

Abstract
Motivation
Alternative polyadenylation (APA) plays a key post-transcriptional regulatory role in mRNA stability and functions in eukaryotes. Single cell RNA-seq (scRNA-seq) is a powerful tool to discover cellular heterogeneity at gene expression level. Given 3′ enriched strategy in library construction, the most commonly used scRNA-seq protocol—10× Genomics enables us to improve the study resolution of APA to the single cell level. However, currently there is no computational tool available for investigating APA profiles from scRNA-seq data.
Results
Here, we present a package scDAPA for detecting and visualizing dynamic APA from scRNA-seq data. Taking bam/sam files and cell cluster labels as inputs, scDAPA detects APA dynamics using a histogram-based method and the Wilcoxon rank-sum test, and visualizes candidate genes with dynamic APA. Benchmarking results demonstrated that scDAPA can effectively identify genes with dynamic APA among different cell groups from scRNA-seq data.
Availability and implementation
The scDAPA package is implemented in Shell and R, and is freely available at https://scdapa.sourceforge.io.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Alternative polyadenylation (APA) is increasingly recognized as an important regulation mechanism for many biological processes (e.g. cell development, differentiation and proliferation) and molecular functions (e.g. mRNA stability, translation efficiency and localization) via dynamically using different polyadenylation sites during maturation of nuclear pre-mRNA (Chen et al., 2017). More and more evidence shows widespread occurrences of cell type-specific APA in eukaryotes (Cao et al., 2019; Hwang et al., 2017; Velten et al., 2015). To profile APA dynamics among different cell types, one traditional way is to dissociate tissues and purify cell types before performing Poly(A)-tag sequencing (Cao et al., 2019), which may introduce unexpected cellular stress and affect the APA profile consequently. An alternative way is using methods like cTag-PAPERCLIP (Hwang et al., 2017), which could directly profile individual cell types from tissues without enzymatic digestion and fluorescent-activated cell sorting. The common limitation of these two strategies is that they cannot deal with unknown cell types or rare cell subpopulations. The ideal way to differentiate APA profiles from different cell types is to perform single cell RNA-seq (scRNA-seq), which can isolate cells of different states and/or types by dissecting their transcriptome profiles (Saliba et al., 2014; Zheng et al., 2017). By integrating a conventional scRNA-seq protocol and a 3′ enriched bulk population RNA-seq protocol, Velten et al. (2015) developed a method BATseq to investigate the APA profile at single cell resolution. However, the low sensitivity and complex steps of BATseq limit its wide application (Chen et al., 2017). Promising experimental protocols for quantifying APA at single cell level are still lacking.
One commonly used scRNA-seq protocol, i.e. 10× Genomics, applying a 3′ selection/enriched strategy in library construction, provides the potential of quantifying APA dynamics at single cell resolution (Chen et al., 2017; Saliba et al., 2014; Ye et al., 2019). In our recent study (Ye et al., 2019), we found out 3′ ends extracted from the 10× Genomics scRNA-seq data are quite adjacent to authentic poly(A) sites or 3′ UTR annotations defined by sequencing experiments, and investigated the role of APA in acute myeloid leukemia using several scRNA-seq datasets from 10× Genomics, demonstrating the high validity and value of existing scRNA-seq data in performing APA analysis. However, currently there is no easy-to-use computational tool available for investigating APA profiles from scRNA-seq data. In this work, we developed a package scDAPA for exploration and visualization of APA dynamics in different cell types and conditions from scRNA-seq data.
2 Implementation
The scDAPA consists of three major steps (Supplementary Fig. S1): (i) 3′ ends extraction and annotation; (ii) dynamic APA detection; and (iii) dynamic APA visualization. First, scDAPA takes the sequence alignment results (a bam/sam file) and cell type information (a cell barcode-cell type data in csv format) as inputs, and extracts valid mapping records (uniquely mapped in genomic region, with valid cell barcode and not PCR duplicates etc.) into different files of distinct cell types. 3′ ends of extracted reads are annotated to genes based on the given genome annotation file (a gff/gtf file). Second, the dynamic APA is detected among different cell types within the same biological samples or among different samples of the same cell type. Briefly, we use a histogram-based method to divide the dispersed 3′ ends into distinct bins with the same width (default 100 bp), and a Site Distribution Difference (SDD) index is calculated to quantify the APA difference between conditions as , where N is the number of bins, A and B denote two different cell groups, and represents the percentage of 3′ ends located in the nth bin of a specific gene in cell group A. Then, the Wilcoxon rank-sum test is applied to measure the significance of differential APA usage. A gene with a SDD value above a given cutoff (e.g. 0.2) and a p-value below a given cutoff (e.g. 0.05) will be recognized a gene with significant differential APA usage (DE-APA gene). For multiple statistical tests, the Benjamini-Hochberg method is employed to control the false discovery rate. Last, scDAPA allows intuitive visualization of APA profiles of candidate genes under different conditions, which presents diverse isoforms of a target gene, and a smooth density plot alongside the gene to show the 3′ ends distribution. The Supplementary Material contains a user manual of the scDAPA package.
3 Application example
We investigated the application of scDAPA on a scRNA-seq dataset of live microglia/macrophages from pooled neuroretinas of normal and light damaged mice generated by the 10× Genomics platform (O’Koren et al., 2019). We extracted the 3′ ends of valid reads from the scRNA-seq dataset and compared them to the latest mouse poly(A) site annotations from PolyA_DB 3/Ensembl (Wang et al., 2018). The distribution of the distances between 3′ ends of the aligned scRNA-seq reads and their nearest poly(A) annotations is similar to that of our previous work (Fig. 1a) (Ye et al., 2019), indicating a high stability of 10× Genomics scRNA-seq data in representing APA site usage. Next, we performed a cell type-to-cell type comparison to detect genes with dynamic APA across different cell groups (DE-APA genes). DE-APA genes were categorized into two groups: APA gene and non-APA gene. Referring to the annotations from PolyA_DB 3/Ensembl, a gene with only one poly(A) site is recognized as a non-APA gene, otherwise it’s recognized as an APA gene. Approximately 98% of 3137 detected non-redundant DE-APA genes are annotated as APA genes (Fisher’s exact test p-value < 2.2 × e−16, Fig. 1b), representing a high confidence of scDAPA in dynamic APA detection using scRNA-seq data.
Fig. 1.
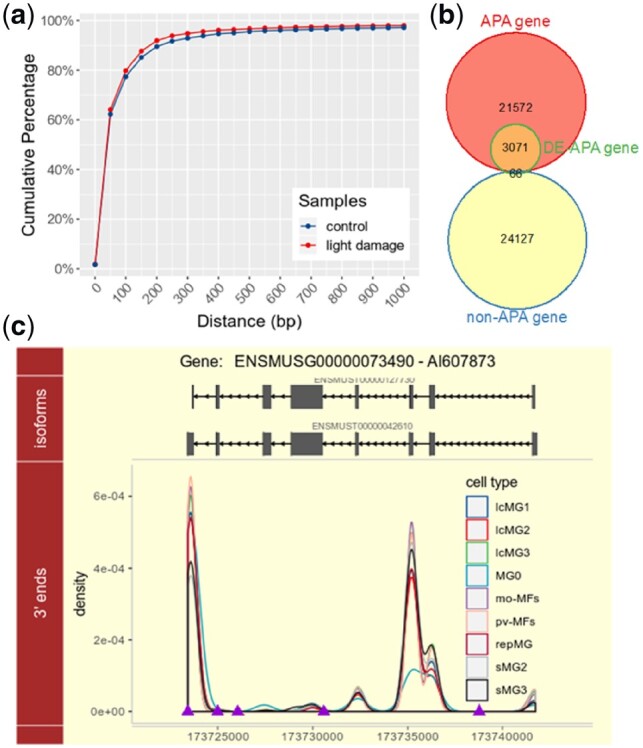
An application example of scDAPA on a scRNA-seq dataset of neuroretinas from mouse. (a) Cumulative distribution of distances between 3′ ends and nearest authentic poly(A) sites; (b) categories of DE-APA genes detected from scRNA-seq data; (c) illustration of a gene AI607873 showing dynamic APA profiles across different cell groups. Top panel, isoforms of gene AI607873 from Ensembl; bottom panel, density distribution of 3′ ends of various cell groups; purple triangles represent the poly(A) site annotation from PolyA_DB 3. (Color version of this figure is available at Bioinformatics online.)
Additionally, in pair-wise cell type comparisons of APA usage, the cell type MG0 (microglia solely came from normal neuroretinas) always shows the highest percent of DE-APA genes compared with other nine cell types (Supplementary Fig. S2), suggesting a dramatically distinct APA preference of the nine cell types (IcMG1∼3, sMG1∼3, mo-MFs, pv-MFs, repMG: microglia mainly came from light damaged neuroretinas) compared to steady state MG0. Among the identified DE-APA genes, e.g. a gene AI607873 prefers using a distal poly(A) site in MG0 compared with other nine cell types (Fig. 1c). Moreover, among the three small microglia clusters (sMG1, sMG2 and sMG3), sMG3 shows the most APA dynamics compared to MG0 (Supplementary Fig. S2). This result is consistent with the previous observation that sMG3 was particularly distinct from MG0, and was found at the final state of a trajectory analysis (O’Koren et al., 2019). Considering that sMG3 is corresponding to the subretinal microglia in light damaged sample (O’Koren et al., 2019), KEGG and REACTOME pathway enrichment analyses for DE-APA genes between MG0 and sMG3 were further performed. We found that these DE-APA genes were significantly over-represented in many signaling pathways, such as NF-kappa B, JAK-STAT and TNF signaling pathways (Supplementary Fig. S3a and b), which are associated with the retinal photoreceptor degeneration (Rashid et al., 2018). These results demonstrate that scDAPA can effectively identify genes with dynamic APA among different cell groups from scRNA-seq data.
4 Conclusion
In summary, we developed a package scDAPA to detect and visualize dynamic APA from scRNA-seq data. We demonstrated its utilities through application to a real dataset. It is believed that scDAPA is a useful tool in studying APA at single cell resolution, and will broadly extend the application scope of scRNA-seq data.
Funding
This research was supported in part by the Fundamental Research Funds for the Central Universities in China [Xiamen University: 20720170076 and 20720190106], and the National Natural Science Foundation of China [61802323, 31801268 and 61573296].
Conflict of Interest: none declared.
Supplementary Material
Contributor Information
Congting Ye, Key Laboratory of the Ministry of Education for Coastal and Wetland Ecosystems, College of the Environment and Ecology, Xiamen University, Xiamen, Fujian 361102, China.
Qian Zhou, Key Laboratory of the Ministry of Education for Coastal and Wetland Ecosystems, College of the Environment and Ecology, Xiamen University, Xiamen, Fujian 361102, China.
Xiaohui Wu, Department of Automation, Xiamen University, Xiamen, Fujian 361005, China; National Institute for Data Science in Health and Medicine, Xiamen University, Xiamen, Fujian 361102, China.
Chen Yu, Department of Ophthalmology, Duke University, Durham, NC 27710, USA.
Guoli Ji, Department of Automation, Xiamen University, Xiamen, Fujian 361005, China; National Institute for Data Science in Health and Medicine, Xiamen University, Xiamen, Fujian 361102, China.
Daniel R Saban, Department of Ophthalmology, Duke University, Durham, NC 27710, USA; Department of Immunology, Duke University, Durham, NC 27710, USA.
Qingshun Q Li, Key Laboratory of the Ministry of Education for Coastal and Wetland Ecosystems, College of the Environment and Ecology, Xiamen University, Xiamen, Fujian 361102, China; Graduate College of Biomedical Sciences, Western University of Health Sciences, Pomona, CA 91766, USA.
References
- Cao J. et al. (2019) Root hair single cell type specific profiles of gene expression and alternative polyadenylation under cadmium stress. Front. Plant Sci., 10, 589.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. et al. (2017) Alternative polyadenylation: methods, findings, and impacts. Genomics Proteomics Bioinformatics, 15, 287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H.-W. et al. (2017) cTag-PAPERCLIP reveals alternative polyadenylation promotes cell-type specific protein diversity and shifts Araf isoforms with microglia activation. Neuron, 95, 1334–1349.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Koren E.G. et al. (2019) Microglial function is distinct in different anatomical locations during retinal homeostasis and degeneration. Immunity, 50, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid K. et al. (2018) Microglia activation and immunomodulatory therapies for retinal degenerations. Front. Cell Neurosci., 12, 176.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba A.-E. et al. (2014) Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res., 42, 8845–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velten L. et al. (2015) Single‐cell polyadenylation site mapping reveals 3′ isoform choice variability. Mol. Syst. Biol., 11, 812.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. et al. (2018) PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res., 46, D315–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C. et al. (2019) Role of alternative polyadenylation dynamics in acute myeloid leukaemia at single-cell resolution. RNA Biol., 16, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G.X.Y. et al. (2017) Massively parallel digital transcriptional profiling of single cells. Nat. Commun., 8, 14049.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.