

Abstract
The cancer stem cell (CSC) concept emerged from the recognition of inherent tumor heterogeneity and suggests that within a given tumor, in analogy to normal tissues, there exists a cellular hierarchy composed of a minority of more primitive cells with enhanced longevity (ie, CSCs) that give rise to shorter-lived, more differentiated cells (ie, cancer bulk populations), which on their own are not capable of tumor perpetuation. CSCs can be responsible for cancer therapeutic resistance to conventional, targeted, and immunotherapeutic treatment modalities, and for cancer progression through CSC-intrinsic molecular mechanisms. The existence of CSCs in colorectal cancer (CRC) was first established through demonstration of enhanced clonogenicity and tumor-forming capacity of this cell subset in human-to-mouse tumor xenotransplantation experiments and subsequently confirmed through lineage-tracing studies in mice. Surface markers for CRC CSC identification and their prospective isolation are now established. Therefore, the application of single-cell omics technologies to CSC characterization, including whole-genome sequencing, RNA sequencing, and epigenetic analyses, opens unprecedented opportunities to discover novel targetable molecular pathways and hence to develop novel strategies for CRC eradication. We review recent advances in this field and discuss the potential implications of next-generation CSC analyses for currently approved and experimental targeted CRC therapies.
Keywords: Colorectal Cancer, Cancer Stem Cells, Omic Technologies, Single-Cell Analyses, Intra-Tumor Heterogeneity, Targeted Therapies
Tumor development and progression have been associated, at the DNA level, with cumulative alterations in oncogenes, tumor suppressor genes, and repair/stability genes.1,2 At the cellular level, it has been recognized for some time that human cancers consist of phenotypically heterogeneous cell populations.3,4 This conclusion has led to the development of the cancer stem cell (CSC) model of cancer initiation and growth.5 The CSC theory suggests that only a subpopulation of cells within a tumor, the CSCs, can proliferate extensively and drive tumorigenic growth5,6 (Figure 1). The CSC theory further postulates that CSCs give rise to the morphologically diverse malignant cell populations contained within a tumor, including nontumorigenic tumor bulk components.6
Figure 1.
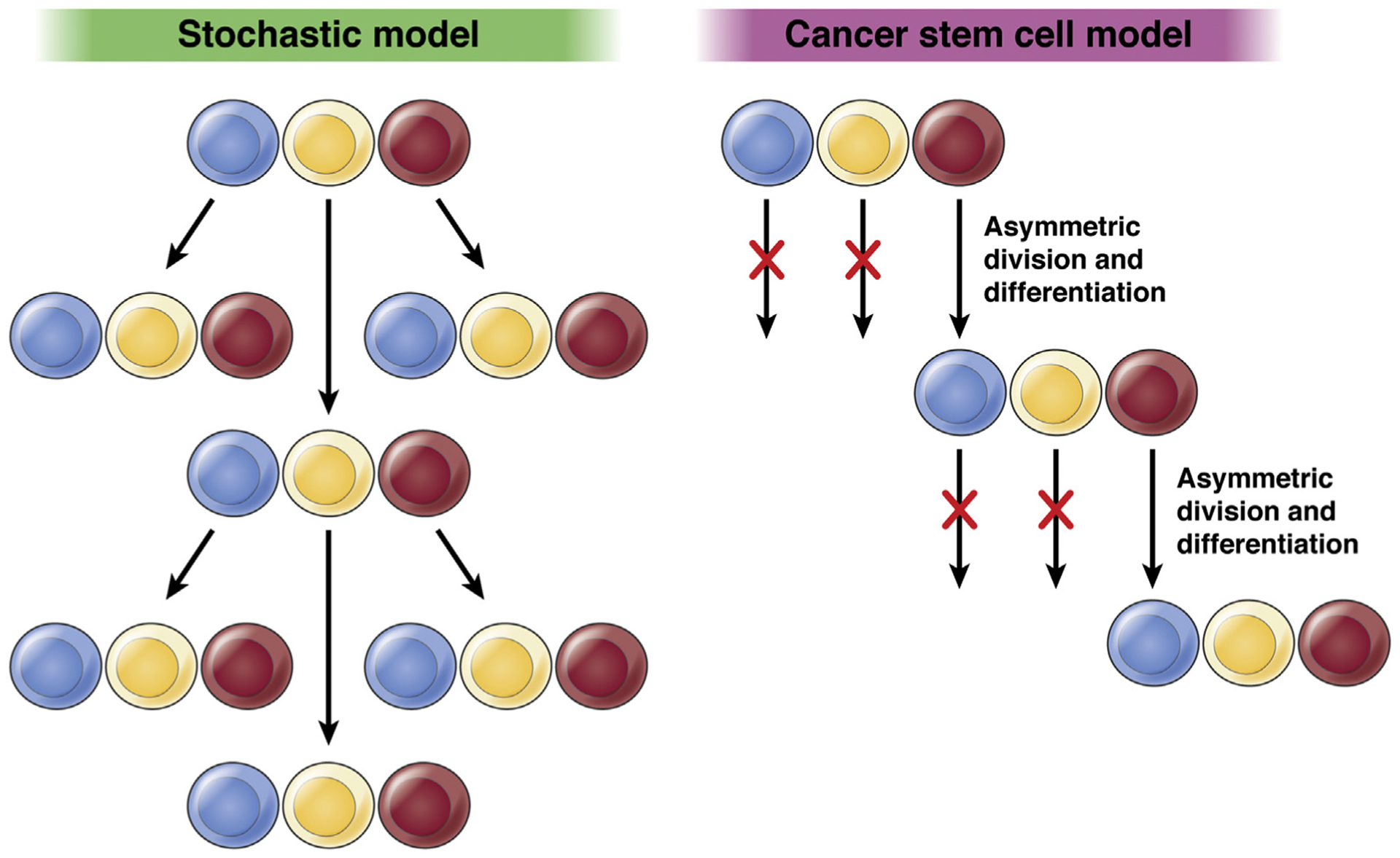
Competing models of cancer initiation and progression. Left: The stochastic model of cancer initiation posits that most cells in a given tumor would be able to initiate a new tumor. Right: The cancer stem cell model postulates that only CSCs initiate tumors. CSCs (depicted in red) are maintained through asymmetric cell division, while simultaneously giving rise to the progeny capable of generating diverse cell clones within the tumor.
The existence of CSCs in human colorectal cancer (CRC) and significant contributions of CRC CSCs to clinical tumor progression, chemoresistance, and therapeutic failure have been suggested by several preclinical studies.7–10 Initially, highly tumorigenic and self-renewing human CRC CSCs were successfully enriched for in 2 separate studies using the CD133 surface marker.8,9 In another study, Dalerba et al7 identified a subset of tumorigenic human colon cancer cells based on an epithelial cell adhesion molecule (EpCAM, also known as ESA)high CD44+ lineage− phenotype. Subsequent stem cell genetic lineage tracing studies in murine cancer models confirmed that physiologic CD133+ stem cells, as well as additional intestinal stem cell populations characterized by Lgr5 or Bmi expression, can serve as cells of origin for CRC development.11–13 As a result, CSC origin is now considered to be a major contributor to CRC heterogeneity, cancer aggressiveness, and therapeutic response.14
Omics Technologies Guide Colorectal Cancer Molecular Classification to Inform Therapy Decisions
The last decade is renowned for the rapid development of various omic technologies that include genomics, transcriptomics, epigenomics, proteomics, metabolomics, and others. Some of these technologies have already been used successfully to generate large data sets that enabled comprehensive molecular characterization of CRC and provided a foundation for several novel molecular-based cancer classifications, with the goal of further refining individualized therapeutic strategies.15–22 Most recently, Guinney et al23 performed highly robust bioinformatic analyses of 18 independent gene expression data sets from 4151 CRC patients and identified 4 consensus molecular subtypes (CMS): microsatellite instability immune (CMS1), with features of microsatellite instability; canonical (CMS2), with activation of WNT and MYC; metabolic (CMS3), with evident metabolic dysregulation; and mesenchymal (CMS4), with prominent transforming growth factor–β activation, stromal invasion, and angiogenesis. This classification provides the most comprehensive molecular CRC characterization to date and has recently been shown to serve as an independent prognostic marker in patients with metastatic CRC who undergo first-line chemotherapy in combination with bevacizumab or cetuximab.24 In addition, the emergence of novel single-cell analysis technologies provided an unprecedented opportunity to examine the cellular diversity within an individual tumor, which might contain clones of cells with more than 1 molecular subtype defined by Guinney et al. These analyses have already enabled a more detailed characterization of molecular and cellular tumor composition and revealed intra-tumor heterogeneity likely responsible for differential therapeutic responses and resistance patterns.
Single-Cell Genomic Analyses, Cancer Stem Cells, and Evolution of Colorectal Cancer
In 1990, Fearon and Vogelstein25 proposed that CRCs develop from colonic adenomas as a result of the successive accumulation of genetic mutations. This linear paradigm of CRC progression has been challenged by recent high-resolution genomic studies that uncovered early nonlinear development of intra-tumor mutational heterogeneity, resulting in simultaneous accumulation of genetically diverse cancer clones subsequent to an initial oncogenic transformation event.26 Using genomic profiling of 349 individual tumor glands, each originating from a single stem cell, Sottoriva et al26 discovered that after the initial transformation, CRC tumors grew predominantly as a single expansion populated by numerous intermixed sub-clones. Based on these observations, they proposed a novel “Big Bang” theory of CRC progression, which posits that after an initial common oncogenic event, the majority of mutations driving tumor growth occur during early tumor expansion and are responsible for the clonal diversity and intra-tumor heterogeneity. Hence, the Big Bang theory points to the existence of a common ancestor during cancer clonal evolution. The question arises, therefore, whether the Big Bang theory is consistent with or even supportive of the CSC theory. According to the latter, tumors are maintained through unlimited CSC self-renewal and differentiation capacity. Moreover, according to the CSC theory, CSCs are capable of fully regenerating original tumor heterogeneity upon transplantation. This functional definition therefore implies that CSCs contain cells representative of all genetic sub-clones within a given tumor (Figure 2A). Moreover, it implies that clones that do not contain CSCs would not self-renew and would ultimately be eliminated from heterogeneous tumors. Thus, observations of long-term co-existence of diverse mutational tumor clones along with heterogeneity for established CSC markers suggests the presence of cells with non-mutationally determined CSC phenotype responsible for the maintenance and persistence of each individual cancer clone in heterogeneous tumors. The Big Bang model is therefore consistent with, and in fact supports, the existence of CSCs as a mutationally heterogeneous, yet with regard to CSC marker expression homogeneous, subpopulation of self-renewing cells, which quite possibly arose from a common ancestor that sustained a first oncogenic mutation and gave rise to diverse clones within a single tumor27 (Figure 2A). It is useful to recall here the original consensus definition of a CSC that was arrived at by Clarke et al28 in 2006, that is, “a cell within a tumor that possesses the capacity to self-renew and to cause the heterogeneous lineages of cancer cells that comprise the tumor.” This consensus view further stipulates that “cancer stem cells can thus only be defined experimentally by their ability to recapitulate the generation of a continuously growing tumor.”28 It is therefore clear that this functional CSC definition makes no assumptions about a particular cell of origin of cancer and its evolutionary trajectory, but indeed allows for, as a specific point-in-time snapshot across an established tumor entity, for cellular conversion of mutationally heterogeneous cells of distinct clones toward CSC phenotype, through intrinsic or acquired plasticity induced by, for example, among other factors, metabolic restraints, microenvironmental signaling factors, and/or therapeutic selection pressures. Thus, the CSC theory accommodates both monoclonal and polyclonal tumor evolution.
Figure 2.

Single-cell omic technologies support the CSC concept. (A) Single-cell genomic analyses showed that after an initial common oncogenic event, the majority of mutations driving tumor growth occur during early tumor expansion and are responsible for the clonal diversity and intra-tumor heterogeneity. This model supports the existence of the CSCs as a subpopulation of self-renewing cells with a common ancestor that sustained a first oncogenic mutation and gave rise to diverse clones within a single tumor. (B) Single-cell triple omics sequencing technique (scTrio-seq) that can assess SCNAs, DNA methylation, and transcriptome information simultaneously from an individual cell enables reconstruction of cancer genetic lineages and their epigenomic and transcriptomic dynamics, thus providing a powerful tool to investigate the contribution of CSCs to CRC progression and therapeutic resistance.
Single-cell genomics also enabled high-resolution analyses of the tumor evolutional hierarchy.29 Wu et al30 employed DNA-based single-cell sequencing to examine the cancer tissues and noncancerous polyps isolated from 2 unrelated patients with sporadic CRC. They demonstrated the existence of common mutations in GPCR, PI3K-Akt, or FGFR signaling pathway genes among different cancer cell clones in individual patients and provided evidence for monoclonal CRC origin with subsequent sub-clonal evolution. This concept was challenged by Yu et al,31 who detected 2 distinct clones in a single colonic adenocarcinoma: the predominant clone driven by antigen presenting cells and TP53 mutations and the minor independent clone characterized by early mutations in CDC27 and PABPC1. However, Wu et al disputed this conclusion upon reanalysis of the study and convincingly demonstrated that the second independent clone likely represented a noncancerous contaminant within the tumor, and colon cancer in this patient originated from a clone defined by antigen presenting cells and TP53 mutations.30
The existence of monoclonal CSC-driven CRC origin is also supported by the single-cell–level examination of somatic copy number alterations (SCNAs). A study of 2 rectal tumors revealed that the majority of SCNAs were shared by all cells within a given tumor and thus represented early events in malignant transformation.32 In addition, the tumors also contained sub-clones with novel focal SCNAs, which defined the intra-tumor clonal heterogeneity. Notably, a follow-up study by the same authors found that both CD45− EpCAMhigh CD44+ CSCs and CD45− EpCAMhigh CD44− differentiated tumor cells had similar SCNA profiles,33 thus supporting the possibility of a monoclonal CSC phenotype.
Single-cell genomic analyses have also been employed to study the clonal evolution of CRC metastases.34,35 Heitzer et al34 performed comprehensive genomic profiling of circulating tumor cells from 6 patients with stage IV CRC. They found that the same CRC-associated copy number changes and driver mutations detected in the primary tumor and metastasis were also observed in corresponding circulating tumor cells. Leung et al,35 using single-cell DNA sequencing, exome sequencing, and targeted deep sequencing of 2 patients with stage IV colon cancer, demonstrated that metastatic clones emerge during the late stages of primary tumor development, and might originate from more than 1 independent cancer sub-clone.
In order to overcome the existing technical challenges of single-cell genomic analyses, such as the need for DNA amplification and resultant technical errors,36 and to enable the functional evaluation of single tumor cells, Roerink et al37 generated organoids derived from single cells isolated from tumors and adjacent normal tissues of 3 patients with CRC. In all tumors, targeted cancer-specific gene sequencing panels and subsequent whole-genome sequencing analyses revealed significant intra-tumor clonal heterogeneity with characteristic mutational signatures. Intriguingly, when subjected to treatment with clinically relevant CRC chemotherapeutic and targeted agents, even organoids derived from the same patient exhibited differential responses independent of their mutational signatures.37 This observation suggests that other factors, such as, for example, the presence of CSCs, might be responsible for the therapeutic resistance of some of the tumor clones and highlights the potential utility of this technology in designing patient-tailored cancer therapies.27
Diagnostic and Prognostic Value of Single-Cell Multi-Omic Analyses
In addition to single-cell genomics, single-cell transcriptome analyses have been used widely to investigate CRC cellular hierarchies, evaluate cell populational heterogeneity and cellular composition, and characterize CSCs.36 Some of these studies have already resulted in the identification of novel molecular signatures that could serve as predictors of patient outcomes. In one of the first such studies, using single-cell polymerase chain reaction gene-expression analysis, Dalerba et al38 found that human colon cancer tissues contained distinct cell sub-populations, transcriptional identities of which were reminiscent of normal colon cellular lineages. Based on these single-cell analyses, they developed a novel 2-gene molecular predictor of clinical patient outcomes with hazard ratios superior to those of pathologic grade determination, highlighting the promising prognostic value of this approach. Subsequently, evaluation of single-cell transcriptomes from 11 primary CRC tumors and matched normal mucosa showed that CRC tumors previously assigned to a single subtype based on bulk transcriptomics could be divided into subgroups with divergent survival probability based on their single-cell signatures.39
To enable a more comprehensive characterization of CRC cells, Bian et al40 developed a single-cell triple-omics sequencing technique (scTrio-seq) that can assess SCNAs, DNA methylation, and transcriptome information simultaneously from an individual cell. In this study, they examined 1900 single cells isolated from the primary and metastatic tumors of 12 patients with stage III/IV CRC and found that DNA methylation profiles were relatively stable within a single genetic lineage. They found that single-cell multi-omic approaches enable reconstruction of cancer genetic lineages and their epigenomic and transcriptomic dynamics, thus providing a powerful tool to investigate the contribution of CSCs to CRC progression and therapeutic resistance (Figure 2B).
Colorectal Cancer Stem Cell Response to Targeted Therapies
Although the majority of CRC patients present with surgically resectable tumors, a high proportion of these patients eventually succumb to metastatic disease originating from residual microscopic malignancy not evident at the time of surgery.41 Adjuvant therapies, including radiotherapy and chemotherapy, are designed to target residual tumor cells; however, their current success is limited by the existence of therapy-resistant cancer cell populations, which may coincide with CSCs.6,10 The development of targeted therapies for advanced CRC has resulted in remarkable improvement in overall survival. Such treatments aim to disrupt cellular pathways essential for tumor growth, survival, and metastasis, and to reduce the toxicity associated with less-specific cytotoxic therapies.42,43
Currently, the following 4 classes of biologic therapies targeted against nonimmune cells have been approved for the treatment of metastatic CRC: monoclonal antibodies against epidermal growth factor receptor (EGFR), inhibitors of vascular endothelial growth factor (VEGF), oral small-molecule inhibitors of intracellular kinases involved in various signaling cascades (regorafenib), and most recently, a small molecule BRAF inhibitor (encorafenib) (Figure 3). Several clinical trials have demonstrated that introduction of targeted therapies with monoclonal antibodies against VEGF (bevacizumab) or EGFR (cetuximab), in addition to 5-fluorouracil (5-FU), results in a substantial increase in the median survival of patients with advanced disease.42,44,45 More recently, 2 large randomized placebo-controlled trials demonstrated that, in patients with progressive CRC after standard cytotoxic and targeted treatments, regorafenib can significantly prolong overall survival compared with placebo.46,47 In patients with metastatic CRC with the BRAF V600E mutation, a recent phase III clinical trial found that a combination of encorafenib and cetuximab with or without the MEK inhibitor binimetinib resulted in significantly longer overall survival and a higher response rate than standard therapy of cetuximab and chemotherapy only.48 These results indicate that specific targeting of cell populations within tumors that have remained viable after radiotherapy and chemotherapy, and even after the first-line targeted therapy, might eventually lead to successful tumor eradication. However, the relatively transient or individually restricted nature of clinical responses to currently available targeted therapies also indicates the urgency for further development of novel therapeutic strategies that specifically target additional therapy-resistant cancer cell populations, which may coincide with CSCs.
Figure 3.
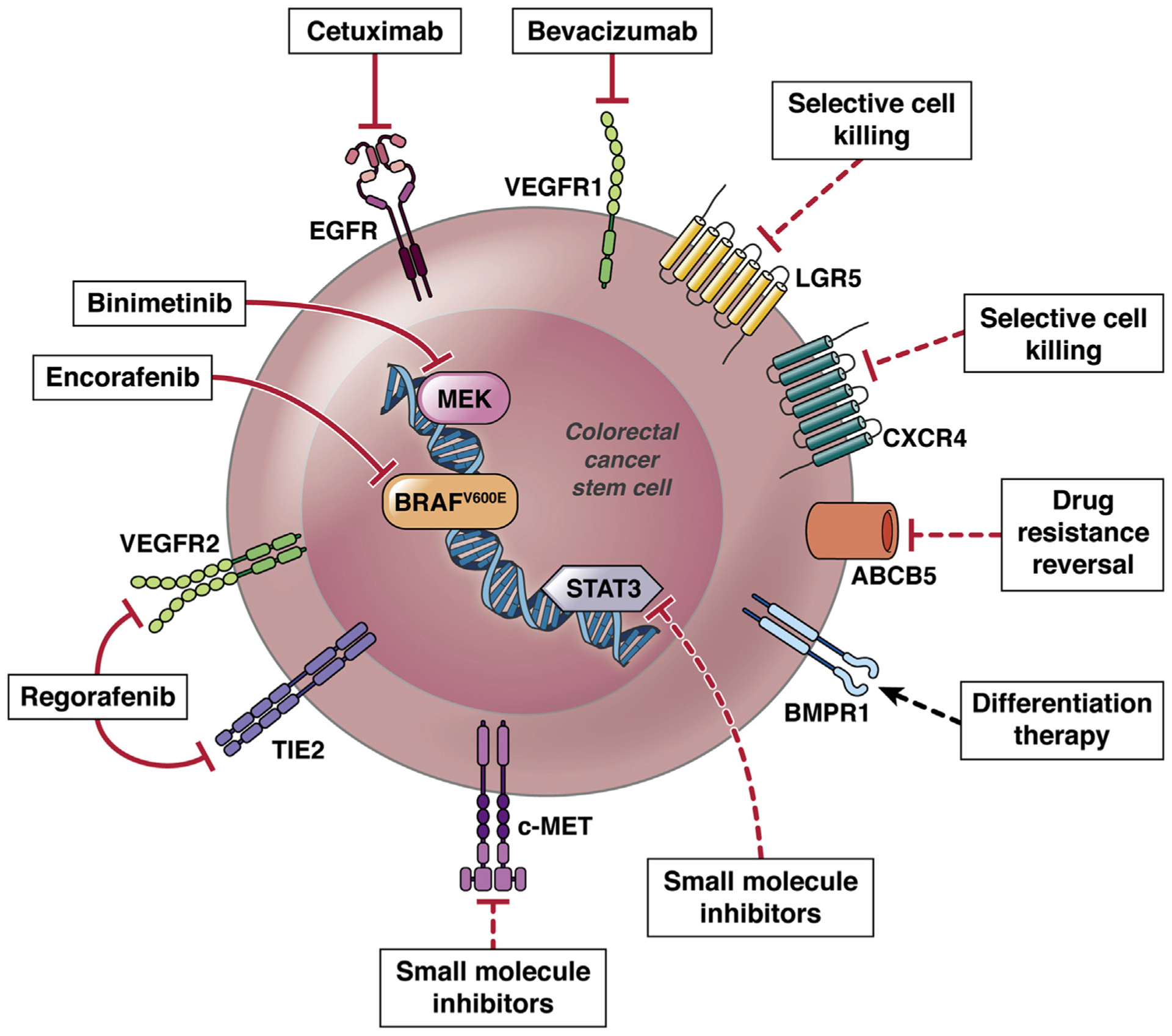
Current approved and investigational therapies targeting CRC CSCs. Clinically approved therapies that target CRC CSCs include monoclonal antibodies against EGFR and VEGFR1, small molecule inhibitors of MEK and mutant BRAF (illustrated with solid line). Additional investigational strategies involve selective CSC killing via cytotoxic drug conjugates, c-MET and STAT3 blockade by small molecule inhibitors, differentiation therapy by administration of BMP4, and MDR reversal though antibody-mediated ABCB5 blockade (illustrated with dotted line).
The EGFR inhibitors cetuximab and panitumumab are the most commonly used targeted agents for the treatment of advanced CRC; however, their clinical success is limited by intrinsic and acquired tumor resistance (reviewed in Chong and Janne49). Based on the established roles of CSCs in CRC resistance to conventional chemotherapies,10,50,51 several groups investigated whether CSCs can also evade EGFR-targeted therapies.52–57 De Angelis et al54 reported that anti-EGFR therapy could counteract CSC expansion induced by chemotherapy in patient-derived CRC spheroids enriched in CSCs. Moreover, cetuximab treatment augmented the apoptotic death of CSC-like CRC cells in the presence of 5-FU through inhibition of 5-FU–induced cell autophagy.57 Expression of mutant EGFR variant on CD44/CD133-positive CSCs was associated with worse overall survival in 70 patients with metastatic CRC.56
CSCs have also been shown to confer CRC resistance to EGFR-targeted therapies by aberrant activation of alternative receptor tyrosine kinases. Using patient-derived CRC xenografts, Luraghi et al52 demonstrated that while in the absence of constitutive RAS activation, CSC proliferation and survival rely on the EGFR pathway, this dependency could be bypassed by wild-type MET activated in paracrine fashion by hepatocyte growth factor (HGF). In another study, Joosten et al,55 found that HGF/MET receptor signaling is promoted by the stem cell CD44 isoform CD44v4–10 and suggested that dual inhibition of MET and CD44 might reverse CRC resistance to EGFR-targeted therapies.
As multiple clinical trials have shown that activating mutations in RAS genes represent negative predictive bio-markers for anti-EGFR therapy,58 anti-angiogenic treatments represent one of the few available targeted options for RAS mutated metastatic CRC.59 Bevacizumab, aflibercept, ramucirumab, and regorafenib are anti-angiogenic agents currently approved by the US Food and Drug Administration for the treatment of metastatic CRC.46,60–62 However, in the majority of patients, the therapeutic response is often short-lived due to up-regulation of compensatory signaling pathways, modulation of the tumor microenvironment, or recruitment of CD11b+Gr1+ myeloid cells.63 CD133-positive CSCs have been shown to confer CRC resistance to bevacizumab through the activation of an anti-apoptotic signaling pathway involving PP2A, p38MAPK, MAPKAPK2, and Hsp27.64 Moreover, in patient-derived CRC xenografts, a discontinuous schedule of bevacizumab significantly increased the number of CSCs, which was attributed to rising proangiogenic factors in the setting of hypoxia.65 These findings provide a plausible explanation for the lack of durable response to anti-angiogenic therapies in CRC and point to the need for CSC ablation for successful cancer eradication.
Immunotherapy and Immune Evasiveness of Colorectal Cancer Stem Cells
Immunotherapy, broadly defined, entails manipulation of the patient’s immune system for the treatment of cancer. Various immune pathways actively promote or antagonize cancer during its development and progression. The antagonist anti-tumor role of the immune system actively counteracts cancer development and shapes the tumors that become clinically apparent through a process known as immunoediting. Cancers that come to clinical attention have managed to escape immune control, either through a relative loss of immune function in the host or through further evolution of cancer that confers the ability to resist immune detection and elimination.66
Recently, much of the effort in anti-cancer immunotherapy has been focused on developing cytotoxic CD8+ T-cell responses against tumors, based on vaccinating the host against tumor antigens, releasing the immune system from suppression, or transferring anti-tumor T cells into the patient. Perhaps the most clinically developed approaches have been checkpoint inhibition and chimeric antigen receptor T cells. Checkpoint inhibition involves targeting molecules on the T cell, such as CTLA-4, PD-1, or its ligand PD-L1, which down-regulate CD8+ T-cell response and promote tolerance. It relies on the ability of T cells to recognize the antigens not present in the host. However, this approach is only effective in approximately 15% of CRCs that have microsatellite instability and high mutation burden.67 To date, success with chimeric antigen receptor T-cell therapy has been confined mainly to hematologic malignancies, where targeting antigens shared between tumor and normal cells does not result in unacceptable toxicity, nevertheless, novel chimeric antigen receptor T-cell therapies against CRC are being investigated68 (Figure 4A).
Figure 4.
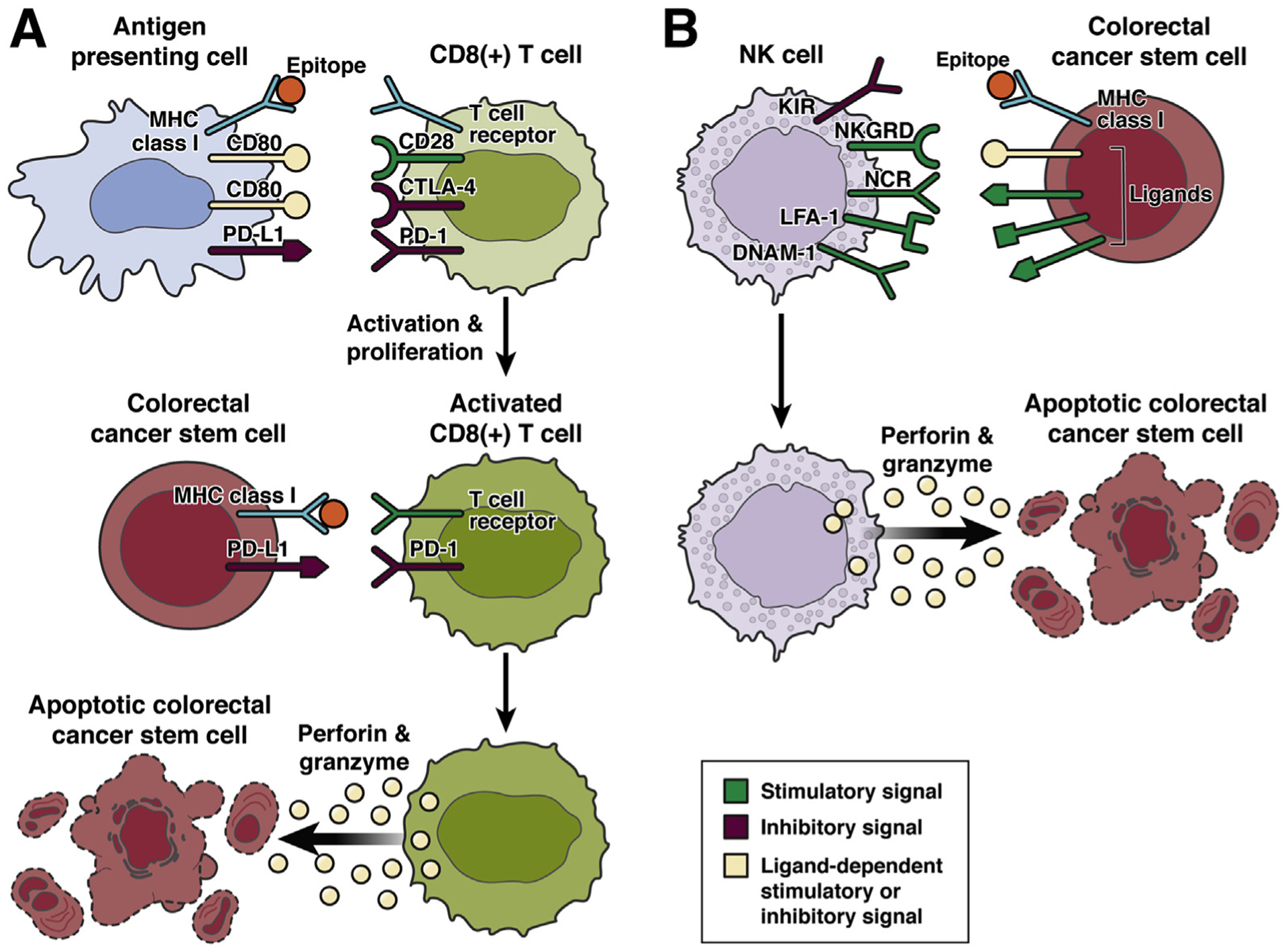
Induction of T cell and NK cell–mediated anti-colorectal stem cell responses. (A) Tumor antigens are phagocytosed by antigen presenting cells (APCs) and lysed into peptide fragments called epitopes that are bound to MHC class I molecules and presented to CD8+ T cells with a T-cell receptor specific for a particular tumor epitope. Induction of immunity also requires the APC to present a co-stimulatory signal such as CD80, which binds to CD28 on the CD8+ T cell. Activation of immune checkpoint receptors on the CD8+ T cell, such as CTLA-4, which is expressed by activated CD8+ T cells and also binds to CD80, or PD-1, which binds to PD-L1 down-regulate activation of CD8+ T-cell immunity. After encountering a tumor epitope in the appropriate context, a CD8+ T cell recognizing the specific tumor epitope becomes activated and proliferates. When an activated CD8+ T cell re-encounters the tumor antigen specifically recognized by its T-cell receptor in the context of MHC class I on a CRC CSC, a cytotoxic response is elicited including the degranulation of perforin and granzyme, which cause apoptosis of the CRC CSC. However, a cytotoxic response can be inhibited by expression of PD-L1 on the tumor cell, which engages PD-1 on the CD8+ T cell. (B) NK cells, as part of the innate immune system, are not specific for particular antigens and do not require activation to mediate cytotoxic anti-CRC CSC responses. Instead cytotoxicity is elicited in the appropriate context as mediated by expression of activating and inhibitory signals on the target cell. Notably, MHC class I is recognized by killer-cell immunoglobulin-like receptors (KIRs) on NK cells and inhibits cytotoxic NK responses. Therefore, cells that down-regulate MHC class I and hence are not subject to adaptive CD8+ T-cell cytotoxicity are instead subject to NK-mediated cytotoxicity. Activating signals for NK cell cytotoxicity include expression by CRC CSCs of ligands for the NK cell receptors NKG2D, natural cytotoxicity triggering receptors, LFA-1, or DNAM-1. NK cell cytotoxicity is also mediated through degranulation of perforin and granzyme from NK cells causing apoptosis of target cells.
Down-regulation of major histocompatibility complex (MHC) class proteins associated with antigen processing and presentation can also facilitate CSC immune escape.66 For CD8+ T cells to recognize and respond to a cell, antigen, in the form of a short protein fragment known as an epitope, must be presented to this T cell in an MHC class I molecule on the surface of another cell. Decreased expression of MHC class I molecules has been shown in some,69,70 but not all,71,72 studies of CRC CSCs. However, the immune system can eradicate cells that down-regulate MHC class I molecules through recognition and destruction by natural killer (NK) cells. Although CRC CSCs might be less susceptible to cytotoxic T cell–mediated eradication, they could be targeted by NK cell–mediated immunotherapy (Figure 4B). Notably, molecules associated with activation and cytotoxicity of NK cells, including ligands for NKG2D,69,70 natural cytotoxicity receptors,70 LFA-1,70 and DNAM-1,70 had their expression retained on CRC CSCs with decreased MHC class I expression. One study showed that CRC CSCs, as opposed to the non–stem cell fraction of the tumor, had increased susceptibility to NK cell–induced cell killing.70 Another mechanism of escape, at least to immunotherapies not involving inhibition of the PD-L1/PD-1 pathway, could be the high expression of PD-L1 on CRC CSCs,73–75 where it might be functionally related to the CRC CSC phenotype.75 The resistance of CSCs to anti-tumor vaccination strategies has been shown in mouse models of human papillomavirus–associated cervical cancer76 and in a human clinical trial of glioblastoma multiforme.77 Strategies to enhance cytotoxic T-cell killing of CSCs may involve counteracting mechanisms that make these cells particularly resistant to immune attack. For instance, a study showing that cervical CSCs were resistant to vaccination also demonstrated that this resistance was mediated by the stem cell transcription factor NANOG, and that targeting NANOG with nanoparticles carrying small interfering RNA enhanced susceptibility to cytotoxic T-cell killing.76
Considerable effort has been directed toward the identification of antigens specifically expressed by CSCs that can be targeted for the development of anti-tumor immunity. In general, tumor antigens have been classified into 3 broad categories: tumor-specific antigens, tumor-associated antigens, and cancer-germline/cancer-testis antigens.78 Tumor-specific antigens are unique to tumors compared with normal cells and often arise from mutations in the tumor. These mutations can either be essential for the tumor (driver mutations) or accumulated due to tumor mutagenesis without playing a functional role (passenger mutations). Tumor-associated antigens are shared with normal cells, typically being either overexpressed in the cancer cells or restricted to similarly differentiated normal cells. Cancer-germline/cancer testis antigens are antigens restricted to tumors as well as cells in immunologically privileged environments (eg, testes) and cells in embryologic development, such that they are more likely to be recognized as non-self by the immune system. Several studies have focused on targeting antigens that are overexpressed in CRC CSCs. The efficacy of anti-tumor cytotoxic T-cell responses against the well-characterized stem markers CD133,79,80 CD44,81 and ALDH182 has been tested in various malignancies. The use of cytotoxic drugs conjugated to antibodies targeting LGR5, a marker shared between stem cells in CRC and the healthy intestine, has shown efficacy in mouse models, indicating the potential utility of LGR5 as a tumor antigen.83,84 Attempts have been made to look for other antigens that are more specific for stem cells, as opposed to the non–stem cell population in CRC. An epitope from ASB4, a potential component of the E3 ubiquitin ligase complex, was identified by looking for CRC CSC tumor epitopes that bind to the MHC class I molecule HLA-A24. The antigen was minimally expressed by a panel of normal adult and fetal tissues. A CD8+ T-cell clone specific for this epitope was found to lyse stem cells but not non–stem cells from CRC and to show efficacy in a mouse model.85 Other groups have looked at generating anti-tumor immunity against CRC CSCs using tumor-associated antigens, such as the antigen COA-1, encoded by the UBXD5 gene69 and centrosome protein 55kd (Cep55/c10orf3),71,72 which are not necessarily specific for CRC CSCs over non–stem cells in the tumor. Nevertheless, the authors were able to demonstrate the expression of these antigens in the stem cell population and to effectively target the CSCs with antigen-specific cytotoxic T cells. Cancer-germline/cancer-testis antigens may represent important antigens for targeting CSCs. In 1 study, the expression of these antigens was compared in stem cell and non–stem cell populations of various cancers, including CRC. Preferential expression was seen in the stem cell population for 18 of the 74 antigens examined.86 Two likely cancer-germline/cancer testis antigens were identified in studies looking for antigens specific for CRC CSCs: Hsp40 family member, DnaJ homolog, subfamily B, member 8 (DNAJB8)87 and olfactory receptor family 7 subfamily C member 1 (OR7C1).88 Cytotoxic T cells generated against both of these antigens showed anti-tumor efficacy in mouse models. Based on results from human patients treated with checkpoint inhibitors, the anti-tumor immune response most often targets tumor-specific antigens generated by mutations within the tumor known as neo-antigens.89–91 As neo-antigens can be either unique to heterogeneous populations within the tumor or shared by all tumor clones, more effective tumor immunity may be developed against the clonal neoantigens, and this has been demonstrated clinically.92 One group, in a proof-of-concept study, identified unique neo-epitopes in CRC patients that were immunogenic and targeted CSCs.93
Some groups have investigated the possibility of promoting anti-tumor immunity against CRC CSCs mediated by cells other than cytotoxic CD8+ T cells. For instance, the rationale for NK cell–mediated anti-stem cell immunity was discussed above.70 Another group explored a strategy using Vγ9Vδ2 T cells, a type of γδ T cells. These cells can be activated by a variety of nonpeptide phosphor-antigens. Amino-bisphosphonates, which are commonly used to inhibit osteoclastic bone resorption, can stimulate biosynthesis of immunogenic phosphor-antigens. The group demonstrated that the amino-bisphosphonate zoledronate sensitizes primary human CRC CSCs to Vγ9Vδ2 T cell killing in vitro.94
In the hierarchical unidirectional model originally derived from hematopoietic stem cells, stem cells have the unique capacity to give rise to daughter cells of all lineages, whereas daughter cells become more and more lineage-restricted as they become more differentiated. CRC CSCs, similar to stem cells in other adult tissue, display plasticity. Differentiated cells can revert to a stem cell phenotype under certain stimuli.95,96 The limitation of targeting CRC CSCs has been demonstrated by studies where Lgr5+ CRC CSCs were ablated. Although this strategy initially controlled tumors, with the cessation of treatment Lgr5+ CSCs were reconstituted, and the tumors re-grew.97,98 The immune microenvironment has been shown to promote the stem cell phenotype, so another potential immunotherapeutic strategy against CSCs may involve modulating the immune system to counteract these stem cell–promoting stimuli. One of the first and most thorough studies of a mechanism by which the immune milieu can promote CRC CSCs investigated the role of interleukin (IL)-22.99 IL-22 was found to induce stemness in CRC cell lines as measured by increased tumorigenic potential, increased tumor sphere formation, and increased expression of several classic CRC CSC markers. It was demonstrated that IL-22 is secreted primarily by CD4+ T cells in the tumor microenvironment and that these T cells could promote tumor sphere formation and expression of stem cell markers when cultured with autologous primary human colon cancer cells. The mechanism by which IL-22 promotes stem cell phenotype is mediated by STAT3. STAT3 was previously shown to be an important regulator of the stem cell compartment in normal intestine,100 and has subsequently been shown to be important in stem cell induction by other immune signals.101,102 STAT3, induced by IL-22, had a direct effect in inducing expression of the stem cell molecule SOX2 by binding to its promoter, but also an indirect effect of inducing expression of NANOG, SOX2, and Pou5F1 (Oct-4) through the expression of disruptor of telomeric silencing-1 (DOT1L), which in turn bound to the promoter of those genes.99 Other immune mediators, such as IL-6,102 IL-8/CXCL8,103 IL-17A,104 IL-32θ,101 IL-33,105 transforming growth factor–β,106 CCL21,107 and CXCL2,108 as well as the lipid mediators prostaglandin E2105,109,110 and leukotriene D4109 have also been shown to promote CRC stemness, although an immune cell source of these molecules was not demonstrated. In addition to specialized CD4+ cells secreting IL-22, macrophages105 and myeloid-derived suppressor cells111 have also been implicated in promoting CRC stemness.
Can Direct Cancer Stem Cell Targeting Improve Patient Outcomes in Colorectal Cancer?
Cancer resistance and recurrence have been attributed to CSCs, which can weather out unfavorable conditions during diverse cancer therapies through entering a state in which they no longer depend on the targetable molecular pathways. Upon withdrawal of treatments and, in some cases, despite the therapies, CSCs re-emerge, adapt, and continue to sustain tumor propagation. This raises the question of whether CSCs can be permanently eradicated, and whether CSC targeting either through cytotoxic agents or modulation of CSC-specific molecular pathways might, as could be anticipated, cure cancer. Several CRC CSC-targeting approaches have been examined thus far with variable success warranting further development.
Selective killing of LGR5-positive CSCs was sufficient to regress tumor growth in mouse and human xenograft CRC models97,98; however, when active CSC ablation was dis-continued, LGR5-positive CSCs re-emerged, leading to tumor regrowth. This phenomenon was attributed to the plasticity of other CRC cell populations that could be reprogrammed into CSCs by molecular signals from the niche. In a metastatic CRC preclinical model, selective elimination of CXCR4+ metastatic CSCs through the nanoparticle-conjugated antitumor drug 5-fluoro-2′-deoxyuridine resulted in reduced tumor intravasation and completely prevented metastases development in up to 83% of mice112 (Figure 3).
In addition, the depletion of CRC CSC pools through induction of differentiation has been examined as a strategy to sensitize tumors to conventional therapies. Lombardo et al113 showed that administration of BMP4 to immuno-compromised mice with CRC CSC-derived tumors could increase the antitumor effects of 5-FU and oxaliplatin through induction of terminal differentiation, apoptosis, and chemosensitization, suggesting that BMP4 might be developed as a therapeutic agent against CSCs in advanced CRC tumors. Veschi et al114 showed that an enhanced variant of BMP7 (BMP7v) promotes CSC differentiation and sensitizes tumor cells to standard chemotherapy, regardless of the mutational, microsatellite instability, and CMS profiles (Figure 3).
Combination therapies that target CSC-specific molecular pathways, as well as the components of the CSC niche in addition to direct CSC killing, might provide a more durable path for sustainable tumor eradication. For example, small molecule inhibitors of STAT3 resulted in inhibition of metastasis and down-regulation of CSC-defining molecules, such as c-Myc, β-catenin, NANOG, and Sox2 in preclinical CRC models, showed promising results in phase I and II clinical trials, and are now being tested in a phase III clinical trial (ClinicalTrials.gov ID: NCT02753127)115 (Figure 3). In addition, targeting CSC niche-derived HGF signaling116 by rilotumumab, a fully human anti-HGF monoclonal antibody, demonstrated improvement in the disease control rate and the duration of response in a phase Ib/II clinical trial in patients with KRAS-wild tumors treated with EGFR inhibitor panitumumab (ClinicalTrials.gov ID: NCT00788957)117 (Figure 3).
The effectiveness of cancer chemotherapy as a strategy to cure metastatic or disseminated malignant disease is frequently impaired by either intrinsic or acquired tumor resistance to multiple, structurally unrelated therapeutic drugs with different mechanisms of action, a phenomenon termed multidrug resistance (MDR) (reviewed in Gottesman et al118). MDR has been attributed to the function of ATP-binding cassette (ABC) transporter family members, which are highly expressed in CSCs.119 Among them, ABCB5 is a clinically relevant MDR mediator,120 which is expressed at high levels on CSCs in several human malignancies, including malignant melanoma,120–124 glioblastoma,125 hepatocellular carcinoma,126 breast cancer,127 lung cancer,128 oral cancer,129 and CRC.10,130 In melanoma, ABCB5 identifies CSCs that correlate with clinical disease progression,122,131,132 which can be specifically targeted through ABCB5 to reverse drug resistance and abrogate tumor growth.122,123 In hepatocellular carcinoma stem cells, ABCB5 also mediates CSC chemoresistance in CD133+EpCAM+ subpopulations and serves as a biomarker of disease recurrence and reduced survival.126 Studies in CRC showed that ABCB5 is specifically overexpressed on therapy-resistant CD133+ CSCs and established that ABCB5 blockade is capable of sensitizing CRC CSCs to 5-FU–induced cell killing.10 Moreover, ABCB5 blockade inhibits tumor growth and invasion through reversing intrinsic ABCB5 molecular functions related to anti-apoptotic signaling epithelial–mesenchymal transition.10,133 In addition, our most recently published studies identified a novel, MDR-independent role of ABCB5 in CRC growth and aggressiveness, through an anti-apoptotic mechanism involving regulation of AXL,133 a receptor tyrosine kinase associated with adverse CRC prognosis134 and known for its anti-apoptotic function in other malignancies.135 These studies suggest that the ABCB5 blockade might represent an additional novel strategy of CSC targeting in CRC, which could lead to significant improvement in patient outcomes (Figure 3).
Challenges to the Hierarchical Cancer Stem Cell Concept Open Additional Novel Therapeutic Opportunities
Recent studies utilizing molecular marker–based cell lineage tracing approaches suggested the possibility of dynamic hierarchical relationships between CSCs and more differentiated bulk tumor cell populations, implying that in the setting of CSC depletion, their pool can be repopulated by more differentiated cancer cells that acquire CSC phenotype in response to the environmental cues.136 In 1 such study, Vermeulen et al116 examined the role of Wnt signaling in defining the CRC CSC phenotype and CRC cell plasticity in primary tumors. Using a TCF/LEF reporter model, the authors showed that CRC CSCs were characterized by high Wnt activity, while cells with low Wnt signaling possessed more differentiated epithelial characteristics. Moreover, the study highlighted the role of tumor surrounding myofibroblasts in promoting Wnt signaling in CRC cells through secretion of HGF and revealed that extrinsic HGF stimulation induced clonogenicity and LGR5 messenger RNA expression in Wnt-low, more differentiated cancer cells. These findings pointed to the possibility of developmental plasticity of tumor bulk populations when directed by the tumor microenvironment. However, as acknowledged by the authors, while the de-differentiated tumor bulk cells possessed enhanced clonogenicity and tumorigenicity, it has not been determined whether these cells also have unlimited replicative potential, which defines CSCs.
Subsequently, Fumagalli et al137 investigated the role of CSCs in driving CRC metastasis using the LGR5 reporter system. They showed that, despite the well-documented essential role of LGR5-positive CRC CSCs in metastatic cancer progression,97 the majority of metastasis-initiating cells in their study were LGR5-negative. Specifically, when examined in an orthotopic murine CRC model, LGR5-negative cells were first to migrate out of the primary tumor, were significantly enhanced within the disseminated cancer cells in the circulation, and were capable of initiating metastatic lesions in the liver. Intriguingly, the clinical progression of these LGR5-negative metastatic lesions was critically dependent on their ability to generate LGR5-positive cells, and selective LGR5-positive cell ablation led to tumor regression. Although this study demonstrates that some of the LGR5-negative cells can give rise to LGR5-positive CRC CSCs, it is also conceivable that this LGR5-negative subpopulation might represent cells hierarchically upstream of LGR5-positive CSCs. Alternatively, LGR5 may have a more restricted capacity than previously anticipated to successfully identify CRC CSC across all CRC models studied to date.
To investigate the developmental plasticity of the differentiated LGR5-negative KRT20-positive CRC cells further, Shimokawa et al98 employed human CRC-derived organoids, in which CRC cells were engineered to express a Caspase-9 construct inserted in the LGR5 locus that can be activated upon chemical dimerization. Using lineage tracing of KRT20-positive CRC cells, they showed that selective ablation of LGR5-positive CSCs enabled KRT20-expressing cells to acquire LGR5 positivity. They posited that in the setting of CSC depletion, migration of KRT20-positive cells into the vacant CSC niche allows their reprogramming by the LGR5-positive CSC-conditioned tumor microenvironment.
The critical role of the tumor microenvironment and CSC niches in the maintenance of the CSC phenotype was further supported by Lenos et al,96 who performed an unbiased marker-free quantitative analysis of colon cancer growth dynamics and response to therapy, using stochastically labeled tumor cells and following their clonal contribution to tumor growth over time. The study proposed that the CRC CSC functional phenotype was independent of marker expression or cell-autonomous clonogenicity, and was defined by the CSC microenvironment. Moreover, the authors suggested that strategies that abolish activating signals from the stromal compartment and their cell-intrinsic molecular pathways are essential for successful cancer therapies.
While these recent studies challenge the existence of an irreversible unidirectional tumor hierarchy in CRC, it is important to note that such an attribute is neither part of nor a logical consequence of the CSC consensus definition.28 Indeed, these findings highlight the importance of multi-faceted, holistic approaches aimed at simultaneous eradication of all cancer cell populations (CSCs or non-CSC bulk populations) and their niches. The specific therapeutic potential of niche-targeted strategies is further illustrated by the clinical success of EGFR- and VEGFR-directed therapies designed to abrogate CSC niche-derived EGF and VEGF signaling. Furthermore, selective killing of tumor bulk populations by cell-cycle targeting radiation and chemotherapeutic agents significantly improves patient outcomes. In addition, novel agents aimed at additional CSC- and CSC niche-specific pathways, such as, for example, inhibition of STAT3 and c-MET have shown significant promise in clinical trials (Figure 3). Indeed, future strategies that enable specific targeting of CSCs and their niches, while sparing normal tissue-resident stem cells by using, for example, tumor-limited drug delivery based on mutation barcoding, might further reduce the considerable toxicity of such therapies.
Conclusions
Despite significant progress, metastatic CRC, remains an largely incurable disease. The CSC concept offers a potential pathophysiologic explanation for cancer therapeutic escape through resistance to cell-cycle targeting agents, such as chemo- and radiation therapy, and suggests that cancer treatments could be improved by elimination of CSCs. This concept, initially proposed at the cellular level, has now been further examined by diverse multi-omic studies. In the not so distant past, all CRCs were treated as 1 pathologic entity, and only the size of the tumor or the presence of metastatic spread guided therapeutic decisions. Subsequent genetic studies uncovered inter-tumor molecular heterogeneity and led to the development of novel targeted biologic agents that have improved patient outcomes significantly. Recent RNA sequencing studies have enabled a more comprehensive CRC molecular classification that can be correlated with patient responses and used to guide treatment decisions. Moreover, most recently, lineage tracing studies have revealed a critical importance of the CRC CSC niche in the development and maintenance of the CSC phenotype and tumor cell plasticity. Such novel single-cell multi-omic and lineage tracing approaches have further delineated and catalogued intra-tumor heterogeneity and enabled the design of some of the most inclusive cancer therapies to date, which simultaneously target all cancer cell populations within a given malignancy. Implementation and further refinement of such approaches might bring us closer to the ultimate goal of cures through successful cancer eradication.
Acknowledgments
Author contributions: Markus H. Frank and Natasha Y. Frank designed the paper, Markus H. Frank, Brian J. Wilson, Jason S. Gold, and Natasha Y. Frank wrote the manuscript.
Funding
Supported by Veterans Affairs Biomedical Laboratory Research and Development 1I01BX000516 and Veterans Affairs Rehabilitation Research and Development 1I01RX000989 Merit Review Awards and a Department of Defense Translational Team Science Award to Natasha Y. Frank, Veterans Affairs Biomedical Laboratory Research and Development I01BX003771 Merit Review Award to Jason S. Gold, as well as National Institutes of Health/National Cancer Institute grants R01CA113796, R01CA158467 and R01CA138231 to Markus H. Frank.
Abbreviations used in this paper:
- ABC
ATP-binding cassette
- CMS
consensus molecular subtype
- CRC
colorectal cancer
- CSC
cancer stem cell
- EGFR
epidermal growth factor receptor
- EpCAM
epithelial cell adhesion molecule
- 5-FU
5-fluorouracil
- HGF
hepatocyte growth factor
- IL
interleukin
- MDR
multidrug resistance
- MHC
major histocompatibility complex
- NK
natural killer
- SCNA
somatic copy number alteration
- VEGF
vascular endothelial growth factor
Footnotes
Conflicts of interest
These authors disclose the following: Natasha Y. Frank and Markus H. Frank are co-inventors of the ABCB5-related US patent 7,928,202 (Targeting ABCB5 for cancer therapy) assigned to Brigham & Women’s Hospital, Boston, Massachusetts, and licensed to Ticeba GmbH (Heidelberg, Germany) and Rheacell GmbH & Co. KG (Heidelberg, Germany). Markus H. Frank serves as a scientific advisor to Ticeba GmbH and Rheacell GmbH & Co. KG. The remaining authors disclose no conflicts.
References
- 1.Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer 2004;4:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004;10:789–799. [DOI] [PubMed] [Google Scholar]
- 3.Bruce WR, Van Der Gaag H. A quantitative assay for the number of murine lymphoma cells capable of proliferation in vivo. Nature 1963;199:79–80. [DOI] [PubMed] [Google Scholar]
- 4.Fidler IJ, Hart IR. Biological diversity in metastatic neo-plasms: origins and implications. Science 1982; 217:998–1003. [DOI] [PubMed] [Google Scholar]
- 5.Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature 2001;414:105–111. [DOI] [PubMed] [Google Scholar]
- 6.Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest 2010;120:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalerba P, Dylla SJ, Park IK, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A 2007;104:10158–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Brien CA, Pollett A, Gallinger S, et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007;445:106–110. [DOI] [PubMed] [Google Scholar]
- 9.Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature 2007;445:111–115. [DOI] [PubMed] [Google Scholar]
- 10.Wilson BJ, Schatton T, Zhan Q, et al. ABCB5 identifies a therapy-refractory tumor cell population in colorectal cancer patients. Cancer Res 2011;71:5307–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009; 457:608–611. [DOI] [PubMed] [Google Scholar]
- 12.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet 2008;40:915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu L, Gibson P, Currle DS, et al. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 2009;457:603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Visvader JE. Cells of origin in cancer. Nature 2011; 469:314–322. [DOI] [PubMed] [Google Scholar]
- 15.The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez-Villamil B, Romera-Lopez A, Hernandez-Prieto S, et al. Colon cancer molecular subtypes identified by expression profiling and associated to stroma, mucinous type and different clinical behavior. BMC Cancer 2012; 12:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlicker A, Beran G, Chresta CM, et al. Subtypes of primary colorectal tumors correlate with response to targeted treatment in colorectal cell lines. BMC Med Genomics 2012;5:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budinska E, Popovici V, Tejpar S, et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol 2013;231:63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marisa L, de Reynies A, Duval A, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med 2013;10:e1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Sousa e Melo F, Wang X, Jansen M, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med 2013;19:614–618. [DOI] [PubMed] [Google Scholar]
- 21.Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med 2013;19:619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roepman P, Schlicker A, Tabernero J, et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int J Cancer 2014;134: 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015; 21:1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lenz H-J, Ou F-S, Venook AP, et al. Impact of consensus molecular subtype on survival in patients with metastatic colorectal cancer: results from CALGB/SWOG 80405 (Alliance). J Clin Oncol 2019;37:1876–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–767. [DOI] [PubMed] [Google Scholar]
- 26.Sottoriva A, Kang H, Ma Z, et al. A Big Bang model of human colorectal tumor growth. Nat Genet 2015; 47:209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clevers H The cancer stem cell: premises, promises and challenges. Nat Med 2011;17:313–319. [DOI] [PubMed] [Google Scholar]
- 28.Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006;66:9339–9344. [DOI] [PubMed] [Google Scholar]
- 29.Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet 2016;17:175–188. [DOI] [PubMed] [Google Scholar]
- 30.Wu H, Zhang XY, Hu Z, et al. Evolution and heterogeneity of non-hereditary colorectal cancer revealed by single-cell exome sequencing. Oncogene 2017; 36:2857–2867. [DOI] [PubMed] [Google Scholar]
- 31.Yu C, Yu J, Yao X, et al. Discovery of biclonal origin and a novel oncogene SLC12A5 in colon cancer by single-cell sequencing. Cell Res 2014;24:701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu M, Liu Y, Di J, et al. Multi-region and single-cell sequencing reveal variable genomic heterogeneity in rectal cancer. BMC Cancer 2017;17:787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu M, Di J, Liu Y, et al. Comparison of EpCAM(high) CD44(+) cancer stem cells with EpCAM(high)CD44(−) tumor cells in colon cancer by single-cell sequencing. Cancer Biol Ther 2018;19:939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heitzer E, Auer M, Gasch C, et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res 2013;73:2965–2975. [DOI] [PubMed] [Google Scholar]
- 35.Leung ML, Davis A, Gao R, et al. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res 2017;27:1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawson DA, Kessenbrock K, Davis RT, et al. Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol 2018;20:1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roerink SF, Sasaki N, Lee-Six H, et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018;556:457–462. [DOI] [PubMed] [Google Scholar]
- 38.Dalerba P, Kalisky T, Sahoo D, et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol 2011;29:1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Courtois ET, Sengupta D, et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet 2017;49:708–718. [DOI] [PubMed] [Google Scholar]
- 40.Bian S, Hou Y, Zhou X, et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 2018;362:1060–1063. [DOI] [PubMed] [Google Scholar]
- 41.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 2009;361:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyerhardt JA, Mayer RJ. Systemic therapy for colorectal cancer. N Engl J Med 2005;352:476–487. [DOI] [PubMed] [Google Scholar]
- 43.Pickering L, Rudman S, Ross PJ, et al. Targeted therapy in colorectal carcinoma: more than a theory. Colorectal Dis 2008;10:209–218; discussion 218–221. [DOI] [PubMed] [Google Scholar]
- 44.Kopetz S, Chang GJ, Overman MJ, et al. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol 2009;27:3677–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bennouna J, Sastre J, Arnold D, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol 2013;14:29–37. [DOI] [PubMed] [Google Scholar]
- 46.Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013; 381:303–312. [DOI] [PubMed] [Google Scholar]
- 47.Li J, Qin S, Xu R, et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2015; 16:619–629. [DOI] [PubMed] [Google Scholar]
- 48.Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E–mutated colorectal cancer. N Engl J Med 2019;381:1632–1643. [DOI] [PubMed] [Google Scholar]
- 49.Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med 2013; 19:1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dylla SJ, Beviglia L, Park IK, et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One 2008;3:e2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Todaro M, Alea MP, Di Stefano AB, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 2007;1:389–402. [DOI] [PubMed] [Google Scholar]
- 52.Luraghi P, Reato G, Cipriano E, et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res 2014;74:1857–1869. [DOI] [PubMed] [Google Scholar]
- 53.Boccaccio C, Luraghi P, Comoglio PM. MET-mediated resistance to EGFR inhibitors: an old liaison rooted in colorectal cancer stem cells. Cancer Res 2014;74:3647–3651. [DOI] [PubMed] [Google Scholar]
- 54.De Angelis ML, Zeuner A, Policicchio E, et al. Cancer stem cell-based models of colorectal cancer reveal molecular determinants of therapy resistance. Stem Cells Transl Med 2016;5:511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joosten SPJ, Mizutani T, Spaargaren M, et al. MET signaling overcomes epidermal growth factor receptor inhibition in normal and colorectal cancer stem cells causing drug resistance. Gastroenterology 2019; 157:1153–1155.e1. [DOI] [PubMed] [Google Scholar]
- 56.Khelwatty SA, Essapen S, Bagwan I, et al. Co-expression and prognostic significance of putative CSC markers CD44, CD133, wild-type EGFR and EGFRvIII in metastatic colorectal cancer. Oncotarget 2019;10:1704–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng Y, Gao S, Gao Y, et al. Anti-EGFR antibody sensitizes colorectal cancer stem-like cells to Fluorouracilinduced apoptosis by affecting autophagy. Oncotarget 2016;7:81402–81409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Modest DP, Ricard I, Heinemann V, et al. Outcome according to KRAS-, NRAS- and BRAF-mutation as well as KRAS mutation variants: pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann Oncol 2016; 27:1746–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016; 27:1386–1422. [DOI] [PubMed] [Google Scholar]
- 60.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350:2335–2342. [DOI] [PubMed] [Google Scholar]
- 61.Tabernero J, Yoshino T, Cohn AL, et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol 2015;16:499–508. [DOI] [PubMed] [Google Scholar]
- 62.Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 2012;30:3499–3506. [DOI] [PubMed] [Google Scholar]
- 63.Ellis LM, Hicklin DJ. Pathways mediating resistance to vascular endothelial growth factor-targeted therapy. Clin Cancer Res 2008;14:6371–6375. [DOI] [PubMed] [Google Scholar]
- 64.Lin SP, Lee YT, Yang SH, et al. Colon cancer stem cells resist antiangiogenesis therapy-induced apoptosis. Cancer Lett 2013;328:226–234. [DOI] [PubMed] [Google Scholar]
- 65.Becherirat S, Valamanesh F, Karimi M, et al. Discontinuous schedule of bevacizumab in colorectal cancer induces accelerated tumor growth and phenotypic changes. Transl Oncol 2018;11:406–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol 2004;22:329–360. [DOI] [PubMed] [Google Scholar]
- 67.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prenen H, Hendlisz A, Shaza L, et al. P330: Results from the completed dose-escalation of the alloSHRINK phase I study evaluating the allogeneic NKG2D-based CAR T-cell therapy CYAD-101 in metastatic colorectal cancer patients. Presented at: The Society for Immunotherapy of Cancer (SITC) Congress; November 6–10, 2019; National Harbor, MD, 2019. [Google Scholar]
- 69.Volonte A, Di Tomaso T, Spinelli M, et al. Cancer-initiating cells from colorectal cancer patients escape from T cell-mediated immunosurveillance in vitro through membrane-bound IL-4. J Immunol 2014; 192:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tallerico R, Todaro M, Di Franco S, et al. Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J Immunol 2013;190:2381–2390. [DOI] [PubMed] [Google Scholar]
- 71.Gao XY, Wang XL. An adoptive T cell immunotherapy targeting cancer stem cells in a colon cancer model. J Buon 2015;20:1456–1463. [PubMed] [Google Scholar]
- 72.Inoda S, Hirohashi Y, Torigoe T, et al. Cytotoxic T lymphocytes efficiently recognize human colon cancer stem-like cells. Am J Pathol 2011;178:1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu Y, Chen M, Wu P, et al. Increased PD-L1 expression in breast and colon cancer stem cells. Clin Exp Pharmacol Physiol 2017;44:602–604. [DOI] [PubMed] [Google Scholar]
- 74.Zhi Y, Mou Z, Chen J, et al. B7H1 Expression and epithelial-to-mesenchymal transition phenotypes on colorectal cancer stem-like cells. PLoS One 2015;10: e0135528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wei F, Zhang T, Deng SC, et al. PD-L1 promotes colorectal cancer stem cell expansion by activating HMGA1-dependent signaling pathways. Cancer Lett 2019;450:1–13. [DOI] [PubMed] [Google Scholar]
- 76.Noh KH, Lee YH, Jeon JH, et al. Cancer vaccination drives Nanog-dependent evolution of tumor cells toward an immune-resistant and stem-like phenotype. Cancer Res 2012;72:1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Irvin DK, Jouanneau E, Duvall G, et al. T cells enhance stem-like properties and conditional malignancy in gliomas. PLoS One 2010;5:e10974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gubin MM, Artyomov MN, Mardis ER, et al. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest 2015;125:3413–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hua W, Yao Y, Chu Y, et al. The CD133+ tumor stem-like cell-associated antigen may elicit highly intense immune responses against human malignant glioma. J Neurooncol 2011;105:149–157. [DOI] [PubMed] [Google Scholar]
- 80.Zhu X, Prasad S, Gaedicke S, et al. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget 2015;6:171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wallach-Dayan SB, Rubinstein AM, Hand C, et al. DNA vaccination with CD44 variant isoform reduces mammary tumor local growth and lung metastasis. Mol Cancer Ther 2008;7:1615–1623. [DOI] [PubMed] [Google Scholar]
- 82.Visus C, Wang Y, Lozano-Leon A, et al. Targeting ALDH(bright) human carcinoma-initiating cells with ALDH1A1-specific CD8(+) T cells. Clin Cancer Res 2011;17:6174–6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gong X, Azhdarinia A, Ghosh SC, et al. LGR5-targeted antibody-drug conjugate eradicates gastrointestinal tumors and prevents recurrence. Mol Cancer Ther 2016; 15:1580–1590. [DOI] [PubMed] [Google Scholar]
- 84.Junttila MR, Mao W, Wang X, et al. Targeting LGR5+ cells with an antibody-drug conjugate for the treatment of colon cancer. Sci Transl Med 2015;7:314ra186. [DOI] [PubMed] [Google Scholar]
- 85.Miyamoto S, Kochin V, Kanaseki T, et al. The antigen ASB4 on cancer stem cells serves as a target for CTL immunotherapy of colorectal cancer. Cancer Immunol Res 2018;6:358–369. [DOI] [PubMed] [Google Scholar]
- 86.Yamada R, Takahashi A, Torigoe T, et al. Preferential expression of cancer/testis genes in cancer stem-like cells: proposal of a novel sub-category, cancer/testis/stem gene. Tissue Antigens 2013;81:428–434. [DOI] [PubMed] [Google Scholar]
- 87.Morita R, Nishizawa S, Torigoe T, et al. Heat shock protein DNAJB8 is a novel target for immunotherapy of colon cancer-initiating cells. Cancer Sci 2014;105:389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morita R, Hirohashi Y, Torigoe T, et al. Olfactory receptor family 7 subfamily C member 1 is a novel marker of colon cancer-initiating cells and is a potent target of immunotherapy. Clin Cancer Res 2016;22:3298–3309. [DOI] [PubMed] [Google Scholar]
- 89.Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van Rooij N, van Buuren MM, Philips D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 2013;31:e439–e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016; 351:1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mennonna D, Maccalli C, Romano MC, et al. T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut 2017;66:454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Todaro M, D’Asaro M, Caccamo N, et al. Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J Immunol 2009;182:7287–7296. [DOI] [PubMed] [Google Scholar]
- 95.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med 2017;23:1124–1134. [DOI] [PubMed] [Google Scholar]
- 96.Lenos KJ, Miedema DM, Lodestijn SC, et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat Cell Biol 2018;20:1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.de Sousa e Melo F, Kurtova AV, Harnoss JM, et al. A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature 2017;543:676–680. [DOI] [PubMed] [Google Scholar]
- 98.Shimokawa M, Ohta Y, Nishikori S, et al. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 2017;545:187–192. [DOI] [PubMed] [Google Scholar]
- 99.Kryczek I, Lin Y, Nagarsheth N, et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness þvia STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014;40:772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Matthews JR, Sansom OJ, Clarke AR. Absolute requirement for STAT3 function in small-intestine crypt stem cell survival. Cell Death Differ 2011;18:1934–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bak Y, Kwon T, Bak IS, et al. IL-32theta inhibits stemness and epithelial-mesenchymal transition of cancer stem cells via the STAT3 pathway in colon cancer. Oncotarget 2016;7:7307–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang T, Song P, Zhong T, et al. The inflammatory cytokine IL-6 induces FRA1 deacetylation promoting colorectal cancer stem-like properties. Oncogene 2019; 38:4932–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shimizu M, Tanaka N. IL-8-induced O-GlcNAc modification via GLUT3 and GFAT regulates cancer stem cell-like properties in colon and lung cancer cells. Oncogene 2019;38:1520–1533. [DOI] [PubMed] [Google Scholar]
- 104.Lotti F, Jarrar AM, Pai RK, et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J Exp Med 2013; 210:2851–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fang M, Li Y, Huang K, et al. IL33 Promotes colon cancer cell stemness via JNK activation and macrophage recruitment. Cancer Res 2017;77:2735–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nakano M, Kikushige Y, Miyawaki K, et al. Dedifferentiation process driven by TGF-beta signaling enhances stem cell properties in human colorectal cancer. Oncogene 2019;38:780–793. [DOI] [PubMed] [Google Scholar]
- 107.Lu LL, Chen XH, Zhang G, et al. CCL21 Facilitates chemoresistance and cancer stem cell-like properties of colorectal cancer cells through AKT/GSK-3beta/Snail signals. Oxid Med Cell Longev 2016; 2016:5874127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen MC, Baskaran R, Lee NH, et al. CXCL2/CXCR2 axis induces cancer stem cell characteristics in CPT-11-resistant LoVo colon cancer cells via Galphai-2 and Galphaq/11. J Cell Physiol 2019;234:11822–11834. [DOI] [PubMed] [Google Scholar]
- 109.Bellamkonda K, Sime W, Sjolander A. The impact of inflammatory lipid mediators on colon cancer-initiating cells. Mol Carcinog 2015;54:1315–1327. [DOI] [PubMed] [Google Scholar]
- 110.Wang D, Fu L, Sun H, et al. Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology 2015;149:1884–1895 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang Y, Yin K, Tian J, et al. Granulocytic myeloid-derived suppressor cells promote the stemness of colorectal cancer cells through exosomal S100A9. Adv Sci (Weinh) 2019;6:1901278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Céspedes MV, Unzueta U, Aviñó A, et al. Selective depletion of metastatic stem cells as therapy for human colorectal cancer. EMBO Mol Med 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lombardo Y, Scopelliti A, Cammareri P, et al. Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology 2011;140:297–309. e6. [DOI] [PubMed] [Google Scholar]
- 114.Veschi V, Mangiapane LR, Nicotra A, et al. Targeting chemoresistant colorectal cancer via systemic administration of a BMP7 variant. Oncogene 2020;39. 987–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Desai A, Yan Y, Gerson SL. Concise reviews: cancer stem cell targeted therapies: toward clinical success. Stem Cells Transl Med 2019;8:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vermeulen L, Felipe De Sousa EM, Van Der Heijden M, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 2010; 12:468–476. [DOI] [PubMed] [Google Scholar]
- 117.Van Cutsem E, Eng C, Nowara E, et al. Randomized phase Ib/II trial of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clin Cancer Res 2014;20:4240–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2002;2:48–58. [DOI] [PubMed] [Google Scholar]
- 119.Fletcher JI, Haber M, Henderson MJ, et al. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer 2010;10:147–156. [DOI] [PubMed] [Google Scholar]
- 120.Frank NY, Pendse SS, Lapchak PH, et al. Regulation of progenitor cell fusion by ABCB5 P-glycoprotein, a novel human ATP-binding cassette transporter. J Biol Chem 2003;278:47156–47165. [DOI] [PubMed] [Google Scholar]
- 121.Frank NY, Margaryan A, Huang Y, et al. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res 2005; 65:4320–4333. [DOI] [PubMed] [Google Scholar]
- 122.Schatton T, Frank MH. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res 2008; 21:39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wilson BJ, Saab KR, Ma J, et al. ABCB5 maintains melanoma-initiating cells through a proinflammatory cytokine signaling circuit. Cancer Res 2014;74:4196–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Civenni G, Walter A, Kobert N, et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res 2011;71:3098–3109. [DOI] [PubMed] [Google Scholar]
- 125.Lee CA, Banerjee P, Wilson BJ, et al. Targeting the ABC transporter ABCB5 sensitizes glioblastoma to temozolomide-induced apoptosis through a cell-cycle checkpoint regulation mechanism. J Biol Chem 2020013778. jbc. RA120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cheung ST, Cheung PF, Cheng CK, et al. Granulinepithelin precursor and ATP-dependent binding cassette (ABC)B5 regulate liver cancer cell chemoresistance. Gastroenterology 2011;140:344–355. [DOI] [PubMed] [Google Scholar]
- 127.Yang JY, Ha SA, Yang YS, et al. p-Glycoprotein ABCB5 and YB-1 expression plays a role in increased heterogeneity of breast cancer cells: correlations with cell fusion and doxorubicin resistance. BMC Cancer 2010; 10:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Govindan R, Ding L, Griffith M, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012;150:1121–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Grimm M, Krimmel M, Polligkeit J, et al. ABCB5 expression and cancer stem cell hypothesis in oral squamous cell carcinoma. Eur J Cancer 2012;48:3186–3197. [DOI] [PubMed] [Google Scholar]
- 130.Kugimiya N, Nishimoto A, Hosoyama T, et al. The c-MYC-ABCB5 axis plays a pivotal role in 5-fluorouracil resistance in human colon cancer cells. J Cell Mol Med 2015;19:1569–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sharma BK, Manglik V, Elias EG. Immuno-expression of human melanoma stem cell markers in tissues at different stages of the disease. J Surg Res 2010; 163:e11–e15. [DOI] [PubMed] [Google Scholar]
- 132.Gazzaniga P, Cigna E, Panasiti V, et al. CD133 and ABCB5 as stem cell markers on sentinel lymph node from melanoma patients. Eur J Surg Oncol 2010; 36:1211–1214. [DOI] [PubMed] [Google Scholar]
- 133.Guo Q, Grimmig T, Gonzalez G, et al. ATP-binding cassette member B5 (ABCB5) promotes tumor cell invasiveness in human colorectal cancer. J Biol Chem 2018;293:11166–11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dunne PD, McArt DG, Blayney JK, et al. AXL is a key regulator of inherent and chemotherapy-induced invasion and predicts a poor clinical outcome in early-stage colon cancer. Clin Cancer Res 2014; 20:164–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Linger RM, Cohen RA, Cummings CT, et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013;32:3420–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Medema JP. Cancer stem cells: the challenges ahead. Nat cell Biol 2013;15:338–344. [DOI] [PubMed] [Google Scholar]
- 137.Fumagalli A, Oost KC, Kester L, et al. Plasticity of Lgr5-negative cancer cells drives metastasis in colorectal cancer. Cell Stem Cell 2020;26:569–578.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]