

Abstract
Background and purpose:
In mammalian cells, several distinct surveillance systems, named cell cycle checkpoints, can interrupt normal cell-cycle progression. The cyclin-dependent kinases are negatively regulated by proteins of cyclin-dependent kinases inhibitors comprising INK4 and Cip/Kip families. Histone deacetylation induced by histone deacetylases (HDACs) inactivates the INK4 and Cip/Kip families lead to cancer induction. HDAC inhibitors (HDACIs) have been indicated to be potent inducers of differentiation, growth arrest, and apoptotic induction. Vorinostat (suberoylanilide hydroxamic acid, SAHA), as an HDACI, is reported to be useful in various cancers. Previously, we reported the effect of trichostatin A on hepatocellular carcinoma and also vorinostat on colon cancer cell lines. The current study was aimed to investigate the effect of vorinostat on p16INK4a, p14ARF, p15INK4b, and class I HDACs 1, 2, and 3 gene expression, cell growth inhibition, and apoptosis induction in pancreatic cancer AsPC-1 and hepatocellular carcinoma LCL-PI 11 cell lines.
Experimental approach:
The AsPC-1 and LCL-PI 11 cell lines were cultured and treated with vorinostat. To determine, viability, apoptosis, and the relative expression level of p16INK4a, p14ARF, p15INK4b, class I HDACs 1, 2, and 3 genes, MTT assay, cell apoptosis assay, and RT-qPCR were performed, respectively.
Findings/Results:
Vorinostat significantly inhibited cell growth, induced apoptosis, increased p16INK4a, p14ARF, p15INK4b, and decreased class I HDACs 1, 2, and 3 gene expression.
Conclusion and implications:
Vorinostat can reactivate the INK4 family through inhibition of class I HDACs 1, 2, and 3 genes activity.
Keywords: Cyclin-dependent kinase inhibitors, Neoplasms, Vorinostat.
INTRODUCTION
Pancreatic cancer and hepatocellular carcinoma (HCC) are currently considered a rapidly progressive disease and a leading cause of cancer-related death worldwide (1,2). Current evidence on histone deacetylation and DNA methylation suggests that these epigenetic changes are involved in tumor formation and progression (3,4).
In mammalian cells, several distinct surveillance systems, named cell cycle checkpoints, can interrupt normal cell-cycle progression. Recent in vitro studies have indicated an important cross-talk between the cell-cycle regulatory system and histone acetylation. Transition through the different phases of the cell cycle is regulated by a group of kinases, the cyclin-dependent kinases (Cdks), and their partner, cyclin. Generally, progression through the checkpoints involves the activation of the Cdks (5). In mammals, the Cdk family includes three cell-cycle-related subfamilies (Cdk1, Cdk4, and Cdk5) and five transcriptional subfamilies (Cdk7, Cdk8, Cdk9, Cdk11, and Cdk20) (6).
The Cdks are negatively regulated by proteins of Cdk inhibitors (CKIs) comprising the INK4 and Cip/Kip families. The INK4 family (p16INK4a, p14ARF, p15INK4b) binds and inhibits Cdk 4 and Cdk 6 whereas the Cip/Kip family, p21, p27, and p57, inhibits cyclin A/Cdk 2, cyclin D/Cdk 4, and cyclin E/ Cdk 2 (7). Histone deacetylation induced by histone deacetylases (HDACs) inactivates the INK4 and Cip/Kip family leads to cancer induction (8). INK4 family is involved in the inhibition of G1 phase progression and is often disrupted in human cancers (9,10). It should be noted that histone acetylation and deacetylation affect chromatin structure and thereby indirectly regulate gene transcription. Histone modification consisting of acetylation and deacetylation is induced by two groups of enzymes, histone acetylases (HATs) and HDACs, respectively. They create a non-permissive chromatin conformation by the removal of acetyl groups from histones (11). These enzymes are encoded by a family of 18 genes grouped into classes I-IV based on their homology to their respective yeast orthologues. Classes, I, II, and IV are referred to as “classical” HDACs which include 11 family members whereas the 7 class III members are called sirtuins (12). The over-expression of class I HDACs 1, 2, and 3 have been reported in various cancers including prostate cancer (13), renal cancer (14), cancer of the stomach, colon, esophagus, ovary, breast, pancreas, lung, and thyroid (15). Further evaluation has indicated that overexpression of HDACs can deacetylate CKIs resulting in cancer induction. This pathway has been demonstrated in gastric cancer (16), pancreatic cancer (17), breast cancer (18), and colon cancer (19). HDAC inhibitors (HDACIs) have been indicated to be potent inducers of differentiation, growth arrest, and apoptotic induction. These compounds inhibit HDAC activity. They have been classified as (1) hydroxamic acids e.g. vorinostat (suberoylanilide hydroxamic acid, SAHA) and trichostatin A (TSA); (2) short-chain fatty acids e.g. butyrates; (3) cyclic peptides e.g. FR901228 and apicidin; (4) cyclic tetrapeptides e.g. trapoxin A; and (5) benzamides e.g. MS-27-275 (20). Vorinostat is a broad inhibitor of HDAC activity that inhibits class I HDACs, HDACs 1, 2, 3, and 8, class II HDACs, HDACs 6 and 10, and HDAC11 (21). Vorinostat is reported to be useful in colorectal cancer, advanced solid tumors, pancreatic cancer, lung cancer, melanoma, and multiple myeloma (22). Previously, we reported the effect of TSA on HCC Hep G2 cell line (23,24) and also the effect of vorinostat on colon cancer SW48 cell line (25). The current study was aimed to investigate the effect of vorinostat on p16INK4a, p14ARF, p15INK4b, and class I HDACs 1, 2, and 3 expressions, cell growth inhibition, and apoptosis induction in pancreatic cancer AsPC-1 and hepatocellular carcinoma LCL-PI 11 cell lines.
MATERIAL AND METHODS
Materials
Human pancreatic cancer AsPC-1 and hepatocellular carcinoma LCL-PI 11 cell lines were provided from the National Cell Bank of Iran-Pasteur Institute and maintained in Dulbecco’s modified eagle’s medium (DMEM) supplemented with fetal bovine serum (FBS) 10% and antibiotics in a humidified atmosphere of 5% CO2 at 37 °C. Vorinostat (Sigma, USA) was dissolved in DMSO as a 10 mmol/L stock solution and diluted in order to different concentrations preparation. Total RNA extraction kit (TRIZOL reagent), reverse transcription-quantitative real-time polymerase chain reaction (RT-qPCR) kits (qPCRMasterMix Plus for SYBR Green I dNTP), and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from Sigma (Sigma St. Louis, MO, USA). Other compounds including materials and kits were purchased as provided for our previous works (26,27).
Cell culture and cell viability
Human pancreatic cancer AsPC-1 and hepatocellular carcinoma LCL-PI 11 cell lines were cultured in DMEM supplemented with 10% FBS and antibiotics at 37 °C in 5% CO2 overnight. Then both cell lines seeded into 96-well plates at a density of 5 × 105 cells per well. The next day, the culture medium was replaced with the medium containing vorinostat at 0, 0.5, 1, 2.5, 5, and 10 μM, except the control groups which received DMSO only, at a concentration of 0.05 %. After treatment times (24 and 48 h), all of the treated and untreated cells were assessed by MTT assay according to standard protocols to determine cell viability. In this regard, the MTT solution was added to each well for 4 h at 37 °C, then this solution was removed and DMSO was added and shaken for 10 min to dissolve all of the crystals. Finally, the optical density was detected by a microplate reader at a wavelength of 570 nM. Each experiment was repeated three times (triplicates).
Cell apoptosis assay
To assess the apoptotic cells, the AsPC-1 and LCL-PI 11 cells were cultured at a density of 5 × 105 cells/well for 24 h and then treated with vorinostat, based on IC50 values indicated in Fig. 1, for 24 and 48 h, respectively. Subsequently, all of the cells were harvested by trypsinization, washed with cold phosphate-buffered saline, and re-suspended in 100 μL binding buffer, and stained with 5 μL of annexin V-FITC (10 mg/mL) and 10 μL of propidium iodide (PI, 50 mg/mL) for 15 min at room temperature in the dark. Then, the samples were analyzed using flow cytometry (Becton Dickinson, Heidelberg, Germany).
Fig. 1.
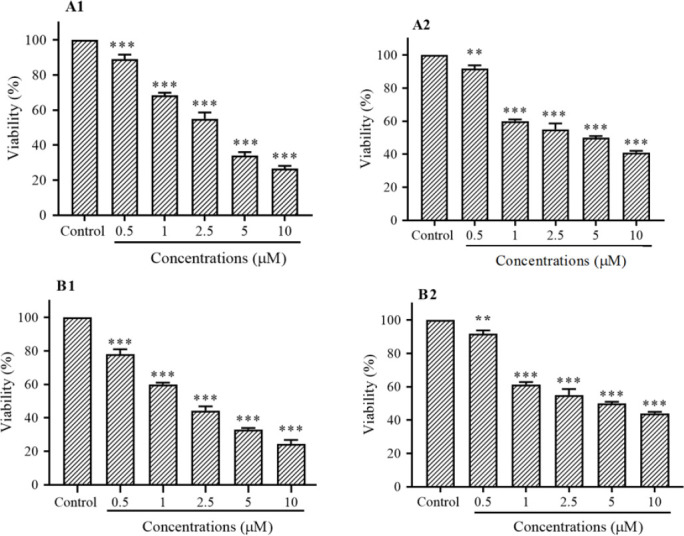
in vitro effects of vorinostat on (A) AsPC-1 and (B) LCL-PI 11 cells viability. Values are the means of three experiments in triplicate. Results are expressed as mean ± SD, n = 3 independent experiments. **P < 0.01 and ***P < 0.001 demonstrate significant differences compared with the untreated control group. A1 and B1, after 24 h vorinostat treatment; A2 and B2, after 48 h vorinostat treatment; HDAC, histone deacetylase.
RT-qPCR
To determine the relative expression level of p16INK4a, p14ARF, p15INK4b, and class I HDACs 1, 2, and 3 genes, RT-qPCR was done. The AsPC-1 and LCL-PI 11 cells were treated with vorinostat, based on IC50 values indicated in Fig. 1, for 24 and 48 h, respectively. After treatment times, RT-qPCR was done as in our previous works (28,29). The primer sequences have been shown in Table 1. The obtained data were standardized using data of the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Table 1.
The primer sequences of p16INK4a, p14ARF, p15INK4b, and class I histone deacetylases 1, 2, and 3 genes.
| Primers | Primer sequences (5’ to 3’) | References |
|---|---|---|
| p14ARF | Forward: GTGGGTTTTAGTTTGTAGTT | 30 |
| Reverse: AAACCTTTCCTACCTAATCT | ||
| p15INK4b | Forward: AAGCTGAGCCCAGGT CTCCTA | 31 |
| Reverse: CCACCGTTGGCCGTAAACT | ||
| p16INK4a | Forward: CCCGCTTTCGTAGTTTTCAT | 32 |
| Reverse: TTATTTGAGCTTTGGTTCTG | ||
| Histone deacetylases 1 | Forward: AACCTGCCTATGCTGATGCT | 33 |
| Reverse: CAGGCAATTCGTTTGTCAGA | ||
| Histone deacetylases 2 | Forward: GGGAATACTTTCCTGGCACA | 33 |
| Reverse: ACGGATTGTGTAGCCACCTC | ||
| Histone deacetylases 3 | Forward: TGGCTTCTGCTATGTCAACG | 33 |
| Reverse: GCACGTGGGTTGGTAGAAGT | ||
| GAPDH | Forward: TCCCATCACCATCTTCCA | 34 |
| Reverse: CATCACGCCACAGTTTCC |
Statistical analysis
The data were analyzed using Graph Pad Prism 8.0. Results are expressed as mean ± standard deviation (SD), n = 3 independent experiments. Statistical comparisons between groups were performed with one-way ANOVA and followed by Turkey’s post-hot test. A significant difference was considered as P < 0.05.
RESULTS
Assessment of cell viability
The viability was assessed by MTT assay. As shown in Fig. 1, vorinostat inhibited cell growth in both cell lines significantly. A significant difference was seen between treated and control groups (Fig. 1). Based on the statistical analysis, a concentration-dependent manner was observed in both cell lines at 24 and 48 h (P < 0.01).
Cell apoptosis assay
To determine apoptotic cells, the AsPC-1 and LCL-PI 11 cells were treated with vorinostat, based on IC50 values. After treatment times, the cells were stained using annexin-V-(FITC) and PI. As shown in Fig. 2, vorinostat induced significant apoptosis in both cell lines after 24 and 48 h of treatment.
Fig. 2.

The apoptotic effect of vorinostat on (A) AsPC-1 and (B) LCL-PI 11 cell lines. Results were obtained from three independent experiments and were expressed as mean ± SD. Vorinostat induced significant apoptosis in both cell lines after 24 and 48 h of treatment. Quadrants (Q) 2 and 3, late and primary apoptosis, respectively, were calculated in (C) this graph. ***P < 0.001 Indicates significant differences compared with the corresponding control group.
Determination of gene expression
The effect of vorinostat on p16INK4a, p14ARF, p15INK4b, and class I HDACs 1, 2, and 3 gene expression were determined by RT-qPCR analysis. The finding demonstrated that vorinostat after 24 and 48 h increased p16INK4a, p14ARF, p15INK4b, and decreased class I HDACs 1, 2, and 3 gene expression significantly in both AsPC-1 and LCL-PI 11 cell lines, Fig. 3. Additionally, the up-regulation of p16INK4a, p14ARF, p15INK4b, and down-regulation of HDACs 1, 2, and 3 gene expression was more significant in LCL-PI 11 cells in comparison with AsPC-1 cells.
Fig. 3.

The relative expression level of class I HDAC1, 2, and 3, p16INK4a, p14ARF, p15INK4b genes in the (A) AsPC-1 and (B) LCL-PI 11 cell lines. Values are the means of three experiments in triplicate. Results are expressed as mean ± SD, n = 3 independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 demonstrate significant differences compared with the untreated control group. A1 and B1, after 24 h vorinostat treatment; A2 and B2, after 48 h vorinostat treatment; HDAC, histone deacetylase.
Discussion
The INK4a/ARF locus encodes a family of tumor suppressor genes (TSGs) which participate in the cell-cycle control pathway (35). Enzymatic deacetylation of the INK4a/ARF family represents major molecular mechanisms controlling gene expression. The activity of these enzymes can repress INK4 p16INK4a, p14ARF, p15INK4b genes expression. The HDAC inhibitor vorinostat has shown significant anticancer activity against both solid and hematologic cancers. This agent inhibits the function of class I HDACs which has been proven to cause cell growth inhibition, differentiation, and apoptosis of various cancer cells (36). In the current study, we indicated that vorinostat could re-activate p16INK4a, p14ARF, p15INK4b gene expression in AsPC-1, and LCL-PI 11 cell lines. We did further evaluation and found that this compound plays its role through inhibition of HDACs 1, 2, and 3 gene expression. Besides, our findings demonstrated that the up-regulation of p16INK4a, p14ARF, p15INK4b, and down-regulation of HDACs 1, 2, and 3 gene expression were more significant in LCL-PI 11 cells in comparison to AsPC-1 cells. Similarly, other researchers have reported that vorinostat reactivates p16INK4a in HCC HepG2, Hep3B, and HuH7 (37), p15 in HCC HepG2, L02, and Huh7 cells (38). As we reported in this study, vorinostat up-regulated INK4a/ARF genes through the down-regulation of HDACs. A similar molecular pathway has been shown by other studies. in vitro study has been demonstrated that vorinostat inhibits HDAC1, 3, and 4 in pancreatic cancer (39). Several studies have shown that vorinostat plays its role through inhibition of class I and II HDACs in cutaneous T-cell lymphoma (40), HDACs class I (HDAC2 and 3) in uterine sarcomas (41), and HDAC1 in breast cancer (42). Other members of the HDAC family such as TSA use the same molecular mechanisms to reactivate cell cycle regulatory INK4 and CIP/KIP genes. It has been shown that TSA inhibits HDAC activity in breast cancer (43). It up-regulates p21/Waf1/CIP1 and induces apoptosis in prostate cancer by inhibiting HDAC activity (44). In cutaneous T-cell lymphoma, TSA relaxes the chromatin structure resulting in the up-regulation of the tumor suppressor genes expression by inhibition of class I and class II HDACs (45). Additionally, HDACIs have been reported to induce growth arrest in colon cancer cells through p15, p16, and p21 reactivation in colon cancer cells HCT116 (46). Similarly, TSA and sodium butyrate activate p15 in HaCa T cells, HCT-116, and HT-29 (47). All reports mentioned above are consistent with our findings. In addition to the class I HDAC pathway, vorinostat plays its role by other classes of HADCs. Numerous studies have been reported that it inhibits HDAC4 and HDAC5 in human cancer cell lines Cal27, A2780, MDA-MB231, and Kyse510 (48). In breast cancer, vorinostat inhibits HDAC6 (49). Another research has been indicated that this agent selectively down-regulates HDAC7 in cutaneous T-cell lymphoma with no effect on the expression of other class I or class II HDACs (50). In summary, we reported that vorinostat up-regulates p16INK4a, p14ARF, p15INK4b through down-regulation of HDACs 1, 2, and 3 genes in LCL-PI 11 and AsPC-1 cells. Whereas, this pathway, HDACs 1, 2, and 3 genes, is not the only pathway of vorinostat in these cell lines. Therefore, the evaluation of the effect of vorinostat on the other HDACs in these cell lines is recommended.
CONCLUSION
In conclusion, our results indicated that vorinostat, an HDACI, can epigenetically down-regulate HDACs 1, 2, and 3 gene expression and reactivate the p16INK4a, p14ARF, p15INK4b genes in LCL-PI 11 and AsPC-1 cells resulting in cell growth inhibition, and apoptosis induction. Thus, this result suggests a dependence of the p16INK4a, p14ARF, p15INK4b genes silencing through histone deacetylation by a mechanism that involves the up-regulation of HDACs 1, 2, and 3 genes. Therefore, the evaluation of the effect of vorinostat on these enzymes and their proteins is recommended.
Conflict of interest statement
The authors declared no conflict of interest in this study.
Authors’ contribution
All authors contributed equally to this work.
Acknowledgments
This work was financially supported by the Adjutancy of Research of Jahrom University of Medical Sciences, Iran under Grant. No. IR.JUMS.REC. 1398.052.
REFERENCES
- 1.Dastjerdi MN, Babazadeh Z, Rabbani M, Gharagozloo M, Esmaeili A, Narimani M. Effects of disulfiram on apoptosis in PANC-1 human pancreatic cancer cell line. Res Pharm Sci. 2014;9(4):287–294. [PMC free article] [PubMed] [Google Scholar]
- 2.Balogh J, Victor III D, Asham EH, Burroughs SG, Boktour M, Saharia A, et al. Hepatocellular carcinoma: a review. J Hepatocell Carcinoma. 2016;3:41–53. doi: 10.2147/JHC.S61146. DOI: 10.2147/JHC.S61146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naghitorabi M, Asl J Mohammadi, Mir Mohammad Sadeghi H, Rabbani M, Jafarian-Dehkordi A, Haghjooye Javanmard S. Quantitative evaluation of DNMT3B promoter methylation in breast cancer patients using differential high resolution melting analysis. Res Pharm Sci. 2013;8(3):167–175. [PMC free article] [PubMed] [Google Scholar]
- 4.Liu K-Y, Wang L-T, Hsu S-H. Modification of epigenetic histone acetylation in hepatocellular carcinoma. Cancers. 2018;10(1):1–13. doi: 10.3390/cancers10010008. DOI: 10.3390/cancers10010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang C, Fu M, Mani S, Wadler S, Senderowicz AM, Pestell RG. Histone acetylation and the cell-cycle in cancer. Front Biosci. 2001;6:610–629. doi: 10.2741/1wang1. DOI: 10.2741/1wang1. [DOI] [PubMed] [Google Scholar]
- 6.Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122–128. doi: 10.1186/gb4184. DOI: 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thullberg M, Bartkova J, Khan S, Hansen K, Rönnstrand L, Lukas J, et al. Distinct versus redundant properties among members of the INK4 family of cyclin-dependent kinase inhibitors. FEBS Lett. 2000;470(2):161–166. doi: 10.1016/s0014-5793(00)01307-7. DOI: 10.1016/s0014-5793(00)01307-7. [DOI] [PubMed] [Google Scholar]
- 8.Shin JY, Kim HS, Park J, Park JB, Lee JY. Mechanism for inactivation of the KIP family cyclin-dependent kinase inhibitor genes in gastric cancer cells. Cancer Res. 2000;60(2):262–265. [PubMed] [Google Scholar]
- 9.Fukai K, Yokosuka O, Imazeki F, Tada M, Mikata R, Miyazaki M, et al. Methylation status of p14ARF, p15INK4b, and p16INK4a genes in human hepatocellular carcinoma. Liver Int. 2005;25(6):1209–1216. doi: 10.1111/j.1478-3231.2005.01162.x. DOI: 10.1111/j.1478-3231.2005.01162.x. [DOI] [PubMed] [Google Scholar]
- 10.Kusy S, Larsen CJ, Roche J. p14ARF, p15INK4b and p16INK4a Methylation status in chronic myelogenous leukemia. Leuk Lymphoma. 2004;45(10):1989–1994. doi: 10.1080/10428190410001714025. DOI: 10.1080/10428190410001714025. [DOI] [PubMed] [Google Scholar]
- 11.Glozak M, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26(37):5420–5432. doi: 10.1038/sj.onc.1210610. DOI: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 12.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: what are the cancer relevant targets? Cancer Lett. 2009;277(1):8–21. doi: 10.1016/j.canlet.2008.08.016. DOI: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 13.Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 2008;98(3):604–610. doi: 10.1038/sj.bjc.6604199. DOI: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fritzsche FR, Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, et al. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer. 2008;8:381–388. doi: 10.1186/1471-2407-8-381. DOI: 10.1186/1471-2407-8-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakagawa M, Oda Y, Eguchi T, Aishima SI, Yao T, Hosoi F, et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18(4):769–774. [PubMed] [Google Scholar]
- 16.Kim JK, Noh JH, Eun JW, Jung KH, Bae HJ, Shen Q, et al. Targeted inactivation of HDAC2 restores p16INK4a activity and exerts antitumor effects on human gastric cancer. Mol Cancer Res. 2013;11(1):62–73. doi: 10.1158/1541-7786.MCR-12-0332. DOI: 10.1158/1541-7786.MCR-12-0332. [DOI] [PubMed] [Google Scholar]
- 17.Stojanovic N, Hassan Z, Wirth M, Wenzel P, Beyer M, Schäfer C, et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene. 2017;36(13):1804–1815. doi: 10.1038/onc.2016.344. DOI: 10.1038/onc.2016.344. [DOI] [PubMed] [Google Scholar]
- 18.Zheng S, Li Q, Zhang Y, Balluff Z, Pan YX. Histone deacetylase 3 (HDAC3) participates in the transcriptional repression of the p16(INK4a) gene in mammary gland of the female rat offspring exposed to an early-life high-fat diet. Epigenetics. 2012;7(2):183–190. doi: 10.4161/epi.7.2.18972. DOI: 10.4161/epi.7.2.18972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hitomi T, Matsuzaki Y, Yokota T, Takaoka Y, Sakai T. p15INK4b in HDAC inhibitor-induced growth arrest. FEBS Lett. 2003;554(3):347–350. doi: 10.1016/s0014-5793(03)01186-4. DOI: 10.1016/s0014-5793(03)01186-4. [DOI] [PubMed] [Google Scholar]
- 20.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92(15):1210–1216. doi: 10.1093/jnci/92.15.1210. DOI: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 21.Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem. 2009;107(4):600–608. doi: 10.1002/jcb.22185. DOI: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh AK, Bishayee A, Pandey AK. Targeting histone deacetylases with natural and synthetic agents: an emerging anticancer strategy. Nutrients. 2018;10(6):731–739. doi: 10.3390/nu10060731. DOI: 10.3390/nu10060731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanaei M, Kavoosi F, Salehi H. Genistein and trichostatin a induction of estrogen receptor alpha gene expression, apoptosis and cell growth inhibition in hepatocellular carcinoma HepG 2 cells. Asian Pac J Cancer Prev. 2017;18(12):3445–3450. doi: 10.22034/APJCP.2017.18.12.3445. DOI: 10.22034/APJCP.2017.18.12.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanaei M, Kavoosi F, Roustazadeh A, Golestan F. Effect of genistein in comparison with trichostatina on reactivation of DNMTs genes in hepatocellular carcinoma. J Clin Transl Hepatol. 2018;6(2):141–146. doi: 10.14218/JCTH.2018.00002. DOI: 10.14218/JCTH.2018.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanaei M, Kavoosi F, Mansoori O. Effect of valproic acid in comparison with vorinostat on cell growth inhibition and apoptosis induction in the human colon cancer SW48 cells in vitro. Exp Oncol. 2018;40(2):95–100. [PubMed] [Google Scholar]
- 26.Sanaei M, Kavoosi F. Effects of 5-aza-2’-deoxycytidine and valproic acid on epigenetic-modifying DNMT1 gene expression, apoptosis induction and cell viability in hepatocellular carcinoma WCH-17 cell line. Iran J Ped Hematol Oncol. 2019;9(2):83–90. DOI: 10.18502/ijpho.v9i2.607. [Google Scholar]
- 27.Sanaei M, Kavoosi F. Effect of DNA methyl transferase in comparison to and in combination with histone deacetylase inhibitors on hepatocellular carcinoma HepG2 cell line. Asian Pac J Cancer Prev. 2019;20(4):1119–1125. doi: 10.31557/APJCP.2019.20.4.1119. DOI: 10.31557/APJCP.2019.20.4.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanaei M, Kavoosi F. Effect of curcumin and trichostatin A on the expression of DNA methyltransfrase 1 in hepatocellular carcinoma cell line Hepa 1-6. Iran J Ped Hematol Oncol. 2018;8(4):193–201. [Google Scholar]
- 29.Sanaei M, Kavoosi F. Effect of zebularine in comparison to and in combination with trichostatin A on CIP/KIP family (p21Cip1/Waf1/Sdi1, p27Kip1, and p57Kip2), DNMTs (DNMT1, DNMT3a, and DNMT3b), class I HDACs (HDACs 1, 2, 3) and class II HDACs (HDACs 4, 5, 6) gene expression, cell growth inhibition and apoptosis induction in colon cancer LS 174T cell line. Asian Pac J Cancer Prev. 2020;21(7):2131–2139. doi: 10.31557/APJCP.2020.21.7.2131. DOI: 10.31557/APJCP.2020.21.7.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakuma K, Chong JM, Sudo M, Ushiku T, Inoue Y, Shibahara J, et al. High-density methylation of p14ARF and p16INK4A in epstein-barr virus-associated gastric carcinoma. Int J Cancer. 2004;112(2):273–278. doi: 10.1002/ijc.20420. DOI: 10.1002/ijc.20420. [DOI] [PubMed] [Google Scholar]
- 31.Li G, Ji Y, Liu C, Li JQ, Zhou YQ. Reduced levels of p15INK4b, p16INK4a, p21cip1 and p27kip1 in pancreatic carcinoma. Mol Med Rep. 2012;5(4):1106–1110. doi: 10.3892/mmr.2012.771. DOI: 10.3892/mmr.2012.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saegusa M, Machida BD, Okayasu I. Possible associations among expression of p14(ARF), p16(INK4a), p21(WAF1/CIP1), p27(KIP1), and p53 accumulation and the balance of apoptosis and cell proliferation in ovarian carcinomas. Cancer. 2001;92(5):1177–1189. doi: 10.1002/1097-0142(20010901)92:5<1177::aid-cncr1436>3.0.co;2-5. DOI: 10.1002/1097-0142(20010901)92:5<1177::aid-cncr1436>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 33.Jin KL, Pak JH, Park JY, Choi WH, Lee JY, Kim JH, et al. Expression profile of histone deacetylases 1, 2 and 3 in ovarian cancer tissues. J Gynecol Oncol. 2008;19(3):185–190. doi: 10.3802/jgo.2008.19.3.185. DOI: 10.3802/jgo.2008.19.3.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanaei M, Kavoosi F. Effect of 5-aza-2’-deoxycytidine in comparison to valproic acid and trichostatin A on histone deacetylase 1, DNA methyltransferase 1, and CIP/KIP family (p21, p27, and p57) genes expression, cell growth inhibition, and apoptosis induction in colon cancer SW480 cell line. Adv Biomed Res. 2019;8:52–59. doi: 10.4103/abr.abr_91_19. DOI: 10.4103/abr.abr_91_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dominguez G, Silva J, Garcia JM, Silva JM, Rodriguez R, Muñoz C, et al. Prevalence of aberrant methylation of p14ARF over p16INK4a in some human primary tumors. Mutat Res. 2003;530(1-2):9–17. doi: 10.1016/s0027-5107(03)00133-7. DOI: 10.1016/s0027-5107(03)00133-7. [DOI] [PubMed] [Google Scholar]
- 36.Richon VM, Zhou X, Rifkind RA, Marks PA. Histone deacetylase inhibitors: development of suberoylanilide hydroxamic acid (SAHA) for the treatment of cancers. Blood Cells Mol Dis. 2001;27(1):260–264. doi: 10.1006/bcmd.2000.0376. DOI: 10.1006/bcmd.2000.0376. [DOI] [PubMed] [Google Scholar]
- 37.Venturelli S, Armeanu S, Pathil A, Hsieh CJ, Weiss TS, Vonthein R, et al. Epigenetic combination therapy as a tumor-selective treatment approach for hepatocellular carcinoma. Cancer. 2007;109(10):2132–2141. doi: 10.1002/cncr.22652. DOI: 10.1002/cncr.22652. [DOI] [PubMed] [Google Scholar]
- 38.Zhou H, Cai Y, Liu D, Li M, Sha Y, Zhang W, et al. Pharmacological or transcriptional inhibition of both HDAC1 and 2 leads to cell cycle blockage and apoptosis via p21Waf1/Cip1 and p19INK4d upregulation in hepatocellular carcinoma. Cell Prolif. 2018;51(3):e12447,1–14. doi: 10.1111/cpr.12447. DOI: 10.1111/cpr.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumagai T, Wakimoto N, Yin D, Gery S, Kawamata N, Takai N, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int J Cancer. 2007;121(3):656–665. doi: 10.1002/ijc.22558. DOI: 10.1002/ijc.22558. [DOI] [PubMed] [Google Scholar]
- 40.Ramalingam SS, Maitland ML, Frankel P, Argiris AE, Koczywas M, Gitlitz B, et al. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(1):56–62. doi: 10.1200/JCO.2009.24.9094. DOI: 10.1200/JCO.2009.24.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hrzenjak A, Moinfar F, Kremser ML, Strohmeier B, Petru E, Zatloukal K, et al. Histone deacetylase inhibitor vorinostat suppresses the growth of uterine sarcomas in vitro and in vivo. Mol Cancer. 2010;9:49–59. doi: 10.1186/1476-4598-9-49. DOI: 10.1186/1476-4598-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gameiro SR, Malamas AS, Tsang KY, Ferrone S, Hodge JW. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget. 2016;7(7):7390–7402. doi: 10.18632/oncotarget.7180. DOI: 10.18632/oncotarget.7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vigushin DM, Ali S, Pace PE, Mirsaidi N, Ito K, Adcock I, et al. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin Cancer Res. 2001;7(4):971–976. [PubMed] [Google Scholar]
- 44.Fortson WS, Kayarthodi S, Fujimura Y, Xu H, Matthews R, Grizzle WE, et al. Histone deacetylase inhibitors, valproic acid and trichostatin-A induce apoptosis and affect acetylation status of p53 in ERG-positive prostate cancer cells. Int J Oncol. 2011;39(1):111–119. doi: 10.3892/ijo.2011.1014. DOI: 10.3892/ijo.2011.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Codd R, Braich N, Liu J, Soe CZ, Pakchung AA. Zn (II)-dependent histone deacetylase inhibitors: suberoylanilide hydroxamic acid and trichostatin A. Int J Biochem Cell Biol. 2009;41(4):736–739. doi: 10.1016/j.biocel.2008.05.026. DOI: 10.1016/j.biocel.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 46.Mariadason JM. HDACs and HDAC inhibitors in colon cancer. Epigenetics. 2008;3(1):28–37. doi: 10.4161/epi.3.1.5736. DOI: 10.4161/epi.3.1.5736. [DOI] [PubMed] [Google Scholar]
- 47.Fang JY. Histone deacetylase inhibitors, anticancerous mechanism and therapy for gastrointestinal cancers. J Gastroenterol Hepatol. 2005;20(7):988–994. doi: 10.1111/j.1440-1746.2005.03807.x. DOI: 10.1111/j.1440-1746.2005.03807.x. [DOI] [PubMed] [Google Scholar]
- 48.Marek L, Hamacher A, Hansen FK, Kuna K, Gohlke H, Kassack MU, et al. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J Med Chem. 2013;56(2):427–436. doi: 10.1021/jm301254q. DOI: 10.1021/jm301254q. [DOI] [PubMed] [Google Scholar]
- 49.Tu Y, Hershman DL, Bhalla K, Fiskus W, Pellegrino CM, Andreopoulou E, et al. A phase I-II study of the histone deacetylase inhibitor vorinostat plus sequential weekly paclitaxel and doxorubicin-cyclophosphamide in locally advanced breast cancer. Breast Cancer Res Treat. 2014;146(1):145–152. doi: 10.1007/s10549-014-3008-5. DOI: 10.1007/s10549-014-3008-5. [DOI] [PubMed] [Google Scholar]
- 50.Dokmanovic M, Perez G, Xu W, Ngo L, Clarke C, Parmigiani RB, et al. Histone deacetylase inhibitors selectively suppress expression of HDAC7. Mol Cancer Ther. 2007;6(9):2525–2534. doi: 10.1158/1535-7163.MCT-07-0251. DOI: 10.1158/1535-7163.MCT-07-0251. [DOI] [PubMed] [Google Scholar]