

Summary
Members of a disintegrin and metalloproteinases with thrombospondin motif (ADAMTS) family have been implicated in various vascular diseases. However, their functional roles in early embryonic vascular development are unknown. In this study, we showed that Adamts18 is highly expressed at E11.5-E14.5 in cells surrounding the embryonic aortic arch (AOAR) and the common carotid artery (CCA) during branchial arch artery development in mice. Adamts18 deficiency was found to cause abnormal development of AOAR, CCA, and the third and fourth branchial arch appendages, leading to hypoplastic carotid body, thymus, and variation of middle cerebral artery. Adamts18 was shown to affect the accumulation of extracellular matrix (ECM) components, in particular fibronectin (Fn), around AOAR and CCA. As a result of increased Fn accumulation, the Notch3 signaling pathway was activated to promote the differentiation of cranial neural crest cells (CNCCs) to vascular smooth muscle cells. These data indicate that Adamts18-mediated ECM homeostasis is crucial for the differentiation of CNCCs.
Subject areas: cell biology, developmental biology
Graphical abstract
Highlights
-
•
Adamts18 is highly expressed during the period of embryonic carotid artery formation
-
•
Adamts18 deficiency leads to abnormal AOAR, CCA, MCA branches and carotid appendages
-
•
Adamts18 deficiency changes the cerebral infarcted areas in mouse tMCAO model
-
•
Adamts18 affects the differentiation of CNCCs to VSMCs by modulating Notch3 pathway
Cell biology; Developmental biology;
Introduction
Extracellular matrix (ECM) is a highly dynamic structure composed of collagens, elastin, fibronectin (Fn), laminins, proteoglycans (e.g.,versican and aggrecan), and polysaccharides. ECM components are constantly modified, degraded, and re-deposited (Theocharis et al., 2016). This process is referred to as remodeling. It has been shown that abnormal ECM remodeling leads to unregulated cell proliferation, differentiation, and adhesion (Lu et al., 2011), resulting in developmental defects (Mongiat et al., 2016; Lu et al., 2011). Metalloproteases, such as matrix metalloproteinases (MMPs), a disintegrin and metalloproteinases (ADAMs), and ADAMs with thrombospondin motifs (ADAMTSs) have the ability to cleave various components of ECM (Lu et al., 2011). However, ADAMTSs differ from MMPs and ADAMs in function, binding and anchoring properties, substrate recognition, and longevity. Investigation of ADAMTS-mediated ECM homeostasis would aid in the understanding of the pathogenesis of certain congenital vascular malformation and related diseases.
ADAMTSs are a group of 19 secreted metalloproteinases with major roles in development, morphogenesis, and tissue repair via the assembly and degradation of ECM (Kelwick et al., 2015; Apte, 2009). Recent studies showed that ADAMTS members participate in cardiovascular physiology and diseases (Santamaria and de Groot, 2020), such as the development of blood vessels and heart valves, atherosclerosis, pulmonary arterial hypertension, and aneurysm. ADAMTS-1, -4, -5, -8, -9, -15, and -20 are involved in the degradation of hyalectans, including aggrecan, versican, and brevican. ADAMTS-1 promotes atherogenesis by cleaving versican and inducing vascular smooth muscle cell (VSMC) migration (Jonsson-Rylander et al., 2005). Adamts1 haploinsufficient mice challenged with angiotensin II have been shown to develop thoracic aortic aneurysm formation (Oller et al., 2017). Loss of ADAMTS-4 is associated with attenuated atherosclerosis in apolipoprotein E-deficient mice on high-fat diet, resulting in a more stable plaque phenotype with reduced lipid content and number of macrophages (Ren et al., 2017). ADAMTS-5 has been shown to modulate vascular proteoglycan turnover and lipoprotein retention in murine atherosclerosis (Didangelos et al., 2012). ADAMTS-5 downregulation and accumulation of aggrecan and versican are evident in human thoracic aortic aneurysm (Cikach et al., 2018). Both ADAMTS-5 and ADAMTS-9 cleave versican and are essential for heart development (Dupuis et al., 2011; Kern et al., 2010). Although much progress has been made in understanding the relationship between ADAMTSs and vascular disease, the roles of ADAMTS members in embryonic vascular morphogenesis remain to be determined.
ADAMTS-18, an orphan ADAMTS, is less understood in its functions and substrate profiles. In humans, ADAMTS-18 mutations have been linked to various malignancies (Jin et al., 2007), developmental eye disorders (Aldahmesh et al., 2013; Peluso et al., 2013; Aldahmesh et al., 2011), reduced bone mineral density (Xiong et al., 2009), and aberrant white matter integrity (Lopez et al., 2012). We have recently created an Adamts18-deficient zebrafish model using morpholino antisense oligonucleotides to generate exon 3 skipped adamts18 mRNA transcripts (Lu et al., 2020b). With this model, Adamts18 was found to be involved in early vascular formations. However, mechanisms of such regulation remain largely unknown. In this study, we used Adamts18 knockout (Adamts18−/−) mice to investigate the role of Adamts18 in vascular development and found that Adamts18 modulates the differentiation fate of cranial neural crest cells (CNCCs) into VSMCs by affecting ECM homeostasis, in particular Fn abundance.
Results
Expression of Adamts18 in mouse branchial arch arteries
The presence of Adamts18 mRNA in mouse branchial arch arteries between E10.5 and postnatal day 1 (P1) was determined by in situ hybridization (Figure 1A). At E10.5, Adamts18 mRNA was detected near the dorsal arch artery (DoA) but was barely seen around the third (third) and fourth (fourth) branchial arch arteries. Adamts18 mRNA levels were increased along with branchial arch artery development. From E11.5 to E14.5, Adamts18 mRNA was abundant in cells (mesenchymal cells and fibroblasts) (Figure S1) surrounding the aortic arch (AOAR), common carotid artery (CCA), thymus primordium, trachea, and esophagus. Adamts18 mRNA levels were decreased at the end of gestation. At E16.5, Adamts18 mRNA was very low in abundance and was detected only in cells of the outer layer of the CCA. At P1, Adamts18 mRNA was undetectable in the CCA. These results suggest that Adamts18 is a phase-specific gene and is highly expressed at E11.5-E14.5 during branchial arch artery development in mice (Figure 1B).
Figure 1.

Expression of Adamts18 in mouse branchial arch arteries
(A) ISH-positive signals are shown as pink dots in cells. Sagittal sections: E10.5 (a), E11.5 (b), E14.5 (i); transverse sections: E12.5 (c), E13 (e, f), E13.5 (g), E14.5 (h, j), E16.5 (k), and P1(l). 3rd, third branchial arch arteries; 4th, fourth branchial arch arteries; DoA, dorsal artery; TR, trachea; E, esophagus; T, thymus primordium; AOAR, aortic arch; L(R)-CCA, left (right) common carotid artery. Scale bar represents 50 μm for E14.5; 100 μm for E10.5, E11.5, E16.5, and P1; and 200 μm for E12.5, E13, and E13.5 images.
(B) Schematic diagram of the spatio-temporal expression pattern of Adamts18 mRNA at different stages of branchial arch artery development in mice.
These experiments were repeated independently at least three times.
Adamts18 deficiency leading to malformation of the aortic arch and the carotid artery system
The effect of Adamts18 on morphological transformation of branchial arch arteries was then examined. From E10.5 to E12.5, the development of the third, fourth, and sixth branchial arch arteries was similar, and these arteries were connected to the dorsal artery normally. There was no significant difference in the pattern of the third and fourth branchial arch arteries to the AOAR and CCA in the embryos of both Adamts18+/+ and Adamts18−/− mice as observed by intracardiac injection of India ink (Figure 2A). After E13.5, both innominate artery (IA) and left common carotid artery (L-CCA) were extended out from the AOAR, but the interval between them was significantly broad in Adamts18−/− mice compared to Adamts18+/+ mice. The CCA was found to bifurcate to become internal and external carotid arteries (ICA-ECA) at the upper end of the larynx in adult Adamts18+/+ mice. However, abnormal carotid bifurcation was observed near the heart apart from the upper end of the larynx in all Adamts18−/− mice (Figure 2B). Cardiovascular casting confirmed that Adamts18 deficiency results in increased interval between IA and L-CCA and reduced length of CCA between the beginning of the AOAR and the branch point of ICA-ECA (Figures 2C–2E).
Figure 2.

Correlation between Adamts18 deficiency and hypogenesis of the carotid artery system
(A and B) Representative images of the arch artery visualized by intracardiac ink injection at E10.5, E11.5, E12, E12.5 (A) and the carotid artery in wild-type (Adamts18+/+) and Adamts18-/- mice (A, E13.5 and E14.5; and B, 8-week-old mice). DOA, dorsal artery; H, heart; third, fourth, and sixth, third, fourth, and sixth branchial arch arteries; TC, thyroid cartilage; ECA, external carotid artery; ICA, internal carotid artery; AOAR, aortic arch; RSCA, right subclavian artery. Scale bar, 2 mm. Numbers of embryos of each genotype per group in (A) are indicated. Numbers of mice used for vascular genotype analysis (B) were 192 for Adamts18+/+ mice (n = 192) and 156 for Adamts18−/− mice (n = 156). Equal numbers of male and female mice (two weeks or older) were used for vascular genotype analysis. All Adamts18−/− mice, regardless of sex, had developmental vascular malformation.
(C) Representative images of cardiovascular system casting in 8-week-old male Adamts18+/+ and Adamts18−/− mice.
(D and E) Determination of the distance between innominate artery and L-CCA (D) and the length of CCA from ascending aortic arch starting point to ICA-ECA branch point (E) in Adamts18+/+ and Adamts18−/− mice. Results are expressed as mean ± SD (n = 4/group); ∗p < 0.05; ∗∗p < 0.01; two-tailed Student's t-test.
Carotid body defect and hypoplastic thymus in Adamts18−/− mice
The carotid body (CB) is situated in the carotid artery bifurcation in close proximity to the superior cervical ganglion (SCG) of the sympathetic trunk (Clarke and de Burgh Daly, 1981). All Adamts18−/− mice had no CB at the bifurcation site. In contrast, CB was found in Adamts18+/+ mice in both left and right sides of the carotid artery (Figure 3A). Consecutive sections of the CB from Adamts18+/+ mice at E18.5 showed that the CB always joins with the SCG of the sympathetic trunk (Figure 3B, upper panel). However, no cells streaming toward the carotid artery and no CB were detected in Adamts18−/− mice. Immunostaining of neurons of the SCG and glomus cells of CB with anti-tyrosine hydroxylase (TH) antibody revealed that TH-positive nerve fibers were densely distributed in both the SCG and CB of E16.5 Adamts18+/+ mice (Figure 3B, lower panel); however, only the SCG of Adamts18−/− embryos displayed the presence of tyrosine hydroxylase.
Figure 3.

Carotid body defect and hypoplastic thymus in Adamts18−/−mice
(A) Representative images of carotid body in 8-week-old mice. Left view (a, c) and right view (b, d) of carotid body in Adamts18+/+ and Adamts18-/- mice. Numbers of male and female mice used for carotid body analysis are the same as those described in Figure 1B.
(B) Histologic examination of carotid body. Upper images: representative images of hematoxylin-eosin (HE)-stained carotid body sections of Adamts18+/+ (a) and Adamts18-/- mice (c) at E18.5. Lower images: immunohistochemical staining of tyrosine hydroxylase (TH) in embryonic (E16.5) Adamts18+/+ (b) and Adamts18-/- mice (d). All sections are consecutive transverse sections. CCA, common carotid artery; ECA, external carotid artery; ICA, internal carotid artery; SCG, superior cervical ganglion; CB, carotid body. Scale bar, 100 μm.
(C) Representative images of thymus in Adamts18+/+ and Adamts18−/− mice at indicated development and growth stages. The green dotted ovals indicate the thymus.
(D) Representative images of modified Masson's Trichrome-stained thorax sections of Adamts18+/+ and Adamts18−/− mice at E14.5 and E16.5. L(R)-CCA, left (right) common carotid artery; TR, trachea. Scale bar, 200 μm.
(E) Thymus weight-to-body weight ratio (%) at different time points. Each dot or square represents one individual; data means are shown with solid horizontal lines.
Data are expressed as mean ± SD. Statistical significance: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; two-tailed Student's t-test.
(F) Ratio of CD4+ cells to CD8+ cells at indicated development stages. Data are expressed as mean ± SD (n = 5/group).
The thymus is derived from the third branchial arch that is attached to the AOAR. From E14.5 to 9 months, the thymus of each Adamts18+/+ mouse was appressed and plumped with a smooth contour, whereas that of each Adamts18−/− mouse was separated with a tipped contour (Figure 3C). The Adamts18+/+ thymus, viewed from the ventral side, was found to cover the AOAR, CCA, and trachea at E14.5 and E16.5; however, Adamts18−/− thymus was ectopic and did not cover the AOAR (Figure 3D). There was a significant decrease in the ratio of the thymus to body weight of Adamts18−/− mice at the time points of 2 weeks, 5 weeks, and 7 to 9 months (Figure 3E). However, the ratio of tissue CD4+ to CD8+ cells was comparable between the two genotypes of mice (Figure 3F). There was no apparent abnormality in the thyroids and parathyroids of Adamts18−/− mice (data not shown).
Alterations of middle cerebral artery branch pattern in Adamts18−/− mice
The middle cerebral artery (MCA) is the major terminal branch of the internal carotid artery (ICA) that supplies the neocortex and basal ganglia. To investigate whether Adamts18 deficiency affects MCA morphogenesis, cerebral vascular casting was performed to show MCA branching pattern. Results showed that Adamts18−/− mice had increased number of deep branches but had decreased number of cortical branches in the MCA (Figures 4A–4C). To determine whether the alteration in MCA branch pattern in Adamts18−/− mice affects cerebral infarction, transient MCA occlusion (tMCAO) was performed. Results showed that the cerebral infarcted areas in 8-week-old Adamts18−/− mice were significantly different from those of Adamts18+/+ littermates (Figure 4D). The cerebral infarcted areas in Adamts18+/+ mice were mainly restricted to the caudate and putamen (basal ganglia) and were not seen in the neocortex, probably because ischemic cortical tissue is salvaged by collateral blood supply and reperfusion (Memezawa et al., 1992). There was a sharp boundary between the cortex and the basal ganglia in Adamts18+/+ mice. In contrast, the cerebral infarcted areas in Adamts18−/− mice were mainly located in the cortex and spread to the basal ganglia, and there was no obvious boundary between the cortex and the basal ganglia. The neurological severity score of Adamts18+/+ mice was significantly higher than that of Adamts18−/− mice due to damage in deep brain regions (Figure 4E). Magnetic resonance imaging after reperfusion at 1.5 h, 7 h, and 24 h (Figure S2) revealed that the cerebral infarcted areas in Adamts18+/+ brains were in the basal ganglia at 1.5 h and that the diffusion of penumbra was impeded by the boundary between the cortex and the basal ganglia. However, the diffusion of penumbra from the cortex to the basal ganglia completely lacked boundary in Adamts18−/− mice. MCA branching pattern and cerebral infarcted areas are illustrated in Figure 4F based on these findings.
Figure 4.

Alterations of middle cerebral artery branch pattern in Adamts18−/− mice
(A) Representative images of intracranial middle cerebral artery (MCA) branches in Adamts18+/+ and Adamts18−/− mice. ECA, external carotid artery; ICA, internal carotid artery; ACA, anterior cerebral artery; PCA, posterior cerebral artery; PPA, pterygopalatine artery; V-BA, vertebral basilar artery. Scale bar represents 2 mm and 5 mm, respectively.
(B and C) Quantification of the number of MCA cortical branches (B) and deep branches (C) in the different genotypes. Second, secondary vascular level; third, third vascular level. Data are expressed as mean ± SD (n = 7–8/group), ∗∗∗p < 0.001; two-tailed Student's t-test.
(D) Representative images of the cerebral infarcted areas in Adamts18+/+ and Adamts18−/− mice. Mouse brains were dissected and stained with triphenyltetrazolium chloride (TTC). White, infarction area.
(E) Quantification of the severity of neurological defect as described in methods. Data are expressed as mean ± SD (n = 7–8/group),∗∗∗p < 0.001; two-tailed Student's t-test.
(F) Diagram of predicted MCA branch and infarction areas. Pentagram represents blocked site; (1) cortical branches of ACA; (2) central branches of ACA; (3) cortical branches of MCA; (4) central branches of MCA.
See also Figure S2.
Expression of ECM-related molecules in cervicothoracic vascular tissue of Adamts18−/− mice
To investigate the underlying mechanisms by which Adamts18 affects the development of AOAR and CCA, the expression of major ECM molecules in cervicothoracic vascular tissue at E14.5 was examined. The mRNA levels of Fn1, Fbn2, and Col1a2 were found to be increased, and those of Lnb1 were decreased in Adamts18−/− cervicothoracic vascular tissue (Figure 5A). A previous study showed that Adamts18 cleaves Fn at its N-terminal end to release a fragment of approximately 30 kDa (Ataca et al., 2020). In Adamts18+/+ mice, Fn was found to distribute in the vascular wall, and increased Fn deposition was observed around the CCA, trachea, and thymus that expressed Adamts18 (Figure 1A) in Adamts18−/− mice (Figures 5B-a,b). Compared with Adamts18+/+ CCA, the level of Fbn1 protein in Adamts18−/− CCA was significantly increased (Figures 5C and 5E). Hart's staining showed that the number of elastic fiber layers was increased in the CCA of neonatal Adamts18−/− mice (Figure 5D). Adult Adamts18−/− mice exhibited significantly reduced carotid artery blood pressure as determined with a Doppler flow meter (Figure 5F). The expression areas of collagen, laminin, aggrecan, and versican in Adamts18−/− CCA at E14.5 or P1 were also shown (Figures S3–S5).
Figure 5.

Expression of ECM molecules in cervicothoracic vascular tissue of Adamts18−/− mice
(A) qRT-PCR results of ECM molecules (Fn1, Fbn1, Fbn2, Col4a1, Col1a1, Col1a2, Col3a1, Lna1, Lna3, Lna5, Lnb1, and Lnc1). The relative quantity of target mRNA was normalized to that of the housekeeping gene Gapdh using the ΔΔCt method.
Each dot or square represents one individual (n = 3–4/group). Data are expressed as mean ± SD. ∗p < 0.05; two-tailed Student's t-test.
(B and C) Representative immunofluorescence images of fibronectin (Fn) (B) and fibrillin-1 (Fbn1) (C) in E14.5 Adamts18+/+ and Adamts18−/− cervicothoracic tissue.
(D) Representative images of Hart's staining at E14.5 (a, b) and P1(c, d).
(E) Quantification of Fbn1 intensity (n = 4/group) was performed with Image-Pro Plus. Scale bar represents 50 μm. AU, arbitrary units. ∗∗p < 0.01; two-tailed Student's t-test.
(F) Carotid blood pressure of Adamts18+/+ (n = 12) and Adamts18−/− mice (n = 11) determined with a Doppler flow meter. Each dot represents one individual mouse. Approximately an equal number of male and female mice aged 8–10 weeks were used. Data are expressed as mean ± SD. ∗p < 0.05; two-tailed Student's t-test.
See also Figures S3–S5 and Tables S1–S4.
Expression of EMT-related molecules in cervicothoracic vascular tissue of Adamts18−/− mice
As the change in the abundance of ECM molecules suggests the occurrence of epithelial-mesenchymal transition (EMT), mRNA levels of several key EMT molecules were determined, including those of transcription factors (e.g., Snail1 and Snail2), cell surface proteins (e.g., Cdh1, Cdh2, Itgav, and Itgb1), and cytoskeletal markers (e.g., Vim, Krt8, Krt18, and Fsp1). Results showed that the expression of Cdh2 was significantly increased and that of Krt8 was decreased in E14.5 Adamts18−/− cervicothoracic tissue. No significant change in mRNA levels of other EMT-related molecules was observed (Table S3).
Expression of DiGeorge syndrome and vascular development-related molecules in cervicothoracic vascular tissue of Adamts18−/− mice
DiGeorge syndrome (DGS) is a human genetic disorder due to a hemizygous microdeletion of chromosome 22q11.2 (TBX1), resulting in abnormal migration and distribution of the neural crest cells originated from the third and fourth pharyngeal sacs and hypoplasia of multiple organs, such as the cardiac outflow tract, AOAR, and thymus (Epstein, 2001). To investigate the effect of Adamts18 deficiency on the expression of DGS-related molecules, mRNA levels of Tbx1, Gbx2, Srf, and Tgfb1 were determined. The mRNA levels of molecules associated with vascular development (e.g., Lims1; transcription factors Ascl1 and Hoxa3; and Hippo pathway molecules Yap and Taz) in cervicothoracic vascular tissues at E14.5 were also assessed (Table S4). Results showed that the expression of CNCC patterning-related gene Gbx2 and Notch signaling pathway-related gene Ascl1 was significantly upregulated.
Activated Notch3 signaling and increased differentiation of CNCCs into VSMCs in Adamts18−/− CCA
Examination of the expression of Notch-related genes showed that mRNA levels of Notch3, Hes5, Hey2, and Acta2 were significantly increased (Figure 6A). By western blotting, protein levels of the Notch3 intracellular domain N3ICD and its downstream targets Hes5 and Hey2 were found to be significantly elevated in Adamts18−/− CCA (Figures 6B and 6C). Immunofluorescence staining showed that the expression of α-Sma (Acta2), a marker of vascular smooth muscle, was significantly increased in all stages of CCA development in Adamts18−/− mice compared to Adamts18+/+ mice (Figures 6D and 6E).
Figure 6.

Activated Notch3 signaling and increased differentiation of cranial neural crest cells (CNCCs) to vascular smooth muscle cells (VSMCs) in Adamts18−/− CCA
(A) qRT-PCR results of Notch-related genes (Jag1, Adam10, Notch3, Hes1, Hes5, Hey1, Hey2, Heyl, and Acta2) in cervicothoracic vascular tissue of E14.5 Adamts18+/+ and Adamts18−/− embryos. The relative quantity of target mRNA was normalized to that of the housekeeping gene Gapdh using the ΔΔCt method. Each dot or square represents one individual mouse. Data are expressed as mean ± SD (n = 4/group). ∗p < 0.05; two-tailed Student's t-test.
(B and C) Levels of Notch3, Hes5, and Hey2 in E14.5 Adamts18+/+ and Adamts18−/− CCA determined by western blotting. The relative quantity of Notch3 (NCID), Hey2, and Hess proteins is normalized to that of Gapdh and expressed as mean ± SD (n = 4). ∗p < 0.05; two-tailed Student's t-test.
(D) Representative immunofluorescence images of Acta2 in E14.5, E16.5, and P1 CCA sections. Scale bar, 50 μm.
(E) Quantification of Acta2 levels at indicated development stages was performed with Image-Pro Plus. AU, arbitrary units. Data are expressed as mean ± SD (n = 3–6/group). ∗p < 0.05; ∗∗∗p < 0.001; two-tailed Student's t-test. These experiments were repeated independently at least three times.
See also Tables S1–S4.
Discussion
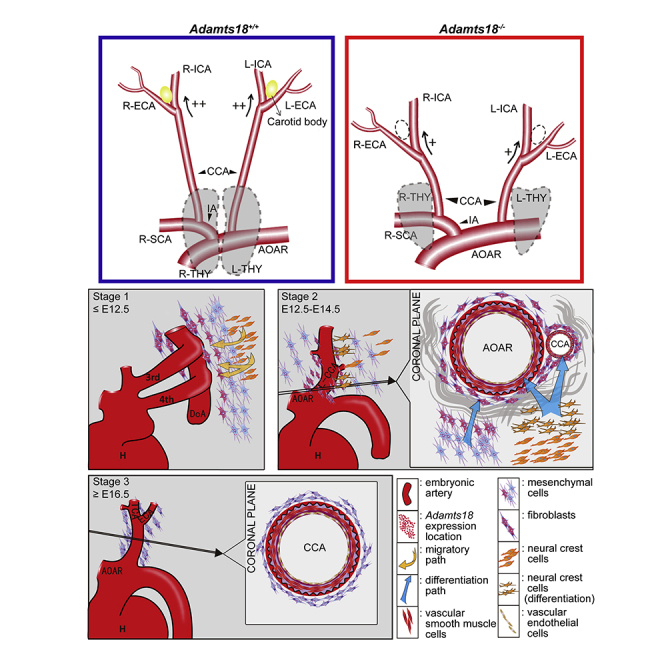
In this study, we demonstrated that Adamts18 is expressed in cells (mesenchymal cells and fibroblasts) around the AOAR and the CCA when CNCCs differentiate into VSMCs in mice. Adamts18 deficiency was found to cause ECM remodeling, in particular, increased Fn accumulation, thus activating the Notch3 signaling pathway (Figure 7). These actions affected the differentiation fate of neural crest cells into VSMCs and the development of the AOAR and the CCA from the third and fourth branchial arch great vessels and their derivatives (e.g., CB and thymus). Our results are complementary to those of Ataca et al. who found that Adamts18 deficiency results in congenital disorders of the lens, lung, and female reproductive tract (Ataca et al., 2016).
Figure 7.

Schematic illustration of the role of Adamts18 in neural crest cell-derived artery development
Adamts18 is a phase-specific gene and is highly expressed at E11.5-E14.5 in cells (mesenchymal cells and fibroblasts) surrounding the embryonic aortic arch (AOAR) and the common carotid artery (CCA) during branchial arch artery development in mice. Adamts18 binds to Fbn1 (Lu et al., 2020a) and Fn and cleaves Fn (Ataca et al., 2020). Adamts18 deficiency results in abnormal ECM remodeling, in particular, increased Fn accumulation, thus activating the Notch3 signaling pathway to promote the differentiation of cranial neural crest cells (CNCCs) to vascular smooth muscle cells.
Adamts18 expression pattern and the development of the AOAR and the CCA
Vasculature formation is triggered by multiple factors or events including growth factors, remodeling and degradation of ECM components by metalloproteinases, and certain membrane receptor signaling pathways. These stimuli affect the growth, differentiation, and movement of VSMCs to generate branched tubular networks (Senger and Davis, 2011). Results of in situ hybridization showed that Adamts18 mRNA was most abundant in the regions where the third and fourth branchial arch arteries develop to become the ascending aorta and carotid artery (Figure 1). Moreover, the stages with high expression of Adamts18 coincide with the periods of morphogenesis of the third and fourth vessels and the development of the AOAR and CCA. These results suggest that Adamts18 plays an important role in the development of the AOAR and CCA and their derivatives in mice.
Adamts18-mediated ECM homeostasis in the differentiation of CNCCs to VSMCs
The origins of VSMCs include neural-crest-derived and non-neural-crest-derived cells. The VSMCs of the mouse ascending aorta and carotid artery have been shown to be derived from CNCCs (Majesky, 2007). As an ECM molecule, Fn is essential for neural-crest-mediated morphogenesis of the AOAR (Majesky, 2007). Fn has been shown to promote the differentiation of neural crest cells to smooth muscle cells by activating Notch signaling (Wang and Astrof, 2016). Adamts18 has been shown to regulate mammary stem cell niche by directly cleaving Fn at its N-terminal end to release a fragment of approximately 30 kDa (Ataca et al., 2020). We hypothesized that the abnormal vascular phenotype in Adamts18−/− mice is associated with altered Fn levels. In this study, we found that Adamts18 deficiency leads to Fn deposition around the CCA (Figure 5). As the deposition regions of Fn completely overlap with the expression regions of Adamts18 mRNA, it is likely that such Fn deposition is due to reduced proteolytic cleavage by Adamts18. It is also possible that the increase in Fn is due to increased Fn1 transcription. In addition, the expression of members (e.g., Hes5 and Hey2) of the Notch3 pathway (Hosseini-Alghaderi and Baron, 2020) was found to be upregulated in cervicothoracic vascular tissue of Adamts18−/− mice (Figure 6), suggesting that Adamts18 regulates the differentiation of CNCCs to VSMCs by activating Notch3 signaling as a result of increased Fn levels.
Mature elastic fibers are composed of elastin (90%) and fibrillin-rich microfibrils (10%). The main constituents of microfibrils are fibrillin-1 and fibrillin-2, which are two structurally related glycoproteins with distinct temporal and spatial expression patterns. Previous studies showed that some ADAMTS (e.g., ADAMTS-6, ADAMTS-10, and ADAMTS-17) proteases can bind to fibrillin-1 or fibrillin-2 or both (Hubmacher et al., 2017; Cain et al., 2016; Hubmacher and Apte, 2011; Kutz et al., 2011). Recently, we found that Adamts18 modulates bronchial fibrillin microfibril formation by binding to fibrillin-1 (Lu et al., 2020a). In this study, the levels of fibrillin-1 were found to be significantly increased in E14.5 CCAs of Adamts18−/− mice, and Adamts18−/− CCA was found to have disordered elastic fibers and reduced carotid blood pressure (Figure 5). These results suggest that Adamts18 is also involved in the regulation of carotid microfibril assembly and synthesis. It is conceivable that the change in the abundance of ECM molecules is secondary to reduced Adamts18-mediated cleavage of matrix proteins, thus affecting cell fates as previously postulated (Nicolas et al., 2020; Muncie and Weaver, 2018).
Adamts18 and EMT
During embryonic morphogenesis, type 1 EMT (type 1 EMT) is crucial and is associated with the relocation of cells from a basement membrane to a fibrillar ECM microenvironment (Zeisberg and Neilson, 2009). Fn is the first molecule to appear when fibrillar ECM is formed, and such appearance is an indication of type 1 EMT. Our data showed that the expression of some markers of type 1 EMT was either increased (e.g., Cdh2) or decreased (e.g., Keratin 8) in Adamts18−/− vascular tissues (Table S3). We postulate that the change in the abundance of these molecules is a secondary response to the changes in ECM molecules instead of excessive EMT in Adamts18−/− CCAs.
Adamts18 and DGS
Adamts18−/− mice exhibit some DGS-like malformations, such as short CCA (premature ICA/ECA branching) and lack of CB at the bifurcation site and increased starting point interval between the IA and L-CCA. The interval between left and right thymus glands was widened (Figure 3). Normally, the two thymuses are clung together in the ventral side of the two starting points. We speculate that the malformation of CB and thymus is caused by the lack of or misplaced localization signals due to vascular malformation. Nevertheless, the possibility of weakened migration and differentiation of neural crest cells to CB and thymus remain to be determined. It has been reported that Hoxa3 homozygous mutant mice also lack the CB and that the malformation of the carotid artery system is due to the defect in the development of the third arch artery (Kameda, 2009). Tbx1 heterozygous mutant mice have been shown to have a high frequency of lacking or hypoplasia of the fourth pharyngeal arch artery at E10.5 and other great artery anomalies at later stages of development (Ryckebusch et al., 2010). In this study, CCA malformation in Adamts18−/− mice was not found to be the consequence of arch artery defect (Figure 2); it was found to be associated with increased accumulation of ECM molecules and overdifferentiation of CNCCs to VSMCs during the process of the AOAR and carotid artery formation and extension after E12.5.
Adamts18 and middle cerebral artery
The MCA is the largest cerebral artery and is the vessel most commonly affected by cerebrovascular accident. MCA occlusion (MCAO) at proximal sites would cause infarctions in the neocortex and basal ganglia. Clinical vessel occlusion followed by resolution of clots and reperfusion can be simulated with the MCAO paradigm by withdrawing the intraluminal thread (the so-called transient MCAO model). In this study, Adamts18−/− mice were found to have alterations in MCA branches, including increased complexity of deep branches and reduced complexity of cortical branches (Figure 4). We hypothesized that MCA alteration in Adamts18−/− mice affects the outcome of ischemic stroke. In the tMCAO model, the infracted areas in Adamts18−/− mice were mostly limited to the cortex rather than the basal ganglia, probably because of the lack of compensatory blood supply in the cortex. This observation confirms MCA alteration in Adamts18−/− mice. Although mutations of several ADAMTS genes (e.g., ADAMTS12 and ADAMTS13) are associated with increased risk of pediatric stroke (Stoll et al., 2016) (Arning et al., 2012), no ADAMTS-18 mutations have been described so far in genome-wide association studies associated with stroke. Further investigations with large human cohorts are needed to uncover the relationship between ADAMTS-18 and stroke.
Limitations of the study
Although developmental abnormalities of the AOAR and CCA in Adamts18−/− mice were shown to be related to increased Notch3 expression and signaling in connection with increased Fn accumulation due to Adamts18 deficiency, we have not identified direct protein substrates of Adamts18. Further investigations are warranted to clarify this matter.
STAR★methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| See Table S1 | ||
| Biological samples | ||
| C57BL/6/129Sv mouse tissues | Lu et al., 2017 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Chlorinated polyvinyl chloride (CPVC) | Shenzhen Jitian Chemical Co., Ltd. | E0905-X |
| Indian ink | Phygene | PH1714 |
| Direct Red | Sigma-Aldrich | CAS:2610-10-8 |
| Experimental models: Organisms/strains | ||
| C57BL/6/129Sv mouse strain | Lu et al., 2017 | N/A |
| Oligonucleotides | ||
| Primers for Figures 5, 6 and Tables S3, S4, see Table S2 | This paper | N/A |
| Software and algorithms | ||
| ImageJ | https://imagej.net/ | RRID:SCR_003070 |
| GraphPad Prism | https://www.graphpad.com/ | RRID:SCR_002798 |
| Other | ||
| Mouse CD4 ELISA Kit | Dogesce | DG94711Q -96T |
| Mouse CD8 ELISA Kit | Dogesce | DG94713Q -96T |
| Hematoxylin-Eosin/HE Staining Kit | Solarbio | G1120 |
| Modified Masson's Trichrome Stain Kit | Solarbio | G1346 |
| Hart’s staining Kit | SenBeiJia | SBJ-0302 |
| RNAscope® 2.5 HD Reagent Kit-RED | ACD | Catalog No. 322350 |
| RNAscope® 2.5 VS Probe- Mm-Adamts18 | ACD | Catalog No. 452259 |
| TaKaRa MiniBEST Universal RNA Extraction Kit | TaKaRa | Code No. 9767 |
| Hifair® Ⅱ 1st Strand cDNA Synthesis SuperMix for qPCR | YEASEN | 11123ES10 |
Resource avaiability
Lead contact
Further information and requests should be directed to and will be fulfilled by the lead contact, Dr. Wei Zhang (wzhang@sat.ecnu.edu.cn)
Materials availability
This study did not generate new animal models and reagents.
Data and code availability
Not applicable.
Experimental model and subject details
Animals
Adamts18-/- and wild-type (Adamts18+/+) mice with the C57BL/6/129Sv background were bred and genotyped as previously described (Lu et al., 2017). Mice were maintained on a 12-h light/dark schedule (lights on at 06:00) in a pathogen-free facility. All procedures for animal experiments were approved by the Institutional Animal Care and Use Committee of the East China Normal University (ECNU). Details of animal (included the age, sex and number) are described in the figure legends.
Post-ischemic stroke model
A 3 × 0.2-mm polyethylene thread attached to a 9-mm 7/0 suture was inserted into the right internal carotid artery and advanced to the bifurcation of the middle cerebral artery. The polyethylene thread was removed 90 min after placement. Motor function and reflexes of the ischemic mice were evaluated using a neurological severity scoring system (NSS), as described previously (Cirillo et al., 2018). All treated mice were sacrificed at 48 h, and all brain tissues were separated and freezed. In order to prepare TTC staining solution, 1% 2,3,5-triphenyltetrazolium chloride was dissolved in phosphate buffer (PBS) and placed in dark. Every pre-cooled mouse brain tissue was cut into 1.5 mm thick coronal sections. The brain slices were covered with the staining solution and stained in a shaker at 37°C for 25 min. The stained brain slices were transferred to 10% neutral buffered formalin and maintained for 24 h, then were imaged with an Mshot microscope.
Method details
Reagents
All reagents were purchased from Sigma–Aldrich (St. Louis, MO, USA) unless otherwise indicated. Primary antibodies used in this study are listed in Table S1.
The list of reagents used in this study can be found in the key resources table.
RNA in situ hybridization
RNA in situ hybridization (ISH) was performed as described previously (Lu et al., 2020a; Zhu et al., 2019). Briefly, mouse embryos were fixed in 10% neutral buffered formalin for 24 h at room temperature (RT) and paraffin-embedded following standard methods. ISH was performed on 5-μm-thick sections using the RNAscope 2.5 HD Reagent Kit-RED (Advanced Cell Diagnostics, Hayward, CA). Specific probes were used to detect target mRNAs as described (Lu et al., 2020a; Zhu et al., 2019).
Ink injection
To assess morphological transformation of branchial arch artery, embryos (E10.5 – E14.5) were dissected and placed in ice cold PBS (n ≥ 3 per genotype for each time point). For clear observation, part of the embryonic tissue was removed, and undiluted India ink was injected into the left ventricle with a finely drawn glass pipette.
Cardiovascular and cerebral artery casting
To prepare casting agent, Chlorinated polyvinyl chloride was dissolved in ethyl acetate to a final concentration of 10% and mixed evenly with appropriate amount of dibutyl phthalate (2.7%), epoxide resin (20%), hardening agent (5%), and oil paint (5 g/100 ml). After anesthetizing the mice, the right atrial appendage were opened and the 4% sodium citrate normal saline (5 ml), ethyl acetate (2 ml), and casting agent was pumped into the left ventricle at a constant rate in this order (All the liquid to be injected should be preheated to 40°C in advance).The vessels (the descending thoracic aorta and bilateral subclavian arteries) that need to be observed were ligated. After baking at 60°C for 1 h, excess tissue was removed, and the vessels were immersed in 40% hydrochloric acid for 24-48 h and then imaged with an Mshot microscope after washed by running tap water.
Isolation and verification of Mouse Embryo Fibroblasts
Embryos from timed matings of Adamts18+/- males and females were dissected out on day 13.5 of gestation. After decapitation and removal of the liver and other viscera, the cervicothoracic vascular tissues were minced, incubated in trypsin/EDTA solution at 37°C, and triturated several times. The dissociated cells were seeded in tissue culture dishes and cultured in Dulbecco's Modified Eagle's Medium (DMEM) with 10% FBS (Durkin et al., 2013). After 4 passages, the MEFs were analyzed by reverse transcription PCR (RT-PCR) to verify Adamts18 expression. Total RNA was isolated from cells using TaKaRa MiniBEST Universal RNA Extraction Kit. Reversed transcription using Hifair® II 1st Strand cDNA Synthesis SuperMix. PCR was conducted with a forward primer (5′-CCTCAAGTTGTCTGCTCCATCA) and a reverse primer (5′-GCTGAAGAAATCCACGCAAGA).
Histology, immunohistochemistry, and immunofluorescence
Mouse embryo tissues were fixed in 10% neutral buffered formalin and embedded in paraffin. After dewaxing and rehydration, hematoxylin and eosin (HE) staining or Hart’s staining or Modified Masson's Trichrome staining was performed on 5-μm sections for histological examination of carotid body, thymus, embryo, and collagen(stained by Sirus Red), elastin fibers in the common carotid artery. For immunohistochemical staining, sections were reacted with anti-Tyrosine Hydroxylase (TH) antibody. For immunofluorescence and immunocytochemistry examination, the sections were incubated with Anti-alpha-smooth muscle actin, anti-fibronectin, anti-fibrillin1, anti-laminin, anti-aggrecan, anti-versican, anti-S100-A4, and anti-vimentin antibodies overnight at 4°C (details of the dilution are described in the Table S1), followed by incubation with the secondary antibody. Sections were counterstained with DAPI (MP, Carlsbad, CA) and imaged with a Leica SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany).
Quantitative real-time RT-PCR analysis
Quantitative real-time RT-PCR (qRT-PCR) was carried out using Hifair® II 1st Strand cDNA Synthesis SuperMix for qPCR (YEASEN 11123ES10) and SuperReal PreMix Plus (SYBR Green; TIANGEN). Primers used are listed in Table S2. The relative quantity of target mRNA was determined using the ΔΔCt method, with Gapdh as the reference gene.
Western blotting
For Western blotting, mouse cervicothoracic vascular tissues (AOAR and CCA) were dissected from mice, weighted, and homogenized in RIPA buffer [50 mM Tris (pH7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS] (Boster Wuhan, China) containing protease and phosphatase inhibitors (Roche, Basel, Switzerland). The samples were centrifuged at 8,000 × g for 15 min at 4°C, and tissue debris was removed. Protein concentration was determined by BCA assay reagent (Pierce, Rockford, IL, USA). Proteins were separated by SDS-PAGE under reducing conditions and then transferred onto a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked in blocking buffer (PBS, 0.5% Tween 20, and 5% non-fat dry milk powder) and then incubated with primary antibody (anti-Notch3 NICD, anti-Hes5, anti-Hey2, and anti-GAPDH) for 1 h at RT (details of the dilution are described in the Table S1). After washing, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at RT. The immunoreactive bands were visualized with enhanced chemiluminescence (ECL) Western blot kit (Millipore, Boston, MA, USA) and quantified by ImageJ (version 1.50i).
Quantification and statistical analysis
Data were analyzed by Student's t test using the software package Prism version 7 (GraphPad, La Jolla, CA, USA). Data are shown as mean ± SD. A p value <0.05 was considered statistically significant.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (NSFC) (grant numbers 81770139, 81570389, 81170481 to W.Z.) and the Shanghai Municipal Natural Science Foundation (16ZR1423700 to S.D.). We thank Dr. Longnian Lin for valuable advice and assistance in this study.
Author contributions
W.Z. and S.D. conceived the study and designed the experiments. S.Y., N.Y., T.L., T. Wu, and L.W. performed experiments and analyzed data. S.D., X.Y., Y.-H.P., T. Wisniewski, and X.C. provided valuable advice. W.Z. and S.D. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: June 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102672.
Contributor Information
Suying Dang, Email: suyingdang@shsmu.edu.cn.
Wei Zhang, Email: wzhang@sat.ecnu.edu.cn.
Supplemental information
References
- Aldahmesh M.A., Alshammari M.J., Khan A.O., Mohamed J.Y., Alhabib F.A., Alkuraya F.S. The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) is caused by mutations in ADAMTS18. Hum. Mutat. 2013;34:1195–1199. doi: 10.1002/humu.22374. [DOI] [PubMed] [Google Scholar]
- Aldahmesh M.A., Khan A.O., Mohamed J.Y., Alkuraya H., Ahmed H., Bobis S., Al-Mesfer S., Alkuraya F.S. Identification of ADAMTS18 as a gene mutated in Knobloch syndrome. J. Med. Genet. 2011;48:597–601. doi: 10.1136/jmedgenet-2011-100306. [DOI] [PubMed] [Google Scholar]
- Apte S.S. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J. Biol. Chem. 2009;284:31493–31497. doi: 10.1074/jbc.R109.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arning A., Hiersche M., Witten A., Kurlemann G., Kurnik K., Manner D., Stoll M., Nowak-Gottl U. A genome-wide association study identifies a gene network of ADAMTS genes in the predisposition to pediatric stroke. Blood. 2012;120:5231–5236. doi: 10.1182/blood-2012-07-442038. [DOI] [PubMed] [Google Scholar]
- Ataca D., Aouad P., Constantin C., Laszlo C., Beleut M., Shamseddin M., Rajaram R.D., Jeitziner R., Mead T.J., Caikovski M. The secreted protease Adamts18 links hormone action to activation of the mammary stem cell niche. Nat. Commun. 2020;11:1571. doi: 10.1038/s41467-020-15357-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataca D., Caikovski M., Piersigilli A., Moulin A., Benarafa C., Earp S.E., Guri Y., Kostic C., Arsenijevic Y., Soininen R. Adamts18 deletion results in distinct developmental defects and provides a model for congenital disorders of lens, lung, and female reproductive tract development. Biol. Open. 2016;5:1585–1594. doi: 10.1242/bio.019711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain S.A., Mularczyk E.J., Singh M., Massam-Wu T., Kielty C.M. ADAMTS-10 and -6 differentially regulate cell-cell junctions and focal adhesions. Sci. Rep. 2016;6:35956. doi: 10.1038/srep35956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cikach F.S., Koch C.D., Mead T.J., Galatioto J., Willard B.B., Emerton K.B., Eagleton M.J., Blackstone E.H., Ramirez F., Roselli E.E. Massive aggrecan and versican accumulation in thoracic aortic aneurysm and dissection. JCI Insight. 2018;3:e97167. doi: 10.1172/jci.insight.97167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo C., Le Friec A., Frisach I., Darmana R., Robert L., Desmoulin F., Loubinoux I. Focal malonate injection into the internal capsule of rats as a model of lacunar stroke. Front. Neurol. 2018;9:1072. doi: 10.3389/fneur.2018.01072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J.A., de Burgh Daly M. A comparative study of the distribution of carotid body type-I cells and periadventitial type-I cells in the carotid bifurcation regions of the rabbit, rat, Guinea-pig and mouse. Cell Tissue Res. 1981;220:753–772. doi: 10.1007/BF00210459. [DOI] [PubMed] [Google Scholar]
- Didangelos A., Mayr U., Monaco C., Mayr M. Novel role of ADAMTS-5 protein in proteoglycan turnover and lipoprotein retention in atherosclerosis. J. Biol. Chem. 2012;287:19341–19345. doi: 10.1074/jbc.C112.350785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L.E., McCulloch D.R., McGarity J.D., Bahan A., Wessels A., Weber D., Diminich A.M., Nelson C.M., Apte S.S., Kern C.B. Altered versican cleavage in ADAMTS5 deficient mice; a novel etiology of myxomatous valve disease. Dev. Biol. 2011;357:152–164. doi: 10.1016/j.ydbio.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin M.E., Qian X., Popescu N.C., Lowy D.R. Isolation of mouse embryo fibroblasts. Bio Protoc. 2013;3:e908. doi: 10.21769/bioprotoc.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein J.A. Developing models of DiGeorge syndrome. Trends Genet. 2001;17:S13–S17. doi: 10.1016/s0168-9525(01)02450-7. [DOI] [PubMed] [Google Scholar]
- Hosseini-Alghaderi S., Baron M. Notch3 in development, Health and disease. Biomolecules. 2020;10:485. doi: 10.3390/biom10030485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubmacher D., Apte S.S. Genetic and functional linkage between ADAMTS superfamily proteins and fibrillin-1: a novel mechanism influencing microfibril assembly and function. Cell Mol. Life Sci. 2011;68:3137–3148. doi: 10.1007/s00018-011-0780-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubmacher D., Schneider M., Berardinelli S.J., Takeuchi H., Willard B., Reinhardt D.P., Haltiwanger R.S., Apte S.S. Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease. Sci. Rep. 2017;7:41871. doi: 10.1038/srep41871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H., Wang X., Ying J., Wong A.H., Li H., Lee K.Y., Srivastava G., Chan A.T., Yeo W., Ma B.B. Epigenetic identification of ADAMTS18 as a novel 16q23.1 tumor suppressor frequently silenced in esophageal, nasopharyngeal and multiple other carcinomas. Oncogene. 2007;26:7490–7498. doi: 10.1038/sj.onc.1210559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson-Rylander A.C., Nilsson T., Fritsche-Danielson R., Hammarstrom A., Behrendt M., Andersson J.O., Lindgren K., Andersson A.K., Wallbrandt P., Rosengren B. Role of ADAMTS-1 in atherosclerosis: remodeling of carotid artery, immunohistochemistry, and proteolysis of versican. Arterioscler. Thromb. Vasc. Biol. 2005;25:180–185. doi: 10.1161/01.ATV.0000150045.27127.37. [DOI] [PubMed] [Google Scholar]
- Kameda Y. Hoxa3 and signaling molecules involved in aortic arch patterning and remodeling. Cell Tissue Res. 2009;336:165–178. doi: 10.1007/s00441-009-0760-7. [DOI] [PubMed] [Google Scholar]
- Kelwick R., Desanlis I., Wheeler G.N., Edwards D.R. The ADAMTS (A disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol. 2015;16:113. doi: 10.1186/s13059-015-0676-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern C.B., Wessels A., McGarity J., Dixon L.J., Alston E., Argraves W.S., Geeting D., Nelson C.M., Menick D.R., Apte S.S. Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biol. 2010;29:304–316. doi: 10.1016/j.matbio.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutz W.E., Wang L.W., Bader H.L., Majors A.K., Iwata K., Traboulsi E.I., Sakai L.Y., Keene D.R., Apte S.S. ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts. J. Biol. Chem. 2011;286:17156–17167. doi: 10.1074/jbc.M111.231571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez L.M., Bastin M.E., Maniega S.M., Penke L., Davies G., Christoforou A., Valdes Hernandez M.C., Royle N.A., Tenesa A., Starr J.M. A genome-wide search for genetic influences and biological pathways related to the brain's white matter integrity. Neurobiol. Aging. 2012;33:1847 e1–14. doi: 10.1016/j.neurobiolaging.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Lu P., Takai K., Weaver V.M., Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011;3:a005058. doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T., Dang S., Zhu R., Wang Y., Nie Z., Hong T., Zhang W. Adamts18 deficiency promotes colon carcinogenesis by enhancing beta-catenin and p38MAPK/ERK1/2 signaling in the mouse model of AOM/DSS-induced colitis-associated colorectal cancer. Oncotarget. 2017;8:18979–18990. doi: 10.18632/oncotarget.14866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T., Lin X., Pan Y.H., Yang N., Ye S., Zhang Q., Wang C., Zhu R., Zhang T., Wisniewski T.M. ADAMTS18 deficiency leads to pulmonary hypoplasia and bronchial microfibril accumulation. iScience. 2020;23:101472. doi: 10.1016/j.isci.2020.101472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T., Zhang T., Wang C., Yang N., Pan Y.H., Dang S., Zhang W. Adamts18 deficiency in zebrafish embryo causes defective trunk angiogenesis and caudal vein plexus formation. Biochem. Biophys. Res. Commun. 2020;521:907–913. doi: 10.1016/j.bbrc.2019.10.202. [DOI] [PubMed] [Google Scholar]
- Majesky M.W. Developmental basis of vascular smooth muscle diversity. Arterioscler. Thromb. Vasc. Biol. 2007;27:1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- Memezawa H., Smith M.L., Siesjo B.K. Penumbral tissues salvaged by reperfusion following middle cerebral artery occlusion in rats. Stroke. 1992;23:552–559. doi: 10.1161/01.str.23.4.552. [DOI] [PubMed] [Google Scholar]
- Mongiat M., Andreuzzi E., Tarticchio G., Paulitti A. Extracellular matrix, a hard player in angiogenesis. Int. J. Mol. Sci. 2016;17:1822. doi: 10.3390/ijms17111822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muncie J.M., Weaver V.M. The physical and biochemical properties of the extracellular matrix regulate cell fate. Curr. Top. Dev. Biol. 2018;130:1–37. doi: 10.1016/bs.ctdb.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas J., Magli S., Rabbachin L., Sampaolesi S., Nicotra F., Russo L. 3D extracellular matrix mimics: fundamental concepts and role of materials chemistry to influence stem cell fate. Biomacromolecules. 2020;21:1968–1994. doi: 10.1021/acs.biomac.0c00045. [DOI] [PubMed] [Google Scholar]
- Oller J., Mendez-Barbero N., Ruiz E.J., Villahoz S., Renard M., Canelas L.I., Briones A.M., Alberca R., Lozano-Vidal N., Hurle M.A. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat. Med. 2017;23:200–212. doi: 10.1038/nm.4266. [DOI] [PubMed] [Google Scholar]
- Peluso I., Conte I., Testa F., Dharmalingam G., Pizzo M., Collin R.W., Meola N., Barbato S., Mutarelli M., Ziviello C. The ADAMTS18 gene is responsible for autosomal recessive early onset severe retinal dystrophy. Orphanet J. Rare Dis. 2013;8:16. doi: 10.1186/1750-1172-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren P., Hughes M., Krishnamoorthy S., Zou S., Zhang L., Wu D., Zhang C., Curci J.A., Coselli J.S., Milewicz D.M. Critical role of ADAMTS-4 in the development of sporadic aortic aneurysm and dissection in mice. Sci. Rep. 2017;7:12351. doi: 10.1038/s41598-017-12248-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckebusch L., Bertrand N., Mesbah K., Bajolle F., Niederreither K., Kelly R.G., Zaffran S. Decreased levels of embryonic retinoic acid synthesis accelerate recovery from arterial growth delay in a mouse model of DiGeorge syndrome. Circ. Res. 2010;106:686–694. doi: 10.1161/CIRCRESAHA.109.205732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria S., de Groot R. ADAMTS proteases in cardiovascular physiology and disease. Open Biol. 2020;10:200333. doi: 10.1098/rsob.200333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger D.R., Davis G.E. Angiogenesis. Cold Spring Harb. Perspect. Biol. 2011;3:a005090. doi: 10.1101/cshperspect.a005090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll M., Ruhle F., Witten A., Barysenka A., Arning A., Strauss C., Nowak-Gottl U. Rare variants in the ADAMTS13 von willebrand factor-binding domain contribute to pediatric stroke. Circ. Cardiovasc. Genet. 2016;9:357–367. doi: 10.1161/CIRCGENETICS.115.001184. [DOI] [PubMed] [Google Scholar]
- Theocharis A.D., Skandalis S.S., Gialeli C., Karamanos N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016;97:4–27. doi: 10.1016/j.addr.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Wang X., Astrof S. Neural crest cell-autonomous roles of fibronectin in cardiovascular development. Development. 2016;143:88–100. doi: 10.1242/dev.125286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong D.H., Liu X.G., Guo Y.F., Tan L.J., Wang L., Sha B.Y., Tang Z.H., Pan F., Yang T.L., Chen X.D. Genome-wide association and follow-up replication studies identified ADAMTS18 and TGFBR3 as bone mass candidate genes in different ethnic groups. Am. J. Hum. Genet. 2009;84:388–398. doi: 10.1016/j.ajhg.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M., Neilson E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R., Pan Y.H., Sun L., Zhang T., Wang C., Ye S., Yang N., Lu T., Wisniewski T., Dang S. ADAMTS18 deficiency affects neuronal morphogenesis and reduces the levels of depression-like behaviors in mice. Neuroscience. 2019;399:53–64. doi: 10.1016/j.neuroscience.2018.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.