

Abstract
Single molecule FRET (smFRET) measurements are uniquely sensitive to protein conformational dynamics, allowing for a parallel investigation of conformations, and transitions across these conformations. Here we describe the smFRET methods used to study the conformational landscape of the ionotropic glutamate receptors (iGluRs) associated with gating, allosteric inhibition, and modulation with small molecule ligands and auxiliary proteins. The extensive single channel functional data of the iGluRs shows the presence of multiple closed and open states necessitating the investigations of the conformational landscape associated with these by single molecule methods such as smFRET. In this chapter, we will describe the smFRET method, tools, and protocols utilized for these investigations.
Keywords: smFRET, ionotropic glutamate receptors, conformational landscape, fluorophores, efficiency histograms
Background
Introduction to single molecule FRET
Förster Resonance Energy Transfer (FRET) serves as a biological ruler to measure the distance between fluorophore pairs which are covalently attached to the site of interest within a biomolecule (Stryer & Haugland, 1967). The power of FRET to study conformational dynamics was significantly enhanced when the molecules could be studied at the single molecule level (Ha et al., 1996). Using FRET at the single molecule mode (smFRET) allows us to perform measurements beyond ensemble averages and assess the diverse heterogenous conformations that biomolecules can populate (Ha et al., 1996) . Additionally, the ability to study protein conformation as a function of time in smFRET allows for simultaneous investigations of the conformational transitions and kinetics between conformations (Cooper et al., 2015; Lerner et al., 2018), which is otherwise not possible in several of the conventional methods of structural investigations. smFRET has been reported in research of fundamental biological processes such as DNA replication, transcription, translation, RNA folding, and ion channels by elucidating sub-millisecond dynamics in these processes (Cooper et al., 2015; MacLean, Durham, & Jayaraman, 2019; Stracy, Uphoff, Garza de Leon, & Kapanidis, 2014; Zhao & Rueda, 2009). In this chapter, we focus on the methodology that we have adapted for performing smFRET measurements on ionotropic glutamate receptors discussing strategies for labeling, protocols for sample preparation, data acquisition and analysis.
Ionotropic Glutamate Receptors (iGluRs)
Ionotropic glutamate receptors (iGluRs) belong to the family of ligand-gated ion channels and are the key mediators of excitatory synaptic transmission in the central nervous system. These receptors play a crucial role in brain development and function by maintaining synaptic plasticity, which is involved in learning and memory formation (Traynelis et al., 2010). The dysfunction of these receptors is implicated in a number of neurological conditions such as epilepsy, seizures, and ischemic stroke (Lai, Zhang, & Wang, 2014; Traynelis et al., 2010). Given their physiological and pathological relevance, it is extremely important to expand our knowledge of receptor activation and modulation. This will give us a better understanding of the structure-function relationship of these receptors for therapeutic applications. AMPA (α-amino-5-methyl-3-hydroxy-4-isoxazole propionate), NMDA (N-methyl-D-aspartate), and kainate receptors are members of the iGluRs and are named based on their selective activation by synthetic agonists. The wealth of structures available for iGluRs through X-ray crystallography and cryo-electron microscopy depicts that these receptors share a common topology. They form large homomeric and heteromeric tetramer complexes depending on their subunit composition. Moreover, each subunit consists of multiple domains. The domains include an extracellular amino-terminal domain (ATD), an extracellular agonist-binding domain (ABD), a transmembrane domain, and an unresolved intracellular carboxy-terminal domain (CTD). The extracellular domains and transmembrane domains are connected to each other through linkers that allow interdomain communication important in channel gating (Sobolevsky, Rosconi, & Gouaux, 2009). During the gating process, the largest conformational changes occur in the extracellular region. Given the excellent foundation of these structures, smFRET allows the investigation of the complete conformational landscape across a distance known to show changes based on these structures. The conformational landscape can be determined under different conditions (in the presence of agonists, antagonists, allosteric modulators) allowing for correlation between conformation and dynamics associated with gating (Bhatia, Carrillo, Durham, Berka, & Jayaraman, 2020; Litwin, Carrillo, Shaikh, Berka, & Jayaraman, 2019; Litwin, Paudyal, Carrillo, Berka, & Jayaraman, 2020; MacLean et al., 2019), allosteric modulations with small molecule ligands (Ramaswamy et al., 2012) and auxiliary proteins (Carrillo et al., 2020), and cooperativity between ligands (Durham et al., 2020).
Experimental design for the application of smFRET on iGluRs
Labeling strategy in iGluRs
In order to address the specific question of interest, the first major hurdle for doing smFRET measurements is choosing the appropriate sites for labeling the protein of interest. The near atomic resolution structures available for these receptors offer us an advantage when selecting sites for smFRET measurement. For labeling strategy, we should first identify sites that (1) reflect the conformational changes associated with the conditions being studied, (2) have minimal cross talk with sites on other subunits (in the context of the tetrameric receptor), (3) are solvent exposed to increase the ease of labeling, (4) have minimal effect on function, and (5) be at the appropriate FRET distance for known donor-acceptor FRET pairs. The points in 1-3 can be addressed by studying the known end-state structures. Characterizing the functionality of each of the constructs and identifying ones with minimal effects is usually the most labor-intensive step. With respect to the ideal distance between the donor and acceptor sites, the relationship between FRET efficiency and distance for a given donor-acceptor pair is dictated by the overlap integral (J) between the donor emission and acceptor absorption which in turn dictates distance at half maximal FRET efficiency (R0) (Roy, Hohng, & Ha, 2008) as shown in equation 1–2.
| 1 |
| 2 |
Where, ΦD is the quantum yield of the donor, η is the refractive index of the media, κ is the orientation factor and R is the distance between the donor and acceptor sites. For smFRET measurements it is preferable to have sites that would give 50% to 90% FRET efficiency (Figure 1), as this region shows large changes in FRET efficiency associated with changes in distance and also holds adequate FRET photons.
Figure 1.
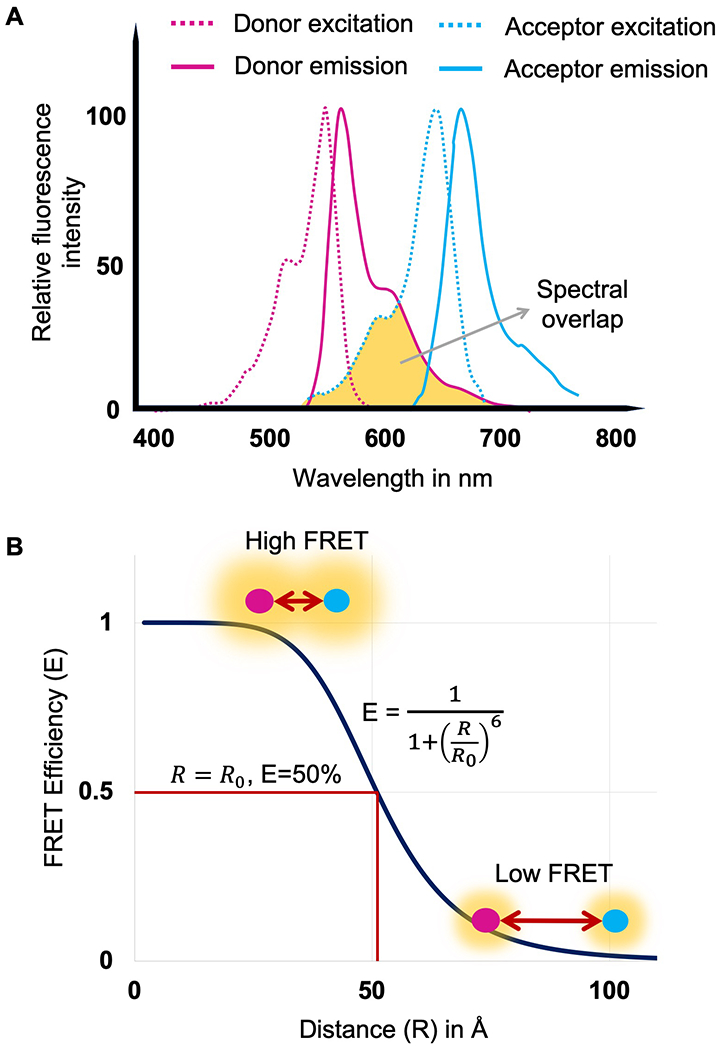
Panel A. Spectral profile of donor (Alexa 555) and acceptor (Alexa 647). Spectral overlap region is shown in yellow and lies between donor emission (solid magenta) and acceptor excitation (dotted blue). Panel B. FRET efficiency as a function of the distance between donor and acceptor fluorophores (R). In the given equation, R0 represents the Forster’s distance at which the FRET efficiency (E) is 50%. Forster’s distance value for Alexa 555 and Alexa 647 is 51Å.
The donor and acceptor fluorophores selected for labeling should have similar high quantum yield value, high photostability, and sufficient spectral separation between donor emission and acceptor excitation (Roy et al., 2008). There are several general schemes available for site-specific labeling of proteins with fluorophore pairs. Given that iGluRs do not have a large number of extracellular accessible cysteines (1-5 in the different subtypes), we have used cysteines as sites for attaching the donor and acceptor fluorophores. In order to introduce cysteines at specific sites, the solvent accessible non-disulfide bonded extracellular cysteines are mutated to serine. This background construct is tested to ensure there is no fluorescence when tagged with thiol reactive fluorophores (e.g. maleimide dyes) and its functionality verified using electrophysiological measurements. Then cysteines are introduced at sites identified for investigating specific conformational changes as discussed above. This construct is then labeled with thiol reactive fluorophores and functionality verified prior to any smFRET investigations. An alternate strategy for attaching fluorophores at specific sites is the use of unnatural amino acids which can be introduced with specific chemistries that are different from the canonical amino acids (Ye et al., 2008). This has the advantage of not requiring additional mutations (Dolino et al., 2015). Figure 2 shows sites in iGluRs that we have used for Alexa 555 maleimide dye as a donor and Alexa 647 maleimide dye as an acceptor that satisfy the constraints listed above and are optimal for smFRET experiments. In the case of homomeric structure of iGluRs, the large size of the extracellular domains allows for minimal cross-talk. Heteromeric receptors such as the NMDA subtype of glutamate receptors as well as the heteromeric GluK2/GluK5 kainate receptors have the advantage of being coded by two different polypeptides and hence have a large number of possible sites that can be studied, such as those close to the transmembrane segments with minimal cross-talk (Figure 2).
Figure 2.
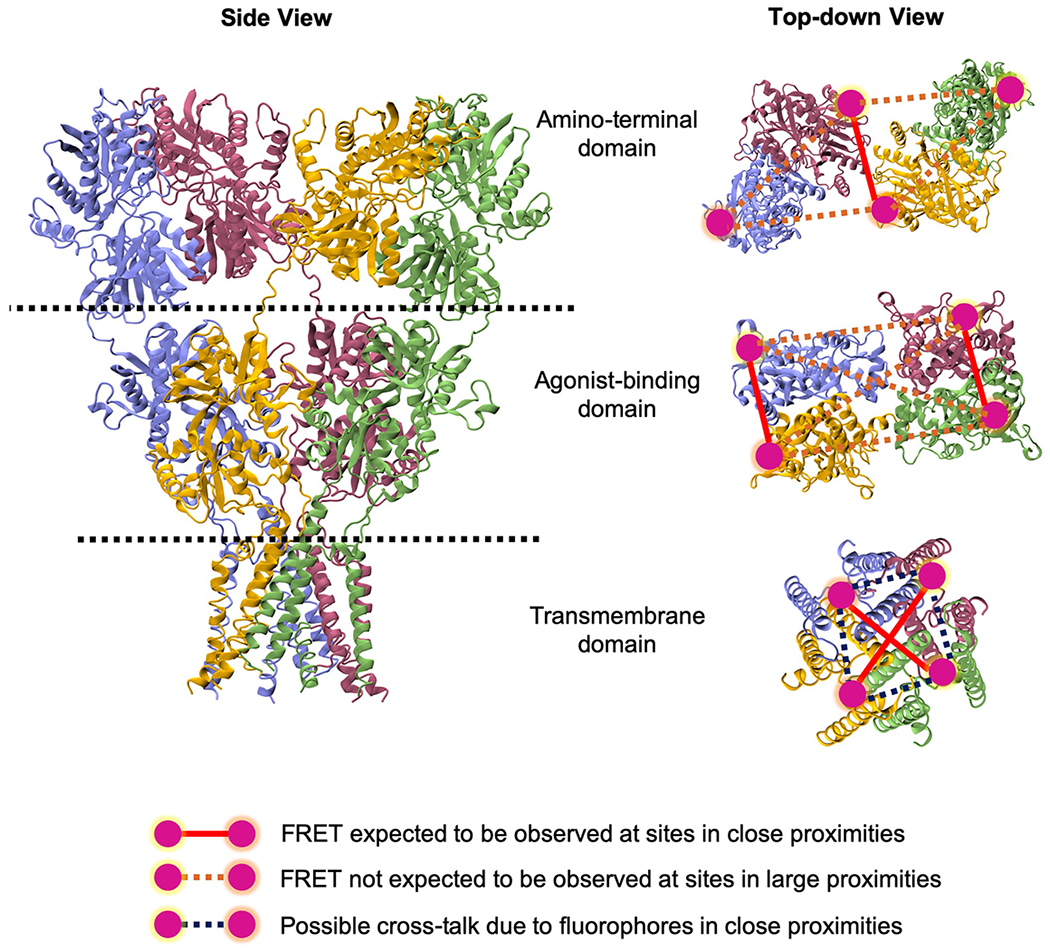
Side view of an iGluR tetramer showing subunit and domain arrangement in left panel. The right panel shows the top-down view of amino-terminal domain, agonist-binding domain and transmembrane domain depicting the sites chosen for labeling for smFRET experiments. In a multidomain protein like iGluR, one domain is labeled at a time at sites highlighted in magenta to study the conformational changes induced in that particular region of a full-length receptor. Structure image was created using Visual Molecular Dynamics (VMD) software.
Immobilization of labeled protein onto microscope slides:
smFRET can be performed either in freely diffusing molecules or using protein attached to a glass slide. For smFRET on the full-length iGluRs, we have used the slide attached mode described here. iGluRs are pulled down on to the glass side using either antibodies specific with an epitope far from the site of investigation to the glutamate receptor being investigated, or by streptavidin pull-down to a streptavidin tag introduced in the receptor far from the site being investigated. In general, the pull-down method for a single molecule detection involves surface passivation and sample immobilization (Aggarwal & Ha, 2014; Litwin, Durham, & Jayaraman, 2019; Roy et al., 2008). The passivation of the surface involves multiple steps, a) ultra-cleaning of slides, b) aminosilanization, and c) PEGylation. First, the slides are cleaned with soap solution using an ultra-bath sonicator to remove any dirt particulate matter. The slides are further cleaned with Tl-1 solution (mixture of ammonium hydroxide and hydrogen peroxide), a strong oxidizing solution, to remove the organic contaminants and hydroxylate the slides to achieve a hydrophilic glass surface. Then, plasma cleaning of slides is done by exposing slides on to a highly energetic oxygen plasma. The high energy results in breaking the organic bonds of the surface contaminants and the reactive oxygen species, which are generated in oxygen plasma, further react with the organic impurities to form gaseous products which are then flushed out of the plasma cleaning chamber by continuous gas flow. The ultra-clean slides are obtained and are treated for aminosilanization. During this process, the slides are treated with 3-aminopropyltriethoxysilane (Vectabond TM reagent) and functionalized with amine group. These slides are further conjugated with the ester group of PEG-N-Hydroxysuccinimide (NHS) esters through multiple PEG treatment schemes, detailed next. First, overnight treatment of slides with mixture of biotin-PEG-NHS (NHS-PEG4-Biotin) and regular PEG (mPEG-succinimidyl carbonate: mPEG-SC) is done. Then, the slides are treated with short chain methyl-PEG NHS ester on the day of experiment for 2-3hrs. Once the slides are passivated with PEG, for sample immobilization, streptavidin is applied to the slides such that it anchors to the biotin group attached to PEG. The streptavidin molecule has four biotin-binding sites and, therefore, it is ideal for selectively pulling down biotinylated antibodies specific to the protein of interest or twinstrep-tagged protein by forming a biotinPEG-Streptavidin-biotin-protein sandwich. Thus, the overall strategy applied in the pull-down method aids us with enhanced specificity and increased resolution, making the analysis easier.
BEFORE YOU BEGIN
Chemical reagents
Protein expression
HEK 293T cells cultured at 37°C and 5% CO2
10% Fetal Bovine Serum (FBS) (no sodium pyruvate), 1unit/mL Penicillin/ and 1ng/mL Streptomycin (P/S) added to Dulbecco’s Modified Eagle’s Medium (DMEM) (GenDepot)
Trypsin
JetPRIME reagent (Polyplus), JetPRIME buffer
Protein purification and labeling
Extracellular Buffer ECB (135mM NaCl, 3mM KCl, 2mM CaCl2, 20mM glucose, and 20mM HEPES, pH 7.5)
Solubilization Buffer (1% Lauryl Maltose Neopentyl Glycol, 2mM Cholesteryl Hydrogen Succinate, Protease Inhibitor in 1xPBS)
Alexa Fluor 555-C2-Maleimide (Invitrogen)
Alexa Fluor 647-C5-Maleimide (Invitrogen)
Slide Preparation
Phosphate Buffered Saline (PBS)
Methanol
Liquinox phosphate-free detergent (Fisher Scientific)
Acetone
Tl-1 solution (4.3% NH4OH and 4.3% H2O2)
Vectabond (Vector Laboratories)
Overnight polyethylene glycol (PEG) solution: 0.25% w/w NHS-PEG4-Biotin (biotinylated PEG), 25% w/w 5 kDa mPEG succinimidyl carbonate (Laysan Bio Inc) in 10mM NaHCO3 (Sigma-Aldrich)
Short-chain PEG solution: (25mM short-chain MS(PEG)4 Methyl-PEG-NHS-Ester Reagent (Thermo Scientific) in 0.1M NaHCO3 (Sigma-Aldrich)
10X Imaging Buffer (10mM nDodecyl-β-D-maltoside (Chem-Impex) detergent and 2mM Cholesteryl Hydrogen Succinate (MP Biomedicals) in 1xPBS)
Streptavidin (Invitrogen) solution (0.2mg/mL in 1xImaging buffer)
Bovine serum albumin (BSA) solution (0.1mg/mL BSA in 1xPBS)
Reactive oxygen species (ROXS) scavenging solution (3.3% w/w glucose (Sigma-Aldrich), 0.1mg/mL pyranose oxidase (Sigma-Aldrich), 0.01mg/mL catalase (Sigma-Aldrich), 1mM ascorbic acid (Sigma-Aldrich), and 1mM methyl viologen (Fisher Scientific), in 1xImaging Buffer, pH maintained 7.5)
MATERIALS AND EQUIPMENT
Eppendorf 5810R centrifuge
Beckman Ultracentrifuge
TLA 100.3 rotor
Ultrasonic Bath Sonicator
Harrick Plasma PDC-32G Plasma Cleaner
PicoQuant MicroTime 200 Fluorescence Lifetime Microscope
532nm (LDH-D-TA-530; Picoquant) and 637nm (LDH-D-C-640; Picoquant) lasers
100x objective (100x1.4 NA; Olympus)
x-y-z piezo stage (P-733.2CD)
SPAD photodiodes (SPCM CD3516H)
550nm (FF01-582/64; AHF) and 650nm (2XH690/70; AHF) emission filters
Silicone Templates (Grace Bio-Labs)
20mm x 20mm microscope cover glasses (Globe Scientific)
Adhesive Hybriwell chamber (Grace Bio-labs)
Tubing connectors (Fisher Scientific)
Origin software (OriginLab)
Matlab (MathWorks)
STEP-BY-STEP METHOD DETAILS
Sample preparation
Use the jet prime transfection protocol to transiently transfect HEK293T cells with 10μg of DNA per 10cm dish and incubate for 24-48 hours at 37°C. Use 2-3 10cm plates for transfection.
Scrape the transfected cells into 50ml conical tubes and spin down the cells in a centrifuge at 1100g at 23°C for 3 minutes. Use ECB to wash and resuspend the cell pellet, transfer it to a 15ml conical vial and spin down. Discard the supernatant and repeat the ECB washing and resuspension two more times.
- Resuspend the cell pellets in 3ml of ECB and label the cells with premixed 600nM of donor fluorophore and 2.4μM of acceptor fluorophore at room temperature for 45-60 minutes. Cover to prevent photobleaching of fluorophores.Note1: Premixing of fluorophores before protein labeling is important because it increases the probability of donor and acceptor being labeled at the site of interest. Labeling with fluorophores before mixing will increase the sites being labeled with donor only or acceptor only depending on whichever is added first.Note2: The amount of fluorophores to be used for labeling should be optimized based on the experimental condition such as labeling for homomeric protein or heteromeric protein.
Following labeling, repeat the cell wash with ECB for 2-3 times. Solubilize the cells with 2ml of solubilization buffer and nutate for 1hour at 4°C. Addition of protease inhibitor to the solubilization buffer will reduce the rapid degradation of the protein due to endogenous protease present in the HEK 293T cells.
Transfer the crude lysate to an ultracentrifuge tube and spin down for 1 hour at 100,000 × g at 4°C and use the supernatant as smFRET sample. The smFRET sample obtained should be applied to the slide immediately for data collection and preserve the remaining sample at 4°C if needed for additional slides.
Slide preparation
Use the bath sonicator to clean the silicone templates with methanol placed in a beaker for 30 minutes. Transfer the silicone templates to a conical tube containing 50ml of methanol, vortex, and store the templates by adding clean methanol to the tube.
Clean 20mm x 20mm microscope cover glasses using bath sonication in a 5% solution of Liquinox phosphate-free detergent for 10 minutes and subsequently by molecular-biology grade water. Repeat the sonication step for cover slides in the presence of acetone followed by cleaning with molecular-biology grade water.
Place the coverslips in a beaker containing Tl-1 solution and incubate at 70°C for 5 minutes. Wash the slides with purified water and dry with a gentle flow of nitrogen.
Place the slides in a metal holder and transfer it to the Harrick Plasma PDC-32G Plasma Cleaner for plasma cleaning. Operate the vacuum pump and wait for the chamber pressure to drop to below 200mTorr. Use oxygen to flush the chamber 3 times and maintain the pressure stability below 200mTorr each time after flushing. Again, perform the plasma cleaning of slides for 2 minutes, release the vacuum, and get the slides.
- Prepare the solution of Vectabond and acetone in a ratio of 1:40 by volume on the day of use and submerge the slides in the solution for 5 minutes. This is the step for aminosilanization. Then dry the slides with a gentle flow of nitrogen and store under vacuum.Note: A new Vectabond bottle once open, can be used for two weeks and must be handled and stored in nitrogen.
A day before each experiment, clean the silicone templates stored in methanol with a nitrogen flow and place it on a slide with the oval area of the template placed at the center of the slide. Add 50μl of overnight PEG solution to the oval area, put the slides on a damp delicate task wipe placed inside a petridish, and incubate overnight in a dark space at room temperature.
The day of experiment, clean the overnight PEG treated slides with water and dry them with nitrogen. Apply 50μl of short-chain PEG solution and incubate for 2-3 hours at room temperature.
Wash the slides with purified water and dry with nitrogen. Mark the position of the oval area at the back of the slide and remove the silicone templates. Apply adhesive Hybriwell chamber to the slide and two press fit tubing connectors at the ports of each chamber.
Prepare streptavidin solution and inject 36μl of the solution through the port into the slide. Incubate the slides for 10 minutes and wash with 60μl of 1xPBS three times.
- For proteins which require selective pull-down using antibodies, apply 60μl of secondary antibody to the slide twice and incubate at 4°C for 20 minutes. Wash with 60μl of 1xPBS two times. Next, apply 60μl of primary antibody to the slides for 2 times and incubate at 4°C for 20 minutes. Repeat the PBS wash two times and add 60μl of BSA solution following 20 minutes of incubation at 4°C. Finally wash the slides with 1xPBS twice. For protein using twin-strep tag pulldown, this step is skipped.Note: Use only sample specific antibody for effective pull-down and avoid nonspecific binding. The binding specificity of antibody to the sample can be checked using western blot
- Use the protein sample directly or dilute sample in a 1:2 to 1:5 proportion with 1X Imaging Buffer, depending on the protein expression. Apply 90μl of protein sample to the slide twice and incubate it at 4°C for 20 minutes. Wash the slide with 60μl of ROXS through the chamber three times.Note: While injecting solution to the slides through connectors, one should make sure that no bubble is trapped in the slide because the presence of bubbles makes for inconsistent labeling of the slides.
The slide is now ready for imaging.
Microscope Setup and Data Collection
A PicoQuant MicroTime 200 Confocal Fluorescence Lifetime Microscope is used with pulsed interleaved excitation (PIE) set at 80MHz pulse rate to excite the fluorophores through 532nm and 637nm lasers for data collection. The slide positioned with a x-y-z piezo scanning stage is observed through an oil-immersed 100x objective lens. The photons emitted through the sample travel back to the objective lens and pass through two 550nm and 650nm emission filters before being detected by two SPAD photodiodes. The emission filters facilitate the visualization of the donor and acceptor channels.
The photostability of fluorophores contributes to the quality of smFRET data. Therefore, during smFRET imaging and data collection the sample slide is flushed with a special imaging buffer, ROXS (reducing and oxidizing system) buffer, to enhance the photostability of fluorophores by minimizing the photobleaching and blinking phenomena (Vogelsang et al., 2008). This buffer is comprised of an oxygen scavenger system (glucose, pyranose oxidase and catalase) and triplet state quenchers, including both an oxidizing agent (methyl viologen) and a reducing agent (ascorbic acid). The oxygen scavenging system removes the oxygen from the solution thus minimizing the photobleaching caused by the excited state reactive oxygen species. The triplet state quenchers (oxidizing and reducing agent) generate radical ions from the triplet state of the fluorophore by electron transfer. After the formation of radical ions, another oxidation (of radical anion) or reduction (radical cation) leads to the recovery of the singlet-ground state fluorophore. The depletion of the triplet state keeps a check on the blinking of the fluorophores.
Scan a 20μm x 20μm area of the sample slide using the confocal microscope for identifying single molecules used for data collection. Check the focus of the microscope prior to imaging
For the molecules selected, record their fluorescence intensity, and only keep those molecules that exhibit a single acceptor and single donor photo-bleaching step, as well as anti-correlation between donor and acceptor.
To confirm the FRET phenomenon observed at the site of interest, perform a controlled experiment with no protein samples, proteins with a single labeled site and proteins with two labeled sites.
- Collect the emission intensities at 1ms resolution.Note: Data can be binned to improve signal to noise ratio at the cost of temporal resolution.
QUANTIFICATION AND STATISTICAL ANALYSIS
Only those molecules that show a single photobleaching step between acceptor and donor as well as an anticorrelation with each other are subjected to further analysis (Figure 3D). The actual FRET efficiencies are then calculated from the donor and acceptor fluorescence intensities from at least 45-50 selected molecules. Further processing of the FRET efficiency traces is required to denoise the raw data and estimate the number of FRET states. First, the data is exported as ASCII files and these files are then used for data processing. Data processing is done in MATLAB software using codes developed by the Landes Research Group (website at http://www.lrg.rice.edu/Content.aspx?id=96) for denoising and step transition and state identification (STASI) (Shuang et al., 2014).
Figure 3.
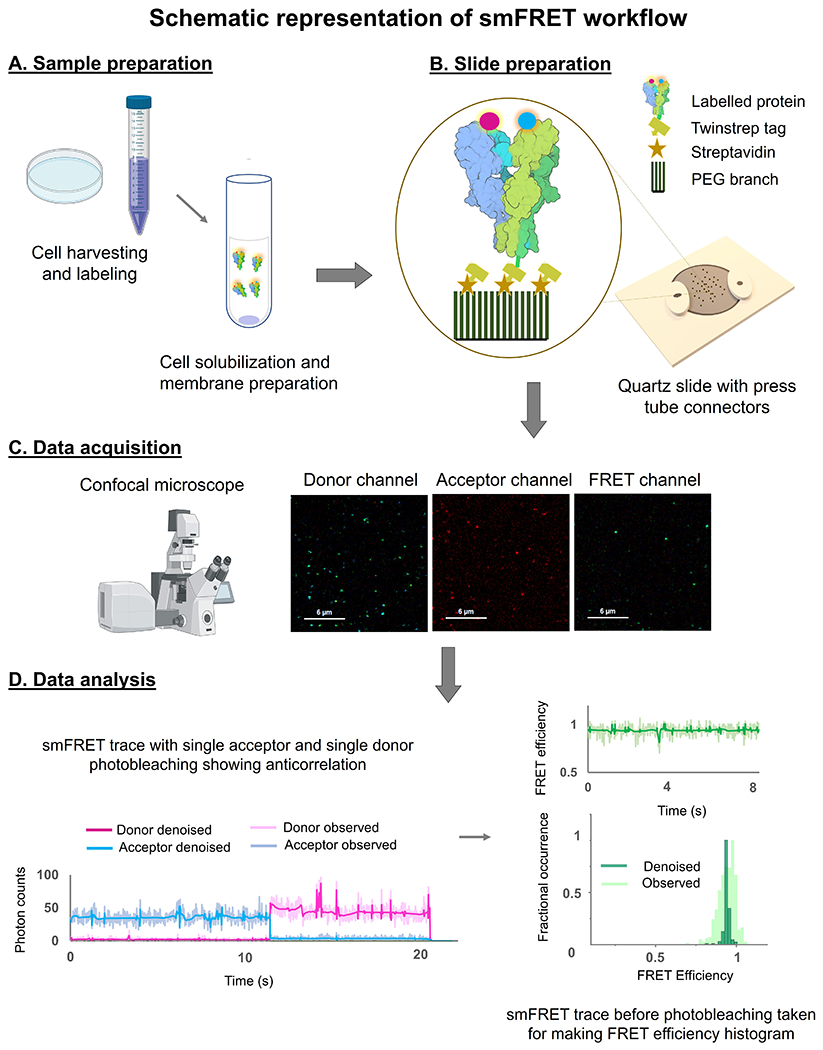
Schematic outline of the smFRET protocol involving A) sample preparation, B) slide preparation, C) data acquisition, and D) data analysis.
Denoising
Denoising of raw FRET data is a critical step in allowing us to identify transitions in the smFRET traces. While there are several methods that can be used to denoise smFRET signals to process the single-molecule data and separate the actual signal from the experimental noise, we use wavelet decomposition method (Taylor, Makarov, & Landes, 2010). Wavelet transform is a mathematical transformation that decomposes the time domain signal into scaling and wavelet functions. This denoising method involves three main steps, 1) Constructing a mathematical function (basis set) to represent the signal in the wavelet domain 2) Setting up a threshold for the coefficients corresponding to the wavelet function to suppress the contribution of quantifiable noise components and 3) Inverting the wavelet transform to obtain denoised time-domain signal. Landes et al also demonstrated the effect of various smFRET specific modifications in the wavelet decomposition method using different combinations of wavelet basis, shrinkage method, noise estimator, and cycle spun wavelet transform (Taylor & Landes, 2011). This group has also provided a complete MATLAB package for accurately denoising the smFRET data and the package is available on the webpage: www.lrg.rice.edu.
Estimation of conformational states
Various methods are available to estimate the number of states from smFRET traces such as Hidden Markov modelling (Andrec, Levy, & Talaga, 2003; McKinney, Joo, & Ha, 2006), change point analysis (Terentyeva et al., 2012; Watkins & Yang, 2005), and STaSI (Cooper et al., 2015; Dolino et al., 2017; Durham et al., 2020; Shuang et al., 2014). The STaSI method developed by Landes’ group has an advantage over other methods as it is an unbiased approach which does not require the prior knowledge of a number of expected states. Moreover, this method can be used on binned data. StaSI first breaks down the smFRET trace into different segments based on the number of transition points and applies student t-test on each identified segment until no further step transitions are detected. After this, StaSI performs a number of iterations on the final output from the step identification process to group the similar states into one state. Minimum descriptive length (MDL) function is then used to determine the optimum number of states. For this, MDL function first measures the goodness of fit and complexity of fit for the model. Then, after multiple iterations, the optimal number of states are identified and corresponds to the global minimum of MDL value. Gaussian curve fitting of raw smFRET histograms using Origin Software can also be performed in addition to StaSI for validating the number of estimated states.
ADVANTAGES
A major advantage of single molecule FRET is the ability of bypassing ensemble averaging. The high sensitivity nature of this technique enables the detection of the conformational landscape of a large complex biomolecule even at low sample concentrations. Despite the probability of background noise, it can detect site specific changes of a biomolecule. This technique also has the ability to measure both intramolecular and intermolecular distances. Kinetic information of the system can be extracted from the dwell time of a biomolecule in its conformational state and allow us to identify the dynamic equilibrium state. This method requires minimal sample purification technique and can be used with a wide range of varying solution conditions. The versatility of this tool and reasonable cost in assembling of this technique makes it prominent for studying several biophysical applications.
LIMITATIONS
Some of the limitations of smFRET techniques are
The fluorophores are larger than the side chains of the amino acids and distances measured are between the fluorophores, hence in order to use absolute distances one must take this into consideration.
κ the orientation factor is assumed to the 2/3, however this error can be addressed by determining the anistropy for the fluorophores attached to the site on the protein.
Fluorophore photobleaching limits the time for observation.
We have found the lowest time resolution for the technique outlined here is 5 milliseconds (limited by the number of photons collected and desired signal to noise ratio).
OPTIMIZATION AND TROUBLESHOOTING
Since cysteine mutations are introduced in the protein for fluorescence labeling, optimal conditions for expression should be determined by using western blotting.
Length of time for labeling with donor and acceptor fluorophores depends on the site being labeled. This can be optimized by looking at the donor and acceptor signals at varying length of labeling time.
Donor and acceptor spots are observed, however there is no FRET. This could imply that the distance based on the structures is not accurate and that the fluorophores are further apart. To determine if this is the reason the fluorophores with higher R0 can be tried.
Summary
The advent of the smFRET technique has enabled the understanding of structural dynamics of a complex biological macromolecule such as glutamate receptor ion channels on a sub-millisecond timescale. Its unique ability to perform measurements on a single molecule level beyond classical ensemble averages has enhanced our knowledge entailing state transitions, energetics involved during switching, subunits arrangement, allosteric communication between subunits, and factors influencing gating mechanism in iGluRs. The minimal protein purification strategy used for smFRET measurements has allowed the studying of biomolecule dynamics in near-native state and with better precision. However, one should take careful considerations during fluorophore selection, data acquisition, and data analysis to reduce the potential artifacts. In due course, if the smFRET-guided measurements are utilized with computational simulations of biomolecules, it will open the platform to investigate the biomolecule dynamics with much better resolution and with greater understanding.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals | ||
| Dulbecco’s Modified Eagle’s Medium (DMEM) | GenDepot, Barker, TX | Cat# CM001-050 |
| Fetal Bovine Serum (FBS) | GenDepot | Cat# F0900-050 |
| Penicillin-Streptomycin (P/S) | Sigma-Aldrich, St. Louis, MO | Cat# P0781 |
| jetPRIME reagent | Polyplus, Berkeley, CA | Cat# 55-133 |
| Alexa 555-C2- maleimide | Invitrogen, Carlsbad, CA | Cat# A20346 |
| Alexa 647-C2-maleimide | Invitrogen, Carlsbad, CA | Cat# A20347 |
| Lauryl Maltose Neopentyl Glycol | Anatrace, Maumee, OH | Cat# NG310 |
| n-Dodecyl-β-D-maltoside | Chem-Impex Int’l Inc., Wood Dale, IL | Cat# 21950 |
| Cholesteryl Hydrogen Succinate | MP Biomedicals, Solon, OH | Cat# 101379 |
| Pierce Protease Inhibitor Mini Tablets | ThermoFisher Scientific, Waltham, MA | Cat# A32953 |
| Liquinox phosphate-free detergent | Fisher Scientific | Cat# NC9906065 |
| Vectabond | Vector Laboratories, Burlingame, CA | Cat# SP-1800 |
| Biotin-terminated Polyethylene glycol (PEG) | NOF Corp., Tokyo, Japan | Cat# BI-050TS |
| mPEG succinimidyl carbonate | Laysan Bio Inc., Arab, AL | Cat# MPEG-SC-5000 |
| Sodium bicarbonate | Sigma-Aldrich | Cat# S6014 |
| MS(PEG)4 Methyl-PEG-NHS-Ester Reagent | Thermo Scientific | Cat # 22341 |
| Streptavidin | Invitrogen | Cat# 434301 |
| Glucose | Sigma-Aldrich | Cat# G8270 |
| Pyranose oxidase | Sigma-Aldrich | Cat# P4234 |
| Catalase | Sigma-Aldrich | Cat# C1345 |
| Ascorbic acid | Sigma-Aldrich | Cat# A7506 |
| Methyl viologen | Fisher Scientific | Cat# AC227320010 |
| Experimental Models: Cell Lines | ||
| HEK 293T cells | ATCC, Manassas VA | Cat# 3216 |
| Recombinant DNA | ||
| pcDNA3.1 | Invitrogen | Cat# V790-20 |
| Software and Algorithms | ||
| Origin Software | Origin Lab | |
| Matlab | MathWorks | |
Acknowledgements
We would like to extend our thanks to Cuauhtémoc U Gonzalez for providing his valuable suggestions and feedback during manuscript preparation. This study was supported by the Houston Area Molecular Biophysics Program Grant No. T32GM00008280-32 to NP and R35GM122528 to VJ. Visual Molecular Dynamics (VMD) software was used to create structure image in Figure 2. BioRender was used to create Figure 3.
REFRENCES
- Aggarwal V, & Ha T, (2014). Single-molecule pull-down (SiMPull) for new-age biochemistry: methodology and biochemical applications of single-molecule pull-down (SiMPull) for probing biomolecular interactions in crude cell extracts. Bioessays, 36(11), 1109–1119. doi: 10.1002/bies.201400090 [DOI] [PubMed] [Google Scholar]
- Andrec M, Levy RM, & Talaga DS, (2003). Direct Determination of Kinetic Rates from Single-Molecule Photon Arrival Trajectories Using Hidden Markov Models. J Phys Chem A, 107(38), 7454–7464. doi: 10.1021/jp035514+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia NK, Carrillo E, Durham RJ, Berka V, & Jayaraman V, (2020). Allosteric Changes in the NMDA Receptor Associated with Calcium-Dependent Inactivation. Biophys J. doi: 10.1016/j.bpj.2020.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo E, Shaikh SA, Berka V, Durham RJ, Litwin DB, Lee G, … Jayaraman V, (2020). Mechanism of modulation of AMPA receptors by TARP-gamma8. J Gen Physiol, 152(1). doi: 10.1085/jgp.201912451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DR, Dolino DM, Jaurich H, Shuang B, Ramaswamy S, Nurik CE, … Landes CF, (2015). Conformational transitions in the glycine-bound GluN1 NMDA receptor LBD via single-molecule FRET. Biophys J, 109(1), 66–75. doi: 10.1016/j.bpj.2015.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolino DM, Chatterjee S, MacLean DM, Flatebo C, Bishop LDC, Shaikh SA, … Jayaraman V, (2017). The structure-energy landscape of NMDA receptor gating. Nat Chem Biol, 13(12), 1232–1238. doi: 10.1038/nchembio.2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolino DM, Cooper D, Ramaswamy S, Jaurich H, Landes CF, & Jayaraman V, (2015). Structural dynamics of the glycine-binding domain of the N-methyl-D-aspartate receptor. J Biol Chem, 290(2), 797–804. doi: 10.1074/jbc.M114.605436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham RJ, Paudyal N, Carrillo E, Bhatia NK, Maclean DM, Berka V, … Jayaraman V, (2020). Conformational spread and dynamics in allostery of NMDA receptors. Proc Natl Acad Sci U S A, 117(7), 3839–3847. doi: 10.1073/pnas.1910950117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T, Enderle T, Ogletree DF, Chemla DS, Selvin PR, & Weiss S, (1996). Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc Natl Acad Sci U S A, 93(13), 6264–6268. doi: 10.1073/pnas.93.13.6264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai TW, Zhang S, & Wang YT, (2014). Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol, 115, 157–188. doi: 10.1016/j.pneurobio.2013.11.006 [DOI] [PubMed] [Google Scholar]
- Lerner E, Cordes T, Ingargiola A, Alhadid Y, Chung S, Michalet X, & Weiss S, (2018). Toward dynamic structural biology: Two decades of single-molecule Forster resonance energy transfer. Science, 359(6373). doi: 10.1126/science.aan1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwin DB, Carrillo E, Shaikh SA, Berka V, & Jayaraman V, (2019). The structural arrangement at intersubunit interfaces in homomeric kainate receptors. Sci Rep, 9(1), 6969. doi: 10.1038/s41598-019-43360-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwin DB, Durham RJ, & Jayaraman V, (2019). Single-Molecule FRET Methods to Study Glutamate Receptors. Methods Mol Biol, 1941, 3–16. doi: 10.1007/978-1-4939-9077-1_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwin DB, Paudyal N, Carrillo E, Berka V, & Jayaraman V, (2020). The structural arrangement and dynamics of the heteromeric GluK2/GluK5 kainate receptor as determined by smFRET. Biochim Biophys Acta Biomembr, 1862(1), 183001. doi: 10.1016/j.bbamem.2019.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean DM, Durham RJ, & Jayaraman V, (2019). Mapping the Conformational Landscape of Glutamate Receptors Using Single Molecule FRET. Trends Neurosci, 42(2), 128–139. doi: 10.1016/j.tins.2018.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney SA, Joo C, & Ha T, (2006). Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys J, 91(5), 1941–1951. doi: 10.1529/biophysj.106.082487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Cooper D, Poddar N, MacLean DM, Rambhadran A, Taylor JN, … Jayaraman V, (2012). Role of conformational dynamics in alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor partial agonism. J Biol Chem, 287(52), 43557–43564. doi: 10.1074/jbc.M112.371815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Hohng S, & Ha T, (2008). A practical guide to single-molecule FRET. Nat Methods, 5(6), 507–516. doi: 10.1038/nmeth.1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuang B, Cooper D, Taylor JN, Kisley L, Chen J, Wang W, … Landes CF, (2014). Fast Step Transition and State Identification (STaSI) for Discrete Single-Molecule Data Analysis. J Phys Chem Lett, 5(18), 3157–3161. doi: 10.1021/jz501435p [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, & Gouaux E, (2009). X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature, 462(7274), 745–756. doi: 10.1038/nature08624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracy M, Uphoff S, Garza de Leon F, & Kapanidis AN, (2014). In vivo single-molecule imaging of bacterial DNA replication, transcription, and repair. FEBS Lett, 588(19), 3585–3594. doi: 10.1016/j.febslet.2014.05.026 [DOI] [PubMed] [Google Scholar]
- Stryer L, & Haugland RP, (1967). Energy transfer: a spectroscopic ruler. Proc Natl Acad Sci U S A, 58(2), 719–726. doi: 10.1073/pnas.58.2.719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JN, & Landes CF, (2011). Improved resolution of complex single-molecule FRET systems via wavelet shrinkage. J Phys Chem B, 115(5), 1105–1114. doi: 10.1021/jp1050707 [DOI] [PubMed] [Google Scholar]
- Taylor JN, Makarov DE, & Landes CF, (2010). Denoising single-molecule FRET trajectories with wavelets and Bayesian inference. Biophys J, 98(1), 164–173. doi: 10.1016/j.bpj.2009.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentyeva TG, Engelkamp H, Rowan AE, Komatsuzaki T, Hofkens J, Li CB, & Blank K, (2012). Dynamic disorder in single-enzyme experiments: facts and artifacts. ACS Nano, 6(1), 346–354. doi: 10.1021/nn203669r [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, … Dingledine R, (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev, 62(3), 405–496. doi: 10.1124/pr.109.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelsang J, Kasper R, Steinhauer C, Person B, Heilemann M, Sauer M, & Tinnefeld P, (2008). A reducing and oxidizing system minimizes photobleaching and blinking of fluorescent dyes. Angew Chem Int Ed Engl, 47(29), 5465–5469. doi: 10.1002/anie.200801518 [DOI] [PubMed] [Google Scholar]
- Watkins LP, & Yang H, (2005). Detection of intensity change points in time-resolved single-molecule measurements. J Phys Chem B, 109(1), 617–628. doi: 10.1021/jp0467548 [DOI] [PubMed] [Google Scholar]
- Ye S, Kohrer C, Huber T, Kazmi M, Sachdev P, Yan EC, … Sakmar TP, (2008). Site-specific incorporation of keto amino acids into functional G protein-coupled receptors using unnatural amino acid mutagenesis. J Biol Chem, 283(3), 1525–1533. doi: 10.1074/jbc.M707355200 [DOI] [PubMed] [Google Scholar]
- Zhao R, & Rueda D, (2009). RNA folding dynamics by single-molecule fluorescence resonance energy transfer. Methods, 49(2), 112–117. doi: 10.1016/j.ymeth.2009.04.017 [DOI] [PubMed] [Google Scholar]