott SA. 2015. Subthreshold membrane currents confer distinct tuning properties that enable neurons to encode the integral or derivative of their input. Front Cell Neurosci 8:452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robba C, Crippa IA, Taccone FS. 2018. Septic encephalopathy. Curr Neurol Neurosci Rep 18:82. [DOI] [PubMed] [Google Scholar]
- Roberts BM, Frye GS, Ahn B, Ferreira LF, Judge AR. 2013. Cancer cachexia decreases specific force and accelerates fatigue in limb muscle. Biochem Biophys Res Commun 435:488–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol B, Gueret G, Pennec J-P, Morel J, Giroux-Metges MA, Talarmin H, and others. 2007. Effects of chronic sepsis on the voltage-gated sodium channel in isolated rat muscle fibers. Crit Care Med 35:351–7. [DOI] [PubMed] [Google Scholar]
- Saif MW, Syrigos K, Kaley K, Isufi I. 2010. Role of pregabalin in treatment of oxaliplatin-induced sensory neuropathy. Anticancer Res 30:2927–33. [PubMed] [Google Scholar]
- Sakai H, Sagara A, Arakawa K, Sugiyama R, Hirosaki A, Takase K, and others. 2014. Mechanisms of cisplatin-induced muscle atrophy. Toxicol Appl Pharmacol 278:190–9. [DOI] [PubMed] [Google Scholar]
- Schefold JC, Bierbrauer J, Weber-Carstens S. 2010. Intensive care unit-acquired weakness (ICUAW) and muscle wasting in critically ill patients with severe sepsis and septic shock. J Cachexia Sarcopenia Muscle 1:147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, Wilson CJ. 2011. The ionic mechanism of gamma resonance in rat striatal fast-spiking neurons. J Neurophysiol 106:2936–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semmler A, Widmann CN, Okulla T, Urbach H, Kaiser M, Widman G, and others. 2013. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry 84:62–9. [DOI] [PubMed] [Google Scholar]
- Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, and others. 2014. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Pain 155:2461–70. [DOI] [PubMed] [Google Scholar]
- Simó M, Rifà-Ros X, Rodriguez-Fornells A, Bruna J. 2013. Chemobrain: a systematic review of structural and functional neuroimaging studies. Neurosci Biobehav Rev 37: 1311–21. [DOI] [PubMed] [Google Scholar]
- Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, and others. 2016. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315:801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittl R, Lampert A, Huth T, Schuy ET, Link AS, Fleckenstein J, and others. 2012. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype Na(V)1.6-resurgent and persistent current. Proc Natl Acad Sci U S A 109:6704–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staff NP, Grisold A, Grisold W, Windebank AJ. 2017. Chemotherapy-induced peripheral neuropathy: a current review. Ann Neurol, 81:772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale MJ. 2002. Cachexia in cancer patients. Nat Rev Cancer 2:862–71. [DOI] [PubMed] [Google Scholar]
- Tofthagen C, Donovan KA, Morgan MA, Shibata D, Yeh Y. 2013. Oxaliplatin-induced peripheral neuropathy’s effects on health-related quality of life of colorectal cancer survivors. Support Care Cancer 21:3307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderVeen BN, Fix DK, Carson JA. 2017. Disrupted skeletal muscle mitochondrial dynamics, mitophagy, and biogenesis during cancer cachexia: a role for inflammation. Oxid Med Cell Longev 2017:3292087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco R, Bruna J, Briani C, Argyriou AA, Cavaletti G, Alberti P, and others. 2014. Early predictors of oxaliplatin-induced cumulative neuropathy in colorectal cancer patients. J Neurol Neurosurg Psychiatry. 85:392–8. [DOI] [PubMed] [Google Scholar]
- Vincent JA, Wieczerzak KB, Gabriel HM, Nardelli P, Rich MM, Cope TC. 2016. A novel path to chronic proprioceptive disability with oxaliplatin: distortion of sensory encoding. Neurobiol Dis 95:54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RG, Brain KL, Wilson RH, Grem JL, Vincent A. 2005. Oxaliplatin induces hyperexcitability at motor and autonomic neuromuscular junctions through effects on voltage-gated sodium channels. Br J Pharmacol 146:1027–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RH, Lehky T, Thomas RR, Quinn MG, Floeter MK, Grem JL. 2002. Acute oxaliplatin-induced peripheral nerve hyperexcitability. J Clin Oncol 20:1767–74. [DOI] [PubMed] [Google Scholar]
- Winocur G, Berman H, Nguyen M, Binns MA, Henkelman M, van Eede M, and others. 2018. Neurobiological mechanisms of chemotherapy-induced cognitive impairment in a transgenic model of breast cancer. Neuroscience 369: 51–65. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Giocomo LM, Boardman I, Hasselmo ME. 2011. Frequency of subthreshold oscillations at different membrane potential voltages in neurons at different anatomical positions on the dorsoventral axis in the rat medial entorhinal cortex. J Neurosci 31:12683–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young GB, Bolton CF, Austin TW, Archibald YM, Gonder J, Wells GA. 1990. The encephalopathy associated with septic illness. Clin Invest Med 13:297–304. [PubMed] [Google Scholar]
- Zajączkowska R, Kocot-Kępska M, Leppert W, Wrzosek A, Mika J, Wordliczek J. 2019. Mechanisms of chemotherapy-induced peripheral neuropathy. Int J Mol Sci 20:1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Z’Graggen Josef W, Lin CS-Y, Howard RS, Beale RJ, Bostock H. 2006. Nerve excitability changes in critical illness polyneuropathy. Brain. 129:2461–70. [DOI] [PubMed] [Google Scholar]
- Zochodne DW, Bolton CF, Wells GA, Gilbert JJ, Hahn AF, Brown JD, and others. 1987. Critical illness polyneuropathy: a complication of sepsis and multiple organ failure. Brain 110:819–41. [DOI] [PubMed] [Google Scholar]