

ABSTRACT
Adenosine-to-inosine (A-to-I) editing is one of the most prevalent post-transcriptional RNA modifications in metazoan. This reaction is catalysed by enzymes called adenosine deaminases acting on RNA (ADARs). RNA editing is involved in the regulation of protein function and gene expression. The numerous A-to-I editing sites have been identified in both coding and non-coding RNA transcripts. These editing sites are also found in various genes expressed in the central nervous system (CNS) and play an important role in neurological development and brain function. Aberrant regulation of RNA editing has been associated with the pathogenesis of neurological and psychiatric disorders, suggesting the physiological significance of RNA editing in the CNS. In this review, we discuss the current knowledge of editing on neurological disease and development.
KEYWORDS: Post-transcriptional RNA modification, A-to-I RNA editing, ADAR, AGS, GluA2, ALS, epilepsy, 5-HT2CR, depression, schizophrenia, bipolar disorder, miRNA, viral RNA
1. Introduction
Eukaryotic RNA transcripts are frequently subjected to various types of post-transcriptional RNA modifications, contributing to the complex regulation of gene expression involved in various biological processes. Increasing evidence proposes that these post-transcriptional RNA modifications play important roles in complex functions of the central nervous system (CNS) [1]. Adenosine (A) is converted to inosine (I) through hydrolytic deamination of the amino group at the C6 position of adenosine, which is catalysed by enzymes termed adenosine deaminases acting on RNA (ADARs) [2] (Fig. 1A). This reaction requires the formation of a double-stranded RNA (dsRNA) structure in the nucleus. Since inosine contains a similar chemical structure with guanosine (G), inosine base pairs with cytidine instead of uridine. Inosines in RNA transcripts are interpreted as guanosines through mRNA-splicing and translation for protein synthesis. As a result, A-to-I modification causes editing of genetic sequences, called A-to-I RNA editing [2]. Up to date, many A-to-I RNA editing sites have been identified in not only protein-coding but also non-coding transcripts expressed in neuronal cells of different organisms. Various studies have demonstrated that these RNA editing sites have important functions and biological significance in neurological development. It has been reported that dysregulation of A-to-I RNA editing is associated with the pathogenesis of various neurological and psychiatric disorders. These findings highlight the biological importance of A-to-I RNA editing in the physiology of the CNS. In this review, we present current knowledge about A-to-I RNA editing in the CNS.
Figure 1.
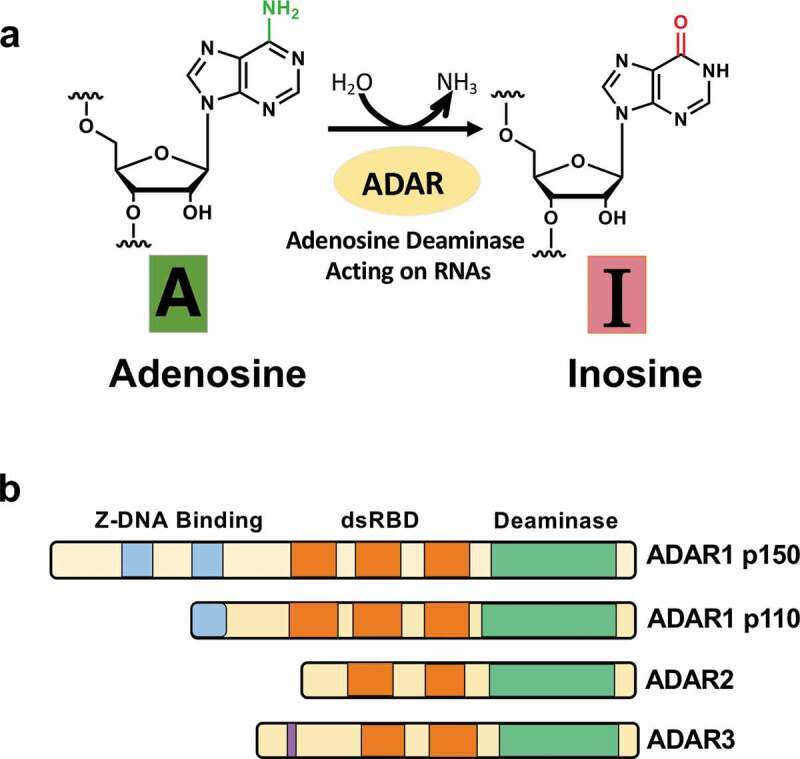
Deamination of adenosine to inosine and ADAR family. (A) The schematic of adenosine-to-inosine deamination reaction catalysed by ADARs. (B) Domain structures of ADAR family genes in the vertebrate. There are three ADAR family members (ADAR1, ADAR2, and ADAR3), and ADAR1 contains two isoforms (p110 and p150). All the enzymes contain a conserved deaminase domain, shown in green, and the double-stranded RNA (dsRNA)-binding domain (dsRBD) that determines substrate specificity, shown in orange. ADAR1 p110 and p150 isoforms are different in their Z-DNA-binding domains, shown in blue. The only ADAR3 contains an arginine-rich domain, shown in purple
1.1. A-to-I RNA editing and ADAR enzymes
The ADAR gene family is widely conserved in metazoans. In vertebrates, three ADAR gene family members have been identified so far: ADAR1 [3], ADAR2 [4], and ADAR3 [5] (Fig. 1B). They share common domain structures. In the N-terminal region, they contain several tandemly repeated double-stranded RNA binding domains (dsRBD) that enable direct binding with dsRNA structures [6]. In the C-terminal region, there is a deaminase domain that catalyzes A-to-I editing.
On the other hand, distinct domains are found between ADARs. ADAR1 has two isoforms derived from alternative transcription promoters: a full-length, interferon-inducible 150-kDa isoform (p150), which is mainly present in the cytoplasm, and a shorter, constitutively expressed 110-kDa isoform (p110), which is localized in the nucleus [7]. While ADAR1 p110 has one full Zβ-domain and trimmed latter part of Zα-domain, ADAR1 p150 has entire the Zβ and Zα-domains. Both ADAR1 p110 and p150 have three dsRBDs. ADAR1p110 is ubiquitously expressed in most of all tissues, while ADAR1 p150 predominantly expressed in the lymph node and spleen [8]. ADAR2 has two dsRBDs, and expresses relatively highly in the brain and testis [9]. ADAR3 has two dsRBDs and Arg-rich single-stranded RNA binding domain. ADAR3 shows brain-specific expression.
The enzymatic activities of ADAR1 and ADAR2 have been verified, while the editing activity of ADAR3 is not shown yet [10,11]. ADAR1 is known to be primarily responsible for RNA editing in repeat elements in non-coding regions of mRNAs. However, ADAR2 is mainly involved in recoding editing in protein-coding genes, most of which are expressed in the CNS.
ADAR1 and ADAR2 are essential genes in mammals. Adar1 null mice show embryonic lethality at E11.5 stage, accompanied by massive apoptosis, defective erythropoiesis, and aberrant innate immune response [12]. Adar2 null mutant mice exhibit postnatal death at several weeks of age following repeated epileptic seizures, caused by neuronal cell death due to the excessive influx of calcium ions through glutamate receptors [13]. Although ADAR3 lacks editing activity, the functional role and its biological importance have been recently characterized. Several papers reported that ADAR3 functions as a negative regulator for RNA editing by competing with ADAR1 or ADAR2 for binding to editing substrates [10]. Mladenova et al. recently reported that Adar3-deficient mice show increased levels of anxiety and deficits in hippocampus-dependent short- and long-term memory formation, suggesting that ADAR3 plays an important role in cognitive processes in mammals [11].
1.2. A-to-I RNA editing sites
In protein-coding regions of mRNA transcripts, RNA editing can occasionally lead to specific changes in codon code that translate mRNA sequences into amino-acids, which generates the multiple, functionally distinct protein variants from a single genetic locus. Therefore, A-to-I RNA editing of mRNA contributes to an increase of proteomic diversity in cells and tissues. In particular, amino-acid alteration causing RNA editing tends to be found and well characterized in the CNS, such as transcripts of voltage- or ligand-gated ion channels and neurotransmitter receptors [14]. Most of the editing sites within protein-coding sequences (CDS) in neuronal gene transcripts are highly conserved in mammals, suggesting the essential their roles in mammalian CNS [15].
On the other hand, A-to-I RNA editing sites are found in non-coding regions of mRNAs far more than those in mRNA CDS regions. It is reported that parts of them are involved in the regulation of gene expression. RNA editing in intronic regions of mRNAs creates or abolishes splicing sites to regulate alternative mRNA spicing [16,17]. In 3ʹ untranslated regions (3ʹ UTRs) of protein-coding transcripts, RNA editing can alter target sequences of micro RNA (miRNA)-mediated translational regulation [18,19] or increase the accessibility to cis-elements for mRNA stability [20], leading to alteration of expression levels of mRNAs or synthesized proteins.
The recent rapid improvement of the next-generation sequencing (NGS) technologies and bioinformatics enables high-throughput transcriptome-wide identification of A-to-I RNA editing in various cultured cell lines or tissues. Up to date, more than hundred thousands of RNA editing sites have been identified in humans [21,22] and more than a few thousands in mice [23,24]. In contrast to the editing sites in CDS regions of mRNAs, most editing sites in non-CDS regions reside in species-specific retrotransposon-originated elements, i.e. SINE-Alu repeats in humans and mainly SINE-B1/B2 repeats in mice. Such repetitive retrotransposon elements are frequently located in intronic or 3ʹ UTRs of transcripts as the forward or inverted orientations. The proximal pair of repeats locates in inverted directions with each other on the same transcript form a long dsRNA structure, which is preferentially recognized and edited by ADAR1 [25,26]. Only a subset of these editing sites in repetitive sequences are functionally annotated: modulation of miRNA-mediated translational suppression [18,19], mRNA stability [20], and regulation of circular RNA production [27]. The functions of the numerous editing sites in repetitive elements are mostly unknown.
2. Neurological and neurodegenerative disease caused by dysregulated RNA editing
2.1. Q/R editing of AMPA glutamate receptor subunit GluA2
An α-amino-3-hydroxy-5-methyl-4-isoxazole- propionic acid (AMPA) receptor is a subtype of ionotropic glutamate receptors, which is involved in the establishment and maintenance of synaptic plasticity in the CNS [28]. The neurotransmitter glutamate released from pre-synaptic densities activates AMPA receptors to allow the influx of Na+ or Ca2+ ions to the post-synaptic densities. Mammalian AMPA receptors consist of a combination of GluA1, GluA2, GluA3, and GluA4 subunits (also known as GluR1 to GluR4 or GluR-A to GluR-D) [29]. The mRNA transcripts encoding the GluA2 subunit are subjected to ADAR2-mediated A-to-I RNA editing that alters a glutamine codon (Q) to an arginine codon (R) (Fig. 2A). This replacement from the neutral amino acid residue to the positively charged one results in significantly reduced Ca2+ permeability of the receptor (Fig. 2A).
Figure 2.
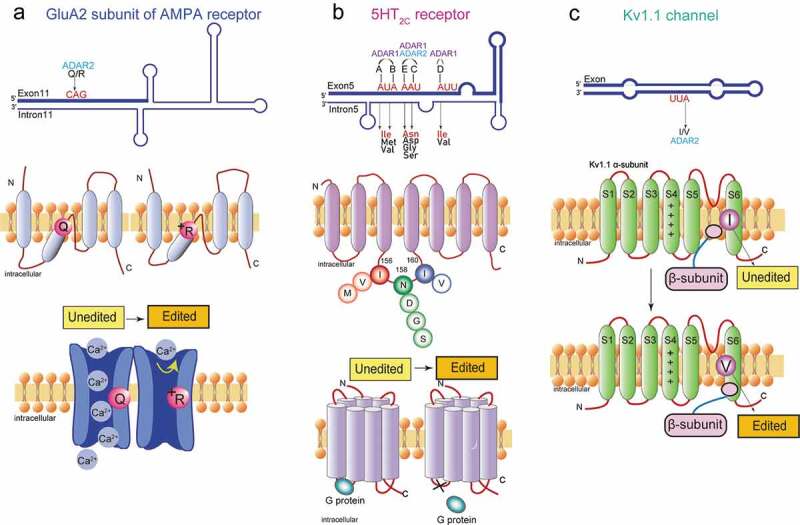
Functional roles of A-to-I RNA editing in the neurotransmitter receptor and the ion channel. (A) The A-to-I editing site (Q/R) in the secondary structure of the GluA2 pre-mRNA regions (top). The schematic of the GluA2 subunit structure and the functional difference in edited and unedited forms (middle and bottom, respectively). RNA editing alters the glutamate residue (Q) (middle left) at the 607th position to a positively charged arginine residue (R) (middle right), which can dramatically reduce calcium permeability (bottom). (B) The five editing sites (A-E) in the secondary structure of the 5HT2CR pre-mRNA regions where exon 5 base-pairs with the intron 5 (top). The schematic drawing of 5-HT2c receptor structure the functional difference in edited and unedited forms (middle and bottom, respectively). RNA editing of 5HT2C receptor converts amino acid isoleucine (I) at 156th position to valine (V), or methionine (M), asparagine (N) at 158th position to aspartic acid (D), serine (S), or glycine (G), and isoleucine (I) at 160th position to valine (V) (middle). Edited 5HT2CR isoforms exhibit reduced G protein coupling efficiency (bottom left) in comparison to the unedited isoform (bottom right). (C) The A-to-I editing site (I/V) in the secondary structure of Kv1.1 mRNAs (top). The schematic drawing of Kv1.1 channel structure and the functional difference in edited and unedited forms (middle and bottom, respectively) The transmembrane voltage sensor (S4), the K+ ion selectivity filter (S5–S6) are indicated in the middle and bottom figure. Editing of Kv1.1 changes the isoleucine (I) (middle) to valine (V) (bottom). which reduces the affinity for the binding of Kv β1.1 and enhances recovery from inactivation
In human and mouse brain tissue, almost all GluA2 mRNA is edited at the Q/R site; therefore, the AMPA receptors containing a GluA2 subunit are essentially Ca2+ impermeable. The GluA2 Q/R site editing plays critical physiological roles in the mammalian CNS. Initially, it was reported that mutant mice harbouring a Q/R-site editing-deficient GluA2 allele, in which complementary genomic sequences required for dsRNA are deleted, leads to postnatal death accompanied by an increase of Ca2+ permeability and neuronal degeneration [30]. Subsequent to this analysis, it was found that Adar2-deficient mice also exhibit death after birth, followed by phenotypes quite similar to those observed in mice with Q/R-site editing-depleted GluA2 [13]. Moreover, this lethal phenotype of ADAR2 null mice was rescued by a genomic point-mutation to alter a glutamine codon to an arginine codon at the GluA2 Q/R site [13]. These studies in mouse models highlight the physiological importance of GluA2 Q/R site editing in brain function and demonstrate that this editing site is the most critical target of the ADAR2 enzyme. These earlier in vivo studies suggest that the deficiency of GluA2 Q/R site editing generates Ca2+ permeable AMPA receptors, leading to excitotoxic neuronal cell death. Some studies showed that this defect of GluA2 RNA editing is observed in neurological neurodegenerative disorders, suggesting this change contributes to the pathogenesis of the diseases (Table 1).
Table 1.
Dysregulated A-to-I editing associated with neurological or neurodegenerative diseases
| Gene | Editing site | Responsible ADAR1/2 | Editing function | Associated disease |
|---|---|---|---|---|
| GRIA2 | GluA2 Q→R | 2 | Change in Ca2+ permeability | Amyotrophic lateral sclerosis (ALS) [32,34,39] Epilepsy [30,45] Developmental epileptic encephalopathy (DEE) [48–50] Astrocytoma [55,56] |
| HT2CR | 5-HT2cR A, B, D sites | 1 | Decrease in 5-HT potency Reduction in G protein-coupling efficiency |
Depression [68,70] Suicide Schizophrenia [69,70] Bipolar disorder [70,71] |
| 5-HT2cR C, E sites | 1, 2 | |||
| KCNA1 | Kv1.1 I→V | 2 | Reduction in the deactivation rate | Episodic Ataxia Type 1 (EA1) [81], Epilepsy [77] |
| MIR21 | pri-miR-21 8 sites |
2 | Inhibit Drosha processing | Astrocytoma [137] |
| MIR221 | pri-miR-221 − 1, +1, +64 | 2 | Inhibit Drosha processing | Astrocytoma [137] |
| MIR222 | pri-miR-222 − 21, +53 | 2 | Inhibit Drosha processing | Astrocytoma [137] |
| MIR376A | pri-miR-376a +9 | 2 | Redirection of target recognition of mature miRNA | Astrocytoma [138] |
2.1.1. Amyotrophic lateral sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS) is a foetal and progressive adult-onset neurodegenerative disorder [31]. The rapid progressive death of both the upper and lower motor neurons leads to terminal atrophy of skeletal muscle and eventually death due to a severe failure of the respiratory system. The genetic annotations of ALS pathology are very limited; only 5 ~ 10% of ALS cases are perceived as familial ALS. More than 90% of ALS cases are with no apparent genetic basis, classified as sporadic ALS.
One proposed model for motor neuron cell death is the excitotoxicity of the AMPA receptor-meditated Ca2+ permeability. Several lines of evidence showed that GluA2 Q/R site editing is remarkably reduced in the spinal motor neurons of sporadic ALS patients compared to control subjects, while the expression level of GluA2 mRNA remains unchanged [32,33]. It is likely that GluA2 RNA editing deficiencies occur in ALS patients due to down-regulation of ADAR2, which was observed in the spinal motor neurons of sporadic ALS patients [34]. To further elucidate the role of ADAR2-mediated GluA2 RNA editing in motor neuron death, ADAR2 was ablated specifically in motor neurons of mice [35]. As a result, the mutant mice developed ALS-like progressive loss of motor neuron function, accompanied by the denervation of skeletal muscles [35]. Furthermore, these mice were shown to exhibit the abnormal activation of calpain, a protease dependent of excessive Ca2+ influx through unedited GluA2-containing AMPA receptors, resulting in cleavage and consequent cytoplasmic mislocalization of TAR DNA-binding protein (TDP-43), which is the reliable pathological hallmark of ALS [36,37]. Moreover, the activated calpain cleaves nucleoporins, which comprise the nucleocytoplasmic transport, leading to defects of the nucleocytoplasmic transport machinery and gene expression [38]. Thus, these studies strongly support the hypothesis that the excitotoxicity of the exaggerated Ca2+ influx by unedited GluA2 causes motor neuron death in patients with sporadic ALS (Fig. 3A).
Figure 3.
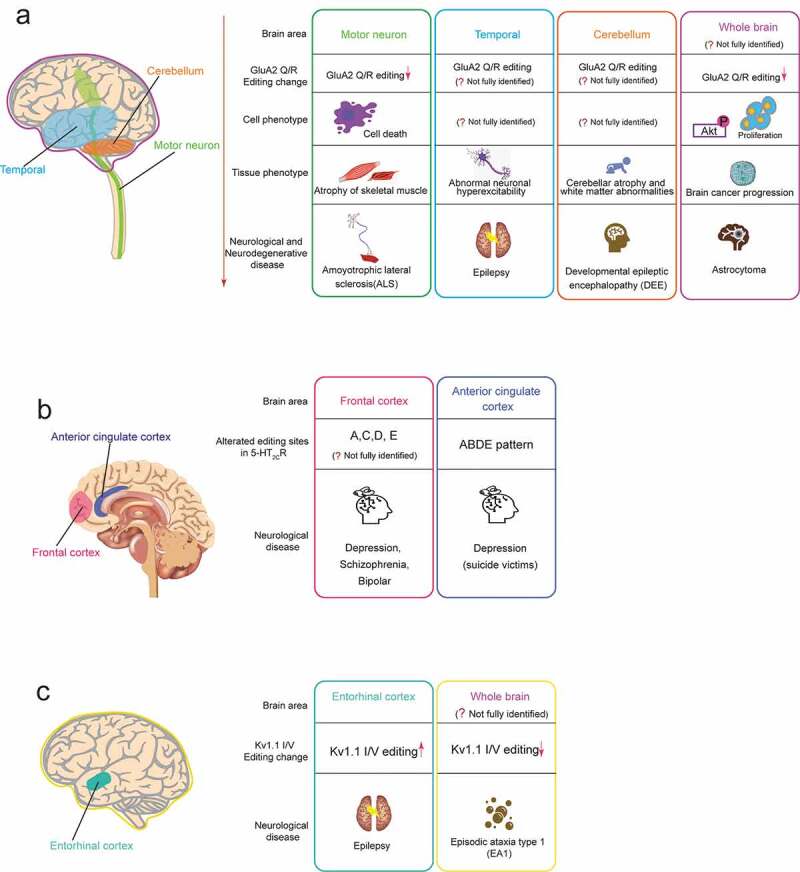
Neurological or neurodegenerative diseases associated with dysregulated A-to-I RNA editing in the neurotransmitter receptor and the ion channel. In each editing gene, the human brain areas affected by dysregulated RNA (including speculated areas) are shown in the left schematic drawing of the human brain, and associated diseases are shown in the right boxes. Each box includes the related brain area, alteration pattern of RNA editing, cell phenotype, tissue phenotype (only for GluA2 Q/R editing), and the associated disease. The colours of the outside line in each box corresponds to those of brain areas, respectively. (A) GluA2 Q/R editing (B) 5-HT2cR editing (C) Kv1.1 I/V editing
Recently, repairment of dying motor neurons from sporadic ALS by reactivating ADAR2 or suppressing excessive Ca2+ influx by AMPA receptors has become a therapeutic strategy for ALS patients [39,40]. To restore the activity of ADAR2 in non-ADAR2-expressing motor neurons, a study used an adeno-associated virus serotype 9 (AAV9) vector modified to promote the cross-blood brain barrier (BBB) to deliver human ADAR2 cDNA to motor neurons of Adar2 knockout mice to restore ADAR2 activity. After injecting the AAV9-ADAR2 vector into the tail vein of Adar2 knockout mice, it was observed that the vector-based ADAR2 gene delivery prevented the progression of the ALS phenotype, including progressive motor dysfunction, and the role of ADAR2 gene transmission was also observed [40]. In addition, AMPA receptor antagonists are a potential treatment for neurological disorders including ALS, but their side effects hinder clinical use [41]. Based on this, a non-competitive AMPA receptor antagonist, perampanel, has been approved as an antiepileptic drug. A study showed that oral administration of perampanel in Adar2 knockout mice can inhibit the progression of ALS phenotype, including progressive motor dysfunction (assessed by stationary time and grip) [39]. In addition, the cytoplasmic mislocalization of TDP-43 was significantly corrected in the treated mice compared to that in the untreated mice. These results suggest that this antiepileptic drug is also a promising medication for ALS treatment.
2.1.2. Epilepsy
Epilepsy is a common neurological disorder characterized by unprovoked recurrent seizures. The causes for epilepsy are multifaceted, resulting from genetic predispositions, CNS injuries and altered epigenetic status. All epileptic manifestations share the common feature of abnormal neuronal hyperexcitability in a subpopulation of neuronal cells, which leads to markedly elevated levels of intracellular Ca2+ ions. Neuronal cells that are exposed to prolonged and non-lethal levels of increased Ca2 ion concentration undergo plastic changes, resulting in epileptogenesis. AMPA glutamate receptors play a pivotal role in the fast excitability neurotransmission and thus have been supposed to contribute to the generation of seizures. Several studies using mouse models mentioned earlier suggest that a deficit of GluA2 Q/R RNA editing is linked to seizure vulnerability [13,30]. This hypothesis is strengthened by the study that the extent of Ca2+ permeability is directly correlated with varying degrees of impairment of GluA2 Q/R RNA editing [42]. Moreover, a conditional mouse mutant where GluA2 editing was inactivated postnatally in selected forebrain regions showed synaptic changes that implied susceptibility to seizure manifestation [43]. These lines of evidence demonstrate that the epileptic phenotype of GluA2 Q/R editing-deficient mouse models could be a direct consequence of altered AMPA-receptor properties in adult brains, but not a result of neurodevelopmental defects.
The molecular mechanism of epileptogenesis arising from the defect of GluA2 Q/R site editing has been unclear. Although AMPA receptors lacking GluA2 subunits are also highly permeable to Ca2+ ions, GluA2 knockout mice exhibit almost normal viability and manifest no phenotype of seizures [44]. This discrepancy is likely attributed to the fact that the nature of the Q/R residue in the GluA2 subunit affects not only Ca2+ permeability but also gating kinetics, channel conductance, channel assembly, and trafficking. In contrast to abundant evidence for the causative relationship between GluA2 Q/R RNA editing and epilepsy in mouse models, the results from studies on GluA2 Q/R editing in clinical specimens of epilepsy patients are inconsistent. A study using needle biopsy samples obtained from hypothalamic hamartoma tissue, which is supposed to be an intrinsic aspect of epilepsy, found loss of nuclear immunostaining of ADAR2 concomitant with decreased RNA editing efficiency at the GluA2 Q/R site [45]. However, several studies reported that down-regulation of GluA2 Q/R site editing was not observed in the surgically excised hippocampus and temporal cortex tissues of epilepsy patients [46,47]. Further studies are needed to truly understand the pathological role of GluA2 Q/R site editing in epilepsy (Fig. 3A).
2.1.3. Developmental epileptic encephalopathy (DEE)
Developmental and epileptic encephalopathy (DEE) is a neurodevelopmental disorder characterized by intractable epileptic seizures, intellectual disability, microcephaly, and developmental delay. Recently, two studies have identified the bi-allelic variants in ADAR2 that are associated with DEE [48,49]. Currently, seven variants are identified, five of which are shown to cause non-synonymous amino acid changes leading to the impairment of RNA-editing activity of ADAR2, strengthening the importance of ADAR2-mediated RNA editing in brain development and function.
As described above, ADAR2 deficient mice exhibit postnatal death due to epileptic seizures caused by under-editing at GluA2 Q/R site [13]. A recent study has identified the de novo heterozygous variants in GRIA2 among individuals with intellectual disability and neurodevelopmental abnormalities [50]. One of the most profound variants, Q607E, occurred at the Q/R site in an individual with a severe DEE. These findings suggest that the epilepsy phenotypes observed in DEE patients arising from bi-allelic ADAR2 variants may be attributable in part to under-editing of the GluA2 Q/R site (Fig. 3A).
2.1.4. Astrocytoma
Astrocytoma is a brain cancer of the astrocytes, which are star-shaped glial cells [51]. Glioblastoma multiforme (GBM) is a grade IV astrocytoma, the most severe grade of malignancy, and a particularly invasive tumour reported to be usually fatal within 18 months [52]. Analysis of GBM tissue samples revealed that the GluA2 Q/R site was significantly under-edited compared to that of control samples [53]. Ozawa et al. clearly demonstrated the causative relationship between under-editing of the Q/R site and astrocytoma malignancy. They showed that GBMs mainly express Ca2+ permeable AMPA receptor subunits (GluA2 and GluA4), suggesting that an alteration of Ca2+ influx has a crucial role in tumour growth. Indeed, they showed that transfection of GBM cells with a GluA2 edited version (GluA2 R) suppressed cell migration and induced apoptosis, whilst the GluA2 unedited version (GluA2 Q) enhanced cell malignant features [54]. Further studies demonstrated that the excessive influx of Ca2+ mediated by AMPA receptors has been shown to activate Akt by phosphorylation, thereby promoting proliferation and mobility [55]. The upregulation of the Akt pathway is reversed when the edited GluA2 R is expressed [55]. These studies suggest that the decrease of RNA editing levels at the GluA2 Q/R site greatly contributes to brain cancer progression (Fig. 3A).
2.2. RNA editing of serotonin receptor (5-HT2CR)
Members of the serotonin (5-hydroxytryptamine or 5-HT) receptor gene family are G-protein coupled receptors that are considered to primarily contribute to physiological and behavioural processes such as circadian rhythms, emotion, appetite, and sexual behaviour. 5-HT 2 C receptor (5-HT2CR), one subtype in the 5-HT receptor family, is predominantly detected in the CNS and the only serotonin receptor that undergoes A-to-I RNA editing [56]. Five editing sites (referred to as A, B, C, D, and E site) are identified in 5-HT2CR pre-mRNAs [57]. Sites A and B are edited by ADAR1, whereas the editing of site D is mediated specifically by ADAR2. Sites C and E are edited by both ADAR1 and ADAR2 (Table 1) [58]. Editing of the five sites located in the second intracellular loop of the receptor would change three amino acids consisting of Ile, Asp, and Ile (INI) at positions 156, 158, and 160, respectively (Fig. 2B) [57]. Theoretically, 24 protein variants can be produced as a result of different combinations of RNA editing sites. Sequencing analysis of cDNA in the 5-HT2CR editing region isolated from rat, mouse, human brain tissues showed that different serotonin receptor isoforms are expressed dependent of specific brain regions, suggesting that differentially edited 5-HT2CR may exert distinct physiological roles in individual brain regions [57,59,60]. The edited mRNA transcripts encoding isoforms with Val-Ser-Val (VSV) or Val-Asn-Val (VNV) are the most highly expressed in the majority of brain regions [57]. Functional comparisons between non-edited (INI) and the fully-edited (VGV) 5-HT2CR isoforms revealed a 40-fold decrease in 5-HT potency as well as reductions in G-protein coupling efficiency and agonist binding [57,59,61]. Moreover, 5-HT2CR RNA editing regulates cell surface expression of the receptors by intracellular trafficking efficacy. The fully edited VGV isoform displays sufficient cell surface expression, whereas the unedited INI isoform is internalized and accumulated in endosomes [62]. Interestingly, the editing efficiencies of these five sites of 5-HT2CR exhibit differential levels during brain development. In the embryonic stage, the predominant isoforms are non-edited INI and D-site-edited INV, while in the adult brain, highly edited isoforms VNV, VNI, and VSV are the most common [63].
To understand the significance of 5-HT2CR RNA editing in vivo, Kawahara et al. generated mutant mice engineered with the expression of one of two significantly different editing isoforms, INI (non-edited) or VGV (fully edited) [64]. Although the unedited model mice (INI) grew naturally, fully edited model mice (VGV) exhibited the abnormal reduction of fat mass, despite the hyperphagia phenotype, due to hyperactivation of the sympathetic nervous system and increased energy expenditure, which led to growth retardation. These results highlight the physiological importance of 5-HT2CR editing in energy metabolism [64]. The accumulating evidence suggests that alteration of editing patterns of 5-HT2CR mRNA may be associated with several psychiatric disorders, including anxiety, depression, bipolar disorder, and schizophrenia [65] (Table 1).
2.2.1. Depression and Schizophrenia
Serotonin signalling has been implicated widely in the aetiology of behavioural and psychiatric disorders and several 5HT receptors, including the 5-HT2CR, are thought to be important targets for pharmacologic treatment. In fact, some antidepressant drugs have been shown to interact with 5-HT2CR [66], and thus it is hypothesized that the 5-HT2CR plays a prominent role in the pathophysiology of several psychiatric disorders including anxiety, depression, bipolar disorder, and schizophrenia. This notion raises an intriguing question on whether different editing patterns at five sites of 5-HT2CR mRNAs are involved in the development of psychological abnormalities. Multiple investigations provide evidence for the alteration of the editing patterns of 5-HT2CR in patients affected by psychiatric disorders; however, there is an apparent discrepancy between these results. This is likely due to problems and confounding variables in studies using post-mortem brain tissues, including a small sample size, a subtle difference in dissected brain regions, and a medication history of subjects. Furthermore, different methods used for analysis and their technical bias contribute to inconsistent results on editing patterns.
Multiple studies reported that no alteration of editing efficiencies in 5-HT2CR transcripts was detected in the frontal cortex regions from individuals diagnosed with major depression [67], schizophrenia, or bipolar disorder compared to that in control subjects [68–70], except for one report that showed a trend for increased RNA editing at the D site in depressive patients [70]. However, individuals who committed suicide exhibited significant changes in editing patterns of 5-HT2cR mRNAs. Two groups reported that there was a significant increase of RNA editing level at the A site of 5-HT2CR in suicide victims regardless of psychiatric diagnosis [68,70]. In contrast, another study that examined suicide victims with a history of major depression showed that elevated levels at the C and E site editing were observed, whereas decreased level at the D site editing was detected [67]. Interestingly, this study also found that mice treated chronically with the antidepressant drug fluoxetine exhibited the editing patterns precisely opposite to those observed in the suicide victims. A different study reported a significant increase in editing levels at the A, C, and D sites in suicide victims diagnosed with major depression, resulting in increased VSV isoform bearing decreased G-protein coupling efficacy (Fig. 3B) [69]. Although the studies mentioned above primarily focus on the dorsolateral prefrontal cortex region of the human brain, an analysis on the anterior cingulate cortex region revealed the region-specific changes in the editing patterns of depressive suicide victims. This study showed a remarkable increase of ABDE editing pattern, which generates VDV isoform compared with non-psychiatric controls, implicating this cortical area in suicide risk (Fig. 3B) [71]. Although there is a discrepancy in editing changes of 5-HT2CR mRNAs in suicide victims, there appears to be a trend of increased editing levels, which is associated with decreased sensitivity of the serotonin receptor. Suicide commitment results from extreme severity of psychiatric disorders; thus, increased 5-HT2CR editing may contribute to the pathology of psychological diseases.
2.3. I/V editing of Kv1.1 channel
Voltage-gated K+ (Kv) channels are critical regulators of neuronal membrane excitability [72]. The KCNA1 gene encodes a voltage-gated delayed potassium channel (Kv1.1) that is phylogenetically related to the Drosophila Shaker channel [73,74]. Kv1.1 channels are composed of four α-subunits, each consisting six transmembrane segments (S1–S6). The transcript encoding Kv1.1 is subjected to the conversion of the codon ATT to ITT through RNA editing in a region encoding the sixth transmembrane domain (S6), resulting in a change of isoleucine to valine (I/V) (Fig. 2C) [75]. The degree of editing was related to the regions of the brain. After editing, the deactivation rate of Kv1.1 channel at negative membrane potential was reduced by 20 times, which may be caused by the reduced affinity for the binding of the inactivating particle of the β-subunits (Fig. 2C) [75]. Editing of KCNA1 mRNAs also reduces the blocking of the Kv1.1 channel by endogenous, highly unsaturated signal lipids that reduce the affinity of the pore residues of these sealers [76]. A four-fold increase in the Kv1.1 editing level was observed in the entorhinal cortex of chronic epileptic animals, and a reduction in the potency of the Kv open channel blocker 4-aminopyridine (4-AP) that induces seizures was observed (Fig. 3C) [77], suggesting that the addition of Kv1.1 transcript editing helped suppress seizures (Table 1). Some studies imply that dysregulation of Kv1.1 transcript editing may be related to Episodic ataxia type 1 symptoms (Table 1).
2.3.1. Episodic ataxia type 1 (EA1)
There is a sporadic neurological disorder called episodic ataxia type 1 (EA1), which can occur concurrently with seizures. Many Kv1.1 mutations are associated with EA1 [78]. Three EA1-related mutations (V404I, I407M, and V408A) have been identified in the RNA duplex region required for Kv1.1RNA editing [79,80]. A study from in vitro and in vivo model systems showed that EA1 mutations lead to a significant reduction of Kv1.1 transcript editing, indicating that these mutations have both direct and indirect influences on EA1 symptoms (Fig. 3C) [81].
3. ADAR1-mediated editing in homoeostasis of innate immunity and related disease caused by ADAR1 mutation
3.1. ADAR1 vs MDA5 for interferon trigger by dsRNA
Although ADAR1 conducts site-selective RNA editing as ADAR2, ADAR1 is known to be predominantly responsible for A-to-I editing in repeat sequences of non-coding mRNA regions that form long RNA duplex structures. Although the functions of these editing sites mostly remain unclear, the primary role of these editing is assumed to suppress the activation of innate immune responses to self-derived dsRNA structures. In RNA viral infection, the presence of cytosolic viral dsRNAs can be sensed as exogenous non-self dsRNA through the innate immune system, leading to triggering the type I interferon signalling as an antiviral defence mechanism. The primary sensing of viral-derived non-self dsRNAs is carried out by pattern recognition receptors known as retinoic acid-inducible I (RIG-I)-like receptors (RLRs) [82]. These include melanoma differentiation-associated protein 5 (MDA5) and RIG-I. Upon viral RNA detection, both MDA5 and RIG-I activate innate immune interferon responses via interaction with mitochondrial antiviral signalling (MAVS) adaptors localized on the mitochondrial membrane.
Mouse models of ADAR1 deficiency provide a clear insight into the importance of ADAR1-mediated A-to-I editing in suppressing sensing of cytosolic self-dsRNAs and subsequent interferon signalling. The deletion of Adar1 (both p110 and p150 isoforms), specifically the deletion of the p150 isoform, or the specific inactivation of the editing activity (E861A mutant, both p110 and p150 are editing deficient) resulted in embryonic lethality between E11.5 and E13.5 [12,83]. The phenotypes in these animals are characterized by defective haematopoiesis, liver disintegration, widespread apoptosis, and profound upregulation of type-I interferon responses. The embryonic lethality and interferon signature phenotypes of Adar1 mutant mice can be rescued by concurrent deletion of either the MDA5 receptor or the MAVS adaptor but not the RIG-I receptor [8,83,84]. Adar1 or p150-specific deficient mice with concurrent deletion of MAVS are recovered until live birth but die after birth. In contrast, Adar1 E861A mutant mice with concurrent depletion of MDA5 are strikingly normal, including their life spans. These results strongly suggest that RNA editing mediated by ADAR1 plays a key role in suppressing MDA5-dependent aberrant dsRNA sensing against cytosolic self-dsRNA structures.
3.2. Aicardi-Goutières syndrome (AGS)
Aicardi-Goutières syndrome (AGS) is a fatal childhood encephalopathy characterized by chronic lymphocytosis in the cerebrospinal fluid and elevated interferon-α, as well as by the main neuroradiological features: cerebral calcification, leukoencephalopathy, and cerebral atrophy [85,86]. Seven different pathogenic genes (AGS1-7) have been identified, with clustering of genes involved in cytosolic DNA metabolism (TREX1, RNASEH2B, RNASEH2C, RNASEH2A, and SAMHD1) and those regulating cytosolic RNA metabolism (ADAR1, classified as AGS6, and IFIH1, also known as MDA5) [86]. The impaired function of any of these proteins ultimately leads to chronic overproduction of type-I interferon. In the case of ADAR1, homozygous or compound heterozygous mutations have been identified in patients with AGS. To date, ten ADAR1 mutations were reported as potential causes of AGS [87,88]. The missense mutations are predominantly found on the surface of the deaminase domain, and some of them are shown to impair editing activity to different extents [84]. Of note, the G1007R mutation in the middle of the deaminase domain leads to almost complete abolishment of editing activity, possibly due to conferring more tight binding to dsRNA substrates and preventing base flipping of target adenosine [87]. One missense mutation outside the deaminase domain is P193A, identified in the first Z-DNA binding domain at the N-terminus of the p150 isoform. This mutation is also shown to reduce the editing activity of ADAR1 p150 [84]. These findings implicate the p150 isoform in the pathogenesis of AGS.
A recent study provides the mechanistic model on the pathogenesis of AGS arising from ADAR1 and MDA5 mutations [89]. This study demonstrated that in ADAR1-deficient 293 T cells, unmodified Alu: Alu dsRNAs formed by inverted Alu repeat elements can be ligands of wild-type MDA5, leading to filament formation on Alu: Alu dsRNAs and thereby activation of an interferon signalling. In contrast, AGS-associated gain-of-function (GOF) MDA5 mutants can recognize Alu: Alu dsRNAs even in the presence of A-to-I editing. These results suggest that ADAR1-mediated RNA editing renders Alu: Alu dsRNAs immunologically inactive against cytosolic dsRNA sensing by MDA5 through disruption of dsRNA structural integrity by introducing mismatches and bulges. In contrast, this shielding effect by editing sites on Alu: Alu dsRNA structures can be accepted by GOF MDA5 mutants, leading to breaching immune tolerance to self-dsRNAs and triggering aberrant antiviral interferon responses.
3.3. Dyschromatosis symmetrica hereditaria (DSH)
DSH is a hereditary pigmentation disorder caused by a heterozygous mutation of ADAR1. More than 180 different mutations in the ADAR1 gene have been identified in patients with DSH [90]. Noticeably, the ethnicity of DSH patients is exclusively clustered in East Asian people. Neurological phenotypes are not observed in most DSH patients, while the case carrying the mutation G1007R, which is also identified in AGS, exhibits intracranial calcification [91]. There is a case that harbours mutations in the coding region included in the p150 transcript only, raising the hypothesis that DSH is caused by abnormalities in ADAR1 p150 isoform [92].
3.4. Bilateral striatal necrosis (BSN) and spastic paraplegia
Bilateral striatal necrosis (BSN), a dystonic or rigid movement disorder arising from abnormalities in the brain, is known to be associated with ADAR1 mutations. The BSN originating from ADAR1 mutations is characterized by the presence of interferon signals [93]. Another hereditary disease associated with an ADAR1 mutation is spastic paraplegia, a neurodegenerative disorder characterized by axonal degeneration and lower limb spasticity [94]. Some mutations of these two diseases, including G1007R and P193A, are overlapped with AGS-associated ADAR1 mutations. Patients in both BSN and spastic paraplegia due to ADAR1 mutations exhibited an up-regulated interferon signature, contributing to the presentation of disease phenotypes.
3.5. Brain disease caused by RNA editing in viral RNA
Exogenous viral RNAs can be recognized by cytosolic dsRNA sensors, leading to the activation of type I interferon response. This interferon signalling pathway promotes the transcription of interferon-stimulated genes (ISGs) to defend the viral infection. ISGs include the interferon-induced long isoform of ADAR1, ADAR1p150. Early observations show that ADAR1 could be expected to serve a protective antiviral function and can edit and unwind dsRNA produced by many types of viruses, such as hepatitis C virus, lymphocytic choriomeningitis virus, and polyomavirus [95]. Indeed, for many viral pathogens, such as the influenza virus, measles virus, and human immunodeficiency virus 1, ADAR1 has been implicated as a proviral factor [95]. The proviral function of ADAR1 has been shown to enhance viral replication and proliferation by either direct editing of viral RNA or inhibiting RNA-activated protein kinase (PKR) [96].
RNA editing appears to be responsible for measles virus hypermutation during persistent infections in humans that cause severe brain disease. Measles virus is an enveloped virus that is usually transmitted through the respiratory tract and causes typical acute childhood infections [97]. Its rare complication is the persistent infection of the CNS, leading to a fatal degenerative neurological disease, subacute sclerosing panencephalitis (SSPE) [98], where measles virus infection is localized in the brain. Viral genomes isolated from patients diagnosed with SSPE were analysed, and it was found that the viral genome contains biased mutations of A-G and U-C. Nearly half of the adenosines were mutated to guanosine due to high levels of A-to-I editing [99]. These mutations inhibit the production of matrix proteins and prolong viral infection of the CNS [100]. A study shows that ADAR1-deficient Hela cells infected with the measles virus exhibit enhanced apoptosis [101]. Increased apoptosis is associated with increased activation of PKR and interferon regulatory transcription factor 3 (proapoptotic protein associated with innate antiviral response). This study demonstrates that ADAR1 is a proviral, antiapoptotic host factor in the case of measles virus infection. Another study found that defective interfering RNA in the measles virus that can form during a negative-stranded RNA virus infection and generate dsRNA hybrids with the template genome [102]. Unedited defective interfering RNA may induce an innate immune response in negative-stranded RNA virus infection, while ADAR1 editing can disrupt the defective interfering RNA with dsRNA structure so that the measles virus cannot be immunodetected [102]. Taken together, these findings suggest that ADAR1 plays a proviral role in the brain disease SSPE by prolonging measles virus infection.
Measles virus infection can trigger innate immune responses by activating the dsRNA binding molecule MDA5 [103], and hypermutations are found in the measles virus due to high levels of A-to-I editing [104]. Combined with evidence that ADAR1-dependent RNA editing inhibits the innate immune system from recognizing endogenous dsRNA by inhibiting the MDA5 pathway, it can be hypothesized that ADAR1 may inhibit measles virus sensing via suppression of MDA5-mediated innate immune response. However, current studies on the proviral role of ADAR1 in the measles virus mainly show an antagonistic effect on PKR activation, and further studies will be required to elucidate the relationship between ADAR1 and MDA5 in measles virus infection.
4. A-to-I RNA editing in normal brain development
4.1. Q/R and R/G editing of AMPA receptor
In addition to Q/R site in the GluA2 subunit. other important recoding sites in AMPA receptors are a substitution of an arginine codon (R) to a glycine codon (G) observed in GluA2, GluA3, and GluA4 subunits. This R/G editing event confers rapid recovery from desensitization, resulting in enhanced synaptic response [105]. Immediately downstream of the R/G editing site, alternative splicing occurs to incorporate one of the mutually-exclusive cassette exons referred to as ‘flip’ and ‘flop’ that encode a portion of ligand-binding domain [105]. It is proposed that the R/G site editing affects flip-flop alternative splicing [106]. This combination of RNA editing and splicing generates a variety of receptors with different kinetics. Both ADAR1 and ADAR2 enzymes are responsible for this R/G editing. The editing levels at the GluA2 Q/R and R/G sites show dynamic changes during brain development from embryos to adults. The presence of an unedited GluA2 has been described early in pig brain embryogenesis [107]. Then the GluA2 Q/R site recoding editing event increases rapidly and efficiently, close to 100% [107]. Similar changes were observed in the maturation of mouse brains [63,108]. In contrast to Q/R site editing, R/G site editing level is increased gradually during brain development [63,108].
4.2. Kainate receptor
Similar to the AMPA-type glutamate receptor, the kainate-type glutamate receptor also undergoes A-to-I RNA editing. Kainate receptor subunits GluK1 and GluK2 encoded by GRIK1 and GRIK2 are subjected to RNA editing. GluK1 can be edited only at the Q/R site, whereas GluK2 can be edited at Q/R, I/V, and Y/C sites [109]. They are targets of both ADAR1 and ADAR2 [110]. The defects of Q/R site editing in GluK1 and GluK2 subunits are demonstrated to increase Ca2+ permeability [110,111]. Interestingly, editing ratios at Q/R sites of GluK1 and GluK2 exhibit a low level in embryonic brains, and then increase to ~ 40% and 80% after birth in most regions of the brain, respectively [112,113]. To investigate the functional significance of GluK2 Q/R editing site in vivo, engineered mice deficient in GluK2 Q/R site editing were studied, and the results suggest that unedited GluK2 may modulate synaptic plasticity and increase vulnerability to seizures [114]. In contrast, GluK1 Q/R site mutant mice do not show developmental alteration or abnormal behaviour, which indicates that editing of the GluK1 subunit is unlikely to play an important role [115]. For I/V and Y/C editing sites, both are located in the first transmembrane domain and also are implicated in the regulation of ionic permeability [111,116]. Electrophysiological data are further required to support this functional hypothesis.
4.3. GABRA3
The GABAA receptors are ligand-gated chloride ion channels that can be constituted from at least 16 subunits. GABAA receptor subunit α3 (GABRA3) is the predominant subunit in the embryonic brain [117]. A-to-I editing on the GABRA3 transcript results in a single amino acid change of the α3 protein at the I/M site [118], which has been shown to alter gating kinetics of the receptor. GABRA3 editing increases during neuron development with the decrease of α3 protein [118]. The ratio of I/M site editing is low (~9%) in mouse embryos, but increases with age, approaching 100% in adult brains [63,118], indicating that I/M site editing plays a vital role in brain development.
4.4. NOVA1
The NOVA1 gene encodes a neuron-specific RNA-binding protein, which is a critical brain-specific regulator of alternative splicing and participates in neuronal activity in the CNS [119]. A-to-I RNA editing in the coding sequence of NOVA1 produces a change in the codon 383 from a serine to a glycine (S/G) [120]. NOVA1 editing levels increase during the embryonic development of mouse and chicken brains, and significant differences are observed between brain regions [120]. Edited and unedited NOVA1 show no difference in splicing regulation activity. Instead, S/G editing affects the stability of NOVA1 proteins by preventing specific degradation by the proteasome, almost doubling the half-life of NOVA1 [120].
5. Other RNA editing associated with neurological functions
5.1. CACNA1D
Voltage-dependent calcium channels (Cav1.3), which is encoded by the CACNA1D gene, mediate the entry of calcium ions into excitable cells and are involved in a variety of calcium-dependent processes [121]. The isoleucine-glutamine (IQ) domain in the Cav1.3 plays a critical role in a robust regulatory function on inhibitory Ca2+-feedback (CDI) on channels. One study found that RNA editing the CACNA1D gene can cause three types of amino acid alterations (I/M, Q/R, and Y/C) in the IQ domain in the Cav1.3. It was demonstrated that ADAR2 is responsible for this RNA editing [122]. Proteomic analysis confirm the presence of the Cav1.3 channel edited in natural brain tissue [122]. Edited Cav1.3 channels exhibit a substantial reduction of Ca2+-feedback [122]. Editing of the Cav1.3 Ca2+ channel is an ideal method for low-pressure Ca2+ inflow and neuronal pacing [122]. This study reveals a mechanism to fine-tune the Cav1.3 channel properties in the CNS.
5.2. CADPS1
Calcium-dependent activator protein for secretion 1 (CAPS1), which is encoded by CADPS1 gene, facilitates the docking and priming of synaptic and dense core vesicles (DCV). A-to-I RNA editing is known to occur in the coding region of CAPS1 mRNA, which results in glutamate at residue 1250 (E1250) to glycine (G) (E/G) in its C-terminal region [123]. Recently, two groups have reported the physiological role of CAPS1 editing. One group generated mutant mice that expressed only edited CAPS1, they found that these mice exhibited leanness due to excessive physical activity and increased energy expenditure, and the dense core vesicle (DCV) exocytosis was increased in chromaffin cells and neurons of these mice [124]. This suggests that RNA editing can regulate in vivo DCV exocytosis to influence physical activity. Another group compared the CAPS1 protein isoforms in primary hippocampal neurons. Their study shows that the increase of the edited CAPS1 isoforms promotes the aggregation and turnover of presynaptic vesicles [125]. In contrast, the unedited CAPS1 isoforms slow the induced release and the increase of spontaneous fusion, and relax the aggregation of synaptic vesicles. Taken together, RNA editing of CAPS1 is a mechanism for fine-tuning the release of neurotransmitters [125]. The CAPS family has been suggested to be involved in developmental disorders that affect physical activity, and CAPS1 RNA editing abnormalities may contribute to the occurrence of some diseases [124].
5.3. Circadian rhythm
A recent study demonstrates that ADAR2-dependent A-to-I editing is involved in circadian rhythm, regulating the rhythmic oscillation of mRNA expression levels [126]. This study shows that Adar2 null mice exhibit complete loss of the RNA editing rhythms, short-period free-running, and gene expression oscillation, suggesting that RNA editing play a key role in post-transcriptional regulation in the circadian clockwork, and ADAR2-mediated rhythms could be important for diverse aspects of physiology including neurological activities.
6. A-to-I editing of miRNA in neurological diseases and normal brain development
A-to-I RNA editing occurs in non-coding transcripts as well as protein-coding ones. miRNAs are small non-coding RNAs with about 22 nucleotides that regulate gene expression through RNAi [127]. The miRNA loaded onto the RNA-induced silencing complex (RISC) binds to semi-complementary sequences in 3ʹ UTRs of target mRNAs, resulting in translation repression and/or mRNA decay. miRNA biogenesis originates from primary miRNA transcripts (pri-miRNA) which fold to form dsRNA structures. They are subjected to processing by the Drosha-DGCR8 complex into precursor miRNAs (pre-miRNA) in the nucleus. Subsequently, they are exported to the cytoplasm and further processed by the Dicer-TRBP complex into mature miRNAs, followed by loading onto the RISC. It is known that some pri-mRNAs can undergo RNA editing. RNA editing can affect various steps of miRNA biogenesis. It is reported that RNA editing blocks pri-miRNA and pre-miRNA cleavage by the Drosha-DGCR8 and Dicer-TRBP complexes, respectively, leading to inhibition of miRNA maturation [128–131]. In addition, A-to-I editing can affect recognition of target mRNAs cases where editing occurs in miRNA seed sequences (+2 to +7 position from the 5ʹend of the miRNA) that are primary determinants for target selectivity [129,130,132]. It is estimated that ~ 16% of pri-mRNAs is edited in the human brain [129]. The biochemical analysis and in silico prediction of editing functions suggest that these RNA editing events may modulate miRNA biogenesis or target recognition of miRNAs. High-throughput sequencing analysis of mature miRNAs across 13 different human tissues found the highest level of RNA editing in the prefrontal cortex, followed by the whole brain. Notably, the same study showed that RNA editing in mature miRNAs is enriched in seed sequences (approximately 70%) [133]. This trend is also confirmed in the mouse brain [134].
One of the representative miRNAs bearing A-to-I editing in seed regions is miR-376a, which is a member of the miR-376 cluster [135]. Members of the miR-376 cluster are transcribed as one transcript bearing multiple hairpin structures that undergo RNA editing at multiple sites prior to processed into individual pre-miRNAs. miR-376a has two major editing sites located at +4 in 5p seed sequence and +44 in 3p seed sequence [135]. These editing sites show high levels of modification frequencies in specific regions of the brain compared to other tissues. Interestingly, edited miR‐376a-5p targets an almost entirely different set of genes than unedited miR‐376a-5p.
RNA editing of some miRNAs can be altered during brain maturation. In the miR-376 cluster, only a low level of mature mmu-miR-376b editing is detected at E15, but increases dramatically after birth and remains at a constant high level later [132]. There is no editing in mature mmu-miR-376 c, another miR-376 family member, before birth, but around 40%-50% editing is detected at the postnatal stage [132]. Inversely, pri-miRNA-376 c is highly edited at E15 and remains highly edited during development. This indicates that editing may interfere with pri-miRNA-376 c maturation before birth [132]. In the miR379-410 cluster, the editing levels of mature mmu-miR-379 are only detected at the adult stage [132]. Mature mmu-miR-381 shows no editing at E15, but two stages (P2 and P21) show an increase in editing. Moreover, pri-miR-381, which has a low editing level at E15, shows a steady increase in the rest stages up to a level higher than its mature miRNA [132]. Although the targets of miR-381 have not been confirmed, unedited miR-381 has at least two predicted target sites in the 3ʹ UTRs of Pumillio-2 (Pum2), which is important for the regulation of dendritic growth by acting as a translation repressor [136]. As editing of miR-381 increases during development, the expression of Pum2 increases, indicating that increased miR-381 editing abolishes the silencing effect against Pum2 mRNA [132].
Dysregulation of A-to-I RNA editing in miRNAs is implicated in the progression and invasion of astrocytoma. Small RNA sequencing of glioblastoma multiforme samples found that miRNAs were significantly hypo-edited, which was positively correlated with the downregulation of ADAR2 [133]. Impairment of ADAR2-mediated RNA editing that inhibits oncogenic miR-222, miR-221, and miR-21 maturation leads to a proliferation and migration of glioblastoma cells [137]. Attenuation of RNA editing in miR-376a-5p promotes glioblastoma invasiveness in vivo [138]. The pro-invasive effect of unedited miR-376a-5p can be attributed to gain the ability to suppress RAP2A, which is demonstrated to be an anti-invasion factor, and loss of the ability to silence AMFR, which is shown to be a pro-invasion factor. Combined with the fact that the decrease of ADAR2-dependent GluA2 Q/R editing contributes to tumour progression as mentioned earlier, these findings support that ADAR2-mediated RNA editing has a tumour-suppressive role in brain cancers.
7. Concluding remarks and outlook
In this review, we summarized the role of A-to-I editing in the CNS. Increasing evidence strengthens the model that RNA editing plays a pivotal role in the CNS and demonstrates that aberrant regulation of RNA editing is associated with the pathogenesis of neurological diseases. However, the molecular mechanisms that change editing levels are still unclear. Recently, many studies reported that RNA binding proteins and RNA helicases act as regulatory factors for RNA editing [139]. Some of these proteins might be involved in the CNS-specific editing profile.
The levels of RNA editing in neurological genes dynamically change in the process of neural differentiation and maturation during brain development from the embryo to the adult. A recent RNA-seq analysis of human brain tissues from 39 foetuses to elderly individuals revealed the systematic and dynamic aspects of RNA editing in brain development [140]. These lines of evidence imply the advantage of post-transcriptional RNA editing over A-to-G gene mutation in organisms because RNA editing can be temporally regulated to precisely control functions or expression levels of neuronal genes during brain development. These multiple layers of regulations mediated by RNA editing might be required to construct a complex organ system such as the brain. At present, the number of identified editing sites has increased significantly through deep sequencing, but the functional effect of these editing events is poorly understood [141]. Among the identified editing sites, only a few have been confirmed to be associated with neurological diseases. In addition to the representative editing sites mentioned in this review, it remains to be elucidated whether the rest of the editing sites are functional and responsible for the pathogenesis of neurological diseases. Further research on the regulatory mechanisms or factors of these editing events can help to develop treatment strategies.
Recently, site-directed A-to-I RNA editing technologies have emerged and can be considered as therapeutic techniques to correct G-to-A mutations associated with various genetic diseases or recover reduced editing levels linked to the pathogenesis of diseases or cancers [142]. Although there are limitations in these tools, the application of site-directed RNA editing in neurological disease has great potential.
Research on differential levels of A-to-I editing in relevance to neurological diseases mainly focuses on the whole brain tissue and specific brain regions. Differences in editing levels between brain regions and cell types, such as neuronal cells and glial cells, are still unclear. In recent years, single-cell RNA sequencing methods have been proven to be an effective method for studying the complexity of RNA editing with single-cell resolution, providing a new direction for exploring the novel physiological role of RNA editing in various neurological diseases [143]. Distinct patterns of RNA editing at the single-cell level may contribute to the identification of novel therapeutic targets and improve our understanding of the pathogenesis of neurological diseases.
Acknowledgments
We are most grateful to Prof. Yukio Kawahara at Osaka University for his critical comments and crucial suggestions to improve this manuscript. We also appreciate Miyoko Nakano for supporting us in preparing the manuscript.
Funding Statement
This work was supported by the following grants: JSPS KAKENHI Grant Numbers JP18H06054 JP19H03159 JP19K16037, MSD Life Science Foundation, The Uehara Memorial Foundation, The Takeda Science Foundation, The Senri Life Science Foundation, THE KATO MEMORIAL TRUST FOR NAMBYO RESEARCH, Princess Takamatsu Cancer Research Fund, Japan Intractable Diseases Research Foundation, and The Tokyo Biochemical Research Foundation.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Flamand MN, Meyer KD.. The epitranscriptome and synaptic plasticity. Curr Opin Neurobiol. 2019;59:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 2016;17(2):83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kim U, Wang Y, Sanford T, et al. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc Natl Acad Sci U S A. 1994;91(24):11457–11461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Melcher T, Maas S, Herb A, et al. A mammalian RNA editing enzyme. Nature. 1996;379(6564):460–464. [DOI] [PubMed] [Google Scholar]
- [5].Chen CX, Cho DS, Wang Q, et al. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. Rna. 2000;6(5):755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Stefl R, Xu M, Skrisovska L, et al. Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure. 2006;14(2):345–355. [DOI] [PubMed] [Google Scholar]
- [7].Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol. 1995;15(10):5376–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pestal K, Funk C, Snyder JM, et al. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity. 2015;43(5):933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Walkley CR, Li JB. Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol. 2017;18(1):205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang Y, Chung DH, Monteleone LR, et al. RNA binding candidates for human ADAR3 from substrates of a gain of function mutant expressed in neuronal cells. Nucleic Acids Res. 2019;47(20):10801–10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mladenova D, Barry G, Konen LM, et al. Adar3 is involved in learning and memory in Mice. Front Neurosci. 2018;12:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang Q, Miyakoda M, Yang W, et al. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem. 2004;279(6):4952–4961. [DOI] [PubMed] [Google Scholar]
- [13].Higuchi M, Maas S, Single FN, et al. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406(6791):78–81. [DOI] [PubMed] [Google Scholar]
- [14].Hood JL, Emeson RB. Editing of neurotransmitter receptor and ion channel RNAs in the nervous system. Curr Top Microbiol Immunol. 2012;353:61–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pinto Y, Cohen HY, Levanon EY. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol. 2014;15(1):R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lev-Maor G, Sorek R, Levanon EY, et al. RNA-editing-mediated exon evolution. Genome Biol. 2007;8(2):R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399(6731):75–80. [DOI] [PubMed] [Google Scholar]
- [18].Borchert GM, Gilmore BL, Spengler RM, et al. Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum Mol Genet. 2009;18(24):4801–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nakano M, Fukami T, Gotoh S, et al. A-to-I RNA editing up-regulates human dihydrofolate reductase in breast cancer. J Biol Chem. 2017;292(12):4873–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stellos K, Gatsiou A, Stamatelopoulos K, et al. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR-mediated post-transcriptional regulation. Nat Med. 2016;22(10):1140–1150. [DOI] [PubMed] [Google Scholar]
- [21].Ramaswami G, Zhang R, Piskol R, et al. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods. 2013;10(2):128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bazak L, Haviv A, Barak M, et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24(3):365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Neeman Y, Levanon EY, Jantsch MF, et al. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA. 2006;12(10):1802–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Danecek P, Nellåker C, McIntyre RE, et al. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol. 2012;13(4):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sakurai M, Ueda H, Yano T, et al. A biochemical landscape of A-to-I RNA editing in the human brain transcriptome. Genome Res. 2014;24(3):522–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bahn JH, Lee JH, Li G, et al. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 2012;22(1):142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ivanov A, Memczak S, Wyler E, et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10(2):170–177 [DOI] [PubMed] [Google Scholar]
- [28].Wright A, Vissel B. The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front Mol Neurosci. 2012;5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dingledine R, Borges K, Bowie D, et al. The glutamate receptor ion channels. Pharmacol Rev. 1999;51(1):7–61 [PubMed] [Google Scholar]
- [30].Brusa R, Zimmermann F, Koh DS, et al. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270(5242):1677–1680 [DOI] [PubMed] [Google Scholar]
- [31].MAJ van den Bos, Geevasinga N, Higashihara M, et al. Pathophysiology and diagnosis of ALS: insights from advances in neurophysiological techniques. Int J Mol Sci. 2019;20(11):2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Takuma H, Kwak S, Yoshizawa T, et al. Reduction of GluR2 RNA editing, a molecular change that increases calcium influx through AMPA receptors, selective in the spinal ventral gray of patients with amyotrophic lateral sclerosis. Ann Neurol. 1999;46(6):806–815. [DOI] [PubMed] [Google Scholar]
- [33].Kawahara Y, Ito K, Sun H, et al. Glutamate receptors: RNA editing and death of motor neurons. Nature. 2004;427(6977):801. [DOI] [PubMed] [Google Scholar]
- [34].Hideyama T, Yamashita T, Aizawa H, et al. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol Dis. 2012;45(3):1121–1128. [DOI] [PubMed] [Google Scholar]
- [35].Hideyama T, Yamashita T, Suzuki T, et al. Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J Neurosci. 2010;30(36):11917–11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aizawa H, Sawada J, Hideyama T, et al. TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol. 2010;120(1):75–84. [DOI] [PubMed] [Google Scholar]
- [37].Yamashita T, Hideyama T, Hachiga K, et al. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat Commun. 2012;3(1):1307. [DOI] [PubMed] [Google Scholar]
- [38].Yamashita T, Aizawa H, Teramoto S, et al. Calpain-dependent disruption of nucleo-cytoplasmic transport in ALS motor neurons. Sci Rep. 2017;7(1):39994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Akamatsu M, Yamashita T, Hirose N, et al. The AMPA receptor antagonist perampanel robustly rescues amyotrophic lateral sclerosis (ALS) pathology in sporadic ALS model mice. Sci Rep. 2016;6(1):28649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yamashita T, Chai HL, Teramoto S, et al. Rescue of amyotrophic lateral sclerosis phenotype in a mouse model by intravenous AAV9-ADAR2 delivery to motor neurons. EMBO Mol Med. 2013;5(11):1710–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Catarzi D, Colotta V, Varano F. Competitive AMPA receptor antagonists. Med Res Rev. 2007;27(2):239–278. [DOI] [PubMed] [Google Scholar]
- [42].Feldmeyer D, Kask K, Brusa R, et al. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci. 1999;2(1):57–64. [DOI] [PubMed] [Google Scholar]
- [43].Krestel HE, Shimshek DR, Jensen V, et al. A genetic switch for epilepsy in adult mice. J Neurosci. 2004;24(46):10568–10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jia Z, Agopyan N, Miu P, et al. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17(5):945–956 [DOI] [PubMed] [Google Scholar]
- [45].Kitaura H, Sonoda M, Teramoto S, et al. Ca(2+) -permeable AMPA receptors associated with epileptogenesis of hypothalamic hamartoma. Epilepsia. 2017;58(4):p. e59-e63. [DOI] [PubMed] [Google Scholar]
- [46].Kortenbruck G, Berger E, Speckmann EJ, et al. RNA editing at the Q/R site for the glutamate receptor subunits GLUR2, GLUR5, and GLUR6 in hippocampus and temporal cortex from epileptic patients. Neurobiol Dis. 2001;8(3):459–468 [DOI] [PubMed] [Google Scholar]
- [47].Krestel H, Raffel S, von Lehe M, et al. Differences between RNA and DNA due to RNA editing in temporal lobe epilepsy. Neurobiol Dis. 2013;56:66–73 [DOI] [PubMed] [Google Scholar]
- [48].Tan TY, Sedmík J, Fitzgerald MP, et al. Bi-allelic ADARB1 variants associated with microcephaly, intellectual disability, and seizures. Am J Hum Genet. 2020;106(4):467–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Maroofian R, Sedmík J, Mazaheri N, et al. Biallelic variants in ADARB1, encoding a dsRNA-specific adenosine deaminase, cause a severe developmental and epileptic encephalopathy. J Med Genet. 2020. DOI: 10.1136/jmedgenet-2020-107048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Salpietro V, Dixon CL, Guo H, et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat Commun. 2019;10(1):3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Galeano F, Tomaselli S, Locatelli F, et al. A-to-I RNA editing: the “ADAR” side of human cancer. Semin Cell Dev Biol. 2012;23(3):244–250. [DOI] [PubMed] [Google Scholar]
- [52].Stupp R, van den Bent MJ, Hegi ME. Optimal role of temozolomide in the treatment of malignant gliomas. Curr Neurol Neurosci Rep. 2005;5(3):198–206. [DOI] [PubMed] [Google Scholar]
- [53].Maas S, Patt S, Schrey M, et al. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci U S A. 2001;98(25):14687–14692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ishiuchi S, Tsuzuki K, Yoshida Y, et al. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. 2002;8(9):971–978. [DOI] [PubMed] [Google Scholar]
- [55].Ishiuchi S, Yoshida Y, Sugawara K, et al. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J Neurosci. 2007;27(30):7987–8001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Giorgetti M, Tecott LH. Contributions of 5-HT(2C) receptors to multiple actions of central serotonin systems. Eur J Pharmacol. 2004;488(1–3):1–9. [DOI] [PubMed] [Google Scholar]
- [57].Burns CM, Chu H, Rueter SM, et al. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387(6630):303–308. [DOI] [PubMed] [Google Scholar]
- [58].Slotkin W, Nishikura K. Adenosine-to-inosine RNA editing and human disease. Genome Med. 2013;5(11):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Niswender CM, Copeland SC, Herrick-Davis K, et al. RNA editing of the human serotonin 5-hydroxytryptamine 2C receptor silences constitutive activity. J Biol Chem. 1999;274(14):9472–9478. [DOI] [PubMed] [Google Scholar]
- [60].Abbas AI, Urban DJ, Jensen NH, et al. Assessing serotonin receptor mRNA editing frequency by a novel ultra high-throughput sequencing method. Nucleic Acids Res. 2010;38(10):e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Price RD, Weiner DM, Chang MS, et al. RNA editing of the human serotonin 5-HT2C receptor alters receptor-mediated activation of G13 protein. J Biol Chem. 2001;276(48):44663–44668 [DOI] [PubMed] [Google Scholar]
- [62].Marion S, Weiner DM, Caron MG. RNA editing induces variation in desensitization and trafficking of 5-hydroxytryptamine 2c receptor isoforms. J Biol Chem. 2004;279(4):2945–2954. [DOI] [PubMed] [Google Scholar]
- [63].Wahlstedt H, Daniel C, Enstero M, et al. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009;19(6):978–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kawahara Y, Grimberg A, Teegarden S, et al. Dysregulated editing of serotonin 2C receptor mRNAs results in energy dissipation and loss of fat mass. J Neurosci. 2008;28(48):12834–12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chagraoui A, Thibaut F, Skiba M, et al. 5-HT2C receptors in psychiatric disorders: A review. Prog Neuropsychopharmacol Biol Psychiatry. 2016;66:120–135. [DOI] [PubMed] [Google Scholar]
- [66].Jenck F, Bös M, Wichmann J, et al. The role of 5-HT2C receptors in affective disorders. Expert Opin Investig Drugs. 1998;7(10):1587–1599. [DOI] [PubMed] [Google Scholar]
- [67].Gurevich I, Tamir H, Arango V, et al. Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron. 2002;34(3):349–356. [DOI] [PubMed] [Google Scholar]
- [68].Niswender CM, Herrick-Davis K, Dilley GE, et al. RNA editing of the human serotonin 5-HT2C receptor. alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharmacology. 2001;24(5):478–491. [DOI] [PubMed] [Google Scholar]
- [69].Dracheva S, Patel N, Woo DA, et al. Increased serotonin 2C receptor mRNA editing: a possible risk factor for suicide. Mol Psychiatry. 2008;13(11):1001–1010 [DOI] [PubMed] [Google Scholar]
- [70].Iwamoto K, Kato T. RNA editing of serotonin 2C receptor in human postmortem brains of major mental disorders. Neurosci Lett. 2003;346(3):169–172. [DOI] [PubMed] [Google Scholar]
- [71].Weissmann D, van der Laan S, Underwood MD, et al. Region-specific alterations of A-to-I RNA editing of serotonin 2c receptor in the cortex of suicides with major depression. Transl Psychiatry. 2016;6(8):e878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Pongs O. Voltage-gated potassium channels: from hyperexcitability to excitement. FEBS Lett. 1999;452(1–2):31–35. [DOI] [PubMed] [Google Scholar]
- [73].Kamb A, Iverson LE, Tanouye MA. Molecular characterization of Shaker, a Drosophila gene that encodes a potassium channel. Cell. 1987;50(3):405–413. [DOI] [PubMed] [Google Scholar]
- [74].Gutman GA, Chandy KG, Grissmer S, et al. International union of pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57(4):473–508. [DOI] [PubMed] [Google Scholar]
- [75].Bhalla T, Rosenthal JJ, Holmgren M, et al. Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat Struct Mol Biol. 2004;11(10):950–956 [DOI] [PubMed] [Google Scholar]
- [76].Decher N, Streit AK, Rapedius M, et al. RNA editing modulates the binding of drugs and highly unsaturated fatty acids to the open pore of Kv potassium channels. Embo J. 2010;29(13):2101–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Streit AK, Derst C, Wegner S, et al. RNA editing of Kv1.1 channels may account for reduced ictogenic potential of 4-aminopyridine in chronic epileptic rats. Epilepsia. 2011;52(3):645–648. [DOI] [PubMed] [Google Scholar]
- [78].Graves TD, Cha Y-H, Hahn AF, et al. Episodic ataxia type 1: clinical characterization, quality of life and genotype-phenotype correlation. Brain. 2014;137(Pt 4):1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Imbrici P, D’Adamo MC, Kullmann DM, et al. Episodicataxia type 1 mutations in the KCNA1 gene impair the fast inactivation properties of the human potassium channels Kv1.4-1.1/Kvβ1.1 and Kv1.4-1.1/Kvβ1.2. Eur J Neurosci. 2006;24(11):3073–3083. [DOI] [PubMed] [Google Scholar]
- [80].Tomlinson SE, Rajakulendran S, Tan SV, et al. Clinical, genetic, neurophysiological and functional study of new mutations in episodic ataxia type 1. J Neurol Neurosurg Psychiatry. 2013;84(10):1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ferrick-Kiddie EA, Rosenthal JJC, Ayers GD, et al. Mutations underlying Episodic Ataxia type-1 antagonize Kv1.1 RNA editing. Sci Rep. 2017;7(1):41095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wu B, Hur S. How RIG-I like receptors activate MAVS. Curr Opin Virol. 2015;12:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Liddicoat BJ, Piskol R, Chalk AM, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349(6252):1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Mannion NM, Greenwood SM, Young R, et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014;9(4):1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Orcesi S, La Piana R, Fazzi E. Aicardi-Goutieres syndrome. Br Med Bull. 2009;89:183–201. [DOI] [PubMed] [Google Scholar]
- [86].Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15(7):429–440. [DOI] [PubMed] [Google Scholar]
- [87].Rice GI, Forte GMA, Mannion NM, et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet. 2012;44(11):1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Schmelzer L, Smitka M, Wolf C, et al. Variable clinical phenotype in two siblings with Aicardi-Goutières syndrome type 6 and a novel mutation in the ADAR gene. Eur J Paediatr Neurol. 2018;22(1):186–189. [DOI] [PubMed] [Google Scholar]
- [89].Ahmad S, Mu X, Yang F, et al. Breaching self-tolerance to Alu Duplex RNA underlies MDA5-mediated inflammation. Cell. 2018;172(4):797–810. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kono M, Akiyama M. Dyschromatosis symmetrica hereditaria and reticulate acropigmentation of Kitamura: an update. J Dermatol Sci. 2019;93(2):75–81. [DOI] [PubMed] [Google Scholar]
- [91].Kondo T, Suzuki T, Ito S, et al. Dyschromatosis symmetrica hereditaria associated with neurological disorders. J Dermatol. 2008;35(10):662–666 [DOI] [PubMed] [Google Scholar]
- [92].Suzuki N, Suzuki T, Inagaki K, et al. Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria. J Invest Dermatol. 2007;127(2):309–311. [DOI] [PubMed] [Google Scholar]
- [93].Livingston JH, Lin JP, Dale RC, et al. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet. 2014;51(2):76–82 [DOI] [PubMed] [Google Scholar]
- [94].Crow YJ, Zaki M, Abdel-Hamid M, et al. Mutations in ADAR1, IFIH1, and RNASEH2B presenting as spastic paraplegia. Neuropediatrics. 2014;45(6):386–393. [DOI] [PubMed] [Google Scholar]
- [95].Samuel CE. ADARs: viruses and innate immunity. Curr Top Microbiol Immunol. 2012;353:163–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Gélinas JF, Clerzius G, Shaw E, et al. Enhancement of replication of RNA viruses by ADAR1 via RNA editing and inhibition of RNA-activated protein kinase. J Virol. 2011;85(17):8460–8466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Moss WJ, Griffin DE. Global measles elimination. Nat Rev Microbiol. 2006;4(12):900–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Oldstone MB. Modeling subacute sclerosing panencephalitis in a transgenic mouse system: uncoding pathogenesis of disease and illuminating components of immune control. Curr Top Microbiol Immunol. 2009;330:31–54. [DOI] [PubMed] [Google Scholar]
- [99].Bass BL, Weintraub H, Cattaneo R, et al. Biased hypermutation of viral RNA genomes could be due to unwinding/modification of double-stranded RNA. Cell. 1989;56(3):331. [DOI] [PubMed] [Google Scholar]
- [100].Ward SV, George CX, Welch MJ, et al. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc Natl Acad Sci U S A. 2011;108(1):331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Toth AM, Li Z, Cattaneo R, et al. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J Biol Chem. 2009;284(43):29350–29356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Pfaller CK, Mastorakos GM, Matchett WE, et al. Measles virus defective interfering RNAs are generated frequently and early in the absence of C protein and can be destabilized by adenosine deaminase acting on RNA-1-Like hypermutations. J Virol. 2015;89(15):7735–7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Berghäll H, Sirén J, Sarkar D, et al. The interferon-inducible RNA helicase, mda-5, is involved in measles virus-induced expression of antiviral cytokines. Microbes Infect. 2006;8(8):2138–2144. [DOI] [PubMed] [Google Scholar]
- [104].Cattaneo R, Schmid A, Eschle D, et al. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell. 1988;55(2):255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Lomeli H, Mosbacher J, Melcher T, et al. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science. 1994;266(5191):1709–1713. [DOI] [PubMed] [Google Scholar]
- [106].Schoft VK, Schopoff S, Jantsch MF. Regulation of glutamate receptor B pre-mRNA splicing by RNA editing. Nucleic Acids Res. 2007;35(11):3723–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Venø M, Bramsen JB, Bendixen C, et al. Spatio-temporal regulation of ADAR editing during development in porcine neural tissues. RNA Biol. 2012;9(8):1054–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Pachernegg S, Münster Y, Muth-Köhne E, et al. GluA2 is rapidly edited at the Q/R site during neural differentiation in vitro. Front Cell Neurosci. 2015;9:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Barbon A, Barlati S. Glutamate receptor RNA editing in health and disease. Biochemistry (Mosc). 2011;76(8):882–889. [DOI] [PubMed] [Google Scholar]
- [110].Egebjerg J, Heinemann SF. Ca2+ permeability of unedited and edited versions of the kainate selective glutamate receptor GluR6. Proc Natl Acad Sci U S A. 1993;90(2):755–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Kohler M, Burnashev N, Sakmann B, et al. Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron. 1993;10(3):491–500. [DOI] [PubMed] [Google Scholar]
- [112].Paschen W, Schmitt J, Gissel C, et al. Developmental changes of RNA editing of glutamate receptor subunits GluR5 and GluR6: in vivo versus in vitro. Brain Res Dev Brain Res. 1997;98(2):271–280. [DOI] [PubMed] [Google Scholar]
- [113].Bernard A, Ferhat L, Dessi F, et al. Q/R editing of the rat GluR5 and GluR6 kainate receptors in vivo and in vitro: evidence for independent developmental, pathological and cellular regulation. Eur J Neurosci. 1999;11(2):604–616. [DOI] [PubMed] [Google Scholar]
- [114].Vissel B, Royle GA, Christie BR, et al. The role of RNA editing of kainate receptors in synaptic plasticity and seizures. Neuron. 2001;29(1):217–227. [DOI] [PubMed] [Google Scholar]
- [115].Sailer A, Swanson GT, Pérez-Otaño I, et al. Generation and analysis of GluR5(Q636R) kainate receptor mutant mice. J Neurosci. 1999;19(20):8757–8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Burnashev N, Zhou Z, Neher E, et al. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. J Physiol. 1995;485(Pt 2):403–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Ortinski PI, Lu C, Takagaki K, et al. Expression of distinct α subunits of GABAA receptor regulates inhibitory synaptic strength. J Neurophysiol. 2004;92(3):1718–1727. [DOI] [PubMed] [Google Scholar]
- [118].Ohlson J, Pedersen JS, Haussler D, et al. Editing modifies the GABA(A) receptor subunit alpha3. Rna. 2007;13(5):698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Buckanovich RJ, Yang YY, Darnell RB. The onconeural antigen Nova-1 is a neuron-specific RNA-binding protein, the activity of which is inhibited by paraneoplastic antibodies. J Neurosci. 1996;16(3):1114–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Irimia M, Denuc A, Ferran JL, et al. Evolutionarily conserved A-to-I editing increases protein stability of the alternative splicing factor Nova1. RNA Biol. 2012;9(1):12–21. [DOI] [PubMed] [Google Scholar]
- [121].Striessnig J, Koschak A. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene knockout models. Channels (Austin). 2008;2(4):233–251. [DOI] [PubMed] [Google Scholar]
- [122].Huang H, Tan B, Shen Y, et al. RNA editing of the IQ domain in Cav1.3 channels modulates their Ca2+-dependent inactivation. Neuron. 2012;73(2):304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Li JB, Levanon EY, Yoon JK, et al. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324(5931):1210–1213 [DOI] [PubMed] [Google Scholar]
- [124].Miyake K, Ohta T, Nakayama H, et al. CAPS1 RNA editing promotes dense core vesicle exocytosis. Cell Rep. 2016;17(8):2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Ulbricht R, Sun S, DelBove C, et al. RNA editing of CAPS1 regulates synaptic vesicle organization, release and retrieval. bioRxiv. 2017;178202 [Google Scholar]
- [126].Terajima H, Yoshitane H, Ozaki H, et al. ADARB1 catalyzes circadian A-to-I editing and regulates RNA rhythm. Nat Genet. 2017;49(1):146–151. [DOI] [PubMed] [Google Scholar]
- [127].Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. [DOI] [PubMed] [Google Scholar]
- [128].Yang W, Chendrimada TP, Wang Q, et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13(1):13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Kawahara Y, Megraw M, Kreider E, et al. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008;36(16):5270–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Kawahara Y, Zinshteyn B, Chendrimada TP, et al. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007;8(8):763–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Iizasa H, Wulff B-E, Alla NR, et al. Editing of epstein-barr virus-encoded BART6 microRNAs controls their dicer targeting and consequently affects viral latency. J Biol Chem. 2010;285(43):33358–33370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ekdahl Y, Farahani HS, Behm M, et al. A-to-I editing of microRNAs in the mammalian brain increases during development. Genome Res. 2012;22(8):1477–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Paul D, Sinha AN, Ray A, et al. A-to-I editing in human miRNAs is enriched in seed sequence, influenced by sequence contexts and significantly hypoedited in glioblastoma multiforme. Sci Rep. 2017;7(1):2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Vesely C, Tauber S, Sedlazeck FJ, et al. ADAR2 induces reproducible changes in sequence and abundance of mature microRNAs in the mouse brain. Nucleic Acids Res. 2014;42(19):12155–12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Kawahara Y, Zinshteyn B, Sethupathy P, et al. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315(5815):1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Fiore R, Khudayberdiev S, Christensen M, et al. Mef2-mediated transcription of the miR379–410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. Embo J. 2009;28(6):697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Tomaselli S, Galeano F, Alon S, et al. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 2015;16(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Choudhury Y, Tay FC, Lam DH, et al. Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J Clin Invest. 2012;122(11):4059–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Quinones-Valdez G, Tran SS, Jun H-I, et al. Regulation of RNA editing by RNA-binding proteins in human cells. Commun Biol. 2019;2(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Hwang T, Park CK, Leung AK, et al. Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci. 2016;19(8):1093–1099 [DOI] [PubMed] [Google Scholar]
- [141].Chalk AM, Taylor S, Heraud-Farlow JE, et al. The majority of A-to-I RNA editing is not required for mammalian homeostasis. Genome Biol. 2019;20(1):268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Montiel-Gonzalez MF, Diaz Quiroz JF, Rosenthal JJC. Current strategies for site-directed RNA editing using ADARs. Methods. 2019;156:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Picardi E, Horner DS, Pesole G. Single-cell transcriptomics reveals specific RNA editing signatures in the human brain. Rna. 2017;23(6):860–865. [DOI] [PMC free article] [PubMed] [Google Scholar]