

SUMMARY
Accumulating evidence indicates that the MDM2 oncoprotein promotes tumorigenesis beyond its canonical negative effects on the p53 tumor suppressor, but these p53-independent functions remain poorly understood. Here, we show that a fraction of endogenous MDM2 is actively imported in mitochondria to control respiration and mitochondrial dynamics independently of p53. Mitochondrial MDM2 represses the transcription of NADH-dehydrogenase 6 (MT-ND6) in vitro and in vivo, impinging on respiratory complex I activity and enhancing mitochondrial ROS production. Recruitment of MDM2 to mitochondria increases during oxidative stress and hypoxia. Accordingly, mice lacking MDM2 in skeletal muscles exhibit higher MT-ND6 levels, enhanced complex I activity, and increased muscular endurance in mild hypoxic conditions. Furthermore, increased mitochondrial MDM2 levels enhance the migratory and invasive properties of cancer cells. Collectively, these data uncover a previously unsuspected function of the MDM2 oncoprotein in mitochondria that play critical roles in skeletal muscle physiology and may contribute to tumor progression.
Graphical Abstract
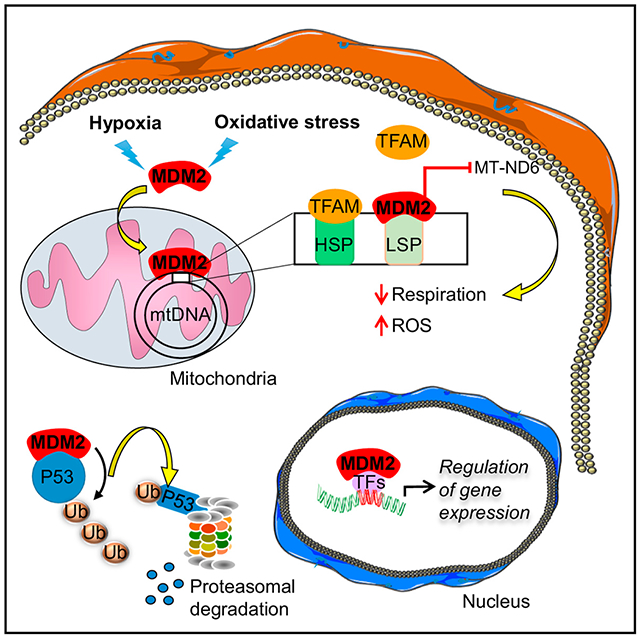
In Brief
Arena et al. show that the proto-oncogene MDM2 is recruited to mitochondria independently of its well-known partner p53. By repressing transcription of a specific subunit of the electron transport chain encoded by the mitochondrial genome, mitochondrial MDM2 controls respiration, influencing skeletal muscle function and cancer cell migration.
INTRODUCTION
The p53 pathway is functionally inactivated in most, if not all, cancers. Somatic mutations in TP53 occur in about 50% of human tumors, and many cancers retaining wild-type TP53 display functional inactivation of the p53 pathway by viral oncoproteins or alterations of p53 regulators, including the Mouse Double Minute 2 (MDM2) oncogene (Wade et al., 2013). MDM2 is a major negative regulator of the p53 pathway through its well-documented E3 ligase activity that targets p53 protein for proteasomal degradation. MDM2 also contributes to the regulation of p53 target genes by directly inhibiting p53 transactivation domains and by ubiquitylating histones at p53-responsive genes (Momand et al., 1992; Oliner et al., 1993; Minsky and Oren, 2004). However, it is now recognized that MDM2 oncogenic activities extend beyond the direct control of p53. Thus, MDM2-mediated ubiquitylation of several transcription factors or transcriptional co-regulators contributes to the regulation of p53-independent programs (Biderman et al., 2012). Recent findings also identified a p53-independent role for chromatin-bound MDM2 in the transcriptional control of genes involved in cell fate and metabolism (Wienken et al., 2016; Riscal et al., 2016). Moreover, the RING domain of MDM2 is required for its binding to the mRNAs of several cancer-related genes to control their stability or translation, including those encoding p53 itself, the X-linked inhibitor of apoptosis (XIAP), N-MYC, and vascular endothelial growth factor (VEGF) (FÁhraeus and Olivares-Illana, 2014). Here, we show that a fraction of MDM2 protein localizes in the mitochondrial matrix independently of p53 and regulates the transcription of the mitochondrial genome to control respiration in both normal and cancer cells.
RESULTS
MDM2 Localizes to the Mitochondrial Matrix
We assessed MDM2 subcellular localization in several cancer cell lines by immunoblotting following biochemical fractionation of various cellular compartments and isolation of purified organelles. As anticipated, MDM2 was detected in fractions enriched in cytosolic or nuclear proteins prepared from p53 null H1299 lung cancer cells. Unexpectedly, significant fractions of both ectopic FLAG-tagged MDM2 and endogenous MDM2 were detected in protein extracts prepared from purified mitochondria. The specificity of MDM2 immunoreactivity was confirmed using extracts prepared from MDM2-depleted H1299 cells, and the quality of the fractionation was verified by measuring the levels of cytosolic tubulin (TUB), nuclear TATA binding protein (TBP), and mitochondrial TIM23 proteins (Figures 1A and 1B). Next, we evaluated the amount of mitochondrial MDM2 in a panel of human cancer cell lines. When detectable by immunoblotting, a variable amount of endogenous MDM2, ranging from 2% to 5% of total MDM2, localized to mitochondria in these cells (Figures 1C, 1D, and S1A). The amount of mitochondrial MDM2 (mtMDM2) did not correlate with the p53 status of these cell lines, suggesting that MDM2 localized to mitochondria in a p53-independent manner. Consistent with this notion, short hairpin RNA (shRNA)-mediated depletion of wild-type (WT) p53 in MCF7 or of mutant p53 in T47D breast cancer cells did not alter the relative proportion of mtMDM2, indicating that neither WT nor mutant p53 was required for the localization of endogenous MDM2 to mitochondria (Figures 1E and S1B). Confocal microscopy analysis showed that FLAG-tagged full-length (FL) MDM2 (residues 1 to 491) exhibited different subcellular localization in H1299 cells. Approximately 35% of H1299 cells displayed a strictly nuclear pattern for ectopic FL-MDM2, 55% of them exhibited both nuclear and cytoplasmic MDM2, and 10% of them displayed a staining pattern that was restricted to the cytoplasm (Figure S1C). Super-resolution microscopy performed at a resolution of 100 nm indicated that approximately 8% of FL-MDM2 co-localized with the mitochondrial marker TFAM (Figure 1F). Mitochondrial localization of ectopic FL-MDM2 in H1299 cells was further confirmed by immunogold labeling and transmission electron microscopy (TEM). Consistent with MDM2 localization in different subcellular compartments, gold particles were detected in the nucleus and the cytoplasm, but also inside the mitochondrial matrix, at the edge of mitochondrial cristae (Figures 1G and S1D). We next performed protease protection assays on purified mitochondria isolated from H1299 cells expressing FL-MDM2. At concentrations of 0.5 and 1 μg/mL Proteinase K (PK), the outer mitochondrial membrane (OMM) protein TOM20 and the inner mitochondrial membrane (IMM) protein TIM23 were progressively degraded. In contrast, MDM2 and the mitochondrial matrix protein TFAM were both resistant to mild protease digestion. Complete PK-mediated digestion of TFAM and MDM2 proteins occurred only after treatment with Triton X-100, indicating that both proteins localized in the same mitochondrial compartment (Figure 1H). These data demonstrate that a fraction of MDM2 protein localizes to the mitochondrial matrix.
Figure 1. MDM2 Localizes in the Mitochondrial Matrix.
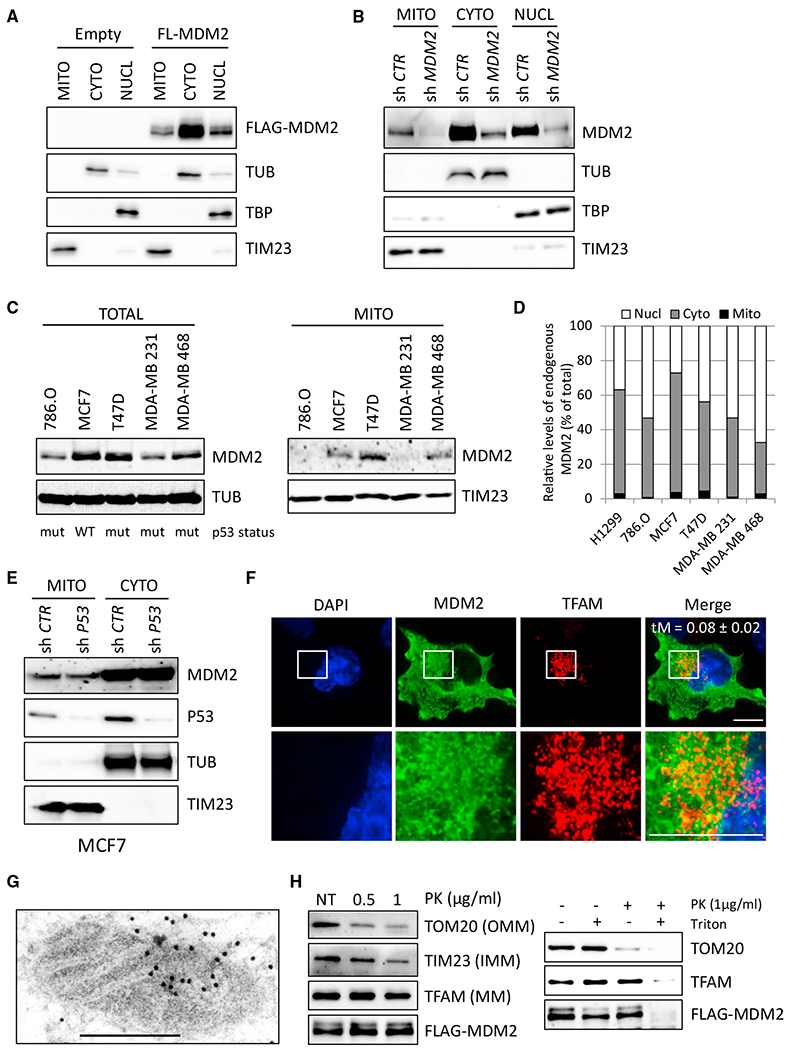
(A) Subcellular localization of FLAG-tagged full-length (aa 1–491) MDM2 (FL-MDM2) in H1299 cells. MDM2 levels were determined by immunoblotting of fractions enriched in mitochondrial (MITO), cytosolic (CYTO), and nuclear (NUCL) proteins with an anti-FLAG antibody. Equal loading was verified with TATA binding protein (TBP), tubulin (TUB), and TIM23 protein levels.
(B) Immunoblot (IB) analysis of endogenous MDM2 subcellular localization in H1299 cells transduced with lentiviruses expressing control or MDM2 shRNAs.
(C) IB analysis of total (left) and mitochondrial (right) endogenous MDM2 levels in different human cancer cell lines. The p53 status of each cell line is indicated: WT or mutant (mut).
(D) Quantitative IB analysis of endogenous MDM2 subcellular localization in different human cancer cell lines. Histobars represent the relative levels of nuclear, cytosolic, and mtMDM2 in the indicated cell lines.
(E) IB analysis of endogenous MDM2 subcellular localization in MCF7 cells transduced with lentiviruses encoding control or p53 shRNAs.
(F) Super-resolution microscopy analysis of FL-MDM2 (green) and TFAM (red) subcellular localization in H1299 cells. Nuclei were stained with DAPI (blue). Scale bars, 10 μM. tM indicates the Manders coefficient representing the colocalization of the signal detected with the anti-FLAG antibody and that detected with the anti-TFAM antibody.
(G) Immunogold staining for transmission electron microscopy (TEM) analysis of MDM2 subcellular localization in H1299 cells expressing FL-MDM2. Scale bar, 500 nM.
(H) Protease protection assays performed on purified mitochondria isolated from H1299 cells expressing FL-MDM2. Left: IB analysis of MDM2, the outer mitochondrial membrane (OMM) protein TOM20, the inner mitochondrial membrane (IMM) protein TIM23, and the mitochondrial matrix (MM) protein TFAM, after incubation of purified mitochondria with 0.5 or 1 μg/mL PK. NT, mock-treated mitochondria. Right: protease protection assays performed in the presence of the permeabilizing agent Triton X-100. MDM2 and TFAM proteins were only digested in presence of Triton X-100, confirming their localization in the mitochondrial matrix.
See also Figure S1.
MDM2 Is Actively Imported in Mitochondria through Mitochondrial Transporters
Mitochondrial proteins often contain an N-terminal leader peptide that is cleaved in the IMM before they can be released in the matrix (Dudek et al., 2013). Analysis of the MDM2 protein sequence failed to identify a canonical mitochondrial localization signal. In order to ascertain which domain of MDM2 was required for its recruitment to mitochondria, we performed subcellular fractionation assays and immunogold staining for TEM in H1299 cells expressing FL-MDM2 or different MDM2 deletion mutants. Deletion of its N-terminal region increased the total levels of MDM2 but did not change its relative subcellular localization, whereas the deletion of the C-terminal region (amino acids [aa] 292–491) impacted on its localization in mitochondria (Figures 2A–2C and S2A–S2C). Smaller deletions of the C-terminal region indicated that the last 61 aa (aa 430–491) of MDM2, and a more central region (aa 291–391) played a significant role in MDM2 localization in mitochondria (Figures S2D and S2E). We next asked whether MDM2 recruitment to this organelle was dependent on mitochondrial transporters. FL-MDM2 co-immunoprecipitated with endogenous TOM20 and TIM23 proteins, two components of the mitochondrial translocator complexes located in the OMM and the IMM, respectively (Figure 2D). Transport through these mitochondrial translocator complexes depends on the membrane potential (ΔΨ) across the IMM (Geissler et al., 2000). Accordingly, the amount of mtMDM2 in H1299 cells decreased upon treatment with carbonyl cyanide4-(trifuoromethoxy) phenylhydrazone (FCCP), a protonophore that disrupts mitochondrial membrane potential (Figure 2E). Moreover, MDM2 co-immunoprecipitated with the mitochondrial 70-kDa heat shock protein (mtHsp70), also known as mortalin, as well as with its direct partner TID1 (Figures 2D and S2F). These two proteins are central components of the mitochondrial protein import motor that play a key role in the import and proper folding of proteins localized in mitochondrial matrix (Syken et al., 1999; Schneider et al., 1994). In H1299 cells, mtHsp70/mortalin and TID1 co-immunoprecipitated with FL-MDM2 but not with the MDM2 1-291 mutant that displayed impaired mitochondrial localization (Figure 2F). Importantly, pharmacological inhibition (by treatment with MKT-077) or shRNA-mediated depletion of mtHsp70/mortalin decreased the basal levels of mtMDM2 (Figures 2G and 2H). Thus, our data indicate that MDM2 is actively imported into the mitochondrial matrix by specific mitochondrial transporters.
Figure 2. MDM2 Is Actively Imported in Mitochondria through Mitochondrial Transporters.
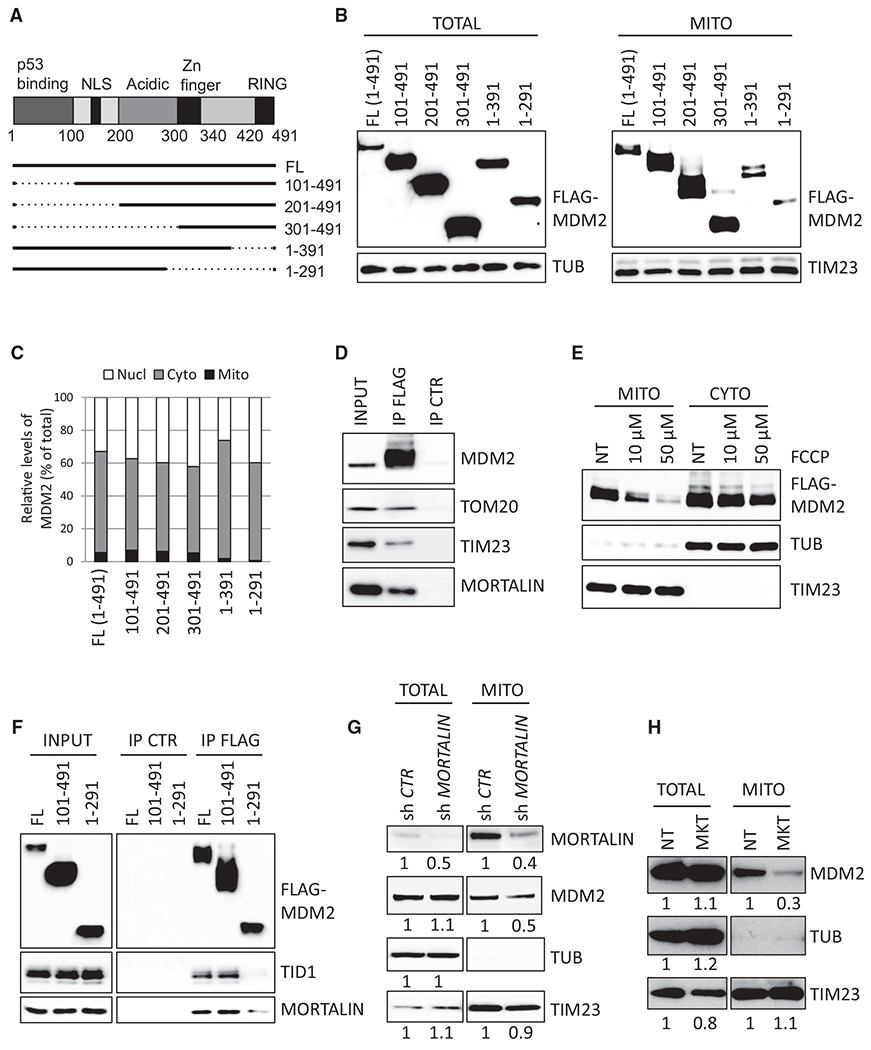
(A) Schematic representation of MDM2 protein structure and of the selected deletion mutants. NLS, nuclear localization signal.
(B) Subcellular localization of FL-MDM2 and of the indicated deletion mutants in H1299 cells, determined by IB using anti-FLAG antibody in whole-cell lysates (TOTAL) and in extracts prepared from purified mitochondria (MITO). Equal loading was verified with TUB and TIM23 protein levels.
(C) Quantitative IB analysis of the subcellular localization of the indicated MDM2 proteins in H1299 cells. Histobars represent the relative levels of nuclear, cytosolic, and mtMDM2.
(D) Co-immunoprecipitation assays showing association between FL-MDM2 and endogenous TOM20, TIM23, and mtHsp70/mortalin proteins in H1299 cells. IP, immunoprecipitation; CTR, control.
(E) Subcellular localization of FL-MDM2 in H1299 cells cultured in the presence of the mitochondrial uncoupler FCCP for 12 hr. NT, untreated. MDM2, TUB, and TIM23 protein levels were determined by IB in fractions enriched in cytosolic or MITO proteins.
(F) Co-immunoprecipitation assays performed in H1299 cells expressing FL-MDM2 or the indicated MDM2 deletion mutants.
(G and H) Quantitative immunoblotting (qIB) analysis of endogenous MDM2 subcellular localization (G) in H1299 cells expressing control or mtHsp70/mortalin shRNAs or (H) in H1299 cells cultured in the presence of the mtHsp70/mortalin pharmacological inhibitor MKT-077 (MKT).
See also Figure S2.
MDM2 Regulates Mitochondrial Network Dynamics and Mitochondrial Ultrastructure
Next, we assessed by confocal microscopy whether modulating mtMDM2 levels influenced the organization of the mitochondrial network. Immunostaining of the ATP5A subunit of the ATP synthase complex or staining with the mitochondrial dye MitoTracker Red indicated that MDM2-depleted cells exhibited a more fragmented mitochondrial network than control cells (Figures 3A, 3B, S3A, and S3B). MDM2 depletion also resulted in enhanced phosphorylation of dynamin-related protein 1 (DRP1) on serine 637, an event previously associated with increased mitochondrial fission (Wang et al., 2012), but had no significant effect on the levels of Mitofusin 2 (MFN2) and Optic Atrophy 1 (OPA1) proteins, which promote fusion (Figure 3C). Strikingly, H1299 cells expressing FL-MDM2 displayed an asymmetrical perinuclear clustering of mitochondria (Figures 3D and 3E). This spatial redistribution of mitochondria was observed only in cells exhibiting cytoplasmic staining of FL-MDM2 but not in cells exhibiting exclusively nuclear MDM2 localization or in cells expressing MDM2 1-291 (Figures 3D, 3E, and S3C), suggesting that the mitochondrial pool of MDM2 was directly implicated in this clustering of mitochondria. To confirm this notion, we generated a chimeric MDM2 protein fused to the N-terminal mitochondrial targeting sequence (MTS) of the E1 subunit of the pyruvate dehydrogenase complex. Biochemical fractionation and immunofluorescence (IF) assays indicated that MTS-MDM2 localized predominantly to mitochondria and that its expression further increased the percentage of cells exhibiting perinuclear clustering of mitochondria (Figures 3D–3E and S3D). A similar phenotype was observed upon expression of MTS-MDM2 in U2OS and MCF7 cells (Figures S3E and S3F), and videomicroscopy indicated that this reorganization of the mitochondrial network occurred as soon as 6 hr after transfecting H1299 cells with a vector encoding MTS-MDM2 (mv1).
Figure 3. MDM2 Regulates Mitochondrial Network Dynamics and Mitochondrial Ultrastructure.
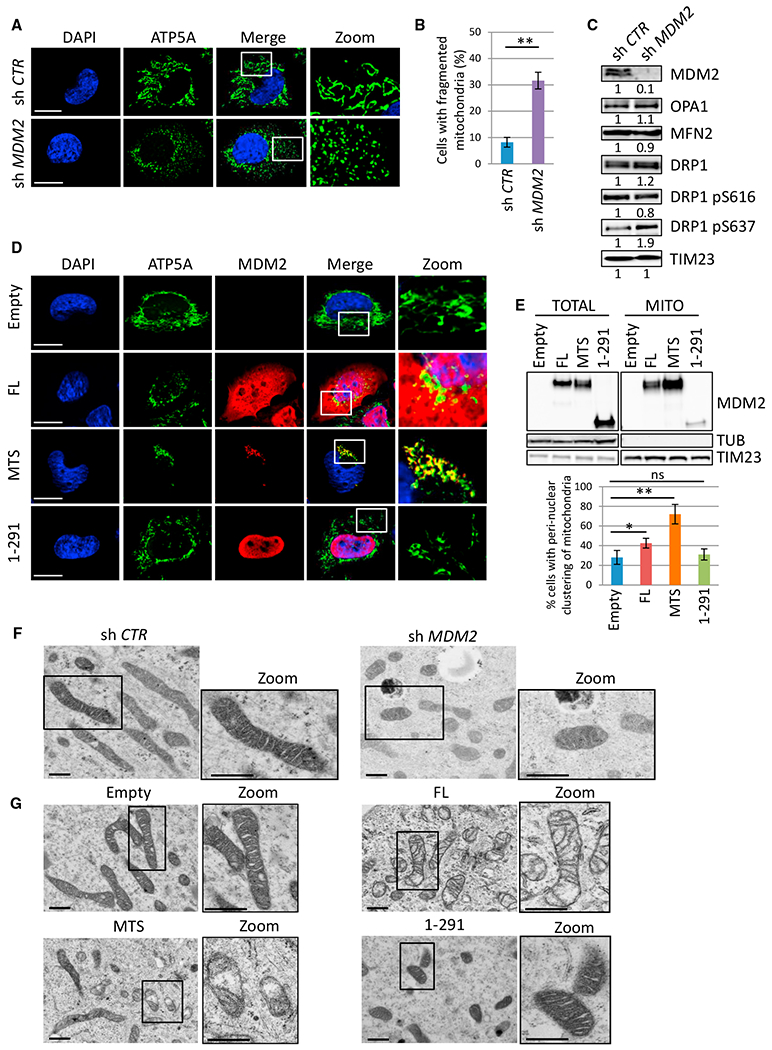
(A) Microphotographs of H1299 cells transduced with lentiviruses encoding control (sh Ctr) or MDM2 (sh MDM2) shRNAs. Cells were stained using an antibody recognizing the ATP5A subunit of the ATP synthase complex (green) and nuclei with DAPI (blue) and then were analyzed by confocal microscopy. Insets show microphotographs at higher magnification (Zoom). Scale bars, 10 μM.
(B) Mitochondrial fragmentation in MDM2-depleted H1299 cells. Histobars represent the frequency of cells exhibiting fragmented mitochondria (mean ± SEM; n = 120 cells from four independent experiments).
(C) IB analysis of total and phosphorylated DRP1 (on Serine 616 or 637), OPA1, MFN2, TIM23 (loading control), and MDM2 protein levels in the same cells as in(A) and (B).
(D) Confocal microscopy analysis of the mitochondrial network in H1299 cells expressing ectopic FL-MDM2 (FL), MTS-MDM2 (MTS), MDM2 1-291 (1-291), or in control cells transfected with the corresponding empty vector (Empty). ATP5A (green), MDM2 (red) and nuclei (blue). Insets show microphotographs at higher magnification (Zoom). Scale bars, 10 μM.
(E) IB analysis (upper panels) of the subcellular localization of the indicated MDM2 proteins in H1299 cells. Histobars (lower panels) represent the percentage of cells exhibiting perinuclear clustering of mitochondria upon expression of the indicated MDM2 proteins in H1299 cells (mean ± SEM; n = 150 cells from five independent experiments).
(F and G) TEM analysis of mitochondrial ultrastructure in (F) H1299 cells expressing control (sh Ctr) or MDM2 (sh MDM2) shRNAs and in (G) H1299 cells expressing FL-MDM2 (FL), MTS-MDM2 (MTS), or MDM2 1-291 (1-291) or in control cells (Empty). Insets show representative microphotographs at higher magnification (Zoom). Scale bars, 500 nM.
*p ≤ 0.05 and **p ≤ 0.01, indicating statistical significance of the observed differences. ns, not significant. See also Figure S3.
To investigate whether alterations in mitochondrial dynamics were also accompanied by changes in ultrastructure, we performed conventional TEM analysis in H1299 cells expressing control or MDM2 shRNAs and in cells expressing FL-MDM2, MTS-MDM2, or the MDM2 1-291 mutant. MDM2-depleted H1299 cells displayed more fragmented mitochondria than control cells but no significant abnormalities of mitochondrial ultrastructure (Figure 3F). In contrast, H1299 cells expressing ectopic FL-MDM2 exhibited loss of mitochondrial matrix electron density, and cristae appeared narrower in these cells, misoriented, and/or reduced in number. These morphological changes were significantly amplified and more frequent upon expression of MTS-MDM2, whereas cells expressing MDM2 1-291 contained mitochondria with an electron-dense matrix and well-organized intact cristae that were indistinguishable from those of control cells (Figure 3G). Importantly, changes of mitochondrial ultrastructure triggered by higher mtMDM2 levels were not associated with increased apoptosis, as shown by the lack of mitochondrial swelling and the absence of detectable cytochrome c release or caspase-3 cleavage (Figures 3G and S3G–S3I).
MtMDM2 Controls Electron Transport Chain Complex I Activity and Respiration
The mitochondrial localization of MDM2 prompted us to investigate its role in respiration. First, we analyzed the consequences of MDM2 deficiency on oxygen consumption and respiratory chain enzymatic activities. State III respiration linked to the electron transport chain (ETC) complex I (CI), measured in the presence of glutamine, malate, and pyruvate (EIII GMP) as substrates, increased significantly upon MDM2 depletion in H1299 cells, as well as after Cre-mediated inactivation of murine Mdm2 in Mdm2flox/flox; p53KO primary mouse embryonic fibroblasts (MEFs) (Figures 4A, 4B, and S4A). In contrast, oxygen consumption driven by complex II (CII), measured in the presence of the CI inhibitor rotenone and succinate (EIII SR) as a substrate, was not affected by MDM2 deficiency (Figure S4B). Of note, increased oxygen consumption in MDM2-depleted H1299 cells was abolished upon incubation with the CI inhibitor metformin, further supporting the role of mtMDM2 in CI-driven respiration (Figure S4C). In line with these results, CI enzymatic activity significantly increased upon shRNA-mediated depletion of MDM2, whereas CII and IV activities remained unchanged (Figure 4C). Next, we measured mitochondrial respiration in cells expressing MTS-MDM2 before these cells exhibited morphological changes in their mitochondrial ultrastructure. CI-driven oxygen consumption was reduced in H1299 cells expressing MTS-MDM2, whereas CII-driven respiration was only marginally affected (Figures 4D and S4D). Consistently, CI activity decreased, whereas that of CII and IV was unaffected in these cells (Figure 4E). Moreover, expression of the MDM2 1-291 that was marginally detected in mitochondria failed to impact on oxygen consumption (Figure 4D). Finally, we evaluated whether the E3 ligase function of MDM2 was implicated in the regulation of mitochondrial respiration. An MDM2 mutant harboring the C464A mutation that abolishes its E3 ligase activity (MDM2-C464A) was efficiently recruited to mitochondria and inhibited CI-driven respiration to an extent similar to that of FL-MDM2 following transient transfection in H1299 cells (Figures S4E and S4F). Inhibition of mitochondrial respiration was also observed in H1299 cells transduced with a lentivirus encoding MTS-MDM2 harboring the C464A mutation (MTS-MDM2-C464A) but not in cells expressing an MDM2 E3 ligase-deficient isoform that predominantly localized to chromatin due to the deletion of its central acidic domain (ΔAD-MDM2-C464A) (Riscal et al., 2016) (Figure S4G).
Figure 4. mtMDM2 Controls ETC Complex I Activity and Respiration.
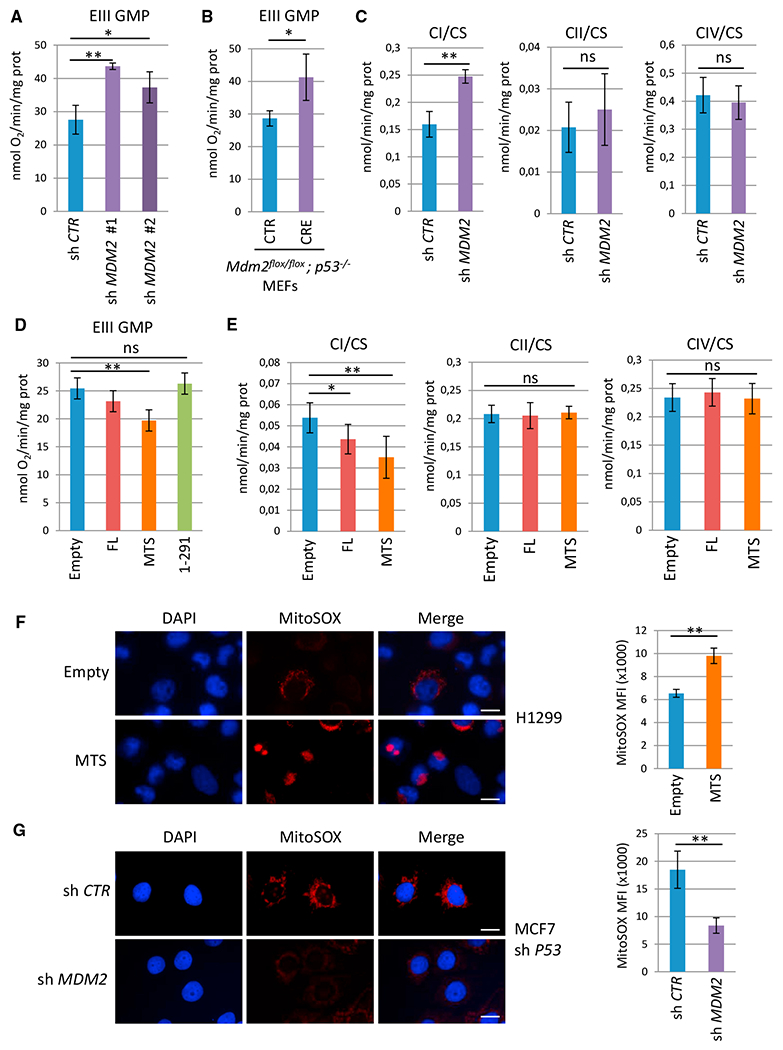
(A) Oxygen consumption in H1299 cells transduced with lentiviruses encoding control or two independent MDM2 shRNAs. ETC CI-driven respiration, in the presence of glutamine, malate, and pyruvate (EIII GMP) as substrates, was measured by using a high-resolution Oxygraph respirometer (mean ± SEM; n = 3).
(B) ETC CI-driven respiration (EIII GMP)in Mdm2flox/flox; p53−/− primary MEFs transduced with control (CTR) or CRE-expressing retroviruses (CRE) (mean ± SEM; n = four independent populations).
(C) Enzymatic activities of the ETC complexes prepared from H1299 cells expressing control or MDM2 shRNAs. ETC CI, CII, and CIV activities were normalized to that of citrate synthase (CS) (mean ± SEM; n = 3).
(D) ETC CI-driven respiration (EIII GMP) in H1299 cells expressing FL-MDM2 (FL), MTS-MDM2 (MTS), or MDM2 1-291 (1-291) and in control cells transfected with the corresponding empty vector (Empty) (mean ± SEM; n = 3).
(E) ETC CI, CII, and CIV activities in H1299 cells expressing FL-MDM2 or MTS-MDM2. Complex activities were normalized to that of CS (mean ± SEM; n = 3).
(F) MitoSOX staining of control (Empty) and H1299 cells expressing ectopic MTS-MDM2.
(G) MitoSOX staining (red) of MCF-7 shp53 cells expressing CTR or MDM2 shRNAs. Nuclei are indicated in blue.
Histobars in (F) and (G) represent the mean fluorescence intensity (MFI) per cell corresponding to the MitoSOX signal (mean ± SEM; n = 60 cells from three independent experiments). Scale bars, 10 μM. *p ≤ 0.05 and **p ≤ 0.01, indicating statistical significance of the observed differences. ns, not significant. See also Figure S4.
Since abnormal ETC CI activity is often associated with oxidative stress, we then measured the effect of mtMDM2 on reactive oxygen species (ROS) levels by IF, using the MitoSox probe that specifically detects mitochondrial superoxide ions. H1299 cells expressing MTS-MDM2 exhibited increased mitochondrial ROS levels when compared to control cells (Figure 4F). Conversely, MCF7-sh p53 cells exhibited decreased ROS levels upon acute depletion of endogenous MDM2 (Figure 4G). Therefore, these data indicate that mtMDM2 interferes with ETC CI-driven respiration, leading to increased mitochondrial ROS production.
MtMDM2 Represses MT-ND6 Transcription
Next, we explored the mechanism by which mtMDM2 controls ETC CI activity. The role of chromatin-bound MDM2 in the transcription of metabolic genes encoded by the nuclear genome (Riscal et al., 2016) prompted us to evaluate whether MDM2 also regulates transcription of the mitochondrial genome independently of p53. Supporting this hypothesis, quantitative chromatin immunoprecipitation (qChIP) assays showed that MTS-MDM2, but not MDM2 1-291, associated with the light strand promoter (LSP) that drives the transcription of the light strand of the mitochondrial genome (Figure 5A). No binding of MTS-MDM2 on the heavy strand promoter (HSP) was detected under the same experimental conditions (Figure S5A). Importantly, binding of endogenous MDM2 to the LSP promoter was also detected in parental but not in MDM2-depleted T47D cells (Figure 5B). Interestingly, these qChIP assays indicated that increased mtMDM2 levels correlated with decreased association of mitochondrial transcription factor A (TFAM) with the LSP, despite total TFAM protein levels remaining constant (Figures 5C and S5B). Consistent with the absence of MDM2 on the HSP, TFAM binding to HSP was not affected by MTS-MDM2 (Figure S5C). The impact of mtMDM2 on TFAM binding to the LSP was confirmed by mTRIP, a single-cell confocal imaging technique combining fluorescence in situ hybridization (FISH) performed with an LSP-specific probe with TFAM IF (Figures 5D and S5D) (Chatre and Ricchetti, 2013). Moreover, atomic force microscopy (AFM) showed that purified recombinant MDM2 protein distributed preferentially to the control region of mtDNA containing the LSP but failed to bind to an irrelevant DNA probe in vitro (Figures 5E and S5E). To further assess the selective inhibitory activity of MDM2 at the LSP, we evaluated the ability of recombinant MDM2 to inhibit mitochondrial transcription in vitro using a dual-promoter template containing both LSP and HSP1 (Uchida et al., 2017). At TFAM concentrations in which both promoters were maximally activated, MDM2 reduced LSP transcription in a dose-dependent manner, whereas it had a much lesser impact on HSP-driven transcription (Figures 5F and S5F).
Figure 5. mtMDM2 Represses MT-ND6 Transcription.
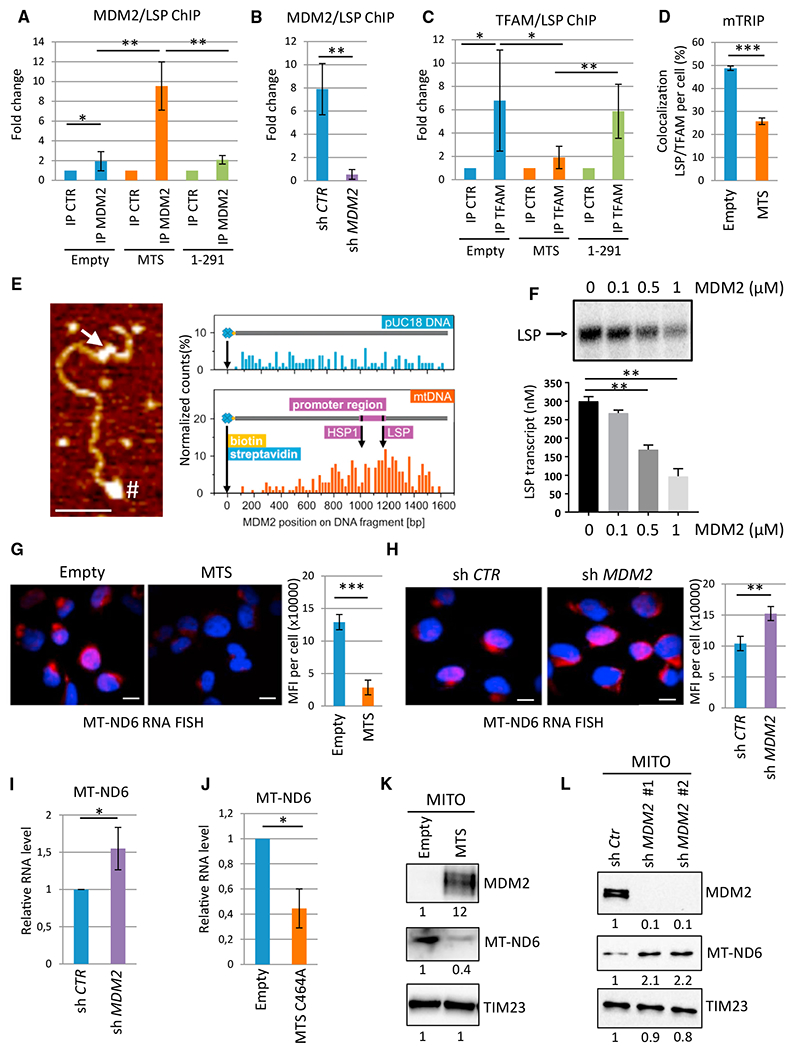
(A) Quantitative chromatin immunoprecipitation (qChIP) experiments showing the relative recruitment of mtMDM2 to the light strand promoter (LSP) in H1299 cells expressing MTS-MDM2 (MTS) or MDM2 1-291 (1-291), or in control cells transfected with the corresponding empty vector (Empty). Results were represented as the relative ratio between the mean value of immunoprecipitated chromatin (calculated as a percentage of the input) with an MDM2 polyclonal antibody (N20) and the one obtained with a control irrelevant antibody (mean ± SEM; n = 3).
(B) qChIP analysis of endogenous MDM2 recruitment to the LSP in T47D transduced with lentiviruses expressing control of MDM2 shRNAs (mean ± SEM; n = 4).
(C) qChIP analysis of TFAM recruitment to the LSP in the same cells as in (A) (mean ± SEM; n = 4).
(D) Confocal microscopy analysis of TFAM (green) recruitment to the LSP (red) following the mTRIP procedure. Histobars represent the percentage of LSP/TFAM co-localization per cell (mean ± SEM; n = 45 cells from three independent experiments).
(E) In vitro binding assay using purified recombinant MDM2 and a 1.6-kb biotinylated DNA probe of the mitochondrial genome encompassing the LSP and the HSP1. Left panel: AFM image showing MDM2 binding to the regulatory region of mtDNA (arrow). # indicates the biotinylated 5′ end of the probe used for orientation. Scale bar, 100 nm. Right panel: quantitative distribution of MDM2 binding along the mtDNA probe ora pUC18-derived DNA probe used as a control for non-specific binding (n = 200 DNA fibers).
(F) Upper panel: Autoradiography of a representative in vitro mitochondrial transcription assay using a dual-promoter template performed in the presence of increasing concentrations of recombinant MDM2. Lower panel: histobars represent the quantification of LSP-driven transcription (mean ± SEM; n = 3).
(G) RNA-FISH/mTRIP analysis of MT-ND6 RNA levels in H1299 cells expressing MTS-MDM2 or in CTR cells transfected with the empty vector (Empty). MT-ND6 probe is indicated in red; Hoechst is indicated in blue. Scale bars, 10 μM. Histobars represent the mean fluorescent intensity (MFI) per cell corresponding to MT-ND6 RNA (mean ± SEM; n = 45 cells from three independent experiments).
(H) RNA-FISH/mTRIP analysis of MT-ND6 RNA levels in H1299 cells transduced with lentiviruses expressing control of MDM2 shRNAs. Scale bars, 10 μM (mean ± SEM; n = 45 cells from three independent experiments).
(I) qRT-PCR analysis of MT-ND6 RNA levels in H1299 cells expressing control or MDM2 shRNAs (mean ± SEM; n = 3).
(J) qRT-PCR analysis of the relative MT-ND6 RNA levels in MDM2-depleted H1299 cells upon transduction with a lentivirus encoding MTS-MDM2-C464A or a control empty virus (mean ± SEM; n = 3).
(K and L) qIB of MDM2, MT-ND6, and TIM23 (loading control) protein levels in purified mitochondria (MITO) prepared (K) from H1299 cells expressing MTS-MDM2 or transfected with the corresponding empty vector and (L) from H1299 cells expressing control (sh Ctr) or two different MDM2 shRNAs.
*p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001, indicating statistical significance of the observed differences. See also Figure S5.
Transcription from the LSP and HSP generates long polycistronic transcripts encoding rRNAs, tRNAs, and structural subunits of respiratory complexes. The LSP-driven transcript encodes only one structural component of the ETC, NADH-de-hydrogenase 6 (MT-ND6), an important subunit of the ETC CI (Montoya et al., 1982). RNA-FISH/mTRIP analyses showed that MT-ND6 RNA levels were lower in H1299 cells expressing MTS-MDM2 than in control cells (Figure 5G). Conversely, shRNA-mediated depletion of endogenous MDM2 resulted in increased MT-ND6 RNA levels (Figure 5H). qRT-PCR analyses showed that MDM2 depletion increased the RNA level of MT-ND6 in H1299 cells but not that of MT-ND2, MT-ND4, and MT-ND5 (Figures 5I and S5G). Moreover, expression of MTS-MDM2-C464A in MDM2-depleted cells still repressed MT-ND6 expression, confirming that the E3 ligase activity of MDM2 was dispensable for the transcriptional repression of MT-ND6 (Figure 5J). Consistent with that repressive function, increased MT-ND6 protein levels were detected both in MDM2-depleted H1299 cells and in Mdm2KO; p53KO MEFs, whereas expression of MTS-MDM2 in H1299 cells decreased MT-ND6 protein levels (Figures 5K, 5L, and S5H). Importantly, the specificity of MT-ND6 immunoblots was confirmed in cell lines harboring a frameshift mutation in MT-ND6 abolishing its expression (Figure S5I) (Perales-Clemente et al., 2010). These data demonstrate that mtMDM2 directly represses the expression of MT-ND6, a key subunit of ETC CI.
Oxidative Stress and Hypoxia Increase mtMDM2 Activities
While screening for MDM2 subcellular localization in H1299 cells exposed to different stress conditions, we found that oxidative stress led to the rapid accumulation of MDM2 in mitochondria. The 4-fold increase of endogenous mtMDM2 levels induced by menadione was abrogated by the addition of the ROS scavenger N-acetyl-cysteine (NAC). MtMDM2 levels also increased upon treatment with rotenone or the pyruvate kinase M2 inhibitor shikonin, likely reflecting the ability of these compounds to induce oxidative stress. In contrast, UV irradiation, the genotoxic agents SN38 and etoposide, or the endoplasmic reticulum (ER)-stress inducer thapsigargin, had no major effect on the amount of mtMDM2 (Figures S6A–S6C). Interestingly, shifting H1299 cells from a 21% O2 to a 1 % O2 atmosphere, or treatment with the hypoxia-mimetic cobalt chloride (CoCl2), led to a 3-fold increase of mitochondrial, but not of total, MDM2 levels (Figures 6A and S6D). Co-immunoprecipitation experiments showed that hypoxia increased the binding of endogenous MDM2 to mtHsp70/mortalin and TID1, suggesting that it stimulated the active import of MDM2 into mitochondria (Figures 6B and S6E). Mild PK digestion of purified mitochondria prepared from hypoxic H1299 cells was sufficient to degrade the OMM protein TOM20, but not MDM2 or TFAM, supporting the notion that both basal and hypoxia-induced mtMDM2 are located in the mitochondrial matrix (Figure S6F). Moreover, qChIP assays indicated that hypoxia increased the recruitment of endogenous MDM2 to the LSP and significantly decreased TFAM association with the LSP (Figure 6C). Accumulation of endogenous MDM2 in mitochondria also occurred in NAC-treated H1299 cells cultured in low-oxygen conditions, indicating that increased recruitment of MDM2 into mitochondria during hypoxia was not an indirect effect of electron leakage through defective ETC that leads to superoxide production (Figure S6G). In line with our findings, increased levels of mtMDM2 during hypoxia or oxidative stress correlated with decreased MT-ND6 protein levels, and hypoxia-induced repression of MT-ND6 was abolished upon MDM2 depletion (Figures 6D and S6C). Importantly, MDM2 deficiency altered neither HIF1α stabilization nor the induction of its target genes Lactate dehydrogenase A (LDHA) and Pyruvate Dehydrogenase Kinase 1 (PDK1) during hypoxia, excluding the possibility that the impact of MDM2 depletion on MT-ND6 regulation was indirectly linked to alterations in HIF-mediated responses (Figures S6H and S6I). Consistent with a previous report, hypoxia induced a rapid perinuclear aggregation of mitochondria in H1299 cells (Al-Mehdi et al., 2012) that was similar to that observed upon expression of MTS-MDM2. Notably, shRNA-mediated depletion of endogenous MDM2 impaired this hypoxia-induced perinuclear clustering of mitochondria (Figure 6E). Thus, we conclude that in cancer cells, both ROS and hypoxia trigger MDM2 recruitment to mitochondria to repress expression of MT-ND6, control respiration, and regulate mitochondrial network dynamics.
Figure 6. mtMDM2 Represses MT-ND6 Expression and Mitochondrial Respiration during Hypoxia.
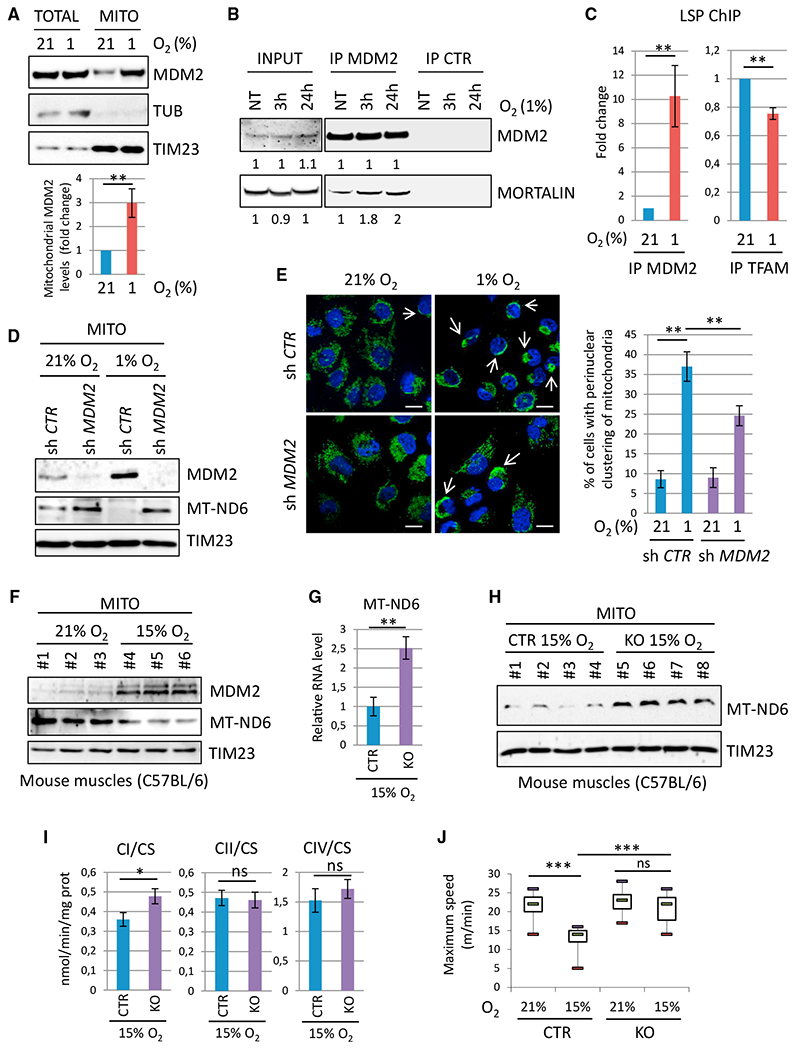
(A) Top: representative IB analysis of endogenous total (TOTAL) or mitochondrial (MITO) MDM2, TUB, and TIM23 (loading controls) proteins in H1299 cells cultured in 21% or 1% O2 for 3 hr. Bottom: histobars represent the relative level of endogenous mtMDM2 determined by qIB (mean ± SEM; n = 5).
(B) Co-immunoprecipitation assays showing increased association between endogenous MDM2 and mtHsp70/mortalin in H1299 cells cultured in 21% or 1% O2 for 3 or 24 hr.
(C) qChIP experiments showing the relative recruitment of endogenous MDM2 (left) and TFAM (right) proteins to the LSP in H1299 cells cultured in 21% or 1% O2 for 3 hr (mean ± SEM; n = 3).
(D) IB analysis of endogenous MDM2, MT-ND6, and TIM23 (loading control) protein levels in purified mitochondria prepared from H1299 cells expressing control (sh Ctr) or MDM2 (sh MDM2) shRNAs cultured in 21% O2 or 1% O2 for 3 hr.
(E) Analysis of the mitochondrial network in H1299 cells expressing control (sh Ctr) or MDM2 (sh MDM2) shRNAs cultured in 21% O2 or 1% O2 for 24 hr. Left: microphotographs acquired by confocal microscopy after staining with anti-ATP5A (green) antibody and DAPI (blue). Arrows indicate perinuclear clustering of mitochondria. Scale bars, 10 μM. Right: histobars represent the percentage of cells exhibiting perinuclear clustering of mitochondria (mean ± SEM; n = 3).
(F) MDM2, MT-ND6, and TIM23 (loading control) protein levels in purified mitochondria isolated from hindlimb muscles of WT C57BL/6 males housed in normoxic (21% O2) or mild hypoxic (15% O2) conditions for 3 hr.
(G) qRT-PCR analysis of MT-ND6 RNA levels in hindlimb muscles of Mdm2CTR(ACTA); p53KO (CTR) and Mdm2KO(ACTA); p53KO (KO) animals housed in 15% O2 for 3 hr (mean ± SEM; n = 6 mice per group).
(H) IB analysis of MT-ND6 and TIM23 (loading control) protein levels in purified mitochondria isolated from the hindlimbs of Mdm2 CTR and KO animals housed in 21% or 15% O2 for 3 hr.
(I) ETC CI, CII, and CIV activities in the hindlimb muscles of Mdm2 CTR and KO animals housed in 15% O2 for 3hr. Enzymatic activities were normalized to that of CS (mean ± SEM; n = 6 mice per group).
(J) Physical endurance of Mdm2 CTR and KO males was determined by forced treadmill running in a 21% or 15% O2 atmosphere. Histobars represent maximal aerobic velocity, expressed in meters per minute (mean ± SEM; n = 10 mice per group).
*p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001, indicating statistical significance of the observed differences. ns, not significant. See also Figure S6.
Mdm2 Inactivation in Striated Muscles Results in Enhanced CI Activity and Increased Muscular Endurance in Mild Hypoxic Conditions
To investigate the relevance of MDM2-mediated control of the ETC CI activity in vivo, we first analyzed the impact of hypoxia in murine skeletal muscles. As soon as 3 hr after exposing WT C57BL/6 mice to an atmosphere composed of 15% O2, mtMDM2 levels in skeletal muscles increased, whereas those of MT-ND6 decreased (Figure 6F). Next, we evaluated the consequences of Mdm2 inactivation in striated muscle cells by crossing Mdm2 conditional knockout (Mdm2 flox) animals (Grier et al., 2002) with Acta1-Cre transgenic (Acta) mice that express the Cre recombinase under the control of the skeletal α-actin promoter (Miniou et al., 1999). To confirm that the phenotypes resulting from Mdm2 inactivation in striated muscles were p53 independent, these animals were also crossed with p53 knockout (KO) mice. We then monitored Cre-driven deletion of the Mdm2flox allele in different tissues harvested from Mdm2flox/flox; ActaCreTg; p53−/− and Mdm2+/flox or +/+; ActaCreTg; p53−/− mice (hereinafter referred to as Mdm2KO(ACTA); p53KO and Mdm2CTR(ACTA); p53KO mice, respectively). In this mouse model, Mdm2 inactivation was restricted to striated skeletal muscles, as evidenced by the strong reduction of Mdm2 mRNA levels in the gastrocnemius, tibialis, and soleus but not in the heart, lungs, liver, and brain (Figure S6J). Mdm2 inactivation had no impact on the mRNA levels of the muscular differentiation markers Myf6, Mef2c, MyoD, and Myogenin or on the overall histological organization of muscle fibers (Figures S6K and S6L). However, skeletal muscles of 12- to 16-week-old Mdm2KO(ACTA); p53KO mice exposed to 15% O2 for 3 hr displayed higher MT-ND6 RNA and protein levels than their age-matched Mdm2CTR(ACTA); p53KO control littermates in the same hypoxic conditions (Figures 6G and 6H). Of note, HIF1α protein and LDHA mRNA levels were identical in the skeletal muscles of control and Mdm2KO(ACTA); p53KO mice exposed to mild hypoxia, confirming that MDM2 deficiency did not impair HIF-mediated transcription (Figures S6M–S6O). Notably, ETC CI activity was significantly higher in the hindlimb muscles of Mdm2KO(ACTA); p53KO mice than in control littermates in these mild hypoxic conditions, whereas ETC CII and IV activities were unaffected (Figure 6I). Next, we evaluated whether MDM2-mediated regulation of CI activity in hypoxic muscles influenced muscular endurance. To answer this question, we measured the locomotor activity of 12- to 16-week-old Mdm2KO(ACTA); p53KO males and control littermates during an acute exercise workload of forced treadmill running in a normoxic (21% O2) or a hypoxic (15% O2) environment. In 21% O2, the physical capacity of Mdm2KO(ACTA); p53KO males was indistinguishable from that of control Mdm2CTR(ACTA); p53KO animals. Strikingly, while these control mice exhibited an expected alteration of their running capacity when shifted to a 15% O2 atmosphere, the physical endurance of Mdm2KO(ACTA); p53KO mice was identical in normoxic and hypoxic conditions (Figure 6J). Collectively, these data indicate that mtMDM2-mediated control of CI activity is critical to regulate muscular endurance in vivo.
MtMDM2 Regulates Migration and Invasion of Cancer Cells
Next, we addressed the relative contribution of the mitochondrial and chromatin-associated pools of MDM2 to the tumorigenic potential of cancer cells. To answer this question, we first assessed the effect of stable expression of MTS-MDM2-C464A or ΔAD-MDM2-C464A on the tumorigenic potential of H1299 cells expressing control or MDM2 shRNAs in xenograft assays in immunocompromised mice. Stable expression of MTS-MDM2-C464A did not enhance the growth rate of control or MDM2-depleted H1299 xenografts, whereas expression of chromatin-bound MDM2 efficiently rescued the tumorigenic potential of MDM2-depleted cells in vivo (Figure S7A). However, expression of MTS-MDM2-C464A clearly increased the clonogenic potential of H1299 cells grown in monolayer cultures (two-dimensional; 2D), as well as their ability to form spheroids in 3-dimensional (3D) anchorage-independent growth conditions (Figures 7A, S7B, and S7C). Of note, MTS-MDM2-C464A H1299 cells in clones that grew in 2D conditions exhibited a more dispersed distribution pattern than that observed in control clones (Figure S7D), suggesting that mtMDM2 influenced cancer cell migration and/or invasion. Videomicroscopy coupled with single-cell tracking analyses confirmed that mtMDM2 enhanced the motility of H1299 cells, as illustrated by the longer distance H1299 cells stably expressing MTS-MDM2 traveled in 7 hr and their enhanced ability to maintain directionality during migration (Figures 7B and S7E). Moreover, MDM2-depleted H1299 cells complemented with MTS-MDM2-C464A exhibited enhanced chemotaxis-induced migration in a Transwell assay compared to shRNA-MDM2 cells transduced with an empty lentivirus, as well as increased capacity to invade a semi-solid matrix in 3D-spheroid invasion assays (Figures 7C and 7D). Interestingly, the enhanced invasive properties of H1299 cells expressing MTS-MDM2-C464A correlated with the increased expression of epithelial-to-mesenchymal transition (EMT) markers, as shown by increased ZEB1, ZEB2, and EPHA2 mRNA levels (Figure 7E). Taken together, these findings indicate that mtMDM2 enhances the migratory and invasive properties of cancer cells.
Figure 7. mtMDM2 Increases Migration and Clonogenic Potential of Cancer Cells.
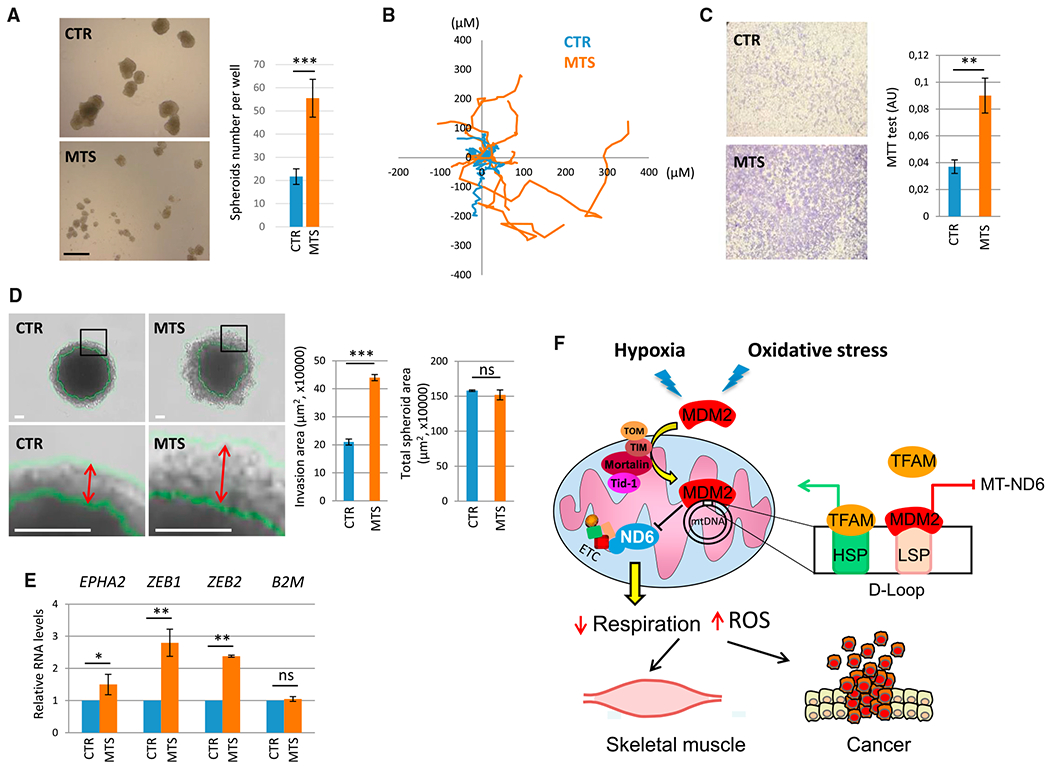
(A) Clonogenic potential in anchorage-independent growth conditions of MDM2-depleted H1299 cells transduced with a lentivirus encoding a shRNA-MDM2-resistant version of MTS-MDM2-C464A (MTS) or with a control empty lentivirus (CTR). Left panel: clones were microphotographed 7 days after seeding a single-cell suspension. Scale bar, 500 μM. Right panel: histobars represent the number of clones per well after 7 days of culture (mean ± SEM; n = 3).
(B) Single-cell tracking analysis of H1299 cells transduced with a lentivirus encoding MTS-MDM2-C464A (MTS) or with an empty lentivirus. Distance (in microns) to origin and the migratory path of ten representative cells for each population are indicated.
(C and D) mtMDM2 increases invasion in anchorage-independent growth conditions.
(C) Left: microphotographs of Transwell assays performed with MDM2-depleted H1299 cells transduced with a lentivirus encoding MTS-MDM2-C464A (MTS) or with an empty lentivirus. Right: histobars represent the number of living cells estimated by a colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay after 16 hr of culture (mean ± SEM; n = 3).
(D) 3D tumor invasion assay. 20,000 MDM2-depleted H1299 cells transduced with a lentivirus encoding MTS-MDM2-C464A (MTS) or with an empty lentivirus (CTR) were seeded into ultra-low attachment plates. 24 hr later, the resulting spheroids were transferred in Matrigel-coated plates and microphotographed after 5 days in culture. Scale bars, 100 μM. Histobars represent the invasion area (histobars at left) or the total surface area (histobars at right) of the spheroids (mean ± SEM; n = 70 from three independent experiments). Double arrows indicate the width of the invasion ring.
(E) qRT-PCR analysis of EPHA2, ZEB1, and ZEB2 mRNA levels in MDM2-depleted H1299 cells transduced with a lentivirus encoding MTS-MDM2-C464A(MTS), or with a control empty lentivirus, after 5 days of culture (mean ± SEM; n = 3).
(F) Schematic representation of mtMDM2 activities in normal muscle and cancer cells. Oxidative stress and hypoxia induce MDM2 localization to mitochondria to repress LSP-driven MT-ND6 transcription, leading to decreased respiration.
*p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 indicate statistical significance of the observed differences. ns = not significant. See also Figure S7.
DISCUSSION
The data presented here point to a novel and unexpected role for MDM2 in mitochondria independent of p53. The mitochondrial localization of MDM2 raises important questions regarding the respective roles of the different components of the p53 pathway in mitochondria and the mechanisms by which they are imported in this organelle. Since neither p53 nor MDM2 contains an MTS, it is likely that their active import in mitochondria through the same mitochondrial TOM/TIM transporters involves binding partners that contain classical MTS such as the TID1 protein (Ahn et al., 2010). It is noteworthy that p53 was previously detected both in the OMM and in the mitochondrial matrix and found to control cell death through its direct interaction with members of the BCL2 family or Cyclophilin D (Marchenko et al., 2000; Mihara et al., 2003; Vaseva et al., 2012). MDM4, another member of the p53 pathway and direct partner of MDM2, has also been detected in mitochondria where it sustains the pro-apoptotic activities of mitochondrial p53 (mtp53) (Mancini et al., 2009). Interestingly, mtp53 was also found to promote the assembly of the F0F1-ATPase and to bind TFAM to increase the expression of the ETC CIV subunit MT-CO1 (Bergeaud et al., 2014; Saleem and Hood, 2013), suggesting that the role of some components of the p53 pathway in mitochondria extends beyond regulation of cell death. Consistent with that notion, increasing mtMDM2 levels did not trigger apoptosis but, instead, led to MT-ND6 repression and decreased CI activity. Collectively, these data suggest that mtMDM2 and mtp53 mediate antagonistic, yet independent, activities to fine-tune mitochondrial respiration. Mechanisms coordinating MDM2 and p53 mitochondrial activities are likely to play important functions in several tissues where mitochondria play a central role in physiological responses to environmental changes, including skeletal muscles. Consistently, p53-TFAM interaction increases upon acute physical exercise and p53 KO animals display decreased muscular endurance due to impaired oxidative metabolism (Park et al., 2009; Saleem et al., 2009; Saleem and Hood, 2013). Strikingly, we found that, in the absence of p53, animals lacking MDM2 in their skeletal muscles exhibited increased exercise endurance that associated with higher MT-ND6 levels and enhanced ETC CI activity. This phenotype was only detected under mild hypoxic conditions, a situation that leads to the rapid accumulation of MDM2 in mitochondria. These results indicate that several components of the p53 pathway, including MDM2, have important physiological functions in muscles. Future studies will be needed to determine whether mitochondrial functions of MDM2 are also at play in other tissues.
Our data also revealed an important role of MDM2 in the regulation of mitochondrial network dynamics. Perinuclear clustering of mitochondria similar to that triggered by mtMDM2 has been described in hypoxic cells (Al-Mehdi et al., 2012), and consistently, we found that hypoxia triggers active MDM2 import into mitochondria. The exact molecular consequences of this clustering remain enigmatic, but it was suggested that it protects cells from apoptosis induced by mitochondrial depolarization (Xiao et al., 2017). Thus, it is possible that increasing mtMDM2 levels is part of a defense mechanism to avoid cell death in mild stress conditions. We previously reported that MDM2 is rapidly recruited to chromatin during oxidative stress to control a transcriptional program implicated in redox homeostasis, a function mediated, at least partly, through aa and glutathione metabolism (Riscal et al., 2016). Therefore, it is tempting to speculate that the mitochondrial and chromatin-associated pools of MDM2 contribute to a global metabolic response that is required to cope with an imbalanced redox state.
The potent oncogenic functions of MDM2 prompted us to evaluate the importance of mtMDM2 activities in cancer cells. The exact role of the ETC in cancer development remains controversial. However, our results are consistent with data showing that increased superoxide production by partial inhibition of the ETC CI, or mutations of MT-ND6, confers a pro-metastatic phenotype (Ishikawa et al., 2008; Li et al., 2015; Porporato et al., 2014). Further work will be required to investigate whether the effects of mtMDM2 on migration are also linked to increased ROS production or to other effects that may derive from altered ETC activity, such as a change in the NAD+/NADH ratio. Nevertheless, our data showing that high mtMDM2 levels enhance the migratory and invasive properties of cancer cells suggest that mitochondrial functions of MDM2 contribute to its oncogenic properties. It also provides a potential molecular explanation for the higher metastatic potential and poor clinical outcome of patients with tumors exhibiting high MDM2 expression (Chen et al., 2012).
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact (laurent.lecam@inserm.fr).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and Treatments
H1299, 786.0, MCF7, T47D, MDA-MB 231, MDA-MB 468, U2OS, 293T and primary MEFs (isolated at E13.5 from Mdm2 flox/flox ; p53−/− mice), were maintained in Dulbecco’s modified Eagle’s medium (DMEM) + Glutamax, supplemented with 10% fetal bovine serum and kept at 37°C in a humidified 5% CO2 incubator. Wild-type (CTR) and ND6-KO (delC13887) mouse 3T3 cells were kindly provided by Dr. Enriquez J.A. and Dr. Acin Perez R. (CNIC, Madrid). The indicated drugs were directly added in the culture medium: FCCP (10 or 50 μM), 17-AAG (5 μM), STA-9090 (200 nM), MKT-077 (10 μM), Menadione (100 μM), H2O2 (10 mM), Shikonin (20 μM), Rotenone (100 nM), SN-38 (2.5 μg/mL), Thapsigargin (100 nM), Etoposide (100 μg/mL), NAC (2 mM), Metformin (2 mM), Staurosporine (1 μM) and Cobalt Chloride (100 μM). UV irradiation (50 mJ/cm2) was used to induce DNA damage. Hypoxia was obtained by placing cells in a 1% oxygen tri-gaz incubator (37°C, 5% CO2, balanced N2).
Mouse Models
To specifically deplete Mdm2 in skeletal muscle, Mdm2+/flox animals (Grier et al., 2002) were intercrossed with Acta1-Cre transgenic mice expressing the Cre recombinase under the control of the skeletal α-actin promoter (Miniou et al., 1999). These mice were then crossed with p53 KO mice (Jacks et al., 1994). 12- to 16-week-old males harboring the Mdm2 flox or the WT alleles, the Acta1-Cre transgene and thep53 null allele were used to generate age-matched experimental groups. Mice genotyping was performed by PCR on genomic DNA prepared from tail biopsies using the Red-N extract kit. Sequence of primers used can be provided upon request.
8-week-old Hsd:AthymicNude-Foxn1nu mice (Envigo) were used for in vivo xenografts assay. Mice were housed under standard conditions with a 12:12 hr light-dark cycle and access to food and water ad libitum. All procedures were approved by the Ethic Committee for Animal Welfare.
METHOD DETAILS
Constructs, Cloning, and Mutagenesis
The following constructs were used for shRNA-mediated knockdown experiments: pLKO.1_Puro shMDM2 #1, pLKO.1_Puro shMDM2 #2 and pMKO.1_puro shp53 (a gift from W. Hahn). Mortalin shRNA and the corresponding control were kindly provided by Prof R. Kruger and Dr. J. Fitzgerald. For inactivation of murine Mdm2 in primary MEFs, a retroviral construct expressing a self-excising CRE recombinase was used (Silver and Livingston, 2001). The pLKO.1_puro Non-Mammalian shRNA Control and retrovirus expressing shRNA targeting the firefly luciferase gene were used as negative controls. For experiments based on expression of ectopic MDM2, cDNAs were all cloned in the pCMV-FLAG vector to generate N-terminally FLAG-tagged proteins. Deletion mutants were generated using the Quikchange site-directed mutagenesis kit (Agilent). The mitochondrial-targeted MDM2 mutant (MTS-MDM2) was generated by removing the N-terminal FLAG tag and replacing it with the sequence encoding the mitochondrial targeting sequence (MTS) of the E1 subunit of the Pyruvate Dehydrogenase (PDHE1) complex (aa 1-30). For complementation experiments, lentiviruses encoding mitochondrial-targeted MDM2 (pLENTI6-MTS-MDM2-C464A) or chromatin-bound MDM2 (ΔAD-MDM2-C464A) harboring silent mutations allowing resistance to MDM2 shRNA were used.
Transfection and Transduction
For expression of ectopic MDM2, cells were transiently transfected with Lipofectamine 2000 according to the manufacturer instructions. Lentiviral and retroviral particles were produced in 293T packaging cells as previously described (Hatchi et al., 2011). Transduction was performed by incubating cells with the viral supernatant overnight, in presence of 8 μg/mL Polybrene.
Subcellular Fractionation Assays
Purified mitochondria and subcellular fractionation assays were mainly performed as previously reported (Frezza et al., 2007). Briefly, cells were resuspended in mitochondria isolation buffer (MIB, 200 mM sucrose, 10 mM Tris/MOPS, 1 mM EGTA/Tris and protease inhibitors), and then lysed using a motor-driven homogenizer operating at 1600 rpm. An aliquot of the resulting extract was used as whole cell lysate (TOTAL). The remaining lysate was centrifuged at 600 g for 10 min at 4°C. The pellet, mainly containing intact nuclei (80%–90%), was washed twice in MIB and resuspended in Laemmli buffer (NUCL). The supernatant was centrifuged again at 600 g for 10 min at 4°C to remove any contaminants coming from the nuclear fraction, and then centrifuged again at 7000 g for 10 min at 4°C to pellet mitochondria. The mitochondrial fraction represented the organelles, washed once in MIB and finally resuspended in Laemmli buffer. The supernatant resulting from the high speed centrifugation, containing cytosoluble proteins, was centrifuged again at 7000 g for 10 min at 4°C to avoid contamination from the mitochondrial fraction and finally diluted in Laemmli buffer 2X (CYTO). For experiments performed on mitochondria isolated from skeletal muscles, mitochondria were prepared from mouse hind limb muscles as previously described (Frezza et al., 2007). Tata-box binding protein (TBP), Tubulin (TUB), and TIM23 were used as markers of nuclear, cytosolic, and mitochondrial proteins, respectively.
Protease Protection Assay
Purified mitochondria were resuspended in MIB and incubated with either 0.5 or 1 μg/mL Proteinase K (PK) for 20 min on ice. 1% Triton X-100 was eventually added to permeabilize the mitochondrial membranes and allow PK to reach the mitochondrial matrix. Samples were then subjected to SDS-PAGE and immunoblotting. Anti- TOM20, TIM23 and TFAM antibodies were used to analyze PK-mediated degradation of proteins located in the outer mitochondrial membrane (OMM), the inner mitochondrial membrane (IMM) and the mitochondrial matrix (MM), respectively.
Oxygen Consumption
To measure oxygen consumption, 5 × 106 cells were resuspended in respiration buffer (EGTA 0.5 mM, MgCl23 mM, K Lactobionate 60 mM, Taurine 20 mM, KH2PO410 mM, HEPES 20 mM, Sucrose 110 mM and BSA 1 mg/mL at pH 7.1) and then permeabilized with digitonin (15 μg/106cells). The respiratory rates were recorded at 37°C in 2 mL glass chambers using a high-resolution Oxygraph-2k respirometer (O2k, OROBOROS Instruments, Innsbruck, Austria). Activation of ATP synthesis was induced by the addition of 1.5 mM ADP. Respiration driven by complex I coupled to ATP synthesis (EIII-GMP) was measured by adding glutamine (2 mM), malate (5 mM) and pyruvate (5 mM). The further addition of Succinate (10 mM) and Rotenone (10 μM) allowed the measurement of respiration driven by complex II (EIII-SR).
Mitochondrial Respiratory Complex Activities
Electron transport chain (ETC) complex activities were determined from total cell lysates or crude homogenates of mixed hind-limb muscles as described previously (Medja et al., 2009). The activities of respiratory chain complexes I, II, and IV were expressed relative to that of the matrix enzyme citrate synthase.
Immunoblotting
Protein extracts were subjected to SDS-PAGE and immunoblotted with the following primary antibodies: anti-p53 (Cell Signaling), -TBP (Santa Cruz), -βTubulin (Sigma-Aldrich), -MDM2 (clones 4B2 and 2A10, Millipore), -TOM20 (BD Biosciences), -TIM23 (BD Biosciences), -TFAM (clone D5C8, Cell Signaling, and Abcam), -FLAG (Sigma-Aldrich), -TID1 (Cell Signaling), -Grp75/Mortalin (Cell Signaling), -Opa1 (Abcam), -MFN2 (Cell Signaling), -DRP1 (Cell Signaling), -pDRP1 (Ser616, Cell Signaling), -pDRP1 (Ser637, Cell Signaling), -AKT1 (Cell Signaling) -HSP90 (Cell Signaling), -Cytochrome C (Cell Signaling), -cleaved Caspase 3 (Cell Signaling), -MT-ND6 (clones FL-174 and V-16, Santa Cruz), -HIF1a (Cell Signaling). HRP-conjugated anti-mouse and anti-rabbit IgG (Cell Signaling) were used as secondary antibodies. Detection was performed by using either the Pierce ECL Western Blotting Substrate or the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific).
Co-Immunoprecipitation Assays
107 cells were lysed in RIPA buffer (10 mM Tris pH 8, 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS) containing protease inhibitors and incubated 1 hr on ice. Immunoprecipitation (IP) was performed by incubating 500 μg of the whole-cell lysates (INPUT) with 2 μg of antibody overnight at 4°C. Irrelevant mouse or rabbit IgGs (Santa Cruz) were used as control. The following day, Dynabeads Protein G were added and incubated for 6 hr at 4°C. The immunocomplexes were then washed 3 times with lysis buffer, resuspended in Laemmli, and processed for immunoblotting.
Immunofluorescence and Staining of Mitochondria
Cells cultured on glass coverslips were fixed in PBS 4% PFA and then washed in PBS. Samples were permeabilized in PBS 0.1% Triton X-100 for 15 min, followed by blocking in PBS 0.1% Triton X-100 + 1% BSA, 1 hr at 37°C. Primary antibodies, including anti-MDM2 (clone 4B2, Calbiochem, 1/50), FLAG (Sigma-Aldrich, 1/500), ATP5A (Abcam, 1/500), TFAM (Cell Signaling, 1/500) and cytochrome c (Cell Signaling, 1/300), were diluted in blocking solution and incubated 2 hr in a humidified chamber. After 3 washes in PBS, samples were incubated with the appropriate Alexa 488- and Alexa 555- conjugated secondary antibodies for 1 hr at room temperature. Coverslips were then washed 3 times in PBS and finally treated with VECTASHIELD Mounting Medium containing DAPI. Representative microphotographs were acquired using a ZEISS ApoTome. For detection of mitochondrial superoxide ions, cells cultured on glass coverslips were incubated with DMEM + 5 mM MitoSOX Red Mitochondrial Superoxide Indicator for 10 min at 37°C. For MitoTracker staining, cells were incubated 1 hr with 200 nM MitoTracker Red FM. Cells were then washed 3 times with DMEM, rinsed in PBS twice and finally processed for immunofluorescence.
mTRIP Procedure
mTRIP analysis was performed as previously described (Chatre and Ricchetti, 2013). Briefly, cells plated on glass coverslips were fixed with 2% PFA and permeabilized with 0.5% Triton X-100. Cells were then incubated in 50% formamide/2XSSC for 30 min at RT, and denatured in 70% formamide/2XSSC for 4 min at 75°C. Hybridization was performed with 40 ng of probe for 16 hr at 37°C. MT-ND6 and LSP probes for FISH were labeled by nick translation of PCR products, incorporating Atto488-dUTP or Atto550-dUTP. Coordinates of MT-ND6 probes were reported previously (Chatre and Ricchetti, 2013). Coordinates of LSP probe are 213-445. 40 ng of labeled probes were mixed with 400 ng of sonicated salmon sperm DNA in hybridization buffer (50% formamide, 10% dextran sulfate, in 2XSSC pH 7.0), denatured at 80°C for 10 min and kept at 37°C for 30 min. After washing the slides in SSC, the DNA was stained with 10 mg/mL Hoechst. For mTRIP-coupled immunofluorescence (LSP-TFAM colocalization), after hybridization and SSC wash, staining with TFAM antibody was performed.
Chromatin Immunoprecipitation
ChIP assays were mainly performed as previously described (Riscal et al., 2016), with some modifications. Briefly, 107 cells were cross-linked in 1% formaldehyde/1% paraformaldehyde for 5 min, followed by addition of 125 mM Glycine to stop the reaction. Cells were then washed in PBS, resuspended in lysis buffer (10 mM Tris pH 8,140 mM NaCl, 0.1% SDS, 0.5% Triton X-100, 0.05% NaDoc, 1 mM EDTA, 0.5 mM EGTA and protease inhibitors) and chromatin was sheared by sonication. qChIPs were carried out by incubating cell lysates (Input) with 20 μL of protein G-Dynabeads and 5 μg of antibody (MDM2, clone N20, Santa Cruz; TFAM, Cell Signaling). The same amount of rabbit IgGs (Santa Cruz) was used for control ChIP experiments. After O/N incubation, washing, reverse cross-linking and treatment with both RNase A and Proteinase K, proteins were removed with phenol/chloroform extraction and DNA was recovered using the NucleoSpin Extract II kit. Input and immunoprecipitated DNA were then analyzed by QPCR using the SYBR Green Master mix on a LightCycler 480 SW 1.5 apparatus (Roche). Results are represented as the mean value of at least 3 independent experiments of immunoprecipitated chromatin (calculated as a percentage of the input) with the indicated antibodies after normalization by a control ChIP performed with rabbit IgGs.
RNA Extraction and Quantitative RT-qPCR
Total RNA was isolated using the TRI-reagent and then reverse transcribed with the SuperScript III Reverse Transcriptase. The resulting cDNAs were quantified by real-time QPCR using the SYBR Green Master mix on a LightCycler 480 SW 1.5 apparatus (Roche). The relative mRNA levels were calculated using the ΔΔCt method and normalized to Tata Binding Protein (TBP) mRNA level.
Electron Microscopy
For immunogold staining, cells were fixed in 4% (w/v) paraformaldehyde (PFA) and 0.1% (w/v) glutaraldehyde in PBS for 1 hr, washed 3 times in PBS and then subjected to dehydration with increasing ethanol concentrations. Samples were then embedded in LR White resin and polymerized by exposure to UV light. Ultrathin sections of the samples were transferred on grids, followed by the hybridization procedure. Sections were first treated with a blocking solution (0.5% glycine, 1% BSA in PBS for 10 min) and then incubated 1 hr with anti-FLAG antibody (1/150, in PBS 1% BSA); grids were washed 3 times, followed by incubation with 10 nm gold-conjugated anti-mouse antibody (1/20 in PBS 1% BSA, 1 hr). Sections were then washed 3 times in PBS and 3 times in distilled water, and finally counterstained in 3% uranyl acetate. Samples were analyzed by using a JEOL JEM 1200 EX2 transmission electron microscope. For ultrastructure analysis, cells were fixed 1 hr in PBS 2.5% glutaraldehyde, washed 3 times in PBS and dehydrated. Samples were then embedded in Epon resin and polymerized at 60°C. Ultrathin sections were transferred on grids and finally counterstained in 2% uranyl acetate. Samples were analyzed using a HITACHI H 7100 transmission electron microscope.
Confocal Acquisition, 3D Reconstruction, and Quantification
Confocal acquisitions were performed using a spinning-disk Perkin-Elmer Ultraview RS Nipkow Disk, an inverted laser-scanning confocal microscope Zeiss Axiovert 200 M with an Apochromat 63X/1.4 NA oil objective and a Hamamatsu ORCA II ER camera (Institut Pasteur, Paris). Microphotographs were acquired using non-saturating settings, and the same imaging parameters were used for all samples. Optical z-slices in 200-nm steps covering the whole depth of the cell were collected, at resolution of 1.024/1.024 pixels. Three-dimensional reconstruction of all the z stacks was achieved using the 3D-volume rendering of IMARIS software (Bitplane). Representative 2D images generated from 3D volume rendering were used for fluorescence quantification using the Integrated Density measurement tool of ImageJ 2.0.0 software (post-acquisition analysis).
Super-Resolution Microscopy
For super-resolution microscopy, images were acquired on a spinning disk system (Roper Scientific France) based on an inverted microscope (Ti-E, Nikon) equipped with a sCMOS camera (Prime 95B, Photometrics), a confocal spinning head (X1, Yokogawa), a 100x 1.4 NA Plan Apo objective lens and a super-resolution module (Gataca systems) based on optical photon reassignment with online processing. Multi-dimensional acquisitions were performed in streaming mode using Metamorph 7.7.6 software (Molecular Devices, France).
Spheroid Clonogenic Assay
H1299 transduced with MTS-MDM2-C464A or control lentiviruses (100 cells per well) were seeded in low attachment multi-well plates and cultured in MEBM (B27 1X, 20 ng/mL EGF, 20 ng/mL bFGF, 4 μg/mL heparin). Spheroids were counted 7 days after seeding.
3D Tumor Spheroid Invasion Assay
H1299 transduced with MTS-MDM2-C464A or control lentiviruses (20,000 cells per well) were seeded into ultra-low attachment (ULA) 96-well round bottom plates. 24 hr later, spheroid formation was visually confirmed before starting the 3D invasion assay. Briefly, 100 μL of matrigel (100 μg/mL) were gently dispensed into the U-bottom wells, followed by centrifugation of the plate (at 300 g for 3 min at 4°C) to ensure central localization of spheroids in each well. Plates were then transferred to 37°C to allow matrigel solidification. 1 hr later, complete growth medium was added and spheroids maintained in culture for 5 days. Plates were scanned and analyzed by Celigo Imaging Cell Cytometer (Nexcelom Bioscience).
Invasion Assay in Boyden Chamber
H1299 cells transduced with MTS-MDM2-C464A or control lentiviruses were cultured in FBS-free DMEM for 16 hr and then seeded in a Matrigel-coated Boyden Chamber with 8 μm pore polyester membrane. 500 μL of DMEM 10% FBS were added in the lower part of the chamber as chemoattractant. After 16 hr, the migrating cells were stained with crystal violet and their number estimated by using a MTT colorimetric assay.
In Vivo Xenografts
Bilateral subcutaneous injections of 8 × 106 H1299 cells were carried out on 8-week-old Hsd: AthymicNude-Foxn1nu mice (Envigo). Volumetric measurements of engrafted tumors were performed every 3 days by the same person using a manual caliper (volume = (length × width2)/2). All animals were sacrificed when the first animal reached the ethical endpoint (volume = 1000 mm3).
2D Clonogenic Assay
5000 H1299 cells transduced with MTS-MDM2 or control lentiviruses were seeded in 10 cm plates and maintained in DMEM 10% FBS. 8 days later, cells were fixed in PFA and stained with crystal violet for 2 hr. Microphotographs of representative clones were taken by bright field microscopy.
2D Cell Migration Assay
Matrigel-coated Ibidi HiQ4 chambers were used for video microscopy of H1299 cells transduced with a lentivirus encoding MTS-MDM2-C464A(MTS) or with an empty control (CTR) lentivirus. Images were acquired in a Nikon Biostation IM-Q (20x objective) every 15 min for 7 hr. Videos were analyzed using the Fiji “manual tracking” plug-in. Displacement (Euclidean distance between the first and last time points) and persistence (ratio of displacement over the total distance traveled) were calculated for 100 randomly selected cells.
Histological Analyses
Muscle biopsies were fixed in 4% (vol/vol) neutral-buffered formalin (VWR Chemicals) for 24 hr. Paraffin-embedded tissues were then sectioned (4 μm) and processed for Hematoxylin and eosin (H&E) staining.
Running Tests
Mice were first trained to run on a treadmill (Colombus Instruments) for 2 sessions (around 15 min/session at constant speed of 10 m/min with no slope). 72 hr later, their maximal aerobic velocity (MAV) was determined by gradually increasing speed (1 m/min increment every 60 s), starting from 10 m/min until exhaustion. For tests performed under mild hypoxic conditions (simulating an altitude of 3000 m), running performances were evaluated in a chamber mimicking moderate hypobaric (−0,3 bars) hypoxia (15%O2) after an acclimatization of 3 hr.
Atomic Force Microscopy
The mtDNA construct used for the localization of MDM2 via AFM was prepared by PCR as described (Uchida et al., 2017) and was a fragment of mtDNA that began 510 bp downstream of LSP and 1000 bp downstream of HSP1 (Sequence of primers used can be provided upon request). The concentration of mtDNA and of the control pUC18 DNA used for the experiments was 0.5 nM; the MDM2 protein concentration was 10 nM. The experiment was conducted by end-labeling the biotin-tagged mtDNA or pUC18 constructs (via biotinylated primers) via adding 20 nM streptavidin (Sigma-Aldrich) to the DNA solution for 5 min in 25 mM NaCl, 25 mM HEPES, 10 mM MgCl2 at pH 7.5. Subsequently, MDM2 was added and incubated for 5 min. The solution was deposited on a freshly cleaved mica surface, washed with MilliQ- water after 10 min and gently dried under a stream of nitrogen. For AFM imaging in air we used a Bruker Bioscope Resolve (Bruker) with PeakForce Tapping mode. Olympus OMCL-AC160TS probes (f = 300 kHz, k = 26 N/m) were used for topographical imaging of the DNA-protein complexes. The scan range was varied from 1 × 1 μm to 3 × 3 μm at 512 × 512 pixels. 200 DNA segments bound by MDM2 were exported with Nanoscope analysis (Bruker) and Gwyddion (http://gwyddion.net/) and further traced and analyzed with FiberApp software.
In Vitro Transcription Assays
Reactions were performed essentially as previously described (Uchida et al., 2017). Briefly, reactions were performed in 1 × reaction buffer (10 mM HEPES pH 7.5,100 mM NaCl, 10 mM MgCl2, 1 mMTCEP and 0.1 μg/μL BSA) with 10 μM 32P-end-labeled RNA primer (pAAA), 500 μM NTPs and 100 nM DNA template. Reactions were performed by incubating template DNA in reaction buffer at 32°C for 5 min and then adding in the following order: MDM2 (0 to 1 μM), TFAM (0.1 or 0.5 μM), TFB2M (0.1 μM) and POLRMT (0.1 μM). Between each addition of protein to the reaction there was an incubation time of 2 min. After addition of POLRMT, the reaction was allowed to incubate at 32°C for 30 min. At each time point 5 μL of the reaction mix were quenched into 5 μL of stop buffer (79.2% formamide, 0.025% bromophenol blue, 0.025% xylene cyanol and 50 mM EDTA final). Products were resolved by denaturing 20% (37:3, acrylamide:bis-acrylamide ratio) PAGE. Proteins were diluted immediately prior to use in 10 mM HEPES, pH 7.5, 1 mM TCEP, and 20% glycerol. The volume of protein added to any reaction was always less than or equal to one-tenth of the total volume.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are expressed as mean ± SEM. To determine statistical significance between groups, comparisons were made using Student’s t or non-parametric Mann-Whitney tests. Analyses of multiple groups were performed using a one-way ANOVA test. Data were analyzed with R software. p values less than 0.05 were considered significant.
Quantification of immunoblots was performed using the ImageJ software, after densitometric analysis of the corresponding bands detected upon revelation with a peroxidase-coupled secondary antibody.
For super-resolution microscopy, quantification of colocalization was performed by using the Fiji Coloc 2 plugin (Manders’ coefficient, with thresholds).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal α-FLAG | Sigma-Aldrich | Cat# F4042 |
| Mouse monoclonal α-βTubulin | Sigma-Aldrich | Cat# T4026, RRID: AB_477577 |
| Mouse monoclonal α-TFIID (TBP) | Santa Cruz Biotechnology | Cat# sc-56795, RRID: AB_786004 |
| Mouse monoclonal α-TIM23 | BD Biosciences | Cat# 611223, RRID: AB_398755 |
| Mouse monoclonal α-MDM2 (4B2C1.11) | Millipore | Cat# OP145, RRID: AB_2235250 |
| Mouse monoclonal α-MDM2 (2A10) | Millipore | Cat# OP115, RRID: AB_564806 |
| Rabbit polyclonal α-MDM2 (N-20) | Santa Cruz Biotechnology | Cat# sc-813, RRID: AB_2250633 |
| Mouse monoclonal α-p53 | Cell Signaling Technology | Cat# 2524, RRID: AB_331743 |
| Rabbit monoclonal α-TFAM (D5C8) | Cell Signaling Technology | Cat# 8076S, RRID: AB_10949110 |
| Rabbit polyclonal α-TFAM | Abcam | Cat# ab131607, RRID: AB_11154693 |
| Mouse monoclonal α-TOM20 | BD Biosciences | Cat# 612278, RRID: AB_399595 |
| Mouse monoclonal α-TID1 | Cell Signaling Technology | Cat# 4775, RRID: AB_2094258 |
| Rabbit monoclonal α-Grp75 (Mortalin) | Cell Signaling Technology | Cat# 3593S, RRID: AB_2120328 |
| Rabbit polyclonal α-ATP5A | Abcam | Cat# ab151229 |
| Rabbit polyclonal α-OPA1 | Abcam | Cat# ab42364, RRID: AB_944549 |
| Rabbit monoclonal α-MFN2 | Cell Signaling Technology | Cat# 11925S |
| Rabbit monoclonal α-DRP1 | Cell Signaling Technology | Cat# 8570, RRID: AB_10950498 |
| Rabbit monoclonal α-Phospho-DRP1 (Ser616) | Cell Signaling Technology | Cat# 4494S, RRID: AB_11178659 |
| Rabbit monoclonal α-Phospho-DRP1 (Ser637) | Cell Signaling Technology | Cat# 6319S, RRID: AB_10971640 |
| Rabbit monoclonal α-HSP90 | Cell Signaling Technology | Cat# 4877S, RRID: AB_2233307 |
| Mouse monoclonal α-Cytochrome C | Cell Signaling Technology | Cat# 12963, RRID: AB_2637072 |
| Rabbit monoclonal α-Akt1 | Cell Signaling Technology | Cat# 2938, RRID: AB_915788 |
| Rabbit polyclonal α-cleaved caspase 3 | Cell Signaling Technology | Cat# 9661 |
| Rabbit polyclonal α-ND6 (FL-174) | Santa Cruz Biotechnology | Cat# sc-20667, RRID: AB_2282522 |
| Rabbit polyclonal α-ND6 (V-16) | Santa Cruz Biotechnology | Cat# sc-20510 |
| Rabbit monoclonal α-HIF1α | Cell Signaling Technology | Cat# 14179, RRID: AB_2622225) |
| HRP-linked α-mouse IgG | Cell Signaling Technology | Cat# 7076, RRID: AB_330924 |
| HRP-linked α-rabbit IgG | Cell Signaling Technology | Cat# 7074, RRID: AB_2099233 |
| Alexa Fluor 488 α-mouse IgG | Thermo Fisher Scientific | Cat# A-11029, RRID: AB_2534088 |
| Alexa Fluor 555 α-mouse IgG | Thermo Fisher Scientific | Cat# A-21425, RRID: AB_2535846 |
| Alexa Fluor 488 α-rabbit IgG | Thermo Fisher Scientific | Cat# A-11034, RRID: AB_2576217 |
| Alexa Fluor 555 α-rabbit IgG | Abcam | Cat# ab150078 |
| 10 nm gold-conjugated α-mouse IgG | Sigma-Aldrich | Cat# G7777 |
| Normal mouse IgG | Santa Cruz Biotechnology | Cat# sc-2025 |
| Normal rabbit IgG | Santa Cruz Biotechnology | Cat# sc-2027 |
| Bacterial and Virus Strains | ||
| ElectroMAX DH10B Cells | Thermo Fisher Scientific | Cat# 18290015 |
| TOP10 chemically competent E. Coli | Thermo Fisher Scientific | Cat# C404010 |
| Rosetta (DE3) competent cells | Novagen | Cat# 70954 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM, high glucose, GlutaMAX | Thermo Fisher Scientific | Cat# 10566016 |
| MEBM Basal Medium | Lonza | Cat# CC-3151 |
| B27 supplement w/o Vitamin A | Thermo Fisher Scientific | Cat# 12587010 |
| Heparin | Fisher Scientific | Cat# 10639163 |
| Human FGF-2IS | Miltenyi Biotech | Cat# 130104922 |
| Human EGF | Miltenyi Biotech | Cat# 130099750 |
| MTT formazan | Sigma-Aldrich | Cat# M2003 |
| 6.5 mm Transwell with 8.0 μm Pore | Corning | Cat# 3464 |
| Matrigel | Corning | Cat# 354248 |
| ULA plates | Corning | Cat# 3473 |
| Protease Inhibitor Complete | Roche | Cat# 11836153001 |
| VECTASHIELD mounting medium +DAPI | Vector Laboratories | Cat# H-1200 |
| Proteinase K | Thermo Fisher Scientific | Cat# 25530049 |
| Triton X-100 | Sigma-Aldrich | Cat# T9284 |
| Glutaraldehyde 25% | Electron Microscopy Science | Cat# 16220 |
| Paraformaldehyde 20% | Electron Microscopy Science | Cat# 15713 |
| Epon resin (EMBed-812 kit) | Electron Microscopy Science | Cat# 14120 |
| Uranyl acetate | BDH Chemicals | Cat# 30550 |
| LR White Embedding Resin | Electron Microscopy Science | Cat# 14381 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat# A2058 |
| FCCP | Sigma-Aldrich | Cat# C2920 |
| MKT-077 | Sigma-Aldrich | Cat# M5449 |
| 17-AAG | Sigma-Aldrich | Cat# A8476 |
| Ganetespib (STA-9090) | Selleck Chemicals | Cat# S1159 |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat# 10004 |
| Mitotracker Red FM | Thermo Fisher Scientific | Cat# M22425 |
| MitoSOX | Thermo Fisher Scientific | Cat# M36008 |
| N-Acetyl-L-cysteine (NAC) | Sigma-Aldrich | Cat# A7250 |
| Staurosporine | Sigma-Aldrich | Cat# S5921 |
| Digitonin | Sigma-Aldrich | Cat# D5628 |
| Metformine | Sigma-Aldrich | Cat# M0605000 |
| L-Glutamine | Thermo Fisher Scientific | Cat# 25030024 |
| ADP | Sigma-Aldrich | Cat# A2754 |
| Malate | Sigma-Aldrich | Cat# M1125 |
| Pyruvate | Sigma-Aldrich | Cat# P5280 |
| Succinate | Sigma-Aldrich | Cat# S9637 |
| Rotenone | Sigma-Aldrich | Cat# R8875 |
| Menadione | Sigma-Aldrich | Cat# M5625 |
| H2O2 | Sigma-Aldrich | Cat# H1009 |
| Shikonin | Sigma-Aldrich | Cat# S7576 |
| SN-38 | Sigma-Aldrich | Cat# H0165 |
| Thapsigargin | Sigma-Aldrich | Cat# T9033 |
| Etoposide | Sigma-Aldrich | Cat# E2600000 |
| Cobalt Chloride | Sigma-Aldrich | Cat# 449776 |
| TRI reagent | Sigma-Aldrich | Cat# T9424 |
| Atto488 NT Labeling kit | Jena Bioscience | Cat# PP-305–488 |
| Atto550 NT Labeling kit | Jena Bioscience | Cat# PP-305–550 |
| Pierce ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat# 32106 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat# 34094 |
| Muscovite Mica V1 25x25mm | Electron Microscopy Science | Cat# 56–25 |
| Streptavidin | Sigma-Aldrich | Cat# S4762 |
| Ulp1 | LifeSensors | Cat# 4010 |
| TFAM | Indigo Biosciences | Cat# MV200 |
| TFB2M | Indigo Biosciences | Cat# MV300 |
| POLRMT | Indigo Biosciences | Cat# MV100 |
| Critical Commercial Assays | ||
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 200521 |
| NucleoBond Xtra Maxi Plus | MACHEREY-NAGEL | Cat# 740416.10 |
| SuperScript III Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18080093 |
| SYBR Green Master Mix | Takara | Cat# RR820L |
| REDExtract-N-Amp Tissue PCR Kit | Sigma-Aldrich | Cat# XNAT-100RXN |
| NucleoSpin Extract II kit | MACHEREY-NAGEL | Cat# 740609.250 |
| In-Fusion HD Cloning Plus | Takara | Cat# 638909 |
| Deposited Data | ||
| Original imaging data | This paper; Mendeley Data | https://doi.org/10.17632/86vx3svs8t.1 |
| Experimental Models: Cell Lines | ||
| Human: H1299 | ATCC | Cat# CRL-5803 |
| Human: 786.O | ATCC | Cat# CRL-1932 |
| Human: MCF7 | ATCC | Cat# HTB-22 |
| Human: T47D | ATCC | Cat# HTB-133 |
| Human: MDA-MB 231 | ATCC | Cat# CRM-HTB-26 |
| Human: MDA-MB 468 | ATCC | Cat# HTB-132 |
| Human: U2OS | ATCC | Cat# HTB-96 |
| Human: 293T | ATCC | Cat# CRL-3216 |
| Murine: 3T3 | Dr. Enriquez lab (CNIC, Madrid) | N/A |
| Murine: 3T3 ND6 delC13887 | Dr. Enriquez lab (CNIC, Madrid) | N/A |
| Murine: Mdm2 flox/flox; p53−/− MEFs | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 | Charles River Laboratory | Cat# 027 |
| C57BL/6 Mdm2 +/flox | Grier et al., 2002 | N/A |
| C57BL/6 Acta1-Cre | Miniou et al., 1999 | N/A |
| C57BL/6 p53 KO | Jacks et al., 1994 | N/A |
| C57BL/6 Mdm2 +/+ (Acta); p53 KO | This paper | N/A |
| C57BL/6 Mdm2 flox/flox (Acta); p53 KO | This paper | N/A |
| Hsd :AthymicNude-Foxn1nu | Envigo | Cat# 6904F |
| Oligonucleotides | ||
| See Table S1 | N/A | N/A |
| Recombinant DNA | ||
| pcDNA3.1 (empty) | Riscal et al., 2016 | N/A |
| pCMV-FLAG MDM2 FL (1-491) | Riscal et al., 2016 | N/A |
| pCMV-FLAG MDM2 101-491 | This paper | N/A |
| pCMV-FLAG MDM2 201-491 | This paper | N/A |
| pCMV-FLAG MDM2 301-491 | This paper | N/A |
| pCMV-FLAG MDM2 1-391 | This paper | N/A |
| pCMV-FLAG MDM2 1-291 | This paper | N/A |
| pCMV-FLAG MDM2 Λ291-381 | This paper | N/A |
| pCMV-FLAG MDM2 Λ331-381 | This paper | N/A |
| pCMV-FLAG MDM2 Λ382-429 | This paper | N/A |
| pCMV-FLAG MDM2 Λ429-491 | This paper | N/A |
| pCMV-HSP90 | This paper | |
| pCMV-MDM2 R181A/K182A/R183A (ΔNLS) | This paper | N/A |
| pCMV-MDM2 MTS | This paper | N/A |
| pCMV-MDM2 C464A | This paper | N/A |
| pLENTI6-MDM2 MTS C464A shMdm2 #2 resistant | This paper | N/A |
| pLENTI6-MDM2 Δ222-302 C464A (DAD C464A) shMdm2 #2 resistant | This paper | N/A |
| pLKO.1_Puro control shRNA (sh Ctr) | Sigma-Aldrich | Cat# SHC002 |
| pLKO.1_Puro shMdm2 #1 | Sigma-Aldrich | Cat# TRCN0000003377 |
| pLKO.1_Puro shMdm2 #2 | Sigma-Aldrich | Cat# TRCN0000003378 |
| pT-REx-DEST30 Mortalin miRNA | R. Kruger and J. Fitzgerald, Hertie-Institute Tubingen | N/A |
| pT-REx-DEST30 neg miRNA control | R. Kruger and J. Fitzgerald, Hertie-Institute Tubingen | N/A |
| pRS-shLuc | Netherlands Cancer Institute (NKI) | N/A |
| pMKO.1 Puro shp53 | Addgene | Cat# 10671 |
| HR-MMP Cre GFP | Silver and Livingston, 2001 | N/A |
| pSUMO-MDM2 | This paper; Arnold et al., 2006 | N/A |
| Software and Algorithms | ||
| Gwyddion | http://gwyddion.net/ | RRID: SCR_015583 |
| FiberApp | Usov I., Mezzenga R., ETH | RRID: SCR_015826 |
| NanoScope Analysis | Bruker Corporation | V1.8 (32bit) |
Highlights.
The MDM2 oncoprotein localizes to the mitochondrial matrix independently of p53
Mitochondrial MDM2 inhibits transcription of the complex I (CI) subunit MT-ND6
MDM2 deficiency in skeletal muscles increases CI activity and muscular endurance
Mitochondrial MDM2 increases cancer cell migration and invasion
ACKNOWLEDGMENTS
The authors thank M. Lacroix, R. Rossignol, G. Lenaers, E.M. Valente, R. Kruger, J. Fitzgerald, J.A. Enriquez, R. Acin-Perez, S. Cristae, J. Hsu, A. Vigneron, and J.E. Sarry for discussions, critical reading of the manuscript, and reagents. We are grateful to members of the histology, animal, and imaging facilities of Montpellier BioCampus and Metamus for technical assistance and to the imaging facilities at Institut Pasteur and Institut Curie that are affiliated with the French National Research Infrastructure (ANR) France-BioImaging (ANR10-INBS-04). This research was supported by grants from the Ligue contre le Cancer (Equipe labelisée 2016), the French National Health Institute for Biomedical Research (INSERM), the French National Cancer Institute (INCa),the General Direction for Care Provision (grant INCa-DGOS-Inserm 6045), and The Laboratory of Excellence EpiGenMed (grant ANR-10-LABX-12-01). G.A. was supported by fellowships from the Région Languedoc Roussillon, the University of Montpellier, and the Fondation de France. R.R. and M.Y.C. were supported by PhD fellowships from the Association pour la Recherche contre le Cancer (ARC) and the French Ministry for Education and Research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, one table, and one movie and can be found with this article online at https://doi.org/10.1016/j.molcel.2018.01.023.
REFERENCES
- Ahn BY, Trinh DLN, Zajchowski LD, Lee B, Elwi AN, and Kim SW (2010). Tid1 is a new regulator of p53 mitochondrial translocation and apoptosis in cancer. Oncogene 29, 1155–1166. [DOI] [PubMed] [Google Scholar]
- Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, and Gillespie MN (2012). Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 5, ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JJ, Bernal A, Uche U, Sterner DE, Butt TR, Cameron CE, and Mattern MR (2006). Small ubiquitin-like modifying protein isopeptidase assay based on poliovirus RNA polymerase activity. Anal. Biochem. 350, 214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeaud M, Mathieu L, Guillaume A, Moll U, Mignotte B, Le Floch N, Vayssière J-L, and Rincheval V (2014). Mitochondrial p53 mediates a transcription-independent regulation of cell respiration and interacts with the mitochondrial F1F0-ATP synthase. Cell Cycle 12, 2781–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biderman L, Manley JL, and Prives C (2012). Mdm2 and MdmX as regulators of gene expression. Genes Cancer 3, 264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatre L, and Ricchetti M (2013). Large heterogeneity of mitochondrial DNA transcription and initiation of replication exposed by single-cell imaging. J. Cell Sci. 126,914–926. [DOI] [PubMed] [Google Scholar]
- Chen H, Xie L, and Liu B (2012). Clinical significance of MDM2 as a tumor biomarker. Chin.-Ger. J. Clin. Oncol. 11, 356–360. [Google Scholar]
- Dudek J, Rehling P, and van der Laan M (2013). Mitochondrial protein import: common principles and physiological networks. Biochim. Biophys. Acta 1833, 274–285. [DOI] [PubMed] [Google Scholar]
- FÁhraeus R, and Olivares-Illana V (2014). MDM2’s social network. Oncogene 33, 4365–4376. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, and Scorrano L (2007). Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2, 287–295. [DOI] [PubMed] [Google Scholar]
- Geissler A, Krimmer T, Bömer U, Guiard B, Rassow J, and Pfanner N (2000). Membrane potential-driven protein import into mitochondria. The sorting sequence of cytochrome b(2) modulates the deltapsi-dependence of translocation of the matrix-targeting sequence. Mol. Biol. Cell 11, 3977–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grier JD, Yan W, and Lozano G (2002). Conditional allele of mdm2 which encodes a p53 inhibitor. Genesis 32, 145–147. [DOI] [PubMed] [Google Scholar]
- Hatchi E, Rodier G, Lacroix M, Caramel J, Kirsh O, Jacquet C, Schrepfer E, Lagarrigue S, Linares LK, Lledo G, et al. (2011). E4F1 deficiency results in oxidative stress-mediated cell death of leukemic cells. J. Exp. Med. 208, 1403–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, and Hayashi J (2008). ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320, 661–664. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, and Weinberg RA (1994). Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4, 1–7. [DOI] [PubMed] [Google Scholar]
- Li L-D, Sun H-F, Liu X-X, Gao S-P, Jiang H-L, Hu X, and Jin W (2015). Down-regulation of NDUFB9 promotes breast cancer cell proliferation, metastasis by mediating mitochondrial metabolism. PLoS ONE 10, e0144441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini F, Di Conza G, Pellegrino M, Rinaldo C, Prodosmo A, Giglio S, D’Agnano I, Florenzano F, Felicioni L, Buttitta F, et al. (2009). MDM4 (MDMX) localizes at the mitochondria and facilitates the p53-mediated intrinsic-apoptotic pathway. EMBO J. 28, 1926–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchenko ND, Zaika A, and Moll UM (2000). Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J. Biol. Chem. 275, 16202–16212. [DOI] [PubMed] [Google Scholar]
- Medja F, Allouche S, Frachon P, Jardel C, Malgat M, Mousson de Camaret B, Slama A, Lunardi J, Mazat JP, and Lombès A (2009). Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 9, 331–339. [DOI] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, and Moll UM (2003). p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 11,577–590. [DOI] [PubMed] [Google Scholar]
- Miniou P, Tiziano D, Frugier T, Roblot N, Le Meur M, and Melki J (1999). Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 27, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minsky N, and Oren M (2004). The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol. Cell 16, 631–639. [DOI] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, and Levine AJ (1992). The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- Montoya J, Christianson T, Levens D, Rabinowitz M, and Attardi G (1982). Identification of initiation sites for heavy-strand and light-strand transcription in human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 79, 7195–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, and Vogelstein B (1993). Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362, 857–860. [DOI] [PubMed] [Google Scholar]
- Park JY, Wang PY, Matsumoto T, Sung HJ, Ma W, Choi JW, Anderson SA, Leary SC, Balaban RS, Kang JG, and Hwang PM (2009). p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ. Res. 105, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales-Clemente E, Fernández-Vizarra E, Acín-Pérez R, Movilla N, Bayona-Bafaluy MP, Moreno-Loshuertos R, Pérez-Martos A, Fernández-Silva P, and Enriquez JA (2010). Five entry points of the mitochondrially encoded subunits in mammalian complex I assembly. Mol. Cell. Biol. 30, 3038–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato PE, Payen VL, Pérez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, et al. (2014). A mitochondrial switch promotes tumor metastasis. Cell Rep. 8, 754–766. [DOI] [PubMed] [Google Scholar]
- Riscal R, Schrepfer E, Arena G, Cissé MY, Bellvert F, Heuillet M, Rambow F, Bonneil E, Sabourdy F, Vincent C, et al. (2016). Chromatin-bound MDM2 regulates serine metabolism and redox homeostasis independently of p53. Mol. Cell 62, 890–902. [DOI] [PubMed] [Google Scholar]
- Saleem A, and Hood DA (2013). Acute exercise induces tumour suppressor protein p53 translocation to the mitochondria and promotes a p53-Tfam-mitochondrial DNA complex in skeletal muscle. J. Physiol. 591, 3625–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem A, Adhihetty PJ, and Hood DA (2009). Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol. Genomics 37, 58–66. [DOI] [PubMed] [Google Scholar]
- Schneider HC, Berthold J, Bauer MF, Dietmeier K, Guiard B, Brunner M, and Neupert W (1994). Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature 371, 768–774. [DOI] [PubMed] [Google Scholar]
- Silver DP, and Livingston DM (2001). Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol. Cell 8, 233–243. [DOI] [PubMed] [Google Scholar]
- Syken J, De-Medina T, and Münger K (1999). TID1, a human homolog of the Drosophila tumor suppressor l(2)tid, encodes two mitochondrial modulators of apoptosis with opposing functions. Proc. Natl. Acad. Sci. USA 96, 8499–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida A, Murugesapillai D, Kastner M, Wang Y, Lodeiro MF, Prabhakar S, Oliver GV, Arnold JJ, Maher LJ, Williams MC, and Cameron CE (2017). Unexpected sequences and structures of mtDNA required for efficient transcription from the first heavy-strand promoter. eLife 6, e27283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, and Moll UM (2012). p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149, 1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade M, Li Y-C, and Wahl GM (2013). MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 13, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, Chang BHJ, Schumacker PT, and Danesh FR (2012). Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 15, 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienken M, Dickmanns A, Nemajerova A, Kramer D, Najafova Z, Weiss M, Karpiuk O, Kassem M, Zhang Y, Lozano G, et al. (2016). MDM2 associates with Polycomb Repressor Complex 2 and enhances stemness-promoting chromatin modifications independent of p53. Mol. Cell 61, 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Deng X, Lim GGY, Zhou W, Saw W-T, Zhou ZD, Lim K-L, and Tan E-K (2017). p62-Mediated mitochondrial clustering attenuates apoptosis induced by mitochondrial depolarization. Biochim. Biophys. Acta 1864,1308–1317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.