

Abstract
Aldosterone is the major mineralocorticoid in the human body controlling blood pressure and salt homeostasis. Overproduction of aldosterone leads to primary aldosteronism, which is the most common form of secondary hypertension with limited treatment options. Production of aldosterone by cytochrome P450 11B2 (CYP11B2, aldosterone synthase) requires two reduction events with the electrons delivered by the iron/sulfur protein adrenodoxin. Very limited information is available about the structural and functional basis of adrenodoxin/CYP11B2 interaction, which impedes the development of new treatment options for primary aldosteronism. A systematic study was carried out to determine if adrenodoxin interaction with CYP11B2 might also have an allosteric component in addition to electron transfer. Indeed, local increases in adrenodoxin concentration promote binding of the substrate 11-deoxycorticosterone and the inhibitor osilodrostat (LCI699) in the active site—over 17 Å away—as well as enhance the inhibitory effect of this latter drug. The CYP11B2 structure in complex with adrenodoxin identified specific residues at the protein–protein interface interacting via five salt bridges and four hydrogen bonds. Comparisons with cholesterol-metabolizing CYP11A1 and cortisol-producing CYP11B1, which also bind adrenodoxin, revealed substantial structural differences in these regions. The structural and functional differences between different P450 interactions with adrenodoxin may provide valuable clues for an orthogonal treatment approach for primary aldosteronism by specifically targeting the interaction between CYP11B2 and adrenodoxin.
Keywords: cytochrome P450, aldosterone, hypertension, CYP11B2, adrenodoxin
Abbreviations: CYP11A1, cytochrome P450 11A1; CYP11B2, cytochrome P450 11B2; LB, lysogeny broth; PDB, Protein Data Bank; SEC, size-exclusion chromatography
The steroid hormone aldosterone is the primary mineralocorticoid in the human body (1). It controls blood pressure by regulating water and sodium retention and potassium release (1). The final steps of aldosterone biosynthesis are catalyzed by cytochrome P450 11B2 (CYP11B2, also known as aldosterone synthase, E.C. 1.14.15.4) (2). CYP11B2 is expressed in the zona glomerulosa, the outermost layer of the adrenal gland. CYP11B2 performs a three-reaction series, starting with 11-deoxycorticosterone 11β-hydroxylation to yield corticosterone. Corticosterone is then hydroxylated at position 18 to form 18-hydroxycorticosterone, and finally position 18 is further oxidized to yield aldosterone (3).
Although aldosterone is an essential hormone in healthy individuals, its overproduction leads to primary aldosteronism, the most common defined cause of secondary hypertension (3). Primary aldosteronism is associated with a higher risk of severe cardiac disease and stroke and leads to a higher mortality compared with hypertension due to unknown causes, called primary hypertension (4, 5). Inhibition of CYP11B2-mediated aldosterone production is thus a promising, yet unrealized, treatment option for primary aldosteronism and the resulting secondary hypertension. An impediment to the development of selective inhibitors is the similarity of CYP11B2 with CYP11B1, which generates cortisol. These two enzymes share 93% sequence identity overall and 100% conservation of active site amino acids (2). Although small molecule inhibitors have been investigated for more than 30 years, they lack selectivity, with those targeted to CYP11B2 also suppressing cortisol production by CYP11B1 (6).
In 2020, the US Food and Drug Administration approved LCI699 (Isturisa or osilodrostat) as a CYP11B1 inhibitor for the treatment of excess cortisol production leading to Cushing’s syndrome. This drug is a second-generation derivative of the breast cancer drug fadrozole (6, 7). LCI699 was actually initially developed as CYP11B2 inhibitor to treat primary aldosteronism (8), but when the relative selectivity was discovered to be opposite of that intended it was repurposed as a CYP11B1 inhibitor to treat Cushing’s. However, LCI699 still retains significant inhibition of both human CYP11B enzymes. This leaves patients with primary aldosteronism without an effective CYP11B2 inhibitor as a treatment option. Further functional and structural insights into the aldosterone biosynthetic system are required to facilitate drug design for primary aldosteronism.
CYP11B2 is a mitochondrial class I cytochrome P450 (9). This means that its biosynthetic activity relies on both the membrane-associated, NADPH-dependent flavoprotein adrenodoxin reductase and the soluble [2Fe-2S] iron–sulfur protein adrenodoxin (also called ferrodoxin 1) (Fig. 1A) (10, 11). Aldosterone synthesis is initiated by the transfer of two electrons from NADPH to adrenodoxin reductase (12). Reduced adrenodoxin reductase transfers one electron at a time to adrenodoxin. Soluble adrenodoxin diffuses to membrane-bound CYP11B2 and transfers one electron at a time to its heme. CYP11B2 requires two electrons for each of the three reactions (Fig. 1B) (13). Thus, biosynthesis of one molecule of aldosterone requires adrenodoxin to deliver six electrons to CYP11B2.
Figure 1.

CYP11B2 catalytic system, reactions, and inhibitors.A, the aldosterone synthase catalytic system is composed of CYP11B2 and FAD-containing adrenodoxin or ferredoxin reductase in the inner mitochondrial membrane plus the soluble iron–sulfur protein adrenodoxin. In the first step NADPH transfers two electrons to the FAD of adrenodoxin reductase. This FAD then transfers one electron at a time to the iron–sulfur cluster of soluble adrenodoxin. Finally, adrenodoxin binds and transfers one electron at a time to CYP11B2 to enable catalysis. B, CYP11B2 converts the substrate 11-deoxycosterone to corticosterone, which is subsequently hydroxylated at position 18 yielding 18OH-corticosterone. A final oxidation at position 18 results in aldosterone. Each of the three reactions requires two electrons, for a total of six electrons. C, the inhibitor fadrozole was initially developed as breast cancer drug and is produced as racemate containing the R and S enantiomer. The (R)-fadrozole derivative LCI699 was initially designed as aldosterone synthase inhibitor, but the selectivity was opposite that desired and this compound is now the first US Food and Drug Administration–approved drug for CYP11B1 inhibition to treat Cushing’s disease.
Relatively few details are known about CYP11B2 interaction with adrenodoxin. When CYP11B2 and adrenodoxin are incubated together and cross-linking reagents added, a number of cross-linked peptides were identified consisting of positively charged residues from the proximal CYP11B2 surface and negatively charged acidic amino acids from the adrenodoxin surface near the iron–sulfur cluster (14). The CYP11B2 residues that were identified are largely conserved in other human mitochondrial P450 enzymes using the same redox system (15), suggesting this interface might be conserved. Another informative study was a structure of the mitochondrial cytochrome P450 11A1 (CYP11A1) fused to adrenodoxin (16). Although the relative orientations of CYP11A1 and adrenodoxin to each other were generally consistent, based on the two different types of information, adrenodoxin may form fewer interactions with CYP11A1 than with CYP11B2 (a detailed tabular comparison can be found in (14)). However, because of limited structural information it is not clear if or how the protein–protein interactions might differ for individual mitochondrial P450 enzyme interactions with the common adrenodoxin redox partner.
In addition to adrenodoxin serving as electron donor, it has been suggested that adrenodoxin may also act as allosteric effector, at least for some P450 enzymes. Experimental evidence suggests that adrenodoxin binding to the proximal surface of CYP11A1 allosterically modulates the position of the intermediate substrate 22R-hydroxycholesterol some 18 Å away in the CYP11A1 active site to ensure reaction processivity (17, 18). For vitamin D3-metabolizing CYP24A1, adrenodoxin increases the population of enzyme that binds substrate (19). These studies raise the possibility of an allosteric effect of adrenodoxin on CYP11B2 substrate and inhibitor binding and catalysis.
Herein is provided a detailed examination of adrenodoxin-induced effects on and interactions with CYP11B2. Complementing ligand binding and catalytic studies, the first structure of an adrenodoxin–CYP11B2 complex identifies interacting residues. These studies provide an enhanced understanding of the aldosterone biosynthetic system and thereby support the development of expanded treatment options for aldosterone-derived hypertension.
Results
Generation and characterization of an adrenodoxin–CYP11B2 fusion protein
In an attempt to encourage more stable complex formation between CYP11B2 and adrenodoxin, a fusion protein was generated consisting of N-terminal adrenodoxin, the linker AAKKTSS, truncated CYP11B2, and a C-terminal His tag. SDS-PAGE analysis revealed a single band at the expected molecular weight of 70 kDa for the purified protein (Fig. S1). The Soret absorbance maximum was 419 nm, consistent with water coordination to the ferric heme iron. The reduced-carbon monoxide difference spectrum had a λmax of 450 nm, which is consistent with a properly incorporated heme prosthetic group. Furthermore, the adrenodoxin–CYP11B2 fusion bound both the 11-deoxycorticosterone substrate (Fig. 1B) and fadrozole and LCI699 inhibitors (Fig. 1C) with the expected respective blue and red shifts in the Soret peak (vide infra). It also converted 11-deoxycorticosterone to corticosterone, then 18-hydroxycorticosterone, and then aldosterone in the presence a 40-fold excess of added adrenodoxin (Fig. 1B and Fig. S2) (vide infra). The extinction coefficient for the adrenodoxin iron–sulfur cluster is much weaker, and the oxidized peaks normally found at 414 and 455 nm were occluded by the stronger heme absorbance. This protein was used in its oxidized form to identify allosteric effects from redox effects. In summary, all indicators are that fusion of adrenodoxin to the N terminus of CYP11B2 does not adversely affect the CYP11B2 structure or function.
Impact of adrenodoxin on CYP11B2 substrate binding
The impact of adrenodoxin on CYP11B2 binding of the substrate 11-deoxycorticosterone was examined using UV-visible spectroscopy. In general, addition of the substrate to CYP11B2 induces a blue shift of the Soret absorbance to 390 nm. This is consistent with the binding of many substrates to P450 enzymes in which a water molecule bound to the active site ferric heme iron is displaced by the incoming substrate. This spectral shift can be used to monitor substrate binding under different conditions.
In the absence of adrenodoxin, the CYP11B2 dissociation constant for 11-deoxycorticosterone (Fig. 2A, black line) was 10.39 μM (Fig. 2C, row 1). When equimolar, 10-fold, and 40-fold amounts of isolated or free adrenodoxin protein was present in solution with isolated CYP11B2 (Fig. 2A), the same spectral shifts occurred but the dissociation constant decreased to 3.94, 3.38, and 3.42 μM, respectively (Fig. 2C, rows 2–4). Thus, the presence of only an equimolar amount of adrenodoxin increases CYP11B2 affinity for its substrate by 2.6-fold. The population of CYP11B2 molecules binding substrate is represented by the maximal absorbance upon saturation with ligand (ΔAmax) and steadily increased from 0.083 for CYP11B2 alone to 0.086, 0.093, and 0.096 in the presence of equimolar, 10-fold, and 40-fold adrenodoxin, respectively (Fig. 2, A and B). Thus, adrenodoxin appears to both increase the population of CYP11B2 molecules binding substrate and increase the substrate affinity.
Figure 2.

Effects of adrenodoxin on CYP11B2 substrate binding. Changes in absorbance observed upon CYP11B2 titrated with increasing 11-deoxycorticosterone substrate in the absence and presence of increasing amounts of adrenodoxin (A) and for the adrenodoxin–CYP11B2 fusion protein (C). For each experiment a CYP11B2 concentration of 1 μM was used. Data shown are technical triplicates. Analysis using a one-site hyperbolic binding equation yields the fitted dissociation constants and maximum absorbance change at saturation (ΔAmax) in the bottom panel (B).
The same 11-deoxycorticosterone binding assays were performed with the adrenodoxin–CYP11B2 fusion enzyme (Fig. 2C), in which the ratio is intrinsically 1:1 for the two proteins, but wherein the local concentration of adrenodoxin adjacent to CYP11B2 is increased and presumably the likelihood of protein–protein interaction as well. In this setting, the dissociation constant for the substrate was 1.88 μM (Fig. 2, B and C, row 5). This is the highest affinity observed, an increase of 5.5-fold increase over the substrate affinity in the absence of adrenodoxin and a 2-fold increase in affinity compared with when the two enzymes were separate polypeptides but coincubated at 1:1. This higher affinity is consistent with adrenodoxin–CYP11B2 interaction allosterically increasing substrate affinity. In addition, the ΔAmax for the fusion binding substrate was 0.097 (Fig. 2B, row 5), which is slightly higher than even the 1:40 CYP11B2:adrenodoxin situation (Fig. 2B, row 4). Correspondence between the trends seen in adrenodoxin increasing CYP11B2 substrate binding and affinity and the exacerbated results for the adrenodoxin–CYP11B2 fusion suggest that fusion enhances the protein–protein interaction.
Impact of adrenodoxin on CYP11B2 catalysis
Although CYP11B2 performs three enzymatic steps to form aldosterone (Fig. S2), the impact of adrenodoxin focused on the first step, conversion of 11-deoxycorticosterone to corticosterone, because the subsequent products are not normally produced in high yields. Reaction times were kept short to minimize the production of the latter products. For CYP11B2 catalysis to occur some adrenodoxin must be present, so the 1:1, 1:10, and 1:40 ratios were used to determine the Michaelis–Menten kinetics for 11-deoxycorticosterone 11β-hydroxylation to corticosterone (Fig. 3A). The amount of substrate turnover (kcat) systematically increased with increasing adrenodoxin concentrations from 7.38 to 14.86 min−1, to 52.86 min−1 for kcat for equimolar, 10-fold, and 40-fold adrenodoxin, respectively (Fig. 3B, rows 1–3). The highest 40-fold excess of adrenodoxin is consistent with literature reports of in vitro CYP11B2 assays that frequently employ such high concentrations to obtain high turnover. The amount of adrenodoxin present in the adrenal gland zona glomerulosa is unknown, but unlikely to be this high. In these experiments, decreases in Km were generally observed upon adding higher concentrations of adrenodoxin. The Km values were 17.05, 7.82, and 10.53 μM for equimolar, 10-fold, and 40-fold adrenodoxin, respectively (Fig. 3B, rows 1–3). Although the Km decreases were not as systematic, both 10-fold and 40-fold adrenodoxin yielded significantly lower Km values. Thus, adrenodoxin clearly both promotes CYP11B2 11β-hydroxylation (kcat) and decreases the Km, although this latter effect may be more complex.
Figure 3.

Effect of adrenodoxin on CYP11B2-mediated catalysis. Michaelis–Menten kinetics for CYP11B2-mediated metabolism of 11-deoxycorticosterone in the presence of 1-fold, 10-fold, and 40-fold excess of adrenodoxin (A) and for the adrenodoxin–CYP11B2 fusion enzyme with 40-fold excess of adrenodoxin (C). Rates were determined for corticosterone formation from 11-deoxycorticosterone. For experiments with 1-fold adrenodoxin, a CYP11B2 concentration of 0.2 μM was used. For experiments with 10-fold and 40-fold adrenodoxin, a reduced CYP11B2 concentration of 0.05 μM was used to prevent substrate depletion. For experiments with the adrenodoxin-CYP11B2 fusion protein, 0.4 μM protein concentration was used. Product profiles did not change under these conditions. Note the differences in the two y-axes. Data are shown in technical triplicates and was fit to the Michaelis–Menten equation to derive the kinetic parameters Km and kcat (B).
The same activity assays were performed with the adrenodoxin–CYP11B2 fusion enzyme (Fig. 3C). In the presence of a 40-fold excess of free adrenodoxin, the fusion protein demonstrated a Km value of 6.56 μM (Fig. 3B, row 4), which is the lowest value observed under any condition herein. The fusion protein exhibited a kcat of 2.17 min−1 (Fig. 3C, row 4), which is also the lowest turnover observed. This latter result is consistent with formation of a relatively stable interaction between the 2Fe-2S surface and CYP11B2, which would be expected to decrease turnover because the 2Fe-2S would be poorly available to accept catalytically necessary electrons from adrenodoxin reductase.
Impact of adrenodoxin on CYP11B2 inhibitor binding
The next logical set of experiments compared the effects of adrenodoxin on substrate binding observed above with its effects on inhibitor binding. The inhibitor LCI699 binds both isolated CYP11B2 and the adrenodoxin–CYP11B2 fusion with a red shift indicative of the inhibitor nitrogen coordinating the heme iron. LCI699 binding to isolated CYP11B2 (Fig. 4A) demonstrated a Kd of 40.12 nM and an Amax was 0.019 (Fig. 4B). Fusing adrenodoxin to CYP11B2 altered inhibitor binding (Fig. 4A), increasing by ∼1.7-fold both the Kd to 72.25 nM and the Amax to 0.032 (Fig. 4B). Thus, the population of CYP11B2-binding inhibitor increased as was observed for substrate, but the affinity decreased, which was the opposite observed for substrate.
Figure 4.

Effects of adrenodoxin on CYP11B2 inhibitor binding and inhibition.A, spectral shifts for LCI699 binding to CYP11B2 alone compared with LCI699 binding to the adrenodoxin–CYP11B2 fusion protein. For each experiment a CYP11B2 concentration of 0.2 μM was used. Data are shown in technical triplicates and were analyzed using the tight-binding or Morrison equation to determine the dissociation constants (B). C, CYP11B2 11-deoxycorticosterone 11β-hydroxylase activity was measured with increasing LCI699 inhibitor concentration in the presence 1-fold, 10-fold, and 40-fold adrenodoxin (46). For experiments with 1-fold adrenodoxin, a CYP11B2 concentration of 0.2 μM was used. For experiments with 10-fold and 40-fold adrenodoxin, a CYP11B2 reduced concentration of 0.05 μM was used to prevent substrate depletion. Data were measured in technical triplicates and fit to the dose–response inhibitor (four-parameter) equation to determine the half-maximal inhibitory concentration (IC50) (D).
Impact of adrenodoxin on CYP11B2 inhibition
Inhibitory studies were performed with different adrenodoxin concentrations to determine the effect on the half-maximal inhibitory concentration (IC50) of LCI699. The same CYP11B2: adrenodoxin ratios (1:1, 1:10, 1:40) were used as for many of the previous studies (Fig. 4C). The LCI699 IC50 with equimolar adrenodoxin was 155.90 nM, but decreased to 24.46 and 35.68 nM, when the ratio was increased to 1:10 and 1:40, respectively (Fig. 4D). Like the adrenodoxin effects on 11-deoxycorticosterone Km, the relationship is complex. However, the presence of higher concentrations of adrenodoxin resulted in a 4- to 6-fold increase in the efficacy of the LCI699 inhibitor. In addition to illustrating the allosteric effects of adrenodoxin on inhibitor action in the distant buried active site, this information suggests one source of the variability between reports in the literature that use different amounts of adrenodoxin.
Structural basis for adrenodoxin interaction with CYP11B2
The above data supported the integrity of heme, substrate, and inhibitor binding to the P450 domain of the adrenodoxin–CYP11B2 fusion protein. Furthermore, trends with the fusion protein mirrored those with high free adrenodoxin. Since the fusion had catalytic activity only upon addition of high concentrations of free adrenodoxin, the logical hypothesis was that the P450 and adrenodoxin domains were stably interacting as desired for X-ray structure determination. As crystallization typically occurs in the presence of higher salt concentrations (>350 mM in this work) and this protein–protein interaction is thought to be electrostatically mediated, there was a concern that the two domains might not be interacting in the crystal. However, in the resulting 2.94 Å resolution (Table 1) structure it was immediately clear from the electron density that both protein domains were interacting (Fig. 5A). In the most complete chain, the last 14 adrenodoxin residues (171–184), the nonnative AAKKTSS linker, and the first two CYP11B2 residues (20, 21) are not visible in the electron density to verify whether the interaction is intermolecular or intramolecular. The distance between the last visible residue of adrenodoxin (T170) and the first visible residue of CYP11B2 (R35) is 17.5 Å, compatible with an intramolecular interaction formed by the 23 disordered intervening residues (Fig. 5A). However, the closest intermolecular interaction between the last visualized adrenodoxin residues and the first visualized CYP11B2 residues is 27 Å, so an intermolecular arrangement cannot be ruled out.
Table 1.
X-ray data collection, refinement, and validation statistics
| Parameters | Adrenodoxin–CYP11B2 fusion protein bound to the inhibitor fadrozole |
|---|---|
| Data collection | |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 140.58, 208.70, 124.92 |
| α, β, γ (°) | 90.00, 114.07, 90.00 |
| Resolution (Å)a | 50.00–2.94 (2.99–2.94) |
| Redundancya | 7.7 (7.0) |
| Rpima | 0.073 (0.612) |
| Mn(I/sd)a | 11.06 (1.2) |
| CC ½a | 0.991 (0.560) |
| Completenessa (%) | 100.0 (99.7) |
| Total Reflectionsa | 535,603 |
| Unique Reflectionsa | 69,937 (3458) |
| Refinement | |
| Resolution (Å) | 48.16–2.93 |
| No. reflections | 63,799 |
| Rwork/Rfree | 0.21/0.24 |
| Number of non-hydrogen atoms/B factor | |
| Protein | 13,182/52.4 |
| Ligand | 51/40.7 |
| Heme | 117/36.9 |
| Iron/sulfur cluster | 12/54.92 |
| RMS deviations | |
| Bond lengths (Å) | 0.012 |
| Bond angles (°) | 1.2 |
| Coordinate error (maximum-likelihood) (Å) | 0.39 |
| Ramachandran plot: preferred/allowed/outliers (%) | 96.19/3.69/0.12 |
Highest resolution shell is shown in parenthesis.
Figure 5.

X-ray protein structure of the adrenodoxin–CYP11B2 fusion protein in complex with (R)-fadrozole (magenta) bound in the active site.A, the proximal face of CYP11B1 (blue ribbons) binds the adrenodoxin domain (brown). B, interactions between CYP11B2 and adrenodoxin include hydrogen bonds and electrostatic interactions between positively charged CYP11B2 amino acids and negatively charged adrenodoxin amino acids.
The CYP11B2 domain has fadrozole in the active site (Fig. 5A). Consistent with a previous CYP11B2 structure (22), electron density in the CYP11B2 active site identified only the (R) enantiomer of fadrozole (Fig. S3), even though the enzyme was cocrystallized with a racemic mixture of S and R fadrozole. As reported previously, the (R)-fadrozole imidazole nitrogen coordinated the heme iron and its benzonitrile was directed toward Arg120.
Adrenodoxin was bound to the proximal surface of CYP11B2 (the CYP11B2 surface nearest the heme-coordinating Cys) with the shortest distance between the CYP11B2 heme prosthetic group and the adrenodoxin iron–sulfur cluster of 17.8 Å (Fig. 5A). Interactions between adrenodoxin and CYP11B2 are mainly mediated on the adrenodoxin side by the meander enclosing the iron–sulfur cluster and the acidic α helix 3 with multiple components of CYP11B2. First, the carbonyl oxygen of Leu50 in the adrenodoxin iron–sulfur meander interacts with the side chain of Arg454 in CYP11B2 (Fig. 5B). Second, three negatively charged residues in the adrenodoxin α3 helix (Asp72, Glu73, and Asp-76) form interactions with positively charged residues of the CYP11B2 K helix (Arg366 and Lys370) and the meander immediately preceding the heme-containing loop (Arg432) (Fig. 5B). Third, CYP11B2 Asn437 and Phe438 further stabilize the interaction by interacting with the carbonyl oxygens of adrenodoxin Ala81 and Asp79, respectively. Fourth, the carbonyl oxygen of adrenodoxin Leu80 forms a hydrogen bond with the side chain of CYP11B2 Gln101 (Fig. 5B).
Discussion
Adrenodoxin enhances CYP11B2 binding of the substrate 11-deoxycorticosterone
Adrenodoxin is required to deliver electrons to CYP11B2 to support its catalysis (2). In the human steroid hormone biosynthetic pathway adrenodoxin is also crucial for CYP11A1-mediated removal of cholesterol’s side chain and CYP11B1-mediated cortisol biosynthesis (23). Studies with CYP11A1 and CYP24A1 suggested that adrenodoxin might also act as an allosteric modulator (17, 19). To investigate the possibility of an allosteric effect on CYP11B2 binding of the substrate 11-deoxycorticosterone, substrate binding studies were performed with increasing amounts of oxidized adrenodoxin present. Adrenodoxin increased the affinity of CYP11B2 for its substrate up to 3.1-fold (Fig. 2C). When performed with the adrenodoxin–CYP11B2 fusion enzyme, the affinity increased 5.5-fold compared with the isolated CYP11B1 enzyme and 2-fold compared with the unfused isolated proteins at the same ratios (Fig. 2C). Thus, adrenodoxin and CYP11B2 colocalization favors CYP11B2 substrate binding in the buried active site. Furthermore, as observed for CYP24A1, both increasing adrenodoxin concentrations in the unfused protein system and protein fusion increased the substrate-bound enzyme population, as indicated by increases in the maximal absorbance change (ΔAmax). As the substrate is sequestered in the CYP11B2 active site, CYP11B2 must open to accept substrate, close after substrate is bound, perform catalysis, then open to release product. Thus a logical explanation for both the increase in enzyme binding substrate at saturation and the decrease in the dissociation constant is that adrenodoxin binding may allosterically promote conformational changes in CYP11B2 to favor substrate binding and promote the enzyme–substrate complex. Similar findings have been reported for increased affinity of CYP11B2 for the intermediate product/substrate corticosterone when adrenodoxin was present (24). The relative amounts of adrenodoxin to CYP11B2 to adrenodoxin in the human adrenal gland is currently unknown. However, it has been reported that, in the adrenal gland, adrenodoxin mRNA is 5.5-folder higher than CYP11B2 mRNA (genome.ucsc.edu). Thus, although the CYP11B2:adrenodoxin protein level is unlikely to be as high as the 1:40 ratio that is often used for in vitro experiments, the current data indicate that only low amounts of adrenodoxin are required to substantially modulate substrate binding.
Comparison of adrenodoxin effects on CYP11B1 and CYP11A1 may provide insights into the flow of the steroidogenesis pathway. A fusion enzyme of adrenodoxin and CYP11A1 did not bind its cholesterol substrate with higher affinity than CYP11A1 alone (16), but our data for CYP11B2 do indicate a significant effect. Cholesterol is available in abundance (25), whereas 11-deoxycorticosterone is present at much lower levels (26). Thus an allosteric effect of adrenodoxin for increased cholesterol binding might not provide any biological advantage for the CYP11A1 reaction but could facilitate CYP11B2 function. Regardless, this demonstrates differences in adrenodoxin effects on even different steroidogenic P450s.
Adrenodoxin availability modulates CYP11B2 catalysis
Following substrate binding, CYP11B2 catalysis requires a number of different reactants: adrenodoxin binding and electron transfer, oxygen binding, a second adrenodoxin binding event and transfer of a second electron, and two protonation steps to finally form the catalytic iron-oxo reactive intermediate. Since adrenodoxin can only deliver one electron at a time, two separate adrenodoxin interactions must occur with CYP11B2 for one reaction cycle. Thus, it is logical that CYP11B2-mediated turnover of 11-deoxycorticosterone is higher with 40-fold adrenodoxin (by 7-fold, Fig. 3B) than with equimolar adrenodoxin. In fact, across the CYP11B2 literature in vitro systems employ CYP11B2:adrenodoxin ratios up to 1:60 to drive all three catalytic cycles to generate the end product aldosterone (27), but again this is likely far outside the physiological conditions in which the enzyme normally operates. The current studies report increases in kcat and decreases in Km even at much lower adrenodoxin concentrations (2-fold for both parameters for 1:10 versus 1:1). The result is that the catalytic efficiency (kcat/Km) increases from 0.43 for equimolar to 1.91 for 1:10 and 5.04 for 1:40. As so many steps are involved in a single reaction cycle, it is difficult to discern from the current studies the origin of these effects, but as substrate binding is one of them, it is possible that at least part of these observations derives from the adrenodoxin-induced increases in substrate affinity and occupancy demonstrated above.
The adrenodoxin–CYP11B2 fusion enzyme is not active when supplemented with only adrenodoxin reductase. This is consistent with a fairly stable complex between the P450 and adrenodoxin domains, which would prevent adrenodoxin reduction by adrenodoxin reductase. However, low amounts of substrate metabolism do occur when relatively high amounts of free adrenodoxin are added (e.g., 40-fold, Fig. 3C). Low turnover was also observed for an adrenodoxin–CYP11A1 fusion protein (16).
The CYP11B2 catalysis in this situation is low, whereas the Km of 6.56 μM for CYP11B2 is also the lowest observed in these studies. In combination with the binding studies above, the low Km suggests that the deficit is not related to substrate binding but rather a later step in the catalytic cycle. Two possibilities were entertained. One is that the fusion protein undergoes a conformational change such that the fused adrenodoxin domain can occasionally release the CYP11B2 domain, thereby making the adrenodoxin iron–sulfur cluster available for reduction by adrenodoxin reductase. Subsequently the adrenodoxin domain can reassociate with the fused CYP11B2 domain for electron transfer. Because no turnover was observed from the fusion protein in the presence of adrenodoxin reductase but without free adrenodoxin, this is less likely, but it is also possible that turnover is so low as to be below the limits of detection. The other logical possibility is that the CYP11B2-fused adrenodoxin domain can be outcompeted by high concentrations of free adrenodoxin, and when this adrenodoxin has already been reduced by adrenodoxin reductase then electron delivery can occur to support catalysis.
Adrenodoxin increases the potency of the drug LCI699
LCI699 was initially developed for treating primary aldosteronism by inhibiting CYP11B2 but is now the first US Food and Drug Administration–approved drug targeting CYP11B1 and is used for Cushing’s disease (8). The impact of adrenodoxin on LCI699 binding was investigated by comparing isolated or free CYP11B2 and the adrenodoxin–CYP11B2 fusion. Although the dissociation constant was 1.8-fold higher for the fusion protein, this was offset by a similar 1.7-fold increase in enzyme population binding the inhibitor at saturation (ΔAmax) (Fig. 4B), yielding almost identical ΔAmax/Kd values. More striking were the effects on the LCI699 IC50. Although the low turnover of the fusion protein precluded its analysis, increasing concentrations of adrenodoxin in the isolated protein system resulted in 4- to 6-fold decreases in the IC50 compared with the equimolar situation (Fig. 4, B and C). It is difficult to compare these results to other literature values. Reported IC50 values have been determined in systems such as kidney cell culture or transfected V79 cells, with unknown or uncontrolled adrenodoxin concentrations, resulting in quite variable results (28, 29). Regardless, observation of this effect underscores the importance of controlling adrenodoxin concentrations when evaluating inhibitor efficacy for CYP11B2, and also the desirability of evaluating potential antihypertensive drugs under physiologically relevant adrenodoxin concentrations.
The structural basis for CYP11B2 interaction with adrenodoxin
The adrenodoxin–CYP11B2 fusion protein X-ray structure revealed adrenodoxin binding to the proximal CYP11B2 surface via a network of electrostatic interactions and hydrogen bonds. The CYP11B2 part of the fusion has an RMSD of 0.38 Å compared with the isolated CYP11B2 structure also bound to fadrozole (Protein Data Bank [PDB] 4FDH (22), Fig. S4A), indicating few changes in the overall structure. Like the isolated CYP11B2 structure, the CYP11B2 part of the fusion protein has a channel from the active site to the substrate exterior passing between helices A', G', and B' and a narrower channel along the N-terminal portion of the H helix. The fusion protein, but not the isolated protein, has another channel that exits the active site between the stretches leading to and from the B' helix, but this could be due to differences in modeling the side chain of Ile112, a residue just at the protein surface whose density is poorly defined in both structures.
The adrenodoxin part of the fusion protein complex could be modeled in its entirety. This is in contrast to the only other P450/adrenodoxin structure, in which only the parts of adrenodoxin associated with the P450 interface could be modeled (16). This could be due to packing because in the current structure adrenodoxin also packs against another symmetry-related copy of the complex. Comparison of the adrenodoxin domain herein with isolated adrenodoxin (PDB 3P1M) yields an RMSD of 0.5296, indicating very little differences in adrenodoxin conformation upon binding CYP11B2 (Fig. S4B).
Three notable aspects of the residues forming the CYP11B2 positively charged interface deserve particular comment. First, almost all of residues forming the CYP11B2 part of the protein–protein interface are in either the K helix (Arg366 and Lys370) or the meander region (Arg432, Asn437, and Phe438) and subsequent loop containing the heme-coordinating Cys (Arg453 and Arg454) (Figs. 5B and 6B). The only CYP11B2 residue that contributes to the adrenodoxin interface but that is not part of this sequential series is Gln101 from the loop between the B and B' helices. Second, although Arg453 does not directly participate in the adrenodoxin interaction, it stabilizes the neighboring Glu449. Glu449 is immediately adjacent to the heme-coordinating Cys450 and might play a role in electron transfer, as has been suggested for CYP11A1 (16). Finally, several of these residues are conserved in CYP11B1 and are key in its function as demonstrated in clinical cases. Patients with single mutations of Arg366, Arg453, or Arg454 to Cys in CYP11B1 are catalytically deficient (30, 31, 32), which could derive from abrogated adrenodoxin interaction and/or electron transfer.
Figure 6.

Comparison of sequences and interacting residues.A, adrenodoxin residues (left column) interacting with CYP11B2 (middle column) and CYP11A1 (right column) with indications of the type of interaction. B, sequence alignment between CYP11A1, CYP11B1, and CYP11B2 highlighting residues participating in adrenodoxin binding (red). The single amino acid difference between CYP11B1 and CYP11B2 in these regions is boxed (position 439). The red asterisk above the sequences indicates the heme-coordinating cysteine residue.
The adrenodoxin negatively charged interface is composed of the loop enclosing the iron–sulfur (Leu50) and α helix 3 (Asp72, Glu73, Asp76, Asp79, Leu80, and Ala81) (Figs. 5B and 6A). The iron-sulfur-enclosing Leu50 forms a hydrogen bond via its backbone carbonyl with the side chain of Arg454. Since the interaction involves the backbone, one might think that mutation at this position would not be problematic. However, deletion mutants in this loop all decreased stability and redox potential, but those at Gly48, Leu50, or Ala51 decreased binding to CYP11A1 (20). The acidic adrenodoxin Asp and Glu residues of the α3 helix have previously been implicated in interactions with several other mitochondrial P450s (21), but the role of Leu80 in the protein–protein interaction is not as clear and may differ between mitochondrial P450s. The current adrenodoxin–CYP11B2 fusion structure reveals a hydrogen bond between the adrenodoxin Leu80 backbone carbonyl oxygen and the side chain of Gln101 in CYP11B2. A similar bond is observed in the adrenodoxin–CYP11A1 fusion structure between adrenodoxin Leu80 and Trp418 (16) (Fig. 6A). In the CYP11B2 fusion structure, the side chain of Leu80 is also in proximity of CYP11B2 Val441 and Phe438 and these hydrophobics may also play a role in adrenodoxin–P450 interactions. Mutation of adrenodoxin Leu80 to lysine nearly abolished 11β-hydroxylase activity in both CYP11B enzymes (14). Conversely, for vitamin D3-metabolizing rat CYP24A1, the mutation L80K led to a tighter binding of adrenodoxin and did not abolish enzyme activity (33). In CYP11B1, mutation of the conserved Val441 to glycine is reported to abolish cortisol production in patients (34) and this could also be due to decreased interactions. Thus, hydrophobic, electrostatic, and hydrogen bonding interactions appear to be important in the adrenodoxin–P450 interaction but vary for different P450 proteins.
The network of interactions observed herein are also usefully compared with those identified from cross-linking experiments. Human CYP11B2 and adrenodoxin were incubated with ethyl-3-[3-(dimethylamino)propyl]carbodiimide or EDC. This zero-length cross-linker reacts with a carboxyl and amino group to form an amide bond between them. The resulting peptides identified by mass spectrometry, revealed cross-links between adrenodoxin Asp and CYP11B2 Lys residues (Arg366 was mutated to Lys to work with the EDC cross-linker). Of the three different cross-links identified, one pairing matches the structure (adrenodoxin Asp76 with CYP11B2 Lys370), another involves residues identified in the interface but with different interactions (adrenodoxin Asp79 with CYP11B2 Arg/Lys366), and the third identifies residues not interacting in the structure (adrenodoxin Asp113 and Lys357). It is possible that these complementary approaches might have captured two different events, one of which is the initial encounter complex and the other being formation of a stable complex for electron transfer. Both states have been detected for the soluble bacterial P450cam with its redox protein putidaredoxin (35). Different amino acid pairing could be relevant for each of these states.
Following formation of the CYP11B2–adrenodoxin complex, electron transfer must occur to support catalysis. The distance between the closest iron of the adrenodoxin iron–sulfur complex and the heme iron is 17.8 Å, which is consistent with the corresponding distance between adrenodoxin and CYP11A1 (17.4 Å) (16) and a complex of the soluble bacterial P450cam with its redox partner protein (16 Å) (35).
Structural comparisons of adrenodoxin binding among human P450 enzymes
Seven of the 57 human P450 enzymes are located in the mitochondria and rely on the class I redox system composed of adrenodoxin and adrenodoxin reductase. Three of these enzymes are involved in steroid hormone biosynthesis: CYP11A1, CYP11B1, and CYP11B2. If the functional differences observed with adrenodoxin interaction are reflections of structural differences in the individual P450–adrenodoxin interactions, then it could be possible to exploit those differences for selective P450 inhibition for various disease states.
The only other structural information available for a human P450 complex with adrenodoxin is for the similarly designed adrenodoxin–CYP11A1 fusion protein (16). CYP11A1 cleaves the side chain cleavage of cholesterol and thus acts upstream of all other steroidogenic P450 enzymes (36). Comparison of the two fusions with adrenodoxin reveals general conservation of the relative protein orientations—adrenodoxin similarly binds on the proximal face of CYP11A1—but there are substantial differences in the detailed interactions. Although positively charged residues (arginine or lysine) conserved among mitochondrial P450s participate in adrenodoxin binding for both P450 enzymes, they do so in different numbers and with different residue pairings (Fig. 6). Although three of the six CYP11B2 residues interacting with adrenodoxin are identical or conservatively substituted (Arg for Lys) in CYP11A1 (Fig. 6B), there are substantially fewer interactions for the latter complex. The adrenodoxin–CYP11B2 complex has five salt bridges, but only two are observed for adrenodoxin–CYP11A1 owing to sequence differences at the other positions. Similarly, the CYP11B2 complex is stabilized by four hydrogen bonds, whereas the CYP11A1 complex demonstrates two such interactions. In addition, the side chains of acidic residues in the adrenodoxin α helix 3 demonstrate different rotamers (Fig. 7A) depending on the P450.
Figure 7.
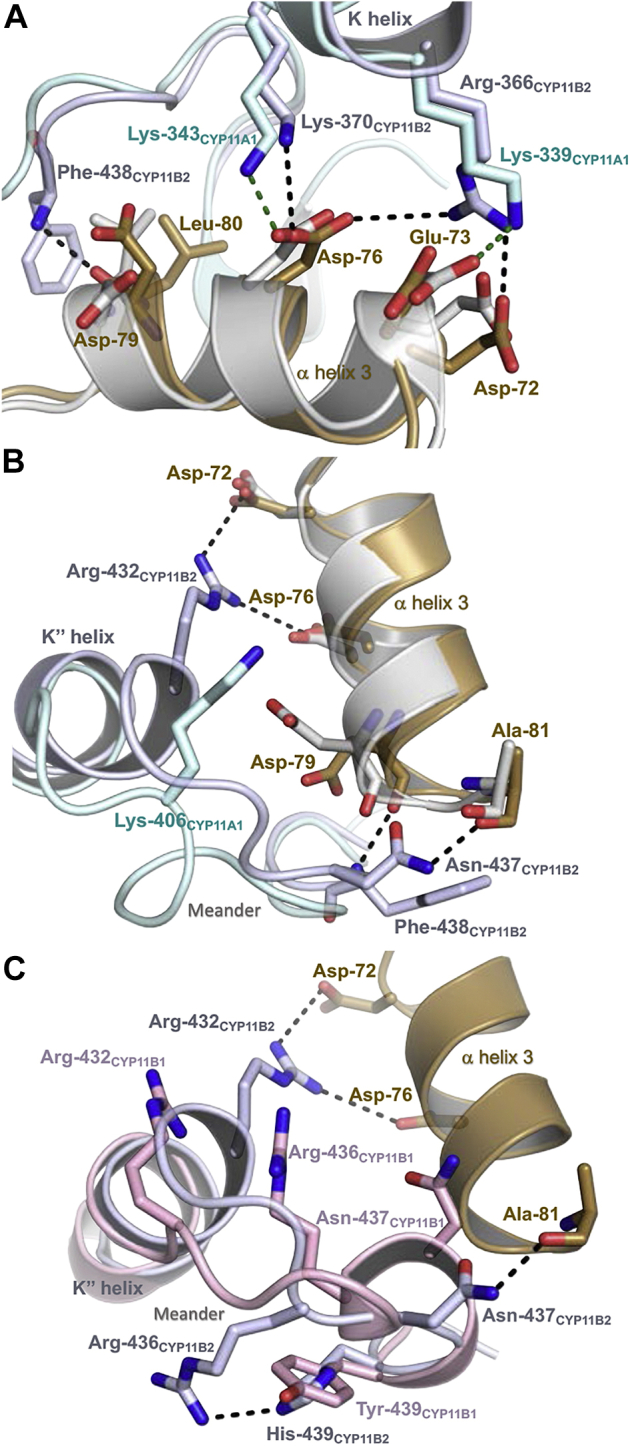
Structure comparisons of the adrenodoxin–CYP11B2 fusion with adrenodoxin–CYP11A1 fusion and with the isolated CYP11B1 enzyme (PDB6M7X). Comparison of adrenodoxin–CYP11B2 fusion with adrenodoxin–CYP11A1 fusion highlights differences in the interactions with adrenodoxin α3 (A) and the P450 meander preceding the heme-coordinating loop (B). Comparison of the adrenodoxin–CYP11B2 fusion with the isolated CYP11B1 also identifies significant conformational differences in the meander (C). The meander region of the adrenodoxin–CYP11A1 fusion differs from that of adrenodoxin–CYP11B2 meander region (B) owing to multiple sequence differences, but this region of the isolated CYP11B1 (light pink) and adrenodoxin–CYP11B2 fusion (C) differ by a single amino acid residue at position 439 (Tyr in CYP11B1, His in CYP11B2). Adrenodoxin–CYP11B2: P450 domain, light blue and fused adrenodoxin domain, brown. Adrenodoxin–CYP11A1 P450 domain, light cyan and fused adrenodoxin domain, gray. CYP11B1 isolated structure, pink.
The most striking structural difference between these two fusion enzymes is the meander region immediately preceding the heme-binding loop (Fig. 7B). In CYP11B2, Arg432, Asn437, and Phe438 interact with adrenodoxin, but in CYP11A1 the meander is in close proximity to adrenodoxin but without such specific interactions. In fact, the positive charge at CYP11B2 Arg432 is substituted by a negative charge because Asp occurs at the corresponding position in CYP11A1 (Fig. 6B). This is consistent with the idea that the meander region could be at least partially responsible for differential adrenodoxin interactions for individual mitochondrial P450s. These findings begin to outline how the same adrenodoxin protein can interact with differences in the proximal surface topology for seven different human mitochondrial P450s. If the individual P450 interactions with adrenodoxin were well understood, differences in the binding modes could potentially be exploited by tailoring small molecules or peptides to the relevant and distinctive part(s) of each P450 surface to prevent adrenodoxin binding and selectively inhibit them (37).
Although no structure is available for the complex of CYP11B1 and adrenodoxin to compare the current results with, each of the CYP11B2 residues interacting with adrenodoxin is also conserved in CYP11B1, which might obviate the idea of selectively inhibiting one of the CYP11B enzymes via this interface. However, two lines of evidence suggest that there may still be structural differences between complexes of adrenodoxin with the two human CYP11B enzymes. First, cross-linking studies suggest slight differences in the mode of interaction between the CYP11B enzymes (14). Second, now that it is known which surface of CYP11B2 interacts with adrenodoxin, one can compare this CYP11B2 interface with the corresponding surface from the one available CYP11B1 structure (PDB 6M7X, (38)). This CYP11B1 structure was also determined for an inhibitor-bound complex, but in the absence of adrenodoxin. However, comparison of these two CYP11B structures reveals significant structural differences in the meander region involved in CYP11B2 binding of adrenodoxin. Although the two CYP11B enzyme sequences are identical in the meander region except for one residue, this region adopts a totally different conformation (Fig. 7C). One of the few amino acid differences between the CYP11B enzymes occurs at position 439 and may be responsible for the differing meander conformations. His439 in CYP11B2 is substituted with Tyr439 in CYP11B1 (Fig. 6B). In CYP11B1 the larger tyrosine side chain is positioned slightly toward the meander backbone and may sterically reposition it, whereas in CYP11B2 His439 interacts with meander residue Arg436, colocalizing these side chains (Fig. 7C). Of interest, in some individuals, a residue swap at this precise position (H439Y) in CYP11B2 leads to increased CYP11B2 activity (39). It is possible that the increased activity might result from enhancing adrenodoxin interaction and/or electron transfer. However, a structure–function analysis of CYP11B1 interaction with adrenodoxin is needed to explore these ideas further.
Conclusions
The current work provides a systematic evaluation of adrenodoxin interactions with human aldosterone synthase, CYP11B2, and finds significant allosteric effect. A local increase of adrenodoxin concentration promotes binding of the substrate 11-deoxycorticosterone and the inhibitor LCI699, as well as enhances the inhibitory effect of this drug. Moreover, the structure of CYP11B2 in complex with adrenodoxin identifies specific interactions. Comparisons with steroidogenic CYP11A1 and CYP11B1 revealed substantial differences in the interacting structures. Overall, the structural and functional differences between different P450 interactions with adrenodoxin may provide valuable clues for an orthogonal treatment approach in which primary aldosteronism could potentially be treated by specifically targeting the interaction between CYP11B2 and adrenodoxin (37).
Experimental procedures
Materials
LCI699 (osilodrostat or Isturisa) (CAS 928134-65-0) was purchased from Selleckchem. Progesterone (CAS 57-83-0), corticosterone (CAS 50-22-6), 11-deoxycorticosterone (CAS 64-85-7), and fadrozole (CAS 102676-31-3) were purchased from Sigma-Aldrich.
Protein expression and purification
Generation of an adrenodoxin–CYP11B2 fusion protein for functional studies
Since the interaction between adrenodoxin and CYP11B2 is normally transient, a fusion protein was constructed to potentially stabilize the protein–protein interaction. The fusion construct was designed as the cDNA for full-length mature human adrenodoxin (residues 60–184 before the mitochondrial import peptide removal, residues 1–125 after removal), fused to a linker sequence coding for AAKKTSS, followed by residues 31 to 503 of the CYP11B2 wildtype protein, and a 4xHis tag. The nucleotide sequence was codon-optimized for E. coli and synthesized (GenScript). The resulting gene was cloned into the pCWori+ vector and transformed into the E. coli strain DH5α already containing the pGro7 vector for expressing the GroEL/ES chaperone system. A single colony from a lysogeny broth (LB) agar plate containing 100 μg/ml carbenicillin for pCW selection and 20 μg/ml chloramphenicol for pGro7 selection was used to initiate a 30-ml LB starter culture grown at 37 °C overnight with the same antibiotics. Expression cultures consisted of 800 ml terrific broth with 2X potassium phosphate buffer (from a 20X stock solution of 4.62 g KH2PO4 and 25 g K2HPO4 per 100 ml buffer), the same antibiotic concentrations, and 8 ml overnight culture in a 2.8-l Fernbach flask. These flasks were incubated at 37 °C and 210 rpm until an absorbance at 600 nm (A600) of 0.4 to 0.6 was reached. Expression of the adrenodoxin–CYP11B2 fusion protein was then induced with 1 mM isopropyl β-D-1-thiogalactopyranoside and the GroEL/ES chaperone with 4 mg/ml arabinose. At this point, cultures were also supplemented with 1 mM δ-aminolevulinic acid heme precursor. Cultures were subsequently grown at 27 °C and 190 rpm for an additional 48 h, after which bacterial cells were collected by centrifugation at 6690g and 4 °C for 15 min. The cell pellet was resuspended in 300 ml lysis buffer (50 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol, 500 mM sodium acetate, 1.5% (w/v) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate [CHAPS], 1.5% (v/v) Tween 20, phenylmethylsulfonyl fluoride [PMSF], 0.1 mM dithiothreitol [DTT], and a SIGMAFAST Protease Inhibitor Tablet [one per 150 ml]), homogenized with a Dounce homogenizer, and treated with lysozyme (60 mg/100 ml) for 1 h at 4 °C. The resuspended cells were disrupted by sonication with an amplitude of 10% (Fisher Scientific Model 100 Ultrasonic Dismembrator with a Qsonica Microson XL2000 Microprobe) for ten pulses of 30 s each while on ice. Membranes and cellular debris were then removed by ultracentrifugation at 142,414g for 50 min at 4 °C. The resulting supernatant was loaded on a Ni-NTA column (25 ml Ni-NTA Superflow, Qiagen) equilibrated with three column volumes of equilibration buffer (50 mM potassium phosphate, pH 7.4, 20% (v/v) glycerol, 500 mM sodium acetate, 1% (w/v) CHAPS, 1% (v/v) Tween 20, 0.1 mM PMSF, 0.1 mM DTT). The column was washed with five column volumes of wash buffer I (50 mM Tris base, pH 7.4, 20% (v/v) glycerol, 500 mM sodium acetate, 50 mM KCl, 20 mM MgCl2, 1% (w/v) CHAPS, 1% (v/v) Tween 20, 4 mM histidine, 0.1 mM PMSF, 0.1 mM DTT) containing 0.1 mM ATP to promote release of GroEL/ES from CYP11B2. The column then was washed with wash buffer II (50 mM Tris base, pH 7.4, 20% (v/v) glycerol, 1% (w/v) CHAPS, 1% (v/v) Tween 20, 4 mM histidine, 0.1 mM PMSF, 0.1 mM DTT) to decrease the salt content and wash out ATP. The adrenodoxin-CYP11B2 fusion protein was then eluted with 80 mM histidine buffer (20 mM Tris base, pH 8.0, 20% (v/v) glycerol, 1% (w/v) CHAPS, 1% (v/v) Tween 20, 80 mM histidine, 0.1 mM PMSF, 0.1 mM DTT). The eluting adrenodoxin–CYP11B2 protein had a maximal absorbance at 423 nm due to histidine binding. Subsequently, anion exchange chromatography was performed using a HiTrap Q HP column (1 ml, GE Healthcare) equilibrated with five column volumes of 20 mM Tris-HCl, pH 8.0, 20% (v/v) glycerol, 1% (w/v) CHAPS, 0.1% (v/v) Tween 20, 0.1 mM PMSF, and 0.1 mM DTT. After loading, the column was washed with ten column volumes of the same equilibration buffer and then eluted with a gradient from 0 to 500 mM NaCl in 50 mM Tris base buffer, pH 7.4, 20% (v/v) glycerol, 1% (w/v) CHAPS, 0.1% (v/v) Tween 20, 0.1 mM PMSF, and 0.1 mM DTT. Fractions with at least equal absorbance at 280 and 419 nm were pooled. Finally, the oligomeric state of the adrenodoxin–CYP11B2 fusion protein was examined by size-exclusion chromatography (SEC) (GE Healthcare HiLoad 16/600 Superdex 200 pg) using 50 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol, 1% (w/v) CHAPS, 0.05% (v/v) Tween 20, and 0.1 mM PMSF. Fractions with at least equal absorbance at 280 and 423 nm were pooled, flash frozen, and stored at −80 °C until use. Purified adrenodoxin–CYP11B2 fusion protein had a Soret peak at 419 nm, consistent with water coordinated to the ferric heme iron. Purity was examined using SDS-PAGE analysis, which yielded a single band at the expected molecular weight of 70 kDa. The ratio of absorbance of the heme Soret peak (419 nm) versus total protein absorbance (280 nm) for this adrenodoxin–CYP11B2 fusion protein was typically ∼1.12.
Recombinant adrenodoxin–CYP11B2 fusion protein generation for crystallography
For X-ray protein crystallography, all purification buffers above were supplemented with 30 μM fadrozole (dissolved in ethanol). The purification by Ni-NTA affinity and anion exchange chromatography was performed as described above, but SEC was run with 50 mM potassium phosphate buffer (pH 7.4), 20% (v/v) glycerol, 500 mM NaCl, and 0.1 mM PMSF to deplete detergents. The resulting purified protein was flash frozen and stored at −80 °C until further use. SDS-PAGE analysis revealed a single band at the expected molecular weight of 70 kDa. The ratio of absorbance of the fadrozole-bound heme Soret peak (423.5 nm) versus total protein absorbance (280 nm) was 0.99.
Recombinant CYP11B2
An E. coli codon-optimized CYP11B2 gene was synthesized (GenScript). This construct coded for N-terminally truncated CYP11B2 with the sequence MAKKTSS preceding the 30th amino acid of the wildtype sequence and a 4xHis tag at the C terminus. The modified gene was cloned into the pCWori+ vector. The resulting construct was transformed into the E. coli strain DH5α already containing the pGro7 vector encoding the GroEL/ES chaperone. A single colony from a LB agar plate containing 100 μg/ml carbenicillin for pCW selection and 20 μg/ml chloramphenicol for pGro7 selection was used to initiate a 30 ml LB starter grown at 37 °C overnight with the same antibiotics. Expression cultures consisted of 800 ml terrific broth with 2X potassium phosphate buffer, the same antibiotics, and 8 ml overnight culture in a 2.8-l Fernbach flask. These flasks were incubated at 37 °C and 210 rpm until an absorbance at 600 nm (A600) of 0.4 to 0.6 was reached. CYP11B2 expression was then induced with 1 mM isopropyl β-D-1-thiogalactopyranoside and the GroEL/ES chaperone with 4 mg/ml arabinose. At this point, cultures were also supplemented with 1 mM δ-aminolevulinic acid heme precursor. Cultures were incubated at 27 °C at 190 rpm for an additional 48 h, after which bacterial pellets were collected by centrifugation at 6690g and 4 °C for 15 min. The cell pellet was resuspended in 300 ml lysis buffer (50 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol, 500 mM sodium acetate, 1.5% (w/v) sodium cholate, 1.5% (v/v) Tween 20, 0.1 mM PMSF, 0.1 mM DTT), homogenized with a Dounce homogenizer, and treated with lysozyme (30 mg/100 ml) for 1 h at 4 °C. The cells were disrupted by sonication with an amplitude of 10% (Fisher Scientific Model 100 Ultrasonic Dismembrator with a Qsonica Microson XL2000 Microprobes) and ten pulses of 30 s while on ice. The cell debris was removed by ultracentrifugation at 142,414g for 50 min at 4 °C. The resulting supernatant was loaded on a Ni-NTA column (25 ml Ni-NTA Superflow, Qiagen) equilibrated with three column volumes of equilibration buffer (50 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol, 500 mM sodium acetate, 1% (w/v) sodium cholate, 1% (v/v) Tween 20, 0.1 mM PMSF, 0.1 mM DTT). The column was first washed with five column volumes of washing buffer I (50 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol, 500 mM sodium acetate, 1% (w/v) sodium cholate, 1% (v/v) Tween 20, 4 mM histidine, 0.1 mM PMSF, 0.1 mM DTT) and then with five column volumes washing buffer II (50 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol, 1% (w/v) sodium cholate, 1% (v/v) Tween 20, 4 mM histidine, 0.1 mM PMSF, 0.1 mM DTT) containing 0.1 mM ATP to promote removal of GroEL/ES from the CYP11B2 protein. CYP11B2 was eluted with 80 mM histidine buffer (20 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol, 1% (w/v) sodium cholate, 1% (v/v) Tween 20, 80 mM histidine, 0.1 mM PMSF, 0.1 mM DTT) and concentrated in a 50-kDa centrifugal device. The eluting CYP11B2 protein had a maximal absorbance at 424.5 nm owing to histidine binding. Subsequent cation exchange chromatography was performed using a HiTrap SP HP column (5 ml, GE Healthcare) equilibrated with five column volumes of 20 mM potassium phosphate, pH 7.4, 20% (v/v) glycerol, 1% (w/v) sodium cholate, 0.1% (v/v) Tween 20, 0.1 mM PMSF, and 0.1 mM DTT. After loading, the column was washed with 15 column volumes of the same equilibration buffer and then eluted with a gradient from 0 to 500 mM NaCl in 50 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol, 1% (w/v) sodium cholate, 0.1% (v/v) Tween 20, 0.1 mM PMSF and 0.1 mM DTT. Fractions with at least equal absorbance at 280 and 421.5 nm were pooled. Finally, the oligomeric state of CYP11B2 was examined by SEC (GE Healthcare HiLoad 16/600 Superdex 200 pg) using 50 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol, 1% (w/v) sodium cholate, 0.1% (v/v) Tween 20, and 0.1 mM PMSF. The resulting purified protein was flash frozen and stored at −80 °C until use. Purity was examined using SDS-PAGE analysis, which yielded a single band at 55 kDa. The ratio of absorbance of the heme Soret peak (421.5 nm) versus total protein absorbance at 280 nm for CYP11B2 purified using this method was typically ∼1.07.
Redox proteins
Recombinant human adrenodoxin (adrenodoxin) and adrenodoxin reductase were expressed and purified as described (38).
Crystallization and structural determination
The adrenodoxin–CYP11B2 fusion protein (15 mg/ml) in 50 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol, 500 mM NaCl, and saturated with fadrozole (Soret maximum at 423.5 nm) was screened for crystal formation with multiple commercial crystallization kits. Initial small crystals were observed after 24 h from 0.75 μl of the above adrenodoxin–CYP11B2/fadrozole stock mixed with 0.75 μl of 0.2 M lithium sulfate, 0.1 M Tris hydrochloride, pH 8.5, and 30% (v/v) PEG 4000 in a sitting drop equilibrated against 200 μl of the same precipitant solution at 20 °C. Since subsequent de novo crystal growth from the same condition was not successful, a seed stock from the original crystals was prepared to promote nucleation. Fresh sitting drop vapor diffusion experiments were conducted with 0.75 μl 15 mg/ml adrenodoxin–CYP11B2 mixed with 0.75 μl of the undiluted seed stock equilibrated against 200 μl of the precipitant solution at 20 °C. Crystals of the same morphology were observed after 48 h, harvested, transferred into the mother liquor with glycerol increased to 18% (v/v) to serve as the cryoprotectant, and cryocooled in liquid nitrogen for transport to the synchrotron. X-ray diffraction data were collected on beamline 21-ID-G of the Argonne Photon Source and processed using HKL2000 (40). The adrenodoxin–CYP11B2 structure in complex with fadrozole was solved by molecular replacement using Phaser (41) and a search model consisting of CYP11B2 bound to fadrozole (PDB 4FDH (22)) and human adrenodoxin (PDB 3P1M). Model building and refinement were performed iteratively with Coot (42) and Phenix.refine (43), respectively. The ligand (R)-fadrozole was generated using elBOW in Phenix using the PDB file (PDB 0T3) (43). The final structure model and structure factors are deposited in the Protein Data Base (PDB 7M8I). Structural comparisons were accomplished and RMSDs calculated using the Secondary Structure Matching algorithm in Coot (44).
Ligand binding assays
The effect of adrenodoxin on CYP11B2 binding of substrates and inhibitors (binding mode and dissociation constant, Kd) was investigated by monitoring changes in UV-visible absorbance during ligand titration. For CYP11B2 binding of the physiological substrate 11-deoxycorticosterone, 1 μM CYP11B2 was diluted in 50 mM potassium phosphate, pH 7.4, and 20% (v/v) glycerol in quartz cuvettes with a 1-cm pathlength. When studying the effect of adrenodoxin, one of three different concentrations of adrenodoxin was added to the sample cuvette: 1 μM (for 1:1 CYP11B2:adrenodoxin), 10 μM (for 1:10), or 40 μM (for 1:40). For all binding experiments, increasing amounts of the 11-deoxycorticosterone substrate (0.63–151.25 μM, dissolved in dimethyl sulfoxide [DMSO]) were titrated into CYP11B2, while DMSO alone was added to the reference cuvette. After each substrate addition, samples were mixed by inverting and incubated for 8 min at room temperature. Difference spectra were then recorded from 300 to 500 nm. The maximal differences in absorbance (local absorbance maximum located between 386.5 and 391 nm minus minimum located between 417.5 and 423 nm) for each ligand concentration were plotted against substrate concentration. The resulting data were fit using the one-site-binding equation (45) in GraphPad Prism version 8.0.0 for Mac OS X (GraphPad Software) to determine values for the maximal absorption change at saturation (ΔAmax) and dissociation constant (Kd).
The inhibitor LCI699 binds CYP11B2 so tightly that modifications of the experimental parameters were required. LCI699 (dissolved in DMSO) was titrated into a reduced concentration of 0.2 μM CYP11B2 or adrenodoxin–CYP11B2 fusion protein in 50 mM potassium phosphate, pH 7.4, 20% (v/v) glycerol, and 0.5% (w/v) CHAPS in quartz cuvettes. To compensate for the lower signal with a 5-fold decrease in CYP11B2 concentration, the path length was increased 5-fold to 5 cm. Otherwise titrations were performed as described above. The resulting plot of changes in absorbance (local absorbance maximum, located between 427.5 and 434 nm, minus minimum, located between 408.5 and 413 nm) versus LCI699 concentration still required using the tight-binding Morrison equation to account for ligand depletion (45).
All binding assays were completed in triplicate, with all values shown for each titration, although the points overlap substantially, so this is not always obvious in the graphs (Figs. 2, A and B, and 4B). Adrenodoxin was used in its oxidized form to identify allosteric effects separate from redox effects.
Determination of kinetic parameters
To determine the kinetic parameters of CYP11B2 for 11β-hydroxylation, in vitro activity assays were conducted with the native initial substrate 11-deoxycorticosterone. To examine the effect of adrenodoxin on catalysis, different experiments were conducted with 1:1, 1:10, and 1:40 ratios with excess adrenodoxin over CYP11B2. The catalytic system was reconstituted in a total reaction volume of 500 μl using 0.2 μM CYP11B2, 0.2 μM adrenodoxin reductase, and 0.2 μM adrenodoxin in 50 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol for assays examining the impact of equimolar adrenodoxin amounts. For examining 10-fold adrenodoxin excess, 0.05 μM CYP11B2, 0.05 μM adrenodoxin reductase, and 0.5 μM adrenodoxin were used. For examining 40-fold adrenodoxin excess, 0.05 μM CYP11B2, 0.05 μM adrenodoxin reductase, and 2 μM adrenodoxin were used. For experiments with the adrenodoxin-CYP11B2 fusion enzyme, 0.4 μM adrenodoxin-CYP11B2 protein, 0.4 μM adrenodoxin reductase, and 16 μM adrenodoxin were used with a reaction time of 1 min. The enzymes were mixed and incubated for 20 min at room temperature. Metabolism of 11-deoxycorticosterone (1–150 μM, dissolved in DMSO; total of 0.5% (v/v) DMSO) was initiated by the addition of 5 mM NADPH and allowed to proceed at 37 °C. For experiments with equimolar adrenodoxin amounts, reactions proceeded for 1 to 5 min. For 10-fold and 40-fold adrenodoxin excess, reaction times were reduced to 30 s to prevent >10% substrate depletion. All reactions were quenched with 500 μl chloroform and 40 μM progesterone (in DMSO) added as an internal standard. Steroids were extracted twice with 500 μl chloroform, dried, and resuspended in 100 μl 20% (v/v) acetonitrile, and 40 μl was injected on a reverse-phase HPLC column (Phenomenex, Luna, 5 μm, C18, 150 × 4.6 mm) using an acetonitrile–water gradient (Phase A: 10% (v/v) acetonitrile, Phase B: 100% acetonitrile) run as follows: 0 to 6 min 20% B (step), 6 to 11 min 40% B (linear gradient), 12 to 22 min 80% B (linear gradient), 23 to 25 min 80% B (step), 26 to 30 min 20% B (step), all at 40 °C and a flow rate of 0.8 ml/min. Steroids were detected by UV absorbance at 240 nm. Retention times of main products, substrate, and internal standard were the following in elution order: 18-hydroxycorticosterone 7.4 min, aldosterone 8.5 min, corticosterone 13.9 min, 11-deoxycorticosterone 17.2 min, and progesterone (internal standard) 21.6 min. Quantification of the steroids was performed determining the ratio of the peak area for the product corticosterone and the peak area of the internal standard progesterone using a calibration curve. Under these conditions, formation of subsequent products (18-hydroxycorticosterone and aldosterone) was <5%. Initial reaction velocities were plotted against the respective 11-deoxycorticosterone concentration and analyzed with the Michaelis–Menten equation using GraphPad Prism version 8.0.0 for Mac OS X (GraphPad Software). All experiments were repeated in triplicate and all values for all reactions shown in the plots (Fig. 3, A and B), although they overlap enough at the lower ratios of adrenodoxin that this is not always obvious.
Determination of the half-maximal inhibitory concentration
The effect of adrenodoxin on the IC50 of LCI699 on the 11β-hydroxylation of 11-deoxycorticosterone by CYP11B2 was determined using 1:1, 1:10, or 1:40 excess of adrenodoxin over CYP11B2. In vitro activity assays were conducted with the physiological substrate 11-deoxycorticosterone (dissolved in DMSO) at the Km determined under these reaction conditions (3.5 μM). The catalytic system was reconstituted in a total reaction volume of 500 μl using 0.05 μM CYP11B2, 0.05 μM adrenodoxin reductase, and 2 μM adrenodoxin in 50 mM potassium phosphate buffer, pH 7.4, 20% glycerol, and 0.5% (w/v) CHAPS for examining 40-fold adrenodoxin excess with a reaction time of 20 s. For 10-fold adrenodoxin excess, 0.05 μM CYP11B2, 0.05 μM adrenodoxin reductase, and 0.5 μM adrenodoxin were used and the reaction time increased to 30 s, which was still under 10% substrate depletion. For equimolar adrenodoxin, 0.2 μM CYP11B2, 0.2 μM adrenodoxin reductase, and 0.2 μM adrenodoxin were used with a reaction time of 30 s. Inhibitor (dissolved in DMSO) concentrations from 0.76 nM to 12.5 μM were used, with the total DMSO concentration maintained at 0.5% (v/v) for all reactions. Metabolism of 11-deoxycorticosterone was initiated by the addition of 5 mM NADPH and proceeded at 37 °C. Reactions were quenched with 500 μl chloroform, and progesterone (dissolved in DMSO) was added as internal standard. Steroids were extracted and analyzed by HPLC as described above. All experiments were repeated in triplicate, and all data points are shown (Fig. 4A). The relative percentage of inhibition compared with the reaction without inhibitor was plotted against the respective inhibitor concentration and analyzed using GraphPad Prism version 8.0.0 for Mac OS X (GraphPad Software) using a dose–response inhibitor (four-parameter) equation.
Data availability
All of the data are included in this article or the supplemental information statement, with the exception of the structure coordinates and structure factors. This latter information has been deposited with the PDB under accession code 7M8I.
Supporting information
This article contains supporting information.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Constructs for expression of adrenodoxin and adrenodoxin reductase were gifts from Dr Richard Auchus at the University of Michigan. Generation of the CYP11B2 and Adx–CYP11B2 fusion constructs, expression, purification, and characterization was supported by startup funds from the University of Michigan. Adx–CYP11B2 coexpression with fadrozole, conducted binding and inhibition studies, protein crystallography and structure determination were supported by a Postdoctoral Fellowship Award of the American Heart Association (AHA, Award No. 19POST34430199). The content of this publication is solely the responsibility of the authors and do not necessarily represent the official views of AHA.
Author contributions
E. E. S. and S. B. -A. conceived the study, performed data analysis, and wrote the manuscript. S. B. -A. conducted experiments.
Edited by F. Peter Guengerich
Supporting information
References
- 1.Connell J.M.C., Davies E. The new biology of aldosterone. J. Endocrinol. 2005;186:1–20. doi: 10.1677/joe.1.06017. [DOI] [PubMed] [Google Scholar]
- 2.Schiffer L., Anderko S., Hannemann F., Eiden-Plach A., Bernhardt R. The CYP11B subfamily. J. Steroid Biochem. Mol. Biol. 2015;151:38–51. doi: 10.1016/j.jsbmb.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 3.Bureik M., Lisurek M., Bernhardt R. The human steroid hydroxylases CYP1B1 and CYP11B2. Biol. Chem. 2002;383:1537–1551. doi: 10.1515/BC.2002.174. [DOI] [PubMed] [Google Scholar]
- 4.Monticone S., D'Ascenzo F., Moretti C., Williams T.A., Veglio F., Gaita F., Mulatero P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2018;6:41–50. doi: 10.1016/S2213-8587(17)30319-4. [DOI] [PubMed] [Google Scholar]
- 5.Funder J.W. Aldosterone, cardiovascular damage and the new biology of mineralocorticoid receptors. J. Hypertens. 2006;24:16. doi: 10.1161/01.HYP.0000203732.03784.3b. [DOI] [PubMed] [Google Scholar]
- 6.Browne L.J., Gude C., Rodriguez H., Steele R.E., Bhatnager A. Fadrozole hydrochloride: A potent, selective, nonsteroidal inhibitor of aromatase for the treatment of estrogen-dependent disease. J. Med. Chem. 1991;34:725–736. doi: 10.1021/jm00106a038. [DOI] [PubMed] [Google Scholar]
- 7.Amar L., Azizi M., Menard J., Peyrard S., Watson C., Plouin P.F. Aldosterone synthase inhibition with LCI699: A proof-of-concept study in patients with primary aldosteronism. Hypertension. 2010;56:831–838. doi: 10.1161/HYPERTENSIONAHA.110.157271. [DOI] [PubMed] [Google Scholar]
- 8.Duggan S. Osilodrostat: First approval. Drugs. 2020;80:495–500. doi: 10.1007/s40265-020-01277-0. [DOI] [PubMed] [Google Scholar]
- 9.Hannemann F., Bichet A., Ewen K.M., Bernhardt R. Cytochrome P450 systems–biological variations of electron transport chains. Biochim. Biophys. Acta. 2007;1770:330–344. doi: 10.1016/j.bbagen.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 10.Muller J.J., Lapko A., Bourenkov G., Ruckpaul K., Heinemann U. Adrenodoxin reductase-adrenodoxin complex structure suggests electron transfer path in steroid biosynthesis. J. Biol. Chem. 2001;276:2786–2789. doi: 10.1074/jbc.M008501200. [DOI] [PubMed] [Google Scholar]
- 11.Lin D., Shi Y.F., Miller W.L. Cloning and sequence of the human adrenodoxin reductase gene. Proc. Natl. Acad. Sci. U. S. A. 1990;87:8516–8520. doi: 10.1073/pnas.87.21.8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lambeth J.D., McCaslin D.R., Kamin H. Adrenodoxin reductase-adrenodexin complex. J. Biol. Chem. 1976;251:7545–7550. [PubMed] [Google Scholar]
- 13.Ewen K.M., Ringle M., Bernhardt R. Adrenodoxin–a versatile ferredoxin. IUBMB Life. 2012;64:506–512. doi: 10.1002/iub.1029. [DOI] [PubMed] [Google Scholar]
- 14.Peng H.M., Auchus R.J. Molecular recognition in mitochondrial cytochromes P450 that catalyze the terminal steps of corticosteroid biosynthesis. Biochemistry. 2017;56:2282–2293. doi: 10.1021/acs.biochem.7b00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Usanov S.A., Graham S.E., Lepesheva G.I., Azeva T.N., Strushkevich N.V., Gilep A.A., Estabrook R.W., Peterson J.A. Probing the interaction of bovine cytochrome P450scc (CYP11A1) with adrenodoxin: Evaluating site-directed mutations by molecular modeling. Biochemistry. 2002;41:8310–8320. doi: 10.1021/bi0255928. [DOI] [PubMed] [Google Scholar]
- 16.Strushkevich N., MacKenzie F., Cherkesova T., Grabovec I., Usanov S., Park H.W. Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc. Natl. Acad. Sci. U. S. A. 2011;108:10139–10143. doi: 10.1073/pnas.1019441108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mast N., Annalora A.J., Lodowski D.T., Palczewski K., Stout C.D., Pikuleva I.A. Structural basis for three-step sequential catalysis by the cholesterol side chain cleavage enzyme CYP11A1. J. Biol. Chem. 2011;286:5607–5613. doi: 10.1074/jbc.M110.188433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katagiri M., Takikawa O., Sato H., Suhara K. Formation of a cytochrome P-450scc-adrenodoxin complex. Biochem. Biophys. Res. Commun. 1977;77:804–809. doi: 10.1016/s0006-291x(77)80049-1. [DOI] [PubMed] [Google Scholar]
- 19.Hartfield K.A., Stout C.D., Annalora A.J. The novel purification and biochemical characterization of a reversible CYP24A1:adrenodoxin complex. J. Steroid Biochem. Mol. Biol. 2013;136:47–53. doi: 10.1016/j.jsbmb.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Zollner A., Hannemann F., Lisurek M., Bernhardt R. Deletions in the loop surrounding the iron-sulfur cluster of adrenodoxin severely affect the interactions with its native redox partners adrenodoxin reductase and cytochrome P450(scc) (CYP11A1) J. Inorg. Biochem. 2002;91:644–654. doi: 10.1016/s0162-0134(02)00463-4. [DOI] [PubMed] [Google Scholar]
- 21.Vickery L.E. Molecular recognition and electron transfer in mitochondrial steroid hydroxylase systems. Steroids. 1997;62:124–127. doi: 10.1016/s0039-128x(96)00170-5. [DOI] [PubMed] [Google Scholar]
- 22.Strushkevich N., Gilep A.A., Shen L., Arrowsmith C.H., Edwards A.M., Usanov S.A., Park H.W. Structural insights into aldosterone synthase substrate specificity and targeted inhibition. Mol. Endocrinol. 2013;27:315–324. doi: 10.1210/me.2012-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beckert V., Bernhardt R. Specific aspects of electron transfer from adrenodoxin to cytochromes P450scc and p45011beta. J. Biol. Chem. 1997;272:4883–4888. doi: 10.1074/jbc.272.8.4883. [DOI] [PubMed] [Google Scholar]
- 24.Reddish M.J., Guengerich F.P. Human cytochrome P450 11B2 produces aldosterone by a processive mechanism due to the lactol form of the intermediate 18-hydroxycorticosterone. J. Biol. Chem. 2019;294:12975–12991. doi: 10.1074/jbc.RA119.009830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birtcher K.K., Ballantyne C.M. Cardiology patient page. Measurement of cholesterol: A patient perspective. Circulation. 2004;110:e296–e297. doi: 10.1161/01.CIR.0000141564.89465.4E. [DOI] [PubMed] [Google Scholar]
- 26.Schiffer L., Barnard L., Baranowski E.S., Gilligan L.C., Taylor A.E., Arlt W., Shackleton C.H.L., Storbeck K.H. Human steroid biosynthesis, metabolism and excretion are differentially reflected by serum and urine steroid metabolomes: A comprehensive review. J. Steroid Biochem. Mol. Biol. 2019;194:105439. doi: 10.1016/j.jsbmb.2019.105439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng H.M., Barlow C., Auchus R.J. Catalytic modulation of human cytochromes P450 17A1 and P450 11B2 by phospholipid. J. Steroid Biochem. Mol. Biol. 2018;181:63–72. doi: 10.1016/j.jsbmb.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roumen L., Sanders M.P., Pieterse K., Hilbers P.A., Plate R., Custers E., de Gooyer M., Smits J.F., Beugels I., Emmen J., Ottenheijm H.C., Leysen D., Hermans J.J. Construction of 3D models of the CYP11B family as a tool to predict ligand binding characteristics. J. Comput. Aided Mol. Des. 2007;21:455–471. doi: 10.1007/s10822-007-9128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Q.Z., Yin L.N., Hartmann R.W. Aldosterone synthase inhibitors as promising treatments for mineralocorticoid dependent cardiovascular and renal diseases. J. Med. Chem. 2014;57:5011–5022. doi: 10.1021/jm401430e. [DOI] [PubMed] [Google Scholar]
- 30.Parajes S., Loidi L., Reisch N., Dhir V., Rose I.T., Hampel R., Quinkler M., Conway G.S., Castro-Feijoo L., Araujo-Vilar D., Pombo M., Dominguez F., Williams E.L., Cole T.R., Kirk J.M. Functional consequences of seven novel mutations in the CYP11B1 gene: Four mutations associated with nonclassic and three mutations causing classic 11beta-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2010;95:779–788. doi: 10.1210/jc.2009-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye Z.Q., Zhang M.N., Zhang H.J., Jiang J.J., Li X.Y., Zhang K.Q. A novel missense mutation, GGC(Arg454)--> TGC(Cys), of CYP11B1 gene identified in a Chinese family with steroid 11beta-hydroxylase deficiency. Chin. Med. J. (Engl.) 2010;123:1264–1268. [PubMed] [Google Scholar]
- 32.Khattab A., Haider S., Kumar A., Dhawan S., Alam D., Romero R., Burns J., Li D., Estatico J., Rahi S., Fatima S., Alzahrani A., Hafez M., Musa N., Razzghy Azar M. Clinical, genetic, and structural basis of congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E1933–E1940. doi: 10.1073/pnas.1621082114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar A., Wilderman P.R., Tu C.J., Shen S.C., Qu J., Estrada D.F. Evidence of allosteric coupling between substrate binding and adx recognition in the vitamin D carbon-24 hydroxylase CYP24A1. Biochemistry. 2020;59:1537–1548. doi: 10.1021/acs.biochem.0c00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curnow K.M., Slutsker L., Vitek J., Cole T., Speiser P.W., New M.I., White P.C., Pascoe L. Mutations in the CYP11B1 gene causing congenital adrenal hyperplasia and hypertension cluster in exons 6, 7, and 8. Proc. Natl. Acad. Sci. U. S. A. 1993;90:4552–4556. doi: 10.1073/pnas.90.10.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hiruma Y., Hass M.A., Kikui Y., Liu W.M., Olmez B., Skinner S.P., Blok A., Kloosterman A., Koteishi H., Lohr F., Schwalbe H., Nojiri M., Ubbink M. The structure of the cytochrome P450cam-putidaredoxin complex determined by paramagnetic NMR spectroscopy and crystallography. J. Mol. Biol. 2013;425:4353–4365. doi: 10.1016/j.jmb.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 36.Chien Y., Rosal K., Chung B.C. Function of CYP11A1 in the mitochondria. Mol. Cell. Endocrinol. 2017;441:55–61. doi: 10.1016/j.mce.2016.10.030. [DOI] [PubMed] [Google Scholar]
- 37.Scott D.E., Bayly A.R., Abell C., Skidmore J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016;15:533–550. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- 38.Brixius-Anderko S., Scott E.E. Structure of human cortisol-producing cytochrome P450 11B1 bound to the breast cancer drug fadrozole provides insights for drug design. J. Biol. Chem. 2019;294:453–460. doi: 10.1074/jbc.RA118.006214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holloway C.D., MacKenzie S.M., Fraser R., Miller S., Barr M., Wilkinson D., Forbes G.H., Friel E., Connell J.M.C., Davies E. Effects of genetic variation in the aldosterone synthase (CYP11B2) gene on enzyme function. Clin. Endocrinol. 2009;70:363–371. doi: 10.1111/j.1365-2265.2008.03383.x. [DOI] [PubMed] [Google Scholar]
- 40.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 41.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krissinel E., Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 45.Godamudunage M.P., Grech A.M., Scott E.E. Comparison of antifungal azole interactions with adult cytochrome P450 3A4 versus neonatal cytochrome P450 3A7. Drug Metab. Dispos. 2018;46:1329–1337. doi: 10.1124/dmd.118.082032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brixius-Anderko S., Scott E.E. Aldosterone synthase structure with Cushing’s Disease drug LCI699 highlights avenues for selective CYP11B drug design. Hypertension. 2021 doi: 10.1161/HYPERTENSIONAHA.121.17615. In press. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the data are included in this article or the supplemental information statement, with the exception of the structure coordinates and structure factors. This latter information has been deposited with the PDB under accession code 7M8I.