

ABSTRACT
It is increasingly recognized that local protein synthesis (LPS) contributes to fundamental aspects of axon biology, in both developing and mature neurons. Mutations in RNA-binding proteins (RBPs), as central players in LPS, and other proteins affecting RNA localization and translation are associated with a range of neurological disorders, suggesting disruption of LPS may be of pathological significance. In this review, we substantiate this hypothesis by examining the link between LPS and key axonal processes, and the implicated pathophysiological consequences of dysregulated LPS. First, we describe how the length and autonomy of axons result in an exceptional reliance on LPS. We next discuss the roles of LPS in maintaining axonal structural and functional polarity and axonal trafficking. We then consider how LPS facilitates the establishment of neuronal connectivity through regulation of axonal branching and pruning, how it mediates axonal survival into adulthood and its involvement in neuronal stress responses.
KEYWORDS: Neurological disorders, local protein synthesis, RNA-binding protein, axonal trafficking, axon branching, axon survival, neuronal stress
Introduction
The nervous system is an interconnected network of billions of individual cells, which is key to its function. As central network building blocks, neurons not only conduct signals to relay information (electrically within and chemically between cells), but also generate, maintain, and adapt inter-neuronal connections to enable dynamic information storage and retrieval (i.e., memory and learning). The sites of connection, synapses or neuroeffector junctions, where the axon terminal of one neuron meets the dendritic spine or soma of another neuron or a target cell, are key for cognition, as well as for control and coordination of the body [1,2]. Aberrant network assembly or progressive network disintegration, due to failure in the establishment or maintenance of synaptic connections, results in neurodevelopmental and neurodegenerative disorders, respectively.
In this review, we focus on the idea that the local synthesis of new proteins (local protein synthesis; LPS) in axons by translation of localized mRNAs is essential for network assembly and its maintenance in adulthood. Evidence that axons can synthesize proteins locally was first reported in axons in the 1960s using metabolic labelling methods [3,4,5], but has only become widely accepted in recent years. Early scepticism sprang from concerns about sample (axonal) purity due to technical difficulties in obtaining axon-only material, and the paucity of ultrastructural evidence for the existence of ribosomes in axons. Technical advances in recent years have overcome these difficulties, enabling the collection of pure axons in vitro [6,7], the use of sophisticated RNA molecular analysis (transcriptomics and translatomics) [8–10] and the acquisition of ultrastructural evidence of ribosome localization in axons [9,11,12,13]. As a consequence, evidence now abounds that thousands of diverse sets of mRNAs reside and are translated in axons of both central nervous system (CNS) and peripheral nervous system (PNS) neurons. However, the exact contribution of axonal translation to function in vivo has been slow to emerge due to the scarcity of approaches that enable precise and controlled inhibition of protein synthesis in axons without affecting cell bodies. The first in vivo experiment where the axonal translation of a specific mRNA was blocked was done in the Xenopus vertebrate visual system [14]. Remarkably, without the translation of a specific intermediate filament protein (Lamin B2), the retinal axons degenerated; hence, the notion that LPS was needed for axon maintenance was born. It is now known that the axonal transcriptome consists of several groups of mRNAs with related functions, which are bound by particular RNA-binding proteins (RBPs) [15]. Meanwhile, research on proteins associated with neurodegenerative diseases has identified an increasing number of disease-associated RBPs, such as Fused in Sarcoma (FUS) and Survival of Motor Neuron (SMN) [16,17,18], providing a parallel strand of evidence linking axon health to RNA regulation. The role of four of these disease-associated RBPs, namely FUS, SMN, Fragile-X Mental Retardation Protein (FMRP), and TAR DNA-binding protein 43 (TDP-43), in local translation in axons and dendrites has recently been reviewed [19]. Here, we discuss the intertwining strands of research on axonal LPS and RBP dysregulation, and in particular, explore the relevance of their combined findings to neurological disorders. We focus on neurological disorders with genetic components, examining to what extent the genetic alterations associated with these diseases (in RBPs as well as other proteins) support a causative role of LPS in pathogenesis or disease progression.
Long-term neural networks rely on cellular specializations
In this section, we briefly examine some specialized features of neurons that underpin neural network assembly and function, particularly the subcellular processes crucial for the in vivo development and maintenance of neuronal processes – dendrites and axons – which, collectively, we refer to here as ‘neurites’. In subsequent sections, we discuss how some of these requirements are met by LPS.
The formation of a large number of synaptic connections between cells with cell bodies that may be far apart requires neurons to be exceptionally structurally and functionally polarized. The average human neocortical neuron forms around seven thousand different synapses with multiple different cells [20], and each synaptic cleft has to be narrow enough to allow rapid and specific signal transmission relying on neurotransmitter diffusion, which results in a breadth of around 20 nm in the central nervous system [21]. Such spatial organization can only be possible if neurons are morphologically polarized: neurons extend long and sometimes branched axons towards the soma or highly branched dendrites of recipient neurons. Axons in particular can reach great lengths, with the longest in the human body being those of motor neurons (up to one meter in length). This length has two further consequences: it limits the speed of macromolecule exchange between axon terminals and the soma, and it places distal parts of axons in different local environments than the soma. Therefore, axons require (i) an efficient active transport mechanism to achieve a stable supply of locally required factors (including mRNAs, proteins, and organelles), which must function efficiently in the spatially confined environment of elongated axon. In practice, the fastest axonal transport mechanisms can reach speeds of around 400 mm/day [22], which is much faster than passive diffusion (especially for molecules with diameters of more than 40 nm, for which the diffusion coefficient drops below 1 μm2/s in nerve cytoplasm [23]). Furthermore, as distal axons can experience very different stimuli than the soma, they need (ii) the ability to independently remodel or change their macromolecular components.
To achieve almost immediate information relay from dendrites to axons at a speed beyond what can be reached by active transport, neurons are electrically excitable. In order for information to be transferred between cells, even fast axonal transport is insufficient: when a hand is withdrawn reflexively from a hot surface, for instance, a signal must travel from the hand to the spinal cord and back to relevant muscles, which is well over a meter of total path length and so would take several days by active transport [22]. In contrast, the unidirectional transmission of changes in membrane potential (action potentials) along axons can reach speeds of over 100 m/s [24], and so can accomplish this information transfer in well under a second. However, excitability comes at an energetic cost. The restoration of dissipated ion gradients following action potentials accounts for the majority of the large neuronal energy expenditure on signalling [25]: it has been estimated that three-quarters of neuronal energy consumption are spent on signalling [26], which is not trivial, considering the central nervous system accounts for 20% of the human body’s energy consumption, but for only 2% of its weight [27]. In addition to membrane potential management, this high energy consumption is accounted for by vesicle recycling, neurotransmitter synthesis, and axonal transport [28]. Therefore, another requirement for neuronal function arises, namely that (iii) high energy consumption must be supported throughout neurites. This requires the continual presence of a population of mitochondria in neurites.
In order for neuronal networks to learn, they must be able to adapt the nature of connections according to various stimuli, as changes in synaptic strength (plasticity) are thought to be important for (efficient) learning and memory [2,29]. This is one of the ways in which neuritic (sub)compartments need to be able to locally change their macromolecular components (ii): as part of synaptic plasticity, components should be changed to alter local synaptic function in response to changes in activity. Furthermore, neurons should be able to add new connections, reduce unused connections, and remove damaged connections. Therefore, synaptic structural plasticity calls for (iv) tightly regulated local ‘death-like’ pathways to remove synapses and even whole axons, as well as for mechanisms to add new synapses.
Lastly, for neuronal networks to store memories long-term, neurons have to be resilient against a range of insults, in order to sustain neural connectivity throughout the organism’s life span. Consequently, neurons are long-lived cells, particularly in comparison with other cell types, such as the intestinal epithelium or red blood cells, which are frequently ‘worn out’ and replenished by reservoirs of stem cells. However, neurons cannot be similarly replaced, as new neurons could not readily integrate into the neuronal network without loss of the information encoded by pre-existing synaptic connections. Notably, adult neurogenesis and subsequent integration of newly formed neurons do in fact occur in the mammalian brain, but only in the olfactory bulb and dentate granule cell layer of the hippocampus, in a process that is modulated by circuit activity [30]. Therefore, the following is required to appropriately maintain neuronal networks: (v) neuronal stress responses should adopt anti-apoptotic strategies to enhance stress tolerance and to avoid cell death, and (vi) neurons must habituate to and mitigate cellular damage accumulated during aging. These unique stress responses have to affect local processes in neurites, including local replenishment and activation of anti-stress factors that involve LPS and post-translational modifications (PTMs), which also become altered with age.
LPS supports multiple axonal functions
LPS enables neurites to autonomously remodel their proteome in response to local stimuli, which means it can provide a way to address some of the requirements outlined above. This is particularly true for the axon [31], which is the longest neurite and contains the largest cytoplasmic volume of any compartment of the mature neuron [32,33].
LPS can be useful to maintain local axonal proteome homeostasis, but its products may also have unique properties that carry functional information. These can arise from their association with local components of signalling cascades or from unique post-translational modifications [34]. For instance, a study in cultured primary hippocampal neurons showed locally produced arginyltransferase 1 (ATE) in the growth cone arginylated adjacent β-actin proteins that were also locally synthesized, and that the arginylation of β-actin in neurites is important for growth cone area size (spreading) and neurite outgrowth [35].
A wide range of mRNAs has been demonstrated to be locally translated, which contribute to a variety of subcellular functions and neuronal specializations beyond synaptic plasticity. In the axon, locally synthesized proteins have been shown to contribute to axon navigation, maintenance and regeneration [36]. Specifically, LPS regulates a range of essential processes in the axon [31], including vesicle trafficking, cytoskeletal remodelling and mitochondrial integrity [37].
Notably, the translatome is not static, which allows it to support a range of functions. Genome-wide analyses have revealed that the axonal translatome changes during the course of development, in step with evolving axon function and behaviour. In mouse retinal ganglion cell (RGC) axons in vivo, for example, the mRNAs translated in early growth stages are associated with axon elongation, followed by branching then synaptogenesis [9]. The context-dependent composition of the axonal translatome is further demonstrated by functional enrichment Gene Ontology (GO) and KEGG pathway analyses of published datasets describing the abundant localized mRNAs and locally synthesized proteins in axons at different developmental stages in different neuronal types [8–10,38–43] (Fig. 1). mRNAs of ribosomal proteins are highly enriched in axons of all stages, as reported by several studies [9,10,40,44]. However, only a subset is bound to ribosomes, according to an axon-TRAP study, and their translation rates decline synchronously after the axonal branching stage [9]. It has been further demonstrated that several ribosomal proteins, particularly the surface components of each subunit, are locally synthesized upon cue stimulation and incorporated on-site into axonal ribosomes [44]. The functional role of this axonal ribosome remodelling is not yet known, but it could extend the lifetime of ribosomes and, perhaps most intriguingly, could ‘tune’ them to translate specific mRNAs [45].
Figure 1.
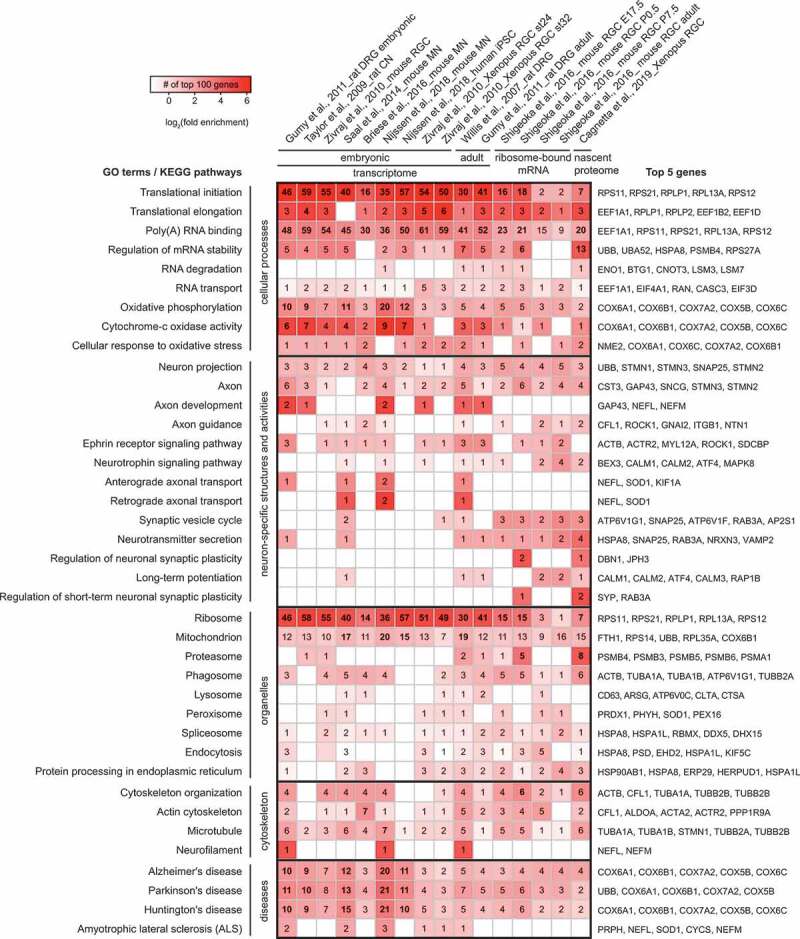
Selective GO terms and KEGG pathways in most abundant axonal transcripts, ribosome-bound mRNAs and nascent proteins
Top 100 annotated genes with most axonal reads in 16 datasets from 9 independent studies (4 microarray, 3 RNA-Seq, 1 Ribo-Seq and 1 nascent proteomic studies) are included in this analysis. The heat map shows the enrichment of GO terms and KEGG pathways relevant to the discussion in this review. The colours of the heat map represent the log2 value of the fold enrichment. The numbers on the heat map indicate the total number of genes among the top 100 genes from each dataset associated with the GO term/KEGG pathway and those with a Benjamini-Hochberg value <0.05 are shown in bold. Human orthologs of the top 2–5 genes associated with each GO/KEGG category ranked by their appearance frequency are indicated next to each row. The enrichment analysis was carried out with DAVID v6.8.
In addition to ribosomal proteins, axonal localization and translation of mRNAs encoding other proteins with roles in LPS are also revealed by the analyses, including those regulating mRNA metabolism (e.g., ubiquitin and proteasome components), those transporting and localizing mRNA (e.g., cytoskeletal proteins and RBPs), those forming part of the translation machinery (e.g., eukaryotic initiation and elongation factors), and those required for energy supply (e.g., mitochondrial proteins). In addition, though mRNAs encoding synaptic components are not strongly enriched, these proteins, including synaptosomal-associated protein 25 (SNAP25) and vesicle-associated membrane protein 2 (VAMP2), are more abundant in the local translatome [9,39]. Furthermore, some components of the oxidative stress response may be locally synthesized to respond to local perturbations of energy supply and mitochondrial function.
Besides housekeeping proteins produced via basal translation (Fig. 1), the stimulus-dependent translatome is also a large constituent of axonal proteome. Stimulus-dependent LPS contributes to a range of axonal functions: it mediates axon guidance and arborization, supports axon maintenance and survival, regulates presynapse formation and synaptic plasticity, and aids the response to stress and injury [31,46,47]. During axon pathfinding in development, asymmetric localization and translation of β-actin mRNAs in the growth cone can be observed in cultured Xenopus RGCs upon 5–10 min gradient stimulation with the guidance cue Netrin-1 or brain-derived neurotrophic factor (BDNF), which facilitates growth cone turning [48,49]. As detected by metabolic labelling, 1-h cue stimulation of developing RGC axons induced a 10–80% increase in the amount of locally synthesized proteins [14]. A recent proteomic study of axonal nascent proteome showed that among 1000 proteins detected in isolated axons, approximately 350 proteins were locally synthesized. The translation rate of over 100 of them changed significantly upon guidance cue stimulation and the pattern of changes varied greatly depending on the types of the cues and lengths of stimulation [39]. In mature neurons, LPS can provide a basis for heterogeneity of synapses made by the same neuron: for instance, LPS enables the activity-mediated upregulation of the key presynaptic kinase CamKII in the Drosophila larval neuromuscular junction [50]. In the model system of Aplysia sensory-motor neuron synapses, presynaptic LPS has been shown to support synaptic plasticity: branch-specific long-term facilitation in response to localized exposure of serotonin requires presynaptic LPS [51], for instance of the peptide neurotransmitter sensorin [52]. Moreover, different aversive stimuli, including acute injury or chronic diseases, elicit distinct landscapes of the local translatome, opening up new opportunities to discover therapeutic targets [8,42,47,53].
RBP dysfunction in neurological disorders indicates compromised LPS may be causative
Considering the range of critical processes in which LPS is involved in neurons, including in axons, it is not surprising that it is disturbed in multiple neurological disorders, and that this disturbance may be part of the pathomechanism(s) of these disorders. Indeed, a bioinformatics search among the highly abundant axonally localized or translated mRNAs identifies a number of genes associated with various neurological disorders (Fig. 2), including amyloid β precursor protein (APP) and ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) related to Alzheimer’s disease (AD) and Parkinson’s disease (PD) susceptibility [9,39,43]. ‘Neurological disorder’ is a broad term referring to any condition in which the function of CNS and/or PNS deteriorates. It covers a wide range of diseases, which place a significant burden on patients and society: neurodevelopmental disorders such as Fragile X syndrome (FXS), autism spectrum disorder (ASD), and schizophrenia, neurodegenerative disorders like AD, PD, and amyotrophic lateral sclerosis (ALS), and acquired disorders, addictions, and injury- or pathogen-induced disorders. Familial neurological disorders are associated with highly or completely penetrant mutations, which can be used not only to develop in vitro or in vivo disease models, but also link the disease to perturbations of certain cellular processes.
Figure 2.

Disease-associated genes enriched in axonal transcriptomes and translatomes
A table shows human orthologs of axonally enriched transcripts or nascent proteins dysregulated in common neurodegenerative or neurodevelopmental diseases among the 100 most abundant genes in each dataset. Dysfunction of the indicated genes either causes or increases susceptibility to the disease, based on the corresponding OMIM disease entries.
Interestingly, structural and functional alterations of RBPs are implicated in neurodevelopmental and neurodegenerative disorders, which strongly points to dysregulation of gene expression as a key feature of diseases. For instance, FXS is caused by loss-of-function mutations in the neuronal RBP FMRP [54]. However, for many neurological disorders in which RBPs can be found mutated, the genetic basis of familial disease variants is less readily interpreted than for FXS.
The case of ALS illustrates the two main reasons why genetic predisposition of a disease does not always readily lead to a hypothesis of pathogenesis [55]. Firstly, the genetic basis of familial ALS (fALS) is heterogeneous. Mutations of genes encoding the RBPs FUS and TDP-43 are prevalent among fALS cases. Since RBPs are key to localization of mRNAs and the regulation of translation, their altered function has, in some cases, been linked to the perturbation of LPS in axons [18,56–59]. However, highly penetrant mutations have also been discovered in other genes, such as in those encoding the following proteins: C9orf72 [60,61], the antioxidant enzyme superoxide dismutase (SOD1) [62], the motor protein kinesin heavy chain isoform 5A (KIF5A) [63], tubulin isoform alpha 4A (TUBA4A) [64], and the actin-associated protein profilin 1 [65]. Secondly, mutant proteins can be expressed in (or lost from) a range of cell types, but the disease phenotype appears restricted to nervous tissues or even certain types of neurons. For instance, FUS and TDP-43 are ubiquitously expressed in all cells [66,67], but their mutations do not affect all tissues or even all neuronal subtypes. Though motor neurons are primarily affected, the extent of degeneration of different motor neuron subtypes varies greatly, with for instance spinal cord motor neurons degenerating relatively early in disease and ocular motor neurons remaining unaffected up to the end stage of the disease [68,69]. However, it should be noted that though the diagnosis of ALS is based on motor symptoms, ALS is increasingly recognized to be associated with a range of non-motor phenotypes in patients: for instance, up to half of ALS patients display some form of cognitive impairment, with 15% meeting the criteria for frontotemporal dementia (FTD) [70]. In fact, ALS shares many pathological features as well as genetic risk factors with frontotemporal dementia (FTD), which like ALS is associated with mutations in and aggregates of TDP-43, and these diseases are considered to be part of the same ‘disease continuum’ of TDP-43 proteinopathies [69]. In such co-occurring ALS/FTD, non-motor neuronal subtypes are also affected: TDP-43 inclusions have been identified in the cortex and hippocampus of both sporadic and C9orf72-associated ALS/FTD patients [71].
Then, the postulation that RBP dysfunction can be causative in multiple neurological disorders, such as ALS, leaves two unanswered questions. Firstly, why do certain mutations in widely expressed RBPs such as FUS exert particularly strong effects on neurons? Secondly, why does RBP dysfunction result in the same phenotype as mutations of other disease-related proteins, such as cytoskeleton-associated proteins?
To begin to answer these questions, the functions of RBPs in neurons require further consideration. Typically, an individual RBP is functionally versatile and some of these functions may be unique to neurons (e.g., due to the presence of neuronally expressed interaction partners). Alternatively, the RBP’s functions may be exceptionally important in neurons. Neuropathology caused by RBP loss-of-function mutations indicates the protein performs an essential role on which neurons rely, whereas for a gain-of-function mutation (such as aggregation), the neuron would be particularly sensitive to this effect. The latter is best illustrated by proposed pathogenesis of neurodegenerative disorders: accumulation of protein deposits containing RBPs is a hallmark of multiple neurodegenerative disorders, such as FUS and TDP-43 aggregates in ALS [72,73,74,75]. Meanwhile, loss-of-function models have also been put forward: functional loss of FUS may affect mRNA stability at dendritic spines and cause axonal transport defects [76]. Therefore, there has been a long debate whether the pathological aggregate is in itself toxic, or whether loss of RBP function is detrimental. However, recent advances in genetic and pathophysiological studies suggest the two theories are not mutually exclusive and their distinction may be blurred, as heterogenous genetics can sometimes converge to shared downstream effects observed in a disease, such as impaired synaptic connectivity.
In the following sections, we provide a summary of evidence and our speculations on how functional alteration of RBPs and other disease-associated proteins may lead to LPS dysregulation in neurites. Using key cellular processes in axonal compartments as examples, we examine potential links between aberrant LPS and observed phenotypes of common neurological disorders, and propose that LPS may serve as a crucial mediator in neuronal health and viability.
Polarity and axonal trafficking
The length and narrowness of axons create specific physical challenges for the transport of cargos, including mRNAs and translational machinery as well as organelles and proteins. Firstly, the narrowness of axons largely limits the distribution of materials by simple diffusion, as it affects flow – the diameters (calibres) of adult axons are typically between 0.1–1 µm for unmyelinated axons [77]. According to Stokes’ law, the opposing force impeding an object’s motion in a viscous fluid is proportional to the object’s size, the fluidic viscosity, and the flow velocity. However, boundary effects (a reduction in flow velocity as fluids approach the wall) play a much more significant role in a narrow cylindrical geometry than a large space (such as a cell body). Therefore, moving cargos encounter greater opposing forces within axons than within the soma, where most of the molecules are relatively far from the plasma membrane [78]. This is best demonstrated by comparing the speed of fast axonal transport (2–5 µm/s) [79] and diffusion coefficient of a GFP molecule in the cytoplasm (7.7–126 µm2/s) [80,81,82]. The second challenge to axonal cargo trafficking is posed by local macromolecular crowding in the axoplasm, which is packed with a dense cytoskeletal network and both static and moving cargos. For instance, membrane-bound and membraneless organelles in axons range from 100 nm to 1–2 µm in diameter, which is close to the average axon calibre of around 1 µm [77]. Local crowded regions in axons may act as physical barriers, resulting in a decrease of cargo velocity or complete stalling.
As a consequence of this limited diffusion, neurons have evolved unique strategies to facilitate the interlinked processes of RNA localization, local translation and axonal transport. These include the establishment of a robust scaffold to maintain axon morphology, and of an active transport network that can counteract drag forces and respond to changes in crowdedness [83,84]. Cytoskeletal elements, motor proteins and adaptor proteins together form the basis of these structures. In addition, RBPs are key for axonal RNA transport through interaction with motor and adaptor proteins. It is now clear that disruption of axonal transport is closely associated with multiple neurological disorders [85,86,87], as are structural and functional impairments of the main axonal cytoskeletal elements [83,87,88].
In this section, we discuss some of the cytoskeleton-related processes compromised in diseased neurons, dysregulation of which results in errors in mRNA localization and therefore LPS (Fig. 3). Interestingly, the interaction between LPS and axonal transport can at times be bidirectional, as a number of studies have revealed axonal localization of mRNAs encoding cytoskeletal building blocks (i.e., neurofilament proteins, β-actin, tubulins) and their associated proteins (e.g., RhoA, cofilin, tau), some of which have been shown to be locally translated [89]. Impaired local synthesis of these cytoskeletal components and modulators would be expected to lead to disrupted axonal trafficking and/or disease progression. However, the concept of a direct link between axonal expression of cytoskeletal proteins and pathogenesis of neurological disorders remains largely hypothetical. To explore this hypothesis, we will next highlight some cytoskeletal components suggested to be locally synthesized.
Figure 3.

Mechanisms to sustain axonal transport related to LPS. Neurofilaments and membrane-associated periodic skeleton regulate axon structure (upper segment); microtubule and motor protein-based active transport maintains cargo trafficking (middle segment); modulation of axonal RBP, RNA and organelle density controls local macromolecular crowdedness (lower segment). Perturbation of these processes can result in defective axonal trafficking, as indicated by pink axon segments
The axonal cytoskeleton maintains axon structure and organization
To maintain structural and functional polarity and sustain transport of cargos of various sizes, it is important that axons are mechanically resilient: axon shafts do not collapse around their circumferences or break during axon elongation or upon deformation by surrounding cells and tissues [90]. The axon diameter is mainly regulated by neurofilaments and actin filaments [91]. Currently, the correlation between axon calibre and neuronal vulnerability in neurodegeneration is still controversial [92], but retaining axonal radial structure and elasticity is undoubtedly important for intra-axonal trafficking and therefore LPS.
Neurofilaments are a type of intermediate filaments most abundant in axon shafts, which structure and organize axons in several ways. Firstly, they are a major determinant of axon calibre, particularly for large axons: a large axon diameter is often associated with a large number of axonal neurofilaments and increased inter-neurofilament spacing [93,94], and loss of neurofilaments results in a reduction in axon calibre and conduction velocity, leading to impairments in axon development, survival, and regeneration [95]. Secondly, neurofilaments interact with axonal organelles and cytoskeletal components. For instance, neurofilaments serve as scaffolds for docking and positioning of endoplasmic reticulum (ER), endosomes, mitochondria and synaptic vesicles in axons [96]. One study in cultured dorsal root ganglion (DRG) neurons demonstrated that Charcot-Marie-Tooth disease (CMT)-associated mutations of the low-molecular-weight neurofilament protein (NF-L) decreased mitochondrial lengths and disrupted mitochondrial fusion and movement in axons [97].
The majority of axonal neurofilament subunits are synthesized in the soma and subsequently transported into axons along microtubules [98]. Accumulation of neurofilaments in the cell bodies and proximal axons, due to an imbalanced expression of neurofilament subunits, altered PTMs of neurofilament proteins, or impaired axon trafficking has been identified as a common feature in multiple neurological disorders, including CMT, ALS, PD and AD [99,100]. There is evidence that mRNAs of neurofilament proteins reside in axons [101,102] and are also locally translated there [103,104]. However, the functions of these locally synthesized proteins are yet to be discovered.
Dynamic and diverse axonal actin structures play important roles throughout development and adulthood, from axon specification, initiation, elongation, guidance, branching to the development of presynaptic terminals [105]. In developing axons, actin filaments are enriched in the peripheral region of growth cones, where they form dynamic lamellipodia and filopodia to facilitate axonal pathfinding [106]. Upon target arrival, actin polymerization is also required for axon arborization [107]. As first observed by super-resolution microscopy, actin is organized in ring structures underneath the plasma membrane in mature axons, which are connected and evenly spaced by spectrin heterotetramers [108]. Such actin ring-spectrin structures together with other interacting proteins form membrane-associated periodic skeletons to support axon architecture by conferring elasticity and stiffness [109]. At the presynapse, actin filaments accumulate at the active zone and associate with synaptic vesicles to promote active zone formation and to regulate synaptic vesicle clustering [110,111]. Conceivably, dysregulation of actin localization and organization can exert a detrimental effect on axon development and survival. Missense mutations in one of the two neuronal actin isoforms, β-actin and γ-actin, have been reported in neurological diseases, including juvenile-onset dystonia [112], late-onset sensory-neural deafness [113] and Baraitser–Winter syndrome [114].
It has been well established that locally synthesized β-actin proteins function in axon steering and branching in developing neurons [48,49,115,116], but the extent of their involvement in mature axons and disease-affected neurons remains to be explored. Early studies demonstrated that whilst β-actin mRNA localizes to axons, γ-actin mRNA is restricted to the soma in developing cortical and adult DRG neurons in cultures [104,117]. However, a recent piece of work challenged this view by showing the localization of γ-actin mRNA in developing cultured motor axons using qRT-PCR and fluorescence in situ hybridization [118]. In the same study, local translation of γ-actin mRNA in growth cones and branch points was also demonstrated by a FRAP assay using reporter constructs [118], suggesting that axonally synthesized actin isoforms may differ between different types of neurons. In addition to actin proteins, actin-associated proteins, such as α-spectrin, were identified in an axonal translatome of mouse retinal neurons [9], suggesting LPS could be involved in the dynamic regulation of axonal actin organization. This could help to provide structural stability and plasticity during axon development and maintenance.
Microtubule-based transport is critical to axonal trafficking
The microtubule cytoskeleton is critical for long-range transport in axons, and therefore for LPS. In this transport system, anterogradely and retrogradely transported cargos, including mRNAs and translational machinery components, are loaded onto motor proteins, which move along polarized microtubule tracks. Conventionally, axonal trafficking is considered to feature two distinct transport modes, namely fast and slow [119]. Fast axonal transport (0.5–5 µm/s) mainly carries organelles and ribonucleoprotein (RNP) granules [79], including complexes carrying disease-related proteins (e.g. APP, Huntingtin) [120,121], whilst slow axonal transport (0.01–0.001 µm/s) carries cytoskeletal components, such as neurofilament proteins [122]. Both modes of axonal transport are carried out by the same microtubule-based motor proteins, anterogradely-moving kinesins and retrogradely-moving dynein. The difference in their average velocity results from the occurrence of prolonged pauses in movement during slow axonal transport [123], which is modulated by dynamic attachment of multiple motors to the cargo [124]. Increasing evidence suggests that fast axonal transport defects are more common in neurological disease-affected neurons, possibly as a result of mutations in proteins mediating fast axonal transport, or trafficking perturbation in cargos undergoing fast axonal transport [125]. Besides determining the speed, cargo attachment to opposing motors allows them to undergo bidirectional transport and frequently change direction, which requires coordination of motor activities, including the duration of individual motor attachment and run lengths in either direction [126,127]. Given the role of axonal transport in delivering structural components, organelles and survival signals, it is not surprising that mutations in motor proteins and their cofactors cause a wide range of neuropathies [128].
Mutations and aberrant post-translational modifications in tubulins lead to multiple neurodevelopmental and neurodegenerative diseases, including ASD, polymicrogyria, ALS, and AD [129,130,131], which could potentially be partly due to errors in local synthesis of these proteins. mRNAs encoding tubulins have been detected in axons in several transcriptomic studies (Fig. 1) [10,40,41]. Moreover, radioactive labelling and proteomic studies have identified several locally synthesized tubulin proteins [34,132,133]. Although these form <1% of the total axonal β-tubulin pool, according to [35S]-Met radioactive capturing analysis [132], this does not disprove the importance of axon-derived tubulins [131], as different tubulin isoforms [134] or PTMs [135] may be enriched in the somatically and axonally synthesized pools, resulting in distinct functionalities. Inhibiting the local synthesis of β2B-tubulin, which mainly localized to the growth cone periphery, resulted in growth cone collapse in cultured DRG neurons [136]. Mutations in β2B-tubulin gene were found in patients diagnosed with polymicrogyria [137,138], but the extent to which axonally expressed β2B-tubulin contributes to the disease needs further research.
Microtubule-associated proteins actively regulate the stability and dynamics of microtubules in axons, and their functional impairments often lead to axonopathy. One of the most extensively studied axonal microtubule-associated proteins is tau, which is important for microtubule stability and implicated in disease [139]. A range of neurological disorders (termed ‘tauopathies’) is characterized by deposition of hyperphosphorylated tau protein in the brain, including AD and frontotemporal dementia (FTD). In axons, tau is reported to facilitate the organization of distal microtubules, which is important for axon trafficking, outgrowth and navigation [140,141]. tau mRNA contains an axonal localization signal and is locally translated [142,143], but the phosphorylation level of axonally synthesized tau is yet to be determined. Intriguingly, functional and pathogenic heterogeneity exists between the six tau splicing isoforms [144,145]. Therefore, characterization of the isoform-specific role of axon-derived tau would provide insights into its functional significance, which is particularly relevant in disease models. In mature healthy neurons, tau proteins are almost exclusively localized to axons, but somatodendritic tau inclusions are frequently found in AD-affected neurons [146]. It is worth noting that, although localized tau synthesis is restricted to axonal compartments, tau mRNA is also localized to dendritic spines. Activation of glutamate receptors triggers local synthesis and hyperphosphorylation of tau in dendrites, leading to somatodendritic accumulation of hyperphosphorylated tau [147]. This has been shown to be a key step in the initiation of tauopathies [148], indicating the importance of correct tau mRNA localization. Besides tau, another axonally synthesized microtubule-associated protein ‘mitogen-activated protein kinase kinase 7ʹ (MKK7) has also been shown to promote microtubule bundling and neurite elongation by correctly positioning Jun ‘N-terminal kinase’ (JNK) signalling in axon shafts [149].
There is also some evidence that LPS of motor proteins contributes to or regulates axonal transport, which further establishes a link between the two processes. Detection of kinesin mRNAs in giant squid axons and dynein light chain mRNAs in rodent axons have been reported over two decades ago [150; 151] and recent axon-TRAP and proteomics-based translatomic studies subsequently revealed that many of the motor protein mRNAs are actively translated, including kinesin-1 proteins (KIF5A, 5B and 5 C) and a kinesin-3 protein KIF1A [9,39,152]. Of these, KIF5A localizes predominantly to axons rather than dendrites in cultured hippocampal cells [153], and KIF1A is a major axonal motor responsible for long-distance transport of synaptic vesicle precursors and neurotrophin-containing dense core vesicles [154,155]. Mutations in or hyperactivation of KIF1A are associated with neurodegenerative disorders, such as hereditary sensory and autonomic Neuropathy Type 2 and hereditary spastic paraplegia [156,157,158]. It will be of interest to determine the role of axonally synthesized kinesins and their link to kinesin-related diseases. In addition, local on-demand production of dynein cofactors has been demonstrated to mediate retrograde transport in healthy and disease-affected axons. Two dynein cofactors are differentially translated upon nerve growth factor (NGF) stimulation or withdrawal in axonal compartments: Lis1, a force-generating component in the dynein complex, and p150Glued, one of the eleven subunits of dynactin. Therefore, a local translation-based mechanism to regulate stimulus-specific retrograde trafficking has been put forward [159].
Neuropathy-related RNP condensation regulates axonal mRNA transport and localization
The mechanism of axonal mRNA localization to support LPS is evolutionally conserved in different cells and organisms: loading of mRNAs onto motor proteins is facilitated by RBPs that recognize localization elements often present at the 3ʹUTR [160,161,162]. Structurally, a majority of RBPs consist of RNA-recognition motifs (RRMs) and intrinsically disordered domains (IDDs), the latter being regions with low sequence complexity and no fixed three-dimensional structure. Gene ontology annotations reveal that a third of human IDD-containing proteins function in RNA-binding [163], illustrating heavy involvement of IDDs in RBP functionalities. IDDs together with RRMs allow RBPs to flexibly and multivalently interact with multiple protein/RNA targets to reversibly form membraneless organelles or granules (a liquid-liquid phase separation, LLPS). This can locally concentrate granule constituents and hence promote physical interactions between these molecules [164]. The strength of their interactions is sensitive to temperature, pH and salt concentration [165], and can be further fine-tuned by various protein PTMs [166], providing additional layers of regulation. However, these useful and unique properties of RBPs are the same feature responsible for their role in the development of neurodegenerative disease. Indeed, structural and functional alterations of a subset of RBPs are over-represented in patients diagnosed with ALS, FTD and AD [167]. When intracellular phase transitions become dysregulated, resulting in hyper-stable RNP granules, proteins and RNA could become irreversibly trapped within the granules, preventing them from performing normal functions, including LPS. Despite being regarded as pathological hallmarks in neurodegenerative diseases, it is under debate whether RNP depositions on their own are pathogenic. It has been proposed that they instead serve as a reporter for the pathogenic dysregulation of cellular processes that often precedes aggregate formation [168]. Therefore, rather than focusing on approaches to ‘dissolve’ these aggregates, it may be more relevant to identify the dysregulated processes that promote hyperstable RNP granule formation.
Previous studies have demonstrated that RBP phase transitions are sensitive to and partly regulated by local protein concentration, RNA concentration and conformation, PTMs, and the availability of chaperones and other binding partners [169]. Consequently, aberrant homoeostasis of any of these factors may enhance the tendency for pathological aggregates to form and persist during disease progression. For instance, RBP:RNA ratio, RNA lengths and secondary structures, and their RBP binding specificity jointly determine the predominant material states and dynamics of RNP granules [170]. As a result, the presence of sub-optimal amounts and species of axonal RNAs may reduce axonal trafficking, exacerbating the disruption of local homeostasis in diseased axons in a negative feedback loop. In addition, the link between aberrant RBP PTMs and neurological disorders has also been recently established. PTMs can effectively alter the strength of intra- and intermolecular interactions by modifying electrostatic charges of amino acids, hydrophobicity and protein structures, for instance, serine/threonine/tyrosine phosphorylation, arginine methylation, and arginine citrullination. Therefore, PTMs are powerful modulators of RBP LLPS and dynamic RNP granule regulation [166], which can be deregulated in disease. For instance, FUS inclusions with unmethylated arginine have been found in FTD patient post-mortem tissue [171,172]. Arginine hypomethylation promotes the formation of cytoplasmic FUS inclusions, and axons expressing hypomethylated FUS showed an increased number of axonal FUS-containing granules accompanied by compromised LPS [58]. This study also showed that the reduced LPS could be effectively restored upon overexpression of a FUS chaperone, Transportin-1, which imports FUS from the cytoplasm into the nucleus and represses FUS aggregate formation [173,174,175]. Changes of LPS in response to FUS hypomethylation and the level of its phase modulator support a close link between PTMs, chaperones, phase separation and LPS in axons.
The neuronal context of spatially confined axonal compartments packed with high density of cytoskeleton and organelles and unique modes of RBP transport may further enhance pathological RNP assembly. Under these conditions, protein and RNA may be concentrated locally, elevating local axoplasmic viscosity and influencing RBP phase behaviour [84]. This can occur in several ways: 1) a regional disruption of axonal transport in response to local stimuli or insults; 2) a burst of LPS, especially of IDD-containing RBPs identified as highly locally translated in axonal translatomic studies, including FUS and hnRNPs [39]; 3) active recruitment of proteins and RNAs by membrane-bound organelles. Recent evidence showed that a proportion of RNP granules ‘hitchhike’ on membrane-bound organelles, such as peroxisomes, mitochondria and endosomes, acting as vehicles for RNP granule trafficking and localization [176,177,178], in contrast to the conventional view that RNP granules undergo long-range trafficking through direct tethering to motor proteins. In vertebrate axons, late endosomes act as platforms to recruit mRNAs and translation machinery to support LPS [179]. Disruption of this process can be disease-causative: CMT2B-associated mutations of Rab7a attenuate LPS in axons, compromise mitochondrial function and eventually result in axon degeneration. In addition, ALS-associated mutations of an adaptor between lysosomes and RNP granules, annexin A11, impair its intra-axonal phase-transitioning ability and its tethering between RNP granules and lysosomes, resulting in perturbed RNA localization in axons [180].
These observations open up an exciting direction for future research into how axons organize local translation into micro-domains and regulate translation specificity in these sub-compartments. As a main driving force for RNP granule formation, LLPS may also contribute to the establishment and stabilization of organelle-RNP compartments, as demonstrated by annexin A11 tethered to lysosomes [180]. The role of such molecular anchors remains to be explored for other organelles. Furthermore, it has been reported that translation only takes place on the surface of late endosomes in Xenopus RGC axons, although both early and late endosomes associate with key components of translational machinery, including mRNA, RBPs and ribosomes [179]. This leads to the question of what activates translation on these RNP-bound organelle platforms. The physical location of the organelles may be a key factor: organelles and RNPs are highly enriched at branch points and axon terminals, where high levels of translation activity often occur [116,181]. It is possible that the local density of organelles and recruited molecules concentrates components required by translation or alters the physical states of the surrounding micro-environment to promote translation. Alternatively, translation activity could be modulated by certain regulatory elements associated with individual organelles, such as miRNAs [182]. Another open question lies in the control of mRNA localization and translation specificity on platforms; recruiting specific RBPs and the subset of mRNAs bound to them could be a way to define the identity of a translation hub. Finally, whether the disruption of micro-domain arrangement and regulation is prevalent in neurological disease-affected neurons remains to be investigated.
Establishment of axon architecture and connectivity
In order for appropriate connectivity between neurons and target cells to be generated and maintained, axonal branches and even whole neurons are at times remodelled. To establish and specify their innervation fields, developing axons from terminal branches with diverse lengths, density and complexity, allowing them to synapse with multiple target cells simultaneously, with excess synapses being pruned at later stages [183]. Local translation is known to have a role in branching of axons. Data from chick embryonic sensory neurons suggest that NGF promotes axon branching by modulating the actin cytoskeleton, in part via stimulation of LPS through phosphoinositide 3 kinase (PI3K) signalling [181]. Furthermore, RNA granules dock at the bases of new branches and invade stable branches, and local synthesis of β-actin at these sites is important for axon arbour dynamics [116]. There is also some preliminary evidence that presynaptic LPS is important in the pruning stage of development, which can intersect with its role in survival signalling. For example, in degeneration-like pruning in the PNS, competition for neurotrophic support is an important driving force [184], and neurotrophin-stimulated LPS is important for this response [185].
In neurological disorders, branching and/or pruning are often compromised. This is perhaps intuitive for neurodevelopmental disorders such as for FXS, but more recent findings imply axonal structure may also be affected in neurodegenerative diseases. The association of these defects with RBPs has been demonstrated for several such disorders, which can to some extent be linked to LPS.
RBP dysregulation compromises axon branching and pruning in neurodevelopmental disorders
In FXS, a clear link between RBP dysregulation and compromised neuronal connectivity exists, which makes it an important case study. We briefly discuss this link, and then outline the evidence that FMRP affects presynaptic translation of proteins important for axonal structure and function. We then indicate the extent to which similar processes are implied in other neurodevelopmental disorders, namely ASD and epilepsy.
In FXS, loss of function of the RBP FMRP results in defects in synaptic formation and plasticity. It is well-known that dendritic spine structure is altered in FXS, with more but longer, potentially immature spines being observed [186]. dfmr (fmrp1 homologue) knockout in Drosophila results in axonal overgrowth and overbranching, which compromises synapse formation [187]. However, decreased connectivity at certain developmental stages has also been reported in FXS models, along with more ‘diffuse’ axon arbours, with a higher connection density along the barrel borders and reduced connectivity at the centre [188]. This is consistent with a pruning defect [186].
Some of the effects of loss of FMRP function are likely due to regulation of LPS being compromised: FMRP is known to be a negative regulator of translation [189], and several observations suggest it locally regulates translation at synapses [190]. Consistent with it having a functionally important role in regulating LPS, FMRP associates with polyribosomes and disruption of this interaction causes particularly severe disease, via the rare I304N mutation in the ribosome-interacting KH-domain [191]. FMRP-mediated regulation of LPS is known to be important in dendrites, where it influences activity-dependent long-term potentiation. For instance, an imaging study showed knockout of fmr1 prevents an increase in levels of the presynaptic protein CamKIIα upon group I metabotropic glutamate receptor stimulation, which was demonstrated to be protein synthesis-dependent by cycloheximide treatment and presumed to be local due to its ten-minute timescale [192]. However, FMRP is increasingly recognized to be important for regulation of presynaptic translation as well [193]. In particular, FMRP-containing granules are found in a subset of axons, most prominently during synapse formation and pruning [194,195], indicating a possible presynaptic role of FMRP in synapse formation [196]. Notably, this association is not limited to early developmental stages: FMRP-containing granules are also found in a subset of mature mammalian axons (but not dendrites), where they associate with ribosomes as well as (a subset of) FMRP mRNA targets [197].
Several key axonal mRNA targets of FMRP have now been identified, which have a range of functions during different developmental stages. In hippocampal neurons, FMRP has been shown to be involved in the LPS-based response to the guidance cue Sema3A during axon extension, including by promoting the local synthesis of the microtubule-associated protein 1B (MAP1B) [198]. Previously, it had been shown that double knock-out of dfmr and futsch (the Drosophila map1b homologue) could rescue synaptic structural defects in the eye and neuromuscular junction [199]. During presynapse formation in mouse cortical neurons, FMRP negatively regulates local translation of the synaptic vesicle fusion protein Munc18-1, as demonstrated in cultured mouse cortical neuron axons that were physically separated from the soma [200]. In Drosophila, it has been shown that FMRP functions in axon maturation in two distinct ways: it inhibits axon growth during late pupal development, and functions in activity-dependent pruning in emerging adult flies, during which time its activity correlates inversely with levels of the profilin homologue chickadee [201]. Though this link has not been demonstrated to be due to regulation of LPS of chickadee (an actin-remodelling protein), chickadee mRNA has been shown to localize to remodelling Drosophila axons, with its mislocalization resulting in remodelling defects [202].
There are implications that perturbed phase separation of FMRP can occur in FXS, though the link to dysregulated LPS is not yet firmly established. Notably, it has recently been found that only certain splicing isoforms of FMRP reduce axonal arbour complexity when overexpressed [203]. This regulation of arbour complexity does not seem to require the RNA-binding domains, including the KH-domain, but does require an intact nuclear export signal as well as the presence of a phosphorylatable serine that regulates translational suppression in FMRP-associated polyribosomes [203,204]. Instead, the I304N (KH-domain) mutant was found to be more prone to fibril formation, indicating that this mutation may affect translation by deregulating FMRP granule phase state rather than simple loss of function of RNA or ribosome binding [203]. In support of this theory of perturbed FMRP phase behaviour in certain disease variants, rare FXS-associated mutations in the fmr1 coding region cause loss of cytoplasmic FMRP1 function through introduction of a nuclear localization signal [205]. This induces nucleolar aggregation of FMRP1 [205], which is consistent with a phase separation behaviour (where increased local concentration makes phase separation and subsequent aggregation more likely). As FMRP has recently been demonstrated to phase separate, which was suggested to be important for activity-dependent translation regulation [206], this raises the interesting idea that perturbation of its phase behaviour may be harmful to local proteomic homoeostasis. Its aggregation would result in cytoplasmic loss of function of FMRP-associated mRNAs, and so could putatively have the same functional consequences as mutations causing nonsense-mediated decay of its frmp mRNA.
There is also evidence that dysregulated RBP activity occurs in other neurodevelopmental disorders that feature altered synaptic connectivity, such as ASD and epilepsy, but the links to altered connectivity and LPS have not been directly established for most of these RBPs. Notably, FXS is comorbid with select variants of these diseases [207]. Epilepsy can arise through acquired brain lesions, but also during the development of the cortex, at the steps of neuronal proliferation, neuronal migration, or synaptic refinement [208]. For instance, tissues from patients with mesial temporal lobe epilepsy recurrently display the aberrant formation of excitatory connections due to sprouting of hippocampal dentate granule cell axons into the dentate inner molecular layer [209]. Deficiencies in several RBPs other than FMRP have been associated with epilepsy, including BRUNOL4/CELF4 [210], RBFOX1 [211], and Pumilio2 [212]. Of these, Pumilio2 is suggested to affect LPS: it is present in dendritic stress granules during metabolic stress [213] and has recently also been reported to influence the transcriptome of the developing axon by somatic retention of certain mRNAs [214]. Other RBPs implicated in epilepsy are known to be regulated by the translation initiation-promoting mTOR/MAPK pathway, pharmacological inhibition of which effectively prevents epileptogenesis [215]. Axon pathology is thought to be at the core of aberrant connectivity in ASD, with changes in axon diameter, myelination and branching being observed in a range of studies [216]. Multiple ASD-associated genetic alterations have been identified as contributing to some of these changes in axon architecture, such as in the gene encoding chromatin remodelling protein ‘chromodomain helicase DNA-binding protein 8ʹ (CDH8) [217] and in the ANK2 gene, which encodes two major ankyrin polypeptides that are important for polarized transport of organelles [218]. However, ASD is also linked to deficiencies in several RBPs, including RBFOX1 [219], CSDE1 [220], and Caprin1 [221]. For CSDE1, a link between its function and aberrant connectivity has been established, though the functional importance of LPS remains to be investigated: knockdown in primary mouse cortical neurons leads to an overgrowth of the neurites and abnormal dendritic spine morphology/synapse formation [220].
RBP variants associated with neurodegenerative diseases also affect axon architecture
Several mutations in RBPs associated with neurodegenerative diseases, with different ages of onset, have also been shown to affect axonal architecture. Here, we review the evidence linking the RBPs SMN, TDP-43, and FUS to axonal structural defects, and consider to what extent these links might be attributable to dysregulation of LPS.
SMN is a ubiquitously expressed RBP, reduction in the levels of which results in selective dysfunction of motor neurons (spinal muscular atrophy; SMA) [222]. SMN localizes to branch points and growth cones in the axons of primary cultured motor neurons [223], and its depletion has been shown to affect motor neuron axon architecture in several model systems. In zebrafish embryos, knockdown of SMN causes defects in motor neuron axonal outgrowth and pathfinding in a cell-autonomous manner, a phenotype that is not seen in other neuronal subtypes [224]. Using a mouse model of SMA, it has been shown that the earliest structural defects occurred at the neuromuscular junction, and included poor terminal arborization and formation of intermediate filament aggregates [225]. In another mouse model of SMA, it has been demonstrated that reduction of SMN levels also results in abnormal synaptogenesis and neurofilament accumulation in retinal neurons [226]. This study also suggested that SMN-deficient retinal neurons displayed a defect in axon outgrowth, as a reduced number of axons in the optic nerve were observed without a decrease in the number of retinal ganglion cells [226].
Several studies indicate that SMN affects LPS of proteins important for the correct establishment of axonal architecture and connectivity. SMN interacts with the RBP HuD [227], with which it is co-transported in axons of mouse primary motor neurons, and knockdown of SMN reduced both axonal HuD and axonal poly(A) mRNA levels, indicating that it has a role in facilitating axonal localization of certain mRNAs [228]. In particular, reduction of SMN levels is associated with reduced axon outgrowth of motor neurons, which correlates with reduced axonal levels of β-actin mRNA, the 3ʹ-UTR of which is bound by SMN’s binding partner hnRNP-R [229]. In the motor neurons of developing zebrafish embryos, hnRNP-R knockdown resulted in reduced axonal outgrowth associated with loss of β-actin mRNA in the growth cone, without motor neuron death or defects in dendrite outgrowth [230]. SMN not only affects LPS by influencing mRNA localization, but also affects LPS rates directly. In particular, it has been demonstrated to regulate axonal translation via the miRNA miR-183: in SMN-deficient neurons, miR-183 levels are increased, which results in reduced local translation of the protein mTOR, a key stimulator of LPS [16]. Furthermore, it has now been shown that SMN deficiency severely disrupts LPS within motor neuron axons and growth cones, and that rescue of localization of the SMN target mRNA encoding ‘cytoskeleton-associated growth-associated protein 43ʹ (GAP43) can rescue axon outgrowth defects in SMA neurons [231].
The ALS-associated protein TDP-43 is increasingly recognized to affect motor neuron axon structure, which may be due to its regulation of axonal mRNA localization. Expression of ALS-associated human variants of TDP-43 in zebrafish embryos caused motor neuron defects, with shorter axons and premature and excessive branching being observed [232]. This effect was phenocopied by knockout of the zebrafish homologue of TDP-43, indicating a loss-of-function mechanism, though a neurotoxic gain-of-function effect associated with TDP-43 mutant aggregation was observed in dissociated spinal cord cultures [232]. It has been suggested that TDP-43 regulates axonal outgrowth in motor neurons by post-transcriptional regulation of cytoplasmic mRNAs, since it was found to be actively transported into axons of primary cultured motor neurons, where it colocalizes with known axonal RBPs [233]. Like for FMRP, loss of function of TDP-43 affects cytoskeletal architecture: knockout affects synaptic growth and bouton shape at the Drosophila neuromuscular junction [234,235], which is associated with reduced levels of Futsch (the Drosophila MAP1B homologue) in distal axons, the mRNA of which is bound by TDP-43 [234]. The structure of the Drosophila mushroom body was similarly affected by overexpression of TDP-43, with smaller axonal lobes being observed [235]. Therefore, it may similarly be speculated that disease-associated variants of TDP-43 affect axonal function through structural alterations associated with changes in LPS of cytoskeletal and/or cytoskeleton-associated proteins.
There is also evidence that ALS-associated mutations in FUS affect axon branching, though the nature of the effect may depend on the neuronal subtype and mutant variant studied. In cultured primary cortical cells, expression of FUS-R521C led to a reduction in the number of primary axonal branches, when compared with wild-type neurons or neurons expressing wild-type FUS [236]. These defects were linked to the interaction of FUS with SMN: mutant FUS interacted more strongly with SMN and perturbed its axonal localization, and overexpression of SMN was able to rescue the branching defects induced by mutant FUS [236]. In human-induced pluripotent stem cells differentiated into motor neurons, mutant variants of FUS (patient-derived or genome-edited) resulted in increased axonal branching [237]. This effect was rescued by suppression of aberrant expression of transcription factor FOS-B, the mRNA of which was detected in axon bundles and is bound by FUS, and which was also found to be abnormally upregulated in ventral horn neurons in autopsy samples of ALS patients [237]. Together with the observation that endogenously expressed FUS is known to affect LPS in axonal growth cones of Xenopus retinal ganglion cells [58], this suggests regulation of LPS by FUS might occur in axons, which could play a role in determining axon architecture.
Axonal survival signalling
After axons establish their innervation fields through branching, pruning and presynapse formation, intricate crosstalk between signalling pathways and metabolic processes involving pro-survival factors and organelles comes into play to support the health and survival of mature axons. Early research proposed axon degeneration occurs as a consequence of cell body death, due to insufficient protein and energy support from the soma [238]. This view was first challenged by the identification of the Wallerian degeneration slow (WldS) protein, which delays degeneration of somaless axons for weeks [239]. WldS was subsequently shown to substitute for activity of the labile protein nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2), an axon survival factor with both foldase and NAD+ synthase activity [240]. However, it has since been demonstrated that NMNAT2 depletion upon axotomy activates a specific axonal degeneration programme via the downstream effector SARM1 [241], and that modulation of this downstream effector’s activity rather than NMNAT2 activity can rescue the lethality of NMNAT2 deprivation [242], indicating axon degeneration upon injury is initiated by specific signalling pathways. Indeed, more evidence has now accumulated that demonstrates that axons rely on multiple axon-initiated pathways for survival [14,15,185] (Fig. 4).
Figure 4.

Selected contributions by LPS to synaptic survival and adaptability. LPS in the presynaptic terminal contributes to a range of processes important for neuronal maintenance, including I. survival signalling, II. remodelling of cytoskeletal elements, and III. maintenance of mitochondria
The most well-established mechanism to promote axon survival relies on the binding of target-derived neurotrophic factors secreted by target cells, including NGF, BDNF, neurotrophin 3 and 4 (NT3 and NT4), to their receptors TrkA, TrkB, TrkC and p75 on axonal membranes [243]. Upon binding to neurotrophins, receptors are internalized, forming signalling endosomes, and subsequently retrogradely transported to the soma by dynein motors [244], where they activate trophic signalling pathways, including PI3K and mitogen-activated protein (MAP) kinase cascades [245,246,247]. This leads to changes in transcriptional profiles of the stimulated neurons through induction of various transcription factors, including cyclic AMP responsive element-binding protein (CREB), which promotes neuronal survival [248,249].
Pruning and apoptosis are respectively triggered by local or global loss of survival signalling via NGF and the TrkA receptor [250], which has downstream effects on both anti-apoptotic signalling and the NMNAT2/SARM1 pathway [238]. Interestingly, several components of these pathways act at least in part on the mitochondria. The anti-apoptotic protein Blcw is found in axons [251], which is part of the Blc-2 family of proteins that represses the mitochondrial permeability transition that is key in apoptotic signalling [252], and its loss in small fibre sensory neurons is associated with mitochondrial abnormalities and primary axonopathy [251]. Furthermore, WldS increases basal mitochondrial mobility and calcium buffering [253]. Therefore, these organelles are a signalling hub in survival signalling, in addition to being important for LPS. Here, we discuss the various intersections between axonal survival signalling, LPS, and mitochondrial function.
Axonal LPS transfers information in survival signalling
The contribution of LPS to soma-independent axonal survival pathways first came to light with the discovery that axonally synthesized Lamin B2 (LB2), an intermediate filament protein, is critical in preventing axonal degeneration but not in axon guidance, which was made using the model system of developing Xenopus RGC neurons [14]. Proteomic screening demonstrated that stimulation with the guidance cue engrailed-1 affected LPS of several hundred proteins, with the most robust increase in axonal synthesis rate occurring for LB2. The localization of lb2 mRNA and its local translation were then respectively confirmed by fluorescence in situ hybridization and by quantitative immunofluorescence in the presence and absence of translation inhibitor anisomycin. To further validate that laminb2 mRNAs are translated in RGC axons in vivo, a grafting experiment was combined with an axon-TRAP assay. First, eye primordia from a donor embryo expressing GFP-tagged ribosomal protein L10a were transplanted to a host wild-type embryo. After exiting the eye, GFP-RPL10a-positive RGC axons innervated the contralateral wild-type brain hemisphere. Next, pulldown of ribosome-bound mRNAs from the host brain lysates, using the GFP-RPL10 as a ribosome tag localizing exclusively to RGC axons, confirmed LB2 was indeed associated with ribosomes in RGC axons. It was then demonstrated that axonally synthesized LB2 is important for axonal survival: electroporation of a translation-blocking antisense morpholino for laminb2 mRNA into distal axons in vivo resulted in axonal death without cell body death after extension into the optic tectum, without retrograde transport of the morpholino being detectable, and expression of exogenous LB2 lacking a nuclear localization signal could almost completely rescue the degenerative phenotype.
LPS of survival-related proteins is now also known to be triggered by neurotrophin signalling. Neurotrophin signalling-related mRNAs have been identified in a range of axons (Fig. 1) [9,39,41]. For instance, NGF derived from target cells is detected by sensory axons during development, stimulating axonal translation of CREB, which is retrogradely trafficked and promotes neuronal survival [254] (Fig. 4). Furthermore, neurotrophins can promote axon survival by stimulating local translation of anti-apoptotic proteins [185]: using compartmentalized cultures of dorsal root ganglion cells stimulated with NGF and BDNF, it was demonstrated in that blcw mRNA is transcribed in response to retrogradely transported neurotrophins, which is then transported to axons and translated into the anti-apoptotic protein Bclw. Neurotrophins may also regulate the local translation of blcw mRNA, in addition to its transcription and transport: cycloheximide addition to the axonal compartment prevented the increase in axonal Blcw observed upon extended neurotrophin stimulation, whilst addition to the somal compartment had no such effect. Importantly, inhibition of local translation prevented neurotrophins’ survival-promoting effects, and was associated with increased activity of caspase 6, which is inhibited by Blcw. Protein transfection of Blcw into axons protected from neurotrophin withdrawal-induced axonal degeneration, further indicating LPS of this protein is particularly key in axonal survival.
Disruption of LPS has, to our knowledge, not yet been shown to be causative in specific diseases associated with disrupted survival signalling. However, it is known that local loss of survival factors can contribute to disease. In TDP-43-associated ALS, for example, there is splicing defect-associated loss of the survival factor stathmin-2 (STMN2), a microtubule-destabilizing factor essential for axonal microtubule integrity, resulting in impairment of neurite growth and neuronal repair after injury [255]. Restoring levels of this survival factor could rescue TDP-43-associated phenotypes in human pluripotent stem cell-derived human motor neurons [255]. Notably, it has been suggested that STMN2 (also known as superior cervical ganglion 10, SCG10) is locally synthesized in response to axonal injury in proximal axons [256] (Fig. 4), and it is prominent in a range of axonal transcriptomes (Fig. 1) [9,10,40]. Furthermore, in a mouse model of SMA, it has been shown that mutation of SMN causes a reduction of muscle cell secretion of C1q/TNF-Related Protein 3 (CTRP3), which in turn regulates axonal LPS via the mTOR pathway, including SMN itself [257].
Axonal mitochondria are closely associated with LPS and axon survival
As uncovered by a series of studies examining local components essential to axon viability, axonal mitochondria have been increasingly recognized to contribute to axonal integrity and survival. Suboptimal mitochondrial activities, which fail to provide sufficient energy, metabolites and calcium buffering, may result in comprised axon survival [258]. Experimentally, it has been demonstrated that the presence of mitochondria in axons of C. elegans protects against degeneration following axotomy [259]. In fact, mitochondrial dysfunctions are known to be associated with several neurodegenerative disorders with prominent axonal phenotypes [260,261], suggesting that axons are particularly sensitive to disturbance to mitochondrial integrity. For instance, mutations of mitochondrial proteins and lamins may cause Charcot-Marie-Tooth type 2B (CMT2B) diseases, an inherited neuropathy characterized by sensory axon degeneration [262,263]. Similarly, CMT2A is commonly caused by mutations in the gene encoding the mitochondrial protein mitofusin-2 (MFN2) and is associated with degenerative changes in axonal mitochondria in patient sural nerve biopsies [264]. MFN2 promotes inter-mitochondrial fusion as well as tethering of ER to mitochondria; compromising of this latter function (rather than altered bioenergetics) may be the main cause of pathologically altered mitochondrial morphology and transport in CMT2A, as has recently been reported in patient-derived fibroblasts as well as mutation-carrying primary mouse motor neurons [265,266].
Mitochondrial function is linked to LPS as well as to axon survival, since mitochondria likely play an active role in LPS as a local energy source [267]. Their localization is affected by local energy demands: globally, signalling energy consumption of neurons and their subcellular compartments correlates with mitochondrial positioning, with dendrites using over half of the energy required for signalling, and containing over half of the mitochondria [25,268]. Furthermore, mitochondria cluster to locations with high rates of LPS: dendritic mitochondria are stably ‘compartmentalized’ to provide ATP for activity-dependent LPS, with mitochondrial filaments of around 30 μm being anchored near spines by tethering to the cytoskeleton [269]; in axons, mitochondria accumulate at branch points, which contributes to actin-dependent branching [116,181].
Importantly, one of the major categories of mRNAs that is localized to and translated in axons in vivo is those related to mitochondrial function [9] (Fig. 1), suggesting that axon-resident mitochondria require a local supply of proteins for their upkeep. A recent publication suggests LPS is important for mitochondrial maintenance at synapses: stimulation of synaptosomes with NMDA and glutamate induced LPS of mitochondrial proteins, which were shown to be incorporated into respiratory complexes by radiolabel tracing, and perturbation of LPS by knockout of fmr1 was associated with morphology defects in synaptosome mitochondria [270]. Therefore, axonal mitochondria potentially both maintain and are maintained by LPS, making LPS of mitochondrial proteins key for continued axon survival: disruption of mitochondrial function may compromise LPS, which then, in turn, compromises mitochondrial function, and vice versa.
Loss of mitochondrial function triggers degenerative pathways, including following compromised LPS of key mitochondrial proteins. Depolarization of the mitochondrial membrane activates the Wallerian degeneration pathway [271], and is a key step in the apoptotic pathway generally as part of the mitochondrial permeability transition [272]. As shown in multiple studies, loss of maintenance of axonal mitochondrial membrane potential is associated with compromised axonal integrity [14,179,273,274]. This can arise as a consequence of attenuation of local mitochondrial protein production, as was demonstrated for LB2: axonal LB2 localizes to mitochondria, and local depletion of LB2 results in a significantly reduced mitochondrial membrane potential and elongated morphology, which is indicative of mitochondrial dysfunction [14] (Fig. 4). Inhibition of LB2 local translation caused axon degeneration by disrupting mitochondrial function and altering mitochondrial trafficking in axons. As phosphorylation of LB2 triggers nuclear membrane fragmentation during cell division [262], LB2 might control mitochondrial membrane cleavage during mitochondrial fission, which could explain the observed elongated mitochondrial morphology and decreased membrane potential in LB2 knockdown axons. laminb2 mRNA is transported into axons by the RNA-binding protein SPFQ [14,275], rare fALS-associated variants of which mislocalize away from axons [276], and on late endosomes [179]. These endosomes localize to the proximity of mitochondria, and are known to act as translation platforms for local synthesis of mitochondrial proteins, a process that is perturbed by mutations associated with Charcot-Marie-Tooth type 2B neuropathy [179].
Neuronal stresses and stress responses
Given their long lengths and large surface areas, neurons are likely to be exposed to environmental insults that, if not dealt with, may perturb intracellular homoeostasis, resulting in impaired neuronal functions and potentially jeopardizing their long-term survival. Some of these insults are unique to the nervous system, such as compartmentalized stresses, excitotoxicity, and neuroinflammation. While many other stressors are shared by other cell types, including ER stress, amino acid deprivation, hypoxia, heat shock, viral infection and oxidative stress, their impact on neurons with specialized morphology and functions is not always comparable to that on other cells and tissues. Neurons therefore have specialized stress responses, which may involve LPS.
Neuronal RNA is susceptible to oxidative damage
Oxidative stress, an imbalance between reactive oxygen species and antioxidant, is considered to be one of the major threats to neuronal survival in the CNS. Calcium signalling, glutamate uptake, high ATP demand, the importance of redox reactions, and low endogenous antioxidant defence in neurons all contribute to the neuronal vulnerability to oxidative stress [277], but the engagement of RNA oxidation in neurodegenerative diseases has been appreciated only recently.
Similar to proteins and DNA, RNA suffers oxidative damage. In fact, it is even more susceptible to oxidation than other cellular components [278,279], due to its storage in the form of membraneless RNP granules, resulting in its direct exposure to cytoplasm, where thousands of other chemical reactions take place, and due to its single-strandedness, which means it provides accessible sites for oxidative enzymatic reactions [278,279].
RNA oxidation can be functional, as it helps to break down damaged RNA in healthy cells [280], but can also compromise translation. Oxidatively damaged RNAs are altered structurally and are translated less efficiently owing to an increased frequency of ribosome stalling because of the failure in ribosome quality control [281]. Furthermore, the overall RNA levels including rRNA and tRNA, are significantly lower upon RNA oxidation, leading to compromised ribosome functioning and reduced availability of mRNA for translation in affected brain areas [282,283]. The consequences of translation attenuation resulting from RNA oxidative stress may be even more severe in axons and dendrites, where local translation takes place. In developing axons, a large proportion of RNA granules were found to localize adjacent to mitochondria as a major source of reactive oxygen species [179,284]. Moreover, neurites and synapses host activities associated with high metabolic rates and oxidative stresses, such as synaptic transmission.
Unsurprisingly, excessive RNA oxidative damage is associated with neurological disorders, mostly independent of genetic inheritance [285]. A high level of RNA oxidation has been detected in brains of AD, PD, and ALS patients, even preceding the development of pathological hallmarks like protein aggregation [286,287,288,289,290]. Furthermore, oxidative damage to RNA increases with aging due to progressive accumulation of free radicals that exceeds the capability of anti-oxidant defences, possibly accounting for the functional decline in aging brains and late onset of many neurodegenerative diseases [291,292]. However, there is currently insufficient evidence to determine whether RNA oxidative damage is disease-causative or a consequence of disease [287].
The compromised activity of the antioxidant enzyme superoxide dismutase 1 (SOD1), responsible for removing superoxide anions, is associated with multiple diseases, highlighting the importance of antioxidative defence system in neuronal health and survival [293,294,295]. Neurons expressing a pathogenic SOD1 mutant show defective axonal transport, distinct axonal transcriptomes and altered mitochondrial morphology and distribution along axons [53,296]. Intriguingly, oxidative stress is found to decrease RBP solubility through cysteine oxidation and to promote the formation of neuronal aggregates, such as stress granules [297]. RBP–RNA interactions may also be weakened due to RNA oxidative damage and RBP structural alterations, potentially enhancing RBP aggregation propensity. Consistently, the addition of mutant SOD1 aggregates effectively triggered the cytoplasmic aggregation of another ALS-associated protein, TDP-43 [298]. As discussed, the tight control of RBP solubility and cytoplasmic viscosity is key to axonal transport and LPS, which plays an important role in axonal mitochondrial functions and axon survival. Therefore, changes in axonal trafficking and the axonal transcriptome, together with perturbations of mitochondrial integrity in SOD1 mutant axons, point towards a hypothesis that SOD1 mutations are associated with impaired axonal protein synthesis, due to the failure of neuronal antioxidative defence.
Neurons form stress granules with distinct properties in response to stress
De novo formation of translationally repressed stress granules (SGs) with diameters of 100 nm to 2 µm is widely observed upon exposure to a range of stressors and across an extensive range of cell types. Historically, the term ‘stress granule’ refers to cytoplasmic RNP granules containing polyadenylated RNA and certain ‘SG markers’, including poly(A)-binding protein (PABP), T cell intracellular antigen 1 (TIA-1), TIA-1-related protein (TIAR) and Ras GTPase-activating protein-binding protein 1 (G3BP1) [299,300]. During stress, RBPs present in SGs may selectively recruit mRNA targets to protect them from degradation, as demonstrated for Zipcode-binding protein 1 (ZBP1) [301]. In addition, mRNA deadenylation, which often precedes mRNA degradation, appears to be inhibited in SGs, implying a connection exists between SGs and RNA stability [302].
Formation of SGs occurs when translation initiation is limited by stress-induced eIF2α phosphorylation, resulting in local accumulation of mRNAs, translation initiation factors, small ribosomal subunits, and associated RBPs [299,303]. Facilitated by the ability of IDD-containing RBPs to phase separate, these factors coalesce into a compact structure, which serves as a stable SG ‘core’ to recruit other SG components as a more dynamic SG ‘shell’ [304]. It is an open question whether classic SG markers like TIA-1 and G3BP act as scaffolding proteins in the SG core or as shuttling components in the shell [304–306]. However, depletion of G3BP1 to inhibit SG formation did not seem to abolish stress-induced translation repression [307], nor did it accelerate mRNA degradation [305], suggesting that the accumulation of SG marker-containing SGs may be a consequence rather than a prerequisite for of cellular stress responses.
In narrow neuronal processes, accumulation of large SGs can pose a great risk to cargo transport and local proteostasis. In addition to SGs acting as ‘roadblocks’, mRNAs and translational machinery may be sequestered by stable SGs from their cytoplasmic pool, disengaging them from mRNA translation. For instance, axonal G3BP1-associated SGs have been shown to act as a negative modulator of LPS by sequestering a subset of mRNAs [308]. In cultured primary neurons, TDP-43/FUS-containing RNP granules are evident in axons in which aggregation-prone FUS mutants or FUS with altered PTMs are present, resulting in perturbed mRNA localization and LPS [56,58]. It is widely accepted that hyper-stable, amyloid-like deposits resulting from chronic stress in neurons are pathological hallmarks of neurodegenerative disorders [304,309,310], and pharmacological inhibition of SG formation and accumulation has been shown to delay neurodegenerative disease progression [311,312]. Therefore, understanding the role played by SG-modulated LPS during disease development may provide further insights into LPS-based therapeutic treatments.
Since SG formation is dispensable for activating the stress response yet may negatively impact on LPS-supported neuronal function, it is possible that neurons strategically prevent the formation of large rigid SGs during the stress response. Efforts to reveal the differences between acute stress-induced RNP granules and pathological aggregates have identified common components, especially RBPs, the mutations and aberrant PTMs of which are disease-relevant [309,313], suggesting a shared molecular origin between early SGs and pathological assemblies. Intriguingly, the formation and expansion of neuronal SGs are reported to be delayed and slow over the prolonged course of neurodegenerative diseases [57,314,315], in contrast to the rapid appearance of SGs in other cell types under stress [316]. This suggests specific factors are in place in neurons to control SG maturation. Indeed, a study combining proximity labelling and mass spectrometry revealed a large population of neuron-specific SG proteins, including neurodegeneration-associated proteins ELAVL2/3/4 [317]. Furthermore, SGs in neurites show different protein compositions compared to somal SGs, suggesting SGs may participate in compartment-specific activities. Notably, chaperones involved in protein folding and transport, as well as autophagy factors, are among the top-ranked neuronal SG proteins [317]. Chaperones have been shown to interact with stress granules to regulate their dynamic assembly and disassembly [318] and their role in clearing pathological aggregates is increasingly being appreciated in neurodegenerative disease studies [319,320].
Neurons utilize compartmentalized stress responses to cope with stress
As long-lived cells, neurons incapable of coping with cellular stresses can come to suffer from chronic stress due to the accumulation of subtle stress-triggered alterations over years, which ultimately can lead to catastrophic consequences. Therefore, neurons must adopt various strategies to cope with distinct stresses.
A cellular stress response that is used widely by neurons as well as other cell types is the unfolded protein response (UPR). The UPR is activated to reduce the misfolded protein load when misfolded proteins come to accumulate in the ER, a process known as ER stress. The first cellular response to alleviate ER stress is to minimize further protein synthesis, which is mediated by the protein kinase RNA-like endoplasmic reticulum kinase (PERK) pathway. Essentially, upon UPR activation, PERK proteins, which are the transmembrane protein kinases of the pancreatic eIF-2α kinase (PEK) family, oligomerize and autophosphorylate. PERK also phosphorylates eIF2α, a component of the ternary translation initiation complex (which consists of eIF2, initiator methionine transfer RNA and guanosine triphosphate (GTP)). p-eIF2α decreases the availability of the ternary complex and thus global protein synthesis by inhibiting the activity of the guanine exchange factor eIF2B, which is responsible for loading GTP onto the ternary complex after each round of translation initiation [321]. Paradoxically, certain mRNAs escape such translation repression and are instead translated more efficiently upon eIF2α phosphorylation, facilitated by upstream open reading frames located at the 5ʹUTR of their mRNAs. One such mRNA is that encoding activating transcription factor 4 (ATF4), which activates the transcription of pro-apoptotic gene CCAAT-enhancer-binding protein homologous protein (CHOP). Protein synthesis repression caused by UPR activation is associated with a wide range of neurodegenerative disorders, including AD, PD, and prion diseases, and restoration of translation activity is neuroprotective in disease models [322].
While the signalling pathway resembles that found in other cell types, the neuronal UPR features the spatiotemporal segregation of specific components, resulting in a compartmentalized stress response unique to neurons. For instance, in a study in which hippocampal axons were exposed to AD-associated peptide Aβ1-42, axonal p-eIF2α levels increased, indicating UPR activation. Unexpectedly, in contrast to the canonical stress response that results in global translational repression, axonal protein synthesis was significantly increased, including axonal ATF4 synthesis. Over the next 24 hours, ATF4 was retrogradely transported to the soma, where it activated CHOP-dependent apoptosis and led to neuron death [323]. The authors demonstrated that inhibition of local synthesis of ATF4 or its retrograde transport upon axonal Aβ1-42 treatment could effectively reverse CHOP activation and cell loss, exemplifying a form of inter-compartmental signalling propagation in neurodegenerative diseases.
Interestingly, while activation of the UPR is extensively associated with human diseases, the pathway itself has evolved to be a robust pro-survival pathway to mitigate cellular stress in adverse situations, particularly when the insult is mild and transient [324]. The UPR also has various physiological functions, such as protein quality control and metabolism [325,326]. Neurons also use the UPR or individual components of the pathway to regulate physiological activities in the absence of classical stress or pathology [327,328]. In developing retinal ganglion cell axons, the increase in LPS upon 10 min of stimulation by the guidance cue Semaphorin 3A (Sema3A) is partly mediated by the PERK pathway [329]. Sema3A stimulation induces PERK activation and eIF2α phosphorylation, but similar to the Aβ1-42-induced response, axonal protein synthesis is also significantly increased. Therefore, it has been proposed that this differential outcome of eIF2α phosphorylation can be explained by Sema3A stimulation eliciting rapid local synthesis and dephosphorylation of eIF2B, generating a higher level of ternary complexes for translation initiation [329]. This unique Sema3A-induced PERK activation in axons provides a first insight into how neurons engage a modified stress response to meet their developmental demands.
Conclusion and further perspectives
In both neurodevelopmental and neurodegenerative disorders, dysfunction of axons and synapses has been proposed to be central to the observed pathology. Neurodevelopmental disorders like FXS and ASD result from failure in the establishment of synaptic connectivity [330]. In contrast, in neurodegenerative disorders, such as AD, Huntington’s disease and prion diseases, synapse loss is among the first pathological signs, and the extent of synapse loss is the best correlate for cognitive decline [331,332,333]. In the case of ALS, the ‘dying-back model’ has been proposed, in which loss of the axon and motor neuron innervation is initiated in the distal compartment [334]. Encouragingly, it has been reported for several animal models of neurological disorders that synaptic dysfunction and concurrent cognitive impairments are reversible during neurodevelopment and at the early stage of neurodegenerative diseases [335,336,337,338], making research into the underlying mechanisms that compromise synapse integrity highly attractive for therapeutic development.
In this review, we have discussed evidence that LPS in neurites is critical to neuronal function, and that it is compromised in neurological disorders. As LPS supports the autonomy of distal compartments, both through the support of homeostasis and as a localizable regulatory response mediator, its dysregulation particularly affects neuritic maintenance and function. Expectedly, failure in LPS regulation may directly contribute to the neurite dysfunction found in many neurological disorders. However, it should be borne in mind that LPS deficiency can also be downstream of disruption in other processes key to neurite survival, such as axonal trafficking. Therefore, a major challenge to thoroughly understanding the role of LPS in neuropathy is to elucidate the causal relationship between LPS perturbation and various disease-associated pathophysiology.
It is not always straightforward to prove an alteration in LPS rather than somatic translation accounts for a disease phenotype. In recent years, several methods have been developed to perform unbiased screens for axonally synthesized proteins in culture [339]: both laser-capture microdissection [10,340] and compartmentalized culture systems, such as modified Boyden Chambers [39,43,341], allow for axon-only samples to be collected. Similarly, microfluidic devices enable the spatial separation of neuronal cell bodies and axons into fluidic isolated compartments connected by 150–600 µm long microgrooves. This not only allows the somatodendritic and axonal material to be collected separately, but also enables specific manipulations to be performed on the axonal compartment without affecting the soma, including methods that selectively label axonal mRNAs and proteins or inhibit mRNA translation locally [44,342]. However, trafficking between the axonal and somal compartments makes this kind of compartmentalized culture experiment less reliable for the investigation of processes that occur on timescales of days. Furthermore, these systems do not recapitulate the range of cues observed in the in vivo context, for instance during synapse formation, which may be important regulators of LPS. These challenges mean the role of compromised LPS in synapse formation and maintenance in neurological disorders is still largely unknown, and further technical advances are being developed to address this.
Subcellular in vivo multi-omics technology has emerged in the past few years as a method of choice to elucidate the role of LPS in the interconnected neuronal context of animal models of disease, as shown by three recent studies. The first two of these studies employed the RiboTag (also known as axon-TRAP) system to identify cell-type-specific ribosome-bound mRNAs in axons [9,152]. The neurons chosen in these studies, RGCs and auditory cortical TE3 neurons, have their axons and somas situated at spatially distinct locations, which can therefore be surgically separated in vivo. As revealed by the RiboTag approach, the repertoire of ribosome-associated mRNAs in mouse RGC axons changes with the developmental stage to support various functional requirements during axon development and maintenance [9]. The study in auditory cortical axons showed that the translatome was altered during the consolidation of associative memory, for instance with mitochondrion-related genes being upregulated and cytoskeleton-related genes being downregulated [152]. In the third study, a method for determining the transcriptome and proteome of growth cones of selectively labelled neurons was developed: in vivo fluorescent labelling of callosal protein neurons of only one hemisphere through in utero electroporation, allowed purification of trans-hemispheric growth cones, by homogenization of the appropriate hemisphere, subcellular fractionation, and use of a modified fluorescence-activated cell sorting setup. This allowed comparison of different neuronal subtypes and highlighted the molecular specialization of the growth cone, where both the mTOR kinase protein and mRNAs containing mTOR-dependent motifs were accumulated [343]. Furthermore, labelling of nascent proteomes in vivo can be achieved by cell-type-specific metabolic labelling using a methionine analogue, azidonorleucine [344,345,346]. Although it is yet to be applied to study the axonal compartment, this technical procedure has shown great compatibility with surgical separation of subcellular compartments in vivo. Assisted by these powerful in vivo methods, similar comparisons of the local translatome in disease models and healthy animals at different developmental stages would provide further insight into the extent to which LPS is disrupted in neurological disorders.
To fully establish a causative link between LPS and neurological disorders, however, methods for in vivo local inhibition of LPS will need to be developed. So far, it has been successfully demonstrated for Xenopus retinal projection that the local introduction of mRNA-specific anti-sense oligonucleotides (morpholinos) can inhibit local mRNA translation [14,116]. However, in vivo manipulation of axonal translation is more technically challenging in less accessible mammalian neurons. Surgical exposure of axon bundles in live animals followed by local compound treatment or dye labelling is sometimes possible for certain peripheral neurons, such as the sciatic nerve in the hind limb [347]. Excitingly, the past decade has witnessed the rapid development of novel optogenetic approaches for neuroscience research conducted on small mammals in vivo [348]. Meanwhile, elegant optogenetic tools to manipulate intracellular organelle positioning [349], protein phase states [350] and translational activities [351] have been designed and refined to yield new discoveries with high spatiotemporal precisions. All these technical advances in optogenetics, although yet to be tested, hold great promise for facilitating the investigation of LPS in animal models in vivo.
In addition to further investigating the complex regulation of axonal LPS in the in vivo context, the role of LPS in other neuronal compartments and non-neuronal cells should also be considered. We have used the axon as an example of the ways LPS can support distal compartments, as it is the most of a highly polarized neurite, but it should be noted that LPS also supports some unique functionalities of dendrites that are disrupted in neurological disorders. For instance, LPS is associated with long-term depression triggered by metabotropic glutamate receptors in dendrites. Loss of FMRP protein enhances this response, resulting in altered synaptic plasticity [352]. Furthermore, there are also other unique features of neuronal tissues that can create unique vulnerabilities, to disruption of LPS as well as to other insults. In particular, neuronal connectivity has here been simply taken to give rise to unique functional requirements that are supported by LPS and compromised in neurological disorders, but the interconnected nature of neurons itself can be a source of vulnerability in some disorders. In neurodegenerative diseases that are associated with protein aggregation, aggregates often the first form in particular regions of the brain, and then ‘spread’ through a characteristic sequence of other brain areas in a prion-like manner, which mirrors the brain’s internal connectivity [353]. Additionally, there is also ample evidence that the function of non-neuronal cells is compromised in neurological disorders, which affects neuronal function, and can again be linked to LPS in some cases. LPS occurs in non-immune glial cells (astrocytes and oligodendrocytes), where it is known to be important to cell function and health, and LPS of key proteins in protrusions of glial cells has found to be reduced in ALS [354]. Furthermore, stresses originated in non-neuronal cell types can strongly affect neuronal cell populations and neurite homoeostasis. Stress within glia themselves may also be detrimental to neuronal survival, as has been shown for activation of the unfolded protein response in astrocytes [355]. Another notable example of such a stress is neuroinflammation: activation of microglia following neuronal damage can result in proinflammatory signalling that can result in neuronal death in several ways [356]. Excitotoxicity due to excessive glutamate signalling is another stress that is associated with signalling between neurons as well as glia: it can occur through astrocyte dysfunction, and is associated with neurodegenerative diseases as well as ischemic stroke [357].
As a final note, this review has limited itself to neurological disorders for which there is an identifiable genetic basis, allowing disease models to be developed relatively easily, and thus does not reflect the full variety of neurological disorders. Some sporadic neurodegenerative cases may be associated with a range of interacting genetic risk factors of low penetrance, or with exposure to environmental factors, or both, and model systems in which these factors can to an extent be replicated would be very informative. Furthermore, some neurological disorders can clearly be considered to be ‘acquired’, such as following traumatic injury, which can be more readily replicated in experimental systems. Intriguingly, for example, it has been shown for substance addiction that LPS and its upstream signalling networks are affected by the altered activity of microRNA networks [358] and specific RNA-binding proteins [359]. It would be interesting to consider the similarities and differences between LPS in these different forms of neurological disorders.
Acknowledgments
The authors thank Prof. Clemens Kaminski for valuable comments on the manuscript, Jianning Kang and Dr Toshiaki Shigeoka for bioinformatics assistance, and Kaiying Zhao and Chan Li for help with Fig. 3.
Funding Statement
This work was supported by a Sir Henry Wellcome Postdoctoral Fellowship from the Wellcome Trust [215943/Z/19/Z, to J.Q.L.], a UKRI Engineering and Physical Sciences Research Council (EPSRC) grant [EP/L015889/1] awarded to the Centre for Doctoral Training in Sensor Technologies and Applications (supporting F.W.v.T.), and Wellcome Trust grants [085314/Z/08/Z and 203249/Z/16/Z], a European Research Council Advanced Investigator Grant [322817] and a Champalimaud Vision Award (to C.E.H.).
Disclosure statement
The authors declare no competing interests.
References
- [1].Bliss TV, Collingridge GL.. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361(6407):31–39. [DOI] [PubMed] [Google Scholar]
- [2].Trettenbrein PC. The demise of the synapse as the locus of memory: a looming paradigm shift? Front Syst Neurosci. 2016;10:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Edstrom A, Sjostrand J. Protein synthesis in the isolated Mauthner nerve fibre of goldfish. J Neurochem. 1969;16:67–81. [DOI] [PubMed] [Google Scholar]
- [4].Giuditta A, Dettbarn WD, Brzin M. Protein synthesis in the isolated giant axon of the squid. Proc Natl Acad Sci U S A. 1968;59:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Koenig E. Synthetic mechanisms in the axon. IV. In vitro incorporation of [3H]precursors into axonal protein and RNA. J Neurochem. 1967;14:437–446. [DOI] [PubMed] [Google Scholar]
- [6].Campenot RB, Lund K, Mok SA. Production of compartmented cultures of rat sympathetic neurons. Nat Protoc. 2009;4:1869–1887. [DOI] [PubMed] [Google Scholar]
- [7].Taylor AM, Blurton-Jones M, Rhee SW, et al. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nijssen J, Aguila J, Hoogstraaten R, et al. Axon-seq decodes the motor axon transcriptome and its modulation in response to ALS. Stem Cell Reports. 2018;11:1565–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shigeoka T, Jung H, Jung J, et al. Dynamic Axonal translation in developing and mature visual circuits. Cell. 2016;166:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zivraj KH, Tung YC, Piper M, et al. Subcellular profiling reveals distinct and developmentally regulated repertoire of growth cone mRNAs. J Neurosci. 2010;30:15464–15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Abbott LC, Sotelo C. Ultrastructural analysis of catecholaminergic innervation in weaver and normal mouse cerebellar cortices. J Comp Neurol. 2000;426(2):316–329. [PubMed] [Google Scholar]
- [12].Koppers M, Cagnetta R, Shigeoka T, et al. Receptor-specific interactome as a hub for rapid cue-induced selective translation in axons. Elife. 2019;8:e48718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Steward O, Ribak CE. Polyribosomes associated with synaptic specializations on axon initial segments: localization of protein-synthetic machinery at inhibitory synapses. J Neurosci. 1986;6:3079–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yoon BC, Jung H, Dwivedy A, et al. Local translation of extranuclear lamin B promotes axon maintenance. Cell. 2012;148:752–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kim E, Jung H. Local mRNA translation in long-term maintenance of axon health and function. Curr Opin Neurobiol. 2020;63:15–22. [DOI] [PubMed] [Google Scholar]
- [16].Kye MJ, Niederst ED, Wertz MH, et al. SMN regulates axonal local translation via miR-183/mTOR pathway. Hum Mol Genet. 2014;23:6318–6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lopez-Erauskin J, Tadokoro T, Baughn MW, et al. ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron. 2018;100(4):816–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Murakami T, Qamar S, Lin JQ, et al. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron. 2015;88:678–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Thelen MP, Kye MJ. The role of RNA binding proteins for local mRNA translation: implications in neurological disorders. Front Mol Biosci. 2019;6:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pakkenberg B, Pelvig D, Marner L, et al. Aging and the human neocortex. Exp Gerontol. 2003;38:95–99. [DOI] [PubMed] [Google Scholar]
- [21].Savtchenko LP, Rusakov DA. The optimal height of the synaptic cleft. Proc Nat Acad Sci. 2007;104:1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ochs S. Rate of fast axoplasmic transport in mammalian nerve fibres. J Physiol. 1972;227:627–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Popov S, Poo MM. Diffusional transport of macromolecules in developing nerve processes. J Neurosci. 1992;12:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hursh JB. Conduction velocity and diameter of nerve fibers. Am J Physiol Heart Circ Physiol. 1939;127:131–139. [Google Scholar]
- [25].Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–777. [DOI] [PubMed] [Google Scholar]
- [26].Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21(10):1133–1145. [DOI] [PubMed] [Google Scholar]
- [27].Mink JW, Blumenschine RJ, Adams DB. Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. Am J Physiol Regul Integr Comp Physiol. 1981;241:R203–R212. [DOI] [PubMed] [Google Scholar]
- [28].Watts ME, Pocock R, Claudianos C. Brain energy and oxygen metabolism: emerging role in normal function and disease. Front Mol Neurosci. 2018;11:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Takeuchi T, Duszkiewicz AJ, Morris RG. The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Song J, Olsen RHJ, Sun J, et al. Neuronal circuitry mechanisms regulating adult mammalian neurogenesis. Cold Spring Harb Perspect Biol. 2016;8(8):a018937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jung H, Yoon BC, Holt CE. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci. 2012;13:308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Muzio MR, Cascella M. Histology: Axon. [Updated 2020 May 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. [Google Scholar]
- [33].Sabry J, O’Connor TP, Kirschner MW. Axonal transport of tubulin in Ti1 pioneer neurons in situ. Neuron. 1995;14:1247–1256. [DOI] [PubMed] [Google Scholar]
- [34].Jung H, Gkogkas CG, Sonenberg N, et al. Remote control of gene function by local translation. Cell. 2014;157:26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang J, Pavlyk I, Vedula P, et al. Arginyltransferase ATE1 is targeted to the neuronal growth cones and regulates neurite outgrowth during brain development. Dev Biol. 2017;430:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Holt CE, Martin KC, Schuman EM. Local translation in neurons: visualization and function. Nat Struct Mol Biol. 2019;26:557–566. [DOI] [PubMed] [Google Scholar]
- [37].Cioni J-M, Koppers M, Holt CE. Molecular control of local translation in axon development and maintenance. Curr Opin Neurobiol. 2018;51:86–94. [DOI] [PubMed] [Google Scholar]
- [38].Briese M, Saal L, Appenzeller S, et al. Whole transcriptome profiling reveals the RNA content of motor axons. Nucleic Acids Res. 2016;44(4):e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cagnetta R, Frese CK, Shigeoka T, et al. Rapid cue-specific remodeling of the nascent axonal proteome. Neuron. 2018;99(1):29–46 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gumy LF, Yeo GS, Tung YC, et al. Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA. 2011;17:85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Saal L, Briese M, Kneitz S, et al. Subcellular transcriptome alterations in a cell culture model of spinal muscular atrophy point to widespread defects in axonal growth and presynaptic differentiation. RNA. 2014;20:1789–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Taylor AM, Berchtold NC, Perreau VM, et al. Axonal mRNA in uninjured and regenerating cortical mammalian axons. J Neurosci. 2009;29:4697–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Willis DE, van Niekerk EA, Sasaki Y, et al. Extracellular stimuli specifically regulate localized levels of individual neuronal mRNAs. J Cell Biol. 2007;178:965–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shigeoka T, Koppers M, Wong HH, et al. On-site ribosome remodeling by locally synthesized ribosomal proteins in axons. Cell Rep. 2019;29:3605–3619 e3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mauro VP, Edelman GM. The ribosome filter hypothesis. Proc Natl Acad Sci U S A. 2002;99:12031–12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sasaki Y. Local translation in growth cones and presynapses, two axonal compartments for local neuronal functions. Biomolecules. 2020;10:668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Terenzio M, Koley S, Samra N, et al. Locally translated mTOR controls axonal local translation in nerve injury. Science. 2018;359:1416–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Leung KM, van Horck FP, Lin AC, et al. Asymmetrical beta-actin mRNA translation in growth cones mediates attractive turning to netrin-1. Nat Neurosci. 2006;9:1247–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yao J, Sasaki Y, Wen Z, et al. An essential role for beta-actin mRNA localization and translation in Ca2+-dependent growth cone guidance. Nat Neurosci. 2006;9:1265–1273. [DOI] [PubMed] [Google Scholar]
- [50].Nesler KR, Starke EL, Boin NG, et al. Presynaptic CamKII regulates activity-dependent axon terminal growth. Mol Cell Neurosci. 2016;76:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Martin KC, Casadio A, Zhu H, et al. Synapse-specific, long-term facilitation of aplysia sensory to motor synapses: a function for local protein synthesis in memory storage. Cell. 1997;91:927–938. [DOI] [PubMed] [Google Scholar]
- [52].Wang DO, Kim SM, Zhao Y, et al. Synapse- and stimulus-specific local translation during long-term neuronal plasticity. Science. 2009;324:1536–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rotem N, Magen I, Ionescu A, et al. ALS along the axons – expression of coding and noncoding RNA differs in axons of ALS models. Sci Rep. 2017;7:44500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pieretti M, Zhang F, Fu Y-H, et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. [DOI] [PubMed] [Google Scholar]
- [55].Cook C, Petrucelli L. Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron. 2019;101(6):1057–1069. [DOI] [PubMed] [Google Scholar]
- [56].Alami NH, Smith RB, Carrasco MA, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81(3):536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].López-Erauskin J, Tadokoro T, Baughn MW, et al. ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron. 2018;100:816–830.e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Qamar S, Wang G, Randle SJ, et al. FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell. 2018;173:720–734.e715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yasuda K, Mili S. Dysregulated axonal RNA translation in amyotrophic lateral sclerosis. Wiley Interdiscip Rev RNA. 2016;7:589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. [DOI] [PubMed] [Google Scholar]
- [63].Nicolas A, Kenna KP, Renton AE, et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron. 2018;97:1268–1283.e1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Smith BN, Ticozzi N, Fallini C, et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014;84:324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wu C-H, Fallini C, Ticozzi N, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].The Human Protein Atlas, v . (2020a). FUS. [Accessed 2020 January 10]. Available from: https://www.proteinatlas.org/ENSG00000089280-FUS/tissue
- [67].The Human Protein Atlas, v . (2020b). TARDBP. [Accessed 2020 January 10]. Available from: https://www.proteinatlas.org/ENSG00000120948-TARDBP/tissue
- [68].Nijssen J, Comley LH, Hedlund E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017;133:863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ragagnin AMG, Shadfar S, Vidal M, et al. Motor neuron susceptibility in ALS/FTD. Front Neurosci. 2019;13:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ringholz GM, Appel SH, Bradshaw M, et al. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65:586–590. [DOI] [PubMed] [Google Scholar]
- [71].Lee SM, Asress S, Hales CM, et al. TDP-43 cytoplasmic inclusion formation is disrupted in C9orf72-associated amyotrophic lateral sclerosis/frontotemporal lobar degeneration. Brain Commun. 2019;1:fcz014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kwiatkowski TJ, Bosco DA, LeClerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. [DOI] [PubMed] [Google Scholar]
- [73].Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- [74].Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ishigaki S, Sobue G. Importance of functional loss of FUS in FTLD/ALS. Front Mol Biosci. 2018;5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Perge JA, Niven JE, Mugnaini E, et al. Why do axons differ in caliber? J Neurosci. 2012;32:626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wortman JC, Shrestha UM, Barry DM, et al. Axonal transport: how high microtubule density can compensate for boundary effects in small-caliber axons. Biophys J. 2014;106:813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Maday S, Twelvetrees AE, Moughamian AJ, et al. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014;84:292–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Di Rienzo C, Piazza V, Gratton E, et al. Probing short-range protein Brownian motion in the cytoplasm of living cells. Nat Commun. 2014;5:5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Elowitz MB, Surette MG, Wolf PE, et al. Protein mobility in the cytoplasm of escherichia coli. J Bacteriol. 1999;181:197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Petrasek Z, Schwille P. Precise measurement of diffusion coefficients using scanning fluorescence correlation spectroscopy. Biophys J. 2008;94:1437–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kevenaar JT, Hoogenraad CC. The axonal cytoskeleton: from organization to function. Front Mol Neurosci. 2015;8:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Sabharwal V, Koushika SP. Crowd control: effects of physical crowding on cargo movement in healthy and diseased neurons. Front Cell Neurosci. 2019;13:470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].De Vos KJ, Grierson AJ, Ackerley S, et al. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. [DOI] [PubMed] [Google Scholar]
- [86].Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14:161–176. [DOI] [PubMed] [Google Scholar]
- [87].Sleigh JN, Rossor AM, Fellows AD, et al. Axonal transport and neurological disease. Nat Rev Neurol. 2019;15:691–703. [DOI] [PubMed] [Google Scholar]
- [88].Breuss M, Keays DA. Microtubules and neurodevelopmental disease: the movers and the makers. Adv Exp Med Biol. 2014;800:75–96. [DOI] [PubMed] [Google Scholar]
- [89].Jung H, Holt CE. Local translation of mRNAs in neural development. Wiley Interdiscip Rev RNA. 2011;2:153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Hammarlund M, Jorgensen EM, Bastiani MJ. Axons break in animals lacking beta-spectrin. J Cell Biol. 2007;176:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Costa AR, Pinto-Costa R, Sousa SC, et al. The regulation of axon diameter: from axonal circumferential contractility to activity-dependent axon swelling. Front Mol Neurosci. 2018;11:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Nguyen MD, Lariviere RC, Julien JP. Reduction of axonal caliber does not alleviate motor neuron disease caused by mutant superoxide dismutase 1. Proc Natl Acad Sci U S A. 2000;97:12306–12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Friede RL, Samorajski T. Axon caliber related to neurofilaments and microtubules in sciatic nerve fibers of rats and mice. Anat Rec. 1970;167:379–387. [DOI] [PubMed] [Google Scholar]
- [94].Hall GF, Chu B, Lee S, et al. The single neurofilament subunit of the lamprey forms filaments and regulates axonal caliber and neuronal size in vivo. Cell Motil Cytoskeleton. 2000;46:166–182. [DOI] [PubMed] [Google Scholar]
- [95].Wang H, Wu M, Zhan C, et al. Neurofilament proteins in axonal regeneration and neurodegenerative diseases. Neural Regen Res. 2012;7:620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Rao MV, Mohan PS, Kumar A, et al. The myosin Va head domain binds to the neurofilament-L rod and modulates endoplasmic reticulum (ER) content and distribution within axons. PLoS One. 2011;6:e17087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Gentil BJ, Minotti S, Beange M, et al. Normal role of the low-molecular-weight neurofilament protein in mitochondrial dynamics and disruption in Charcot-Marie-tooth disease. Faseb J. 2012;26:1194–1203. [DOI] [PubMed] [Google Scholar]
- [98].Yuan A, Rao MV, Nixon RA. Neurofilaments at a glance. J Cell Sci. 2012;125:3257–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Dale JM, Garcia ML. Neurofilament phosphorylation during development and disease: which came first, the phosphorylation or the accumulation? J Amino Acids. 2012;2012:382107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Didonna A, Opal P. The role of neurofilament aggregation in neurodegeneration: lessons from rare inherited neurological disorders. Mol Neurodegener. 2019;14:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Sotelo-Silveira JR, Calliari A, Kun A, et al. Neurofilament mRNAs are present and translated in the normal and severed sciatic nerve. J Neurosci Res. 2000;62:65–74. [DOI] [PubMed] [Google Scholar]
- [102].Weiner OD, Zorn AM, Krieg PA, et al. Medium weight neurofilament mRNA in goldfish Mauthner axoplasm. Neurosci Lett. 1996;213:83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lee SK, Hollenbeck PJ. Organization and translation of mRNA in sympathetic axons. J Cell Sci. 2003;116:4467–4478. [DOI] [PubMed] [Google Scholar]
- [104].Zheng JQ, Kelly TK, Chang B, et al. A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J Neurosci. 2001;21:9291–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Papandreou MJ, Leterrier C. The functional architecture of axonal actin. Mol Cell Neurosci. 2018;91:151–159. [DOI] [PubMed] [Google Scholar]
- [106].Omotade OF, Pollitt SL, Zheng JQ. Actin-based growth cone motility and guidance. Mol Cell Neurosci. 2017;84:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Armijo-Weingart L, Gallo G. It takes a village to raise a branch: cellular mechanisms of the initiation of axon collateral branches. Mol Cell Neurosci. 2017;84:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Xu K, Zhong G, Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 2013;339:452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Zhang Y, Abiraman K, Li H, et al. Modeling of the axon membrane skeleton structure and implications for its mechanical properties. PLoS Comput Biol. 2017;13:e1005407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Dubey S, Bhembre N, Bodas S, et al. The axonal actin-spectrin lattice acts as a tension buffering shock absorber. Elife. 2020;9:e51772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Nelson JC, Stavoe AK, Colon-Ramos DA. The actin cytoskeleton in presynaptic assembly. Cell Adh Migr. 2013;7:379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Procaccio V, Salazar G, Ono S, et al. A mutation of beta-actin that alters depolymerization dynamics is associated with autosomal dominant developmental malformations, deafness, and dystonia. Am J Hum Genet. 2006;78:947–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Zhu M, Yang T, Wei S, et al. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am J Hum Genet. 2003;73:1082–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Riviere JB, van Bon BW, Hoischen A, et al. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-winter syndrome. Nat Genet. 2012;44:440–444, S441–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Donnelly CJ, Park M, Spillane M, et al. Axonally synthesized beta-actin and GAP-43 proteins support distinct modes of axonal growth. J Neurosci. 2013;33:3311–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Wong HH-W, Lin JQ, Ströhl F, et al. RNA docking and local translation regulate site-specific axon remodeling in vivo. Neuron. 2017;95:852–868.e858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Bassell GJ, Zhang H, Byrd AL, et al. Sorting of β-actin mRNA and protein to neurites and growth cones in culture. J Neurosci. 1998;18(1):251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Moradi M, Sivadasan R, Saal L, et al. Differential roles of alpha-, beta-, and gamma-actin in axon growth and collateral branch formation in motoneurons. J Cell Biol. 2017;216:793–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Tytell M, Black MM, Garner JA, et al. Axonal transport: each major rate component reflects the movement of distinct macromolecular complexes. Science. 1981;214:179–181. [DOI] [PubMed] [Google Scholar]
- [120].Block-Galarza J, Chase KO, Sapp E, et al. Fast transport and retrograde movement of huntingtin and HAP 1 in axons. Neuroreport. 1997;8(9):2247–2251. [DOI] [PubMed] [Google Scholar]
- [121].Brunholz S, Sisodia S, Lorenzo A, et al. Axonal transport of APP and the spatial regulation of APP cleavage and function in neuronal cells. Exp Brain Res. 2012;217(3–4):353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hoffman PN, Lasek RJ. The slow component of axonal transport. Identification of major structural polypeptides of the axon and their generality among mammalian neurons. J Cell Biol. 1975;66:351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Wang L, Ho CL, Sun D, et al. Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat Cell Biol. 2000;2:137–141. [DOI] [PubMed] [Google Scholar]
- [124].Conway L, Wood D, Tuzel E, et al. Motor transport of self-assembled cargos in crowded environments. Proc Natl Acad Sci U S A. 2012;109(51):20814–20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Hinckelmann MV, Zala D, Saudou F. Releasing the brake: restoring fast axonal transport in neurodegenerative disorders. Trends Cell Biol. 2013;23:634–643. [DOI] [PubMed] [Google Scholar]
- [126].Hendricks AG, Perlson E, Ross JL, et al. Motor coordination via a tug-of-war mechanism drives bidirectional vesicle transport. Curr Biol. 2010;20:697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Welte MA. Bidirectional transport along microtubules. Curr Biol. 2004;14:R525–537. [DOI] [PubMed] [Google Scholar]
- [128].Liu YT, Laura M, Hersheson J, et al. Extended phenotypic spectrum of KIF5A mutations: from spastic paraplegia to axonal neuropathy. Neurology. 2014;83:612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Clark JA, Yeaman EJ, Blizzard CA, et al. A case for microtubule vulnerability in amyotrophic lateral sclerosis: altered dynamics during disease. Front Cell Neurosci. 2016;10:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Lasser M, Tiber J, Lowery LA. The role of the microtubule cytoskeleton in neurodevelopmental disorders. Front Cell Neurosci. 2018;12:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Matamoros AJ, Baas PW. Microtubules in health and degenerative disease of the nervous system. Brain Res Bull. 2016;126:217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Eng H, Lund K, Campenot RB. Synthesis of beta-tubulin, actin, and other proteins in axons of sympathetic neurons in compartmented cultures. J Neurosci. 1999;19:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Moccia R, Chen D, Lyles V, et al. An unbiased cDNA library prepared from isolated Aplysia sensory neuron processes is enriched for cytoskeletal and translational mRNAs. J Neurosci. 2003;23:9409–9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Breuss MW, Leca I, Gstrein T, et al. Tubulins and brain development – the origins of functional specification. Mol Cell Neurosci. 2017;84:58–67. [DOI] [PubMed] [Google Scholar]
- [135].Park JH, Roll-Mecak A. The tubulin code in neuronal polarity. Curr Opin Neurobiol. 2018;51:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Preitner N, Quan J, Nowakowski DW, et al. APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell. 2014;158:368–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Cushion TD, Dobyns WB, Mullins JGL, et al. Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain. 2013;136(2):536–548. [DOI] [PubMed] [Google Scholar]
- [138].Jaglin XH, Poirier K, Saillour Y, et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet. 2009;41:746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Weingarten MD, Lockwood AH, Hwo SY, et al. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Biswas S, Kalil K. The microtubule-associated protein tau mediates the organization of microtubules and their dynamic exploration of actin-rich lamellipodia and filopodia of cortical growth cones. J Neurosci. 2018;38(2):291–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Johnson GV, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. 2004;117:5721–5729. [DOI] [PubMed] [Google Scholar]
- [142].Aronov S, Aranda G, Behar L, et al. Axonal tau mRNA localization coincides with tau protein in living neuronal cells and depends on axonal targeting signal. J Neurosci. 2001;21(17):6577–6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Aronov S. Visualization of translated tau protein in the axons of neuronal P19 cells and characterization of tau RNP granules. J Cell Sci. 2002;115(19):3817–3827. [DOI] [PubMed] [Google Scholar]
- [144].Dujardin S, Begard S, Caillierez R, et al. Different tau species lead to heterogeneous tau pathology propagation and misfolding. Acta Neuropathol Commun. 2018;6:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Zempel H, Dennissen FJA, Kumar Y, et al. Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture. J Biol Chem. 2017;292:12192–12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Kubo A, Misonou H, Matsuyama M, et al. Distribution of endogenous normal tau in the mouse brain. J Comp Neurol. 2019;527:985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Kobayashi S, Tanaka T, Soeda Y, et al. Local somatodendritic translation and hyperphosphorylation of tau protein triggered by AMPA and NMDA receptor stimulation. EBioMedicine. 2017;20:120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Zempel H, Mandelkow E. Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014;37:721–732. [DOI] [PubMed] [Google Scholar]
- [149].Feltrin D, Fusco L, Witte H, et al. Growth cone MKK7 mRNA targeting regulates MAP1b-dependent microtubule bundling to control neurite elongation. PLoS Biol. 2012;10:e1001439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Chun J-T, Gioio AE, Crispino M, et al. Differential compartmentalization of mRNAs in squid giant axon. J Neurochem. 2002;67(5):1806–1812. [DOI] [PubMed] [Google Scholar]
- [151].Gioio AE, Chun JT, Crispino M, et al. Kinesin mRNA is present in the squid giant axon. J Neurochem. 1994;63:13–18. [DOI] [PubMed] [Google Scholar]
- [152].Ostroff LE, Santini E, Sears R, et al. Axon TRAP reveals learning-associated alterations in cortical axonal mRNAs in the lateral amygdala. Elife. 2019;8:e51607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004;43:513–525. [DOI] [PubMed] [Google Scholar]
- [154].Gabrych DR, Lau VZ, Niwa S, et al. Going too far is the same as falling short(dagger): kinesin-3 family members in hereditary spastic paraplegia. Front Cell Neurosci. 2019;13:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Okada Y, Yamazaki H, Sekine-Aizawa Y, et al. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. [DOI] [PubMed] [Google Scholar]
- [156].Chiba K, Takahashi H, Chen M, et al. Disease-associated mutations hyperactivate KIF1A motility and anterograde axonal transport of synaptic vesicle precursors. Proc Natl Acad Sci U S A. 2019;116(37):18429–18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Kaur S, Van Bergen NJ, Verhey KJ, et al. Expansion of the phenotypic spectrum of de novo missense variants in kinesin family member 1A (KIF1A). Hum Mutat. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Riviere JB, Ramalingam S, Lavastre V, et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet. 2011;89:219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Villarin JM, McCurdy EP, Martinez JC, et al. Local synthesis of dynein cofactors matches retrograde transport to acutely changing demands. Nat Commun. 2016;7:13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Jambhekar A, Derisi JL. Cis-acting determinants of asymmetric, cytoplasmic RNA transport. Rna. 2007;13:625–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Shahbabian K, Chartrand P. Control of cytoplasmic mRNA localization. Cell Mol Life Sci. 2012;69:535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Xing L, Bassell GJ. mRNA localization: an orchestration of assembly, traffic and synthesis. Traffic. 2013;14:2–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].March ZM, King OD, Shorter J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016;1647:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Feng Z, Chen X, Wu X, et al. Formation of biological condensates via phase separation: characteristics, analytical methods, and physiological implications. J Biol Chem. 2019;294:14823–14835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Alberti S, Gladfelter A, Mittag T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell. 2019;176(3):419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Bah A, Forman-Kay JD. Modulation of intrinsically disordered protein function by post-translational modifications. J Biol Chem. 2016;291(13):6696–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. [DOI] [PubMed] [Google Scholar]
- [168].Elbaum-Garfinkle S. Matter over mind: liquid phase separation and neurodegeneration. J Biol Chem. 2019;294:7160–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Gomes E, Shorter J. The molecular language of membraneless organelles. J Biol Chem. 2019;294:7115–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Polymenidou M. The RNA face of phase separation. Science. 2018;360:859–860. [DOI] [PubMed] [Google Scholar]
- [171].Dormann D, Madl T, Valori CF, et al. Arginine methylation next to the PY-NLS modulates transportin binding and nuclear import of FUS. Embo J. 2012;31:4258–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Suarez-Calvet M, Neumann M, Arzberger T, et al. Monomethylated and unmethylated FUS exhibit increased binding to transportin and distinguish FTLD-FUS from ALS-FUS. Acta Neuropathol. 2016;131:587–604. [DOI] [PubMed] [Google Scholar]
- [173].Guo L, Kim HJ, Wang H, et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell. 2018;173:677–692 e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Hofweber M, Hutten S, Bourgeois B, et al. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell. 2018;173:706–719.e713. [DOI] [PubMed] [Google Scholar]
- [175].Yoshizawa T, Ali R, Jiou J, et al. Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell. 2018;173:693–705.e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Baumann S, Konig J, Koepke J, et al. Endosomal transport of septin mRNA and protein indicates local translation on endosomes and is required for correct septin filamentation. EMBO reports. 2014;15(1):94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Lesnik C, Golani-Armon A, Arava Y. Localized translation near the mitochondrial outer membrane: an update. RNA Biol. 2015;12:801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Yarmishyn AA, Kremenskoy M, Batagov AO, et al. Genome-wide analysis of mRNAs associated with mouse peroxisomes. BMC Genomics. 2016;17:1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Cioni J-M, Lin JQ, Holtermann AV, et al. Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell. 2019;176(1–2):56–72 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Liao YC, Fernandopulle MS, Wang G, et al. RNA granules hitchhike on lysosomes for long-distance transport, using annexin A11 as a molecular tether. Cell. 2019;179:147–164 e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Spillane M, Ketschek A, Merianda TT, et al. Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep. 2013;5:1564–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Corradi E, Dalla Costa I, Gavoci A, et al. Axonal precursor miRNAs hitchhike on endosomes and locally regulate the development of neural circuits. Embo J. 2020;39(6):e102513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Gibson DA, Ma L. Developmental regulation of axon branching in the vertebrate nervous system. Development. 2011;138:183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Riccomagno MM, Kolodkin AL. Sculpting neural circuits by axon and dendrite pruning. Annu Rev Cell Dev Biol. 2015;31:779–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Cosker KE, Pazyra-Murphy MF, Fenstermacher SJ, et al. Target-derived neurotrophins coordinate transcription and transport of Bclw to prevent axonal degeneration. J Neurosci. 2013;33(12):5195–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Pfeiffer BE, Huber KM. The state of synapses in fragile X syndrome. Neuroscientist. 2009;15:549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Pan L, Zhang YQ, Woodruff E, et al. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol. 2004;14:1863–1870. [DOI] [PubMed] [Google Scholar]
- [188].Bureau I, Shepherd GMG, Svoboda K. Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J Neurosci. 2008;28(20):5178–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Li Z, Zhang Y, Ku L, et al. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29:2276–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Banerjee A, Ifrim MF, Valdez AN, et al. Aberrant RNA translation in fragile X syndrome: from FMRP mechanisms to emerging therapeutic strategies. Brain Res. 2018;1693:24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Feng Y, Absher D, Eberhart DE, et al. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997;1:109–118. [DOI] [PubMed] [Google Scholar]
- [192].Kao DI, Aldridge GM, Weiler IJ, et al. Altered mRNA transport, docking, and protein translation in neurons lacking fragile X mental retardation protein. Proc Natl Acad Sci U S A. 2010;107:15601–15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60(2):201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Akins MR, Leblanc HF, Stackpole EE, et al. Systematic mapping of fragile X granules in the mouse brain reveals a potential role for presynaptic FMRP in sensorimotor functions. J Comp Neurol. 2012;520(16):3687–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Christie SB, Akins MR, Schwob JE, et al. The FXG: a presynaptic fragile X granule expressed in a subset of developing brain circuits. J Neurosci. 2009;29(5):1514–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Hörnberg H, Holt C. RNA-binding proteins and translational regulation in axons and growth cones. Front Neurosci. 2013;7:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Akins MR, Berk-Rauch HE, Kwan KY, et al. Axonal ribosomes and mRNAs associate with fragile X granules in adult rodent and human brains. Hum Mol Genet. 2016;26:192–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Li C, Bassell GJ, Sasaki Y. Fragile X mental retardation protein is involved in protein synthesis-dependent collapse of growth cones induced by semaphorin-3A. Front Neural Circuits. 2009;3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Zhang YQ, Bailey AM, Matthies HJG, et al. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. [DOI] [PubMed] [Google Scholar]
- [200].Parvin S, Takeda R, Sugiura Y, et al. Fragile X mental retardation protein regulates accumulation of the active zone protein Munc18-1 in presynapses via local translation in axons during synaptogenesis. Neurosci Res. 2019;146:36–47. [DOI] [PubMed] [Google Scholar]
- [201].Tessier CR, Broadie K. Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development. 2008;135:1547–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Medioni C, Ramialison M, Ephrussi A, et al. Imp promotes axonal remodeling by regulating profilin mRNA during brain development. Curr Biol. 2014;24:793–800. [DOI] [PubMed] [Google Scholar]
- [203].Zimmer SE, Doll SG, Garcia ADR, et al. Splice form-dependent regulation of axonal arbor complexity by FMRP. Dev Neurobiol. 2017;77:738–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Ceman S, O’Donnell WT, Reed M, et al. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet. 2003;12(24):3295–3305. [DOI] [PubMed] [Google Scholar]
- [205].Okray Z, de Esch CEF, Van Esch H, et al. A novel fragile X syndrome mutation reveals a conserved role for the carboxy-terminus in FMRP localization and function. EMBO Mol Med. 2015;7:423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Tsang B, Arsenault J, Vernon RM, et al. Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc Nat Acad Sci. 2019;116:4218–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Kidd SA, Lachiewicz A, Barbouth D, et al. Fragile X syndrome: a review of associated medical problems. Pediatrics. 2014;134:995–1005. [DOI] [PubMed] [Google Scholar]
- [208].Bozzi Y, Casarosa S, Caleo M. Epilepsy as a neurodevelopmental disorder. Front Psychiatry. 2012;3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Godale CM, Danzer SC. Signaling pathways and cellular mechanisms regulating mossy fiber sprouting in the development of epilepsy. Front Neurol. 2018;9:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Yang Y, Mahaffey CL, Bérubé N, et al. Complex seizure disorder caused by Brunol4 deficiency in mice. PLoS Genet. 2007;3:e124–e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Lal D, Trucks H, Møller RS, et al. Rare exonic deletions of the RBFOX1 gene increase risk of idiopathic generalized epilepsy. Epilepsia. 2013;54:265–271. [DOI] [PubMed] [Google Scholar]
- [212].Follwaczny P, Schieweck R, Riedemann T, et al. Pumilio2-deficient mice show a predisposition for epilepsy. Dis Model Mech. 2017;10:1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Vessey JP, Vaccani A, Xie Y, et al. Dendritic localization of the translational repressor Pumilio 2 and its contribution to dendritic stress granules. J Neurosci. 2006;26:6496–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Martínez JC, Randolph LK, Iascone DM, et al. Pum2 shapes the transcriptome in developing axons through retention of target mRNAs in the cell body. Neuron. 2019;104:931–946.e935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Pernice HF, Schieweck R, Kiebler MA, et al. mTOR and MAPK: from localized translation control to epilepsy. BMC Neurosci. 2016;17:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Zikopoulos B, Barbas H. Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front Hum Neurosci. 2013;7:609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Xu Q, Liu YY, Wang X, et al. Autism-associated CHD8 deficiency impairs axon development and migration of cortical neurons. Mol Autism. 2018;9:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Yang R, Walder-Christensen KK, Kim N, et al. ANK2 autism mutation targeting giant ankyrin-B promotes axon branching and ectopic connectivity. Proc Nat Acad Sci. 2019;116:15262–15271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Weyn-Vanhentenryck SM, Mele A, Yan Q, et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014;6:1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Guo H, Li Y, Shen L, et al. Disruptive variants of CSDE1 associate with autism and interfere with neuronal development and synaptic transmission. Sci Adv. 2019;5:eaax2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Ohashi R, Takao K, Miyakawa T, et al. Comprehensive behavioral analysis of RNG105 (Caprin1) heterozygous mice: reduced social interaction and attenuated response to novelty. Sci Rep. 2016;6:20775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Burghes AHM, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Jablonka S, Bandilla M, Wiese S, et al. Co-regulation of survival of motor neuron (SMN) protein and its interactor SIP1 during development and in spinal muscular atrophy. Hum Mol Genet. 2001;10:497–505. [DOI] [PubMed] [Google Scholar]
- [224].McWhorter ML, Monani UR, Burghes AHM, et al. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol. 2003;162:919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Kariya S, Park G-H, Maeno-Hikichi Y, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Liu H, Beauvais A, Baker AN, et al. Smn deficiency causes neuritogenesis and neurogenesis defects in the retinal neurons of a mouse model of spinal muscular atrophy. Dev Neurobiol. 2011;71:153–169. [DOI] [PubMed] [Google Scholar]
- [227].Hubers L, Valderrama-Carvajal H, Laframboise J, et al. HuD interacts with survival motor neuron protein and can rescue spinal muscular atrophy-like neuronal defects. Hum Mol Genet. 2010;20:553–579. [DOI] [PubMed] [Google Scholar]
- [228].Fallini C, Zhang H, Su Y, et al. The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(A) mRNA in primary motor neuron axons. J Neurosci. 2011;31:3914–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Rossoll W, Jablonka S, Andreassi C, et al. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163:801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Glinka M, Herrmann T, Funk N, et al. The heterogeneous nuclear ribonucleoprotein-R is necessary for axonal β-actin mRNA translocation in spinal motor neurons. Hum Mol Genet. 2010;19:1951–1966. [DOI] [PubMed] [Google Scholar]
- [231].Fallini C, Donlin-Asp PG, Rouanet JP, et al. Deficiency of the survival of motor neuron protein impairs mRNA localization and local translation in the growth cone of motor neurons. J Neurosci. 2016;36:3811–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Kabashi E, Lin L, Tradewell ML, et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19:671–683. [DOI] [PubMed] [Google Scholar]
- [233].Fallini C, Bassell GJ, Rossoll W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet. 2012;21:3703–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Godena VK, Romano G, Romano M, et al. TDP-43 regulates Drosophila neuromuscular junctions growth by modulating Futsch/MAP1B levels and synaptic microtubules organization. PloS One. 2011;6:e17808–e17808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Lin MJ, Cheng CW, Shen CK. Neuronal function and dysfunction of Drosophila dTDP. PLoS One. 2011;6:e20371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Groen EJ, Fumoto K, Blokhuis AM, et al. ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet. 2013;22:3690–3704. [DOI] [PubMed] [Google Scholar]
- [237].Akiyama T, Suzuki N, Ishikawa M, et al. Aberrant axon branching via Fos-B dysregulation in FUS-ALS motor neurons. EBioMedicine. 2019;45:362–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Pease SE, Segal RA. Preserve and protect: maintaining axons within functional circuits. Trends Neurosci. 2014;37:572–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Coleman MP, Conforti L, Buckmaster EA, et al. An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci U S A. 1998;95(17):9985–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Brazill JM, Li C, Zhu Y, et al. NMNAT: it’s an NAD + synthase… It’s a chaperone… It’s a neuroprotector. Curr Opin Genet Dev. 2017;44:156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Gilley J, Orsomando G, Nascimento-Ferreira I, et al. Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. 2015;10:1974–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Gilley J, Ribchester RR, Coleman MP. Sarm1 deletion, but not Wlds, confers lifelong rescue in a mouse model of severe axonopathy. Cell Rep. 2017;21:10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4(4):299–309. [DOI] [PubMed] [Google Scholar]
- [244].Yamashita N, Kuruvilla R. Neurotrophin signaling endosomes: biogenesis, regulation, and functions. Curr Opin Neurobiol. 2016;39:139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. [DOI] [PubMed] [Google Scholar]
- [246].Kuruvilla R, Ye H, Ginty DD. Spatially and functionally distinct roles of the PI3-K effector pathway during NGF signaling in sympathetic neurons. Neuron. 2000;27:499–512. [DOI] [PubMed] [Google Scholar]
- [247].Watson FL, Heerssen HM, Bhattacharyya A, et al. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci. 2001;4:981–988. [DOI] [PubMed] [Google Scholar]
- [248].Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11–14. [DOI] [PubMed] [Google Scholar]
- [249].Finkbeiner S, Tavazoie SF, Maloratsky A, et al. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. [DOI] [PubMed] [Google Scholar]
- [250].Geden MJ, Romero SE, Deshmukh M. Apoptosis versus axon pruning: molecular intersection of two distinct pathways for axon degeneration. Neurosci Res. 2019;139:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Courchesne SL, Karch C, Pazyra-Murphy MF, et al. Sensory neuropathy attributable to loss of Bcl-w. J Neurosci. 2011;31(5):1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Sharpe JC, Arnoult D, Youle RJ. Control of mitochondrial permeability by Bcl-2 family members. Biochim Biophys Acta, Mol Cell Res. 2004;1644:107–113. [DOI] [PubMed] [Google Scholar]
- [253].Avery MA, Rooney TM, Pandya JD, et al. WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr Biol. 2012;22(7):596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Cox LJ, Hengst U, Gurskaya NG, et al. Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat Cell Biol. 2008;10(2):149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Klim JR, Williams LA, Limone F, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Shin JE, Geisler S, DiAntonio A. Dynamic regulation of SCG10 in regenerating axons after injury. Exp Neurol. 2014;252:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Rehorst WA, Thelen MP, Nolte H, et al. Muscle regulates mTOR dependent axonal local translation in motor neurons via CTRP3 secretion: implications for a neuromuscular disorder, spinal muscular atrophy. Acta Neuropathol Commun. 2019;7:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Court FA, Coleman MP. Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci. 2012;35(6):364–372. [DOI] [PubMed] [Google Scholar]
- [259].Rawson RL, Yam L, Weimer RM, et al. Axons degenerate in the absence of mitochondria in C. elegans. Curr Biol. 2014;24:760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Delettre C, Lenaers G, Griffoin JM, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. [DOI] [PubMed] [Google Scholar]
- [261].Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Dauer WT, Worman HJ. The nuclear envelope as a signaling node in development and disease. Dev Cell. 2009;17:626–638. [DOI] [PubMed] [Google Scholar]
- [263].Lu J, Sharma LK, Bai Y. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res. 2009;19:802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Verhoeven K, Claeys KG, Züchner S, et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot–Marie–Tooth type 2. Brain. 2006;129:2093–2102. [DOI] [PubMed] [Google Scholar]
- [265].Bernard-Marissal N, van Hameren G, Juneja M, et al. Altered interplay between endoplasmic reticulum and mitochondria in Charcot–Marie–Tooth type 2A neuropathy. Proc Nat Acad Sci. 2019;116(6):2328–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Larrea D, Pera M, Gonnelli A, et al. MFN2 mutations in Charcot-Marie-Tooth disease alter mitochondria-associated ER membrane function but do not impair bioenergetics. Hum Mol Genet. 2019;28:1782–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Mandal A, Drerup CM. Axonal transport and mitochondrial function in neurons. Front Cell Neurosci. 2019;13:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Wong-Riley MT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. [DOI] [PubMed] [Google Scholar]
- [269].Rangaraju V, Lauterbach M, Schuman EM. Spatially stable mitochondrial compartments fuel local translation during plasticity. Cell. 2019;176:73–84.e15. [DOI] [PubMed] [Google Scholar]
- [270].Kuzniewska B, Cysewski D, Wasilewski M. et al. Mitochondrial protein biogenesis in the synapse is supported by local translation. EMBO Rep. 2020;21(8):e48882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Loreto A, Hill CS, Hewitt VL, et al. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol Dis. 2020;134:104678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Lemasters JJ, Nieminen A-L, Qian T, et al. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta Bioenerg. 1998;1366:177–196. [DOI] [PubMed] [Google Scholar]
- [273].Hillefors M, Gioio AE, Mameza MG, et al. Axon viability and mitochondrial function are dependent on local protein synthesis in sympathetic neurons. Cell Mol Neurobiol. 2007;27:701–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Roque CG, Wong HH, Lin JQ, et al. Tumor protein Tctp regulates axon development in the embryonic visual system. Development. 2016;143:1134–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Cosker KE, Fenstermacher SJ, Pazyra-Murphy MF, et al. The RNA-binding protein SFPQ orchestrates an RNA regulon to promote axon viability. Nat Neurosci. 2016;19(5):690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Thomas-Jinu S, Gordon PM, Fielding T, et al. Non-nuclear pool of splicing factor SFPQ regulates axonal transcripts required for normal motor development. Neuron. 2017;94:322–336.e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Cobley JN, Fiorello ML, Bailey DM. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018;15:490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Aas PA, Otterlei M, Falnes PØ, et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003;421(6925):859–863. [DOI] [PubMed] [Google Scholar]
- [279].Simms CL, Zaher HS. Quality control of chemically damaged RNA. Cell Mol Life Sci. 2016;73:3639–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Weimann A, Belling D, Poulsen HE. Quantification of 8-oxo-guanine and guanine as the nucleobase, nucleoside and deoxynucleoside forms in human urine by high-performance liquid chromatography-electrospray tandem mass spectrometry. Nucleic Acids Res. 2002;30:E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Yan LL, Zaher HS. How do cells cope with RNA damage and its consequences? J Biol Chem. 2019;294:15158–15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Ding Q, Dimayuga E, Keller JN. Oxidative stress alters neuronal RNA- and protein-synthesis: implications for neural viability. Free Radic Res. 2007;41:903–910. [DOI] [PubMed] [Google Scholar]
- [283].Ding Q, Markesbery WR, Chen Q, et al. Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci. 2005;25:9171–9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem. 2015;30:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Broedbaek K, Ribel-Madsen R, Henriksen T, et al. Genetic and environmental influences on oxidative damage assessed in elderly Danish twins. Free Radic Biol Med. 2011;50(11):1488–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Chang Y, Kong Q, Shan X, et al. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS One. 2008;3(8):e2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Kong Q, Shan X, Chang Y, et al. RNA oxidation: a contributing factor or an epiphenomenon in the process of neurodegeneration. Free Radic Res. 2008;42:773–777. [DOI] [PubMed] [Google Scholar]
- [288].Nunomura A, Chiba S, Kosaka K, et al. Neuronal RNA oxidation is a prominent feature of dementia with Lewy bodies. Neuroreport. 2002;13:2035–2039. [DOI] [PubMed] [Google Scholar]
- [289].Nunomura A, Perry G, Pappolla MA, et al. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci. 1999;19:1959–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Shan X, Chang Y, Lin CL. Messenger RNA oxidation is an early event preceding cell death and causes reduced protein expression. Faseb J. 2007;21:2753–2764. [DOI] [PubMed] [Google Scholar]
- [291].Liu J, Head E, Gharib AM, et al. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha-lipoic acid. Proc Natl Acad Sci U S A. 2002;99:2356–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].Nie B, Gan W, Shi F, et al. Age-dependent accumulation of 8-oxoguanine in the DNA and RNA in various rat tissues. Oxid Med Cell Longev. 2013;2013:303181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- [294].Rotunno MS, Bosco DA. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. 2013;7:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Zemlan FP, Thienhaus OJ, Bosmann HB. Superoxide dismutase activity in Alzheimer’s disease: possible mechanism for paired helical filament formation. Brain Res. 1989;476:160–162. [DOI] [PubMed] [Google Scholar]
- [296].Vande Velde C, McDonald KK, Boukhedimi Y, et al. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS One. 2011;6:e22031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Cohen TJ, Hwang AW, Unger T, et al. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. Embo J. 2012;31(5):1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Cohen TJ, Hwang AW, Restrepo CR, et al. An acetylation switch controls TDP-43 function and aggregation propensity. Nat Commun. 2015;6(1):5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Kedersha NL, Gupta M, Li W, et al. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;147:1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Tourriere H, Chebli K, Zekri L, et al. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol. 2003;160:823–831. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [301].Stohr N, Lederer M, Reinke C, et al. ZBP1 regulates mRNA stability during cellular stress. J Cell Biol. 2006;175:527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Gowrishankar G, Winzen R, Dittrich-Breiholz O, et al. Inhibition of mRNA deadenylation and degradation by different types of cell stress. Biol Chem. 2006;387:323–327. [DOI] [PubMed] [Google Scholar]
- [303].Kedersha N, Chen S, Gilks N, et al. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell. 2002;13:195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Wolozin B, Ivanov P. Stress granules and neurodegeneration. Nat Rev Neurosci. 2019;20:649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Bley N, Lederer M, Pfalz B, et al. Stress granules are dispensable for mRNA stabilization during cellular stress. Nucleic Acids Res. 2015;43(4):e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Wheeler JR, Matheny T, Jain S, et al. Distinct stages in stress granule assembly and disassembly. Elife. 2016;5:e18413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Mokas S, Mills JR, Garreau C, et al. Uncoupling stress granule assembly and translation initiation inhibition. Mol Biol Cell. 2009;20:2673–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Sahoo PK, Lee SJ, Jaiswal PB, et al. Axonal G3BP1 stress granule protein limits axonal mRNA translation and nerve regeneration. Nat Commun. 2018;9:3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Maziuk B, Ballance HI, Wolozin B. Dysregulation of RNA binding protein aggregation in neurodegenerative disorders. Front Mol Neurosci. 2017;10:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].St George-Hyslop P, Lin JQ, Miyashita A, et al. The physiological and pathological biophysics of phase separation and gelation of RNA binding proteins in amyotrophic lateral sclerosis and fronto-temporal lobar degeneration. Brain Res. 2018;1693:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Kim HJ, Raphael AR, LaDow ES, et al. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Radford H, Moreno JA, Verity N, et al. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015;130:633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Wolozin B. Physiological protein aggregation run amuck: stress granules and the genesis of neurodegenerative disease. Discov Med. 2014;17:47–52. [PMC free article] [PubMed] [Google Scholar]
- [314].Janssens J, Wils H, Kleinberger G, et al. Overexpression of ALS-associated p.M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol Neurobiol. 2013;48:22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [315].Vanderweyde T, Yu H, Varnum M, et al. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci. 2012;32:8270–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Kedersha N, Cho MR, Li W, et al. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol. 2000;151:1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Markmiller S, Soltanieh S, Server KL, et al. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell. 2018;172:590–604 e513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Protter DSW, Parker R. Principles and properties of stress granules. Trends Cell Biol. 2016;26:668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [319].Hay DG, Sathasivam K, Tobaben S, et al. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet. 2004;13:1389–1405. [DOI] [PubMed] [Google Scholar]
- [320].Wyttenbach A, Carmichael J, Swartz J, et al. Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington’s disease. Proc Natl Acad Sci U S A. 2000;97:2898–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. [DOI] [PubMed] [Google Scholar]
- [322].Halliday M, Mallucci GR. Review: modulating the unfolded protein response to prevent neurodegeneration and enhance memory. Neuropathol Appl Neurobiol. 2015;41:414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [323].Baleriola J, Walker CA, Jean YY, et al. Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell. 2014;158(5):1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [324].Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [325].Han J, Kaufman RJ. Physiological/pathological ramifications of transcription factors in the unfolded protein response. Genes Dev. 2017;31:1417–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Lindholm D, Korhonen L, Eriksson O, et al. Recent insights into the role of unfolded protein response in ER stress in health and disease. Front Cell Dev Biol. 2017;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Dalton RP, Lyons DB, Lomvardas S. Co-opting the unfolded protein response to elicit olfactory receptor feedback. Cell. 2013;155:321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [328].Di Prisco GV, Huang W, Buffington SA, et al. Translational control of mGluR-dependent long-term depression and object-place learning by eIF2alpha. Nat Neurosci. 2014;17:1073–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [329].Cagnetta R, Wong HH-W, Frese CK, et al. Noncanonical modulation of the eIF2 pathway controls an increase in local translation during neural wiring. Mol Cell. 2019;73(3):474–489 e475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Bagni C, Zukin RS. A synaptic perspective of fragile X syndrome and autism spectrum disorders. Neuron. 2019;101(6):1070–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [331].Mallucci GR. Prion neurodegeneration: starts and stops at the synapse. Prion. 2009;3:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [332].Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends Neurosci. 2010;33:513–523. [DOI] [PubMed] [Google Scholar]
- [333].Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. [DOI] [PubMed] [Google Scholar]
- [334].Fischer LR, Culver DG, Tennant P, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. [DOI] [PubMed] [Google Scholar]
- [335].Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480(7375):63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [336].Mallucci GR, White MD, Farmer M, et al. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron. 2007;53:325–335. [DOI] [PubMed] [Google Scholar]
- [337].Marzo A, Galli S, Lopes D, et al. Reversal of synapse degeneration by restoring wnt signaling in the adult hippocampus. Curr Biol. 2016;26:2551–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [338].Sydow A, Van der Jeugd A, Zheng F, et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 2011;31:2511–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [339].Kim E, Jung H. Local protein synthesis in neuronal axons: why and how we study. BMB Rep. 2015;48:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [340].Farias J, Holt CE, Sotelo JR, et al. Axon microdissection and transcriptome profiling reveals the in vivo RNA content of fully differentiated myelinated motor axons. RNA. 2020;26:595–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [341].Maciel R, Bis DM, Rebelo AP, et al. The human motor neuron axonal transcriptome is enriched for transcripts related to mitochondrial function and microtubule-based axonal transport. Exp Neurol. 2018;307:155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [342].Batista AFR, Martinez JC, Hengst U. Intra-axonal synthesis of SNAP25 is required for the formation of presynaptic terminals. Cell Rep. 2017;20(13):3085–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [343].Poulopoulos A, Murphy AJ, Ozkan A, et al. Subcellular transcriptomes and proteomes of developing axon projections in the cerebral cortex. Nature. 2019;565:356–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [344].Alvarez-Castelao B, Schanzenbacher CT, Hanus C, et al. Cell-type-specific metabolic labeling of nascent proteomes in vivo. Nat Biotechnol. 2017;35:1196–1201. [DOI] [PubMed] [Google Scholar]
- [345].Alvarez-Castelao B, Schanzenbacher CT, Langer JD, et al. Cell-type-specific metabolic labeling, detection and identification of nascent proteomes in vivo. Nat Protoc. 2019;14(2):556–575. [DOI] [PubMed] [Google Scholar]
- [346].Erdmann I, Marter K, Kobler O, et al. Cell-selective labelling of proteomes in Drosophila melanogaster. Nat Commun. 2015;6:7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [347].Gibbs KL, Kalmar B, Sleigh JN, et al. In vivo imaging of axonal transport in murine motor and sensory neurons. J Neurosci Methods. 2016;257:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [348].Deubner J, Coulon P, Diester I. Optogenetic approaches to study the mammalian brain. Curr Opin Struct Biol. 2019;57:157–163. [DOI] [PubMed] [Google Scholar]
- [349].van Bergeijk P, Adrian M, Hoogenraad CC, et al. Optogenetic control of organelle transport and positioning. Nature. 2015;518:111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [350].Shin Y, Berry J, Pannucci N, et al. Spatiotemporal control of intracellular phase transitions using light-activated optoDroplets. Cell. 2017;168:159–171 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [351].Lu H, Mazumder M, Jaikaran ASI, et al. A yeast system for discovering optogenetic inhibitors of eukaryotic translation initiation. ACS Synth Biol. 2019;8:744–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [352].Huber KM, Gallagher SM, Warren ST, et al. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [353].Davis AA, Leyns CEG, Holtzman DM. Intercellular spread of protein aggregates in neurodegenerative disease. Annu Rev Cell Dev Biol. 2018;34:545–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [354].Barton SK, Gregory JM, Chandran S, et al. Could an impairment in local translation of mRNAs in glia be contributing to pathogenesis in ALS? Front Mol Neurosci. 2019;12:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [355].Smith HL, Freeman OJ, Butcher AJ, et al. Astrocyte unfolded protein response induces a specific reactivity state that causes non-cell-autonomous neuronal degeneration. Neuron. 2020;105(5):855–866.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [356].Brown GC, Vilalta A. How microglia kill neurons. Brain Res. 2015;1628(Part B):288–297. [DOI] [PubMed] [Google Scholar]
- [357].Lewerenz J, Maher P. Chronic glutamate toxicity in neurodegenerative diseases—what is the evidence? Front Neurosci. 2015;9:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [358].Most D, Workman E, Harris RA. Synaptic adaptations by alcohol and drugs of abuse: changes in microRNA expression and mRNA regulation. Front Mol Neurosci. 2014;7:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [359].Oliver RJ, Brigman JL, Bolognani F, et al. Neuronal RNA-binding protein HuD regulates addiction-related gene expression and behavior. Genes Brain Behav. 2018;17:e12454–e12454. [DOI] [PMC free article] [PubMed] [Google Scholar]