

Abstract
A possible role for structure-specific recognition protein 1 (SSRP1) in replication-associated repair processes has previously been suggested based on its interaction with several DNA repair factors and the replication defects observed in SSRP1 mutants. In this study, we investigated the potential role of SSRP1 in association with DNA repair mediated by homologous recombination (HR), one of the pathways involved in repairing replication-associated DNA damage, in mammalian cells. Surprisingly, over-expression of SSRP1 reduced the number of hprt+ recombinants generated via HR both spontaneously and upon hydroxyurea (HU) treatment, whereas knockdown of SSRP1 resulted in an increase of HR events in response to DNA double-strand break formation. In correlation, we found that the depletion of SSRP1 in HU-treated human cells elevated the number of Rad51 and H2AX foci, while over-expression of the wild-type SSRP1 markedly reduced HU-induced Rad51 foci formation. We also found that SSRP1 physically interacts with a key HR repair protein, Rad54 both in vitro and in vivo. Further, branch migration studies demonstrated that SSRP1 inhibits Rad54-promoted branch migration of Holliday junctions in vitro. Taken together, our data suggest a functional role for SSRP1 in spontaneous and replication-associated DNA damage response by suppressing avoidable HR repair events.
Keywords: branch migration, dna damage repair, homologous recombination, Rad54, ssrp1
Homologous recombination (HR) is one of the DNA repair mechanisms crucial for the cellular response to various kinds of stress. HR engages a strand exchange process involving a homologous DNA partner to repair the damaged DNA in an error-free manner [Thompson and Schild, 2001]. Replication machinery is frequently arrested in response to DNA damage [for review, see Hyrien, 2000] and many lesions, including DNA gaps, double-strand breaks (DSBs), and interstrand crosslinks, formed at the stalled replication forks are repaired by HR in mammalian cells [Johnson and Jasin, 2000; Lundin et al., 2002], thereby maintaining genomic stability and integrity. Deregulation of the HR repair pathway has been shown to be associated with carcinogenesis.
The RAD52 group of proteins, which play an important role in HR and DSB repair, has been extensively studied on both genetic and biochemical levels [Krogh and Symington, 2004]. Rad51 and Rad54 are two key members of this group. The Rad51 protein forms a nucleoprotein filament on single-stranded DNA, which promotes the search for a homologous DNA partner followed by DNA strand exchange with this partner [Bianco et al., 1998]. Rad54 is an evolutionarily conserved SWI/SNF2 protein [Thoma et al., 2005; Heyer et al., 2006]. In vitro, Rad54 has a spectrum of activities suggesting its multiple functions at various stages of HR: it physically interacts with the Rad51-ssDNA nucleoprotein filament [Mazin et al., 2000; Van Komen et al., 2000], displaces Rad51 from dsDNA [Solinger et al., 2002] and stimulates DNA strand exchange promoted by Rad51 [Petukhova et al., 1998]. In addition, Rad54 protein can translocate along dsDNA using the energy of ATP hydrolysis [Amitani et al., 2006], remodel chromatin [Alexeev et al., 2003; Zhang et al., 2007] and promote branch migration (BM) of Holliday junctions (HJs) [Bugreev et al., 2006b]. Rad54 mutants in mouse [Wesoly et al., 2006], chicken [Bezzubova et al., 1997] and yeast [Osman et al., 2000] show strong deficiency in HR and are sensitive to different DNA damaging agents.
In mammals, the facilitates chromatin transcription (FACT) complex was identified as a heterodimer composed of an high mobility group (HMG)-containing protein, structure-specific recognition protein 1 (SSRP1), and p140/hSpt16, a human homolog of the yeast Spt16/Cdc68 [Orphanides et al., 1999]. Genetic and biochemical studies suggest that FACT is a chromatin factor that is involved in regulating multiple cellular processes. In addition to its role in transcription regulation, FACT has been shown to be involved in replication by physically associating with DNA polymerase α in Saccharomyces cerevisiae [Wittmeyer and Formosa, 1997] and the human replicative helicase complex, MCM [Tan et al., 2006]. Immunodepletion of Xenopus DUF complex (DNA unwinding factor), the homolog of human FACT, makes the egg extracts defective in replication [Okuhara et al., 1999], indicating that this factor is important for the replication process. Recent studies have shown an interaction between the FACT complex and two other proteins involved in HR and other DNA damage repair processes, PARP1 and RPA [Huang et al., 2006; VanDemark et al., 2006].
Human SSRP1 was first cloned on the basis of its ability to bind to cisplatinated DNA [Bruhn et al., 1992]. The murine homolog of SSRP1, T160, co-localizes with the toroidal DNA structures typical of mid and late S phase, suggesting its function in an advanced stage of replication [Hertel et al., 1999]. There has been some speculation that HMG proteins, including SSRP1/T160, may be involved in DNA repair processes such as HR and non-homologous end joining (NHEJ) [Shirakata et al., 1991; Stros and Muselikova, 2000; Stros et al., 2000], but this remains to be confirmed. It has also been shown that SSRP1 is associated with both ultraviolet (UV)-induced and hydroxyurea (HU)-induced DNA damage [Schlesinger and Formosa, 2000; Keller and Lu, 2002]; these types of damage are known to stall the replication forks, and further stimulate the HR repair process [Li and Heyer, 2008].
Given that the FACT complex interacts with the repair factors, and its depletion causes replication defects, we investigated the independent role of the SSRP1 component of the FACT complex in the replication-associated DNA damage response. Our study showed that over-expression of SSRP1 reduces HR activity in response to spontaneous and HU-induced DNA damage. Depletion of SSRP1 resulted in an induction of HR as well as increased formation of H2AX and Rad51 foci. Interestingly, we also found that SSRP1 can physically interact with Rad54 and functionally inhibit the BM activity of HJs promoted by Rad54 in vitro. Altogether, these results demonstrate that SSRP1 may play an important role in the HR-mediated DNA damage response.
MATERIALS AND METHODS
CELL LINES AND CULTURE CONDITIONS
All cell lines (H1299, SPD8, SW480SN.3) used in our study were maintained in DMEM with 10% fetal calf serum and a penicillin/streptomycin mixture (100 μg/ml). SPD8 (Chinese hamster) and SW480SN.3 (human colon cancer) cells were used for recombination assays. For SW480.SN3 cells, media was supplemented with hygromycin (0.05 mM) in order to maintain the SCneo vector [Delacote et al., 2002]. The SPD8 cells were grown in media supplemented with 6-thioguanine (6-TG) in order to maintain a homogenous population of hprt-deficient cells. The generation of H1299 inducible cell line has been described previously [Li et al., 2007]. All cell lines were kept at 37°C in a 5% CO2/95% air incubator.
DRUGS AND siRNA OLIGOMERS
For this study, we used G418 (Cellgro), hygromycin (Cellgro), thymidine (Sigma), HU (Sigma), 6-TG (Sigma), hypoxanthine (Sigma), Doxycycline (Sigma), and azaserine (Sigma). The SSRP1 siRNA sequences were synthesized by Dharmacon. A cocktail consisting of two siRNA sequences (oligo 1 — 5′-gaatggccatgtctacaag-3′; oligo 3 — 5′-gaggagtgggatcgcaagg-3′) was used for transient transfection experiments.
PLASMID CONSTRUCTION AND TRANSFECTION
The pcDNA3-Flag-2xSSRP1 and SSRP1 pcDNA3-Flag-2x N- (1–160 aa) and C-terminus (450–709 aa) constructs have been described previously [Landais et al., 2006]. The green fluorescent proteins (GFP)-SSRP1 and N- and C-SSRP1 were generated in our lab using the pEGFP-C1 vector (Clontech). The constructs used for generating GST- and His-fusion proteins have been described previously [Li et al., 2005]. Cells were transfected with SSRP1 constructs (2 mg or else specified in the text) using TransFectin™ reagent (Bio-Rad); cells were harvested 48–96 h posttransfection (as indicated in figure legend or text). Empty vector DNA was used as a negative control for comparison. The H1299 inducible cell line expressing SSRP1 siRNA has been described previously [Li et al., 2007]. Cells were transfected with siRNA using SilentFect lipid reagent (Bio-Rad).
MUTAGENESIS
The plasmids carrying Flag-2xSSRP1, GST-SSRP1, and GST-C-SSRP1 were used as templates for mutagenesis experiments and the point mutants generated have been referred as Flag-SS552A, SSRP1S552A, and GST-C-S552A, respectively. The mutants carrying a serine to alanine point mutation at position 552 of SSRP1 were generated by site-directed mutagenesis using the QuickChange kit (Stratagene). The primers used for generating the S552A mutation were hSSRP1S552A-forw: 5′-cccaagaggcccatggctgcatacatgctgtgg-3′; hSSRP1S552A-rev: 5′-ccacagcatgtatgca gccatgggcctcttggg-3′.
RECOMBINATION ASSAYS
The recombination assays were performed as described for SPD8 and SW480SN.3 cells [Arnaudeau et al., 2001; Mohindra et al., 2002]. In brief, 1 × 106 cells were plated and transfected the following day, using TransFectin™/SilentFect™, according to the manufacturer’s instructions (Bio-Rad). Twenty-four hours posttransfection, SPD8 cells were incubated with HU (20 μM) for 24 h, or 48 h posttransfection, SW480SN.3 cells were transfected with 30 ng of pCMV3nls-I-SceI expression vector. The cells were supplemented with fresh medium after 5 h of I-SceI induction. After a 48 h recovery period from HU/I-SceI induction, cells were trypsinized, counted, and plated at 0.3 × 106 cells (SPD8) per plate in triplicate in medium containing HAsT (50 μM hypoxanthine, 10 μm l-azaserine, and 5 μm thymidine), or 0.2 × 106 cells (SW480SN.3) per plate in medium containing G418 (1 mg/ml). In addition, 500 cells were plated in duplicate for assessing the clonogenic survival of the cells. The colonies obtained on cloning and selection plates were fixed after 7–14 days, respectively, using methylene blue in methanol (4 mg/ml) and the colonies were counted. All experiments were repeated independently three times. The statistical significance of differences was determined using Student’s unpaired t-test.
WESTERN BLOT ANALYSES AND ANTIBODIES
The cell lysates were prepared using a lysis buffer consisting of 50 mM Tris–HCl (pH 8.0), 0.5% Nonidet P-40, 1 mM EDTA, 150 mM NaCl, and 1 mM phenylmethylsulfonyl fluoride. Protein samples (50 μg or else specified) were run on a SDS–PAGE gel, followed by transfer to the PVDF membranes that were then immunoblotted with the appropriate antibodies. The following antibodies were used in this study: Monoclonal mouse anti-α-tubulin (Sigma); monoclonal mouse anti-Flag (M2, Sigma); polyclonal rabbit anti-GST (a gift from Dr. Richard Goodman); polyclonal rabbit anti-phospho H2AX (ser139, Upstate); polyclonal rabbit anti-Rad51 (H-92, Santa Cruz), and monoclonal mouse anti-hRad54 (clone 4E-3/1, Upstate). Monoclonal mouse anti-SSRP1 antibody has been described previously [Keller et al., 2001; Landais et al., 2006]. Proteins were detected by ECL reagents (Santa Cruz Biotech).
For Far-Western blot, ~150 ng purified protein samples (GST-0, GST-hRad54, and His-SSRP1) were immunoblotted on a PVDF membrane. This was followed by blocking with 5% non-fat milk (prepared in TBS-T) and the blot was incubated with purified His-SSRP1 protein (6 μg) for 5 h followed by thorough washing with TBS-T buffer and probing first with anti-SSRP1 followed by anti-GST antibody. Proteins were detected by ECL reagents (Santa Cruz Biotech).
PURIFICATION OF THE RECOMBINANT PROTEINS
GST-fusion proteins were bound to glutathione-agarose beads (Sigma) and eluted with glutathione, followed by dialysis against BC100 buffer (20 mM Tris–HCl (pH 7.9), 0.1 mM EDTA, 15% glycerol, 100 mM KCl, 1 mM dithioreitol, and protease inhibitors including 0.2 mM phenylmethylsulfonyl fluoride, 4 mM pepstatin A). His-SSRP1 was expressed and purified from bacteria using Ni-NTA-agarose (Qiagen), as described elsewhere [Keller and Lu, 2002]. GST-tagged hRad54 protein was purified as described previously [Mazina and Mazin, 2004].
IMMUNOFLUORESCENCE ANALYSES
Cells were transiently transfected with Flag-SSRP1 expression vector for 24 h prior to HU treatment. The SSRP1 siRNA inducible H1299 cell line was kept under Doxycycline (5 μg/ml) treatment for 3 days to turn on the siRNA expression prior to HU treatment. The cells were fixed in 4% paraformaldehyde for 20 min, washed with PBS (2–3 times), permeabilized with PBS-T (PBS with 0.1% Triton-X) for 10 min and blocked with a solution containing PBS and 8% BSA. Conditions followed for primary antibodies: phosphor H2AX—1:1,000 dilution for 1 h at 37°C; Rad51—1:1,000 dilution for 16 h at 4°C; Flag—1:600 dilution for 1 h at room temperature; SSRP1 monoclonal—1:100 dilution for 1 h at room temperature. Appropriate fluorochrome-conjugated secondary antibodies were used at a dilution of 1:300 along with DAPI (1:300) for 30–45 min prior to mounting the slides with the gel mount (Biomeda), and fluorescence was visualized using an Axiovert 200 M Zeiss microscope. Nuclei containing more than 10 foci were scored as positive. The statistical significance of differences was determined using Student’s unpaired t-test.
BRANCH MIGRATION ASSAYS
The scheme of BM of synthetic HJ is shown in Figure S4. Sequences of oligonucleotides (IDT, Inc.) are shown in Supplementary Table S1. Oligonucleotides were purified and labeled using [γ-32P] ATP and T4 polynucleotide kinase, as described previously [Mazin et al., 2003]. The 3′-tailed DNAs (170/171) and the 32P-labeled forked DNA (71/169*) were obtained by annealing indicated oligonucleotides as described previously [Bugreev et al., 2006a]. The 3′-tailed DNA at a concentration of 48 nM (molecules) was mixed with 32P-labeled forked DNA 71/169 (32 nM, molecules) in BM buffer containing 25 mM Tris acetate, pH 7.5, 2 mM ATP, 3 mM magnesium acetate, 2 mM DTT, 100 μg/ml BSA, and the ATP regenerating system (15 mM phosphocreatine and 10 U/ml creatine phosphokinase). The mixture was incubated at 37°C for 10 min, and then at 30°C for 10 min to allow the forked and tailed DNA substrates to anneal. As a result, the HJ 170/171/71/169 was formed. It contained a single mismatch (A → G), which blocks a spontaneous BM. This HJ was diluted with BM buffer to a final concentration of 16 nM (molecules) and pre-incubated with the indicated amounts of GST-tagged wild-type SSRP1, SSRP1S552A, or N-SSRP1 truncated mutant (1–242 aa) protein at 30°C for 5 min. Then hRad54 (50 nM) was added and the reaction was carried out at 30°C for 20 min. The reaction was deproteinized by treatment with proteinase K (1.6 mg/ml) and 1.2% SDS for 30 min at 37°C, mixed with a 0.10 volume of loading buffer (70% glycerol, 0.1% bromophenol blue) and analyzed by electrophoresis through an 8% non-denaturing polyacrylamide (29:1) gel in 1× TBE buffer (89 mM Tris–borate, pH 8.3, and 1 mM EDTA) at 135 V for 1.5 h. Gels were dried on DEAE-81 paper (Whatman) and quantified using a Storm PhosphorImager (Molecular Dynamics).
RESULTS
OVER-EXPRESSION OF SSRP1 SUPPRESSES SPONTANEOUS AND HU-INDUCED HR
Pob3 (S. cerevisiae homolog of SSRP1) mutants are sensitive to HU [Schlesinger and Formosa, 2000]. HU is a commonly known replication inhibitor that arrests cells in S phase and stalls the replication fork [Arnaudeau et al., 2001; Lundin et al., 2002] by depleting the dNTP pool. Consequently, DSBs are generated and HR repair process is triggered as a damage response [Lundin et al., 2003]. The sensitivity to HU of the Pob3 mutants could be the consequence of an inability to repair HU-induced DSBs, resulting in cell death.
To assess whether SSRP1 could be involved in the HU-induced HR repair pathway, we measured the HR frequency under spontaneous and HU-induced conditions after over-expression of the human SSRP1 in the Chinese hamster SPD8 cell line. SPD8 cells carry a 5 Kb duplication in the hprt gene that renders the gene inactive; the function of the hprt gene can be restored via HR [Helleday et al., 1998]. These cells were kept under the selection of 6-TG for 3–4 passages prior to the recombination assay so that a homogenous population of hprt-deficient cells could be achieved and any spurious results could be ruled out. We found that the over-expression of Flag-SSRP1 in SPD8 cells (Fig. 1A) led to a minor but statistically significant reduction in HR events in response to spontaneous damage (P<0.05, t-test) (Fig. 1B). The cloning efficiency of the SSRP1 over-expressing cells was higher or in the same range as the control cells (data not shown), ruling out the possibility that the HR reduction upon SSRP1 over-expression was due to cell death or reduced cell proliferation.
Fig. 1.
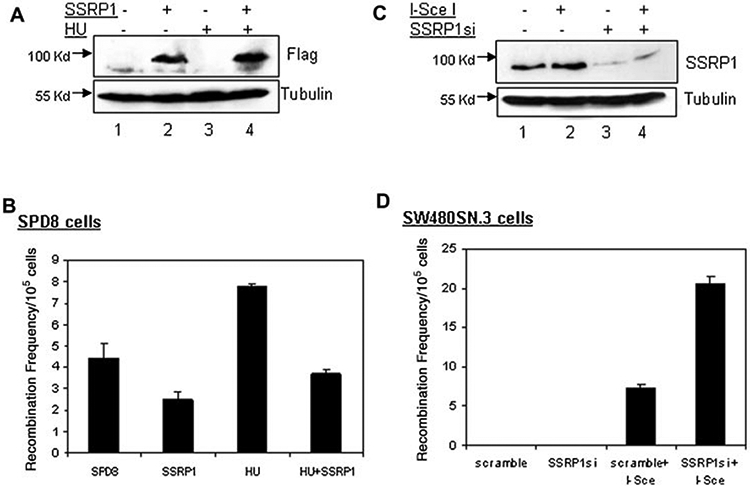
Over-expression of SSRP1 suppresses homology-based DSB repair while depletion of SSRP1 elevates the HR frequency. A: Western blot analysis to show ectopic expression of Flag-2xSSRP1 in SPD8 cells. The cell lysates (50 μg) used for the western blot analysis were extracted from the same population that was used for the recombination assays. Immunoblotting was carried out using the antibodies indicated on the right. B: Ectopic Flag-2xSSRP1 reduces spontaneous and HU-induced HR in Chinese hamster SPD8 cells. The cells were allowed to over-express the protein for 24 h prior to HU treatment (20 μM). Error bars represent the standard deviations from three independent experiments. C: Western blot analysis of endogenous SSRP1 levels when cells were treated with SSRP1 or scrambled siRNA using silentFect™ reagent (Bio-Rad). The cell lysates (50 μg) were prepared from the same population that was used for the recombination assays. Immunoblotting was carried out using the antibodies indicated on the right. D: SW480SN.3 cells transfected with I-SceI expression vector showed an increase of HR when SSRP1 was knocked down using siRNA against it. Error bars represent the standard deviations from three independent experiments.
Also, SSRP1 over-expressing cells, when exposed to HU, displayed significantly lower frequency of HR events than the empty vector-transfected cells treated with HU (P<0.01, t-test) (Fig. 1B). These observations suggest that SSRP1 can possibly suppress HR-mediated repair processes in response to both spontaneous and HU-induced DNA damage.
SSRP1 DEPLETION ELEVATES THE LEVEL OF HR
To further investigate the role of SSRP1 in HR, we employed the human SW480SN.3 cell line as an additional model system for HR and depleted SSRP1 by siRNA expression (Fig. 1C) in these cells. This cell line contains a stably integrated copy of the commonly used recombination reporter vector, SCneo. The SCneo construct has two inactive cassettes of the neomycin resistant gene separated by a hygromycin gene, and the second copy of the neomycin gene carries a restriction site for I-SceI endonuclease [Mohindra et al., 2002]. The expression of I-SceI endonuclease generates DSBs that can subsequently be repaired by HR allowing restoration of a functional neoR gene [Delacote et al., 2002]. Another advantage of this system is that it gives readout of HR events taking place by conservative HR processes (via strand invasion) but rules out non-conservative repair processes (via single-strand annealing). The non-conservative repair process by single-strand annealing between two repeated sequences within the SCneo substrate cannot generate a functional neoR gene and thus, their products are not scored for by selection with G418 [Delacote et al., 2002]. The scoring of recombinants in SW480SN.3 cells showed an elevation of HR events for the cells that had been siRNA depleted of the SSRP1 protein (P<0.05, t-test) (Fig. 1D). An increase in the number of I-SceI-induced HR events in an SSRP1-deficient background suggested that SSRP1 can inhibit the HR process either directly or indirectly.
THE C-TERMINUS OF SSRP1 CONTRIBUTES TOWARD HR SUPPRESSION
To determine the specific region of the protein that is important for SSRP1-mediated HR suppression, we performed recombination assays using truncated forms of SSRP1: the N-terminus (1–160 aa) and the C-terminus (472–709 aa) (see Supplementary Fig. 1). In vitro studies have revealed that the C-terminus of SSRP1 is required for its DNA-binding activity, and this process is mediated by the HMG domain [Gariglio et al., 1997; Li et al., 2005]. We used the N-terminus and the C-terminus of SSRP1 fused with a Flag or GFP tag. The immunofluorescence studies done with GFP-C/N terminus SSRP1 fusion proteins showed that the C-terminus SSRP1 localizes in the nucleus while the N-terminus SSRP1 predominantly resides in the cytoplasm (Fig. 2A).
Fig. 2.

The C-terminus of SSRP1 mediates suppression of HR activity. A: Immunofluorescence analysis of the localization of GFP-fused N- or C-terminus of SSRP1 in SPD8 cells. B: Western blot analysis to show the over-expression of the Flag-2x C- or N-terminus of SSRP1 in SPD8 cells. The cell lysates (50 μg) were prepared from the same population that was used for the recombination assays. Immunoblotting was carried out using the antibodies indicated on the right. C,D: Measurement of the recombination frequency in SPD8 cells over-expressing the Flag-2x C- or N-terminus of SSRP1.
Next, we scored for the recombination events in SPD8 cells that transiently expressed the Flag-N- or C-terminus fusion proteins (Fig. 2B). Similar to the full-length SSRP1 (Fig. 1B), ectopic expression of the SSRP1 C-terminus fragment also led to reduction of HR events, though to a lesser extent (Fig. 2C). In contrast, the N-terminus of SSRP1 failed to cause any HR suppression. This finding indicated that the C-terminus, but not the N-terminus, of SSRP1 may contribute toward SSRP1-mediated suppression of HR events (Fig. 2C,D).
SSRP1 DEPLETED CELLS DISPLAY MORE DNA DAMAGE AT STALLED REPLICATION SITES
Generation of DSBs causes replication arrest, which in turn induces the assembly of the HR repair protein Rad51 into discrete nuclear foci [Tashiro et al., 1996; Lundin et al., 2003]. To determine whether HR stimulation in SSRP1 depleted cells was correlated with intranuclear Rad51 foci formation, an inducible cell line (H1299) expressing SSRP1 siRNA [Li et al., 2007] was analyzed for Rad51 foci formation following treatment with the replication inhibitor HU. Compared to controls (cells expressing scramble siRNA), we observed that there was an accumulation of Rad51 foci in SSRP1 depleted cells, which further increased significantly in response to HU treatment (P<0.05, t-test) (Fig. 3A,B). Increased Rad51 foci in SSRP1 depleted cells could be associated with an increased Rad51-dependent HR activity.
Fig. 3.

SSRP1 depleted cells are inflicted with more DNA damage in response to hydroxyurea. Visualization of Rad51 (A), H2AX (C) foci in inducible SSRP1 siRNA expressing H1299 cells by immunofluorescence staining. The magnified cells are circled (white dotted lines) on the respective overlay image. The siRNA expressing cells were induced with Doxycycline for 3 days prior to a 24 h treatment with HU (200 μM). The mean percentage of SSRP1 depleted cells containing Rad51 (B) and H2AX foci (D) in response to HU treatment was calculated.
We also studied HU-induced H2AX foci formation in SSRP1 siRNA inducible cells. Consistent with the above results, SSRP1 depleted cells also displayed an elevated level of H2AX foci in comparison to scramble siRNA expressing cells (Fig. 3C,D) in response both to spontaneous and to HU-induced DNA damage. An elevated level of H2AX foci in an SSRP1-depleted background is most likely a consequence of an increase in the number of DSBs, which subsequently can be repaired by a Rad51-dependent HR mechanism.
We next addressed whether SSRP1 over-expression also affects HU-induced Rad51 foci formation. We investigated Rad51 foci levels in cells transiently transfected with the plasmid over-expressing wild-type SSRP1. Fewer HU-induced Rad51 foci were observed in wild-type SSRP1 transfected cells than the mock cells (Fig. 4). A reduction in the level of Rad51 foci formation indicated that there are fewer Rad51-mediated repair events occurring in the HU-treated SSRP1 over-expressing cells.
Fig. 4.

SSRP1 over-expressing cells suppress the formation of Rad51 foci induced by HU. The SPD8 cells were transiently transfected with Flag-SSRP1 for 24 h followed by a 24 h treatment with HU (1 mM), and then subjected to IF procedure.
SSRP1 SPECIFICALLY INTERACTS WITH hRad54
To determine whether SSRP1 is directly associated with the HR repair pathway, we first looked for its interaction with Rad51 and H2AX. However, using co-immunoprecipitation studies (Supplementary Figs. 2 and 3), we found no evidence for interaction between SSRP1 and RAD51 or H2AX.
Next, we explored a possible association between SSRP1 and Rad54, one of the interacting partners of Rad51 that stabilizes and stimulates its DNA pairing activity during HR process [Petukhova et al., 1998; Mazin et al., 2000]. Interestingly, the co-immunoprecipitation analyses showed an in vivo interaction between SSRP1 and Rad54 in H1299 cells. The cells were treated with 200 μM HU for 24 h and anti-SSRP1 monoclonal antibodies were used to immunoprecipitate SSRP1. We detected a weak interaction between Rad54 and SSRP1 at the endogenous levels in untreated H1299 cell lysates; this interaction significantly increased in HU-treated cells (Fig. 5A, lanes 2 and 3 upper panel), suggesting this interaction to be critical for replication-induced DNA damage. In an IgG control experiment, no bands were detected (Fig. 5A, lane 1 upper panel), demonstrating the specificity of the antibodies used as well as the specificity of the interaction. SSRP1-Rad54 interaction was further confirmed by co-immunoprecipitating endogenous Rad54 with ectopic Flag-SSRP1 fusion protein in H1299 cells (Fig. 5B, lane 4).
Fig. 5.
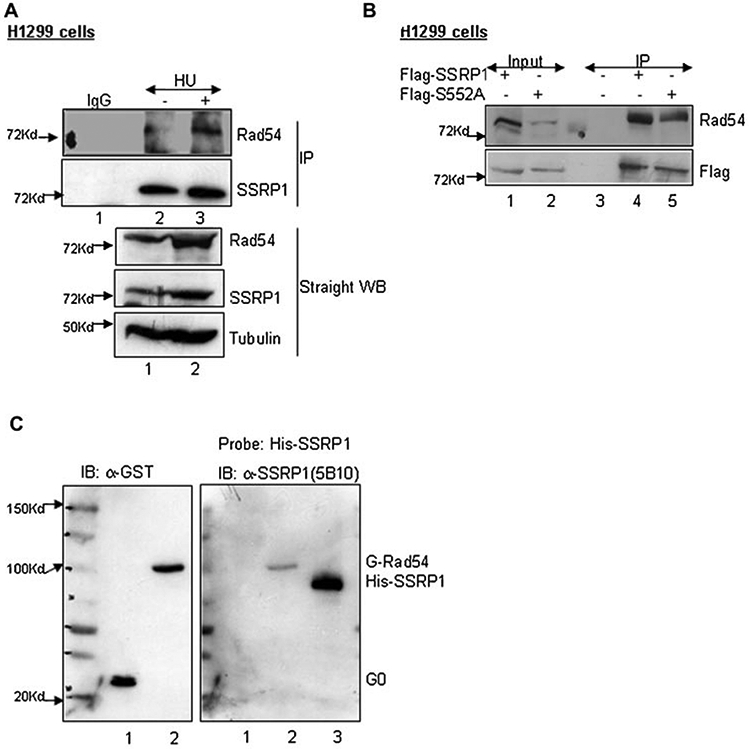
SSRP1 associates with Rad54 in vitro and in vivo. A: Co-immunoprecipitation of endogenous Rad54 with SSRP1. H1299 cells were treated with 200 μM of HU for 24 h before the cells were lysed with lysis buffer containing 200 mM NaCl and a 500 μg aliquot of the lysate was precleared with Pro-G beads before being subjected to immunoprecipitation with anti-SSRP1 (mouse monoclonal) or anti-IgG (mouse monoclonal) as indicated. Total cell lysates (100 μg) were also run on the same gel. Immunoblotting was carried out using the antibodies indicated on the right. B: Endogenous Rad54 co-immunoprecipitates with over-expressed full-length SSRP1 or SSRP1 mutant carrying S552A mutation. H1299 cells were transfected with Flag-2xSSRP1 or Flag-2xS552A constructs (4 μg) and a 400 μg aliquot of lysate was used for immunoprecipitation with the anti-flag antibody followed by probing of the blots with antibodies as indicated on the right. An aliquot of the same lysate was used as input (40 μg). C: Far-Western analysis of full-length hRad54 and SSRP1. PVDF membrane, to which purified GST-hRad54 was transferred following SDS–PAGE, was incubated with His-SSRP1 protein and then probed for the presence of bound SSRP1 by Western blotting using an anti-SSRP1 antibody (right panel). Purified His-SSRP1 was also loaded in one of the lanes to show the specificity of SSRP1 antibody (lane 3, right panel). The left panel is the same blot that was later probed with anti-GST antibody to show the relative amounts of purified GST-0 and GST-hRad54 used.
To test for a direct interaction between SSRP1 and Rad54, a Far-Western analysis was carried out using purified SSRP1 and hRad54 protein. GST-hRad54 protein was run on a SDS–gel along with GST-0 as a negative control (Fig. 5C, lanes 1 and 2 left panel). The gel was transferred onto a membrane and probed with His-SSRP1 fusion protein [Keller and Lu, 2002]. In addition, another membrane was probed with a buffer lacking the SSRP1 protein. After thorough washes, monoclonal anti-SSRP1 antibodies, followed by secondary anti-mouse antibodies, were used to probe the blot to detect the position of the bands that bound SSRP1 protein in the first step. As shown in lane 2 of Figure 5C (right panel), SSRP1 protein bound to the purified GST-hRad54 protein but not to GST-0 (lane 1, right panel); no band was seen at the position of GST-hRad54 on the control blot that had been incubated with the buffer alone (data not shown). The blots were later probed with anti-GST antibody to show the even levels of GST-0 and GST-hRad54 (Fig. 5C, left panel). These data confirmed that there is a direct physical association between SSRP1 and Rad54, further suggesting a more specific role for SSRP1 in the HR repair process.
SSRP1 INHIBITS hRad54-MEDIATED BRANCH MIGRATION
We next sought to determine the functional consequences of SSRP1-Rad54 interaction in the context of the HR repair pathway. It has been previously shown that Rad54 promotes branch-migration of HJs [Bugreev et al., 2006b], a key intermediate step in HR and DSB repair. Thus, we questioned whether SSRP1 might be functionally involved in Rad54-mediated processing of HR intermediates. To achieve this aim, we used synthetic HJ substrates, which were prepared by annealing DNA oligonucleotides (Fig. S4). The BM studies showed that Rad54-promoted BM of HJs was significantly inhibited by the full-length GST-SSRP1, in vitro (Fig. 6A,C).
Fig. 6.

SSRP1 protein inhibits branch migration promoted by hRad54. A: Full-length GST-tagged wild-type SSRP1 (0, 100, 200, 300, 400, 500, and 500 nM in lanes 2–8, respectively), or SSRP1S552A (60, 120, 180, 240, 300, and 300 nM in lanes 9–14, respectively) or (B) N-SSRP1 truncated mutant (1–242 aa) protein (0, 100, 200, 300, 400, 500, and 500 nM in lanes 2–8, respectively) were incubated with HJs (16 nM) for 5 min at 30°C and then hRad54 protein (50 nM) was added to the reaction mixtures, followed by incubation for 20 min at 30°C. In lane 1 (A,B), hRad54 and SSRP1 were omitted; in lane 8 (A,B) and 14 (A), hRad54 was omitted. The DNA products of branch migration were deproteinized and analyzed as described in Materials and Methods Section. The coomassie gel shows the purified GST-tagged N-SSRP1, full-length wild-type SSRP1 and SSRP1S552A mutant proteins. C: Graphical representation of the results from panels A and B.
It could be questioned that the inhibition of BM activity of Rad54 is a consequence of masking DNA by SSRP1 protein and not due to direct interaction between SSRP1 and Rad54 proteins. We and others have previously shown that the C-terminus of SSRP1 retains the ability to bind to DNA [Yarnell et al., 2001; Li et al., 2005]. Additionally, in agreement with a recent structural study of the HMG-1 domain of Drosophila melanogaster SSRP1 [Kasai et al., 2005], our structural model for the HMG domain of human SSRP1 predicted that serine 552 (corresponding to Thr 560 in D. melanogaster) (Fig. S5A) is a key residue for SSRP1 DNA binding activity (Fig. S5B). A site-directed serine-to-alanine substitution of serine 552 in the HMG domain of the C-terminus was sufficient to impair the DNA binding activity of GST-C-SSRP1 protein (Fig. S5C,D). However, the co-immunoprecipitation study and BM assay showed that similar to the wild-type full-length SSRP1, Rad54 not only immunoprecipitated with SSRP1S552A full-length protein (lane 5, Fig. 5B) but was also able to inhibit the BM activity of Rad54 (Fig. 6A,C), suggesting that the SSRP1-mediated inhibition of the BM activity is a consequence of direct interaction between SSRP1 and Rad54.
Consistent with our cellular studies, we also found that the GST-N-SSRP1 (1–242 aa, Fig. S1) failed to inhibit Rad54-mediated BM under the same conditions (Fig. 6B,C). The inhibitory effect of SSRP1 on Rad54-promoted BM of HJs suggests a role for SSRP1 as a negative regulator of HR activity.
DISCUSSION
Conserved complexes homologous to human FACT have been found in all eukaryotes suggesting an indispensable role for this core complex in multiple cellular processes which require DNA to be made accessible to various factors. Although FACT complex was isolated as a chromatin-specific transcript elongation factor [Orphanides et al., 1999], the recent findings support the notion that in conjunction with pathway-specific cofactors, the FACT components, Spt16 and SSRP1, may also participate in processes such as, replication, recombination, and repair. We have previously reported that in response to UV-induced DNA damage, FACT complex physically interacts and alters the specificity of casein kinase II (CKII), further facilitating a selective phosphorylation of p53 over other substrates [Keller et al., 2001; Keller and Lu, 2002]. Our group has also demonstrated a role for SSRP1 in regulating gene transcription in an Spt16-dependent and independent manner [Li et al., 2007]; although, this study was conducted in the absence of any DNA damage but it can be hypothesized that a different subset of genes might be regulated by SSRP1 under stress conditions. Therefore, this study was aimed to explore the contribution of the SSRP1 subunit in regulating the cellular response to other kinds of cellular stress.
In this study, we show that SSRP1 plays a vital role in the prevention of spontaneous and replication-associated DNA damage by avoiding inappropriate HR events. Our results demonstrate that the SSRP1 over-expressing cells exhibit fewer H2AX, Rad51 foci and decreased levels of HR, in response to both spontaneous and HU-induced DNA damage. We also found that the depletion of SSRP1 is coupled with an accumulation of DNA DSB breaks (increased H2AX foci), increased Rad51 foci formation as well as stimulation of the HR repair process. Taken together, these findings suggest that SSRP1 serves as a protective barrier against DNA damage and any modulation of SSRP1 levels directly or indirectly affects the HR repair process involved in cellular recovery following DSB-induced DNA damage. Our observations are consistent with the previous study showing that Pob3 (yeast version of SSRP1) is required for normal replication and loss of its function resulted in replication defects and a poor recovery from HU-induced DNA damage [Schlesinger and Formosa, 2000].
The SSRP1 protein contains two conserved domains that contribute to the multiple functions of SSRP1. The N-terminal region of SSRP1 has been shown to directly interact with its partner Spt16 to form the heterodimeric FACT complex [Keller et al., 2001], while the C-terminus encompassing the HMG domain has been shown to bind to DNA non-specifically [Li et al., 2005], as well as to specific structures, such as cisplatinated DNA or cruciform DNA [Gariglio et al., 1997; Yarnell et al., 2001]. As anticipated, we found that over-expression of the C-terminus, but not the N-terminus, of SSRP1 was required for suppression of HR activity. Interestingly, despite the considerably high expression levels of the SSRP1 C-terminus, the level of HR suppression was not as pronounced as seen when the full-length SSRP1 was over-expressed (compare Fig. 2C with Fig. 1B). One plausible explanation for this result could be that even though the C-terminus of SSRP1 is critical for the HR suppression, the N-terminus and mid-region (1–440 aa) together may act as an accessory fragment that modulates or stabilizes the DNA binding affinity of the C-terminus. In favor of this hypothesis are results from our laboratory demonstrating that the endogenous C-terminus does not tightly associate with chromatin after apoptotic cleavage of SSRP1, while the N-terminal fragment covering 1–440 aa residues remains strongly bound to chromatin [Landais et al., 2006].
Identification of the interacting partners in the HR repair process and understanding their influence on each other functionally is crucial for gaining insight into the mechanism of the process. To reflect on the mechanistic role of SSRP1 in the HR process, we studied its physical interaction with several HR proteins including Rad51 and Rad54, and also with the DNA DSB marker H2AX. This study shows for the first time that SSRP1 can interact directly with an HR protein, hRAD54, in vitro and in vivo. Although, it has been demonstrated that hRAD51 and hRAD54 physically interact, it was intriguing to find that SSRP1 binds specifically to Rad54 but not the other RAD52 family member, Rad51. As a next step, it would be exciting to explore whether the SSRP1-Rad54 assembly takes place in one sub-complex or whether they constitute a part of a bigger complex. The existence of different sub-complexes at different stages of the HR repair process may explain the interaction seen between Rad54-Rad51, and SSRP1-Rad54, as well as the lack of interaction between Rad51-SSRP1 [Sugawara et al., 2003; Wolner et al., 2003]. It is very possible that the Rad54-Rad51 interaction might be a prerequisite for several steps but not for the entire recombinational repair process [Clever et al., 1997].
The unique role of Rad54 has been demonstrated in stimulating DNA strand exchange activity of Rad51 [Petukhova et al., 1998] and also in promoting BM of HJs [Bugreev et al., 2006b] formed at the stalled replication forks during the HR repair process. Tan et al. [2006] demonstrated that the human heterodimeric complex of SSRP1 and Spt16 proteins can stimulate DNA unwinding activity of the MCM helicase on nucleosomal template [Tan et al., 2006]. However, they found that the FACT complex does not alter the MCM helicase activity on the naked DNA template [Tan et al., 2006]. Our in vitro assay showed a significant inhibition of Rad54 BM activity by purified full-length SSRP1 protein suggesting this function of SSRP1 to be Rad54-specific. Our findings support a model in which SSRP1 binds specifically to Rad54 and functionally inhibits its BM activity. This inhibition of Rad54 may assist in inhibiting inappropriate recombination at the sites where recombination is possible but, not necessarily desirable. Further studies are required for understanding the mechanism and functional consequences of SSRP1-Rad54 interaction in vivo. It would be interesting to investigate the effect of SSRP1-Rad54 interaction in the context of chromatin remodeling; it is possible that the chromatin-associated SSRP1 protein may influence the Rad54-dependent chromatin modulating activity during search for homologous partner [Zhang et al., 2007].
In summary, our study demonstrates a novel role for SSRP1 in recombination-mediated response to DNA damage provoked upon replication inhibition. Although we have focused on the HR repair process, our study does not eliminate the prospect of SSRP1 being a potential participant in other repair mechanisms. Hence, our work opens up a wide avenue of research for a better understanding the role of SSRP1 in DNA repair mechanisms.
Supplementary Material
ACKNOWLEDGMENTS
We thank Thomas Helleday for providing us with the SPD8 and SW480SN.3 cell lines and the I-SceI expression plasmid. We would also like to thank Mary MacPartlin and Irina G. Minko for proofreading the manuscript, Maureen Hoatlin, Alexandra Sobeck, and Igor Landais for active discussion on this work.
Grant sponsor: Hua Lu; Grant numbers: CA095441, CA93614, CA079721; Grant sponsor: Alexander Mazin; Grant number: CA100839.
Abbreviations used:
- 6-TG
6-thioguanine
- BM
branch migration
- DSB
double-strand break
- FACT
facilitates chromatin transcription
- HAsT
hypoxanthine-l-azaserine-thymidine
- hprt
hypoxanthine guanine phosphoribosyl transferase
- HR
homologous recombination
- HJs
Holliday junctions
- HMG
high mobility group
- HU
hydroxyurea
- NHEJ
non-homologous end joining
- SSRP1
structure-specific recognition protein 1
- UV
ultraviolet
Footnotes
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- Alexeev A, Mazin A, Kowalczykowski SC. 2003. Rad54 protein possesses chromatin-remodeling activity stimulated by the Rad51-ssDNA nucleoprotein filament. Nat Struct Biol 10:182–186. [DOI] [PubMed] [Google Scholar]
- Amitani I, Baskin RJ, Kowalczykowski SC. 2006. Visualization of Rad54, a chromatin remodeling protein, translocating on single DNA molecules. Mol Cell 23:143–148. [DOI] [PubMed] [Google Scholar]
- Arnaudeau C, Lundin C, Helleday T. 2001. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol 307:1235–1245. [DOI] [PubMed] [Google Scholar]
- Bezzubova O, Silbergleit A, Yamaguchi-Iwai Y, Takeda S, Buerstedde JM. 1997. Reduced X-ray resistance and homologous recombination frequencies in a RAD54−/− mutant of the chicken DT40 cell line. Cell 89:185–193. [DOI] [PubMed] [Google Scholar]
- Bianco PR, Tracy RB, Kowalczykowski SC. 1998. DNA strand exchange proteins: A biochemical and physical comparison. Front Biosci 3:D570–D603. [DOI] [PubMed] [Google Scholar]
- Bruhn SL, Pil PM, Essigmann JM, Housman DE, Lippard SJ. 1992. Isolation and characterization of human cDNA clones encoding a high mobility group box protein that recognizes structural distortions to DNA caused by binding of the anticancer agent cisplatin. Proc Natl Acad Sci USA 89:2307–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugreev DV, Mazina OM, Mazin AV. 2006a. Analysis of branch migration activities of proteins using synthetic DNA substrates. Nat Protoc 217. DOI: 2010.1038/nprot.2006.2217. [Google Scholar]
- Bugreev DV, Mazina OM, Mazin AV. 2006b. Rad54 protein promotes branch migration of Holliday junctions. Nature 442:590–593. [DOI] [PubMed] [Google Scholar]
- Clever B, Interthal H, Schmuckli-Maurer J, King J, Sigrist M, Heyer WD. 1997. Recombinational repair in yeast: Functional interactions between Rad51 and Rad54 proteins. EMBO J 16:2535–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacote F, Han M, Stamato TD, Jasin M, Lopez BS. 2002. An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res 30:3454–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariglio M, Ying GG, Hertel L, Gaboli M, Clerc RG, Landolfo S. 1997. The high-mobility group protein T160 binds to both linear and cruciform DNA and mediates DNA bending as determined by ring closure. Exp Cell Res 236:472–481. [DOI] [PubMed] [Google Scholar]
- Helleday T, Arnaudeau C, Jenssen D. 1998. A partial hprt gene duplication generated by non-homologous recombination in V79 Chinese hamster cells is eliminated by homologous recombination. J Mol Biol 279:687–694. [DOI] [PubMed] [Google Scholar]
- Hertel L, De Andrea M, Bellomo G, Santoro P, Landolfo S, Gariglio M. 1999. The HMG protein T160 colocalizes with DNA replication foci and is down-regulated during cell differentiation. Exp Cell Res 250:313–328. [DOI] [PubMed] [Google Scholar]
- Heyer WD, Li X, Rolfsmeier M, Zhang XP. 2006. Rad54: The Swiss Army knife of homologous recombination? Nucleic Acids Res 34:4115–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JY, Chen WH, Chang YL, Wang HT, Chuang WT, Lee SC. 2006. Modulation of nucleosome-binding activity of FACT by poly(ADP-ribosyl)ation. Nucleic Acids Res 34:2398–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrien O 2000. Mechanisms and consequences of replication fork arrest. Biochimie 82:5–17. [DOI] [PubMed] [Google Scholar]
- Johnson RD, Jasin M. 2000. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 19:3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai N, Tsunaka Y, Ohki I, Hirose S, Morikawa K, Tate S. 2005. Solution structure of the HMG-box domain in the SSRP1 subunit of FACT. J Biomol NMR 32:83–88. [DOI] [PubMed] [Google Scholar]
- Keller DM, Lu H. 2002. p53 serine 392 phosphorylation increases after UV through induction of the assembly of the CK2.hSPT16.SSRP1 complex. J Biol Chem 277:50206–50213. [DOI] [PubMed] [Google Scholar]
- Keller DM, Zeng X, Wang Y, Zhang QH, Kapoor M, Shu H, Goodman R, Lozano G, Zhao Y, Lu H. 2001. A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol Cell 7:283–292. [DOI] [PubMed] [Google Scholar]
- Krogh BO, Symington LS. 2004. Recombination proteins in yeast. Annu Rev Genet 38:233–271. [DOI] [PubMed] [Google Scholar]
- Landais I, Lee H, Lu H. 2006. Coupling caspase cleavage and ubiquitin-proteasome-dependent degradation of SSRP1 during apoptosis. Cell Death Differ 13:1866–1878. [DOI] [PubMed] [Google Scholar]
- Li X, Heyer WD. 2008. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 18:99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Keller DM, Scott JD, Lu H. 2005. CK2 phosphorylates SSRP1 and inhibits its DNA-binding activity. J Biol Chem 280:11869–11875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zeng SX, Landais I, Lu H. 2007. Human SSRP1 has Spt16-dependent and -independent roles in gene transcription. J Biol Chem 282:6936–6945. [DOI] [PubMed] [Google Scholar]
- Lundin C, Erixon K, Arnaudeau C, Schultz N, Jenssen D, Meuth M, Helleday T. 2002. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol Cell Biol 22:5869–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin C, Schultz N, Arnaudeau C, Mohindra A, Hansen LT, Helleday T. 2003. RAD51 is involved in repair of damage associated with DNA replication in mammalian cells. J Mol Biol 328:521–535. [DOI] [PubMed] [Google Scholar]
- Mazin AV, Bornarth CJ, Solinger JA, Heyer WD, Kowalczykowski SC. 2000. Rad54 protein is targeted to pairing loci by the Rad51 nucleoprotein filament. Mol Cell 6:583–592. [DOI] [PubMed] [Google Scholar]
- Mazin AV, Alexeev AA, Kowalczykowski SC. 2003. A novel function of Rad54 protein. Stabilization of the Rad51 nucleoprotein filament. J Biol Chem 278:14029–14036. [DOI] [PubMed] [Google Scholar]
- Mazina OM, Mazin AV. 2004. Human Rad54 protein stimulates DNA strand exchange activity of hRad51 protein in the presence of Ca2+. J Biol Chem 279:52042–52051. [DOI] [PubMed] [Google Scholar]
- Mohindra A, Hays LE, Phillips EN, Preston BD, Helleday T, Meuth M. 2002. Defects in homologous recombination repair in mismatch-repair-deficient tumour cell lines. Hum Mol Genet 11:2189–2200. [DOI] [PubMed] [Google Scholar]
- Okuhara K, Ohta K, Seo H, Shioda M, Yamada T, Tanaka Y, Dohmae N, Seyama Y, Shibata T, Murofushi H. 1999. A DNA unwinding factor involved in DNA replication in cell-free extracts of Xenopus eggs. Curr Biol 9:341–350. [DOI] [PubMed] [Google Scholar]
- Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D. 1999. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400:284–288. [DOI] [PubMed] [Google Scholar]
- Osman F, Adriance M, McCready S. 2000. The genetic control of spontaneous and UV-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Curr Genet 38:113–125. [DOI] [PubMed] [Google Scholar]
- Petukhova G, Stratton S, Sung P. 1998. Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature 393:91–94. [DOI] [PubMed] [Google Scholar]
- Schlesinger MB, Formosa T. 2000. POB3 is required for both transcription and replication in the yeast Saccharomyces cerevisiae. Genetics 155:1593–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakata M, Huppi K, Usuda S, Okazaki K, Yoshida K, Sakano H. 1991. HMG1-related DNA-binding protein isolated with V-(D)-J recombination signal probes. Mol Cell Biol 11:4528–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinger JA, Kiianitsa K, Heyer WD. 2002. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol Cell 10:1175–1188. [DOI] [PubMed] [Google Scholar]
- Stros M, Muselikova E. 2000. A role of basic residues and the putative intercalating phenylalanine of the HMG-1 box B in DNA supercoiling and binding to four-way DNA junctions. J Biol Chem 275:35699–35707. [DOI] [PubMed] [Google Scholar]
- Stros M, Cherny D, Jovin TM. 2000. HMG1 protein stimulates DNA end joining by promoting association of DNA molecules via their ends. Eur J Biochem 267:4088–4097. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Wang X, Haber JE. 2003. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol Cell 12:209–219. [DOI] [PubMed] [Google Scholar]
- Tan BC, Chien CT, Hirose S, Lee SC. 2006. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J 25:3975–3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro S, Kotomura N, Shinohara A, Tanaka K, Ueda K, Kamada N. 1996. S phase specific formation of the human Rad51 protein nuclear foci in lymphocytes. Oncogene 12:2165–2170. [PubMed] [Google Scholar]
- Thoma NH, Czyzewski BK, Alexeev AA, Mazin AV, Kowalczykowski SC, Pavletich NP. 2005. Structure of the SWI2/SNF2 chromatin-remodeling domain of eukaryotic Rad54. Nat Struct Mol Biol 12:350–356. [DOI] [PubMed] [Google Scholar]
- Thompson LH, Schild D. 2001. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat Res 477:131–153. [DOI] [PubMed] [Google Scholar]
- Van Komen S, Petukhova G, Sigurdsson S, Stratton S, Sung P. 2000. Superhelicity-driven homologous DNA pairing by yeast recombination factors Rad51 and Rad54. Mol Cell 6:563–572. [DOI] [PubMed] [Google Scholar]
- VanDemark AP, Blanksma M, Ferris E, Heroux A, Hill CP, Formosa T. 2006. The structure of the yFACT Pob3-M domain, its interaction with the DNA replication factor RPA, and a potential role in nucleosome deposition. Mol Cell 22:363–374. [DOI] [PubMed] [Google Scholar]
- Wesoly J, Agarwal S, Sigurdsson S, Bussen W, Van Komen S, Qin J, van Steeg H, van Benthem J, Wassenaar E, Baarends WM, Ghazvini M, Tafel AA, Heath H, Galjart N, Essers J, Grootegoed JA, Arnheim N, Bezzubova O, Buerstedde JM, Sung P, Kanaar R. 2006. Differential contributions of mammalian Rad54 paralogs to recombination, DNA damage repair, and meiosis. Mol Cell Biol 26:976–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmeyer J, Formosa T. 1997. The Saccharomyces cerevisiae DNA polymerase alpha catalytic subunit interacts with Cdc68/Spt16 and with Pob3, a protein similar to an HMG1-like protein. Mol Cell Biol 17:4178–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolner B, van Komen S, Sung P, Peterson CL. 2003. Recruitment of the recombinational repair machinery to a DNA double-strand break in yeast. Mol Cell 12:221–232. [DOI] [PubMed] [Google Scholar]
- Yarnell AT, Oh S, Reinberg D, Lippard SJ. 2001. Interaction of FACT, SSRP1, and the high mobility group (HMG) domain of SSRP1 with DNA damaged by the anticancer drug cisplatin. J Biol Chem 276:25736–25741. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Fan HY, Goldman JA, Kingston RE. 2007. Homology-driven chromatin remodeling by human RAD54. Nat Struct Mol Biol 14:397–405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.