

SUMMARY
Candida albicans is a fungal component of the human gut microbiota and an opportunistic pathogen. C. albicans transcription factors (TFs), Wor1 and Efg1, are master regulators of an epigenetic switch required for fungal mating that also control colonization of the mammalian gut. We show that additional mating regulators, WOR2, WOR3, WOR4, AHR1, CZF1, and SSN6, also influence gut commensalism. Using Calling Card-seq to record Candida TF DNA-binding events in the host, we examined the role and relationships of these regulators during murine gut colonization. By comparing in-host transcriptomes of regulatory mutants with enhanced versus diminished commensal fitness, we also identify a set of candidate commensalism effectors. These include Cht2, a GPI-linked chitinase whose gene is bound by Wor1, Czf1, and Efg1 in vivo, that we show promotes commensalism. Thus, the network required for a C. albicans sexual switch is biochemically active in the host intestine and repurposed to direct commensalism.
Graphical abstract
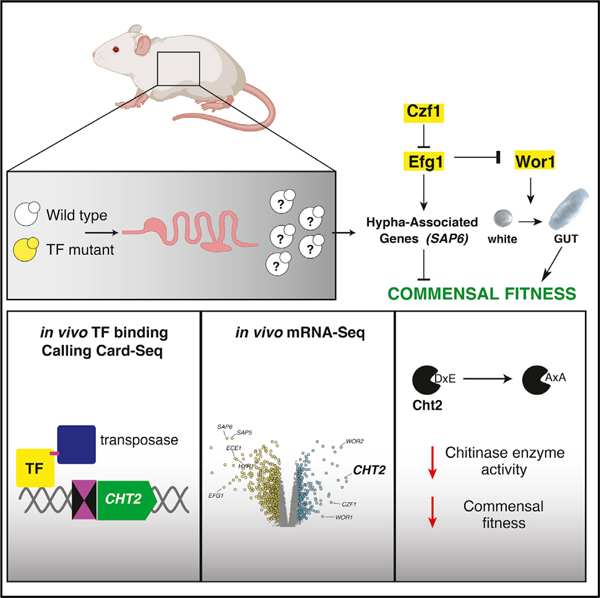
In brief
Candida albicans is a fungal pathobiont of the mammalian gut. Witchley, Basso, et al. show that transcription factors (TFs) that control fungal mating also control commensalism. Using Calling Card-seq and RNA-seq to determine TF DNA-binding and regulatory activity during host colonization, they identify chitinase Cht2 as a commensalism effector.
INTRODUCTION
Candida albicans is the major fungal commensal and opportunistic pathogen of humans. As a pathogen, C. albicans is responsible for syndromes ranging from simple vaginitis and oral thrush to devastating infections of the blood and internal organs. Life-threatening disease is restricted primarily to patients with underlying risk factors (Odds, 1987; Pfaller and Diekema, 2007), whereas virtually all humans encounter commensal C. albicans as part of normal gut, skin, and genitourinary microbiota (Drell et al., 2013; Hoffmann et al., 2013; Odds, 1988). Observations in gnotobiotic mice suggest that gut colonization confers immune benefits to the host by triggering the expansion of CD4+Th17 cells, a critical defense at mucosal barriers (Atarashi et al., 2015; Shao et al., 2019). Likewise, analysis of human CD4+Th17 cells suggests that commensal C. albicans may entrain host immune responses to exogenous fungal pathogens (Bacher et al., 2019). Given the fundamental roles played by this pathobiont in human health and disease, it is important to understand the mechanisms that underpin its interactions with mammalian hosts.
Our group previously identified two C. albicans transcription factors (TFs), Efg1 and Wor1, with opposing roles in the regulation of gut commensalism (Figure 1A) (Pande et al., 2013). Efg1 inhibits commensalism, such that an efg1 null mutant strongly outcompetes wild-type (WT) C. albicans in a mouse model of gut colonization (Pande et al., 2013; Pierce and Kumamoto, 2012; Witchley et al., 2019). In contrast, Wor1 promotes commensalism, and a wor1 mutant is rapidly outcompeted by WT (Pande et al., 2013). Consistent with a special role for Wor1 in the gastrointestinal niche, WOR1 gene expression—which is barely detectable under in vitro conditions—is upregulated ~10,000-fold in the host digestive tract (Pande et al., 2013). Furthermore, forced expression of WOR1 (WOR1OE) via a heterologous promoter confers enhanced commensal fitness (Pande et al., 2013). Interestingly, the fitness advantage of WOR1OE does not emerge until after 5 to 10 days of host colonization, and it coincides with a morphological transition from the standard, oval-shaped ‘‘white’’ cell type to a larger, elongated gastrointestinally induced transition (‘‘GUT’’) cell type (Pande et al., 2013). Compared with white cells, the metabolism of GUT cells is optimized for nutrients available in the mammalian digestive tract. We hypothesize that WT C. albicans undergoes an analogous transition while in the host, but reverts to white morphology when excreted because of the loss of signals required for WOR1 expression.
Figure 1. C. albicans sexual regulators control commensal fitness.
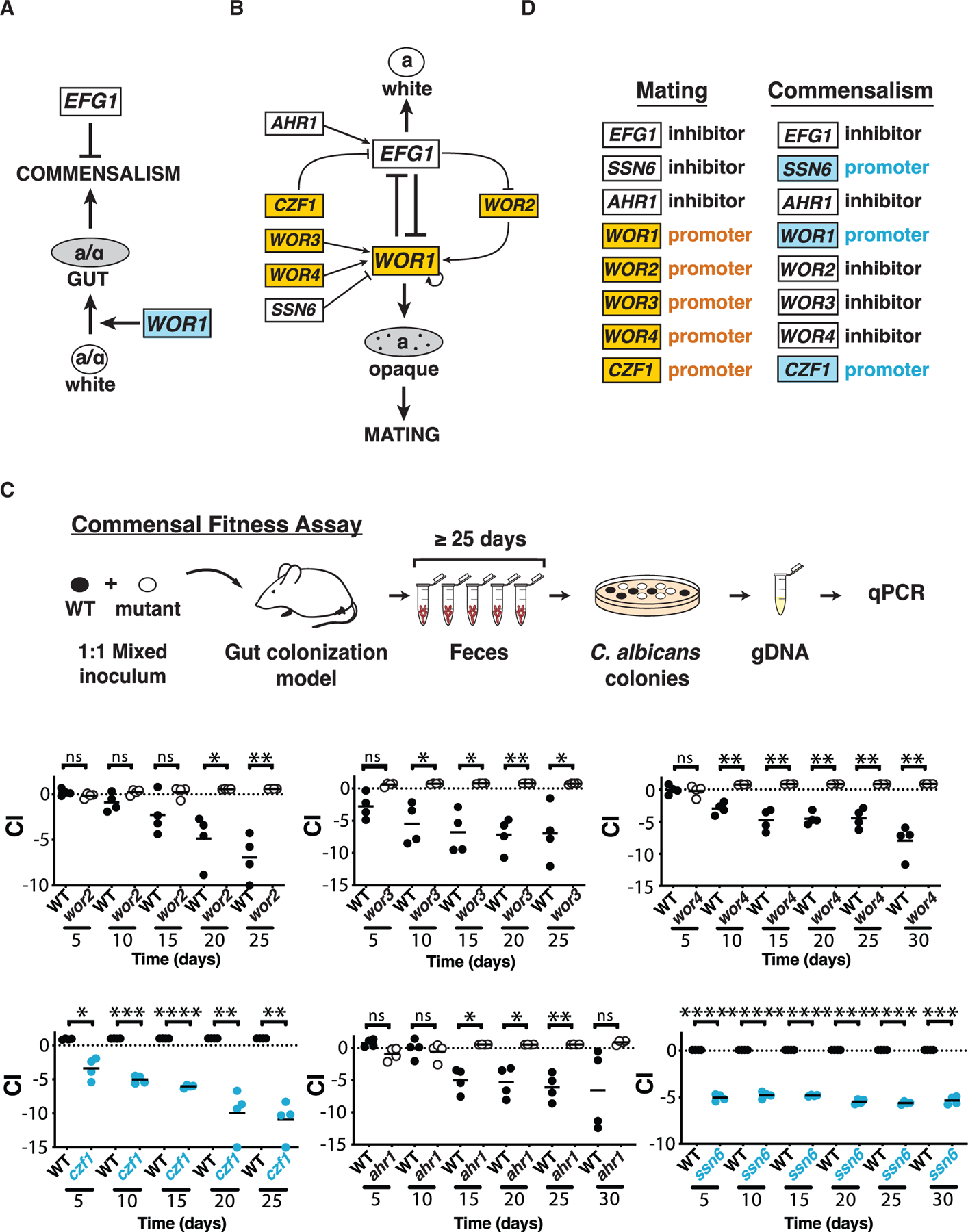
(A) Regulation of gut commensalism. Efg1 inhibits (bar) and Wor1 promotes (arrow) fungal commensalism (Pande et al., 2013). Wor1 promotes the white-to-gut developmental switch that confers enhanced fitness in the mammalian digestive tract.
(B) Regulation of mating. Efg1 is the central inhibitor and Wor1 the central activator of mating and the white-to-opaque switch. Six additional transcription factors interact with Efg1 and Wor1 via the depicted regulatory circuit (Hernday et al., 2013, 2016, 2010; Huang et al., 2006; Lohse et al., 2010; Tuch et al., 2010; Wang et al.; Zordan et al., 2006, 2007).
(C) WOR2, WOR3, WOR3, CZF1, AHR1, and SSN6 regulate the commensal fitness of C. albicans. Each of the indicated null mutants was tested in 1:1 competitions with WT in a mouse model of stable gut colonization. Mutants with loss-of-fitness phenotypes are shown in blue, and those with gain-of-fitness in white (with black outline). Statistical comparisons were performed using the paired Student’s t test; ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Results for independent isolates of these mutants are presented in Figure S2.
(D) Comparison of the roles of eight fungal transcription factors in mating versus commensalism.
Besides their roles in commensalism, Efg1 and Wor1 are also ‘‘master regulators’’ of C. albicans mating (Huang et al., 2006; Srikantha et al., 2006; Zordan et al., 2006, 2007). Wor1 promotes and Efg1 opposes the white-to-’’opaque’’ morphological transition, an epigenetic switch that confers sexual competency in this species. Most C. albicans strains are incapable of mating because they express a1-α2, a potent transcriptional inhibitor of WOR1 that is jointly encoded by two heterologous alleles of the mating type-like locus, MTLa and MTLα. To mate, diploid a/α white cells must lose one allele of MTL and switch to the opaque cell type, before opaque a and α cells fuse to generate tetraploid a/α progeny. In addition to Wor1 and Efg1, other TFs control sexual competency through a complex, self-reinforcing regulatory circuit that has been deduced from genetic epistasis experiments (Figure 1B; Du et al., 2015; Hernday et al., 2013, 2016; Lohse and Johnson, 2016; Lohse et al., 2010). Furthermore, chromatin immunoprecipitation (ChIP) experiments indicate that Wor1, Efg1, Wor2, Wor3, Wor4, Czf1, Ahr1, and Ssn6 engage in extensive cross-regulation, with each TF binding to its own promoter and to those for most or all of the other regulators in the circuit (Figures S1A and S1B; Hernday et al., 2013, 2016; Lohse et al., 2013, 2010; Lohse and Johnson, 2016; Srikantha et al., 2006; Vinces and Kumamoto, 2007; Wang et al., 2011a, 2011b; Zordan et al., 2006, 2007).
Given that an antagonistic relationship between Wor1 and Efg1 is conserved between mating and commensalism, we hypothesized that additional components of the mating circuit may participate in the regulation of commensalism. To test this idea, we performed a systematic analysis of wor2, wor3, wor4, czf1, ahr1, and ssn6 null mutants in a murine gut colonization model. Remarkably, all six TFs exert significant effects on commensal fitness, although the activities of several regulators differ between sex and commensalism. To validate these relationships in Candida colonizing the murine gut, we adapted a transposon-based Calling Card-seq method to record the in-host regulatory targets of DNA-binding proteins. Finally, by comparing the transcriptomes of regulatory mutants with enhanced versus diminished commensal fitness, we identified a set of candidate commensalism effectors. These include a glycophosphatidylinositol (GPI)-linked fungal chitinase gene, CHT2, which is directly targeted by Wor1, Czf1, and Efg1 in commensally propagated strains and is required for normal commensal fitness in animals. Our work establishes that a transcriptional circuit that determines C. albicans mating competency under in vitro conditions is repurposed in the host to control its ability to flourish as a gut commensal.
RESULTS
Regulators of the mating circuit control commensal fitness
To determine whether sexual regulators besides Wor1 and Efg1 play roles in commensalism, we generated null mutants affecting CZF1, WOR2, WOR3, WOR4, AHR1, and SSN6. Homozygous disruptants of each open reading frame (ORF) were created in SN152, the same genetic background used for our previous studies of wor1, efg1, and WOR1OE (Pande et al., 2013). The competitive fitness of each mutant was determined using a mouse gastrointestinal colonization model depicted in Figure 1C. Immunocompetent BALB/c mice were treated with broad spectrum antibiotics (ampicillin and gentamicin in drinking water) for one week, followed by gavage with a 1:1 mixture of WT C. albicans and a TF mutant. Antibiotics were continued, and the relative abundance of fungal strains recovered from host feces over ≥25 days was determined by qPCR. The competitive index (CI) of each strain was calculated as the log2 ratio of its relative abundance after recovery from the host (R) compared with its abundance in the inoculum (I).
To our surprise, all six tested regulators exert significant influence on the fitness of C. albicans as a gut commensal. Results for two independent isolates of each mutant except ssn6 are shown in Figures 1C and S2. wor2, wor3, wor4, and ahr1 exhibit hypercompetitive phenotypes (white data points, Figures 1C, S2A–S2C, and S2E), suggesting that Wor2, Wor3, Wor4, and Ahr1 function as inhibitors of commensalism. Conversely, czf1 and ssn6 exhibit diminished fitness compared with WT (blue data points, Figures 1C and S2D), suggesting that Czf1 and Ssn6 promote commensalism in the gut. We note that the requirement for Ssn6 in gut colonization may not be specific, because ssn6 also exhibits growth defects under multiple in vitro conditions (Figure S2F). The remaining mutants do not exhibit generalized growth defects and a consistent pattern of growth differences under anaerobic conditions (Figure S2G).
The finding that all eight (including Wor1 and Efg1) tested mating factors exert substantial effects on gut colonization suggested that sex and commensalism may be controlled by a common regulatory circuit. In support of this hypothesis, Wor1, Efg1, Czf1, and Ahr1 play consistent roles in sex and commensalism, with each factor serving either to inhibit or to promote both processes (Figure 1D). In contrast, the activities of Wor2, Wor3, Wor4, and Ssn6 seem to be divergent (Figure 1D).
Genetic epistasis reveals functional relationships among commensal regulators
To clarify the relationships among selected sexual regulators in the control of commensalism, we turned to genetic epistasis analysis. This classical technique is used to infer the nature of regulatory pathways based on the phenotypes of strains that combine two mutations whose individual phenotypes are distinct. For example, imagine a process that is promoted by factor A and inhibited by factor B. If A and B participate in a linear regulatory pathway (Figure 2A, linear), then the loss-of-function phenotype of the more downstream regulator will predominate (be epistatic) in a mutant that lacks both factors. Conversely, if A and B are related by parallel or branched regulatory pathways (Figure 2A, parallel and branched), then the phenotype of a double mutant typically differs from (often assuming an intermediate phenotype between) those of either single mutant.
Figure 2. Epistasis analysis suggests a branched regulatory pathway.
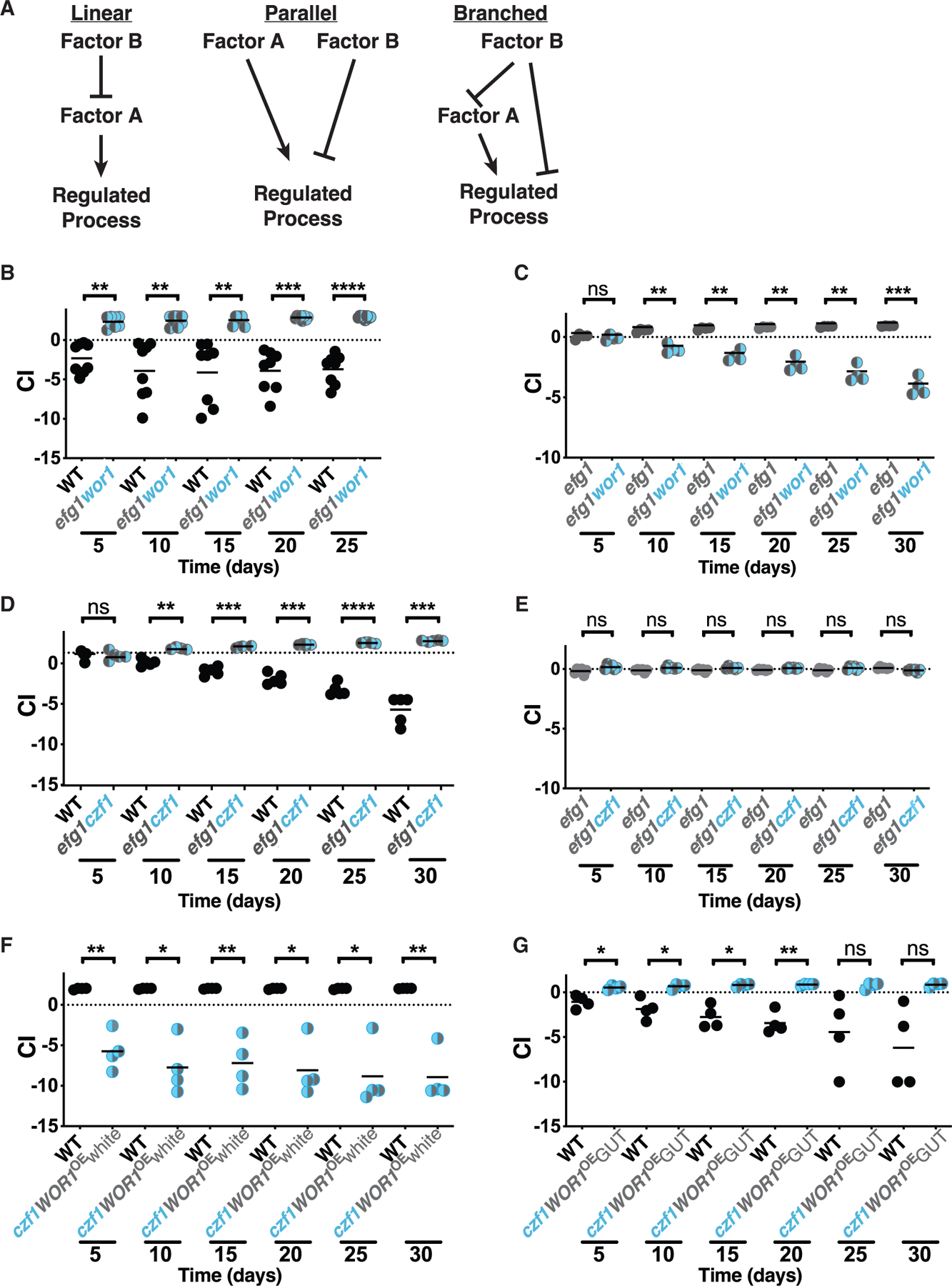
(A) Examples of linear, parallel, and branched regulatory pathways. If factor A and factor B play opposite roles in a linear regulatory pathway, the double mutant ab will exhibit the same phenotype as a null mutant affecting the more downstream regulator (mutant a, in this example). If factor A and factor B participate in a parallel or branched regulatory pathway, then the phenotype of the double mutant is likely to be intermediate between those of (A) and (B).
(B–G) Competitive fitness of double mutants in the gut colonization model. B. WT (ySN425) v. efg1wor1 (ySN1126).
(C) efg1 (ySN1338) v. efg1wor1 (ySN1126).
(D) WT (ySN425) v. efg1czf1 (ySN1373).
(E) efg1 (ySN1338) v. efg1czf1 (ySN1373).
(F) WT (ySN425) v. czf1WOR1OE (ySN1147).
(G) WT (ySN425) v. czf1GUT (ySN1486). Statistical analysis was performed as in Figure 1C. Results for additional efg1 versus efg1wor1 and WT versus efg1czf1 competition experiments are presented in Figures S2H and S2I, respectively.
We began with Efg1 (a commensalism inhibitor) and Wor1 (a commensalism promoter). To create an efg1wor1 double mutant, both copies of the WOR1 ORF were disrupted in an existing efg1 null mutant (note that the C. albicans genome is diploid). As shown in Figure 2B, efg1wor1 outcompetes WT in the gut colonization model. Nevertheless, it seemed to us that the enhanced fitness of efg1wor1 is less pronounced than our previous observations of efg1 single mutants (Pande et al., 2013; Witchley et al., 2019). To clarify the relative fitness of efg1 and efg1wor1, we compared the two strains directly in the same animals. As shown in Figures 2C and S2H, efg1 consistently outcompetes efg1wor1 in direct competition. The finding that an efg1wor1 double mutant manifests an intermediate phenotype between the strong gain-of-fitness phenotype of efg1 and the moderate loss-of-fitness phenotype of wor1 suggests that Wor1 and Efg1 regulate commensalism by a nonlinear pathway.
We next turned to Efg1 and Czf1 (a commensalism promoter). As shown in Figures 2D and S2I, an efg1czf1 double mutant is hypercompetitive in the gut colonization model, similar to the phenotype of efg1. However, unlike the previous result with efg1wor1, efg1czf1 and efg1 exhibit identical commensal fitness in a direct competition experiment (Figure 2E). EFG1 is thus epistatic to CZF1, which suggests that Efg1 lies downstream of Czf1 in a linear regulatory pathway.
Because Wor1 and Czf1 both promote commensalism and share similar (loss-of-fitness) null mutant phenotypes, we paired a WOR1OE gain-of-fitness allele with a czf1 null allele for epistasis analysis. In competition with WT, czf1WOR1OE exhibits reduced commensal fitness (Figure 2F), similar to the phenotype of czf1 (Figure 1C). These results would seem to place CZF1 downstream of WOR1 in a linear regulatory pathway. However, as described in the introduction, Wor1 promotes commensalism by fostering the white-to-GUT switch after 5 to 10 days of exposure to the host digestive tract (Pande et al., 2013). Because czf1WOR1OE is outcompeted within 5 days (Figure 2F), there may be insufficient time to manifest any fitness advantage conferred by WOR1OE. To determine whether czf1WOR1OE white cells are able to undergo the white-to-GUT switch, we gavaged three animals with czf1WOR1OE white cells. Despite successful colonization of the murine GI tract (median titer of ~5 × 107-CFUs/g feces), czf1WOR1OE failed to produce any fully GUT colonies during 30 days of observation. However, interestingly, when homozygous czf1 deletions are introduced into a WOR1OE GUT strain, the GUT phenotype is maintained. Moreover, these czf1WOR1OE GUT cells are able to outcompete WT white cells in the gut colonization model (Figure 2G), albeit less robustly than a WOR1OE GUT strain (Pande et al., 2013). Taken together, these results suggest that Czf1 promotes the white-to-GUT switch but is not required for maintenance of the GUT phenotype and that Czf1 and Wor1 regulate commensalism by a nonlinear pathway.
Calling Card-seq records TF binding events in C. albicans
To further explore the commensal function of these regulators, we sought a method to determine their DNA-binding activities during active colonization of the host. ChIP is a commonly used biochemical technique for identifying the binding sites of TFs. ChIP relies on extracts prepared from cells that are typically propagated under in vitro conditions that foster the activity of the relevant TF. Because there are no known laboratory conditions that accurately mimic the mammalian gastrointestinal tract, where commensalism occurs, we developed an alternative strategy that can be performed in cells that are propagated in a host.
Originally developed for use in S. cerevisiae and later in mammalian cells, the Calling Card-seq technique utilizes PiggyBac transposase (PBase) fused to a TF of interest to deposit PiggyBac transposon (PB) into a TTAA sequence near the binding site of the TF (Wang et al., 2011a, 2012). Transposition is mediated in living cells, and the binding sites are determined by subsequent DNA sequencing of transposon insertion sites across a cell population. Multiple modifications were required to adapt the technique for use in C. albicans (Figures 3A and S3 and STAR methods). First, until recently (Bijlani et al., 2019), there have been no stable, autonomously replicating plasmids available for use in C. albicans. Therefore, we integrated one or two copies of PB into a neutral locus in the C. albicans genome. Second, because C. albicans uses a non-universal genetic code (CTG encodes serine rather than leucine in this species; Santos et al., 2011), we synthesized a codon-optimized version of PBase. Finally, we created a custom C. albicans strain to facilitate the detection and selection of transposon excision and reinsertion events. Starting with Arg−His−Leu− reference strain SN152, a copy of PB flanked by 5′ and 3′ halves of the ARG4 gene was introduced to a neutral chromosomal locus. Precise excision of PB from the donor site results in prototrophy for arginine. In addition, an intact copy of HIS1 was embedded within PB itself, such that cells that have undergone transposon excision followed by reinsertion elsewhere in the genome are prototrophic for both arginine and histidine. For experiments in animals, we created a second Calling Card strain that contains two copies of PB, one marked with HIS1 and the second marked with LEU2.
Figure 3. Determination of transcription factor binding sites using Calling Card-seq.
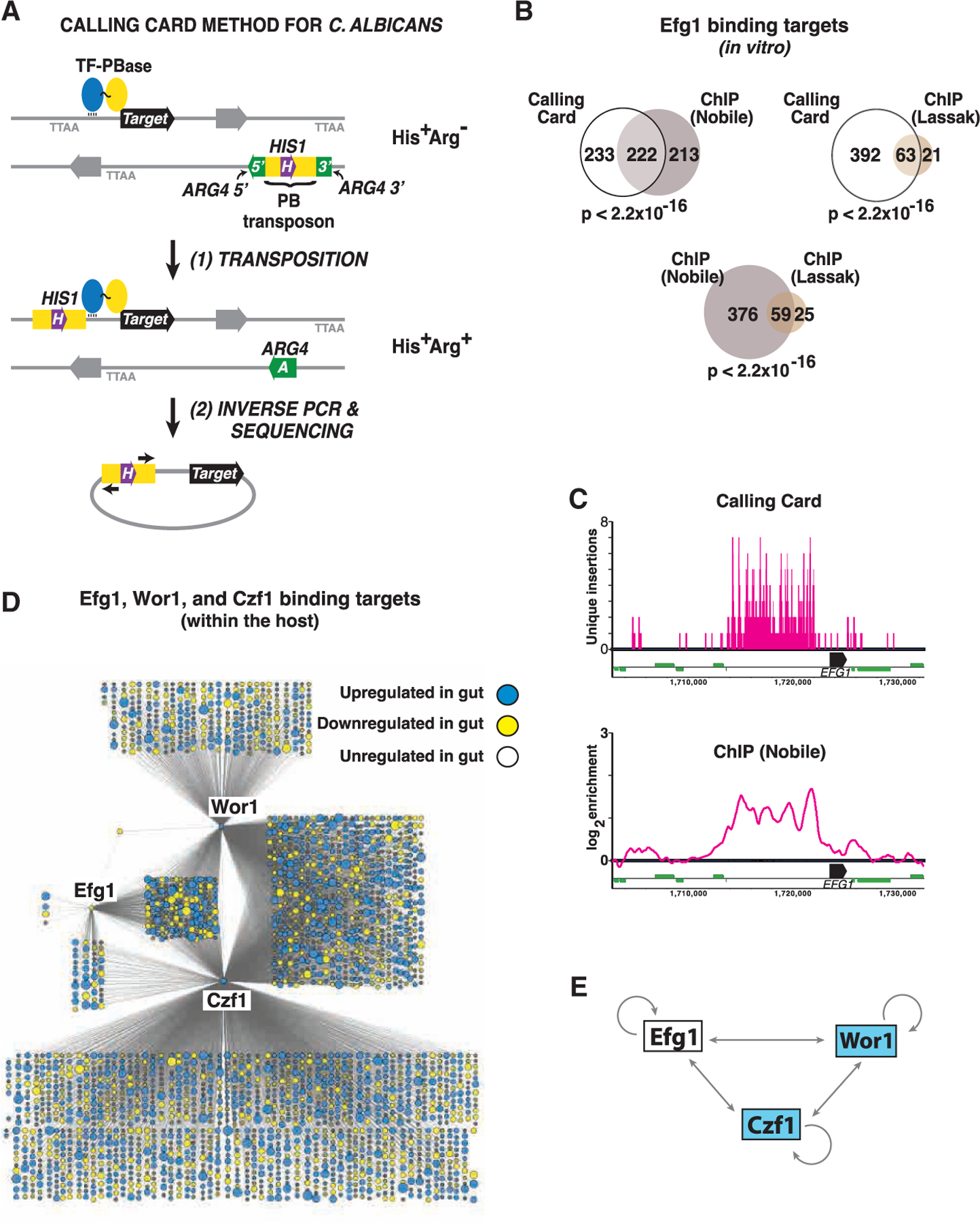
(A) Cartoon of the method. Test strains contain 1 or 2 copies of PB express a TF-PBase fusion protein. Propagation of strains in the laboratory or in a mammalian host allows for PBase-mediated excision of PB from a neutral donor site and reinsertion into a TTAA sequence abutting the binding site of the TF. Transposon excision from the donor site repairs a split ARG4 gene. Because PB is marked with HIS1, cells that have undergone transposon excision followed by reinsertion elsewhere in the genome will exhibit an Arg+His+ phenotype (or Arg+His+Leu+ in cells that contain two copies of PB).
(B) Similar Efg1 binding sites are determined by in vitro CCS and ChIP. Venn diagrams depict the overlap between 455 Efg1 binding targets identified by CCS and targets reported in two previous ChIP studies (Lassak et al., 2011; Nobile et al., 2012); note that only 435 of the 446 Efg1 binding targets reported by Nobile et al. exist in the current C. albicans genome assembly (Assembly 22). Significance of target overlap was determined by Fisher’s exact test.
(C) Comparison of results obtained using in vitro CCS and ChIP for Efg1 binding to the EFG1 locus. CCS results are depicted in the upper panel. Note that a maximum of 2 events can be mapped to a given TTAA sequence depending on the direction of insertion, and vertical bars >1 represent independent insertion events in closely spaced TTAA sequences. ChIP binding peaks reported by Nobile et al. (Nobile et al., 2012) are depicted on the lower panel. Black horizontal bars represent EFG1 ORF, thinner green bars represent neighboring ORFs.
(D) Regulatory networks of Efg1, Wor1, and Czf1 during commensal growth in the host. Each node corresponds to a direct binding target of Efg1, Wor1, and/or Czf1 mapped by in vivo CCS, with node size reflecting the fold-change in expression of the corresponding ORF when WT C. albicans is propagated in the gut compared to in vitro (YPD, 30°C; Witchley et al., 2019). Blue indicates ≥2-fold upregulation in the gut and yellow ≥2-fold downregulation (adjusted p < 0.05 using a linear fit model).
(E) Schematic of cross regulation among Efg1, Wor1, and Czf1 within the host digestive tract. Results obtained with in vivo CCS suggest that, unlike under in vitro conditions, a/α cells express all three regulators within the mammalian gut.
To validate the modified Calling Card system, we deployed it to map the binding sites of Efg1-PBase under in vitro conditions. A strain expressing untethered PBase (not fused to a TF) under the regulation of the EFG1 promoter was used as a control; note that PBase contains its own nuclear localization signal (Keith et al., 2008). Transposition was permitted for seven days, while the strains were propagated on solid culture medium (Lees 2% GlcNAc/agar pH 6.8) at 25°C. Transposition reintegrants were then selected by replica-plating onto medium lacking arginine and histidine. Genomic DNA from Arg+His+ colonies was used for inverse PCR and sequencing library preparation, as described in the STAR methods. After mapping of transposon insertion sites to the genome, background signal associated with untethered PBase was subtracted from the Efg1-PBase signal. Finally, high confidence Efg1-PBase binding sites were defined as promoters with at least 5 independent transposon insertion events (i.e., at different TTAA sequences) within a 1,000 bp window and p < 0.05 using a Poisson or hypergeometric test.
The modified Calling Card-seq technique identified 455 high confidence DNA-binding targets for Efg1-PBase (Table S1). Two groups have previously used ChIP to identify Efg1 binding targets in a/α strains. Lassak et al. reported 84 Efg1 binding sites in C. albicans yeast propagated in liquid YEPD medium at 30°C (Lassak et al., 2011), and Nobile et al. reported 435 Efg1 binding sites in C. albicans biofilms propagated in Spider medium at 37°C (Nobile et al., 2012). Notably, the set of targets identified using Calling Card-seq exhibits significant overlap with both sets of ChIP-defined targets (63 in common with the Lassak dataset, p < 2.2 × 10−16, and 222 in common with the Nobile dataset, p < 2.2 × 10−16). Moreover, the significance of overlap between Calling Card-defined targets and either set of ChIP-defined targets is approximately the same as those of ChIP-defined targets with each other (59 common targets, p < 2.22 × 10−16) (Figure 3B). GO-term analysis of the 55 targets identified by all three studies reveals significant enrichment for processes known to be regulated by Efg1, including ‘‘regulation of filamentous growth’’ (p = 1.2 × 10−5), ‘‘cell adhesion’’ (p = 2.6 × 10−5), ‘‘regulation of growth’’ (p = 2.9 × 10−5), ‘‘regulation of single-species biofilm formation’’ (p = 2.9 × 10−5), and ‘‘regulation of phenotypic switching’’ (p = 4.2 × 10−5). Moreover, the three studies reveal similar ‘‘footprints’’ of multiple Efg1 binding events at some large promoters, including the ~10-kb promoter of EFG1 itself (Figure 3C; Lassak et al., 2011; Nobile et al., 2012).
In addition to targets that were also detected using ChIP, Calling Card-seq identified 227 potential Efg1 binding targets that have not previously been described (Table S1). GO-term analysis of this gene set indicates enrichment of terms such as ‘‘interspecies interaction between organisms’’ (p = 8.3 × 10−3, FDR = 2%), ‘‘septum digestion after cytokinesis’’ (p = 0.021, FDR = 1%), and ‘‘symbiotic process’’ (p = 0.027, FDR = 0.67%). Consistent with a role for Efg1 in the regulation of at least some of these targets, 8 of the Calling Card-seq-specific targets (RBE1, EAP1, RCT1, PGA54, HWP2, CHT2, SUN41, and PTP3) have previously been reported to exhibit abnormal expression in efg1 mutants (Harcus et al., 2004; Li and Palecek, 2003; Nobile et al., 2012; Sohn et al., 2003).
Recording of TF binding events during mammalian gut commensalism
To determine the activities of Efg1, Wor1, and Czf1 during commensalism, we utilized strains containing Efg1-PBase, Wor1-PBase, or Czf1-PBase. Each strain was introduced into an independent group of BALB/c mice (n = 4–5 animals per strain), and colonization was allowed to progress for 15 (Efg1-PBase and Czf1-PBase) or 20 (Wor1-PBase) days; the Wor1-PBase strain was provided 5 extra days because of our previous observation that the pro-commensalism activity of WOR1 (in a WOR1OE strain) begins after ~5 days of host colonization (Pande et al., 2013). Animals were euthanized periodically over the experimental time course, and yeasts recovered from ceca (the gut compartment containing the highest burden of C. albicans) were plated onto medium lacking arginine, histidine, and leucine to select for transposon excision and reintegration events. A strain that expresses untethered PBase (PBaseOE) was propagated in vitro as a negative control. Genomic DNA extraction, inverse PCR, and preparation of sequencing libraries were performed as described in the STAR methods.
After subtraction of background signal (signal from untethered PBase) and after thresholding for high confidence hits, Calling Card-seq revealed 2,745 C. albicans genes that are targeted by at least one of the TFs in commensally propagated cells (Table S2). Direct binding targets Wor1, Czf1, and Efg1 are depicted as nodes in the graphic presented in Figure 3D. Notably, each of the three TFs binds to its own promoter, and those of the other two profiled TFs during host colonization (Figures 3D and 3E). Similar results for Efg1 have been reported by Nobile et al., based on ChIP analysis of in vitro-propagated cells (Nobile et al., 2012); however, the DNA-binding activities of Wor1 and Czf1 have not previously been studied in a/α cells. Indeed, it would be difficult to analyze Wor1 using ChIP because of its very low abundance in this cell type under in vitro conditions. 178 genes are targeted by all three DNA-binding proteins (Figure 3D; Table S2); this set of coregulated genes includes the ones encoding 24 TFs, in addition to Wor1, Czf1, and Efg1. Importantly, eight of the additional TFs play demonstrated roles in commensal regulation: Wor2, Wor3, Wor4, and Ahr1 (shown in Figure 1C) and Tec1, Brg1, and Ume6 (Witchley et al., 2019) inhibit gut colonization, whereas Crz2 has been reported to enhance the establishment of colonization (Znaidi et al., 2018).
In addition to direct binding targets, the graphic in Figure 3D includes transcriptomics (RNA-seq) information from a previous comparison of WT C. albicans propagated in the murine digestive tract versus under standard in vitro culture conditions (YEPD, 30°C; Witchley et al., 2019). For genes exhibiting differences of ≥2-fold between the two conditions (p < 0.05 using a linear fit model; Ritchie et al., 2015), node size reflects the relative magnitude of regulation, and node color reflects whether expression is increased (blue) or decreased (yellow) in the gut. Overall, Wor1, Czf1, and Efg1 directly bind to almost half of the 2,201 genes with gut-associated expression changes.
Comparative transcriptomics of circuit-altered strains uncovers commensalism effectors
Our previous unbiased genetic screens for commensalism factors have exclusively identified transcriptional regulators (Pande et al., 2013; Witchley et al., 2019), likely reflecting the outsized effects of regulatory proteins that control the expression of hundreds or thousands of genes. We hypothesized that TF mutants with contrasting commensal phenotypes might be useful tools for the discovery of commensalism effectors. In particular, we predicted that comparing the transcriptomes of mutants with enhanced versus diminished commensal fitness would reveal effectors of commensal interactions with the host.
To test this hypothesis, we used RNA-seq to profile RNA expression in efg1, efg1wor1, WOR1OE GUT cells, wor1, czf1, and isogenic WT controls. To capture gene expression under commensal conditions, each strain was gavaged into a separate group of BALB/c mice (n = 5–6 per strain). After 10 days of colonization, strains were recovered from host large intestines, and RNAs were extracted for RNA-seq. For comparison, the strains were also profiled under in vitro conditions (YEPD, 30°C).
Shown in Figure 4A is a volcano plot of gene expression in commensally propagated hyperfit strains (efg1, WOR1OE GUT cells, efg1wor1) versus strains with reduced commensal fitness (wor1, czf1). For each gene, relative RNA abundance (log2[abundance in hyperfit/abundance in hypofit]) is plotted on the x axis, and significance (−log10[adjusted p value]) is plotted on the y axis; all RNA-seq results are presented in Table S3. In total, 312 genes were significantly (p < 0.05) upregulated by at least 2-fold in the hyperfit strains, whereas 590 were downregulated. We hypothesized that genes that inhibit commensalism would be downregulated in hyperfit strains. In support of this idea, GO-term analysis of the downregulated gene set reveals significant enrichment of hypha-associated genes (hyphal cell wall, p = 6.48 × 10−7). In previous work, we have shown that hypha-specific cell wall and secreted factors diminish the fitness of C. albicans in the gut colonization model, likely because of host restriction of this invasive cell type (Witchley et al., 2019).
Figure 4. Transcriptomic comparisons between mutants with enhanced versus diminished commensal fitness reveals commensalism effectors.
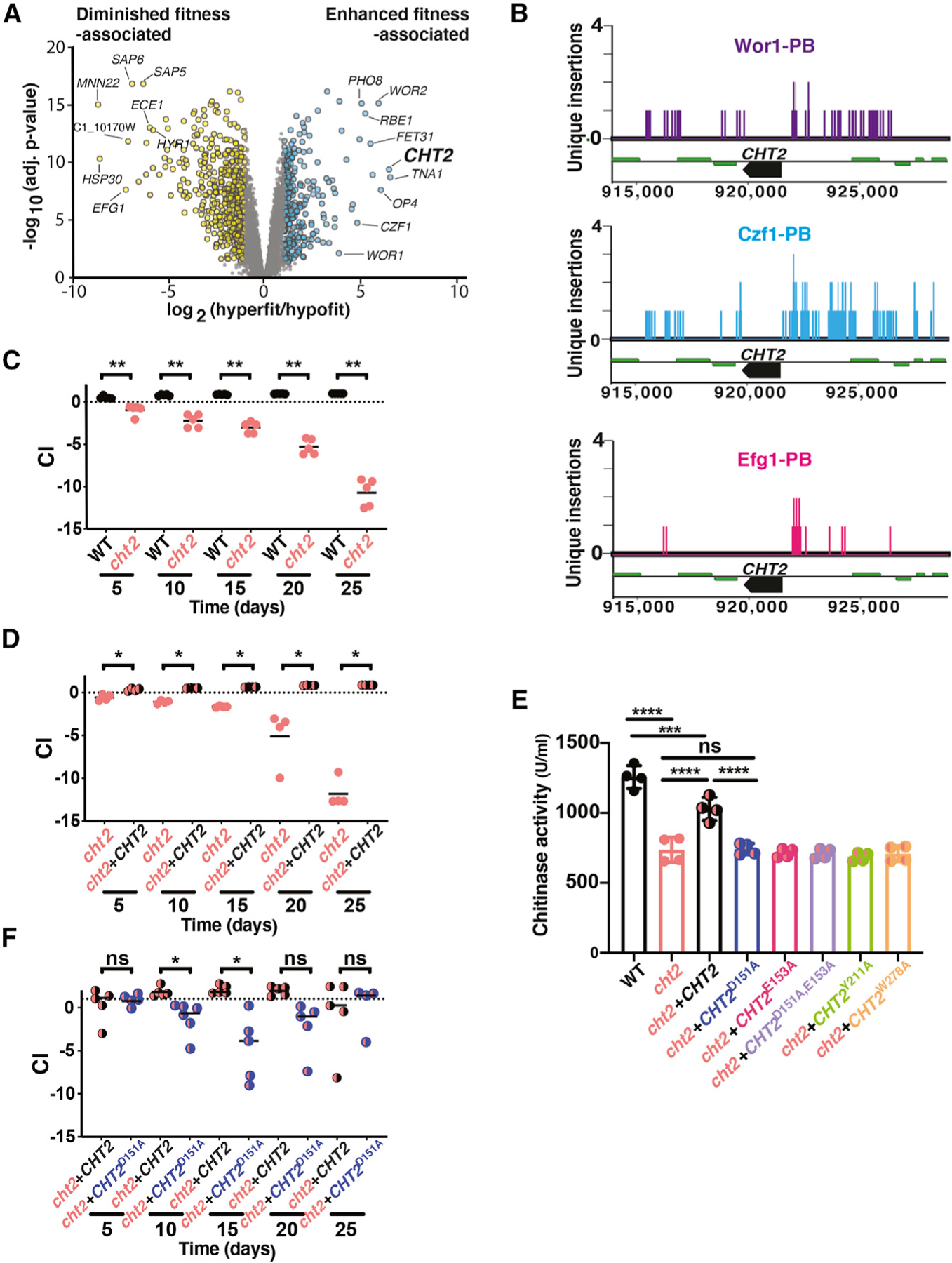
(A) Volcano plot of the log2 transformed ratio of gene expression among mutants with enhanced commensal fitness (efg1, WOR1OE GUT cells, efg1wor1) compared with those with commensal defects (wor1, czf1; x axis) versus significance (y axis).
(B) In vivo CCS reveals direct binding of Wor1, Czf1, and Efg1 to the CHT2 promoter in commensally growing cells.
(C) CHT2 is required for gut commensalism. 1:1 competition between WT (SN250) and cht2 (SN2162) in the gut colonization model. Statistical analysis was performed as in Figure 1C.
(D) The DxE motif and residues in the predicted chitin binding pocket are required for chitinase activity of Cht2. Cell pellets of WT (SN250), a cht2 homozygous null mutant (SN2162), and derivatives of cht2 to which a single copy of WT CHT2 (SN2225), CHT2D151A (SN2172), CHT2E153A (SN2174), CHT2D151A,E152A (SN2176), CHT2Y211A (SN2178), or CHT2W278A (SN2180) was restored to its endogenous locus were tested using a fluorescently labeled endochitinase substrate (4-methylumbelliferyl β-D-N,N′,N′′-triacetylchitotriose). Significance was calculated using ordinary one-way ANOVA; ns, not significant, ***p < 0.001, ****p < 0.0001.
(E) A WT copy of CHT2 complements the commensal fitness defect of cht2. 1:1 competition between cht2 (SN2161) and cht2+CHT2 (SN2170).
(F) The chitinase activity of Cht2 is required for full commensal fitness. 1:1 competition between cht2+CHT2 (SN2225) and cht2+CHT2D151A (SN2227).
We hypothesized that genes encoding positive effectors of commensalism would be upregulated in the hyperfit strains. GO-term analysis of this gene set revealed enrichment of genes associated with translation (ribosome, p = 4.51 × 10−8) and sugar transport (monosaccharide transmembrane transporter activity, p = 0.00119), among other processes. Focusing on the most strongly upregulated transcripts in Figure 4A, we noted that CHT2 is upregulated by more than 40-fold (p = 4.3 × 10−10) in hyperfit strains (Figure 4A; Table S3). CHT2 encodes a GPI-linked chitinase that is predicted to be localized at the fungal cell surface, where it could potentially interact with the host and/or co-colonizing microbes. Moreover, CHT2 is among the common binding targets of Efg1, Wor1, and Czf1 identified by in vivo Calling Card-seq (Figure 4B; Table S2) and was also identified by in vitro Calling Card-seq analysis of Efg1-PBase (Table S1) but not by previous Efg1 ChIP studies (Lassak et al., 2011; Nobile et al., 2012).
To determine whether CHT2 plays a role in C. albicans commensalism, we generated two independent isolates of a cht2 null mutant. As shown in Figure S4, both isolates of cht2 resemble WT under standard in vitro conditions (YPD agar, Figure S4A), under cell wall stress-inducing conditions (Congo red, Calcofluor white, O.04% SDS, Figure S4A), and under microaerophilic and anaerobic conditions (Figure S4B). In contrast, cht2 exhibits a striking competitive deficit in the gut colonization model (Figures 4C and S4C). Restoration of one copy of CHT2 to the null mutant suppresses the commensalism defect (Figure 4D), supporting linkage between CHT2 and commensal fitness. Together, these results suggest that Cht2 is a positive effector of commensalism.
Cht2 chitinase activity promotes commensalism
Fungal chitinases degrade chitin, a linear polymer of β(1,4)-N-acetyl-D-glucosamine (GlcNAc) that provides tensile strength to the fungal cell wall. Chitinases have previously been implicated in cell wall remodeling, autolysis, nutrient acquisition, and virulence (Yang and Zhang, 2019), but not in commensalism. Among the four chitinase genes in the C. albicans genome (CHT1, CHT2, CHT3, CHT4; Dünkler et al., 2005), CHT2 is the only one to be induced in commensally hyperfit strains (Table S3).
To determine whether chitinase activity is required for the commensal role of Cht2, we generated a series of mutant CHT2 alleles that substitute alanine (A) for conserved residues that are critical for the enzymatic function of Cts1, a well-characterized homolog in Saccharomyces cerevisiae (Figure S4D; Dünkler et al., 2005; Hurtado-Guerrero and van Aalten, 2007). CHT2D151A, CHT2E153A, and CHT2D151A+E153A target aspartate (D) and/or glutamate (E) residues of the DxE motif at the predicted active site, and CHT2Y211A and CHT2W278A target tyrosine (Y) or tryptophan (W) residues that line the predicted chitin binding pocket. A set of isogenic strains was created by integrating one copy of each point mutant allele into its endogenous locus in a cht2 null background. Total cellular chitinase activity was then determined for the mutants and WT, cht2, and cht2+CHT2 controls (Figure 4E). Compared with WT, a cht2 null strain exhibits a ~2-fold reduction in chitinase activity, with residual activity likely attributable to the remaining cellular chitinases (Cht1, Cht3, Cht4). Restoration of one copy of CHT2 to the null mutant yields intermediate activity between that of WT (CHT2/CHT2) and cht2, suggesting that enzymatic activity reflects the gene copy number. By comparison, all five CHT2 point mutants exhibit a similar level of chitinase activity to cht2, suggesting that the encoded proteins are enzymatically inactive.
We next tested the commensal fitness of strains bearing one copy of CHT2D151A or CHT2D151A+E153A in competition with a strain bearing one copy of WT CHT2. As shown in Figures 4F and S4E, the strains expressing enzymatically inactive protein are outcompeted over the first 15 days of colonization, suggesting that chitinase activity contributes to the commensal function of Cht2. However, unlike a cht2 null mutant (Figures 4C and S4C), the CHT2 point mutants recover at subsequent time points. The superior fitness of cht2+CHT2D151A and cht+−CHT2D151A+E153A compared with cht2 suggests Cht2 may promote commensalism using features in addition to chitinase activity.
DISCUSSION
In this work, we establish roles for 8 key regulators of C. albicans mating in colonization of the mammalian gut. Null mutants affecting WOR2, WOR3, WOR4, AHR1, CZF1, and SSN6 exhibit altered fitness in a mouse model of commensal colonization, as we have previously demonstrated for mutants affecting WOR1 and EFG1 (Pande et al., 2013). Moreover, genetic epistasis analysis of double mutants indicates that core relationships among Czf1, Efg1, and Wor1 are preserved between sex and host colonization. The repurposing of a complex regulatory pathway for two very different outcomes was unexpected. However, our in vivo analysis of TF DNA-binding activity using Calling Card-seq supports activity of all tested regulators in commensally growing yeasts.
Based on our analysis of C. albicans single and double null mutants in the murine gut, we propose a model for the regulation of commensalism that is depicted in Figure 5. CZF1 is proposed to sit atop the regulatory cascade, where it promotes commensalism by (directly or indirectly) inhibiting EFG1, a known inhibitor of commensalism (Liang et al., 2019; Pande et al., 2013; Pierce and Kumamoto, 2012; Witchley et al., 2019). The pro-commensal activity of CZF1 is deduced from the fitness defect of czf1 (Figures 1C and S2D), and the position of CZF1 upstream of EFG1 in a linear regulatory pathway is deduced from the enhanced fitness of efg1czf1, which precisely matches the phenotype of efg1 (Figures 2D and 2E). EFG1 is proposed to inhibit commensalism by a branched regulatory pathway: The first branch draws upon Efg1’s well-documented role as a transcriptional activator of hypha-associated genes (Lo et al., 1997; Stoldt et al., 1997); such genes include SAP6, which we have previously shown to inhibit commensal fitness in the gut (Witchley et al., 2019). In this study, we show that Efg1 binds to the SAP6 promoter (Table S2) and activates SAP6 expression by more than 50-fold (p = 2.0 × 10−11) (Table S3) in commensally propagated cells. The second regulatory role of Efg1 is proposed to occur by (direct or indirect) repression of WOR1 and/or the white-to-GUT switch, which are required for normal commensal fitness (Pande et al., 2013). The proposed branched regulatory network is consistent with observations that efg1wor1 and czf1WOR1OE GUT strains exhibit distinct commensal phenotypes from those of the corresponding single mutants. For example, although efg1 exhibits a strongly enhanced fitness phenotype and wor1 exhibits a fitness defect (Pande et al., 2013), the efg1wor1 strain exhibits an intermediate level of enhanced fitness compared with WT (Figure 2B) and is outcompeted by efg1 (Figures 2C and S2H). These observations can be rationalized if both forms of Efg1-mediated inhibition (activation of SAP6 and repression of WOR1 and the white-to-GUT switch) are reversed in an efg1 mutant, but only SAP6 activation is reversed in the efg1wor1 strain (because the white-to-GUT switch does not occur in the absence of WOR1). Finally, the observed requirement for CZF1 for the white-to-GUT switch can be rationalized if switching requires at least transient repression of EFG1.
Figure 5. Model of commensal regulation in Candida albicans.
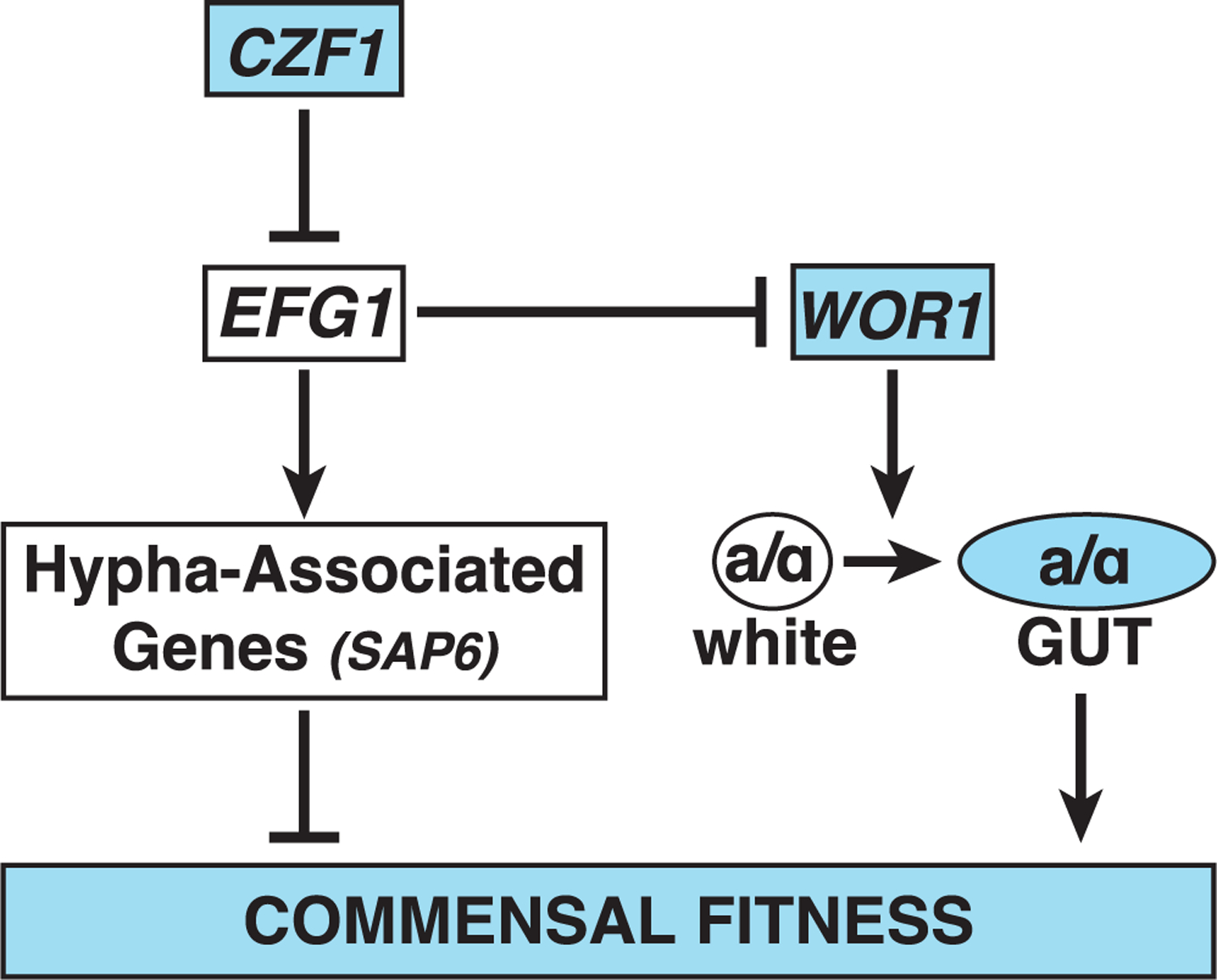
The depicted regulatory circuit is consistent with the results of epistasis analysis of mutants affecting WOR1, EFG1, and CZF1 in a mouse model of gut commensalism, in vivo CCS analysis of Wor1-PBase, Efg1-PBase, and Czf1-PBase, and published observations. Czf1 is proposed to promote gut commensalism primarily via inhibition of EFG1. Efg1 inhibits gut commensalism by two mechanisms: (1) activation of hypha-associated genes, including SAP6, which itself inhibits commensal fitness (Witchley et al., 2019) and (2) inhibition of WOR1 and/or the white-to-gut switch to a commensal-specific cell type (Pande et al., 2013).
We note that the core genetic logic of the proposed commensalism pathway resembles that of the sexual circuit (Figure 1B), with Czf1 inhibiting Efg1, Efg1 inhibiting Wor1 and/or GUT cell formation, and Wor1 promoting a cell type switch that favors the regulated process. Differences between the circuits include (1) dual inhibitory roles for Efg1 in commensalism but only one in mating and (2) differing roles for Ssn6, Wor2, Wor3, and Wor4 in activation versus inhibition of the respective processes (Figure 1D).
To gain insight into the DNA-binding activity of three core regulators during commensal colonization, we adapted Calling Card-seq (Wang et al., 2011a, 2012) for use in C. albicans (Figure 3A). Under in vitro growth conditions, the modified technique identified 455 direct binding targets of Efg1 (Figure 3B; Table S1), approximately half of which had previously been reported in ChIP-based investigations (Lassak et al., 2011; Nobile et al., 2012). The significant overlap among targets identified by Calling Card-seq and ChIP (p < 2.2 × 10−16 for each comparison) supports the validity of the Calling Card-seq method. Furthermore, Calling Card-seq performed in colonized animals identified 11 additional regulators of commensalism (WOR2, WOR3, WOR4, AHR1, TEC1, BRG1, UME6, CRZ2, WOR1, CZF1, and EFG1) (Table S3) among the common targets of Efg1, Czf1, and Wor1. In addition to common targets, in vitro Calling Card-seq and the previous ChIP investigations of Efg1 each identified a number of study-specific binding targets (Table S1; Figure 3B; Lassak et al., 2011; Nobile et al., 2012). Besides random experimental ‘‘noise,’’ it is worthwhile to consider other variables that may have influenced the results. For example, although cells for all three studies were propagated in vitro, the cell culture conditions and the structure of the cell population (biofilm versus planktonic yeast versus yeast colonies) were distinct in each case. If the expression or DNA-binding activity of Efg1 is sensitive to environmental cues, differences in cell state, etc., then at least some of the discrepant findings may reflect bone fide differences in Efg1’s genomic localization. Technical features of ChIP and Calling Card-seq could also affect the results. Nobile et al. and Lassak et al. fused epitope tags (7xMyc-Efg1 and hemagglutinin, respectively) to Efg1 to facilitate immunoprecipitation. However, epitope tags may sterically hinder protein-DNA or protein-protein interactions that are required for binding to certain targets. Furthermore, differences in epitope accessibility and/or antibody affinity for the epitope can alter the ability to detect some targets. In Calling Card-seq, PBase fusion to the TF of interest may similarly alter TF binding to some targets. Moreover, because TTAA sequences are required for PB transposon insertion, Calling Card-seq may underrepresent targets that contain few if any of these sequences.
Compared with other genome-wide methods, the major advantage of Calling Card-seq is that it enables the study of TF binding activity under physiologically relevant conditions such as in hosts. For example, the Efg1-PBase, Czf1-PBase, and Wor1-PBase strains described in this study are suitable for use in existing mouse models of C. albicans skin colonization, cellulitis, oroesophageal infection, vaginitis, gut colonization, gut inflammation, and disseminated infection (Cassone and Sobel, 2016; Igyártó et al., 2011; Kadosh et al., 2016; Koh, 2013; Kvaal et al., 1999; MacCallum and Odds, 2005; Solis and Filler, 2012; Xie et al., 2013).
We utilized TF mutants with strong gain- or loss-of-commensal fitness phenotypes as tools for discovery of commensalism effectors. Our transcriptomic analysis of commensally propagated hyperfit (efg1, efg1wor1, WOR1OE GUT) and hypofit (wor1 and czf1) strains revealed consistent downregulation of hypha-associated genes in hyperfit strains, including SAP6 (Figure 4A; Table S3). Among strongly upregulated genes, we focused on CHT2, which encodes a GPI-linked chitinase that is predicted to be localized at the fungal cell surface. CHT2 was also identified by in vivo Calling Card-seq as a direct binding target of Efg1, Wor1, and Czf1 (Figure 4B; Table S2) and is among the Calling Card Seq-specific Efg1 binding targets under in vitro conditions (Table S1; Lassak et al., 2011; Nobile et al., 2012). Consistent with a role for Cht2 as a commensalism effector, the cht2 null mutant exhibits a substantial competitive defect (Figure 4C). Chitinase activity seems to contribute to effector activity, given that strains expressing catalytically dead alleles (CHT2D151A or CHT2D151AE153A) are defective at early times during gut colonization (Figures 4F and S4F). Chitin is a conserved structural component of the fungal cell wall that can trigger host innate immune responses (Reese et al., 2007). Although the precise mechanism by which Cht2 promotes C. albicans fitness in the gut remains to be determined, an intriguing possibility is that Cht2 may alter host detection of commensally propagating yeasts or may modulate the resulting immune response.
Mating and the white-to-opaque epigenetic switch are among the most highly studied processes in C. albicans. Nevertheless, there is little evidence for sexual exchange among naturally circulating strains (Bougnoux et al., 2008; Legrand et al., 2004), and mating is inferred to be exceedingly uncommon. In contrast, gut colonization with C. albicans is ubiquitous among humans and other mammals, and propagation within the commensal niche may provide ongoing selection for circuit components that are shared between mating and commensalism. Recently, seven of the regulators profiled in this study (Efg1, Czf1, Wor1, Wor2, Wor3, Wor4, and Ssn6) were reported to possess prion-like domains that promote the formation of phase-separated condensates within the nucleus (Frazer et al., 2020). In future, it will interesting to determine whether the ability to form distinct condensates or alternative mechanisms permit one set of transcriptional regulators to orchestrate two very different cellular outcomes (mating versus host colonization).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Suzanne M. Noble (suzanne.noble@ucsf.edu).
Materials availability
C. albicans strains and plasmids generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and code availability
The raw files used for mRNA-seq analysis are available through GEO accession number GSE150855. The raw data for Calling Card experiments are available through GEO accession number GSE150856. Custom code used for analysis of CCS data is available at https://github.com/jwitch/CallingCard.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains
The yeast strains used for this study are described in Table S4, plasmids in Table S5, and primers in Table S6. All media were prepared as previously described in Homann et al. (Homann et al., 2009) for YPD, Spider, Calcofluor white, and 0.04% SDS, in Lee et al. (Lee et al., 1975) for Lee’s media, and in Bruno et al. (Bruno et al., 2006) for Congo red concentration (50 μg/mL). From glycerols, strains were streaked to YPD and grown for 2 days at 30°C. Cells were resuspended in distilled water to an OD of 1 and then diluted as appropriate for the assay. For single colony growth analysis, 100–200 CFUs were plated in appropriate media. For spot plating, strains were diluted five-fold and 3 μL of each dilution spotted to designated media.
Studies in animals
All procedures involving animals were approved by the UCSF Institutional Animal Care and Use Committee. The mouse model of C. albicans commensalism was performed as previously described (Chen et al., 2011; Pande et al., 2013) with minor modifications. Groups of 8- to 10-week-old (18–21 gram) female BALB/c mice were housed 1 to 2 per cage and treated with penicillin 1,500 un/mL and streptomycin 2 mg/mL in drinking water starting 7 days before gavage and continuously for the remainder of the experiment. Feces were plated on Sabouraud agar, LB agar, and MRS agar to monitor for fungal and bacterial growth.
For commensal competition experiments, groups of 4–8 mice were gavaged with 108 CFUs of a 1:1 mix of two fungal strains in 0.9% saline. C. albicans was recovered from the inoculum and host feces at specified intervals, and strain quantification was performed as described previously (Noble et al., 2010).
For mRNA-seq experiments, 5 or 6 mice were colonized with a single strain and individually housed. After 10 days, animals were humanely euthanized, and large intestines were dissected for RNA analysis.
METHOD DETAILS
mRNA-seq library preparation and analysis
RNA extraction for mRNA-seq was performed as previously described (Witchley et al., 2019), with modifications. For in vitro expression comparisons, three replicates were used for each strain per condition. Luminal contents of large intestines were resuspended in a mixture of 500 μL Buffer A (200 mM NaCl and 20 mM EDTA), 500 μL acid phenol-chloroform, 168 μL 25% SDS, and ~400 μL 0.5 mm glass beads. Samples were vortexed for 5 cycles of 2 min, interrupted with 1-min rests on ice. Samples were centrifuged at max speed in a microcentrifuge for 10 min, and supernatants were extracted four times with equal volumes of acid phenol-chloroform (Ambion), followed by four rounds of extraction with equal volumes of phenol-chloroform-isoamyl alcohol (Ambion). After precipitation and resuspension of pellets in RNase-free water, RNA was further purified using the MEGAclear transcription clean-up kit (Ambion) for wor1 (ySN1351) or Sera-Mag beads in a homemade PEG solution (https://openwetware.org/wiki/SPRI_bead_mix) following the RNAclean XP protocol for ySN425, efg1 (ySN1011), czf1 (ySN1145), WOR1OE GUT (ySN1045), and efg1wor1 (ySN1126). DNase I (NEB) treatment was performed on at least 5 µg RNA for 10 min at 37°C. After DNase treatment, a final acid phenol-chloroform extraction was performed. RNA was then precipitated and resuspended in RNase-free water.
NEBNext Ultra Directional RNA Library Prep Kit for Illumina was used in combination with NEBNext Poly(A) mRNA Magnetic Isolation Module and NEBNext Multiplex Oligos for Illumina to generate mRNA-seq libraries. The protocol provided by NEB was followed using 1 µg total RNA and 14 cycles of PCR, with the exception that Ampure XP bead mix was replaced with Sera-Mag Speed Beads (Thermo Fisher Scientific) in a homemade PEG solution (Rohland and Reich, 2012). Library fragment size was determined using High Sensitivity DNA chips on a 2100 Bioanalyzer (Agilent). Library quantification was performed by qPCR with a library quantification kit from KAPA Biosystems (KK4824). Sequencing was performed on a HiSeq4000 device (Illumina).
Sequencing reads were mapped to the current haploid C. albicans transcriptome (Assembly 22 default coding, downloaded from candidagenome.org on November 19, 2018), and transcript abundances were determined using kallisto (Bray et al., 2016). Statistical comparisons of transcript abundances between different samples were performed on estimated counts generated by kallisto using limma as previously described (Ritchie et al., 2015). Transcriptomic analysis was performed by binning RNA expression results of strains with enhanced versus diminished commensal fitness into ‘‘hyperfit’’ and ‘‘hypofit’’ groups. Voom implemented in limma was used to model the observational variance in log-counts, and this model was subsequently used in limma’s empirical Bayes method to calculate adjusted p values for differences in expression between the two groups, using condition (YEPD 30°C, YEPD+-serum 37°C or large intestine) and batch (date of processing) as factors.
GO-term analysis was performed using the Candida Genome Database GO-term finder. Differentially expressed genes were considered to be those with a fold change greater than 2 and an adjusted p < 0.05. Upregulated or downregulated genes were analyzed for process, function, and component. The raw files are available through GEO accession number GSE150855.
Generation of C. albicans strains for Calling Card-seq
Because stable, autonomously replicating plasmids have not been available for use in C. albicans until quite recently (Bijlani et al., 2019), we devised a strategy to integrate recombinant Piggyback transposons, genes encoding TF-PBase fusion proteins, and codon-optimized PBASE into the genome. First, we commissioned synthesis of a PBase gene (PBASE) that is codon-optimized for C. albicans (GenScript, Piscataway, NJ). Next, we engineered a series of plasmids to target codon-optimized PBASE to the 3′ end of a TF ORF of interest; integration results in the expression of a TF-PBase fusion protein via the natural promoter for the TF. Plasmid pSN427 (which is used to create TF-specific integrating constructs) contains the following insert: PmeI restriction site—sequence encoding an 18 amino acid linker—PBASE—FLP—SAT1—PmeI restriction site. pSN427 was generated by homologous recombination in S. cerevisiae of the following three PCR products: (1) codon-optimized PBASE, amplified with primers that include linker homology and a PmeI site at the 5′ end and homology to FLP-SAT1 cassette at the 3′ end; (2) linearized pRS316 vector, amplified with primers containing linker homology on one end and FLP-SAT1 cassette homology on the other; and (3) FLP-SAT1, amplified from pSFS2A using primers with homology to PBASE at the 5′ end and pRS316 at the 3′ end.
To create custom integration constructs that target PBASE to the WOR1 (pSN459), EFG1 (pSN467), and CZF1 (pSN465) ORFs, homologous recombination in S. cerevisiae was used to fuse the following three inserts: (1) linker—PBASE—FLP—SAT1 fragment from PmeI-digested pSN427; (2) PmeI restriction site—terminal ~300 bp of TF ORF sequence; and (3) 300 bp of 3′ UTR for the TF—PmeI site. To create a PBaseOE control strain, an analogous method was used to create pSN422, which targets PBASE to replace one copy of the TDH3 ORF. pSN422 contains the following insert: PmeI—~300 bp of TDH3 5′ UTR—linker—PBASE—FLP—SAT1—~300 bp of TDH3 3′ UTR-PmeI.
Inserts from the PBaseOE (pSN422) and TF-PBase plasmids (pSN459, pSN467, pSN465) were liberated with PmeI and used to transform C. albicans strains containing either one (ySN1873) or two (ySN1877) copies of PB transposon (at the neutral leu2Δ locus). After 1 day of propagation on YPD/agar containing 200 μg/mL nourseothricin (YPD+Nat) at 30°C, Nat-resistant colonies were streaked to fresh YPD+Nat medium and incubated overnight at 30°C. Finally, single colonies were patched to YPD+Nat, SD-histidine, SD-histidine-arginine, and SD-histidine-leucine-arginine plates. 5′ and 3′ junctions of integrants were verified by colony PCR using primers described in Table S6. Glycerol stocks were prepared from patches exhibiting His+ (1 transposon) or His+Leu+ (2 transposons) and Arg− (transposon still present at the donor site) phenotypes. In addition, we screened for NatS versions of the strains after overnight propagation in YPD+maltose liquid culture at 30°C, which allows for spontaneous excision of the FLP-SAT1 cassette.
To validate the modified method under in vitro conditions, Efg1-PBase (SN1988) and EFG1 promoter-PBase (SN2012) strains (containing one copy of PB) were propagated for 2 days on YEPD, followed by replating of ~50,000 CFUs onto Lee’s GlcNAc pH 6.8 agar (hypha-inducing conditions where EFG1 is well-expressed) in 100 mm petri plates. Strains were incubated at 37°C for 7 days to allow for transposition to occur. Cells were then replica plated to SD-histidine-arginine and incubated for 2 days at 30°C. His+Arg+ cells were collected by washing with water, followed by extraction of genomic DNA from cell pellets.
Calling Card-seq mouse experiments
WOR1-PBASE (ySN1933), EFG1-PBASE (ySN1943), and CZF1-PBASE (ySN1941) strains containing two copies of PB were streaked from frozen glycerol stocks onto YPD plates and incubated for 16 h at 30°C. Cells were scraped from the plates, diluted to 5 × 105 CFU/mL, and 100 μL each cell suspension was spread onto several SD-histidine-leucine plates, with incubation for 48 h at 30°C. A disposable yellow loop was used to scrape colonies into 0.9% saline. 108 CFUs was gavaged into each antibiotic-treated mouse. One gavage volume was plated to SD-histidine-leucine-arginine plates to monitor for transposition events that had already occurred during preparation of the inocula (these proved to be minimal; Witchley and Noble, data not shown). To estimate the background rate of nonspecific transposition events, the PBaseOE strain (ySN1904) was grown in liquid YPD or YPD+100 μM BPS (iron chelator related to TFs not reported here) overnight and then plated to selection media.
After 5, 10, 15, and/or 20 days, animals were euthanized and dissected using sterile technique to recover the ceca. 1–2 animals per group were euthanized after 5, 10, or 15 days for Efg1-PBase, 10 or 15 days for Czf1-PBase, and 10, 15 and 20 days for Wor1-PBase. Each organ was homogenized in 3 mL PBS pH 7.4. 100 μL was diluted 1:10 in PBS to determine CFUs/gram cecum, and the remainder was plated 500 μL per 100 mm SD-histidine-leucine-arginine plate. Cells were incubated for 2 days at 30°C, and colonies were recovered for DNA extraction and sequencing library preparation. All reads from the same strain recovered from animals were pooled before data analysis.
Calling Card sequencing library preparation and analysis
Sequencing libraries were prepared as previously described (Wang et al., 2011a, 2011b, 2012) with modifications. Genomic DNA was digested MspI, RsaI and Taq1α, in independent reactions. Next, the DNA was precipitated with isopropanol and 5 M ammonium acetate, washed with 70% ethanol, and resuspended in 30 μL nuclease-free water. To circularize the pooled restriction fragments, the DNA was resuspended to 2–3 ng/µL in a 400 μL ligation reaction with T4 ligase. The ligation reaction was incubated at 15°C for greater than 16 h then precipitated with ethanol and 3 M sodium acetate, washed with 70% ethanol, and resuspended in 30 μL nuclease-free water. 15 μL of ligated DNA was used in a PCR reaction with Phusion polymerase and custom read one primer SNO2980 and one of 96 barcoded paired end primer 2. The PCR program used was as follows: step 1, 94°C 2 min; step 2, 94°C 30 s; step 3, 94°C 30 s; go to step 2 for 30 cycles; and hold at 10°C. PCR clean-up was performed using 1.0× homemade SPRI bead mix (Rohland and Reich, 2012). PCR product quantification was performed using Quant-iT PicoGreen dsDNA assay kit (P11496). After quantification, all reactions were pooled in equal quantities. Size selection was performed on the Center for Advanced Technology (UCSF) Sage Science BluePippin with 2% agarose dye-free cassettes with marker V1 using 250–500 base pair elution criteria. A subsequent buffer exchange to TE was performed with SPRI bead mix (Rohland and Reich, 2012). Libraries were visualized using High Sensitivity DNA chips on an Agilent Bioanalyzer. Single end 50 base pair sequencing was performed on an Illumina HiSeq 4000 using custom read 1 sequencing primer SNO3189.
Sequenced reads were aligned to C. albicans diploid genome Assembly 22 using bowtie (Langmead et al., 2009). Custom Calling Card-seq analysis code provided by the Mitra lab was adapted for use in C. albicans as follows: To control for possible jackpots in PCR amplification, all reads mapping to a specific genomic integration position were collapsed to a single instance of integration at that position. To limit random sequencing noise, a minimum of 50 reads per integration site was required to include the site. Transposon insertions were grouped into clusters within 1000 base pair windows. Insertion clusters for a given TF-PBase fusion protein were compared with background signal (i.e., clusters associated with untethered PBaseOE), and significant differences at each position were determined using Poisson and hypergeometric distributions. Clusters were annotated according to the names of the two closest genes using bedtools closest feature. For interpretation, the data were collapsed by assigning clusters to the closest promoters and subtracting all untethered PBase transposition events from the TF-PBase insertions. If insertions occurred between two genes transcribed in opposite directions away from each other, both genes were assigned to these insertions. High confidence TF binding targets were defined as genes associated with at least 5 independent insertions above background and p < 0.01 (Poisson or hypergeometric distribution) for at least one of the clusters upstream of the target gene. These data are presented in Figures 3B and 3D and Tables S1 and S2. The raw data are available through GEO accession number GSE150856.
For visualization in MochiView (Homann and Johnson, 2010), duplicate reads were first removed using a two-step process: duplicates in each BAM (binary sequence alignment map) file were flagged using Picard MarkDuplicates (Broad Institute, MIT); then, if the number of duplicate reads was greater than 50, a single read was kept in the BAM file as the ‘‘Unique Insertion,’’ else the reads were discarded. The genomeCoverageBed function of bedtools (Quinlan and Hall, 2010) was used to collect sequencing depth information in a bedGraph file format. The bedGraph files were then converted to fit the MochiView file format (Homann and Johnson, 2010). Formatted files were imported into MochiView as a Location/Data set. A bar graph style was used for visualization owing to the nature of the data and the MochiView file format. These data are presented in Figures 3C and 4B.
Chitinase activity assay
Overnight cultures of C. albicans strains were diluted in YEPD to OD600 = 0.1 and allowed to grow to log phase (OD600 = 1). One mL of each culture was centrifuged at 3000 rpm for 10 min. The supernatant was removed and saved, and pellets were washed and resuspended in 1 mL of 0.9% sterile saline. Ten µL of the supernatant and cell suspension were tested separately for enzymatic activity using the fluorometric chitinase activity kit (CS1030) from Sigma-Aldrich according to the manufacturer’s instructions. The plate was incubated for 1 h at 37°C. Fluorescence was measured with a Tecan plate reader with an excitation wavelength of 360 nm and an emission of 450 nm.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters and statistical test type (value of n, statistical significance, dispersion, and precision measures) are reported in the figure legends and in method details, above. Significance is generally defined as p < 0.05. The Student’s t test was used to assess the results of commensal competition experiments and chitinase assays, using GraphPad Prism 9.0. Statistical analysis of RNA-seq and Calling Card-seq experiments are included with the description of the methods, above. The Fisher’s exact test was used to determine the significance of overlap between in vitro binding targets of Efg1 identified by CCS versus ChIP, using the built-in fisher.test function of RStudio.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acid phenol chloroform | Ambion | AM9722 |
| Phenol chloroform isoamyl alcohol | Ambion | AM9732 |
|
Critical Commercial Assays | ||
| MEGAclear transcription clean-up kit | Ambion | AM1908 |
| NEB DNase I | New England Biolabs | M0303 |
| NEBNext Ultra Directional RNA Library Prep Kit for Illumina | New England Biolabs | E7420 |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | E7490 |
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | E7335, E7500 |
| High sensitivity DNA kit for Bioanalyzer | Agilent | 5067-4626 |
| Library quantification kit | KAPA Biosystems | KK4824 |
| Fluorometric chitinase activity kit | Sigma-Aldrich | CS1030 |
|
Deposited Data | ||
| RNA-Seq data | this study | GEO: GSE150855 |
| Card-See data | this study | GEO: GSE150856 |
|
Experimental Models: Organisms/Strains | ||
| Mouse: BALB/c | Charles River Laboratories | Stock No: 028 |
| SN152 | Noble et al. Euk Cell 2005 | leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN235 | this study | leu2Δleu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN250 | Noble et al. Euk Cell 2005 | leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN425 | Noble et al. Euk Cell 2005 | leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ::C.d.ARG4, iro1Δ/IRO1, MTLa/α |
| SN1011 | Homann et al. 2009 | efg1Δ::ST8-C.m.LEU2/efg1Δ::ST8-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1338 | this study | efg1Δ::ST8-C.m.LEU2/efg1Δ::ST8-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4::ST61-C.d.ARG4Δ, iro1Δ/IRO1, MTLa/α |
| SN1351 | this study | wor1Δ::ST63-C.m.LEU2/wor1Δ::ST15-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1352 | this study | wor1Δ::ST63-C.m.LEU2/wor1Δ::ST15-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1142 | Homann et al. 2009 | wor2Δ::ST27-C.m.LEU2/wor2Δ::ST27-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1143 | Homann et al. 2009 | wor2Δ::ST27-C.m.LEU2/wor2Δ::ST27-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1111 | this study | wor3Δ::C.m.LEU2/wor3Δ::C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1433 | this study | wor3Δ::C.m.LEU2/wor3Δ::ST35-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1493 | this study | wor4Δ::ST67-C.m.LEU2/wor4Δ::ST5-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1494 | this study | wor4Δ::ST67-C.m.LEU2/wor4Δ::ST6-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1144 | Homann et al. 2009 | czf1Δ::ST42-C.m.LEU2/czf1Δ::ST42-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1145 | Homann et al. 2009 | czf1Δ::ST42-C.m.LEU2/czf1Δ::ST42-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1187 | Homann et al. 2009 | ahr1Δ::ST42-C.m.LEU2/ahr1Δ::ST42-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1188 | Homann et al. 2009 | ahr1Δ::ST45-C.m.LEU2/ahr1Δ::ST45-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1488 | this study | ssn6Δ::C.m.LEU2/ssn6Δ::ST18-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN928 | Pande et al 2013 | SAT1-PTDH3-WOR1/wor1Δ::C.d.ARG4, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1448 | this study | SAT1-PTDH3-WOR1/wor1Δ::C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1507 | this study | FRT-FLP-SAT1-FRT-PTDH3-WOR1/wor1Δ::C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1045 | Pande et al 2013 | SAT1-PTDH3-WOR1/wor1Δ::C.d.ARG4, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α (mouse passage) |
| SN1465 | this study | SAT1-PTDH3-WOR1/wor1Δ::C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α (mouse passage) |
| SN1251 | this study | FRT-FLP-SAT1-FRT-PTDH3-WOR2/wor2Δ::C.d.ARG4, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1245 | this study | FRT-FLP-SAT1-FRT-PTDH3-WOR3/wor3Δ::C.d.ARG4, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1533 | this study | FRT-FLP-SAT1-FRT-PTDH3-WOR4/wor4Δ::C.d.ARG4, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1447 | this study | FRT-FLP-SAT1-FRT-PTDH3-CZF1/czf1Δ::C.d.ARG4, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1445 | this study | FRT-FLP-SAT1-FRT-PTDH3-CZF1/czf1Δ::C.d.ARG4, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1522 | this study | FRT-FLP-SAT1-FRT-PTDH3-AHR1/ahr1Δ::C.d.ARG4, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1525 | this study | FRT-FLP-SAT1-FRT-PTDH3-AHR1/ahr1Δ::C.d.ARG4, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1126 | this study | efg1Δ::C.m.LEU2/efg1Δ::C.d.HIS1, wor1Δ::C.d.ARG4/wor1Δ::FRT-FLP-SAT1-FRT, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1373 | this study | efg1Δ::C.m.LEU2/efg1Δ::C.d.HIS1, czf1Δ::C.d.ARG4/czf1Δ::FRT-FLP-SAT1-FRT, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1341 | this study | wor3Δ::C.m.LEU2/wor3Δ::C.d.HIS1, wor1Δ::C.d.ARG4/wor1Δ::FRT-FLP-SAT1-FRT, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1377 | this study | wor3Δ::C.m.LEU2/wor3Δ::C.d.HIS1, czf1Δ::C.d.ARG4/czf1Δ::FRT-FLP-SAT1-FRT, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1340 | this study | wor2Δ::C.m.LEU2/wor2Δ::C.d.HIS1, wor1Δ::C.d.ARG4/wor1Δ::FRT-FLP-SAT1-FRT, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1375 | this study | wor2Δ::C.m.LEU2/wor2Δ::C.d.HIS1, czf1Δ::C.d.ARG4/czf1Δ::FRT-FLP-SAT1-FRT, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1245 | this study | wor3Δ::C.m.LEU2/wor3Δ::C.d.HIS1, FRT-FLP-SAT1-FRT-PTDH3-WOR2/wor2Δ::C.d.ARG4, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1656 | this study | FRT-FLP-SAT1-FRT-PTDH3-WOR1/wor1Δ::C.m.LEU2, FRT-PTDH3-WOR2/wor2Δ::C.d.HIS1 leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1657 | this study | FRT-FLP-SAT1-FRT-PTDH3-WOR1/wor1Δ::C.m.LEU2, FRT-PTDH3-WOR2/wor2Δ::C.d.HIS1 leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1147 | this study | SAT1-PTDH3-WOR1/wor1Δ::C.d.ARG4, czf1Δ::C.m.LEU2/czf1Δ::C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1468 | this study | SAT1-PTDH3-WOR1/wor1Δ::C.d.HIS1, czf1Δ::C.m.LEU2/czf1Δ::C.d.ARG4, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1873 | this study | leu2Δ:: 5′ UTR homology-split C.d.ARG4 3′-PiggyBac transposon LTR-C.d.HIS1-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1877 | this study | leu2Δ::split C.d.ARG4 3′-PiggyBac transposon LTR-C.d.HIS1-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′/leu2Δ::split C.d.ARG4 3′-PiggyBac transposon LTR-C.m.LEU2-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1903 | this study | leu2Δ::split C.d.ARG4 3′-PiggyBac transposon LTR-C.d.HIS1-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′/leu2Δ::split C.d.ARG4 3′-PiggyBac transposon LTR-C.m.LEU2-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′, TDH3/tdh3::P_TDH3-C.a.PBASE-FRT-FLP-SAT1-FRT |
| SN1933 | this study | leu2Δ::split C.d.ARG4 3′-PiggyBac transposon LTR-C.d.HIS1-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′/leu2Δ::split C.d.ARG4 3′-PiggyBac transposon LTR-C.m.LEU2-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′, WOR1-PBASE-FRT/WOR1 leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2012 | this study | leu2Δ:: 5′ UTR homology-split C.d.ARG4 3′-PiggyBac transposon LTR-C.d.HIS1-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′/leu2Δ efg1Δ::PBASE-FRT/EFG1 leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1988 | this study | leu2Δ:: 5′ UTR homology-split C.d.ARG4 3′-PiggyBac transposon LTR-C.d.HIS1-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′/leu2Δ, EFG1-PBASE-FRT/EFG1 leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1943 | this study | leu2Δ::split C.d.ARG4 3′-PiggyBac transposon LTR-C.d.HIS1-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′/leu2Δ::split C.d.ARG4 3′-PiggyBac transposon LTR-C.m.LEU2-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′ EFG1-PBASE-FRT/EFG1 leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN1941 | this study | leu2Δ::split C.d.ARG4 3′-PiggyBac transposon LTR-C.d.HIS1-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′/leu2Δ::split C.d.ARG4 3′-PiggyBac transposon LTR-C.m.LEU2-multiplex PE primer 2-PiggyBac transposon LTR-split C.d.ARG4 5′ CZF1-PBASE-FRT/CZF1 leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2157 | this study | cht2Δ::ST94-C.m.LEU2/cht2Δ::ST46-C.d.HIS1, leu2Δ/leu2Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2161 | this study | cht2Δ::FRT-FLP-SAT1-FRT/cht2Δ::FRT-FLP-SAT1-FRT, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2162 | this study | cht2Δ::FRT/cht2Δ::FRT, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2170 | this study | CHT2/cht2Δ::FRT, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2172 | this study | CHT2 D151A/cht2Δ::FRT, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2174 | this study | CHT2 E153A/cht2Δ::FRT, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2175 | this study | CHT2 D151A,E153A/cht2Δ::FRT, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2178 | this study | CHT2 Y211A/cht2Δ::FRT, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2180 | this study | CHT2 W278A/cht2Δ::FRT, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2225 | this study | CHT2/cht2Δ::FRT, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ::ST4-C.d. ARG4/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2227 | this study | CHT2 D151A/cht2Δ::FRT, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ::ST3-C.d. ARG4/arg4Δ, iro1Δ/IRO1, MTLa/α |
| SN2230 | this study | CHT2 D151A,E153A/cht2Δ::FRT, leu2Δ::C.m.LEU2/leu2::C.d.HIS1Δ, ura3Δ/URA3, his1Δ/his1Δ, arg4Δ::ST1-C.d. ARG4/arg4Δ, iro1Δ/IRO1, MTLa/α |
|
Oligonucleotides | ||
| Oligonucleotides are described in Table S6 | N/A | N/A |
|
Recombinant DNA | ||
| pSFS2A | ||
| pSN42 | Noble et al. 2005 | Gene replacement with LEU2 marker |
| pSN52 | Noble et al. 2005 | Gene replacement with HIS1 marker |
| pSN69 | Noble et al. 2005 | Gene replacement with ARG4 marker |
| pSN209 | Pande et al. 2013 | Replacement of WOR1 promoter with (SAT1 gene and) TDH3 promoter |
| pSN377 | this study | Replacement of WOR1 promoter with (Flipper cassette and) TDH3 promoter |
| pSN301 | this study | Replacement of WOR1 ORF with FLP-SAT1 construct |
| pSN314 | this study | Replacement of WOR2 promoter with (Flipper cassette and) TDH3 promoter |
| pSN311 | this study | Replacement of WOR3 promoter with (Flipper cassette and) TDH3 promoter |
| pSN360 | this study | Replacement of WOR4 promoter with (Flipper cassette and) TDH3 promoter |
| pSN352 | this study | Replacement of CZF1 promoter with (Flipper cassette and) TDH3 promoter |
| pSN329 | this study | Replacement of CZF1 ORF with FLP-SAT1 construct |
| pSN378 | this study | Replacement of AHR1 promoter with (Flipper cassette and) TDH3 promoter |
| pSN445 | this study | Transposon integration into C.a.LEU2 locus |
| pSN447 | this study | Transposon integration into C.a.LEU2 locus |
| pSN427 | this study | Plasmid that can be used to create additional plasmids with gene homology for TF-PiggyBac transposase fusions |
| pSN422 | this study | Replacement of TDH3 ORF with PiggyBac transposase for overexpression |
| pSN459 | this study | Fusion of PiggyBac transposase to WOR1 ORF |
| pSN467 | this study | Fusion of PiggyBac transposase to EFG1 ORF |
| pSN465 | this study | Fusion of PiggyBac transposase to CZF1 ORF |
| pSN503 | this study | CHT2 allele B addbback to endogenous locus |
| pSN504 | this study | CHT2 allele B D151A addbback to endogenous locus |
| pSN505 | this study | CHT2 allele B E153A addbback to endogenous locus |
| pSN506 | this study | CHT2 allele B D151A,E153A addbback to endogenous locus |
| pSN507 | this study | CHT2 allele B Y211A addbback to endogenous locus |
| pSN508 | this study | CHT2 allele B W278A addbback to endogenous locus |
|
Software and Algorithms | ||
| NumPy 1.14.3 | Oliphant, 2015 | RRID: SCR_008633 |
| Pandas | Reback et al., 2021 | RRID:SCR_018214 |
| SciPy | Virtanen et al., 2020 | RRID:SCR_008058 |
| tabulate | Astanin et al., 2021 | https://pypi.org/project/tabulate/ |
| pybedtools | Dale et al., 2011 | RRID:SCR_021018 |
| BedTools | Quinlan and Hall, 2010 | RRID:SCR_006646 |
| Picard | http://broadinstitute.github.io/picard/ | RRID:SCR_006525 |
| Htslib | Li et al., 2009 | RRID:SCR_002105 |
| samtools | Li et al., 2009 | RRID:SCR_002105 |
| Pysam | https://github.com/pysam-developers/pysam | RRID:SCR_021017 |
| Kallisto | Bray et al., 2016 | RRID:SCR_016582 |
| Limma | Ritchie et al., 2015 | RRID:SCR_010943 |
| Bowtie | Langmead and Salzberg., 2012 | RRID:SCR_005476 |
| MochiView | Homann and Johnson, 2010 | RRID:SCR_000259 |
| Custom code for Calling Card analysis in Candida albicans | this study | https://github.com/jwitch/CallingCard |
|
Other | ||
| Sera-Mag beads in a homemade PEG solution | Thermo-Fisher | (https://openwetware.org/wiki/SPRI_bead_mix) |
Highlights.
Eight regulators of C. albicans mating also control its fitness as a gut commensal
Calling Card-seq revealed DNA-binding activity of these regulators in colonized hosts
Analysis of highly regulated target genes reveals effectors of fungal commensalism
ACKNOWLEDGMENTS
We are grateful to Rob Mitra, Zongtai Qi, and Xuhua Chen for generously sharing plasmids and software for Calling Card-seq. We thank Hiten Madhani, Rob Mitra, and Anita Sil for helpful comments on the manuscript. We thank Theresa Chu for assistance with C. albicans strain construction. This work was supported by the United States National Institutes of Health (NIH) grant R01AI108992, a Burroughs Wellcome Award in the pathogenesis of Infectious Disease, and a Pew Foundation scholarship. J.N.W. was supported by NIH grant T32AI060537 and a Discovery Fellows Grant from University of California, San Francisco (UCSF).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.chom.2021.03.019.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Setoyama H, Nagamori T, et al. (2015). Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 163, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher P, Hohnstein T, Beerbaum E, Röcker M, Blango MG, Kaufmann S, Röhmel J, Eschenhagen P, Grehn C, Seidel K, et al. (2019). Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against Candida albicans. Cell 176, 1340–1355.e15. [DOI] [PubMed] [Google Scholar]
- Bijlani S, Thevandavakkam MA, Tsai HJ, and Berman J (2019). Autonomously replicating linear plasmids that facilitate the analysis of replication origin function in Candida albicans. mSphere 4, e00103–e00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougnoux ME, Pujol C, Diogo D, Bouchier C, Soll DR, and d’Enfert C (2008). Mating is rare within and between clades of the human pathogen Candida albicans. Fungal Genet. Biol 45, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Bruno VM, Kalachikov S, Subaran R, Nobile CJ, Kyratsous C, and Mitchell AP (2006). Control of the C. albicans cell wall damage response by transcriptional regulator Cas5. PLoS Pathog 2, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassone A, and Sobel JD (2016). Experimental models of vaginal candidiasis and their relevance to human candidiasis. Infect. Immun 84, 1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Pande K, French SD, Tuch BB, and Noble SM (2011). An iron homeostasis regulatory circuit with reciprocal roles in Candida albicans commensalism and pathogenesis. Cell Host Microbe 10, 118–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale RK, Pedersen BS, and Quinlan AR (2011). Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics 27, 3423–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drell T, Lillsaar T, Tummeleht L, Simm J, Aaspõllu A, Väin E, Saarma I, Salumets A, Donders GG, and Metsis M (2013). Characterization of the vaginal micro- and mycobiome in asymptomatic reproductive-age Estonian women. PLoS One 8, e54379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Li X, Huang G, Kang Y, and Zhu L (2015). The zinc-finger transcription factor, Ofi1, regulates white-opaque switching and filamentation in the yeast Candida albicans. Acta Biochim. Biophys. Sin. (Shanghai) 47, 335–341. [DOI] [PubMed] [Google Scholar]
- Dünkler A, Walther A, Specht CA, and Wendland J (2005). Candida albicans CHT3 encodes the functional homolog of the Cts1 chitinase of Saccharomyces cerevisiae. Fungal Genet. Biol 42, 935–947. [DOI] [PubMed] [Google Scholar]
- Frazer C, Staples MI, Kim Y, Hirakawa M, Dowell MA, Johnson NV, Hernday AD, Ryan VH, Fawzi NL, Finkelstein IJ, and Bennett RJ (2020). Epigenetic cell fate in Candida albicans is controlled by transcription factor condensates acting at super-enhancer-like elements. Nat. Microbiol 5, 1374–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcus D, Nantel A, Marcil A, Rigby T, and Whiteway M (2004). Transcription profiling of cyclic AMP signaling in Candida albicans. Mol. Biol. Cell 15, 4490–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernday AD, Lohse MB, Fordyce PM, Nobile CJ, DeRisi JL, and Johnson AD (2013). Structure of the transcriptional network controlling white-opaque switching in Candida albicans. Mol. Microbiol 90, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernday AD, Lohse MB, Nobile CJ, Noiman L, Laksana CN, and Johnson AD (2016). Ssn6 defines a new level of regulation of white-opaque switching in Candida albicans and is required for the stochasticity of the switch. mBio 7, e01565–e01515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernday AD, Noble SM, Mitrovich QM, and Johnson AD (2010). Genetics and molecular biology in Candida albicans. Methods Enzymol 470, 737–758. [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Dollive S, Grunberg S, Chen J, Li H, Wu GD, Lewis JD, and Bushman FD (2013). Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS One 8, e66019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann OR, Dea J, Noble SM, and Johnson AD (2009). A phenotypic profile of the Candida albicans regulatory network. PLoS Genet 5, e1000783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann OR, and Johnson AD (2010). MochiView: versatile software for genome browsing and DNA motif analysis. BMC Biol 8, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Wang H, Chou S, Nie X, Chen J, and Liu H (2006). Bistable expression of WOR1, a master regulator of white-opaque switching in Candida albicans. Proc. Natl. Acad. Sci. USA 103, 12813–12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado-Guerrero R, and van Aalten DM (2007). Structure of Saccharomyces cerevisiae chitinase 1 and screening-based discovery of potent inhibitors. Chem. Biol 14, 589–599. [DOI] [PubMed] [Google Scholar]
- Igyártó BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, Edelson BT, Zurawski SM, Malissen B, Zurawski G, Berman J, and Kaplan DH (2011). Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 35, 260–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadosh D, Najvar LK, Bocanegra R, Olivo M, Kirkpatrick WR, Wiederhold NP, and Patterson TF (2016). Effect of antifungal treatment in a diet-based murine model of disseminated candidiasis acquired via the gastrointestinal tract. Antimicrob. Agents Chemother 60, 6703–6708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith JH, Fraser TS, and Fraser MJ Jr. (2008). Analysis of the piggyBac transposase reveals a functional nuclear targeting signal in the 94 C-terminal residues. BMC Mol. Biol 9, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh AY (2013). Murine models of Candida gastrointestinal colonization and dissemination. Eukaryot. Cell 12, 1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvaal C, Lachke SA, Srikantha T, Daniels K, McCoy J, and Soll DR (1999). Misexpression of the opaque-phase-specific gene PEP1 (SAP1) in the white phase of Candida albicans confers increased virulence in a mouse model of cutaneous infection. Infect. Immun 67, 6652–6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassak T, Schneider E, Bussmann M, Kurtz D, Manak JR, Srikantha T, Soll DR, and Ernst JF (2011). Target specificity of the Candida albicans Efg1 regulator. Mol. Microbiol 82, 602–618. [DOI] [PubMed] [Google Scholar]
- Lee KL, Buckley HR, and Campbell CC (1975). An amino acid liquid synthetic medium for the development of mycelial and yeast forms of Candida albicans. Sabouraudia 13, 148–153. [DOI] [PubMed] [Google Scholar]
- Legrand M, Lephart P, Forche A, Mueller FM, Walsh T, Magee PT, and Magee BB (2004). Homozygosity at the MTL locus in clinical strains of Candida albicans: karyotypic rearrangements and tetraploid formation. Mol. Microbiol 52, 1451–1462. [DOI] [PubMed] [Google Scholar]
- Li F, and Palecek SP (2003). EAP1, a Candida albicans gene involved in binding human epithelial cells. Eukaryot. Cell 2, 1266–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SH, Anderson MZ, Hirakawa MP, Wang JM, Frazer C, Alaalm LM, Thomson GJ, Ene IV, and Bennett RJ (2019). Hemizygosity enables a mutational transition governing fungal virulence and commensalism. Cell Host Microbe 25, 418–431.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HJ, Köhler JR, DiDomenico B, Loebenberg D, Cacciapuoti A, and Fink GR (1997). Nonfilamentous C. albicans mutants are avirulent. Cell 90, 939–949. [DOI] [PubMed] [Google Scholar]
- Lohse MB, Hernday AD, Fordyce PM, Noiman L, Sorrells TR, Hanson-Smith V, Nobile CJ, DeRisi JL, and Johnson AD (2013). Identification and characterization of a previously undescribed family of sequence-specific DNA-binding domains. Proc. Natl. Acad. Sci. USA 110, 7660–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MB, and Johnson AD (2016). Identification and characterization of Wor4, a new transcriptional regulator of white-opaque switching. G3 (Bethesda) 6, 721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MB, Zordan RE, Cain CW, and Johnson AD (2010). Distinct class of DNA-binding domains is exemplified by a master regulator of phenotypic switching in Candida albicans. Proc. Natl. Acad. Sci. USA 107, 14105–14110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCallum DM, and Odds FC (2005). Temporal events in the intravenous challenge model for experimental Candida albicans infections in female mice. Mycoses 48, 151–161. [DOI] [PubMed] [Google Scholar]
- Nobile CJ, Fox EP, Nett JE, Sorrells TR, Mitrovich QM, Hernday AD, Tuch BB, Andes DR, and Johnson AD (2012). A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 148, 126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble SM, French S, Kohn LA, Chen V, and Johnson AD (2010). Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat. Genet 42, 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odds FC (1987). Candida infections: an overview. Crit. Rev. Microbiol 15, 1–5. [DOI] [PubMed] [Google Scholar]
- Odds FC (1988). Candida and Candidosis, A Review and Bibliography, 2nd Edition (W.B. Saunders; ). [Google Scholar]
- Oliphant TE (2015). Guide to NumPy (CreateSpace independent publishing platform; ). [Google Scholar]
- Pande K, Chen C, and Noble SM (2013). Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat. Genet 45, 1088–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller MA, and Diekema DJ (2007). Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev 20, 133–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JV, and Kumamoto CA (2012). Variation in Candida albicans EFG1 expression enables host-dependent changes in colonizing fungal populations. mBio 3, e00117–e00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reback CJ, McKinney W, jbrockmendel, Van den Bossche J, Augspurger T, Cloud P, gfyoung, Hawkins S, Sinhrks, Roeschke M, et al. (2021). pandas-dev/pandas: Pandas 1.2.3 https://zenodo.org/record/4572994.
- Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, and Locksley RM (2007). Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature 447, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohland N, and Reich D (2012). Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res 22, 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MA, Gomes AC, Santos MC, Carreto LC, and Moura GR (2011). The genetic code of the fungal CTG clade. C. R. Biol 334, 607–611. [DOI] [PubMed] [Google Scholar]
- Shao TY, Ang WXG, Jiang TT, Huang FS, Andersen H, Kinder JM, Pham G, Burg AR, Ruff B, Gonzalez T, et al. (2019). Commensal Candida albicans positively calibrates systemic Th17 immunological responses. Cell Host Microbe 25, 404–417.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn K, Urban C, Brunner H, and Rupp S (2003). EFG1 is a major regulator of cell wall dynamics in Candida albicans as revealed by DNA microarrays. Mol. Microbiol 47, 89–102. [DOI] [PubMed] [Google Scholar]
- Solis NV, and Filler SG (2012). Mouse model of oropharyngeal candidiasis. Nat. Protoc 7, 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikantha T, Borneman AR, Daniels KJ, Pujol C, Wu W, Seringhaus MR, Gerstein M, Yi S, Snyder M, and Soll DR (2006). TOS9 regulates white-opaque switching in Candida albicans. Eukaryot. Cell 5, 1674–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoldt VR, Sonneborn A, Leuker CE, and Ernst JF (1997). Efg1p, an essential regulator of morphogenesis of the human pathogen Candida albicans, is a member of a conserved class of bHLH proteins regulating morphogenetic processes in fungi. EMBO J 16, 1982–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuch BB, Mitrovich QM, Homann OR, Hernday AD, Monighetti CK, De La Vega FM, and Johnson AD (2010). The transcriptomes of two heritable cell types illuminate the circuit governing their differentiation. PLoS Genet 6, e1001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinces MD, and Kumamoto CA (2007). The morphogenetic regulator Czf1p is a DNA-binding protein that regulates white opaque switching in Candida albicans. Microbiology (Reading) 153, 2877–2884. [DOI] [PubMed] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. (2020). SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Mayhew D, Chen X, Johnston M, and Mitra RD (2011a). Calling Cards enable multiplexed identification of the genomic targets of DNA-binding proteins. Genome Res 21, 748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Mayhew D, Chen X, Johnston M, and Mitra RD (2012). Calling cards” for DNA-binding proteins in mammalian cells. Genetics 190, 941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Song W, Huang G, Zhou Z, Ding Y, and Chen J (2011b). Candida albicans Zcf37, a zinc finger protein, is required for stabilization of the white state. FEBS Lett 585, 797–802. [DOI] [PubMed] [Google Scholar]
- Witchley JN, Penumetcha P, Abon NV, Woolford CA, Mitchell AP, and Noble SM (2019). Candida albicans morphogenesis programs control the balance between gut commensalism and invasive infection. Cell Host Microbe 25, 432–443.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Tao L, Nobile CJ, Tong Y, Guan G, Sun Y, Cao C, Hernday AD, Johnson AD, Zhang L, et al. (2013). White-opaque switching in natural MTLa/αlpha isolates of Candida albicans: evolutionary implications for roles in host adaptation, pathogenesis, and sex. PLoS Biol 11, e1001525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, and Zhang K (2019). Chitin synthesis and degradation in fungi: biology and enzymes. In Targeting chitin-containing organisms, Yang Q and Fukamizo T, eds. (Springer; ). [DOI] [PubMed] [Google Scholar]
- Znaidi S, van Wijlick L, Hernandez-Cervantes A, Sertour N, Desseyn JL, Vincent F, Atanassova R, Gouyer V, Munro CA, Bachellier-Bassi S, et al. (2018). Systematic gene overexpression in Candida albicans identifies a regulator of early adaptation to the mammalian gut. Cell. Microbiol 20, e12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan RE, Galgoczy DJ, and Johnson AD (2006). Epigenetic properties of white-opaque switching in Candida albicans are based on a self-sustaining transcriptional feedback loop. Proc. Natl. Acad. Sci. USA 103, 12807–12812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan RE, Miller MG, Galgoczy DJ, Tuch BB, and Johnson AD (2007). Interlocking transcriptional feedback loops control white-opaque switching in Candida albicans. PLoS Biol 5, e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw files used for mRNA-seq analysis are available through GEO accession number GSE150855. The raw data for Calling Card experiments are available through GEO accession number GSE150856. Custom code used for analysis of CCS data is available at https://github.com/jwitch/CallingCard.