

ABSTRACT
T-cell receptor (TCR) redirected T cells are considered as the next generation of care for the treatment of numerous solid tumors. KRAS mutations are driver neoantigens that are expressed in over 25% of all cancers and are thus regarded as ideal targets for Adoptive Cell Therapy (ACT). We have isolated four KRAS-specific TCRs from a long-term surviving pancreatic cancer patient vaccinated with a mix of mutated KRAS peptides. The sequence of these TCRs could be identified and expressed in primary cells. We demonstrated stable expression of all TCRs as well as target-specific functionality when expressing T cells were co-incubated with target cells presenting KRAS peptides. In addition, these TCRs were all partially co-receptor independent since they were functional in both CD4 and CD8 T cells, thus indicating high affinity. Interestingly, we observed that certain TCRs were able to recognize several KRAS mutations in complex with their cognate Human leukocyte antigen (HLA), suggesting that, here, the point mutations were less important for the HLA binding and TCR recognition, whereas others were single-mutation restricted. Finally, we demonstrated that these peptides were indeed processed and presented, since HLA-matched antigen presenting cells exogenously loaded with KRAS proteins were recognized by TCR-transduced T cells. Taken together, our data demonstrate that KRAS mutations are immunogenic for CD4 T cells and are interesting targets for TCR-based cancer immunotherapy.
KEYWORDS: Immunotherapy, Adoptive Cell Therapy, TCR, KRAS
Introduction
Cancer is one of the leading causes of mortality worldwide, being responsible for 9.6 million deaths in 20181, of which 8.6 million are attributable to solid tumors. The prognosis for these is often worse than their liquid counterparts. In addition to recent advances in prevention, screening and diagnosis of cancer, the landscape of cancer treatment has recently been reshaped by the advent of immunotherapy, offering improved survival in several solid cancers and establishing itself as a new therapeutic modality.2 As an important component of immunotherapy, Adoptive Cell Therapy (ACT) is considered a new treatment opportunity for solid tumors.3 It relies on the use of genetically modified T cells that permit the guidance of these effector cells to the cancer through the introduction of a synthetic T Cell Receptor (TCR) or a Chimeric Antigen Receptor (CAR). The most advanced ACT is a CAR recognizing the B-cell antigen marker CD19 that has been successfully used in the treatment of several B-cell malignancies.4,5 Despite their efficacy, CAR molecules solely recognize surface molecules and are thus limited in their target repertoire while increasing the risk of recognition of healthy tissues.6 TCRs, on the other hand, recognize peptides loaded onto the HLA complex and can thus potentially detect peptides from any protein in the cell.
Cancer development and growth require the mutations of numerous proteins to prevent apoptosis while promoting aberrant proliferation. Such mutated proteins, also called neoantigens, are ideal targets for TCR-based ACT because redirected T cells could then specifically target the tumor while drastically reducing the risk of unwanted cross-reactivity.7 In this line, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) is considered an ideal target. Indeed, KRAS is a proto-oncogene, coding for a GTPase that controls cell proliferation.8,9 Ras is a critical player in the maintenance of normal proliferation and survival, and KRAS mutations are defined as driver mutations, i.e. they are responsible for both the initiation and the maintenance of cancer.10 Thus, these mutations (often on codon 12 (72.5% of cases) or codon 13 (20% of cases)),11 are implicated in the emergence of various malignancies, most frequently in non-small cell lung cancer (NSCLC), colorectal cancer (CRC), and pancreatic ductal adenocarcinoma (PDAC). As an example, PDAC still has limited therapeutic options with a 5-year survival rate below 5%. Additionally, it is estimated that a quarter of all patients with cancer carry such mutations, including half of those with metastatic cancer.12 Finally, KRAS mutations are associated with the emergence of treatment resistance, as seen with mutated KRAS conferring resistance to cetuximab (anti-EGFR) treatment in CRC.13,14 These observations explain why several T cell- based ACT approaches have been proposed to target KRAS in solid tumors.15 As an example, Cafri and colleagues identified both CD8 and CD4 memory T cells from patient blood that recognized two KRAS mutations and isolated two KRAS-specific, HLA class II-restricted TCRs.13 HLA class II-restricted TCRs recognizing KRAS G12V mutant peptide have also been identified in two patients with endometrial cancer patient and lung cancer.16,17 We have previously shown, in the first neoantigen vaccine trial performed, that KRAS can be successfully targeted by vaccination with mutant ras peptides to induce T-cell responses.18 A series of follow-up clinical studies confirmed this,19–22 however, due to frequent late-stage diagnosis, where the clinical benefit of vaccines is limited, more potent, faster working therapy such as ACT is required in patients with metastatic disease. Therapy targeting KRAS mutations is thus regarded as very promising, even if the RAS family is considered difficult to attack with small-molecule inhibitors, due to redundancy in signaling pathways,23 and that clinical trials with TCR redirected T cells have yet to show convincing efficacy.14
Several approaches have been developed to create antigen-specific TCR molecules. One relies on directed evolution or the use of transgenic mice24,25 to create high-affinity TCRs. However, such molecules have been shown to lead to fatal outcomes, mainly due to off-tumor toxicity.25,26 A second approach, used by our group and other labs, relies on the use of tumor-reactive TCRs isolated from patients responding to immunotherapy.27–29 These TCRs are considered safer since they have not been modified and have already passed selection in a thymus.
Here we report the isolation and the pre-clinical evaluation of four TCRs (Radium-10, −11, −12 and −13) recognizing several KRAS mutations presented by different HLA molecules. The TCRs were identified in a pancreatic cancer patient several years after vaccination (CTN Ras 9801021). As the KRAS mutational status of the patients included in this protocol was unknown, the vaccine consisted of a mixture of seven peptides covering different KRAS mutations. The patient presented here responded immunologically against all seven peptides in the vaccine and displayed long term immunological memory. We investigated the TCR specificity in functional assays showing that all these TCRs were efficiently expressed and active in both CD4 and CD8 cells, suggesting their high affinity and partial co-receptor independency. In order to validate that these KRAS peptides-HLA were bona fide complexes, we tested the TCR reactivity against APCs which were loaded with full-length protein and measured T-cell activation. In summary, we are reporting on the effectiveness of several MHC class II-restricted TCRs to recognize some of the most prominent neoantigens in solid tumors: KRAS mutations. Taken together, our data suggest that KRAS TCRs are valid candidates for future clinical development.
Material and methods
Cell lines, media and reagents
Epstein Barr Virus-transformed lymphoblastoid cell lines (EBV-LCLs) were used as target cells, and were generated by immortalization of B cells from peripheral blood mononuclear cells (PBMCs) of patients and donors using EBV supernatant from the marmoset cell line B95.8.30 The TCR-negative cell line, J76 was a kind gift from M. Heemskerk (Leiden University Medical Center, the Netherlands). All cell lines were passaged for fewer than 6 months after their purchase. Cell lines were tested for mycoplasma contamination using a PCR based detection kit (Venor®GeM, Minerva Biolabs, Berlin, Germany). Cell lines were cultured in RPMI-1640 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with gentamicin and 10% Fetal calf serum (FCS) (Gibco, Thermo Fisher Scientific), and Hek-P were grown in DMEM (GE Healthcare, Chicago, IL, USA) supplemented with 10% HyClone FCS (GE Healthcare) and 1% antibiotic-antimitotic (penicillin/streptomycin, p/s, GE Healthcare). All T cells were grown in X–Vivo 15 (Lonza, Basel, Switzerland) supplemented with 5% CST serum replacement (Thermo Fisher Scientific) and 100 U/ml IL-2 (Proleukin, Novartis, Basel, Switzerland), denoted complete medium hereafter, unless otherwise stated.
Patient T cell clones
Vaccine-specific T cell responses in patient samples in the clinical study CTN Ras 98010 were tested as previously described in 3H-thymidine incorporation assays.21 The Stimulation Index (SI) was defined as proliferation with peptide divided by proliferation without peptide. SI ≥ 2 was considered a positive response and counts per minute (cpm) of all tested conditions were used and plotted. Between February and April 2006, blood samples from 5‐year survivors were collected and analyzed for long‐term immunological response using the same assay. T cells were cloned by limiting dilution as previously described, then expanded using irradiated PBMCs as feeder cells, PHA (Oxoid Ltd, Basingstoke, UK) and IL-2, then screened for peptide specificity in proliferation assays.22,31 The resulting T-cell clones were characterized with respect to peptide responses and HLA restriction in 3 H-thymidine incorporation assays.31 Briefly, the addition of blocking antibodies B7/21 (anti-HLA-DP), SPV-L3 (anti-HLA-DQ), and B8.11 (anti-HLA-DR) was used to determine the HLA-restriction of the T cell clones.31 Autologous EBV-LCLs were used as the antigen presenting cell line.
Patient lymphocytes were genotyped for HLA class II by the National tissue typing laboratory and found to be HLA-DRB1*04:04/04:01, -DQB1*03:02 (see Supplementary Table S1).
TCR cloning
KRAS-specific T-cell clones were cultured and total RNA was prepared. TCR cloning was performed using a modified 5ʹ-RACE method. Briefly, cDNA was synthesized using an oligo-dT primer and tailed at the 5ʹ-end with a stretch of cytosines. A polyguanosine primer and a constant domain-specific primer were used to amplify TCR chains.32 Upon sequence identification, a TCR-2A construct consisting of the TCRa connected to the TCRb via a 2A ribosome skipping sequence, was designed and ordered as a synthetic fragment containing unique cloning sites, NotI and XhoI for direct cloning into pENTR (Invitrogen, Carlsbad, CA, USA). Radium-10, Radium-11 and Radium-12 were further tagged with a truncated CD34 peptide via a second 2A sequence inserted instead of the STOP codon of the TCRb chain. Retroviral constructs were prepared by recombining these pENTR constructs into pMP71, Gateway-modified.32
Retroviral particle production
Viral particles were used to transduce T cells and produced as previously stated.32 Briefly, 1.2 × 106 Hek-P cells were seeded in a 6-well plate. Transfection was performed using Extreme-gene 9 (Roche) with a mix of DNA including the retroviral packaging vectors and the expression vector in an equimolar ratio. After 24 hours, the medium was replaced with 1% HyClone FCS-containing DMEM, and the cells were transferred to a 32°C incubator. Supernatants were harvested after 24 h and 48 h of incubation.
Transduction of human T cells
The study was approved by the Regional Committee for Medical Research Ethics (Oslo, Norway) (REC approval no: 2013/624, 2016/2247). PBMCs were isolated from Buffy coats by density gradient centrifugation. PBMCs were then incubated for 2 days in a 24 well-plate coated with CD3 and CD28 at 106 cells/mL. A 24-well plate was coated with 50 µg/mL of retronectin during for 3 hours at room temperature before being washed with PBS and blocked with a solution of 1 mg/mL of FBS for 30 minutes. One mL of virus solution was deposited in each well and topped with 500 µL of activated T-cells at a concentration of 0.3 × 105 cells/mL. The plate was then incubated at 37°C, 5% CO2 for 30 minutes, sealed, and then spun down at 750 g, 32°C for 60 minutes, before being returned to the incubator. The same spinoculation step was repeated the following day. Cells were then collected, spun down, washed and resuspended in complete X–Vivo 15 medium for 2 days. The expression of the TCR was then checked and the cells expanded using the procedure previously described.29
In vitro functional assay, antibodies, and flow cytometry
All antibodies were purchased from eBioscience (Thermo Fisher Scientific), except where noted. TCR expression was determined as follows: J76 or T cells were washed in 500 µL of FCS and spun down. The supernatant was discarded and J76 or T cells were incubated with 5 µL of anti-Vβ3 FITC (Radium-13), anti- Vβ13.1-PE (Radium-10), anti-Vβ20-PE (Radium-11) (Beckman Coulter, Marseille, France), anti-TCRαβ-PE (Radium-12, J76), anti-CD3 BV 605 and anti-CD34 APC (Radium-10, Radium-11 and Radium-12). KRAS peptides, 306: KLVVVGAGDVGKSALTI, 313: KLVVVGADGVGKSALTI, 319: KLVVVGAVGVGKSALTI, 322: KLVVVGACGVGKSALTI, 484: KLVVVGAAGVGKSALTI, 516: KLVVVGASGVGKSALTI, 321: KLVVVGARGVGKSALTI, and 382: KLVVVGAGGVGKSALTI, as well as unrelated hTERT peptide (negative control): 720 PGLLGASVLGLDDIH were synthesized by ProImmune Ltd, Oxford, UK. KRAS proteins were obtained from Sino Biological, Wayne, PA US. For cytokine production assays, T cells were stimulated for 16 hours with APCs, loaded or not with various concentrations (as indicated) of KRAS peptides or KRAS proteins, at an effector to target (E:T) ratio of 1:2, and in the presence of BD GolgiPlug and BD Golgistop at recommended concentrations. For intracellular staining, cells were stained using the PerFix-nc kit according to the manufacturer’s instructions (Beckman Coulter Inc, Brea, CA, USA). The following antibodies were used: CD4-BV421 (BioLegend, San Diego, CA, USA), CD8-PE-Cy7, IFN-γ-FITC, and TNF-α-PE (BD Biosciences). Cells were acquired on a BD FACSCanto flow cytometer and the data analyzed using FlowJo software (Treestar Inc., Ashland, OR, USA).
Bioluminescence-based cytotoxicity assay
Luciferase-expressing EBV-LCL tumor cells were counted and resuspended at a concentration of 3 × 105 cells/mL. Cells were given D-Luciferin (75 µg/ml; Perkin Elmer, Waltham, MS, USA) and were placed in 96-well white flat-bottomed plates at 100 µl cells/well in triplicates. The EBV-LCLs were then loaded with different 10 µM KRAS mutated peptides as indicated. Effector T cells were added at a 20:1 E:T ratio. In order to determine spontaneous and maximal killing, wells with target cells only or with target cells in 1% Triton™ X-100 (Sigma-Aldrich, St Louis, MO, USA) were seeded. Cells were left at 37°C and the bioluminescence (BLI) was measured with a luminometer (VICTOR Multilabel Plate Reader, Perkin Elmer) as relative light units (RLU) at indicated time points. Target cells incubated without any effector cells were used to determine baseline spontaneous death RLU at each time point. Triplicate wells were averaged and lysis percentage was calculated using the following equation: % specific lysis = 100x(spontaneous cell death RLU – sample RLU)/(spontaneous death RLU – maximal killing RLU). Metrics were then computed using Igor Pro 8.1 (Wavemetrics, Portland, OR, USA). Sigmoid curves (no Hill equation) were fitted for every set of points (using Igor Pro 8.1) for visualization, with standard deviation as weighting factor.
Statistical analysis
Statistics were made with 2-way ANOVA (multiple comparison). All statistical analyses were performed using GraphPad Prism v.8 software (GraphPad Software, San Diego, CA, USA).
Results
Isolation and expression of KRAS TCRs
TCR clones were isolated and identified from the peripheral blood of a 60-year old pancreatic cancer patient (patient 705) showing a strong vaccine response and long term survival following therapy.21 The treatment consisted of several injections of a mix of seven 17-mer KRAS mutated peptides: 306 (G13D), 313 (G12D), 319 (G12V), 322 (G12C), 484 (G12A), 516 (G12S) and 321 (G12R). Importantly, these mutations are commonly found in different cancer types (Figure 1(a)). Eighty-six T-cell clones were subsequently isolated, and their activity and specificity were assessed. Four of these clones (27, 48, 57, 61) were found to be particularly potent and specifically recognized KRAS mutated peptides (Figure 1(b)): Radium-10, −11, −12 and −13. Upon determination of their HLA specificity, it was found that Radium-10, Radium-11 and Radium-13 recognized the peptides presented on HLA-DR, whereas Radium-12 recognized the peptides presented on HLA- DQ (Figure 1(c)). The patient was genotyped and found to be HLA-DR*0404/0401 and HLA-DQ*0302 (Supplementary Table S1). Further analysis showed that the HLA-DR*0404 allele was the HLA-DR restriction of the clones. We identified the coding sequence of their respective TCRs and prepared 2A-based constructs, as previously described.32 Radium- −12 displayed a Vβ chain for which no commercial antibodies exist. In contrast, Radium-10, −11, and −13 displayed Vβ chains for which commercial antibodies were available, but not optimal for use in primary T cells. We cloned Radium-10, −11-, and −12 as 2A-based fusions with a truncated CD34 (tCD34) in order to facilitate their detection, whereas the Radium-13 TCR was cloned without the tCD34.34 We then validated TCR expression, first in the TCR-negative Jurkat cell line J76 (Figure (2b)), then in primary human T cells (Figure 2(c)), by retroviral transduction. As shown, all four constructs displayed an expression between 30–65% in J76 cells and over 70% of CD34 in primary T cells, demonstrating efficient and stable TCR heterodimer formation.
Figure 1.

KRAS mutations are shared neoantigens. A. Summary of the peptides used to assess KRAS Radium TCRs specificities. These peptides represent various KRAS mutations found in the population. Mutation frequencies are based on.33
B. T cell clones 27, 48, 57 and 61 from pancreatic cancer patient 705 were tested against autologous EBV-LCLs loaded with several KRAS mutant peptides contained in the vaccine and KRAS WT peptide. Peptide-specific T cell proliferation was measured in 3H-thymidine incorporation assays and displayed as stimulation index (SI). An SI ≥2 was considered a positive response. C. The HLA restriction of the T-cell clones was identified by blocking the T-cell proliferation with anti-HLA-DR, -DQ, and -DP antibodies. Examples for each clone are shown (yellow, red, blue). For clone 57 (green), the corresponding Radium-12 TCR transduced T cells were used due to insufficient material for testing.
Figure 2.
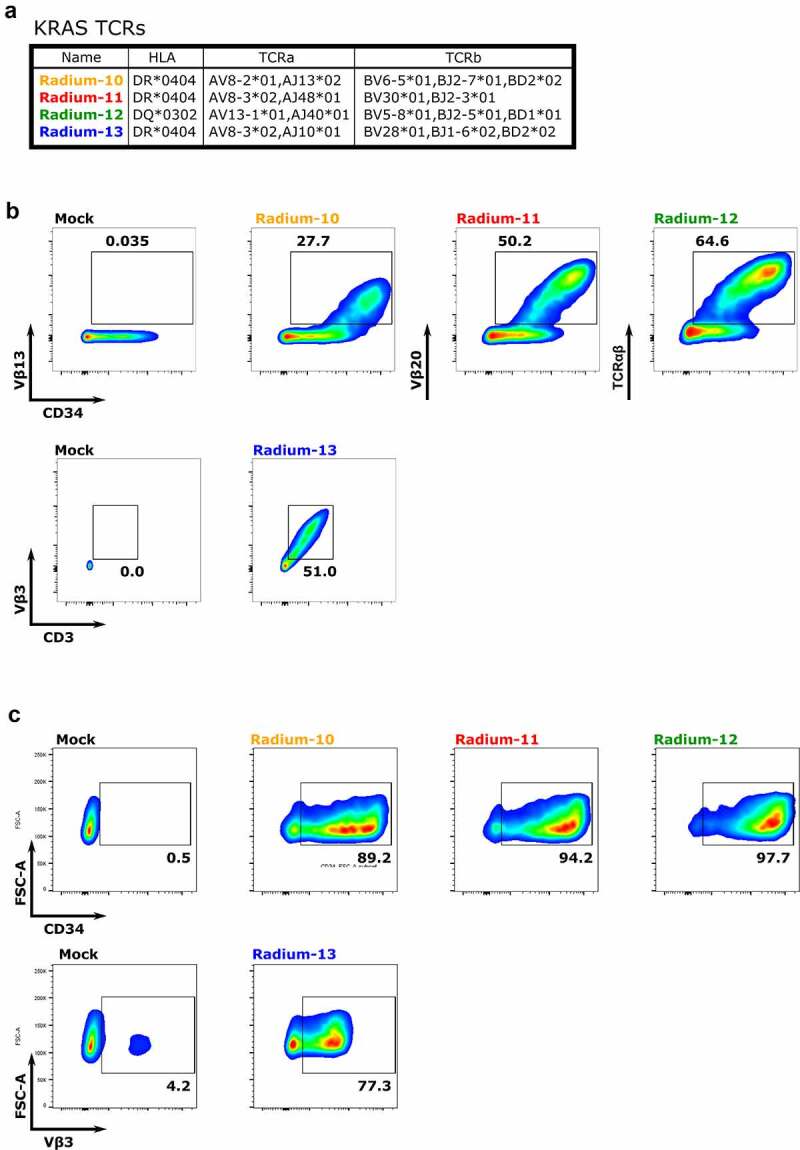
Isolated KRAS Radium TCRs are effectively expressed. A. Summary of KRAS TCR sequences. B. Expression of Radium-10, Radium-11, Radium-12 and Radium-13 after retroviral transduction into J76 cells, detected by anti-CD34 antibody, anti-CD3, and anti-Vβ specific for each TCR chain or anti-TCRαβ antibodies, as indicated. Radium-13 did not have a tCD34 tag. Data shown are representative flow diagrams of two independent experiments. C. TCR detection in primary T cells by staining with anti-CD34 or anti-Vβ3 antibodies. Data shown are representative flow diagrams from two or three independent experiments
KRAS TCRs recognized several mutated peptides
The specificity of the retrovirally transduced KRAS TCRs was then assessed upon measuring the production of intracellular cytokines (Interferon-γ (IFNγ) and Tumor Necrosis Factor α (TNFα)). This was performed by co-culturing the transduced effector T-cells with an autologous Epstein Barr Virus-transformed lymphoblastoid cell line (EBV-LCL), previously loaded with the indicated 17-mer peptides used in the vaccination trial (p306 G13D, p313 G12D, p319 G12V, p322 G12C, p484 G12A, p516 G12S, and p321 G12R), the wild type KRAS peptide (p382 KRAS WT), or an unrelated peptide (peptide 720 from the hTERT protein35), see Figure 1(a) for sequences. As depicted in Figure 3(a) (see Supp Figure S1 for gating strategy), all our TCRs reacted with at least one peptide, supporting the KRAS-specific reactivity of the isolated clones. Radium-11 and −13 TCRs demonstrated a similar pattern of expression with a great flexibility, since they recognized most of the 17-mer peptides, including KRAS WT. This suggests that these TCRs, although KRAS specific, are not sensitive to amino acid variations in the core of the peptide. The Radium-12 TCR was also flexible in its recognition since it reacted against 5 different mutated peptides (p319, p322, p484, p321 and p516), but did not demonstrate activity against the KRAS WT. Finally, Radium-10, although restricted to the same HLA as Radium-11 and −13, was the strictest TCR in terms of flexibility, and only reacted against p313 G12D. Importantly, for all TCRs, cytokine secretion was identified in both CD4+ and CD8 + T cells, indicating a co-receptor independency, often related to high TCR affinity.
Figure 3.
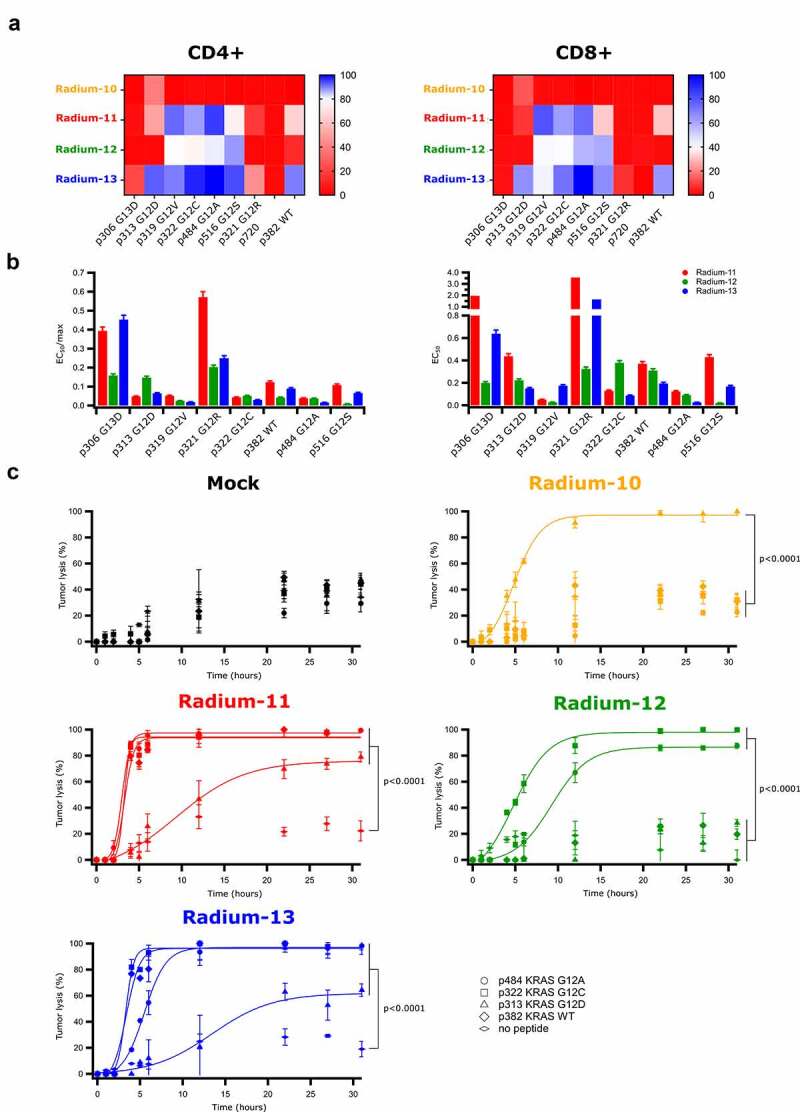
KRAS Radium TCRs recognize several KRAS mutations and promote specific killing of tumor targets. A. Heat maps representing the percentage of CD4+ and CD8+ KRAS Radium transduced T cells secreting cytokines (IFN-γ and TNF-α) upon co-culture with EBV-LCL cell lines loaded overnight with 10 µM of KRAS or hTERT peptides. Data shown are representative of two independent experiments. B. Bar graphs representing the ratio between the half peptide concentrations extracted from Supplementary Figure 1 and the maximum percentage of cytokine secreting cells obtained from Figure 2A. C. Cytotoxicity obtained by BLI-assay of effector T cells co-cultured with luciferase-expressing EBV-LCLs loaded or not with 10 µM of KRAS peptides. Data represent mean ± SD of triplicates
We then performed kinetic analysis in order to quantify our KRAS TCR response. Peptide titration supported the results previously obtained and showed that for the most strongly recognized mutated peptides (p319, p322, p484 and p516), the EC50 of Radium-11, 12 and 13, obtained from saturation fitting modeling, was below 1 µM (Supp. Figure S2a). The ratio defined by the EC50 over the maximal percentage of cells secreting cytokines (from Figure 3(a)) showed that for both CD4+ and CD8+ cells, Radium-11, −12 and −13 demonstrated the highest affinity for p319, p322 and p484 (Figure 3(b)). In order to investigate whether the KRAS peptides could be recognized at lower concentrations, we performed additional experiments where one of the highest affinity peptide for each TCR induced a strong response was titrated down to 0.001 µM (Supp. Figure S2b). Cytokine responses were induced down to 1–0.1 µM of peptide, depending on the TCR. A non-HLA matched EBV-LCL was included as a negative control. These results confirmed the HLA-restriction of the most strongly recognized mutated KRAS peptide responses in the lower micromolar range.
KRAS TCR expressing T cells kill target cells
To confirm the high potency of our four KRAS-specific TCRs we studied their capacity to induce T-cell killing. A co-culture of redirected T cells with HLA-matched, luciferase-expressing EBV-LCLs loaded with the indicated peptides was performed. We here demonstrated that the cytotoxic capacity of KRAS TCRs aligned with the cytokine release activity (Figure 3(c) and Supp. Figure S3). As shown, Radium-10 TCR T cells displayed a killing activity restricted to target cells loaded with p313, whereas Radium-11 and 13 expressing cells killed targets loaded with p313, p322, and p484 mutated peptides, as well as targets loaded with WT peptide. Finally, Radium-12 displayed a cytotoxic activity restricted to p322 and p484 mutated peptides. Taken together, these data show that KRAS TCRs, although isolated from CD4 T cells, can provide cytotoxic capacity to both CD4+ and CD8+ TCR expressing cells.
KRAS TCRs are able to recognize exogenously loaded KRAS proteins
Although an immune response was detected in the patient after vaccination, it could result from TCR recognition of the vaccine peptides only. Indeed, whether the KRAS peptides were bona fide peptides had not been demonstrated. Thus, in order to verify that the peptides could be generated through antigen processing and presented on HLA class II molecules, we performed a co-culture experiment using autologous EBV-LCL cells, either exogenously loaded with three KRAS proteins, or with their corresponding peptides as controls, namely G12A, G12C, and WT. As shown in Figure 4, KRAS TCR-redirected T cells could detect these peptides generated from exogenous protein loading, since Radium-11, −12 and −13 TCR could recognize the protein-loaded target cells. Of note, Radium-11 TCR was particularly potent in the recognition of all three KRAS proteins, whereas Radium-12 TCR only recognized mutated KRAS proteins (G12C and G12A), and Radium-13 displayed a high affinity for G12A and WT proteins. As expected, Radium-10 did not react against any of the proteins, since it is strictly restricted to the G12D mutation for which we were unable to acquire the protein. From these data, we concluded that APCs were able to degrade KRAS protein and load HLA-II molecules with peptides similar to those used for vaccination.
Figure 4.
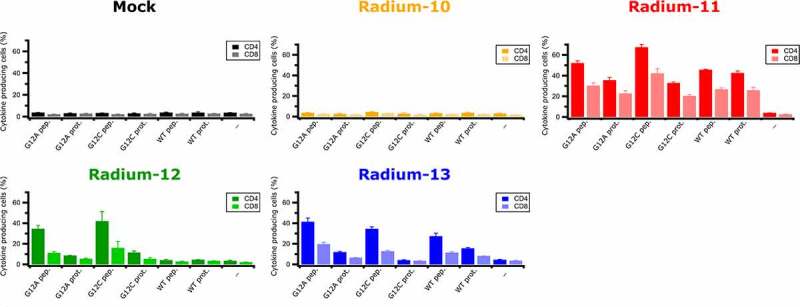
KRAS Radium TCRs recognize endogenously processed full length KRAS proteins. Intracellular IFNγ and TNFα production in Radium-10, −11, −12, −13 TCR transduced CD4+ and CD8+ effector T cells co-cultured with EBV-LCL cells loaded or not with 5 µM of KRAS proteins or 5 µM of peptides. Data represent mean ± SD of triplicates
Discussion
With more than 90% of cancer patients dying from solid tumors,36 there is critical need for new therapies. Recent developments in ACT have yet to address this issue. CAR therapy, despite its effectiveness for hematological malignancies, has not proved efficient against solid tumors, and has raised several concerns regarding toxicity.37 On the other hand, TCR therapy, owing to its ability to recognize any processed antigen and its low activation threshold, has shown impressive results against metastatic solid tumors without any therapeutic options.38 Moreover, when targeting tumor specific neoantigens, the risk of cross-reactivity with healthy tissues is low.
We report here the in vitro evaluation of four TCRs isolated from CD4+ T cells which recognize key driver mutations of the proto-oncogene KRAS that are highly expressed in hard-to-treat cancers. We demonstrated that stable expression of these TCRs in primary T cells led to specific recognition of cognate peptide-HLA complex, which induced inflammatory cytokine production and efficient killing of target cells. Importantly, the peptides recognized on the HLAs were either the vaccination peptides, or derived from exogenously loaded protein. The latter indicates that these peptides are naturally processed, and furthermore, suggests that the KRAS mutation present in the tumor may already have been presented to the patient’s immune system before vaccination.
HLA-II restricted TCRs are reported to exhibit a lower affinity compared to those restricted to HLA-I.39 We observed that the present KRAS TCRs were able to activate both CD4+ and CD8+ T cells. This indicates a partial co-receptor independency that could be beneficial in clinical settings, since bulk TCR-modified T cells could be used to combat the KRAS+ tumor, providing the benefit of both T-cell cytotoxicity and helper functions. Furthermore, loss of HLA class I and transporter for antigen presentation (TAP) is frequently observed in cancers like PDAC, thus HLA class II-restricted TCRs could potentially be more successful than those restricted to HLA class I.40 Cancer cell-specific expression of MHC class II has been demonstrated to regulate T-cell infiltration and sensitivity to immune checkpoint blockade in preclinical models.41
Radium-10 and Radium-12 TCRs, despite lower activity in comparison with Radium-11 and −13, did not recognize WT peptides or proteins and thus can be considered as the safest candidates with respect to cross-reactivity. Radium-11 and Radium-13, which displayed the highest affinity for most of the KRAS vaccination peptides and also recognized the WT KRAS, have undergone thymic selection in the patient and were therefore present at high levels in the circulation without any reported side effects.21 One might therefore suggest that these TCRs, although able to react against WT KRAS, are safe. This is also in line with other published studies detecting T-cell reactivity against WT KRAS as well as the mutant epitope.13,16,17
It is tempting to speculate that HLA-II loading of this KRAS peptide might be related to abnormal events such as cancer development or an inflammatory response, whereas in steady state, KRAS might simply not be loaded.42
Isolation of memory T cells recognizing the KRAS G12D mutation has already been reported13 and a recent vaccine trial in four patients confirmed that the majority of neoantigen-specific T cells induced were CD4+, including those specific for mutant KRAS.43 Our study corroborates these results and further demonstrates the potency of HLA-II restricted TCR therapies. We believe that upon complementary studies, these KRAS-specific TCRs could be valuable tools for T-cell based immunotherapies for a variety of advanced solid tumors.
Supplementary Material
Acknowledgments
The authors would like to thank our colleagues from the Translational Research Unit for their critical discussions. We are grateful to Kari Lislerud for expert technical assistance with the cloning of the T cells. We also acknowledge the flow cytometry core facility at the Institute for Cancer Research (OUS) for their support. We thank Prof. Michael Nishimura (Loyola University Chicago Stritch School of Medicine, USA) for the CD34t containing constructs. We thank Gibco/Life Technologies AS (Oslo, Norway) for supplying CTS™ Dynabeads™ CD3/CD28.
Funding Statement
This work was supported by the Research Council of Norway (BIA 269589 and BIOTEK 244388) and the South-Eastern Norway Regional Health Authority (Innovation grant).
Disclosure of potential conflicts of interest
GG and GK are shareholders in Zelluna Immunotherapy AS. SP is currently employed by Zelluna Immunotherapy AS. All other authors declare no potential conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A.. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–10. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Weber JS, O’Day S, Urba W, Powderly J, Nichol G, Yellin M, Snively J, Hersh E. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol. 2008;26(36):5950–5956. doi:. [DOI] [PubMed] [Google Scholar]
- 3.Garber K. Driving T-cell immunotherapy to solid tumors. Nat Biotechnol. 2018;36(3):215–219. doi: 10.1038/nbt.4090. [DOI] [PubMed] [Google Scholar]
- 4.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. 2018;9(3):282. doi: 10.1038/s41419-018-0278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishra M, Jiang H, Chawsheen HA, Gerard M, Toledano MB, Wei Q. Nrf2-activated expression of sulfiredoxin contributes to urethane-induced lung tumorigenesis. Cancer Lett. 2018;432:216–226. doi: 10.1016/j.canlet.2018.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129(5):865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 9.Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170(1):17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li S, Balmain A, Counter CM. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat Rev Cancer. 2018;18(12):767–777. doi: 10.1038/s41568-018-0076-6. [DOI] [PubMed] [Google Scholar]
- 11.Marzinotto S, Sessa F, Franzoni A, Anselmi A, Gastaldo LR, Mason S, Damante G, Beltrami CA, Mariuzzi L. KRAS codons 12 and 13 mutation analysis: a comparative study between direct sequencing and a new sensitive real-time PCR assay. Sequencing. 2012;2011:e895709. doi: 10.1155/2011/895709 [DOI] [Google Scholar]
- 12.Birkeland E, Wik E, Mjøs S, Hoivik EA, Trovik J, Werner HMJ, Kusonmano K, Petersen K, Raeder MB, Holst F, et al. KRAS gene amplification and overexpression but not mutation associates with aggressive and metastatic endometrial cancer. Br J Cancer. 2012;107(12):1997–2004. doi: 10.1038/bjc.2012.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cafri G, Yossef R, Pasetto A, Deniger DC, Lu Y-C, Parkhurst M, Gartner JJ, Jia L, Ray S, Ngo LT, et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nature commun. 2019;10(1):449. doi: 10.1038/s41467-019-08304-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375(23):2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang QJ, Yu Z, Griffith K, Hanada K, Restifo NP, Yang JC. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol Res. 2016;4(3):204–214. doi: 10.1158/2326-6066.CIR-15-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, Gartner JJ, Prickett TD, Cafri G, Robbins PF, et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight. 2018;3(19). doi: 10.1172/jci.insight.122467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veatch JR, Jesernig BL, Kargl J, Fitzgibbon M, Lee SM, Baik C, Martins R, Houghton AM, Riddell SR. Endogenous CD4(+) T cells recognize neoantigens in lung cancer patients, including recurrent oncogenic KRAS and ERBB2 (Her2) driver mutations. Cancer Immunol Res. May 1 2019. doi: 10.1158/2326-6066.cir-18-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gjertsen MK, Bakka A, Breivik J, Saeterdal I, Solheim BG, Soreide O, Thorsby E, Gaudernack G. Vaccination with mutant ras peptides and induction of T-cell responsiveness in pancreatic carcinoma patients carrying the corresponding RAS mutation. Lancet. 1995;346(8987):1399–1400. doi: 10.1016/s0140-6736(95)92408-6. [DOI] [PubMed] [Google Scholar]
- 19.Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N, et al. Kirsten ras mutations in patients with colorectal cancer: the “RASCAL II” study. Br J Cancer. 2001;85(5):692–696. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gjertsen MK, Saeterdal I, Saeboe-Larssen S, Gaudernack G. HLA-A3 restricted mutant ras specific cytotoxic T-lymphocytes induced by vaccination with T-helper epitopes. J Mol Med. 2003;81(1):43–50. doi:. [DOI] [PubMed] [Google Scholar]
- 21.Wedén S, Klemp M, Gladhaug IP, Møller M, Eriksen JA, Gaudernack G, Buanes T. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int J Cancer. 2011;128(5):1120–1128. doi: 10.1002/ijc.25449. [DOI] [PubMed] [Google Scholar]
- 22.Gjertsen MK, Buanes T, Rosseland AR, Bakka A, Gladhaug I, Søreide O, Eriksen JA, Møller M, Baksaas I, Lothe RA, et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J Cancer. 2001;92(3):441–450. doi: 10.1002/ijc.1205. [DOI] [PubMed] [Google Scholar]
- 23.Chatani PD, Yang JC. Mutated RAS: targeting the “Untargetable” with T cells. Clin Cancer Res. 2020;26(3):537–544. doi: 10.1158/1078-0432.CCR-19-2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sim MJW, Lu J, Spencer M, Hopkins F, Tran E, Rosenberg SA, Long EO, Sun PD. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc Natl Acad Sci U S A. 2020;117(23):12826–12835. doi: 10.1073/pnas.1921964117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inderberg EM, Walchli S. Long-term surviving cancer patients as a source of therapeutic TCR. Cancer Immunol Immunother. 2020;69(5):859–865. doi: 10.1007/s00262-019-02468-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inderberg EM, Walchli S, Myhre MR, Trachsel S, Almasbak H, Kvalheim G, Gaudernack G. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology. 2017;6(4):e1302631. doi: 10.1080/2162402X.2017.1302631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tateishi M, Saito I, Yamamoto K, Miyasaka N. Spontaneous production of epstein-barr virus by b lymphoblastoid cell lines obtained from patients with sjögren’s syndrome. possible involvement of a novel strain of epstein-barr virus in disease pathogenesis. Arthritis Rheum. 1993;36(6):827–835. doi: 10.1002/art.1780360614. [DOI] [PubMed] [Google Scholar]
- 31.Gjertsen MK, Bjorheim J, Saeterdal I, Myklebust J, Gaudernack G. Cytotoxic CD4+ and CD8+ T lymphocytes, generated by mutant p21-ras (12Val) peptide vaccination of a patient, recognize 12Val-dependent nested epitopes present within the vaccine peptide and kill autologous tumour cells carrying this mutation. Int J Cancer. 1997;72(5):784–790. doi:. [DOI] [PubMed] [Google Scholar]
- 32.Walchli S, Loset GA, Kumari S, Johansen JN, Yang W, Sandlie I, Olweus J. A practical approach to T-cell receptor cloning and expression. PLoS One. 2011;6(11):e27930. doi: 10.1371/journal.pone.0027930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47(D1):D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Norell H, Zhang Y, McCracken J, Martins Da Palma T, Lesher A, Liu Y, Roszkowski JJ, Temple A, Callender GG, Clay T, et al. CD34-based enrichment of genetically engineered human T cells for clinical use results in dramatically enhanced tumor targeting. Cancer Immunol Immunother. 2010;59(6):851–862. doi: 10.1007/s00262-009-0810-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inderberg-Suso EM, Trachsel S, Lislerud K, Rasmussen AM, Gaudernack G. Widespread CD4+ T-cell reactivity to novel hTERT epitopes following vaccination of cancer patients with a single hTERT peptide GV1001. Oncoimmunology. 2012;1(5):670–686. doi: 10.4161/onci.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Houts PS, Lenhard RE, Varricchio C. ACS cancer facts and figures. Cancer Pract. 2000;8(3):105–108. doi: 10.1046/j.1523-5394.2000.83001.x. [DOI] [Google Scholar]
- 37.Hou B, Tang Y, Li W, Zeng Q, Chang D. Efficiency of CAR-T therapy for treatment of solid tumor in clinical trials: a meta-analysis. Dis Markers. 2019;2019:3425291. doi: 10.1155/2019/3425291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y-C, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. 2018;24(6):724–730. doi: 10.1038/s41591-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stone JD, Harris DT, Kranz DM. TCR affinity for p/MHC formed by tumor antigens that are self-proteins: impact on efficacy and toxicity. Curr Opin Immunol. 2015;33:16–22. doi: 10.1016/j.coi.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pandha H, Rigg A, John J, Lemoine N. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clin Exp Immunol. 2007;148(1):127–135. doi: 10.1111/j.1365-2249.2006.03289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson AM, Bullock BL, Neuwelt AJ, Poczobutt JM, Kaspar RE, Li HY, Kwak JW, Hopp K, Weiser-Evans MCM, Heasley LE, et al. Cancer cell–intrinsic expression of MHC Class II regulates the immune microenvironment and response to Anti–PD-1 therapy in lung adenocarcinoma. J Immunol. 2020;204(8):2295–2307. doi: 10.4049/jimmunol.1900778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM, Werbeck ND, Briem H, Boemer U, Weiske J, et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc Natl Acad Sci U S A. 2019;116(7):2551–2560. doi: 10.1073/pnas.1812963116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cafri G, Gartner JJ, Zaks T, Hopson K, Levin N, Paria BC, Parkhurst MR, Yossef R, Lowery FJ, Jafferji MS, et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J Clin Invest. 2020;130(11):5976–5988. doi: 10.1172/JCI134915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.