

ABSTRACT
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common dementia among the elderly. The pathophysiology of AD is characterized by two hallmarks: amyloid plaques, produced by amyloid β (Aβ) aggregation, and neurofibrillary tangle (NFT), produced by accumulation of phosphorylated tau. The regulatory roles of non-coding RNAs (ncRNAs), particularly long noncoding RNAs (lncRNAs), have been widely recognized in gene expression at the transcriptional and posttranscriptional levels. Mounting evidence shows that lncRNAs are aberrantly expressed in AD progression. Here, we review the lncRNAs that implicated in the regulation of Aβ peptide, tau, inflammation, cell death, and other aspects which are the main mechanisms of AD pathology. We also discuss the possible clinical or therapeutic utility of lncRNA detection or targeting to help diagnose or possibly combat AD.
KEYWORDS: LncRNAs, Alzheimer’s disease, Aβ, tau
Introduction
Alzheimer’s disease (AD) was initially described as a ‘peculiar severe disease process of the cerebral cortex’, by Dr. Alois Alzheimer in 1906 at a conference in Tubingen, Germany [1]. AD is now characterized as a progressive degenerative disease with cognitive dysfunction, memory loss, and emotional disorder [2]. More than 50 million people are currently affected by dementia worldwide, and AD is the most common form of dementia and may contribute to up to 70% of the dementia cases [3]. The majority of AD cases are sporadic with a late-onset (LOAD), which usually occurs in people older than 65 years; whereas a small portion of AD cases are familial with early-onset AD (EOAD), which commonly affects people under 65 years of age and has a genetic predisposition [4].
Long non-coding RNAs (lncRNAs) comprise a class of non-coding RNAs (ncRNAs) that are longer than 200 nucleotides (nt) in length, and account for one of the largest proportions of the non-coding transcriptome in number of transcripts. Given their diverse structural and biochemical features, lncRNAs impact AD pathogenesis via a variety of regulatory mechanisms, including transcriptional, posttranscriptional and translational regulation [5].
AD pathophysiology
The pathophysiology of AD is characterized by two hallmarks: amyloid plaques resulting from abnormal proteolytic processing of amyloid precursor protein (APP) in the extra neuronal environment; and neurofibrillary tangle (NFT) resulting from accumulation of hyperphosphorylated tau protein in neurons [6]. Amyloid plaques are mainly comprised of amyloid β (Aβ) peptide, particularly Aβ42 isoform, which is a hydrophobic peptide that is considered to have pronounced toxicity [7]. Given its insoluble properties, Aβ peptide has a tendency to aggregate and forms the core of the amyloid plaque by acquiring the configuration of a β-pleated sheet [8–11]. Aβ production is a consequence of the sequential proteolytic cleavage of amyloid precursor protein (APP) by a β-secretase (BACE1) and γ-secretase [12], wherein APP is a type I transmembrane glycoprotein of with three major isoforms arising from alternative splicing [13]. Under normal conditions APP is cleaved by α-secretase, leading to the release of a soluble molecule called sAPPα, and a fragment of the carboxyl terminal portion (CTF83) [14,15]. In contrast to Aβ, sAPPα functions in neuronal protection against toxicity [16,17], and is found at a lower level in AD patients compared to controls. When APP is cleaved by BACE1, a shorter amino-terminal portion (sAPPβ) and a longer carboxyl terminal fragment (CTF99) are generated [18,19]. APP CTFs are further processed by γ-secretase to produce Aβ, which is secreted, aggregated and accumulated in extracellular plaques, leading to the formation of amyloid neurotic plaques [20]. In addition to Aβ production, dysregulation of Aβ metabolism also contributes substantially to AD pathology, including Aβ degradation and transportation [3,21].
Neurofibrillary lesions are hallmarks of AD and other tauopathic neurodegenerative diseases. NFTs are mainly aggregations of hyperphosphorylated microtubule-associated protein tau, which mainly exists in the cytoplasm of the neuronal axon, synaptic regions and cerebrospinal fluid (CSF) [22]. Hyperphosphorylated tau forms aberrant aggregations with cytoskeletal proteins and shows a lower grade of interaction with microtubules, leading to dysfunction of axonal transport [23]. The phosphorylation levels of tau are significantly increased in AD patients, compared to age-matched controls, supporting that tau phosphorylation contributes to AD pathology. In addition to the amyloid and tau hypothesis of AD, many other hypotheses including inflammatory mechanism, mitochondrial dysfunction, oxidative stress and cholinergic hypothesis have also been raised [6,24,25]. Altogether, AD is a complex and multifaceted disease influenced by genetics, environment, and lifestyle.
Long-noncoding RNAs
LncRNAs are a class of endogenous regulatory RNA molecules longer than 200 nucleotides. These transcripts typically contain no obvious open reading frame (ORF) and thus lack protein-coding capacity [26–28]. LncRNAs have been discovered predominantly by high-throughput sequencing technologies, including microarrays and next-generation sequencing (NGS) [29]. The estimated number of lncRNAs ranges from around 59,000 to 102,000 listed in the NONCODE database [30,31,32]. Like protein-coding genes, lncRNAs are commonly transcribed by RNA polymerase II and are often post-transcriptionally modified by 5ʹ capping, 3ʹ polyadenylation, and RNA splicing. However, lncRNAs have features distinct from protein-coding genes, such as being shorter in length and having lower sequence conservation across species [33]. LncRNAs are involved in a variety of biological functions including development, differentiation, and metabolism [34,35]. Moreover, lncRNAs affect many cellular processes, including chromatin and DNA modification, RNA transcription, pre-RNA splicing, mRNA stability, and translation [36,37].
LnRNAs involved in Alzheimer’s disease (AD)
Mammalian genomes encode tens of thousands of lncRNAs, and up to 40% of these lncRNAs are specifically expressed in the brain [34]. Aberrant lncRNA expression has been associated with many neurodegenerative diseases [38–41]. Particularly, several hundreds of lncRNAs are found differentially expressed in 3xTg-AD model mice, compared to age-matched control animals [42]. In addition, 99 lncRNAs are down-regulated and 150 lncRNAs are up-regulated in the hippocampus of APP/PS1 transgenic mice, compared to controls [43]. Transcriptome analyses on post-mortem human brains also identify that levels of multiple lncRNAs are altered significantly in AD patient brains [38,44,45]. Collectively, lncRNAs are differentially expressed in AD patients or animal models, therefore can serve as biomarkers even potential treatment target for AD.
LncRNA and mRNA transcription
Transcription begins with the binding of RNA polymerase to the promoter region of a gene with the assistance of transcription factors (TFs). Gene expression changes have been identified during AD progression in a cell-type specific manner. Particularly, AD risk genes, such as ApoE, BACE1 and TREM2, show aberrant expression levels during AD progression [46,47]. Gene expression control primarily occurs at the level of transcription [46]. Importantly, an increasing number of lncRNAs have been identified as transcriptional regulators. LncRNAs, participate in AD pathology via modulating RNA transcription, are summarized below.
NDM29
Neuroblastoma differentiation marker 29 (NDM29), an ncRNA transcribed by RNA pol III in human, drives the neuroblastoma (NB) cell differentiation process towards a non-malignant neuron-like phenotype [48–50]. Notably, NDM29 expression is enhanced in the cerebral cortex of AD patients, as compared to age-matched controls, demonstrating that NDM29 is linked to AD pathology. Additionally, NDM29 biosynthesis is sensitive to pro-inflammatory molecules, such as interleukin 1α (IL-1α) and TNFα (tumour necrosis factor α); both are considered landmarks of AD onset. NDM29 significantly increases both APP mRNA and protein levels, and subsequently enhances production of the two major Aβ isoforms, Aβ42 and Aβ40. Aβ42 (42 residues long) contains two more residues than Aβ40 (40 residues long) on the C-terminus. Aβ42, but not Aβ40, is the major component of amyloid plaques in AD brains [51]. NDM29 preferentially enhances the ratio of Aβ42/Aβ40 [52], leading to accumulation of toxic Aβ.
LRP1-AS
Low-density lipoprotein (LDL)-related protein (LRP1) belongs to the LDL receptor family. LRP1 is abundantly expressed in the brain. LRP1 and its ligand apolipoprotein E (ApoE) have been identified in senile plaques in AD brains [53], suggesting a role of LRP1 in Aβ accumulation. In addition, numerous studies have demonstrated that LRP1 is deeply involved in APP trafficking and Aβ processing. Receptor-associated protein (RAP)-mediated blocking of LRP1 function increases APP levels on the cell surface and decreases Aβ production [54]. Transmembrane domain on the C-terminus of LRP1 reduces Aβ production by competing with APP for cleavage mediated by β- and γ-secretase [55]. LRP1 also facilitates heparan sulphate proteoglycan (HSPG) dependent Aβ uptake into cells [56]. Moreover, LRP1 also modulates ApoE levels and lipid metabolism by altering the stability of ApoE mRNA [57]. Altogether, LRP1 participates in AD pathogenesis via a variety of mechanisms.
LRP1-AS, a 1387 nt lncRNA, is transcribed from the opposite strand of mouse LRP1 gene. There is a 395 bp overlap of exon2 of LRP1-AS and exon5 and exon6 of LRP1, and this location of LRP1/LRP1-AS has been maintained throughout evolution. RNAi silencing of LRP1-AS leads to increased LRP1 expression, whereas overexpression of LRP1-AS results in decreased LRP1 levels. Thus, LRP1-AS negatively regulates LRP1 expression at both RNA and protein levels [58]. In addition to direct sequence pairing, LRP1-AS also reduces LRP1 transcription via decreasing LRP1 promoter activity induced by Hmgb2 and Srebp1. Srebp1 is a transcription factor that regulates LRP1 transcription, and Hmgb2 interacts with Srebp1 and forms a transcriptional complex to regulate LRP1 levels. Conflicting conclusions have been obtained concerning the link between LRP1 levels and AD pathogenesis. Some groups reported that LRP1 levels are increased in the AD brain [59], whereas others reported that LRP1 levels are decreased compared to age-matched controls. Importantly, the levels of LRP1-AS are significantly increased [58]. Altogether LRP1-AS could serve as a diagnostic marker and a potential therapeutic target for AD treatment.
LncRNA and pre-mRNA splicing
Alternative splicing, allowing a single gene coding for multiple proteins, is an important regulatory mechanism for gene expression. Dysregulation of mRNA splicing is considered a key feature of AD, particularly genes related to hallmarks of AD such as Aβ burden and neurofibrillary tangles [60]. LncRNAs can regulate alternative splicing through interacting with specific splicing factors or form RNA-RNA duplex with mRNAs. LncRNAs, participate in AD pathology via modulating RNA splicing, are summarized below.
51A
Sortilin-related receptor (SORL1, also known as LR11), a member of the low-density lipoprotein receptor (LDLR) family, is abundantly and specifically expressed in neurons [61,62]. SORL1 levels are significantly reduced in neurons in AD brains [63,64], suggesting a role in AD pathogenesis. Importantly, SORL1 affects APP trafficking and proteolytic processing by directly interacting with APP, leading to reduced Aβ production in the brain. By contrast, reduction of SORL1 expression preferentially guides APP into the β-secretase cleavage pathway, instead of the retromer recycling pathway, resulting in increased production of neurotoxic Aβ [65].
51A, a lncRNA around 300 nt in length, is transcribed by RNA polymerase III from the antisense configuration to intron 1 of the SORL1 gene in human. In contrast to SORL1, lncRNA 51A levels are upregulated in post-mortem AD brains. Mechanistically, 51A pairs with intron 1 of SORL1 pre-mRNA, where an alternative splicing event occurs, and leads to reduced production of canonical SORL1 protein and increased production of an alternative variant. Consequently, as an upstream regulator of APP processing and Aβ production, 51A promotes Aβ secretion [66].
GDNFOS
Glial cell line-derived neurotrophic factor (GDNF) was initially reported to promote the survival of dopamine neurons and dopamine uptake in the midbrain [67]. Given its role in protecting dopamine neurons and rescuing motor neurons, it has the potential to be developed as a therapeutic agent for Parkinson’s disease (PD) [68], wherein the major pathological landmark is progressive loss of dopaminergic neurons. Additionally, GDNF is also involved in extensively regulating neuronal populations, neurite branching, and synaptic plasticity, but is not specific to dopamine neurons [69]. Two GDNF mRNAs are produced by alternative splicing of exon2 of the GDNF gene, pre-(α) long pro-GDNF and pre-(β) short pro-GDNF [70,71], resulting in the production of two secreted proteins in neurons. Notably, secretion of long GDNF is constitutive, whereas short GDNF is activity-dependent [72]. In AD patients, the levels of GDNF are significantly up-regulated in cerebrospinal fluid (CSF), whereas levels are down-regulated in serum compared to control patients [73]. However, this conclusion remains controversial, as another study showed that GDNF mRNA is significantly increased in the middle temporal gyrus of AD brains, whereas the peptide was decreased in comparison with age-matched normal brains [69].
GDNFOS is a cis-natural antisense transcript, transcribed from the opposite strand of the GDNF gene in human. Three GDNFOS isoforms have been identified – GDNFOS1, GDNFOS2, and GDNFOS3. Both GDNFOS1 and GDNFOS2 are lncRNAs, with no obvious ORF. GDNFOS1 is reverse complementary to the 5ʹ-UTR of the GDNF, and GDNFOS2 has no pairing sequence to the GDNF transcript. GDNFOS3 transcript contains an ORF and has potential for protein translation [69]. GDNFOS1 is abundantly expressed in the brain but has lower expression than GDNF isoform containing exon 1 and exon 4 transcripts. GDNFOS1 levels are similar between AD and control brains, however, alteration of the GDNFOS isoform ratio may contribute to AD pathogenesis [69]. Given that levels of GDNFOS and GDNF are closely correlated, where GDNF is proved to be involved in neurite branching and synaptic plasticity [69]. Therefore, it is fair to speculate that GDNFOS may play a role in regulating synaptic plasticity. Clearly, more evidence is required to demonstrate the implication of GDNFOS in AD pathogenesis.
LncRNA and mRNA stability
The steady-state level of an mRNA is determined by the rate of synthesis and degradation; therefore, mRNA degradation is an essential point to control gene expression. Previous study has demonstrated that destabilization of mRNAs encoding for synaptic transmission proteins contributes to synaptic function loss in AD pathology [74]. Additionally, AD risk genes exhibit aberrant mRNA decay rate in AD patients [75]. LncRNAs, participate in AD pathology via modulating mRNA stability, are summarized below.
BACE1-AS
The extracellular plaque deposition of the Aβ peptide is one of the hallmark pathologies in AD patients. Aβ production requires sequential proteolytic cleavage of APP by β-secretase and γ-secretase. Beta-site APP-cleaving enzyme 1 (BACE1) is a protease that increases the amount of β-secretase cleavage products [76]. BACE1 is highly expressed in the brain, and both the expression and enzymatic activity are enhanced in AD brains [77,78]. BACE1 deficiency substantially reduces the Aβ burden in APP transgenic mice [79].
BACE1-AS and BACE1 mRNA are two transcripts from the same locus in chromosome 11 (11q 23.3) in human; BACE1 mRNA is transcribed from the sense strand and BACE1-AS from the antisense strand. BACE1-AS pairs to BACE1 and forms an RNA duplex, resulting in an altered structure of BACE1 and enhanced mRNA stability, therefore BACE1-AS not only increases the mRNA levels but also the protein levels of BACE1 [80]. BACE1-AS is heavily involved in Aβ metabolism, as deficiency of BACE1-AS not only reduces BACE1 levels, but further suppresses production of Aβ40 and Aβ42 [80]. BACE1-AS levels are sensitive to multiple cell stressors, such as serum starvation, Aβ42, and hydrogen peroxide (H2O2) [80]. RNAi-mediated BACE1-AS inhibition reduces BACE1-mediated APP cleavage in human SH-SY5Y cell [81]. BACE1-AS is also stabilized by HuD, a neuronal RNA-binding protein that is implicated in AD pathogenesis [82]. BACE1-AS is increased not only in APP transgenic mice, but also in AD brains [80]; the ratio of BACE1-AS to BACE1 is particularly enhanced. BACE1-AS and the ratio of BACE1-AS to BACE1 have even been proposed as a new biomarker of AD diagnosis. Knockdown of BACE1 or BACE1-AS transcript by continuous infusion of siRNAs into the third ventricle of APP transgenic mice, not only downregulates BACE1 protein levels, but also significantly reduces insoluble Aβ [83]. These findings raise the possibility of BACE1-AS as a promising treatment regime for AD patients.
BDNF-AS
Brain-derived neurotrophic factor (BDNF) is involved in synaptic function, neurogenesis and cognitive function [84]. In addition, BDNF protects neurons from injuries, prevents neuronal function loss and facilitates the regeneration of damaged neurons [85,86]. BDNF is also linked to the core pathological features of AD and may serve as a biomarker for disease diagnosis. BDNF regulates APP processing by provoking the non-amyloidogenic processing pathway, therefore reducing the production of Aβ [87,88]. Pretreatment of BDNF protects cells against the toxicity induced by Aβ42 in cultured neurons and in mouse brain [89]. Interestingly, Aβ42 also promotes BDNF production in astrocytes, which in turn rescues neurite degeneration in neuroblastoma cells [90]. The AD mouse model produces more aggregated and insoluble Aβ42 and shows significant reduction of BDNF levels [91]. Moreover, BDNF administration reverses the neurodegenerative changes in AD mouse brains, such as synapse loss, learning and memory impairment [92], and therefore merits exploration as a potential therapy for AD.
BDNF-AS is a natural antisense transcript to BDNF. Human BDNF-AS gene is located on the positive strand of chromosome 11, and the transcription start site is approximately 200 kb downstream from the BDNF gene promoter. BDNF-AS negatively modulates BDNF levels in vitro and in vivo [93]. Aβ treatment not only decreases BDNF, but also promotes BDNF-AS levels in PC12 cells. Silencing BDNF-AS significantly increases BDNF levels, and alleviates cell apoptosis and ROS intensity induced by Aβ treatment; cell viability is also enhanced [94]. Other study also reported that BDNF-AS-mediated decrease of BDNF down-regulates levels of activity-regulated cytoskeleton-associated protein (ARC), which is an immediate-early gene and involved in synaptogenesis and synaptic plasticity [95].
EBF3-AS
Early B cell factor 3 (EBF3, also known as olf) is expressed in the olfactory receptor neurons and their precursors [96]. As a DNA binding TF, it is involved in apoptosis, cell cycle arrest and neurogenesis [97,98]. Notably, the expression of EBF3 is elevated in the hippocampus of AD mice.
EBF3-AS is an 842 nt non-coding RNA that is transcribed from the opposite strand of the protein-coding gene EBF3; its levels are up-regulated in the hippocampus of APP/PS1 mice. LncRNA EBF3-AS deficiency not only reduces EBF3 levels but also inhibits apoptosis induced by okadaic acid (OA) or Aβ in human SH-SY5Y cell. These lines of evidence indicate that EBF3-AS may serve as a new therapeutic target for the treatment of AD [99].
Sox2OT
Sox2 is a key TF for the regulation of stem cell pluripotency and neuronal genesis [100]. Sox2 overlapping transcript (Sox2OT) is a lncRNA transcribed from the intron of the Sox2 gene [101]. Sox2OT is roughly 3.4 kb in length and is an evolutionarily conserved transcript between human and mouse [102]. Sox2OT plays a key role in the induction and/or maintenance of Sox2 expression [103]. Moreover, Sox2OT is expressed in the cerebral cortex of the developing mouse brain, where it promotes neuronal differentiation and neurogenesis by repressing Sox2 [104]. Furthermore, Sox2OT is differentially expressed in the AD model mouse and is involved in early and late disease stages, suggesting a role of Sox2OT as a biomarker of AD [105].
SNHG1
Small nucleolar RNA host gene 1 (SNHG1), a lncRNA located at 11q12.3 in human, promotes neuroinflammation and neuronal toxicity in PD [106,107]. In AD pathology, Aβ treatment increases the expression of SNHG1, while repression of SNHG1 in Aβ treated cells attenuates the effect of Aβ on cell viability, and mitochondrial membrane potential (MMP). This is achieved via targeting kringle containing transmembrane protein 1 (KREMEN1), a transmembrane receptor that has an intrinsic pro-apoptotic activity, via a mechanism of SNHG1 mediated miR-137 sponge, that selectively targets the untranslated region of KREMEN1 in SH-SY5Y and human primary neuron (HPN) cells [108].
NAT-RAD18
NAT-RAD18 is a lncRNA that is 509 nt in length, also a natural antisense transcript against Rad18. It contains an ORF of 225 bases, a 5ʹ-untranslated region of 93 bases and a poly(A) containing 3ʹ-untranslated region of 188 bases. NAT-RAD18 shows 78% homology with Rad18 genomic sequence and the reverse-complement sequence of Rad18 transcript in adult rat brain [109].
NAT-RAD18 is specifically expressed in neurons, as demonstrated by co-localization of NAT-Rad18 with NeuN, which is a neuronal marker. At the cellular level, the expression of Rad18 was counterbalanced by that of NAT-Rad8 in both mRNA and protein level. Expression of NAT-RAD18 is up-regulated, whereas Rad18 is down-regulated in response to cell stressor Aβ40 [109]. Rad18, a member of the structural maintenance of chromosome (SMC) family, is responsible for repair of multiple types of DNA damage [110], and its dysregulation makes cells more sensitive to DNA lesions [111]. Defective DNA repair has been linked to neurodegenerative disorders; therefore, it is worth further determining the function of NAT-Rad18 in AD pathogenesis.
NEAT1
Nuclear paraspeckles assembly transcript 1 (NEAT1) is a lncRNA that is enriched in the nucleus and serves as an essential architectural component of paraspeckle nuclear bodies [112,113]. NEAT1 regulates gene expression by influencing the nuclear retention of structured or edited RNAs, such as RNA containing inverted Alu repeat element [114].
Levels of NEAT1 are significantly reduced in AD mouse brains and AD patients [115]. Mechanistically, NETA1 knockdown decreases Aβ degradation via altering the levels of endocytosis-related genes, such as CAV2, TGFB2 and TGFBR1 in the human astrocytic U251 cell line [115]. Aβ also induces production of NEAT1, and knockdown of NEAT1 attenuates apoptosis and p-tau levels caused by Aβ. Additionally, NEAT1 acts as a sponge for miR-107, which is known to reduce Aβ-induced injuries, leading to the aggravation of Aβ-induced neuronal damage in human SH-SY5Y and SK-N-SH cells [116]. These findings show that NEAT1 regulates AD pathogenesis via a variety of mechanisms.
MALAT1
LncRNA MALAT1 (also known as NEAT2), a long intergenic non-coding RNA, is around 7 kb in human and 6.7 kb in mouse. MALAT1 is a highly conserved lncRNA that is preferentially localized to nuclei [117]. MALAT1 is abundantly expressed at a level comparable to, or even higher than, housekeeping protein-coding genes, such as GAPDH or β-actin [118]. MALAT1 is also highly expressed in neurons, and it regulates synaptogenesis via modulating levels of neuroligin1 and synaptic cell adhesion molecule 1 (SynCAM1) [119].
Lower MALAT1 levels are detected in the CSF collected from AD patients, compared to controls [120], suggesting that CSF MALAT1 could serve as a diagnostic marker. Nonetheless, lncRNA MALAT1 inhibits neuronal apoptosis and inflammation and promotes neurite outgrowth in AD. Additionally, lncRNA MALAT1 reversely regulates the expression of miR-125b, which increased Prostaglandin-Endoperoxide Synthase 2 (PTGS2) and Cyclin Dependent Kinase 5 (CDK5) expression levels and decreased Forkhead Box Q1 (FQXQ1) expression in AD pathologies, all three are predicted regulatory targets by miR-125 [121].
LncRNA and protein translation
Translational control is another important step in gene expression. The global reduction of polysomal mRNA translation and impaired ribosome function have been identified in AD patient brains [122,123]. LncRNAs, which are involved in AD pathology via modulating protein translation, are summarized below.
BC200
Synaptic plasticity is widely accepted as an essential neurochemical foundation of learning and memory [124]. Dendritic protein synthesis is critical to maintaining long-term synaptic plasticity [125–130]. Synaptic impairment is related to AD-related memory deficits [131]. BC200 is a 200 nt lncRNA that is structurally subdivided into three domains. The 5ʹ region of BC200 contains Alu repetitive elements, the central part is rich with A-residues, and the 3ʹ region is unique to BC200. Cytoplasmic BC200 is almost exclusively expressed in neurons, and transported to dendritic processes [132]. BC200 represses translation by directly interacting with dendritic mRNA, and this repression is reversed by Poly(A)-binding protein (PABP), a protein component of the BC200 ribonucleoprotein (RNP) complexes [133,134]. Protein synthesis at the synapses of neurons contribute to neuronal plasticity; therefore, BC200 is speculated to regulate synaptic plasticity at translational level.
BC200 expression is decreased in cortical tissue in normal ageing, between the ages of 49 and 86, in addition, its levels are significantly increased in AD brains compared with age-matched normal brains. Importantly, the extent of increase is associated with the severity of the disease [135]. Mislocalization and overexpression of BC200 RNA results in dendritic regression and clogging in AD neurons. Moreover, the distribution of BC200 appeared reduced in neuropil areas in AD brains, compared with normal brains. Importantly, this altered BC200 distribution is associated with dendritic loss in AD neurons [135]. Alteration of BC200 levels is a potential drug target to rescue memory loss in AD [135–137].
BC200 also regulates BACE1 expression: knockdown of BC200 RNA significantly suppresses BACE1 levels and overexpression of BC200 increases BACE1 levels. This may lead to altered Aβ metabolism [138]. Additionally, reduction of BC200 promotes cell viability and decreases cell apoptosis via targeting BACE1 [138]. However, more evidence is required to demonstrate an interaction between BC200 and local protein translation and its implications in AD pathogenesis.
LoNA
Ribosomes and protein biosynthesis are altered in the cerebral cortex in AD brains. Levels of ribosomal RNA (rRNA), the major component of ribosomes, particularly rRNA 28 S/18 S, are decreased during the progression of AD [139,140]. Mechanistically, the nuclear organizer region (NOR) surface, where rDNA is located in the nucleolus, is reduced in AD patients [141]. Additionally, rDNA promoter is hyper-methylated [142], suggesting that epigenetic silencing occurs in AD patients. We have previously identified a long nucleolar noncoding RNA (LoNA), which is transcribed by RNA polymerase II and specifically enriched in nucleoli. The mature form of mouse LoNA is 1.5 kb in length, it contains two exons but no obvious ORF. Structurally it contains a poly(A) signal at the 3ʹ end, but no cap at the 5ʹ end. LoNA regulates rRNA transcription by binding directly to nucleolin (NCL) and reducing its activity. In addition to that, LoNA also alters rRNA methylation via interacting with fibrillarin (FBL). Consequently, LoNA regulates protein translation by modulating essential components of the ribosome and its assembly. Protein translation primarily takes place at neuronal soma but also occurs at synapses. Local protein translation has an important role in synaptic development and plasticity [130,143,144]. We have demonstrated that LoNA regulates synaptic plasticity by altering ribosomal RNA and protein levels at synapses. Subsequently, the levels of synaptic proteins, including PSD95, Synaptophysin and Snap25, are significantly increased in LoNA deficient mice. Moreover, polysome binding mRNAs of these genes are also enhanced in N2a cells lacking LoNA. In support of this, the dendrite spine density is profoundly increased in LoNA deficient mice, as demonstrated by Golgi staining. Conversely, LoNA administered mice show reduced levels of synaptic proteins, and LoNA expressing N2a cells exhibit decreased polysome binding mRNAs of same synaptic genes. Importantly, LoNA administered mice display severe LTP deficits, and behaviourally, these mice exhibit impaired cognitive functions as demonstrated by the Morris water maze task [145].
During the early stages of AD pathology, silencing of the rDNA appears to account for AD-related ribosomal deficiency [142]. Moreover, the ratio of rRNA 28 S/18 S is significantly suppressed in AD. Notably, LoNA levels are significantly increased in the hippocampus of AD mice, compared to age-matched controls, accompanied by decreased rRNA levels. Knockdown of LoNA not only partially restores rRNA levels but also rescues the cognitive deficits in AD mice [145], potentially providing an approach for AD treatment. A table (Table 1) summarizes lncRNAs discussed in this review, their biological function and roles in AD pathology have provided.
Table 1.
Key features of AD associated lncRNAs
| LncRNA | Species | Length(nt) | 5ʹCap | Poly(A) tail | Splicing pattern | Protein binding partners | References |
|---|---|---|---|---|---|---|---|
| LoNA | M | 1516 | No | Yes | CS | NCL,FBL | [145] |
| LRP1-AS | H/M | 645/1387 | Yes | Yes | CS,ES | Hmgb2 | [58] |
| BACE1-AS | H/M | 840/2025 | Yes | Yes | CS,A5’SS,A3’SS | HuD | [82] |
| Sox2OT | H/M/R | 3132/2998/722 | Yes | Yes | CS,ES,A5’SS,MECE | FUS,YY1 | [103–105,146] |
| NEAT1 | H/M | 3756/3190 | Yes | Yes | IR,A5’SS,A3’SS | NONO,SFPQ,PSF,Ezh2 | [113,116,147,148] |
| MALAT1 | H/M | 8545/6983 | Yes | No | IR,A5’SS,A3’SS,MECE | SRSF1,SFPQ | [121,149] |
| SNHG1 | H/M | 1137/476 | Yes | Yes | IR,ES,A5’SS,A3’SS,MECE | MATR3,Ezh2 | [108,150,151] |
| BDNF-AS | H | 1437 | Yes | Yes | IR,ES,A5’SS,A3’SS,MECE | PABPC1 | [93,152] |
| BC200 | H | 200 | No | No | N/A | PABP | [133,134,138] |
| 51A | H/M | 300 | No | No | N/A | unknow | [66] |
| GDNFOS | H | 1550 | Yes | Yes | IR,ES,MECE | unknow | [69] |
| EBF3-AS | H/M | 842 | Yes | Yes | CS | unknow | [99] |
| NAT-RAD18 | R | 509 | No | No | N/A | unknow | [109] |
| NDM29 | H | 1584 | No | No | N/A | unknow | [51,52] |
H:human, M:mouse, R:rat, ES: exon skipping, IR: intron retention, A5’SS: alternative 5ʹ splice sites, A3’SS: alternative 3ʹ splice sites, CS: constitutive splicing, MECE: mutually exclusive exon
Discussion and perspectives
Many lncRNAs have been discovered by sensitive, precise high-throughput genomic transcriptome sequencing, and up to 40% of these lncRNA are preferentially expressed in mammalian brains [28]. These lncRNAs also exhibit exquisitely spatiotemporally specific expression patterns, suggesting that they are biologically meaningful. As the most common type of dementia, AD is a tremendous challenge around the world. Given the advancements in transcriptome-wide profiling, numerous lncRNAs have been discovered to be associated with AD. Furthermore, levels of multiple lncRNAs are dysregulated in AD patients [42,153]. Indeed, some of the lncRNAs have demonstrated their roles in AD pathology. Although most of these studies focus on the effect of lncRNAs on Aβ metabolism, deeper and more insightful investigations are necessary. Schematic mechanisms of AD associated LncRNAs are summarized in Fig. 1.
Figure 1.
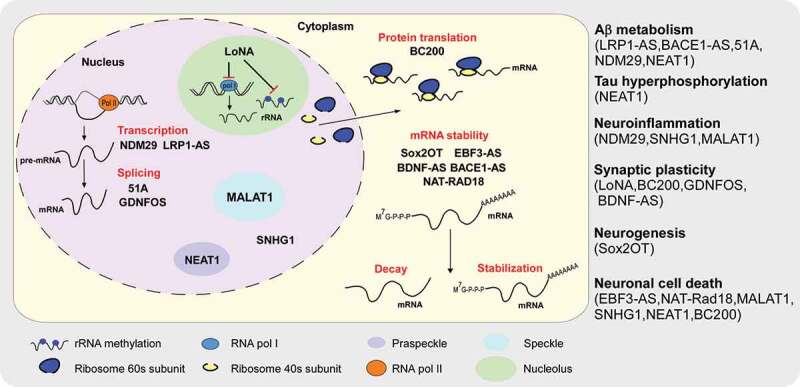
Schematic of LncRNAs and their regulatory mechanisms in AD. LncRNAs modulate cellular process such as mRNA transcription (NDM29 and LRP1-AS), mRNA splicing (51A and GDNFOS), mRNA stability (Sox2OT, EBF3-AS, BDNF-AS, BACE1-AS and NAT-RAD18), and protein translation (LoNA and BC200). LncRNAs participate in AD pathology via having impact on Aβ metabolism (LRP1-AS, BACE-AS, 51A, NDM29 and NEAT1), tau hyperphosphorylation (NEAT1), neuroinflammation (NDM29, SNHG1 and MALAT1), synaptic plasticity (LoNA, BC200, GDNFOS and BDNF-AS), neurogenesis (Sox2OT), and neuronal cell death (EBF3-AS, NAT-Rad18, MALAT1, SNHG1, NEAT1 and BC200)
In addition to deepening the understanding of AD pathology, there is also an increasing focus on the need to develop novel biomarkers to facilitate early clinical diagnosis of diseases. In the past, the diagnosis of AD has relied heavily on dementia symptoms, when the disease typically has reached a late stage. Biomarkers that show abnormal concentrations in the pre-clinical stage of AD thus allow early diagnosis of AD. LncRNAs have been considered desirable candidates for AD biomarkers. Indeed, the plasma lncRNA BACE1-AS has shown potentials as a biomarker for AD diagnosis [154]. CSF biomarkers are, however, regarded as preferential biomarkers over plasma, as the brain (interstitial fluid) is in a direct contact with the CSF, which may more accurately reflect the metabolism and pathology in the brain. Some of the exosome vectored miRNAs derived from CSF have demonstrated the possibilities of serving as biomarkers [155], however, more investigations on lncRNAs and their potential roles in AD diagnosis are still lacking.
LncRNAs could also become a novel therapeutic target for AD treatment. Antisense oligonucleotide (ASO)-based lncRNA knockdown has demonstrated a promising therapeutic effect in patients with Angelman syndrome, a single-gene disorder characterized by intellectual disability [156,157]. Although no ASOs targeting lncRNA have been developed in the treatment of neurodegenerative diseases, ASOs targeting mRNAs indeed have been approved by Food and Drug Administration (FDA) for the treatment of Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA) [158]. Additionally, ASOs also have demonstrated promising efficacy in treatment of Huntington’s disease (HD) by targeting huntingtin gene (HTT), in treatment of amyotrophic lateral sclerosis (ALS) by targeting SOD1 and C9ORF72, in treatment of AD by targeting MAPT (TAU) [159]. Multiple lncRNAs have shown differential expression in AD and control animals/patients, and most of them are tightly related to Aβ metabolism, thus lncRNAs could also serve as a novel therapeutic target for AD treatment.
Funding Statement
This research was supported by the National Natural Science Foundation of China [31871082, 91849101, 81601221], The Strategic Priority Research Program of the Chinese Academy of Sciences [XDB39000000], Key Research Program of Frontier Sciences of CAS [QYZDB-SSW-SMC035], the Innovative Program of Development Foundation of Hefei Center for Physical Science and Technology [2018CXFX005], the Fundamental Research Funds for the Central Universities, China Postdoctoral Science Foundation [2019M662178] and Anhui Provincial Natural Science Foundation [2008085QC117].
References
- [1].Alzheimer A. Über einen eigenartigen schweren erkrankungsproze β der hirnrincle. Neurol Cent. 1906;25:1134. [Google Scholar]
- [2].Sanabria-Castro A, Alvarado-Echeverria I, Monge-Bonilla C. Molecular pathogenesis of Alzheimer’s disease: an update. Ann Neurosci. 2017;24(1):46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci. 2009;11(2):111–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Campion D, Dumanchin C, Hannequin D, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664–670. DOI: 10.1086/302553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Idda ML, Munk R, Abdelmohsen K, et al. Noncoding RNAs in Alzheimer’s disease. Wiley Interdiscip Rev RNA. 2018;9(2):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430(7000):631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mohandas E, Rajmohan V, Raghunath B. Neurobiology of Alzheimer’s disease. Indian J Psychiatry. 2009;51:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jucker M, Walker LC. Neurodegeneration: amyloid-β pathology induced in humans. Nature. 2015;525:193–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].McGowan E, Pickford F, Kim J, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. [DOI] [PubMed] [Google Scholar]
- [11].Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9:768–778. [DOI] [PubMed] [Google Scholar]
- [12].Sala Frigerio C, De Strooper B. Alzheimer’s disease mechanisms and emerging roads to novel therapeutics. Annu Rev Neurosci. 2016;39:57–79. [DOI] [PubMed] [Google Scholar]
- [13].Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–706. DOI: 10.1038/349704a0 [DOI] [PubMed] [Google Scholar]
- [14].Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. [DOI] [PubMed] [Google Scholar]
- [15].Lichtenthaler SF, Haass C; Lichtenthaler SF, Haass C . Amyloid at the cutting edge: activation of alpha-secretase prevents amyloidogenesis in an Alzheimer disease mouse model. J Clin Invest. 2004;113:1384–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77(4):1081–1132. [DOI] [PubMed] [Google Scholar]
- [17].Furukawa K, Sopher BL, Rydel RE, et al. Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem. 1996;67(5):1882–1896. DOI: 10.1046/j.1471-4159.1996.67051882.x [DOI] [PubMed] [Google Scholar]
- [18].Velliquette RA, O’Connor T, Vassar R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer’s disease pathogenesis. J Neurosci. 2005;25:10874–10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Siegel GS, Chauhan N, Karczmar AG. Links between amyloid and tau biology in Alzheimer’s disease and their cholinergic aspects. In: Karczmar AG, editor. Exploring the vertebrate central cholinergic nervous system. Boston: Springer; 2007. p. 597–603. [Google Scholar]
- [20].Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–781. [DOI] [PubMed] [Google Scholar]
- [21].Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Caillet-Boudin ML, Buée L, Sergeant N, et al. Regulation of human MAPT gene expression. Mol Neurodegener. 2015;10:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kuret J, Congdon EE, Li G, et al. Evaluating triggers and enhancers of tau fibrillization. Microsc Res Tech. 2005;67:141–155. [DOI] [PubMed] [Google Scholar]
- [24].Mangialasche F, Solomon A, Winblad B, et al. Alzheimer’s disease: clinical trials and drug development. Lancet Neurol. 2010;9(7):702–716. [DOI] [PubMed] [Google Scholar]
- [25].Aisen PS, Cummings J, Jack CR Jr, et al. On the path to 2025: understanding the Alzheimer’s disease continuum. Alzheimers Res Ther. 2017;9(1):60. DOI: 10.1186/s13195-017-0283-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Knauss JL, Sun T. Regulatory mechanisms of long noncoding RNAs in vertebrate central nervous system development and function. Neuroscience. 2013;235:200–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Harrow J, Frankish A, Gonzalez JM, et al. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res. 2012;22(9):1760–1774. DOI: 10.1101/gr.135350.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–1789. DOI: 10.1101/gr.132159.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Khorkova O, Hsiao J, Wahlestedt C. Basic biology and therapeutic implications of lncRNA. Adv Drug Deliv Rev. 2015;87:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jandura A, Krause HM. The new RNA world: growing evidence for long noncoding RNA functionality. Trends Genet. 2017;33(10):665–676. [DOI] [PubMed] [Google Scholar]
- [31].Iyer MK, Niknafs YS, Malik R, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47(3):199–208. DOI: 10.1038/ng.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhao Y, Li H, Fang S, et al. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016;44(D1):D203–D208. DOI: 10.1093/nar/gkv1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62. [DOI] [PubMed] [Google Scholar]
- [34].Briggs JA, Wolvetang EJ, Mattick JS, et al. Mechanisms of long non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron. 2015;88(5):861–877. [DOI] [PubMed] [Google Scholar]
- [35].Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–641. [DOI] [PubMed] [Google Scholar]
- [36].Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10(3):155–159. [DOI] [PubMed] [Google Scholar]
- [37].Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23(13):1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhou X, Xu J. Identification of Alzheimer’s disease-associated long noncoding RNAs. Neurobiol Aging. 2015;36(11):2925–2931. [DOI] [PubMed] [Google Scholar]
- [39].Ni Y, Huang H, Chen Y, et al. Investigation of long non-coding RNA expression profiles in the substantia Nigra of Parkinson’s disease. Cell Mol Neurobiol. 2017;37(2):329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lyu Y, Bai L, Qin C. Long noncoding RNAs in neurodevelopment and Parkinson’s disease. Animal Model Exp Med. 2019;2(4):239–251. DOI: 10.1002/ame2.12093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Johnson R. Long non-coding RNAs in Huntington’s disease neurodegeneration. Neurobiol Dis. 2012;46(2):245–254. [DOI] [PubMed] [Google Scholar]
- [42].Lee DY, Moon J, Lee ST, et al. Distinct expression of long non-coding RNAs in an Alzheimer’s disease model. J Alzheimers Dis. 2015;45(3):837–849. DOI: 10.3233/JAD-142919 [DOI] [PubMed] [Google Scholar]
- [43].Fang M, Zhang P, Zhao Y, et al. Bioinformatics and co-expression network analysis of differentially expressed lncRNAs and mRNAs in hippocampus of APP/PS1 transgenic mice with Alzheimer disease. Am J Transl Res. 2017;9(3):1381–1391. [PMC free article] [PubMed] [Google Scholar]
- [44].Annese A, Manzari C, Lionetti C, et al. Whole transcriptome profiling of late-onset Alzheimer’s disease patients provides insights into the molecular changes involved in the disease. Sci Rep. 2018;8(1):4282. DOI: 10.1038/s41598-018-22701-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cao M, Li H, Zhao J, et al. Identification of age- and gender-associated long noncoding RNAs in the human brain with Alzheimer’s disease. Neurobiol Aging. 2019;81:116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mathys H, Davila-Velderrain J, Peng Z, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570(7761):332–337. DOI: 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Decourt B, Sabbagh MN. BACE1 as a Potential Biomarker for Alzheimer’s Disease. J Alzheimers Dis. 2011;24:53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pagano A, Castelnuovo M, Tortelli F, et al. New small nuclear RNA gene-like transcriptional units as sources of regulatory transcripts. Plos Genet. 2007;3(2):174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Castelnuovo M, Massone S, Tasso R, et al. An Alu-like RNA promotes cell differentiation and reduces malignancy of human neuroblastoma cells. Faseb J. 2010;24(10):4033–4046. DOI: 10.1096/fj.10-157032 [DOI] [PubMed] [Google Scholar]
- [50].Gavazzo P, Vella S, Marchetti C, et al. Acquisition of neuron-like electrophysiological properties in neuroblastoma cells by controlled expression of NDM29 ncRNA. J Neurochem. 2011;119(5):989–1001. [DOI] [PubMed] [Google Scholar]
- [51].Gu L, Guo Z. Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J Neurochem. 2013;126(3):305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Massone S, Ciarlo E, Vella S, et al. NDM29, a RNA polymerase III-dependent non coding RNA, promotes amyloidogenic processing of APP and amyloid beta secretion. Bba-Mol Cell Res. 2012;1823(7):1170–1177. [DOI] [PubMed] [Google Scholar]
- [53].Rebeck GW, Reiter JS, Strickland DK, et al. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron. 1993;11(4):575–580. [DOI] [PubMed] [Google Scholar]
- [54].Ulery PG, Beers J, Mikhailenko I. et al. Modulation of beta-amyloid precursor protein processing by the low density lipoprotein receptor-related protein (LRP). Evidence that LRP contributes to the pathogenesis of Alzheimer’s disease. J Biol Chem. 2000;275(10):7410–7415. [DOI] [PubMed] [Google Scholar]
- [55].von Einem B, Schwanzar D, Rehn F, et al. The role of low-density receptor-related protein 1 (LRP1) as a competitive substrate of the amyloid precursor protein (APP) for BACE1. Exp Neurol. 2010;225(1):85–93. DOI: 10.1016/j.expneurol.2010.05.017 [DOI] [PubMed] [Google Scholar]
- [56].Kanekiyo T, Cirrito JR, Liu CC, et al. Neuronal clearance of amyloid-β by endocytic receptor LRP1. J Neurosci. 2013;33(49):19276–19283. DOI: 10.1523/JNEUROSCI.3487-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Liu Q, Zerbinatti CV, Zhang J, et al. Amyloid precursor protein regulates brain apolipoprotein e and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007;56(1):66–78. DOI: 10.1016/j.neuron.2007.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yamanaka Y, Faghihi MA, Magistri M, et al. Antisense RNA controls LRP1 sense transcript expression through interaction with a chromatin-associated protein, HMGB2. Cell Rep. 2015;11(6):967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Matsui T, Ingelsson M, Fukumoto H, et al. Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res. 2007;1161:116–123. [DOI] [PubMed] [Google Scholar]
- [60].Raj T, Li YI, Wong G, et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat Genet. 2018;50(11):1584-+. DOI: 10.1038/s41588-018-0238-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Motoi Y, Aizawa T, Haga S, et al. Neuronal localization of a novel mosaic apolipoprotein E receptor, LR11, in rat and human brain. Brain Res. 1999;833(2):209–215. [DOI] [PubMed] [Google Scholar]
- [62].Hermans-Borgmeyer I, Hampe W, Schinke B, et al. Unique expression pattern of a novel mosaic receptor in the developing cerebral cortex. Mech Dev. 1998;70(1–2):65–76. DOI: 10.1016/S0925-4773(97)00177-9 [DOI] [PubMed] [Google Scholar]
- [63].CR S, Offe K, Gearing M, et al. Loss of apolipoprotein E receptor LR11 in Alzheimer disease [published correction appears in arch neurol. 2007 Apr;64(4):557]. Arch Neurol. 2004;61(8):1200–1205. DOI: 10.1001/archneur.61.8.1200 [DOI] [PubMed] [Google Scholar]
- [64].Offe K, SE D, JT S, et al. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J Neurosci. 2006;26(5):1596–1603. DOI: 10.1523/JNEUROSCI.4946-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Reitz C. The role of intracellular trafficking and the VPS10d receptors in Alzheimer’s disease. Future Neurol. 2012;7(4):423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ciarlo E, Massone S, Penna I, et al. An intronic ncRNA-dependent regulation of SORL1 expression affecting Abeta formation is upregulated in post-mortem Alzheimer’s disease brain samples. Dis Model Mech. 2013;6(2):424–433. DOI: 10.1242/dmm.009761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lin LF, Doherty DH, Lile JD, et al. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260(5111):1130–1132. [DOI] [PubMed] [Google Scholar]
- [68].Gash DM, Zhang Z, Ovadia A, et al. Functional recovery in parkinsonian monkeys treated with GDNF. Nature. 1996;380(6571):252–255. DOI: 10.1038/380252a0 [DOI] [PubMed] [Google Scholar]
- [69].Airavaara M, Pletnikova O, Doyle ME, et al. Identification of novel GDNF isoforms and cis-antisense GDNFOS gene and their regulation in human middle temporal gyrus of Alzheimer disease. J Biol Chem. 2011;286(52):45093–45102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Matsushita N, Fujita Y, Tanaka M, et al. Cloning and structural organization of the gene encoding the mouse glial cell line-derived neurotrophic factor, GDNF. Gene. 1997;203(2):149–157. [DOI] [PubMed] [Google Scholar]
- [71].Grimm L, Holinski-Feder E, Teodoridis J, et al. Analysis of the human GDNF gene reveals an inducible promoter, three exons, a triplet repeat within the 3ʹ-UTR and alternative splice products. Hum Mol Genet. 1998;7(12):1873–1886. DOI: 10.1093/hmg/7.12.1873 [DOI] [PubMed] [Google Scholar]
- [72].Lonka-Nevalaita L, Lume M, Leppanen S, et al. Characterization of the intracellular localization, processing, and secretion of two glial cell line-derived neurotrophic factor splice isoforms. J Neurosci. 2010;30(34):11403–11413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Straten G, Eschweiler GW, Maetzler W, et al. Glial cell-line derived neurotrophic factor (GDNF) concentrations in cerebrospinal fluid and serum of patients with early Alzheimer’s disease and normal controls. J Alzheimers Dis. 2009;18(2):331–337. [DOI] [PubMed] [Google Scholar]
- [74].Alkallas R, Fish L, Goodarzi H, et al. Inference of RNA decay rate from transcriptional profiling highlights the regulatory programs of Alzheimer’s disease. Nat Commun. 2017;8(1):909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Beyer N, Coulson DTR, Heggarty S, et al. ZnT3 mRNA levels are reduced in Alzheimer’s disease post-mortem brain. Mol Neurodegener. 2009;4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Vassar R, Bennett BD, Babu-Khan S, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. DOI: 10.1126/science.286.5440.735 [DOI] [PubMed] [Google Scholar]
- [77].Fukumoto H, Cheung BS, Hyman BT, et al. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59(9):1381–1389. [DOI] [PubMed] [Google Scholar]
- [78].Yang LB, Lindholm K, Yan R, et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9(1):3–4. DOI: 10.1038/nm0103-3 [DOI] [PubMed] [Google Scholar]
- [79].Luo Y, Bolon B, Kahn S, et al. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001;4(3):231–232. DOI: 10.1038/85059 [DOI] [PubMed] [Google Scholar]
- [80].Faghihi MA, Modarresi F, Khalil AM, et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14(7):723–730. DOI: 10.1038/nm1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Liu T, Huang YY, Chen JL, et al. Attenuated ability of BACE1 to cleave the amyloid precursor protein via silencing long noncoding RNA BACE1-AS expression. Mol Med Rep. 2014;10(3):1275–1281. DOI: 10.3892/mmr.2014.2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kang MJ, Abdelmohsen K, Hutchison ER, et al. HuD regulates coding and noncoding RNA to induce APP–>Abeta processing. Cell Rep. 2014;7(5):1401–1409. DOI: 10.1016/j.celrep.2014.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Modarresi F, Faghihi MA, Patel NS, et al. Knockdown of BACE1-AS nonprotein-coding transcript modulates beta-amyloid-related hippocampal neurogenesis. Int J Alzheimers Dis. 2011;2011:929042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Binder DK, Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. 2004;22(3):123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Huang W, Meng F, Cao J, et al. Neuroprotective role of exogenous brain-derived neurotrophic factor in hypoxia-hypoglycemia-induced hippocampal neuron injury via regulating Trkb/MiR134 signaling. J Mol Neurosci. 2017;62(1):35–42. [DOI] [PubMed] [Google Scholar]
- [86].Li GD, Bi R, Zhang DF, et al. Female-specific effect of the BDNF gene on Alzheimer’s disease. Neurobiol Aging. 2017;53:192.e11–192.e19. [DOI] [PubMed] [Google Scholar]
- [87].Diniz BS, Teixeira AL. Brain-derived neurotrophic factor and Alzheimer’s disease: physiopathology and beyond. Neuromolecular Med. 2011;13(4):217–222. [DOI] [PubMed] [Google Scholar]
- [88].Poon WW, Blurton-Jones M, Tu CH, et al. β-Amyloid impairs axonal BDNF retrograde trafficking. Neurobiol Aging. 2011;32(5):821–833. DOI: 10.1016/j.neurobiolaging.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Arancibia S, Silhol M, Mouliere F, et al. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol Dis. 2008;31(3):316–326. DOI: 10.1016/j.nbd.2008.05.012 [DOI] [PubMed] [Google Scholar]
- [90].Kimura N, Takahashi M, Tashiro T, et al. Amyloid beta up-regulates brain-derived neurotrophic factor production from astrocytes: rescue from amyloid beta-related neuritic degeneration. J Neurosci Res. 2006;84(4):782–789. [DOI] [PubMed] [Google Scholar]
- [91].Peng S, Garzon DJ, Marchese M, et al. Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease. J Neurosci. 2009;29(29):9321–9329. DOI: 10.1523/JNEUROSCI.4736-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Nagahara AH, Merrill DA, Coppola G, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15(3):331–337. DOI: 10.1038/nm.1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Modarresi F, Faghihi MA, Lopez-Toledano MA, et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol. 2012;30(5):453–459. DOI: 10.1038/nbt.2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Guo CC, Jiao CH, Gao ZM. Silencing of LncRNA BDNF-AS attenuates Aβ25-35-induced neurotoxicity in PC12 cells by suppressing cell apoptosis and oxidative stress. Neurol Res. 2018;40(9):795–804. [DOI] [PubMed] [Google Scholar]
- [95].Bohnsack JP, Teppen T, Kyzar EJ, et al. The lncRNA BDNF-AS is an epigenetic regulator in the human amygdala in early onset alcohol use disorders. Transl Psychiatry. 2019;9(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wang MM, Reed RR. Molecular cloning of the olfactory neuronal transcription factor Olf-1 by genetic selection in yeast. Nature. 1993;364(6433):121–126. [DOI] [PubMed] [Google Scholar]
- [97].Chao HT, Davids M, Burke E, et al. A syndromic neurodevelopmental disorder caused by De Novo variants in EBF3. Am J Hum Genet. 2017;100(1):128–137. DOI: 10.1016/j.ajhg.2016.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Zhao LY, Niu Y, Santiago A, et al. An EBF3-mediated transcriptional program that induces cell cycle arrest and apoptosis. Cancer Res. 2006;66(19):9445–9452. DOI: 10.1158/0008-5472.CAN-06-1713 [DOI] [PubMed] [Google Scholar]
- [99].Gu C, Chen C, Wu R, et al. Long noncoding RNA EBF3-AS PROMOTES NEURON APOPTOSIS in Alzheimer’s disease. DNA Cell Biol. 2018;37(3):220–226. DOI: 10.1089/dna.2017.4012 [DOI] [PubMed] [Google Scholar]
- [100].Zhang S, Cui W. Sox2, a key factor in the regulation of pluripotency and neural differentiation. World J Stem Cells. 2014;6(3):305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Amaral PP, Neyt C, Wilkins SJ, et al. Complex architecture and regulated expression of the Sox2ot locus during vertebrate development. RNA. 2009;15(11):2013–2027. DOI: 10.1261/rna.1705309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Fantes J, Ragge NK, Lynch SA, et al. Mutations in SOX2 cause anophthalmia. Nat Genet. 2003;33(4):461–463. DOI: 10.1038/ng1120 [DOI] [PubMed] [Google Scholar]
- [103].Askarian-Amiri ME, Seyfoddin V, Smart CE, et al. Emerging role of long non-coding RNA SOX2OT in SOX2 regulation in breast cancer. PLoS One. 2014;9(7):e102140. DOI: 10.1371/journal.pone.0102140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Knauss JL, Miao N, Kim SN, et al. Long noncoding RNA Sox2ot and transcription factor YY1 co-regulate the differentiation of cortical neural progenitors by repressing Sox2. Cell Death Dis. 2018;9(8):799. DOI: 10.1038/s41419-018-0840-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Arisi I, D’Onofrio M, Brandi R, et al. Gene expression biomarkers in the brain of a mouse model for Alzheimer’s disease: mining of microarray data by logic classification and feature selection. J Alzheimers Dis. 2011;24(4):721–738. DOI: 10.3233/JAD-2011-101881 [DOI] [PubMed] [Google Scholar]
- [106].Cao B, Wang T, Qu Q, et al. Long noncoding RNA SNHG1 promotes neuroinflammation in Parkinson’s disease via regulating miR-7/NLRP3 pathway. Neuroscience. 2018;388:118–127. [DOI] [PubMed] [Google Scholar]
- [107].Chen Y, Lian YJ, Ma YQ, et al. LncRNA SNHG1 promotes alpha-synuclein aggregation and toxicity by targeting miR-15b-5p to activate SIAH1 in human neuroblastoma SH-SY5Y cells. Neurotoxicology. 2018;68:212–221. [DOI] [PubMed] [Google Scholar]
- [108].Wang H, Lu B, Chen J. Knockdown of lncRNA SNHG1 attenuated Aβ25-35-induced neuronal injury via regulating KREMEN1 by acting as a ceRNA of miR-137 in neuronal cells. Biochem Biophys Res Commun. 2019;518(3):438–444. [DOI] [PubMed] [Google Scholar]
- [109].Parenti R, Paratore S, Torrisi A, et al. A natural antisense transcript against Rad18, specifically expressed in neurons and upregulated during beta-amyloid-induced apoptosis. Eur J Neurosci. 2007;26(9):2444–2457. [DOI] [PubMed] [Google Scholar]
- [110].Harvey SH, Sheedy DM, Cuddihy AR, et al. Coordination of DNA damage responses via the Smc5/Smc6 complex. Mol Cell Biol. 2004;24(2):662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Tateishi S, Sakuraba Y, Masuyama S, et al. Dysfunction of human Rad18 results in defective postreplication repair and hypersensitivity to multiple mutagens. Proc Natl Acad Sci U S A. 2000;97(14):7927–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Clemson CM, Hutchinson JN, Sara SA, et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell. 2009;33(6):717–726. DOI: 10.1016/j.molcel.2009.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Yamazaki T, Souquere S, Chujo T, et al. Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol Cell. 2018;70(6):1038–53 e7. DOI: 10.1016/j.molcel.2018.05.019 [DOI] [PubMed] [Google Scholar]
- [114].Chen LL, Carmichael GG. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol Cell. 2009;35(4):467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Wang Z, Zhao Y, Xu N, et al. NEAT1 regulates neuroglial cell mediating Abeta clearance via the epigenetic regulation of endocytosis-related genes expression. Cell Mol Life Sci. 2019;76(15):3005–3018. DOI: 10.1007/s00018-019-03074-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Ke S, Yang Z, Yang F, et al. Long noncoding RNA NEAT1 aggravates Aβ-induced neuronal damage by targeting miR-107 in Alzheimer’s disease. Yonsei Med J. 2019;60(7):640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Hutchinson JN, Ensminger AW, Clemson CM, et al. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007;8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zhang B, Arun G, Mao YS, et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012;2(1):111–123. DOI: 10.1016/j.celrep.2012.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Bernard D, Prasanth KV, Tripathi V, et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. Embo J. 2010;29(18):3082–3093. DOI: 10.1038/emboj.2010.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Yao J, Wang XQ, Li YJ, et al. Long non-coding RNA MALAT1 regulates retinal neurodegeneration through CREB signaling. EMBO Mol Med. 2016;8(9):1113. DOI: 10.15252/emmm.201606749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Ma P, Li Y, Zhang W, et al. Long non-coding RNA MALAT1 inhibits neuron apoptosis and neuroinflammation while stimulates neurite outgrowth and its correlation with MiR-125b mediates PTGS2, CDK5 and FOXQ1 in Alzheimer’s disease. Curr Alzheimer Res. 2019;16(7):596–612. DOI: 10.2174/1567205016666190725130134 [DOI] [PubMed] [Google Scholar]
- [122].Langstrom NS, Anderson JP, Lindroos HG, et al. Alzheimer’s disease-associated reduction of polysomal mRNA translation. Brain Res Mol Brain Res. 1989;5(4):259–269. [DOI] [PubMed] [Google Scholar]
- [123].QX D, WR M, QH C, et al. Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci. 2005;25(40):9171–9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Takeuchi T, Duszkiewicz AJ, Morris RG. The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci. 2013;369(1633):20130288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127(1):49–58. [DOI] [PubMed] [Google Scholar]
- [126].Bradshaw KD, Emptage NJ, Bliss TVP. A role for dendritic protein synthesis in hippocampal late LTP. Eur J Neurosci. 2003;18(11):3150–3152. [DOI] [PubMed] [Google Scholar]
- [127].Pfeiffer BE, Huber KM. Current advances in local protein synthesis and synaptic plasticity. J Neurosci. 2006;26(27):7147–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Schuman EM, Dynes JL, Steward O. Synaptic regulation of translation of dendritic mRNAs. J Neurosci. 2006;26(27):7143–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Steward O, Schuman EM. Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci. 2001;24:299–325. [DOI] [PubMed] [Google Scholar]
- [130].Richter JD, Klann E. Making synaptic plasticity and memory last: mechanisms of translational regulation. Gene Dev. 2009;23(1):1–11. [DOI] [PubMed] [Google Scholar]
- [131].Nistico R, Pignatelli M, Piccinin S, et al. Targeting synaptic dysfunction in Alzheimer’s disease therapy. Mol Neurobiol. 2012;46(3):572–587. [DOI] [PubMed] [Google Scholar]
- [132].Tiedge H, Chen W, Brosius J. Primary structure, neural-specific expression, and dendritic location of human BC200 RNA. J Neurosci. 1993;13(6):2382–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Kondrashov AV, Kiefmann M, Ebnet K, et al. Inhibitory effect of naked neural BC1 RNA or BC200 RNA on eukaryotic in vitro translation systems is reversed by poly(A)-binding protein (PABP). J Mol Biol. 2005;353(1):88–103. [DOI] [PubMed] [Google Scholar]
- [134].Muddashetty R, Khanam T, Kondrashov A, et al. Poly(A)-binding protein is associated with neuronal BC1 and BC200 ribonucleoprotein particles. J Mol Biol. 2002;321(3):433–445. DOI: 10.1016/S0022-2836(02)00655-1 [DOI] [PubMed] [Google Scholar]
- [135].Mus E, Hof PR, Tiedge H. Dendritic BC200 RNA in aging and in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007;104(25):10679–10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Albert MS. Cognitive and neurobiologic markers of early Alzheimer disease. Proc Natl Acad Sci U S A. 1996;93(24):13547–13551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Saxton J, Lopez OL, Ratcliff G, et al. Preclinical Alzheimer disease: neuropsychological test performance 1.5 to 8 years prior to onset. Neurology. 2004;63(12):2341–2347. DOI: 10.1212/01.WNL.0000147470.58328.50 [DOI] [PubMed] [Google Scholar]
- [138].Li H, Zheng L, Jiang A, et al. Identification of the biological affection of long noncoding RNA BC200 in Alzheimer’s disease. Neuroreport. 2018;29(13):1061–1067. [DOI] [PubMed] [Google Scholar]
- [139].Ding Q, WR M, Cecarini V, et al. Decreased RNA, and increased RNA oxidation, in ribosomes from early Alzheimer’s disease. Neurochem Res. 2006;31(5):705–710. [DOI] [PubMed] [Google Scholar]
- [140].Silva AM, Payao SL, Borsatto B, et al. Quantitative evaluation of the rRNA in Alzheimer’s disease. Mech Ageing Dev. 2000;120(1–3):57–64. [DOI] [PubMed] [Google Scholar]
- [141].Dönmez-Altuntaş H, Akalin H, Karaman Y, et al. Evaluation of the nucleolar organizer regions in Alzheimer’s disease. Gerontology. 2005;51(5):297–301. [DOI] [PubMed] [Google Scholar]
- [142].Pietrzak M, Rempala G, Nelson PT, et al. Epigenetic silencing of nucleolar rRNA genes in Alzheimer’s disease. PLoS One. 2011;6(7):e22585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Riba A, Di Nanni N, Mittal N, et al. Protein synthesis rates and ribosome occupancies reveal determinants of translation elongation rates. Proc Natl Acad Sci U S A. 2019;116(30):15023–15032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Martin KC, Ephrussi A. mRNA Localization: gene Expression in the Spatial Dimension. Cell. 2009;136(4):719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Li DF, Zhang J, Wang M, et al. Activity dependent LoNA regulates translation by coordinating rRNA transcription and methylation. Nat Commun. 2018;9(1):1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Chen L, Zhang JJ, Chen Q, et al. Long noncoding RNA SOX2OT promotes the proliferation of pancreatic cancer by binding to FUS. Int J Cancer. 2020;147(1):175–188. DOI: 10.1002/ijc.32827 [DOI] [PubMed] [Google Scholar]
- [147].Jiang L, Shao CW, Wu QJ, et al. NEAT1 scaffolds RNA-binding proteins and the Microprocessor to globally enhance pri-miRNA processing. Nat Struct Mol Biol. 2017;24(10):816-+. DOI: 10.1038/nsmb.3455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Wang SS, Zuo H, Jin JJ, et al. Long noncoding RNA Neat1 modulates myogenesis by recruiting Ezh2. Cell Death Dis. 2019;10(7):505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Tripathi V, Ellis JD, Shen Z, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39(6):925–938. DOI: 10.1016/j.molcel.2010.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Yang TW, Sahu D, Chang YW, et al. RNA-binding proteomics reveals MATR3 interacting with lncRNA SNHG1 to enhance neuroblastoma progression. J Proteome Res. 2019;18(1):406–416. DOI: 10.1021/acs.jproteome.8b00693 [DOI] [PubMed] [Google Scholar]
- [151].Xu M, Chen XX, Lin K, et al. The long noncoding RNA SNHG1 regulates colorectal cancer cell growth through interactions with EZH2 and miR-154-5p. Mol Cancer. 2018;17(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Su R, Ma J, Zheng J, et al. PABPC1-induced stabilization of BDNF-AS inhibits malignant progression of glioblastoma cells through STAU1-mediated decay. Cell Death Dis. 2020;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Magistri M, Velmeshev D, Makhmutova M, et al. Transcriptomics profiling of Alzheimer’s disease reveal neurovascular defects, altered amyloid-beta homeostasis, and deregulated expression of long noncoding RNAs. J Alzheimers Dis. 2015;48(3):647–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Feng L, Liao Y-T, He J-C, et al. Plasma long non-coding RNA BACE1 as a novel biomarker for diagnosis of Alzheimer disease. BMC Neurol. 2018;18(1):4. DOI: 10.1186/s12883-017-1008-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Gui Y, Liu H, Zhang L, et al. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget. 2015;6(35):37043–37053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Bird LM. Angelman syndrome: review of clinical and molecular aspects. Appl Clin Genet. 2014;7:93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Meng L, Ward AJ, Chun S, et al. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518(7539):409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Wurster CD, Ludolph AC. Antisense oligonucleotides in neurological disorders. Ther Adv Neurol Diso. 2018;11:1756286418776932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Scoles DR, Minikel EV, Pulst SM. Antisense oligonucleotides. Neurol-Genet. 2019;5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]