

ABSTRACT
RNA-binding proteins are a critical group of multifunctional proteins that precisely regulate all aspects of gene expression, from alternative splicing to mRNA trafficking, stability, and translation. Converging evidence highlights aberrant RNA metabolism as a common pathogenic mechanism in several neurodevelopmental and neurodegenerative diseases. However, dysregulation of disease-linked RNA-binding proteins results in widespread, often tissue-specific and/or pleiotropic effects on the transcriptome, making it challenging to determine the underlying cellular and molecular mechanisms that contribute to disease pathogenesis. Understanding how splicing misregulation as well as alterations of mRNA stability and localization impact the activity and function of neuronal proteins is fundamental to addressing neurodevelopmental defects and synaptic dysfunction in disease. Here we highlight recent exciting studies that use high-throughput transcriptomic analysis and advanced genetic, cell biological, and imaging approaches to dissect the role of disease-linked RNA-binding proteins on different RNA processing steps. We focus specifically on efforts to elucidate the functional consequences of aberrant RNA processing on neuronal morphology, synaptic activity and plasticity in development and disease. We also consider new areas of investigation that will elucidate the molecular mechanisms RNA-binding proteins use to achieve spatiotemporal control of gene expression for neuronal homeostasis and plasticity.
KEYWORDS: RNA-binding proteins, neurobiology/neurological disease, splicing in neurodevelopment, mRNA stability and localization in synaptic function, Local translation, FMRP, RBFOX1, SMN, TDP-43, FUS
Introduction
The mammalian central nervous system requires a high degree of spatial and temporal control of gene expression for neuronal function, development, and plasticity. Each neuron within the brain possesses the extraordinary ability to modify synaptic function and connectivity in response to stimuli. Furthermore, the size, polarity and structural complexity of neurons present unique challenges for fulfilling critical functions in neurodevelopment and synapse plasticity. While regulation at the level of transcription is fundamentally important for establishing and maintaining the structural and functional complexity of the brain, neuronal development and synaptic function also require robust post-transcriptional mechanisms to direct neuronal cell-type specification and enable rapid adaptation to changing neuronal demands. Post-transcriptional regulation of RNA, including mRNA alternative splicing, stability, and trafficking, allows for precise spatiotemporal and activity-dependent control of gene expression in neurons [1]. RNA-binding proteins serve as critical trans-acting factors that regulate every step in the RNA life-cycle and profoundly impact neurodevelopment and synaptic function (Fig. 1).
Figure 1.
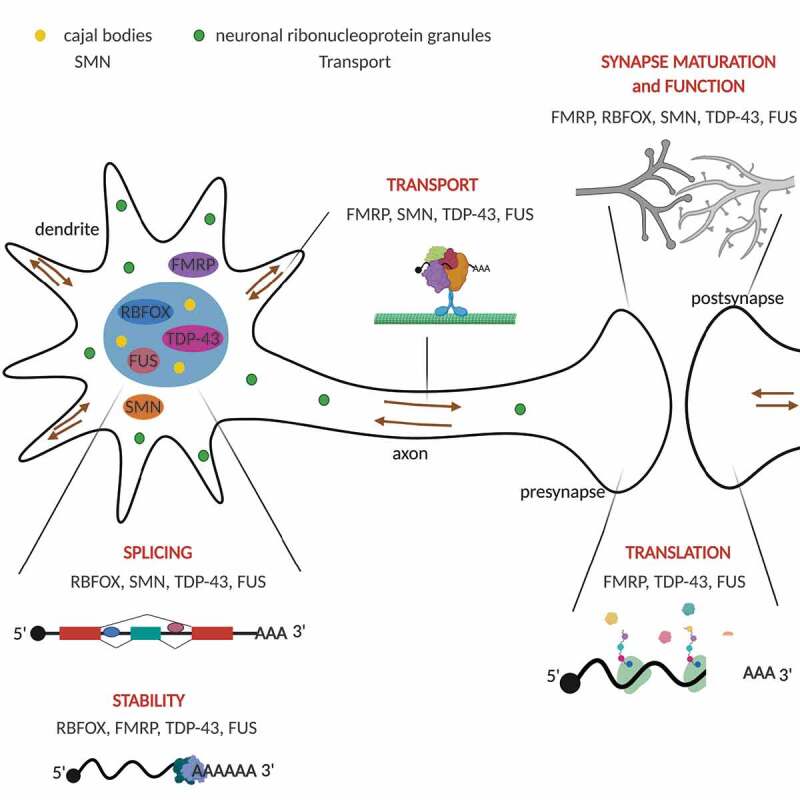
Dysfunction of RNA-binding proteins has profound effects on the neuronal transcriptome. RNA-binding proteins precisely regulate mRNA processing, stability, transport and translation to meet critical functions in neurodevelopment, synaptic function, and plasticity. In this review, we discuss how the RNA-binding proteins, RBFOX1, FMRP, SMN, TDP-43 and FUS, regulate myriad aspects of RNA metabolism and impact neurodevelopment, synapse homeostasis, and the neuronal cytoskeleton. The figure depicts the nuclear localization and functions of RBFOX1, TDP-43 and FUS, whereas SMN and FMRP are mainly localized in the cytoplasm. SMN also is a component of Cajal bodies in the nucleus. RNA-binding proteins are components of neuronal ribonucleoprotein granules that function in the bidirectional transport, localization and/or translation of mRNAs in the dendrites and axon
RNA processing events in the brain create a functionally diverse proteome and also provide a method to adjust transcript levels and dynamics in response to stimuli. RNA-binding proteins regulate alternative splicing of transcripts, and thus contribute to vertebrate transcriptome diversity. In neurons, axons and dendrites have distinct transcriptomes and rely on RNA-binding proteins to regulate transport and translation of mRNA during development and in response to activity-dependent synaptic plasticity. The importance of these multifunctional proteins in neuronal development and function is highlighted by the fact that numerous RNA-binding proteins have been linked to neurodevelopmental and neurodegenerative disorders. Transcriptomic studies in human brain tissue indicate that widespread alterations in RNA processing are a common feature of neurodevelopmental and neurodegenerative diseases, and are often linked to the dysregulation of RNA binding proteins [2-5]. Recognizing how splicing misregulation and alterations of mRNA stability and localization impact the activity and function of neuronal proteins is fundamental to understanding the pathogenesis of neurodevelopmental defects and synaptic dysfunction in disease. In this review, we focus on recent exciting work that combines advances in RNA-sequencing and transcriptomic analysis with cell biology, genetics and high-resolution imaging strategies to elucidate the functional implications of RNA-binding protein-dependent post-transcriptional regulation for neuronal development, synaptic function and plasticity.
Neurodevelopment
Autism spectrum disorders: FMRP, RBFOX1
Autism spectrum disorder (ASD), a common but clinically and genetically heterogeneous neurodevelopmental condition, primarily affects social interactions and is associated with restricted behaviours and interests [6,7]. Intellectual disability, atypical language development, and epilepsy are often co-occurring conditions [6]. The genetics of ASD are complex, but approximately 6-10% of individuals with ASD have a known genetic syndrome with Mendelian inheritance pattern [7]. For example, silencing/disruption of the FMR1 gene (discussed below) causes Fragile X syndrome and is the most common monogenic cause of autism [7]. In addition, rare chromosomal abnormalities and copy number variations (CNVs) contribute genetic risk in 5-10% of ASD cases [7]. Genetic studies in patients with autism have identified either chromosomal translocations or CNVs involving RBFOX1, which encodes a highly conserved member of the RNA-binding Fox (RBFOX) family of RNA-binding proteins, and suggest that RBFOX1 is an autism susceptibility gene [8,9]. Thus, reduction and/or alterations in the expression of Fragile X mental retardation protein (FMRP), encoded by FMR1, and RBFOX1, both critical RNA-binding proteins in neurodevelopment, are strongly implicated in ASD.
Despite a high degree of genetic complexity, evidence from structural and functional neuroimaging studies, information processing, and molecular genetics all suggest that ASD is characterized by atypical neural connectivity both at the systems and cellular level [6]. Shared molecular features in ASD include splicing changes and altered stability of transcripts enriched for synaptic transmission and function, suggesting disruption of common cellular pathways involved in neuronal connectivity [3]. In this section, we discuss functions of FMRP and RBFOX1, highly conserved RNA-binding proteins which share overlapping RNA targets. In particular, we highlight the roles of FMRP in mRNA transport and translation regulation that are required for synapse development and neuronal plasticity. Furthermore, we discuss the functional implications of RBFOX1-mediated alternative splicing and mRNA stabilization in neurodevelopment and establishing neural connectivity. Lastly, we present a recently appreciated function of RBFOX and other RNA-binding proteins in the regulation of neural microexons and discuss their relevance in ASD.
Fragile X mental retardation protein (FMRP)
Fragile X syndrome (FXS) is the most prevalent inherited cause of intellectual impairment, developmental delay, and ASD. Patients with FXS commonly exhibit additional features associated with autism such as hyperactivity and epilepsy [10]. FXS arises from microsatellite repeat expansion of cytosine-guanine-guanine (CGG) triplets in the 5ʹ untranslated region (UTR) of the Fragile X mental retardation 1 (FMR1) gene. While the functional FMR1 gene contains 5-45 CGG repeats, the disease-linked FMR1 in FXS has more than 200 CGG repeats and is transcriptionally silenced as a result of these repeat expansions. Some mechanisms proposed to silence transcription of FMR1 include i) abnormal DNA methylation of CpG islands proximal to and within CGG repeats [11-13], ii) deacetylation and dimethylation of histones prior to DNA methylation [14,15], and iii) the formation of FMR1CGG mRNA::DNA duplexes that induce FMR1 promoter silencing [16]. Transcriptional silencing of the FMR1 gene by the aforementioned mechanisms alters FMR1 mRNA levels leading to diminished synthesis of the RNA-binding protein, Fragile X mental retardation protein (FMRP). In male FXS patients with normal length CGG repeats, recent advances in sequencing technologies have identified rare missense mutations at conserved positions in coding regions of FMRP [17]; Fig. 2. The point mutations result in FXS by interfering with FMRP’s RNA-binding function, as discussed below and more in-depth by a recent review [17]. Therefore, loss of FMRP expression and/or loss of function, due to CGG repeats or pathogenic mutations in the FMR1 gene, directly cause FXS phenotypes and associated clinical features of autism.
Figure 2.
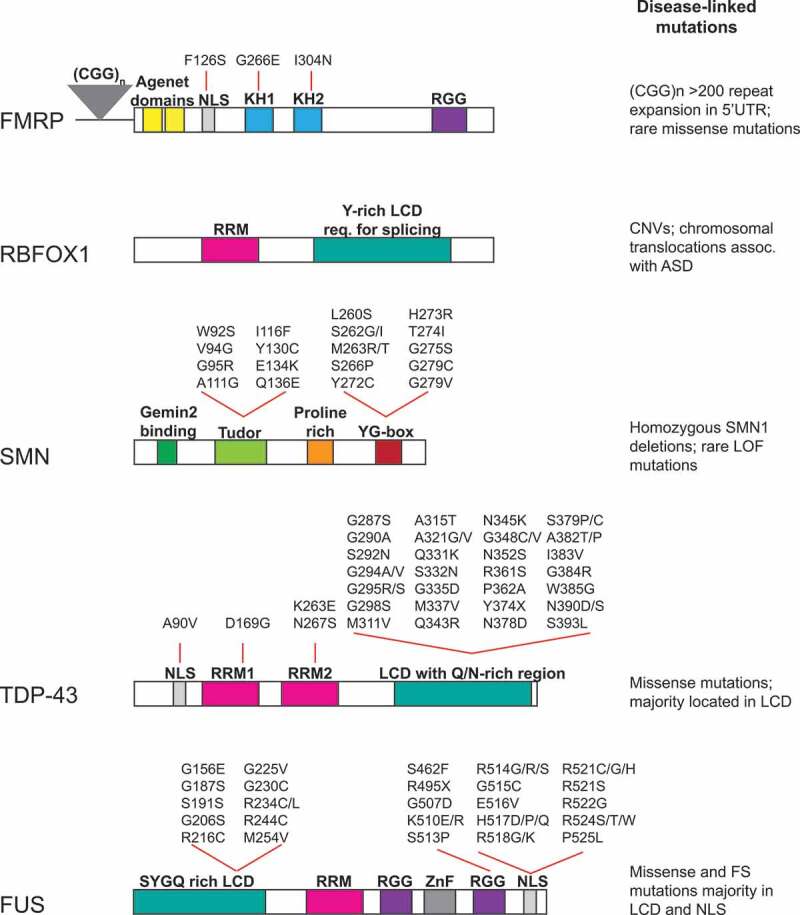
Schematic of RNA-binding protein domain structure, mutations, and disease mechanism. RBFOX1, FMRP, SMN, TDP-43 and FUS functional domains and locations of disease-linked mutations are highlighted. Abbreviations used: Nuclear localization signal (NLS); hnRNP K protein homology (KH); arginine-glycine-glycine box (RGG); RNA recognition motif (RRM); Low complexity domain (LCD); Zinc finger (ZnF); Tyrosine- and glycine-rich region (YG-box); Frameshift (FS); Loss of function (LOF)
FMRP is predominantly expressed in granule layers of the hippocampus and cerebellum during early stages of neurodevelopment [18]. FMRP comprises two Agenet protein interaction domains, nuclear localization/export signals, and three functionally conserved RNA-binding domains, namely the two hnRNP K protein homology (KH1 and KH2) motifs and an arginine-glycine-glycine (RGG) box [17,19,20]; Fig. 2. FXS-linked missense mutations (e.g. Gly266Glu, Ile304Asn) present within the conserved KH1 and KH2 domains, respectively, impair RNA binding and association of FMRP with polyribosomes [17]. These clinical genetic data underscore the importance of the KH domains for FMRP’s functions. FMRP binds preferentially to WGGA, GACR and TAY motifs in the coding region of target mRNAs and the 3ʹ UTR of a subset of these mRNAs [21-24]; Table 1. The severe neurodevelopmental disorders linked to the reduction of FMRP and/or its loss of function suggest that FMRP performs multiple essential RNA regulatory functions [10,17]; here we focus on FMRP-dependent mRNA trafficking and translation regulation, which play critical roles in neurodevelopment and sustaining synapse function and plasticity.
Table 1.
RNA-binding protein consensus motifs and target transcript characteristics
| RNA-bindingprotein |
Binding motif(s) |
Predominant regions of binding within target transcripts |
| FMRP | WGGA, GACR and TAY; G-quadruplex | Coding regions and 3’ UTR in a subset of transcripts |
| RBFOX1 | (U)GCAUG | Introns and 3’ UTR |
| TDP-43 | UG repeats; G-quadruplex |
Introns; 3’ UTR and 5’UTR |
| FUS | GGUG; G-quadruplex |
Introns; 3’UTR |
FMRP mediates activity-dependent synapse development and remodelling by regulating mRNA transport and translation
Neurons use several mechanisms to establish and maintain distinct mRNA pools in the dendrites and axon, allowing these cellular compartments to respond rapidly to local environmental cues [1]. Neuronal RNA transport granules, dynamic ribonucleoprotein complexes that contain mRNA, RNA-binding proteins, and molecular motors, regulate mRNA trafficking and local translation in subcellular compartments [25-27]. Loss of translational control is a recurring mechanism in FXS and ASD, and the inability to increase translation of specific localized mRNAs in response to physiologic stimuli plays a critical role in disease pathophysiology [1,10]. FMRP localizes to the soma and within dendrites, spines, and axonal growth cones as discrete cytoplasmic granules [28,29]. Work from many laboratories has established that FMRP is a component of neuronal RNA transport granules [25-27]. Consistent with these data, live-cell imaging studies have revealed that FMRP-positive neuronal granules are actively transported into the dendrites in a microtubule-dependent manner within minutes of neuronal stimulation [30,31]. FMRP granules colocalize with specific mRNAs, including Fmr1 and Map1b, and FMRP is required for transport of Map1b, CAMKIIa, GABA-AR, and SAPAP4 mRNAs into dendrites and spines in response to metabotropic glutamate receptor (mGluR) activation [28,30,31]. Activity-induced transport of FMRP granules and mRNA is dependent upon intracellular calcium signalling [28]. Subsequent post-translational modifications of FMRP, such as phosphorylation and SUMOylation, allow for its dissociation from target mRNAs, followed by local mRNA translation [32,33]. These data suggest mGluR stimulation and downstream calcium signalling activate FMRP-dependent mRNA transport and an uncoupling mechanism that subsequently enables local translation.
To further delineate the function of FMRP in activity-induced mRNA trafficking, additional studies have suggested a model in which FMRP acts as an adaptor to facilitate the association between molecular motors and FMRP target mRNAs. Biochemical studies show that FMRP interacts with KIF5, KIF3C and cytoplasmic dynein [26,27,34]. Overexpression of the kinesin light chain (KLC) cargo binding domain or C-terminal region of FMRP that interacts with kinesin, both of which are expected to act in a dominant negative fashion according to this model, disrupt activity-dependent transport of FMRP and CAMKIIa mRNA into the dendrites. In the absence of FMRP, the steady-state levels of target mRNAs are not dramatically altered, but kinesin heavy chain immunoprecipitation from Fmr1 knockout (KO) mouse brains shows reduced pull-down of some, but not all, FMRP target mRNAs compared to wild type brains [30]. Thus, FMRP regulates the dendritic localization of mRNA in response to metabotropic glutamate receptor stimulation and may also enhance the association of a subset of target mRNAs with kinesin motors in order to facilitate their activity-dependent transport.
FMRP-dependent mRNA trafficking plays an important role in development of dendritic spines and is highly relevant to the FXS disease phenotype. Dendritic spines show immature morphology, with more long, thin spines than mature mushroom-shaped spines in the cortex of FXS patients [35]. In developing neurons, FMRP is present at dendritic spine protrusions that make contact with synapses. Upon stimulation, wild type neurons exhibit a marked rise in spine-synapse contact sites. In contrast, Fmr1 KO neurons exhibit an excess of long protrusions or immature filopodia-like spines which fail to make contact with synapses at baseline, and are also unable to increase synaptic contacts in response to stimulation [29]. Overexpression of a dominant negative FMRP construct not only disrupts activity-dependent localization of CAMKIIa mRNA into the dendrites, but also recapitulates these key morphologic defects seen in FXS patients and animal models [30,35]. While these studies focus on FMRP-mediated mRNA localization as a prerequisite for dendritic development and activity-dependent remodelling, they also strongly imply that FMRP’s role in translation regulation is highly intertwined in these developmental processes.
FMRP is essential for translation homeostasis
Elevated baseline global protein synthesis is a key pathologic phenotype in the brains of FXS patients and Fmr1 deficient mice, suggesting that FMRP is a translational repressor [10]. As a result, in the absence of FMRP there is diminished capacity to further increase translation in a spatiotemporal and/or activity-dependent manner (i.e. reduced “signal-to-noise” ratio) [10]. FMRP has been proposed to regulate translation via several different mechanisms, including at translation initiation, elongation, and through interactions with microRNAs (miRNAs). FMRP represses translation initiation through its interactions with cytoplasmic FMRP-interacting protein 1 (CYFIP1) at the 5ʹ end of mRNA; in turn, CYFIP1 blocks the association of eIF4G and eIF4E translation initiation factors and ribosome tethering. In response to mGluR signalling, eIF4E is released from the FMRP/CYFIP1 complex and translation repression is released [36]. FMRP also associates with the translation apparatus and stalls ribosome transit on target mRNAs, thus inhibiting translation elongation; FMRP phosphorylation-state may regulate this form of translation regulation [23,37,38]. The FMRP Ile304Asn mutation abolishes its ability to associate with polyribosomes [39]. FMRP loss of function may also de-repress translation indirectly through dysregulation of diacylglycerol-mediated lipid signalling [40] or mTORC1 signalling pathways [10,23]. In addition, FMRP cooperates with RNA-induced silencing complex (RISC) and miRNAs to repress translation through the RNA interference pathway [41-43]. Functional interactions between FMRP and miRNA125a, miRNA-125b, and miRNA-132 result in distinct effects on dendritic spine morphology; these effects are mediated by regulating the translation of mRNAs encoding PSD-95 and NMDA receptor subunit NR2A [42,43]. As a whole, these studies show how FMRP regulates mRNA trafficking and translation through a variety of mechanisms in the brain to affect dendritic morphology and synapse development in FXS.
RBFOX1
RBFOX1, a large (1.7 Mb) gene located on chromosome 16, was first implicated in autism because chromosomal translocations and CNVs resulting in disruption or loss of RBFOX1 were identified in patients with ASD [8,9]. Mutations and deletions in RBFOX1 also have been linked with epilepsy and intellectual disability, which may be comorbid with ASD [44]. More recently, weighted gene co-expression network analysis in cerebral cortex from patients with ASD identified RBFOX1 as a central hub for regulating co-expressed neuronal and synaptic mRNA transcripts in autism. These human tissue studies also demonstrated reduced levels of RBFOX1 and dysregulation of RBFOX1-dependent alternative splicing in ASD brains [3].
RBFOX1, also known as Ataxin-2 binding protein 1 [A2BP1], is exclusively expressed in neurons, heart, and muscle, and has two mammalian paralogs, RBFOX2 (also known as RBM9), and RBFOX3/NeuN [45]. RBFOX proteins include a single, highly conserved RNA recognition motif (RRM domain) that binds (U)GCAUG hexanucleotide with remarkable specificity. In addition, a tyrosine-rich low complexity domain (LCD) is required for RBFOX1-mediated splicing [45-47] (Fig. 2; Table 1). Crosslinking immunoprecipitation and RNA-seq (CLIP-seq) and functional studies in knockout mouse models demonstrate Rbfox1 plays a critical role in alternative splicing and stabilization of transcripts encoding proteins important for neurotransmission [48-50]. Rbfox1 itself is alternatively spliced to generate nuclear and cytoplasmic isoforms, and the ratio of these isoforms is regulated by cellular depolarization [51]. The presence of Rbfox1 CLIP sites in both introns and 3ʹUTR further suggest Rbfox1 has distinct roles, regulating splicing in the nucleus, and mRNA stability in the cytoplasm. Moreover, the functional significance of nuclear and cytoplasmic Rbfox1 isoforms (Rbfox1_N and Rbfox1_C, respectively) in neuronal development and establishing connectivity is becoming clear through recent exciting work.
Rbfox1-dependent alternative splicing regulates critical neuronal functions
As a key splicing regulator, Rbfox1 profoundly influences synaptic function, neuronal excitability and maturation[48-53]. Rbfox1 and other RBFOX family members either promote or repress exon inclusion in target pre-mRNAs by binding UGCAUG elements in flanking introns. While UGCAUG motifs downstream of the alternatively spliced exon enhance exon inclusion, upstream motifs suppress exon inclusion [45]. During development, these Rbfox-dependent alternative splicing events are necessary for structural and electrophysiological maturation of neurons [52,53]. Rbfox1/Rbfox2/Rbfox3 triple knockout (tKO) mouse embryonic stem cell-derived motor neurons exhibit immature electrical properties and splicing profile characteristic of an embryonic developmental state. Accordingly, Rbfox tKO neurons show impaired axon initial segment assembly (AIS) because they fail to activate a developmentally regulated switch in the alternative splicing of Ankyrin G and several other genes encoding AIS components [52]. Likewise, Rbfox1 is necessary for establishing cortical interneuron inhibitory activity and contributes to cell-type specific alternative splicing programs in developing somatostatin- and parvalbumin-positive cortical interneurons [53]. Interestingly, the AIS and interneuron defects in Rbfox tKO motor neurons and Rbfox1 KO interneurons, respectively, could be rescued by expressing Rbfox1_N but not the cytoplasmic isoform [52,53], highlighting the importance of Rbfox1-dependent splicing regulation for these developmental processes. However, the molecular mechanisms underlying Rbfox1-mediated cell-type specific alternative splicing are not known and will be an intriguing area of future investigation.
Despite some overlap in RNA targets of RBFOX RNA-binding proteins, Rbfox1 appears to alternatively splice a unique subset of transcripts important for neuronal excitation and synaptic function [48,53]. In cellular models, chronic depolarization triggers Rbfox1-dependent alternative splicing of NMDA receptor 1 (Grin1 transcript), and promotes inclusion of an exon that regulates agonist binding [51]. As a result, Rbfox1 modulates membrane physiology and potentially deleterious effects of prolonged stimulation. Moreover, pan-neuronal loss of Rbfox1 (in nestin-cre;Rbfox1flox/flox conditional knockout mice), but not Rbfox2, causes increased seizure susceptibility, highlighting the functional consequences of Rbfox1 loss in vivo [48]. Mice with interneuron-specific loss of Rbfox1 are even more severely affected and die before 2 months of age, most likely from lethal seizures [53]. Molecularly, Rbfox1 loss results in differential splicing of multiple transcripts encoding ion channels and functional components of synaptic transmission [48,53]. Individual-nucleotide resolution crosslinking immunoprecipitation (iCLIP) confirmed that many of these alternatively spliced transcripts, including Grin1, voltage-gated potassium channel Kv4.3 (Kcnd3), L-type calcium channel subunit (Cacna1d) and synaptosomal-associated protein 25 (Snap25), are direct Rbfox1 targets [48]. Several of these RNA targets are also known to be affected in ASD [3]. Overall, these studies suggest that Rbfox1 plays a critical role in shaping excitatory and inhibitory synaptic function and neuronal connectivity.
Cytoplasmic Rbfox1: roles in mRNA stability and decay
Emerging evidence suggests that cytoplasmic Rbfox1 (Rbfox1_C) also plays an important role in regulating the stability of transcripts encoding synaptic proteins, including mRNAs affected in ASD. Rbfox knockdown and isoform-specific rescue experiments in primary mouse hippocampal neurons reveal that Rbfox1_C, but not the nuclear isoform, is required to maintain the expression level of hundreds of target mRNA transcripts [54]. Furthermore, RNA-seq of wildtype and Rbfox1 cKO mouse adult hippocampus combined with Rbfox1 iCLIP analysis of neuronal nuclear and cytoplasmic fractions have shown that Rbfox1_C directly binds one or more GCAUG clusters in the 3ʹUTR of target mRNAs [49,54]. These mRNAs are particularly enriched for ASD susceptibility genes as well as Gene Ontology (GO) terms of synaptic transmission and calcium signalling [54]. Neuron-specific vSNARE Vamp1, specifically expressed at inhibitory pre-synaptic terminals, is one of the most downregulated genes in the absence of Rbfox1. Rbfox1_C binding to multiple (U)GCAUG sites in the Vamp1 3ʹUTR rescues transcript levels both by increasing Vamp1 stability and also by blocking microRNA-9 (miR-9) mediated decay [49]. From a physiologic perspective, Vamp1 downregulation impairs inhibitory synaptic transmission and provides another example of how Rbfox1 regulates synaptic function at the cellular level and excitatory/inhibitory balance of electrical circuits to influence overall neuronal network function [49,54].
RBFOX1 and other RNA-binding proteins regulate neural-specific microexons implicated in ASD
As discussed above, alternative splicing and expression of genes important for neurodevelopment, maintaining synapse function, and plasticity are significantly altered in autism [3]. Recent work of great interest describes the frequent misregulation of neuronal microexons in the brains of patients with ASD [4]. Furthermore, new evidence suggests that dysregulation of RBFOX1 and other RNA-binding proteins contributes to the misusage of these neural microexons. Neural microexons are highly evolutionarily conserved exons of 3-27 nucleotides that typically preserve the reading frame within corresponding genes [4]. Neural microexons encode regions on protein surfaces that are adjacent to folded domains, suggesting that they could alter protein-protein interaction networks in neurogenesis and synapse biology. In this section, we discuss the emerging roles of neural microexons, their regulation by RNA-binding proteins, and possible functional significance.
Of the microexons that partake in alternative splicing events, approximately one-third of the neural-regulated microexons are significantly misregulated in idiopathic ASD patients [4]. RBFOX1 and additional RNA-binding proteins have been implicated in regulating the alternative splicing and inclusion of neural microexons. Li et al. 2015 found that RBFOX1 binding sites are predominantly located within 3ʹ flanking introns that surround short microexons [55]. The same group also noted that the inclusion of microexons by RBFOX and other potential splicing factors could result in protein sequence changes, thereby leading to alterations in protein-protein interaction. A distinct subset of neural microexons is tightly and frequently controlled by the RNA-binding protein and splicing factor, serine/arginine repetitive matrix protein 4 (SRRM4). Moreover, neuronal hyperexcitability downregulates the levels of SRRM4, leading to the skipping of microexons that overlaps with microexon splicing defects in ASD patients [56]. These studies suggest that RBFOX1, SRRM4, and additional RNA-binding proteins are important regulators of neural microexons.
While the functions of the neural-specific microexons are not well understood, recent work reports a critical role for a novel microexon identified in the neuronal isoform of eIF4G1. Expression of the eIF4G1 microexon controls the translation of synaptic proteins that are essential for synaptic plasticity, learning, and social behaviour [57]. Of note, the eIF4G1 microexon is ranked as one of the strongest microexons whose splicing is disrupted in brains of ASD patients. Interestingly, the eIF4G1 microexon promotes interactions with FMRP and other proteins within cytoplasmic ribonucleoprotein (RNP) granules, leading to ribosome stalling and reduced translation. This finding is consistent with the notion that microexons encode surface-accessible domains that can mediate protein-protein interactions. Now there is also evidence to support its functional relevance in activity-dependent regulation. The activity-dependent exclusion of the eIF4G1 microexon dissociates protein-protein interactions within RNP granules and as a result, translation of synaptic proteins is enhanced for fulfilling neurotransmission and cognitive functions [57]. Taken together, these data suggest that the spatiotemporal control of microexons is essential for remodelling of the neuronal proteome and for modulating synapse function. An in-depth understanding of microexons and their functional implications would provide insights into the pathogenesis of ASD.
Spinal muscular atrophy: SMN1
Spinal muscular atrophy (SMA) is the most frequent cause of inherited infant mortality. The most common form of SMA, characterized by lower motor neuron loss with progressive muscle weakness and atrophy, is caused by reduced expression of Survival Motor Neuron (SMN), an essential actor in the assembly of ribonucleoprotein (RNP) complexes [58,59]. Several studies have shown that SMN expression, which is particularly enriched in the brain and spinal cord, is highest during embryonic development and drops sharply postnatally, suggesting an absolute requirement for SMN in motor neuron and/or neuromuscular junction (NMJ) development and maintenance [60]. Consistent with this notion, SMN plays an essential, cell autonomous role in motor neuron axon outgrowth, branching, and pathfinding during zebrafish development [61]. In SMA mouse models, restoring SMN expression embryonically or early postnatally, but not at later time points, is effective in rescuing motor neuron defects. Similarly, in human studies restoration of SMN within the first six weeks of life is associated with marked clinical improvement [60,62]. Given this critical window of prenatal and neonatal SMN expression, SMA may be considered a disease of motor neuron development.
SMA, an autosomal recessive disorder, arises from large deletions, or less commonly missense mutations, of the survival motor neuron 1 (SMN1) gene [58,63] (Fig. 2). A C >T difference in the nucleotide sequence of the closely related SMN2 gene results in exon 7 exclusion in >80% of transcripts and expression of a truncated, unstable form of SMN [64,65]. Therefore, only a minority of SMN2-derived transcripts results in full-length SMN protein, and SMN2 gene is unable to compensate for SMN1 loss of function [65]. Nevertheless, SMA patients who possess multiple copies of the SMN2 gene exhibit less severe disease phenotype, suggesting that increasing the efficiency of exon 7 inclusion in SMN2 pre-mRNA or other methods to increase SMN protein levels would be promising targets for SMA therapy [60,66]. Indeed, antisense oligonucleotides to enhance SMN2 exon 7 inclusion have proven to be highly effective in treating SMA patients and increasing their survival [67]. Clinical trials of an adeno-associated virus nine (AAV9) mediated gene replacement therapy to restore SMN levels are also promising, and additional approaches to increase functional SMN are being investigated in preclinical studies [68]. These exciting new therapeutic strategies have been reviewed recently in detail [69].
Despite great bench-to-bedside clinical success of SMA therapeutics, the precise cellular mechanisms that determine how reduced SMN expression leads to motor neuron loss are not completely understood. This is largely because SMN1 loss has pleiotropic effects that disrupt multiple RNA processing steps. Detailed studies over many years have elucidated SMN protein’s essential role in bringing together a multiprotein complex (the SMN complex) that assembles and maintains critical spliceosome components [reviewed in [70]]. SMN self-oligomerizes to interact with Gemins, uridine-rich small nuclear RNAs (U snRNAs), and Sm proteins. Subsequently, the SMN complex incorporates Sm proteins onto U snRNAs to form core spliceosomal small nuclear ribonucleoproteins (U snRNPs) [71-75]. The ability of SMN to perform these roles requires several functional domains: a lysine-rich region responsible for binding to Gemin2; a conserved YG-box required for SMN oligomerization; and a Tudor domain which mediates association with dimethylarginines commonly found in Sm proteins (Fig. 2 [70]). Loss of SMN globally alters snRNA and snRNP stoichiometry, especially in the brain and spinal cord, and leads to widespread pre-mRNA splicing defects in SMA mouse models [76,77]. Injection of purified U snRNPs into SMN-deficient zebrafish embryos rescues motor neuron defects [78]. Of note, the minor (U12-dependent) spliceosome is preferentially affected, and the Stasimon gene, which is required for motor circuit development and function, is spliced by the minor spliceosome [77,79,80]. Moreover, Stasimon expression rescues motor axon abnormalities and synaptic function in SMN-deficient animal models [79]. In SMA motor neurons specifically, the splicing dysregulation affects transcripts critical for neuromuscular junction maintenance and synaptic pruning [81]. Thus, disruption of motor neuron-specific developmental splicing programs is one major contribution to motor neuron loss in SMA. However, SMN loss and disruption of snRNP assembly, particularly U1 snRNP, impact several additional steps of RNA processing [82-85]. U1 snRNP is required not only for pre-mRNA splicing, but also for suppression of premature cleavage and polyadenylation. Thus, U1 snRNP promotes full-length transcription elongation in the sense-coding direction [82-84].
Furthermore, a broader role for the SMN complex as a ribonucleoprotein (RNP) assemblysome is now recognized, which has raised the possibility that additional motor neuron-specific functions of the SMN protein in RNP assembly exist [60,85,86]. The long distance between the motor neuron soma and NMJ poses a unique challenge, and therefore, motor neurons are highly dependent upon axonal transport [87]. Indeed, previous observations suggest that SMN may also regulate assembly and/or function of neuronal RNA transport granules, a type of cytoplasmic mRNA-protein (mRNP) complex ([88-90]. SMN co-localizes and co-transports with several RNA-binding proteins that are components of neuronal RNA transport granules, including HuD and IMP1/ZBP [90-92]. β-actin and neuritin/cpg15 mRNAs have been isolated by immunoprecipitation from neuronal granules containing SMN and HuD [92]. Importantly, Smn deficient neurons show both reduced axonal β-actin and Gap43 mRNA and protein levels as well as impaired motor axon length and growth cone area [88,93]. Recent work using in situ trimolecular fluorescence complementation method has now revealed that Smn is necessary for assembly of neuronal transport granules in motor neurons, and SMA patient fibroblasts also show failure of β-actin mRNA/IMP1 association. These defects can be rescued by restoring SMN protein levels [94]. Together, these studies establish that SMN protein also has an important non-canonical role in cytoplasmic mRNP granule assembly and transport critical for motor axon development. However, the relative contribution of the many SMN-dependent RNA processing functions to SMA pathogenesis is difficult to ascertain.
Neurodegeneration
ALS/FTD: RNA-binding proteins with low-complexity domains
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a debilitating neurodegenerative disease characterized by the selective loss of motor neurons and muscle atrophy. In contrast, frontotemporal dementia (FTD) is defined by neurocognitive decline and changes in social behaviour, personality and speech due to degeneration of the frontal and temporal cortex. ALS and FTD are thought to exist on a disease continuum because they have shared clinical, genetic and pathological features [95,96]. About 50% of ALS patients present with cognitive impairment, whereas 30% of FTD patients develop motor deficits. While ~90% of ALS cases are sporadic, the remaining cases are familial and genetically linked to mutations in genes encoding RNA-binding proteins, including transactive DNA response binding protein of 43 kDa (TDP-43) and fused in sarcoma (FUS) (Fig. 2) [96-99]. In this section, we discuss the mechanisms by which TDP-43 and FUS modulate the neuronal transcriptome. Specifically, we focus on recent advances that elucidate the roles of TDP-43 and FUS in alternative splicing, mRNA stability, transport and translation, as relevant for neurodevelopment, neuronal cytoskeleton, and synaptic plasticity.
Transactive DNA response binding protein of 43 kDa (TDP-43)
Pathologic aggregates composed of TDP-43 define the molecular histopathology of the most common form of FTD and the vast majority (>95%) of ALS cases. Cytoplasmic inclusions of insoluble ubiquitinated, hyperphosphorylated TDP-43 with concomitant loss of nuclear TDP-43 defines the pathology of ALS and FTD [95]. Autosomal dominant missense mutations in TARDBP, the gene encoding TDP-43 have been linked to ~5% of familial ALS cases, and TDP-43 mutations are also found in rare sporadic cases of ALS and FTD [97]. Despite great progress towards understanding the myriad functions of TDP-43 in RNA processing, whether neurodegeneration associated with TDP-43 pathology results from loss-of-function mechanisms, gain-of-function mechanisms, or both, remains unclear.
TDP-43, a highly conserved DNA/RNA binding protein and a member of the heterogenous nuclear ribonucleoprotein (hnRNP) family, was initially identified as a transcriptional repressor linked to TAT-induced HIV-1 transcription [100]. TDP-43 is ubiquitously expressed and essential during embryonic development. TDP-43 is composed of a N terminal domain capable of dimerizing, two conserved RNA recognition motifs (RRMs), and a less conserved, disordered low complexity domain (LCD) comprising a prion-like glutamine-asparagine-rich (Q/N) domain and a glycine rich region at the C terminal [101,102]. The LCD is a hotspot for the majority of ALS-linked TDP-43 mutations (Fig. 2). Consistent with roles in both the nucleus and cytoplasm, TDP-43 has nuclear localization and export signals to enable its mobility between the nucleus and cytoplasm [103]. Comprehensive analyses of TDP-43 interaction with RNAs by multiple laboratories have revealed the importance of TDP-43 in regulating splicing and stability of mRNAs [5,104,105]. CLIP-seq and RNA immunoprecipitation approaches show that TDP-43 binds to (UG)n consensus binding motifs within introns of thousands of pre-mRNAs and regulates their alternative splicing and stability (Table). In particular, TDP-43 preferentially binds and stabilizes transcripts with very long introns (>100 kb), many of which encode synaptic proteins. Importantly, TDP-43 also binds to UG-rich clusters within untranslated regions and exons and affects the stability of these mRNA targets as well. Thus, TDP-43 depletion profoundly affects the neuronal transcriptome, particularly affecting genes involved in synaptic activity and neuronal survival.
TDP-43 regulates mRNA stability and represses cryptic exon inclusion
TDP-43 has a well-established role in regulating the stability of pre-mRNAs and mRNAs, particularly those encoding proteins required for neuronal maintenance, synaptic activity, and cytoskeletal function [5,104,106,107]. Work from many laboratories has shown that TDP-43 is able to regulate transcript stability through several distinct mechanisms. TDP-43 modulates the stability of its own mRNA and other transcripts through interactions at the 3ʹUTR. TDP-43 autoregulates its expression by binding to the 3ʹ UTR and destabilizes its mRNA for targeted degradation; this is highly dependent on the RRM1 and an amino acid sequence (aa 321-366) within the C terminal domain [108]. TDP-43 also differentially regulates the stability of other transcripts via binding to the 3ʹ UTR and in some cases, recruits deadenylases. While high levels of TDP-43 downregulate tau mRNA expression, TDP-43 promotes stabilization of the hNFL (neurofilament) and add2 (adducin) mRNAs, which encode cytoskeletal proteins [109-112].
Recent studies highlight a central mechanism of TDP-43 dependent transcript stabilization [113]. TDP-43 prevents the aberrant splicing of pre-mRNAs by binding to introns and repressing inclusion of cryptic exons, which are non-conserved regions of the genome flanked by microsatellite repeats. Strikingly, TDP-43 has an evolutionarily conserved function in recognizing cryptic exons even though the cryptic exons themselves are species and cell-type specific, and impair distinct molecular pathways in different cell types [113,114]. The inclusion of cryptic exons introduces frameshifts, premature stop codons and premature polyadenylation sites, which can subject the mRNA to nonsense mediated decay (NMD). When splicing repression is restored in TDP-43 depleted neurons, both suppression of cryptic exons and neuronal survival are rescued [113]. Thus, TDP-43 is necessary for repression of cryptic exons and is a principal mechanism of TDP-43 dependent transcript stabilization. Inclusion of cryptic exons is also a feature of ALS-FTD patient brains with TDP-43 pathology, but precisely how these transcriptomic alterations lead to disease is unclear. Two new independent studies begin to elucidate how changes at the level of RNA stability may impact the axon and neuronal cytoskeleton [106,107]. Both laboratories find that TDP-43 represses cryptic polyadenylation site within intron 1 of the stathmin-2 mRNA, a microtubule binding protein that is essential for axonal growth and regeneration. Depletion or mislocalization of TDP-43 introduces premature polyadenylation and non-functional stathmin-2 mRNA, which significantly suppresses axonal regeneration following injury [106,107]. Collectively these studies suggest that there are differential mechanisms by which TDP-43 regulates the stability of its target transcripts and in different cell types. Additional efforts are needed to further explore how the dysregulation of specific TDP-43 target transcripts impact neuronal function and survival. These insights will shed light on the cellular consequences of TDP-43 dysregulation in ALS and FTD pathogenesis.
Role of TDP-43 in mRNA transport
Like FMRP, TDP-43 also regulates the trafficking and localization of specific mRNAs in the dendrites and axon. While TDP-43 is predominantly localized to the nucleus, it is a component of neuronal RNA transport granules [115,116]. Neuronal granules composed of wild type TDP-43 are dynamic and exhibit bidirectional transport in the axon and dendrites, whereas granules composed of mutant TDP-43 show reduced motility and distinct biophysical properties [116-119]. These data suggest ALS-linked TDP-43 mutations alter the transport of specific mRNAs to distal compartments. Indeed, TDP-43 cotransports with specific mRNAs in the axon and dendrites, including Neurofilament (Nefl) mRNA, and localizes Rac1 mRNA to dendritic spines in the context of neuronal activity [117-120]. TDP-43 granules that co-transport with the neurofilament (nefl) mRNA display net anterograde movement in the distal axon, which is disrupted in the presence of TDP-43 ALS-linked mutations such as M337V, A315T and G298S [118]. TDP-43 binds UG repeats in the 3ʹUTR of Nefl and other mRNAs, but may also be able to recognize G-quadruplexes within the 3ʹ UTR of its target mRNAs. TDP-43 M337V mutations reduce the transport of G-quadruplex mRNAs, such as PSD-95 and CAMKII, in the distal neurites [121]. These data suggest that TDP-43 may also bind and transport mRNAs with specific secondary structures even though they lack canonical TDP-43 sequence recognition motifs. Taken together, these data highlight an important role for TDP-43 in mRNA trafficking and localization, including several transcripts that encode cytoskeletal or cytoskeletal associated proteins (e.g. Nefl, Map1b) [118,122]. However, the functional significance of mRNA transport defects in ALS/FTD remains unclear, and further work is needed to delineate the functional implications of TDP-43 dependent mRNA trafficking in cytoskeletal maintenance and axonal homeostasis.
Translation regulation of specific mRNAs by TDP-43
In addition to multiple functions in RNA stability and splicing, recent studies indicate that TDP-43 also regulates translation. Global proteome analysis and translating ribosome affinity purification (TRAP) assay reveal that TDP-43 interacts with translational components and associates with polyribosomes in actively translating cells [123,124]. TDP-43 can either enhance or repress translation of associated mRNAs. One recent study reported that TDP-43 enhances translation of camta1 (calmodulin binding transcription activator 1) and mig12 (Mid1 interacting protein 1) through interactions with the 5ʹ UTRs and 3ʹ UTRs of these transcripts [124]. In contrast, TDP-43 overexpression in a Drosophila model of ALS represses translation of futsch/map1b mRNA at neuromuscular junctions and reduces the number of synaptic boutons [122]. This recent work also highlights that TDP-43 may have differential mechanisms for enhancing or repressing the translation of specific mRNAs, possibly related to distinct 5ʹ and 3ʹ UTR properties. A potential mechanism of TDP-43 mediated translation repression is through an interaction with a ribosomal scaffolding protein, receptor activated C kinase 1 (RACK1). In sporadic ALS spinal cord samples, TDP-43 cytoplasmic inclusions contain RACK1, suggesting translation misregulation could be a contributing factor in motor neuron degeneration [125].
TDP-43 has been linked to the localization and translation regulation of specific mRNAs for regulating synaptic plasticity. The Rac1 mRNA is translationally repressed by co-operative interaction between TDP-43, FMRP and its interacting protein, CYFIP1 [126]. FMRP overexpression results in a reduction in the density of dendritic spines which can be rescued by TDP-43 depletion, suggesting that TDP-43 represses translation of specific mRNAs with the help of FMRP to regulate spinogenesis and neuronal plasticity [126]. Follow-up studies reported that TDP-43 is required for silencing of the Rac1 mRNA during transport until the granules are captured at dendritic spines for local translation [120]. Collectively, these studies indicate that TDP-43 modulates the localization and translation of specific mRNAs essential for maintaining synaptic plasticity. Future efforts are needed to expand the precise mechanisms by which TDP-43 granules traffic its target mRNAs for uncoupling and local translation at local compartments in neurons.
Fused in Sarcoma (FUS)
Autosomal dominant mutations in FUS account for 4-5% of familial ALS cases [96,98,99]. R521C, R521H and R521G in exon 15 of the FUS gene are the most common mutations across FUS-linked familial ALS cases [98,99]. Similar to TDP-43, there is nuclear loss of mutant FUS concomitant with cytoplasmic ubiquitinated FUS aggregates in spinal cord motor neurons. Interestingly, TDP-43 inclusions were not detected in the neurons containing FUS pathology [98]. FUS mutations also have not been found in the context of FTD, although a subset of cases contain FUS inclusions that are negative for TDP-43 [127]. Therefore, the mechanisms driving TDP-43 and FUS pathogenesis appear to occur independently.
FUS is a highly conserved 526 amino acid protein composed of a glycine, serine and tyrosine (Q/G/S/Y) rich N terminal low complexity domain, an RNA recognition motif, three interspersed arginine-glycine-glycine (RGG) rich motifs, a zinc finger domain, and a nuclear localization signal at the C terminal end (Fig. 2). FUS is highly expressed and tightly controlled in the central nervous system during development and homozygous loss leads to embryonic lethality [128]. FUS, like TDP-43, is mainly localized in the nucleus and regulates transcription and pre-mRNA splicing, but also shuttles to the cytoplasm where it modulates stability, transport, and translation of mRNAs linked to neuronal function.
FUS regulates alternative splicing and stabilizes transcripts involved in synapse maturation
FUS plays a critical role in alternative splicing of pre-mRNA, and FUS depletion results in widespread alterations in RNA metabolism. This is in part because FUS binds to highly conserved introns within genes that encode other RNA-binding proteins and regulates their alternative splicing [129,130]. RGG motifs within FUS are required to recognize GGUG motifs in introns and 3ʹ UTRs of target transcripts, though FUS may also recognize stable secondary structures such as G quadruplexes [129-134] (Fig. 2; Table). Remarkably, there is no significant overlap between FUS and TDP-43 splicing changes or RNA binding sites. Like TDP-43, however, FUS preferentially stabilizes transcripts with very long introns (>100 kb); these genes are enriched for pathways linked to neurotransmission, exocytosis, vesicle-mediated transport and membrane organization. Of note, FUS maintains mRNA stability of specific mRNAs that regulate dendritic spine morphology and synaptic transmission [135,136]. FUS and ELAVL proteins cooperatively bind the long 3ʹ UTR of SynGAPα2 (synaptic GTPase activating protein alpha 2) mRNA and stabilize its levels in the postsynaptic density [135]. Depletion of FUS alters the expression of SynGAPα2, which leads to abnormal spine morphology. FUS also regulates the stability of the GluA1 mRNA which encodes an AMPA receptor subunit by recruiting factors to prevent deadenylation [136]. Interestingly, FUS depletion alters the levels of GluA1 (AMPAR subunit 1) mRNA in the soma and not dendritic localization. This evidence suggests that FUS stabilizes mature mRNAs for their efficient translation in the soma, but in other cases FUS is required for dendritic localization of specific mRNAs (discussed next). In a newly described mechanism for FUS-mediated RNA stabilization, FUS forms a complex with U1 snRNP and RNAPII to prevent alternative polyadenylation of nascent transcripts, suggesting that mutations in FUS could lead to misusage of alternative polyadenylation sites in ALS [137]. Taken together, these studies indicate that FUS alternatively splices and stabilizes transcripts enriched for genes encoding RNA-binding proteins as well as synaptic proteins essential for neurotransmission.
Transport and localization of FUS linked to synapse maturation and function
In neurons, FUS is mainly nuclear but is also a component of RNA transport granules in the soma and dendrites. The translocation of FUS translocation into dendrites is facilitated by the actin-based motor, myosin Va [138]. RNA transport granules containing FUS exhibit both stationary behaviour and directed motility in dendrites [139]. In response to metabotropic glutamate receptor 5 (mGluR5) stimulation, FUS granules transiently show enhanced localization in dendrites, which dissipates upon return to basal activity [139]. FUS is essential for development and spine maturation in neurons. Indeed, Fujii et al. reported that FUS deficient neurons display irregular dendritic branches, long filopodia and decreased spine number in the absence of FUS protein. Deficiency of FUS interferes with activity-dependent localization of Nd1-L mRNA to dendritic tips and results in immature spine morphology. This defect can be rescued by overexpressing the Nd1-L protein to stabilize actin and restore spine maturation [140]. Therefore, FUS plays a critical part in the spatiotemporal transport of mRNAs encoding actin-stabilizing proteins that are necessary for dendritic spine development and maturation.
Interestingly, the localization of FUS appears to be developmentally regulated, which may have implications for its role in synapse development versus in ALS. While FUS is preferentially localized at post synapses during development, it is predominantly present within presynapses in mature human motoneurons [141]. Super-resolution microscopy reveals that FUS also localizes at presynapses in close proximity to active zone proteins and synaptic vesicles [128]. Furthermore, mutant FUS forms abnormal aggregates at synapses in ALS derived motoneurons, suggesting it could interfere with neurotransmission at the NMJ [141]. While these data are intriguing, FUS’s exact presynaptic function is yet to be determined. Specifically, evidence to support whether FUS transports mRNA to presynapses for local translation are lacking. Future investigations will also need to address whether defects in mRNA trafficking and local translation are relevant for motor neuron degeneration and ALS pathogenesis.
Function of FUS in protein translation
FUS promotes translation of localized mRNAs within APC-RNP granules in fibroblast protrusions [142], but there are little data as to whether FUS also regulates local translation in neurons. However, a recent study raises the possibility that FUS plays a role in intra-axonal translation [143]. Alterations in translation regulatory pathways, including mTOR and glutamate signalling, are present in the spinal cords of mutant FUS [R521C and R521H] transgenic mice. FUS mutant spinal cords also show depletion of mRNAs encoding ion channels, transporters and ribosomal proteins. Mutant FUS accumulates within the axons, and the authors speculate that defects in local protein synthesis may account for synapse loss and motor deficits. Additional studies also conclude that ALS-linked mutations in FUS impair protein translation [144,145]. Whether FUS also plays a role in local translation in neurons and how this function may contribute to ALS pathogenesis remain open areas of research. It is likely that FUS functions similarly to other RNA-binding proteins that regulate both the trafficking and translation of mRNAs in the dendrites or axon.
Challenges, emerging themes and areas for future work
RNA-binding proteins control every aspect of the RNA life cycle, ranging from co-transcriptional processing and splicing in the nucleus to mRNA stability, transport, and translation in the cytoplasm (Fig. 1). Disruptions of RNA-binding proteins initiate a cascade of transcriptomic changes that are central to the pathogenesis of several neurodevelopmental and neurodegenerative diseases. Advances in high throughput sequencing and molecular genetics have dramatically expanded our knowledge of the mechanisms RNA-binding proteins use to influence post-transcriptional gene regulation. From these studies, several themes have emerged. First is that alterations or mutations of different RNA-binding proteins often perturb overlapping functional classes of genes and cellular pathways relevant for neurodevelopment, synaptic function, and plasticity (Fig. 3). Second, given their myriad functions in RNA processing, dysregulation of disease-linked RNA-binding proteins result in widespread and often pleiotropic effects on the transcriptome. Therefore, it has been difficult to dissect how each of these different functions contributes to disease pathogenesis and to develop appropriate therapies. Nevertheless, one challenge and key area for future work are to understand exactly how altering splicing, stability, and/or localization of specific transcripts impact the activity and cellular function(s) of neuronal proteins. This is a fundamental area that will allow us to elucidate the cellular and molecular mechanisms that lead to abnormal neurodevelopment and neurodegeneration in disease. Combining cell biology with RNA biology is now increasingly important to understand the biological implications of specific splice variants on neuronal differentiation, morphology, and synaptic function. A second related question is: how do RNA-binding proteins exert cell-type specific RNA regulatory functions in different populations of neurons? Efforts to address these questions will contribute to a deeper understanding of the physiologic and developmental functions of RNA-binding proteins in neurons.
Figure 3.
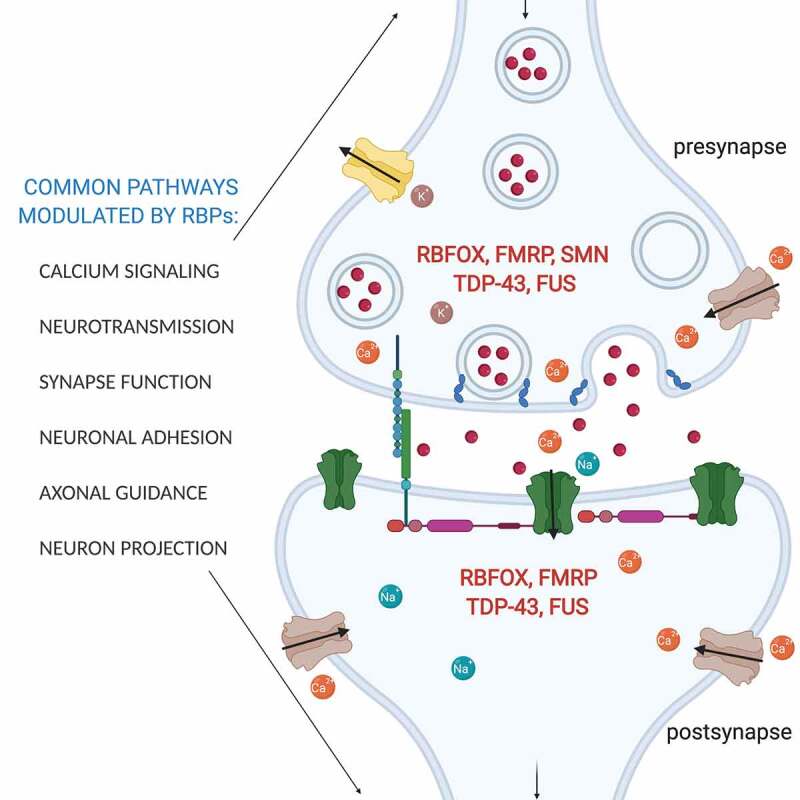
Shared functional pathways at the pre- and post-synapse that are influenced by RBFOX1, FMRP, SMN, TDP-43 and FUS. RNA-binding proteins can regulate mRNA targets at the level of alternative splicing, stability, transport, or translation to modulate several common pathways in neurons. Examples of mRNA targets for each RNA-binding proteins are highlighted: RBFOX1 (Snap25, Grin1, Vamp1, Kcnd3), FMRP (CamKIIa, Map1b), SMN (beta-actin, neuritin), TDP-43 (Rac1, Nefl, Map1b), and FUS (Mapt, GluA1, synGAPa2, Nd1-L)
Another critical area of future work is to understand at the molecular level how post-translational modifications and mutations of RNA-binding proteins affect their function and biophysical properties. Elegant studies from several laboratories have shown that many RNA-binding proteins and RNAs assemble via weak multivalent interactions to form membraneless compartments called ribonucleoprotein (RNP) granules. These RNP granules form via liquid-liquid phase separation (LLPS), a phenomenon driven by the demixing of biomolecules into a condensed liquid phase that has a distinct composition from the surrounding nucleoplasm or cytoplasm. Examples of cellular RNP granules include stress granules and neuronal RNA transport granules [117,146]. RNA transport granules traffic mRNAs in a translationally silent state and have been proposed to disassemble and release the mRNAs for local translation at synaptic terminals. Though the molecular mechanisms that control phase separation of RNP granules in neurons are not well understood, recent work demonstrates that posttranslational modifications of FMRP modulate the phase separation and function of FMRP RNP granules [147]. While phosphorylation of FMRP promotes phase separation and translational inhibition of RNA granules, methylation of FMRP decreases phase separation propensity and dissembles the RNP granules, promoting translation in vitro [147]. In another example, hypomethylation of FUS impairs phase separation causes formation of stable fibrillary condensates, and diminishes local protein synthesis in axon terminals [148]. Thus, striking a balance between different posttranslational modifications appears to be important for regulating the function of RNP granules.
The interplay between phase separation and disease-linked mutations in RNA-binding proteins is another area of ongoing work. Both TDP-43 and FUS undergo LLPS to form dynamic phase-separated liquid droplets. ALS-linked mutations drive FUS and TDP-43 RNP granules to transition from liquid to solid-like droplets and form amyloid-like aggregate with irregular morphology [101,117,145,146]. A fundamental question here is whether perturbing the biophysical properties of RNP granules disrupts RNA processing functions. And if yes, is this relevant in disease? A recent study addresses the first question and shows that ALS-linked TDP-43 mutations alter the helical stabilization of the low complexity domain which disrupts not only phase separation, but also affects TDP-43 splicing function [149,150]. In the future, efforts aimed at determining how the biophysical properties of phase-separated RNA-binding proteins impact their biological functions will clarify whether aberrant phase transitions are relevant in disease pathogenesis.
In summary, many questions and scientific challenges lie ahead in order to fully understand the cellular and molecular consequences of transcriptomic alterations in neurological diseases that arise from dysregulation of RNA-binding proteins. It is likely, however, that the increasingly powerful and elegant tools available to RNA biologists, cell biologists and neuroscientists will be key to defining the causative mechanisms of disease and in developing useful therapeutic solutions.
Acknowledgements
This research was supported by the Amyotrophic Lateral Sclerosis Association and National Institutes of Health under Award Number to K08-NS094744 to PPG.
Funding Statement
This work was supported by the Amyotrophic Lateral Sclerosis Association; National Institute of Neurological Disorders and Stroke [K08-NS094744].
Declaration of interest statement
Authors have no potential conflicts of interest or disclosures.
References
- 1.Holt CE, Schuman EM.. The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron. 2013;80(3):648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raj T, Li YI, Wong G, et al.. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat Genet. 2018;50(11):1584–1592. DOI: 10.1038/s41588-018-0238-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voineagu I, Wang X, Johnston P, et al.. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–384. DOI: 10.1038/nature10110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irimia M, Weatheritt R, Ellis JD, et al.. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159(7):1511–1523. DOI: 10.1016/j.cell.2014.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tollervey JR, Curk T, Rogelj B, et al.. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14(4):452–458. DOI: 10.1038/nn.2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lai M-C, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383(9920):896–910. [DOI] [PubMed] [Google Scholar]
- 7.Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev. 2012;22(3):229–237. [DOI] [PubMed] [Google Scholar]
- 8.Martin CL, Duvall JA, Ilkin Y, et al.. Cytogenetic and molecular characterization of A2BP1 / FOX1 as a candidate gene for autism. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(7):869–876. DOI: 10.1002/ajmg.b.30530 [DOI] [PubMed] [Google Scholar]
- 9.Sebat J, Lakshmi B, Malhotra D, et al.. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. DOI: 10.1126/science.1138659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richter JD, Bassell GJ, Klann E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci. 2015;16(10):595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutcliffe JS, Nelson DL, Zhang F, et al.. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1(6):397–400. DOI: 10.1093/hmg/1.6.397 [DOI] [PubMed] [Google Scholar]
- 12.Oberle I, Rousseau F, Heitz D, et al.. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252(5009):1097–1102. DOI: 10.1126/science.252.5009.1097 [DOI] [PubMed] [Google Scholar]
- 13.Hornstra IK, Nelson DL, Warren ST, et al.. High resolution methylation analysis of the FMR1 gene trinucleotide repeat region in fragile X syndrome. Hum Mol Genet. 1993;2(10):1659–1665. [DOI] [PubMed] [Google Scholar]
- 14.Kumari D, Lokanga R, Yudkin D, et al.. Chromatin changes in the development and pathology of the Fragile X-associated disorders and Friedreich ataxia. Biochim Biophys Acta. 2012;1819(7):802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coffee B, Zhang F, Warren ST, et al.. Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat Genet. 1999;22(1):98–101. [DOI] [PubMed] [Google Scholar]
- 16.Colak D, Zaninovic N, Cohen MS, et al.. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 2014;343(6174):1002–1005. DOI: 10.1126/science.1245831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quartier A, Poquet H, Gilbert-Dussardier B, et al.. Intragenic FMR1 disease-causing variants: a significant mutational mechanism leading to Fragile-X syndrome. Eur J Hum Genet. 2017;25(4):423–431. DOI: 10.1038/ejhg.2016.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinds HL, Ashley CT, Sutcliffe JS, et al.. Tissue specific expression of FMR–1 provides evidence for a functional role in fragile X syndrome. Nat Genet. 1993;3(1):36–43. DOI: 10.1038/ng0193-36 [DOI] [PubMed] [Google Scholar]
- 19.Ashley CT Jr., Wilkinson KD, Reines D, et al.. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262(5133):563–566. [DOI] [PubMed] [Google Scholar]
- 20.Siomi H, Choi M, Siomi MC, et al.. Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMR1 that causes fragile X syndrome. Cell. 1994;77(1):33–39. [DOI] [PubMed] [Google Scholar]
- 21.Ascano M Jr., Mukherjee N, Bandaru P, et al.. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492(7429):382–386. DOI: 10.1038/nature11737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson BR, Chopra P, Suhl JA, et al.. Identification of consensus binding sites clarifies FMRP binding determinants. Nucleic Acids Res. 2016;44(14):6649–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darnell JC, Van Driesche S, Zhang C, et al.. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–261. DOI: 10.1016/j.cell.2011.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suhl JA, Chopra P, Anderson BR, et al.. Analysis of FMRP mRNA target datasets reveals highly associated mRNAs mediated by G-quadruplex structures formed via clustered WGGA sequences. Hum Mol Genet. 2014;23(20):5479–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol. 2009;10(6):430–436. [DOI] [PubMed] [Google Scholar]
- 26.Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004;43(4):513–525. [DOI] [PubMed] [Google Scholar]
- 27.Ling S-C, Fahrner PS, Greenough WT, et al.. Transport of Drosophila fragile X mental retardation protein-containing ribonucleoprotein granules by kinesin-1 and cytoplasmic dynein. Proc Natl Acad Sci U S A. 2004;101(50):17428–17433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antar LN. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24(11):2648–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antar LN, Li C, Zhang H, et al.. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32(1–2):37–48. [DOI] [PubMed] [Google Scholar]
- 30.Dictenberg JB, Swanger SA, Antar LN, et al.. A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev Cell. 2008;14(6):926–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kao D-I, Aldridge GM, Weiler IJ, et al.. Altered mRNA transport, docking, and protein translation in neurons lacking fragile X mental retardation protein. Proc Natl Acad Sci U S A. 2010;107(35):15601–15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Narayanan U, Nalavadi V, Nakamoto M, et al.. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J Neurosci. 2007;27(52):14349–14357. DOI: 10.1523/JNEUROSCI.2969-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khayachi A, Gwizdek C, Poupon G, et al.. Sumoylation regulates FMRP-mediated dendritic spine elimination and maturation. Nat Commun. 2018;9(1):757. DOI: 10.1038/s41467-018-03222-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davidovic L, Jaglin XH, Lepagnol-Bestel A-M, et al.. The fragile X mental retardation protein is a molecular adaptor between the neurospecific KIF3C kinesin and dendritic RNA granules. Hum Mol Genet. 2007;16(24):3047–3058. DOI: 10.1093/hmg/ddm263 [DOI] [PubMed] [Google Scholar]
- 35.Irwin SA. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10(10):1038–1044. [DOI] [PubMed] [Google Scholar]
- 36.Napoli I, Mercaldo V, Boyl PP, et al.. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell. 2008;134(6):1042–1054. DOI: 10.1016/j.cell.2008.07.031 [DOI] [PubMed] [Google Scholar]
- 37.Ceman S, O’Donnell WT, Reed M, et al.. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet. 2003;12(24):3295–3305. DOI: 10.1093/hmg/ddg350 [DOI] [PubMed] [Google Scholar]
- 38.Darnell JC, Fraser CE, Mostovetsky O, et al.. Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 2005;19(8):903–918. DOI: 10.1101/gad.1276805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng Y, Absher D, Eberhart DE, et al.. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997;1(1):109–118. DOI: 10.1016/S1097-2765(00)80012-X [DOI] [PubMed] [Google Scholar]
- 40.Tabet R, Moutin E, Becker JAJ, et al.. Fragile X Mental Retardation Protein (FMRP) controls diacylglycerol kinase activity in neurons. Proc Natl Acad Sci U S A. 2016;113(26):E3619–3628. DOI: 10.1073/pnas.1522631113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plante I, Davidovic L, Ouellet DL, et al.. Dicer-derived microRNAs are utilized by the fragile X mental retardation protein for assembly on target RNAs. J Biomed Biotechnol. 2006;2006(4):64347. DOI: 10.1155/JBB/2006/64347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edbauer D, Neilson JR, Foster KA, et al.. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron. 2010;65(3):373–384. DOI: 10.1016/j.neuron.2010.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muddashetty RS, Nalavadi V, Gross C, et al.. Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGluR signaling. Mol Cell. 2011;42(5):673–688. DOI: 10.1016/j.molcel.2011.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhalla K, Phillips HA, Crawford J, et al.. The de novo chromosome 16 translocations of two patients with abnormal phenotypes (mental retardation and epilepsy) disrupt the A2BP1 gene. J Hum Genet. 2004;49(6):308–311. DOI: 10.1007/s10038-004-0145-4 [DOI] [PubMed] [Google Scholar]
- 45.Kuroyanagi H. Fox-1 family of RNA-binding proteins. Cell Mol Life Sci. 2009;66(24):3895–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin Y, Suzuki H, Maegawa S, et al.. A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 2003;22(4):905–912. DOI: 10.1093/emboj/cdg089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ying Y, Wang X-J, Vuong CK, et al.. Splicing Activation by Rbfox Requires Self-Aggregation through Its Tyrosine-Rich Domain. Cell. 2017;170(2):312–323 e310. DOI: 10.1016/j.cell.2017.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gehman LT, Stoilov P, Maguire J, et al.. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat Genet. 2011;43(7):706–711. DOI: 10.1038/ng.841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vuong CK, Wei W, Lee J-A, et al.. Rbfox1 Regulates Synaptic Transmission through the Inhibitory Neuron-Specific vSNARE Vamp1. Neuron. 2018;98(1):127–141 e127. DOI: 10.1016/j.neuron.2018.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weyn-Vanhentenryck SM, Mele A, Yan Q, et al.. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014;6(6):1139–1152. DOI: 10.1016/j.celrep.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee J-A, Tang -Z-Z, Black DL. An inducible change in Fox-1/A2BP1 splicing modulates the alternative splicing of downstream neuronal target exons. Genes Dev. 2009;23(19):2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacko M, Weyn-Vanhentenryck SM, Smerdon JW, et al.. Rbfox Splicing Factors Promote Neuronal Maturation and Axon Initial Segment Assembly. Neuron. 2018;97(4):853–868 e856. DOI: 10.1016/j.neuron.2018.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wamsley B, Jaglin XH, Favuzzi E, et alet al.. Rbfox1 Mediates Cell-type-Specific Splicing in Cortical Interneurons. Neuron. 2018;100(4):846–859 e847. DOI: 10.1016/j.neuron.2018.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee J-A, Damianov A, Lin C-H, et al.. Cytoplasmic Rbfox1 Regulates the Expression of Synaptic and Autism-Related Genes. Neuron. 2016;89(1):113–128. DOI: 10.1016/j.neuron.2015.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li YI, Sanchez-Pulido L, Haerty W, et al.. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015;25(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quesnel-Vallieres M, Dargaei Z, Irimia M, et al.. Misregulation of an Activity-Dependent Splicing Network as a Common Mechanism Underlying Autism Spectrum Disorders. Mol Cell. 2016;64(6):1023–1034. DOI: 10.1016/j.molcel.2016.11.033 [DOI] [PubMed] [Google Scholar]
- 57.Gonatopoulos-Pournatzis T, Niibori R, Salter EW, et al.. Autism-Misregulated eIF4G Microexons Control Synaptic Translation and Higher Order Cognitive Functions. Mol Cell. 2020;77(6):1176–1192 e1116. DOI: 10.1016/j.molcel.2020.01.006 [DOI] [PubMed] [Google Scholar]
- 58.Wee CD, Kong L, Sumner CJ. The genetics of spinal muscular atrophies. Curr Opin Neurol. 2010;23(5):450–458. [DOI] [PubMed] [Google Scholar]
- 59.Kolb SJ, Kissel JT. Spinal Muscular Atrophy. Neurol Clin. 2015;33(4):831–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48(6):885–896. [DOI] [PubMed] [Google Scholar]
- 61.McWhorter ML, Monani UR, Burghes AH, et al.. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol. 2003;162(5):919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramos DM, d’Ydewalle C, Gabbeta V, et al.. Age-dependent SMN expression in disease-relevant tissue and implications for SMA treatment. J Clin Invest. 2019;129(11):4817–4831. DOI: 10.1172/JCI124120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lefebvre S, Bürglen L, Reboullet S, et al.. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–165. DOI: 10.1016/0092-8674(95)90460-3 [DOI] [PubMed] [Google Scholar]
- 64.Monani UR, Lorson CL, Parsons DW, et al.. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8(7):1177–1183. DOI: 10.1093/hmg/8.7.1177 [DOI] [PubMed] [Google Scholar]
- 65.Lorson CL, Hahnen E, Androphy EJ, et al.. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96(11):6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lefebvre S, Burlet P, Liu Q, et al.. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16(3):265–269. DOI: 10.1038/ng0797-265 [DOI] [PubMed] [Google Scholar]
- 67.Finkel RS, Mercuri E, Darras BT, et al.. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. 2017;377(18):1723–1732. DOI: 10.1056/NEJMoa1702752 [DOI] [PubMed] [Google Scholar]
- 68.Mendell JR, Al-Zaidy S, Shell R, et al.. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. 2017;377(18):1713–1722. DOI: 10.1056/NEJMoa1706198 [DOI] [PubMed] [Google Scholar]
- 69.Groen EJN, Talbot K, Gillingwater TH. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat Rev Neurol. 2018;14(4):214–224. [DOI] [PubMed] [Google Scholar]
- 70.Ibrahim F, Nakaya T, Mourelatos Z. RNA dysregulation in diseases of motor neurons. Annu Rev Pathol. 2012;7(1):323–352. [DOI] [PubMed] [Google Scholar]
- 71.Liu Q, Fischer U, Wang F, et al.. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90(6):1013–1021. [DOI] [PubMed] [Google Scholar]
- 72.Fischer U, Liu Q, Dreyfuss G. The SMN–SIP1 Complex Has an Essential Role in Spliceosomal snRNP Biogenesis. Cell. 1997;90(6):1023–1029. [DOI] [PubMed] [Google Scholar]
- 73.Chari A, Golas MM, Klingenhäger M, et al.. An assembly chaperone collaborates with the SMN complex to generate spliceosomal SnRNPs. Cell. 2008;135(3):497–509. DOI: 10.1016/j.cell.2008.09.020 [DOI] [PubMed] [Google Scholar]
- 74.Yong J, Kasim M, Bachorik JL, et al.. Gemin5 delivers snRNA precursors to the SMN complex for snRNP biogenesis. Mol Cell. 2010;38(4):551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lorson CL, Strasswimmer J, Yao J-M, et al.. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19(1):63–66. DOI: 10.1038/ng0598-63 [DOI] [PubMed] [Google Scholar]
- 76.Zhang Z, Lotti F, Dittmar K, et al.. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133(4):585–600. DOI: 10.1016/j.cell.2008.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gabanella F, Butchbach MER, Saieva L, et al.. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One. 2007;2(9):e921. DOI: 10.1371/journal.pone.0000921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Winkler C, Eggert C, Gradl D, et al.. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev. 2005;19(19):2320–2330. DOI: 10.1101/gad.342005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lotti F, Imlach W, Saieva L, et al.. An SMN-dependent U12 splicing event essential for motor circuit function. Cell. 2012;151(2):440–454. DOI: 10.1016/j.cell.2012.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Imlach WL, Beck E, Choi B, et al.. SMN is required for sensory-motor circuit function in Drosophila. Cell. 2012;151(2):427–439. DOI: 10.1016/j.cell.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Z, Pinto AM, Wan L, et al.. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc Natl Acad Sci U S A. 2013;110(48):19348–19353. DOI: 10.1073/pnas.1319280110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaida D, Berg MG, Younis I, et al.. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468(7324):664–668. DOI: 10.1038/nature09479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Berg MG, Singh L, Younis I, et al.. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012;150(1):53–64. DOI: 10.1016/j.cell.2012.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Almada AE, Wu X, Kriz AJ, et al.. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013;499(7458):360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.So BR, Wan L, Zhang Z, et al.. A U1 snRNP–specific assembly pathway reveals the SMN complex as a versatile hub for RNP exchange. Nat Struct Mol Biol. 2016;23(3):225–230. DOI: 10.1038/nsmb.3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Donlin-Asp PG, Bassell GJ, Rossoll W. A role for the survival of motor neuron protein in mRNP assembly and transport. Curr Opin Neurobiol. 2016;39:53–61. [DOI] [PubMed] [Google Scholar]
- 87.Maday S, Twelvetrees AE, Moughamian AJ, et al.. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014;84(2):292–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rossoll W, Jablonka S, Andreassi C, et al.. Smn, the spinal muscular atrophy–determining gene product, modulates axon growth and localization of β-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163(4):801–812. DOI: 10.1083/jcb.200304128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang H, Xing L, Rossoll W, et al.. Multiprotein complexes of the survival of motor neuron protein SMN with Gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci. 2006;26(33):8622–8632. DOI: 10.1523/JNEUROSCI.3967-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fallini C, Zhang H, Su Y, et al.. The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(A) mRNA in primary motor neuron axons. J Neurosci. 2011;31(10):3914–3925. DOI: 10.1523/JNEUROSCI.3631-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fallini C, Rouanet JP, Donlin-Asp PG, et al.. Dynamics of survival of motor neuron (SMN) protein interaction with the mRNA-binding protein IMP1 facilitates its trafficking into motor neuron axons. Developmental Neurobiology. 2014;74(3):319–332. DOI: 10.1002/dneu.22111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Akten B, Kye MJ, Hao LT, et al.. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci U S A. 2011;108(25):10337–10342. DOI: 10.1073/pnas.1104928108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fallini C, Donlin-Asp PG, Rouanet JP, et al.. Deficiency of the Survival of Motor Neuron Protein Impairs mRNA Localization and Local Translation in the Growth Cone of Motor Neurons. J Neurosci. 2016;36(13):3811–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Donlin-Asp PG, Fallini C, Campos J, et al.. The Survival of Motor Neuron Protein Acts as a Molecular Chaperone for mRNP Assembly. Cell Rep. 2017;18(7):1660–1673. DOI: 10.1016/j.celrep.2017.01.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Neumann M, Sampathu DM, Kwong LK, et al.. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. DOI: 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 96.Ling S-C, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sreedharan J, Blair IP, Tripathi VB, et al.. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–1672. DOI: 10.1126/science.1154584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vance C, Rogelj B, Hortobagyi T, et al.. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. DOI: 10.1126/science.1165942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kwiatkowski TJ Jr., Bosco DA, LeClerc AL, et al.. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. DOI: 10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- 100.Ou SH, Wu F, Harrich D, et al.. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69(6):3584–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Prasad A, Bharathi V, Sivalingam V, et al.. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front Mol Neurosci. 2019;12:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276(39):36337–36343. [DOI] [PubMed] [Google Scholar]
- 103.Ayala YM, Zago P, D’Ambrogio A, et al.. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121(22):3778–3785. DOI: 10.1242/jcs.038950 [DOI] [PubMed] [Google Scholar]
- 104.Polymenidou M, Lagier-Tourenne C, Hutt KR, et al.. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–468. DOI: 10.1038/nn.2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sephton CF, Cenik C, Kucukural A, et al.. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem. 2011;286(2):1204–1215. DOI: 10.1074/jbc.M110.190884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Klim JR, Williams LA, Limone F, et al.. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22(2):167–179. DOI: 10.1038/s41593-018-0300-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Melamed Z, López-Erauskin J, Baughn MW, et al.. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22(2):180–190. DOI: 10.1038/s41593-018-0293-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ayala YM, De Conti L, Avendaño-Vázquez SE, et al.. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30(2):277–288. DOI: 10.1038/emboj.2010.310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Strong MJ, Volkening K, Hammond R, et al.. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. 2007;35(2):320–327. DOI: 10.1016/j.mcn.2007.03.007 [DOI] [PubMed] [Google Scholar]
- 110.Costessi L, Porro F, Iaconcig A, et al.. TDP-43 regulates β-adducin(Add2) transcript stability. RNA Biol. 2014;11(10):1280–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gu J, Wu F, Xu W, et al.. TDP-43 suppresses tau expression via promoting its mRNA instability. Nucleic Acids Res. 2017;45(10):6177–6193. DOI: 10.1093/nar/gkx175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fukushima M, Hosoda N, Chifu K, et al.. TDP-43 accelerates deadenylation of target mRNAs by recruiting Caf1 deadenylase. FEBS Lett. 2019;593(3):277–287. [DOI] [PubMed] [Google Scholar]
- 113.Ling JP, Pletnikova O, Troncoso JC, et al.. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349(6248):650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jeong YH, Ling JP, Lin SZ, et al.. Tdp-43 cryptic exons are highly variable between cell types. Mol Neurodegener. 2017;12(1):13. DOI: 10.1186/s13024-016-0144-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Elvira G, Wasiak S, Blandford V, et al.. Characterization of an RNA granule from developing brain. Mol Cell Proteomics. 2006;5(4):635–651. DOI: 10.1074/mcp.M500255-MCP200 [DOI] [PubMed] [Google Scholar]
- 116.Fallini C, Bassell GJ, Rossoll W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet. 2012;21(16):3703–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gopal PP, Nirschl JJ, Klinman E, et al.. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc Natl Acad Sci U S A. 2017;114(12):E2466–E2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Alami NH, Smith R, Carrasco M, et al.. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81(3):536–543. DOI: 10.1016/j.neuron.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu-Yesucevitz L, Lin AY, Ebata A, et al.. ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J Neurosci. 2014;34(12):4167–4174. DOI: 10.1523/JNEUROSCI.2350-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chu JF, Majumder P, Chatterjee B, et al.. TDP-43 Regulates Coupled Dendritic mRNA Transport-Translation Processes in Co-operation with FMRP and Staufen1. Cell Rep. 2019;29(10):3118–3133 e3116. [DOI] [PubMed] [Google Scholar]
- 121.Ishiguro A, Kimura N, Watanabe Y, et al.. TDP-43 binds and transports G-quadruplex-containing mRNAs into neurites for local translation. Genes Cells. 2016;21(5):466–481. [DOI] [PubMed] [Google Scholar]
- 122.Coyne AN, Siddegowda BB, Estes PS, et al.. Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J Neurosci. 2014;34(48):15962–15974. DOI: 10.1523/JNEUROSCI.2526-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Freibaum BD, Chitta RK, High AA, et al.. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res. 2010;9(2):1104–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Neelagandan N, Gonnella G, Dang S, et al.. TDP-43 enhances translation of specific mRNAs linked to neurodegenerative disease. Nucleic Acids Res. 2019;47(1):341–361. DOI: 10.1093/nar/gky972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Russo A, Scardigli R, La Regina F, et al.. Increased cytoplasmic TDP-43 reduces global protein synthesis by interacting with RACK1 on polyribosomes. Hum Mol Genet. 2017;26(8):1407–1418. DOI: 10.1093/hmg/ddx035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Majumder P, Chu JF, Chatterjee B, et al.. Co-regulation of mRNA translation by TDP-43 and Fragile X Syndrome protein FMRP. Acta Neuropathol. 2016;132(5):721–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nolan M, Talbot K, Ansorge O. Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta Neuropathol Commun. 2016;4(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schoen M, Reichel JM, Demestre M, et al. Super-Resolution Microscopy Reveals Presynaptic Localization of the ALS/FTD Related Protein FUS in Hippocampal Neurons. Front Cell Neurosci. 2015;9:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ishigaki S, Masuda A, Fujioka Y, et al.. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep. 2012;2(1):529. DOI: 10.1038/srep00529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nakaya T, Alexiou P, Maragkakis M, et al.. FUS regulates genes coding for RNA-binding proteins in neurons by binding to their highly conserved introns. RNA. 2013;19(4):498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lerga A, Hallier M, Delva L, et al.. Identification of an RNA binding specificity for the potential splicing factor TLS. J Biol Chem. 2001;276(9):6807–6816. DOI: 10.1074/jbc.M008304200 [DOI] [PubMed] [Google Scholar]
- 132.Imperatore JA, McAninch DS, Valdez-Sinon AN, et al.. FUS Recognizes G Quadruplex Structures Within Neuronal mRNAs. Front Mol Biosci. 2020;7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Orozco D, Edbauer D. FUS-mediated alternative splicing in the nervous system: consequences for ALS and FTLD. J Mol Med (Berl). 2013;91(12):1343–1354. [DOI] [PubMed] [Google Scholar]
- 134.Lagier-Tourenne C, Polymenidou M, Hutt KR, et al.. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15(11):1488–1497. DOI: 10.1038/nn.3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yokoi S, Udagawa T, Fujioka Y, et al.. 3′UTR Length-Dependent Control of SynGAP Isoform α2 mRNA by FUS and ELAV-like Proteins Promotes Dendritic Spine Maturation and Cognitive Function. Cell Rep. 2017;20(13):3071–3084. DOI: 10.1016/j.celrep.2017.08.100 [DOI] [PubMed] [Google Scholar]
- 136.Udagawa T, Fujioka Y, Tanaka M, et al.. FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat Commun. 2015;6(1):7098. DOI: 10.1038/ncomms8098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Masuda A, Kawachi T, Takeda J-I, et al.. tRIP-seq reveals repression of premature polyadenylation by co-transcriptional FUS-U1 snRNP assembly. EMBO reports. 2020;21(5). DOI: 10.15252/embr.201949890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yoshimura A, Fujii R, Watanabe Y, et al.. Myosin-Va facilitates the accumulation of mRNA/protein complex in dendritic spines. Curr Biol. 2006;16(23):2345–2351. DOI: 10.1016/j.cub.2006.10.024 [DOI] [PubMed] [Google Scholar]
- 139.Fujii R, Okabe S, Urushido T, et al.. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol. 2005;15(6):587–593. DOI: 10.1016/j.cub.2005.01.058 [DOI] [PubMed] [Google Scholar]
- 140.Fujii R, Takumi T. TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci. 2005;118(Pt 24):5755–5765. [DOI] [PubMed] [Google Scholar]
- 141.Deshpande D, Higelin J, Schoen M, et al. Synaptic FUS Localization During Motoneuron Development and Its Accumulation in Human ALS Synapses. Front Cell Neurosci. 2019;13:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yasuda K, Zhang H, Loiselle D, et al.. The RNA-binding protein Fus directs translation of localized mRNAs in APC-RNP granules. J Cell Biol. 2013;203(5):737–746. DOI: 10.1083/jcb.201306058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lopez-Erauskin J, Tadokoro T, Baughn MW, et al.. ALS/FTD-Linked Mutation in FUS Suppresses Intra-axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron. 2018;100(4):816–830 e817. DOI: 10.1016/j.neuron.2018.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kamelgarn M, Chen J, Kuang L, et al.. ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc Natl Acad Sci U S A. 2018;115(51):E11904–E11913. DOI: 10.1073/pnas.1810413115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Murakami T, Qamar S, Lin J, et al.. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron. 2015;88(4):678–690. DOI: 10.1016/j.neuron.2015.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ryan VH, Fawzi NL. Physiological, Pathological, and Targetable Membraneless Organelles in Neurons. Trends Neurosci. 2019;42(10):693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tsang B, Arsenault J, Vernon RM, et al.. Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc Natl Acad Sci U S A. 2019;116(10):4218–4227. DOI: 10.1073/pnas.1814385116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Qamar S, Wang G, Randle SJ, et al.. FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-π Interactions. Cell. 2018;173(3):720–734 e715. DOI: 10.1016/j.cell.2018.03.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Conicella AE, Dignon GL, Zerze GH, et al.. TDP-43 α-helical structure tunes liquid–liquid phase separation and function. Proc Natl Acad Sci U S A. 2020;117(11):5883–5894. DOI: 10.1073/pnas.1912055117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Conicella AE, Zerze GH, Mittal J, et al.. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure. 2016;24(9):1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]