


Abstract
Adoption of CRISPR–Cas systems, such as CRISPR–Cas9 and CRISPR–Cas12a, has revolutionized genome engineering in recent years; however, application of genome editing with CRISPR type I—the most abundant CRISPR system in bacteria—remains less developed. Type I systems, such as type I-E, and I-F, comprise the CRISPR-associated complex for antiviral defense (‘Cascade’: Cas5, Cas6, Cas7, Cas8 and the small subunit) and Cas3, which degrades the target DNA; in contrast, for the sub-type CRISPR–Cas type I-D, which lacks a typical Cas3 nuclease in its CRISPR locus, the mechanism of target DNA degradation remains unknown. Here, we found that Cas10d is a functional nuclease in the type I-D system, performing the role played by Cas3 in other CRISPR–Cas type I systems. The type I-D system can be used for targeted mutagenesis of genomic DNA in human cells, directing both bi-directional long-range deletions and short insertions/deletions. Our findings suggest the CRISPR–Cas type I-D system as a unique effector pathway in CRISPR that can be repurposed for genome engineering in eukaryotic cells.
INTRODUCTION
CRISPR–Cas (clustered regularly interspaced short palindromic repeats-CRISPR-associated) systems are RNA-based adaptive immune systems that protect bacteria and archaea from viruses, plasmids, and other foreign genetic elements (1,2). The CRISPR–Cas systems, especially Class 2 type II systems [CRISPR–Cas9 (1,2)] and type V [CRISPR–Cas12a (3)] have been used widely as a tool in genome editing to introduce mutations efficiently into a target DNA of interest in eukaryotic chromosomal DNA. On the other hand, the application of Class 1 CRISPR–Cas systems, which consist of multiple-subunit effector proteins, remains limited, despite Class 1 CRISPR systems being the most abundant CRISPR systems in bacteria and archaea (4).
Class 1 CRISPR systems comprise 3 types (I, III and IV) and 18 subtypes (4). Among them, CRISPR–Cas type I systems are most prevalent; more than 80% of CRISPR–Cas systems belong to type I systems (4). Type I systems comprise a short gRNA molecule, a target recognition module, and a DNA cleavage module (4). The short gRNA molecule is composed of a CRISPR RNA (crRNA) spacer, and crRNA repeats adjacent to both 5′- and 3′-sides of the spacer (1–4). In nature, gRNAs are transcribed from the CRISPR array in precursor form, and are subsequently processed into their mature forms, typically about 30–50 nucleotides in length (4–7). After processing of the crRNA repeat by Cas6, a mature crRNA is incorporated into the target recognition module, which consists of Cas5, Cas6, Cas7 and Cas8, the so-called ‘Cascade’ (CRISPR-associated complex for antiviral defense) (8,9). Cascade complexes recognize short (typically 3–5 base) sequence elements called protospacer adjacent motifs (PAMs) in target DNAs (10–12). Components of Cascade differ in type I sub-families, varying from three Cas effectors (Cas5, Cas7 and Cas8) in type I-C (13) to five Cas effectors (Cas5, Cas6, Cas7, Cas8 and Cas11—formerly referred to as Cse2) in type I-E (14). In type I systems, the Cascade disrupts local DNA pairing to form R-loop structures, in which base-pairing between crRNA and the complementary target strand displaces the non-target DNA strand (14–18). This binding and unwinding of the double-stranded DNA by the Cas effectors and crRNA is required for DNA cleavage and destruction by type-specific Cas effector nucleases Cas3 (18–21). Cas3 and Cas3′ are known as DNA cleavage modules in the type I system. Both proteins possess a central helicase motif, and, most importantly, Cas3 also has a nuclease domain, the so-called HD domain. Recent studies have shown that Cas3 is recruited to the R-loop formed by Cascade, and that it nicks non-target strand DNAs, processively degrading DNA in the region upstream of the PAM-proximal side (18,22–24).
Recently, application of Class 1 type I-A, -B, -E and -F CRISPR–Cas system to genome editing and gene regulation has been reported in both bacteria and eukaryotes (23–31). In prokaryotes, repurposing of endogenous CRISPR–Cas systems to genome editing or gene regulation has been reported (25,30). In eukaryotic cells, type I-E and type I-F systems have been utilized to regulate gene expression by fusing transcriptional activators/repressors to Cas proteins in plant (29) and human (31) cells, respectively. The type I-E system has also been applied to genome editing. Cameron et al. (28) fused Cas protein with FokI nuclease and induced mutation at the target site. Dolan et al. (23) and Morisaka et al. (24) successfully induced mutations in human cells by applying Cas3 protein as a nuclease and found that type I-E CRISPR–Cas3 induces unidirectional long-range deletion towards the 5′ upstream region of the PAM sequence in human cells, which differed from the small indels induced by Cas9 and Cas12a. Type I systems are also expected to have an advantage in genome-editing specificity compared with Cas9 and Cas12a because they recognize longer target sequences than Cas9 and Cas12a (23,24,28).
Here, we analyzed a CRISPR genomic locus encoding a previously assigned, but uncharacterized, Class 1 CRISPR–Cas type I-D from Microcystis aeruginosa. Although biochemical studies on CRISPR–Cas type I-D Cas6 (32) and Cas7 (33) and the Cascade complex (34,35) have been reported, the genome editing activity of the type I-D system has not yet been clarified in mammal cells. The type I-D system contains a Cas3 effector protein, but lacks a nuclease domain; instead, Cas10d—a unique effector protein of the type I-D system not found in other CRISPR systems—possesses a typical nuclease domain. In addition, one of major Cascade factors for PAM recognition, Cas8 homologous protein (36) is missing in the type I-D system. Therefore, the mode of interference through PAM recognition to target DNA degradation in the type I-D system must differ from that of other type I systems. Recently, we have successfully applied the type I-D system to plant genome editing (37), but this system has not yet been applied to mammalian genome editing.
We explore the function of novel Cas effector proteins from the type I-D system identifying Cas10d protein as the functional nuclease in this system. Of particular importance, we found that Cas10d acts as the functional nuclease effector protein and in PAM recognition through interaction with other effectors in the Cascade complex, including Cas3d. Furthermore, using the type I-D system with an engineered gRNA generated mutations with small insertions/deletions (indels) but also long-range and bi-directional DNA deletions in human cells; the type I-D system-induced mutation patterns differ from those of other CRISPR systems. We also analyzed a unique feature of off-target mutation against a mismatch position in the type I-D gRNA using luc reporter assay, as well as long-range PCR analysis to evaluate off-target effects on the human genome. Our findings suggest novel genome editing tools for eukaryotic cells using the CRISPR effector module pathway of the CRISPR–Cas type I-D system.
MATERIALS AND METHODS
Vector construction
Details of all plasmid DNAs used in this study are described in Supplementary Figure S13. PCR amplification for cloning gene fragments was done using PrimeSTAR Max (TaKaRa). Cloning for assembly was done using Quick ligation kit (NEB), NEBuilder HiFi DNA Assembly (NEB), and Multisite gateway Pro (Thermo Fisher Scientific).
Gene fragments corresponding to human codon-optimized cas effector genes; cas3d, cas5d, cas6d, cas7d, and cas10d, were synthesized with the SV40 nuclear localization signal (NLS) at their N-termini (gBlocks®) (Integrated DNA Technologies), assembled, and cloned into the pEFs vector (38) separately to yield pEFs-HA-SV40NLS-Cas3d, pEFs-Strept-SV40NLS-Cas5d, pEFs-myc-SV40NLS-Cas6d, pEFs-FLAG-SV40NLS-Cas7d, and pEFs-6xHis-SV40NLS-Cas10d. The tags fused to each Cas protein were as follows; HA-tag to Cas3d, Strep-tag to Cas5d, Myc-tag to Cas6d, FLAG-tag to Cas7d, and 6x His-tag to Cas10d. All cas genes were combined into an all-in-one vector, pEFs-All, by fusing with sequences encoding 2A self-cleavage peptides. pEFs_Cas3d-Cas10d and pEFs_Cas6d-Cas5d-Cas7d expression vectors were also constructed by linking to the 2A self-cleavage peptide. To evaluate different promoters to express Cas5d and Cas6d, the human EFs promoter was replaced with the CAG promoter in pEFs-Strept-SV40NLS-Cas5d, and pEFs-Myc-SV40NLS-Cas6d to yield pCAG-Strept-SV40NLS-Cas5d or pCAG-Myc-SV40NLS-Cas6d. To construct the Cas-expressing vector with bipartite NLS (bpNLS), the DNA fragment (myc-bpNLS-Cas10d-bpNLS-6xHis) was first synthesized (IDT) and cloned into the pEFs vector, resulting in pEFs-Myc-bpNLS-Cas10d-bpNLS-6xHis vector. The cas10d gene fragment in pEFs-Myc-bpNLS-Cas10d-bpNLS-6xHis vector was replaced by cas3d, cas5d, cas6d and cas7d gene fragments, respectively, to yield pEFs-Myc-bpNLS-Cas3d-bpNLS-6xHis, pEFs-Myc-bpNLS-Cas5d-bpNLS-6xHis, pEFs-Myc-bpNLS-Cas7d-bpNLS-6xHis and pEFs-Myc-bpNLS-Cas6d-bpNLS-6xHis. The resulting vectors were used for the construction of pEFs-Myc-bpNLS-Cas3d-FLAG-NLS, pEFs-Myc-bpNLS-Cas5d-FLAG-NLS, pEFs-Myc-bpNLS-Cas6d-FLAG-NLS, pEFs-Myc-bpNLS-Cas7d-FLAG-NLS, pEFs-Myc-bpNLS-Cas10d-FLAG-NLS by replacing bpNLS-6xHis with FLAG-NLS.
To construct the mutated Cas10d (H177A) expression vectors, the dCas10d (H177A) fragment was synthesized (IDT) and the wild-type Cas10d fragment in pEFs-Myc-bpNLS-Cas10d-bpNLS-6xHis was replaced by dCas10d (H177A) to yield pEFs-myc-bpNLS-dCas10d (H177A)-bpNLS-6xHis. To construct mutated Cas7 expression vectors with myc-tag and 3xSV40NLS [Cas7d (K58A), Cas7d (K62A), Cas7d (K67A), Cas7d (R68A), Cas7d (K69A), Cas7d (T75A), Ca7d (E148Q), Cas7d(F66R/K67A/R68M/K69R), Cas7d (K67S/R68Q), Cas7d (K67A/R68A), Cas7d (K67S/R68Q/K69Q) and Cas7d (F66K/K67S/R68Q)], the Cas7d fragment was first cloned into the pEFs2 vector, which has Myc-tag and 3x SV40NLS at its N-terminus to yield pEFs2-Myc-3xSV40NLS-Cas7d. Then, each mutation was introduced by PCR amplification to yield pEFs2-Myc-3xSV40NLS-Cas7d (K58A), pEFs2-Myc-3xSV40NLS-Cas7d (K62A), pEFs2-Myc-3xSV40NLS-Cas7d (K67A), pEFs2-Myc-3xSV40NLS-Cas7d (R68A), pEFs2-Myc-3xSV40NLS-Cas7d (K69A), pEFs2-Myc-3xSV40NLS-Cas7d (T75A), pEFs2-Myc-3xSV40NLS-Ca7d (E148Q), pEFs2-Myc-3xSV40NLS-Cas7d(F66R/K67A/R68M/K69R), pEFs2-Myc-3xSV40NLS-Cas7d (K67S/R68Q), pEFs2-Myc-3xSV40NLS-Cas7d (K67A/R68A), pEFs2-Myc-3xSV40NLS-Cas7d (K67S/R68Q/K69Q) and pEFs2-Myc-3xSV40NLS-Cas7d (F66K/K67S/R68Q). The Cas7d fragment in pEFs2-Myc-3xSV40NLS-Cas7d was replaced with Cas3d, Cas5d, Cas6d, or Cas10d fragment to yield pEFs2-Myc-3xSV40NLS-Cas3d, pEFs2-Myc-3xSV40NLS-Cas5d, pEFs2-Myc-3xSV40NLS-Cas6d, pEFs2-Myc-3xSV40NLS-Cas10d. To construct mutated Cas10d or Cas3d expression vectors that have mutations in ATP-binding domains, Cas3d (D179A), Cas10d (D293A), or Cas10d (K407A) fragments were synthesized (IDT) and the wild-type Cas10d fragment in pEFs2–3xNLS-SV40NLS-Cas10d was replaced by each respective fragment to yield pEFs2-myc-3x SV40NLS-Cas3d (D179A), pEFs2-myc-3x SV40NLS-Cas10d (D293A) and pEFs2-myc-3x SV40NLS-Cas10d (K407A).
For crRNA expression, a DNA fragment containing a repeat-spacer-repeat sequence was cloned under the human U6 promoter in pEX-A2J1 (Eurofins Genomics) to yield pAEX-hU6crRNA. To construct pAEX-hU6crRNA_mature, two repeat sequences were replaced by the predicted processed repeat sequences. The mature crRNA was predicted based on a report by Shao and Li (6) and the stability of the secondary structure analyzed using the RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). For insertion of gRNA sequence, two oligonucleotides containing a target sequence were annealed and cloned into the gRNA expression vector using Golden Gate cloning using the restriction enzyme BsaI (NEB).
Luc reporter assay plasmids: for the luc reporter assay, NanoLUxxUC expression vectors were constructed. First, NLUxxUC_Block1 and NLUxxUC_Block2 DNA fragments were synthesized (IDT). NLUxxUC_Block1 includes the first 351 bp of the NanoLUC gene sequence and Multi Cloning Site. An XbaI site was attached to the 5′ end of the NLUxxUC_Block1. NLUxxUC_Block2 includes 465 bp of the 3′ end of the NanoLUC gene. An XhoI site was attached to the 3′ end of the NUxxUC_Block2. These fragments were assembled and replaced with EGxxFP fragment in the pCAG-EGxxFP vector obtained from addgene (#50716) to yield the pCAG-nLUxxUC vector. Each split type of the NLUxxUC reporter (pCAG-nLUxxUC_Block1, pCAG-nLUxxUC_Block2) was constructed by removing the NLUxxUC_Block1 fragment by XhoI and BamHI digestion, or by removing NLUxxUC_Block2 fragment by XbaI and EcoRI digestion from the pCAG-NLUxxUC vector, respectively. Each digested vector was assembled with a Multi Cloning Site to yield pCAG-NLUxxUC_Block1_MCS and pCAG-NLUxxUC_MCS_Block2 vector (Supplementary Figure S13).
Cell culture and transfection
Human embryonic kidney cell line 293T (HEK293T, RIKEN BRC) was cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific), GlutalMAX™ Supplement (Thermo Fisher Scientific), 100 units/ml penicillin and 100 μg/ml streptomycin in a 60 mm dish at 37°C with 5% CO2 incubation. HEK293T cells were seeded into six-well plates (Corning) the day before transfection. Cells were transfected using TurboFect Transfection Reagent (Thermo Fisher Scientific) following the manufacturer's recommended protocol. The repeat-spacer-repeat crRNA was used for the transfection unless otherwise specified. For each well of a six-well plate, a total of 4 μg plasmid DNA, extracted using NucleoSpin® Plasmid Transfection-grade kit (Macherey-Nagel), was used. Transfected cells were harvested after 48 h for mutation analysis.
Purification of recombinant Cas proteins
Recombinant Cas10d and Cas3d proteins were expressed in human HEK293T cells and purified as mentioned below. At first, human HEK293T cells were seeded in 150-mm culture dishes a day before transfection. Then, human HEK293T cells were transfected with pEFs-Myc-bpNLS-Cas10d-bpNLS-6xHis or pEFs-Myc-bpNLS-Cas3d-bpNLS-6xHis or pEFs-Myc-bpNLS-Cas10d (H177A)-bpNLS-6xHis vector using Lipofectamine 2000 (Thermo Fisher Scientific). Two days after transfection, the cells were collected and suspended in buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM DTT, 10% glycerol). Cells were disrupted by sonication and cell debris was removed by the centrifugation at 12,000 rpm for 15 min at 4°C. The supernatant was collected and mixed with Ni-NTA Agarose (QIAGEN) pre-equilibrated with a buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM DTT, 10% glycerol). The mixture was then incubated with rotating in 4°C for 1 h. The Ni-NTA resin was washed with 5 column volumes of buffer 1 (120 mM HEPES pH 7.5, 150 mM KCl, 1 mM DTT, 10% glycerol, 10 mM imidazole), followed by washing with 5 column volumes of buffer 2 (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM DTT, 10% glycerol, 20 mM imidazole). The 6× His-tagged proteins were eluted with 5 column volumes of elution buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM DTT, 10% glycerol, 300 mM imidazole). The eluate was then concentrated by ultrafiltration using Apollo device (M & S Techno Systems). The proteins were further purified by gel-filtration chromatography on pre-equilibrated Superdex 200 Increase 10/30GL (Cytiva) using a Biologic DuoFlow 10 system (Bio-Rad). Fractions were analyzed by SDS-PAGE. The fractions with expected proteins were collected and used for in vitro assay.
Protein identification by mass spectrometry
Excised gel bands were digested with Trypsin (APRO Science) overnight at 37°C. The digested samples were desalted using SPE C-Tip (Nikkyo Technos). The desalted samples were concentrated and resuspended in 0.1% (v/v) formic acid. The recovered peptides were analyzed using Q Exactive Plus (Thermo Fisher Scientific) coupled with a capillary HPLC system (EASY-nLC 1200, Thermo Fisher Scientific) to acquire tandem mass spectrometry (MS/MS) spectra. Data derived from MS/MS spectra were used to search the protein database SWISS-Prot using the MASCOT server (http://www.matrixscience.com) and identify proteins using the program Scaffold viewer (http://www.proteomesoftware.com/products/scaffold).
In vitro ssDNA nuclease assay
M13mp18 single-strand DNA (ssDNA) (NEB) was used as a substrate for the ssDNA nuclease assay. For the time course study of ssDNA nuclease activity, 700 nM of Cas10d or Cas3d was added to Cut smart buffer (NEB) supplemented with 2 mM ATP, 100 μM CoCl2, 100 μM NiCl2 and 3 nM M13mp18 single-strand DNA) and incubated for 0, 0.5, 1 or 2 h at 37°C. The reaction products were purified by phenol/chloroform extraction and the supernatant analyzed by agarose gel electrophoresis. The DNAs were visualized by GelRed® Nucleic Acid Gel Strain (Biotium). To study the concentration dependence of ssDNA nuclease activity, various amounts of Cas10d (0, 50, 125, 250, 500, 700 μM) were added to the buffer mentioned above. To study the effects of divalent cations, 25 mM EDTA or the following concentrations of each divalent cation were added to a buffer [10 mM Tris–HCl (pH 7.5), 60 mM KCl, 10% glycerol, 3 nM M13mp18 single-strand DNA, 700 nM Cas10d] and incubated for 2 h at 37°C; 10, 30, 60 mM of MgCl2 or 50, 100, 150 μM of NiCl2 or 50, 100, 150 μM CoCl2. The range of dication concentration was chosen based on data from Type I-E Cas3 (39). The reaction products were purified by phenol/chloroform extraction and visualized as mentioned above. Experiments were repeated three times independently.
In vitro dsDNA nuclease assay
Plasmid DNA, pNEB193 (NEB) was linearized by BamHI and used as a substrate for in vitro double-strand DNA (dsDNA) nuclease assay. Linearized DNA (70 ng) was mixed with 700 nM of each Cas protein in Cut smart buffer solution (NEB) supplemented with 2 mM ATP, 100 μM CoCl2 and 100 μM NiCl2, and incubated at 37°C for 0, 0.5, 1 or 2 h. The reaction mixture was subjected to phenol/chloroform extraction. Aqueous fractions were mixed with 6× loading dye and separated by electrophoresis through 1% agarose gel. The DNAs were visualized using GelRed ® Nucleic Acid Gel Strain (Biotium). The experiments were repeated three times independently.
In vitro helicase assay
Oligo DNA (100 nt) labeled with fluorescent dye ATTO532 at the 5′ terminal was used as a ssDNA in this experiment. Partial duplex DNA was prepared by annealing the ATTO532-labeled oligonucleotide DNA (100 nt) to oligo DNA (40 nt) with the complementary sequence. Oligonucleotide DNA sequences are listed in Supplementary Table S1. Annealing was performed by mixing ATTO532-conjugated oligo DNA and complementary oligo DNA at molecular ratio of 1:1.5, with the reaction taking place under the following conditions: 98°C 5 min, cooling to 85°C at a speed of 2°C/s, 85°C 1 min, cooling to 25°C at a speed of 0.1°C/s, 25°C 1 min. The annealed partial duplex DNA (2 μM) was used as a dsDNA for the helicase assay. Cas10d or Cas3d, or both proteins, were added to the reaction buffer (1 mM Tris–HCl, 25 mM KCl, 15% glycerol, 200 μM ATP, 100 μM MgCl2) (20). ssDNA and partial duplex DNA without Cas proteins were used as negative controls. The helicase reaction was performed at 37°C for 2 h in a buffer mentioned above with or without ATP. The reaction was stopped by adding 6× gel loading dye (NEB). The reaction products were analyzed by 8% non-denaturing polyacrylamide electrophoresis. The run was carried out at a constant current of 15 mA for 1 hour. Fluorescence was visualized by Typhoon FLA9500 (Cytiva). The experiments were repeated three times independently.
ATPase assay
The ATPase reactions were performed at 37°C in CutSmart reaction buffer (NEB) supplemented with 2 mM ATP, 100 μM CoCl2, 100 μM NiCl2 and 700 nM Cas10d or Cas3d. To investigate the ssDNA-dependent ATPase activity, various amounts of ssDNA (M13mp18) or dsDNA (pNEB193) (0, 0.5, 1, 3, 5, 10 nM) were added to the reaction mixture. Reactions were performed at 37°C for 2 h and stopped by adding 27 mM EDTA. In the time-course study of ATPase activity, each reaction was stopped by adding 27 mM of EDTA solution at fixed time intervals (0, 5, 10, 20, 30, 60 min). The detection of liberated phosphate was performed using BIOMOL® Green Reagent kit (Cosmo Bio Co. Ltd) according to the manufacturer's protocol. The amount of free-liberated phosphate was calculated based on the calibration curve that was established using the phosphate solutions provided by the manufacturer. The amount of liberated phosphate at 0 nM of DNA or at 0 min of reaction time was subtracted as a background from the measured amount of free-liberated phosphate in each experiment. The reaction rate (min–1) was calculated from the slope of the time course. ATPase experiments were performed three times independently.
Luc reporter assay
Human HEK293T cells were used for the luc reporter assay. Cells (2.0 × 104 cells/well) were seeded onto 96-well plates (Corning) the day before transfection, and transfected as mentioned above. A total of 200 ng plasmid DNAs including (i) pGL4.53 vector encoding Fluc gene (Promega) used as an internal control, (i) two kinds of pCAG-nLUxxUC vectors (pCAG-nLUxxUC_Block1 and pCAG-nLUxxUC_Block2) interrupted with target DNA fragment (Supplementary Table S3), (iii) plasmid DNAs encoding type I-D CRISPR–Cas components (Supplementary Figure S13, pEFs vectors) and (iv) pAEX-hU6gRNA for the gRNA expression vector were used in each well of a 96-well plate. For the luc reporter assay of EGFP gRNAs, a non-split type LUC vector was used; the pCAG-nLUxxUC vector interrupted with target DNA fragment was used instead of pCAG-nLUxxUC_Block1 and pCAG-nLUxxUC_Block2 vectors. NanoLuc and Fluc luciferase activities were measured 2 days after transfection using the Nano-Glo® Dual-Luciferase® Reporter Assay System (Promega). The NanoLuc/Fluc ratio was calculated for each sample. The NanoLuc/Fluc ratio of the sample transfected with non-targeting gRNAs was used as a control, and the relative activity to the control was calculated for each sample to evaluate the gRNA activity. The experiments were repeated more than three times independently.
Western blotting
At 2 days post-transfection, total proteins were extracted from HEK293T cells using RIPA Lysis and Extraction Buffer (Thermo Scientific) supplemented with Protease Inhibitor Cocktail for Use with Mammalian Cell and Tissue Extracts (Nakalai Tesque) according to the manufacturer's protocol. For the isolation of nuclear and cytoplasmic proteins, NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific) was used. The extracted proteins were quantified using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). After the quantification of protein concentrations, the samples were mixed with 4× Laemmli Sample buffer (Bio-Rad) and 2.5% β-mercaptoethanol, followed by heat treatment at 95°C for 5 min. The denatured proteins were loaded onto 12% SDS-PAGE gel with Tris–glycine–SDS buffer [0.25 M Tris, 1.92 M glycine, 1% (w/v) SDS] and separated by SDS-PAGE for 60 min at 150 V. The proteins were transferred to Immobilon-P Polyvinylidene fluoride (PVDF) membranes (Millipore) in Tris-Glycine-SDS buffer containing 20% methanol for 2 h at 50 V. The blot was washed by TTBS (25 mM Tris, 137 nM NaCl, 2.68 nM KCl) for 5 min three times and blocked at room temperature in Blocking One (Nacalai Tesque) for 60 min. The primary antibody reactions were performed at room temperature for 60 min. After washing the membranes in TTBS for 5 min three times, the secondary antibody, Anti-Mouse IgG (H+L), HRP Conjugate (Promega, 1:10 000 in Blocking one), was added to the membrane. After incubation for 60 min at room temperature, the membrane was washed with TTBS 5 min three times. Signals were detected using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific) according to the protocol provided by the manufacturer. Images were captured using an ImageQuant LAS4000 mini (GE Healthcare Bioscience). Primary antibodies used in this study are as follows: anti-DDDDK-tag mAb (1:10 000) (MBL, Japan), anti-HA-tag mAb (1:10 000) (MBL), anti-His-tag mAb (1:10 000) (MBL), anti-Myc-tag mAb (1:10 000) (MBL), anti-Strep-tag mAb (1:1000) (MBL), anti-β-actin mAb (1:10 000) (MBL). The experiments were repeated three times independently.
Immunoprecipitation
HEK293T cells were transfected using TurboFect Transfection Reagent (Thermo Fisher Scientific) with each Cas expression vector in the following combinations: (i) pEFs-FLAG-SV40NLS-Cas7d with other pEFs2-Myc-3xSV40NLS-Cas-vectors (Cas3d, 5d, 6d, 10d) and crRNA; (ii) pEFs-Myc-bpNLS-Cas3d-FLAG-SV40NLS with other pEFs-Myc-3xSV40NLS-Cas vectors (Cas5d, 6d, 7d, 10d) and crRNA or (iii) pEFs2–3xNLS-SV40NLS-Cas3d and pEFs-Myc-bpNLS-Cas5d-FLAG-SV40NLS or pEFs-Myc-bpNLS-Cas6d-FLAG-SV40NLS or pEFs-Myc-bpNLS-Cas10d-FLAG-SV40NLS or pEFs-FLAG-Cas7d. Protein extraction was performed according to the protocol described in Katoh et al. (40) at 48 h post-transfection. Briefly, the medium was replaced with Lysis buffer [20 mM HEPES, 150 mM NaCl, 0.1% (w/v) Triton X-100, 10% (w/v) glycerol] containing Protease Inhibitor Cocktail for Use with Mammalian Cell and Tissue Extracts (Nacalai Tesque) and incubated for 5 min on ice. The cell lysates were mixed by pipetting and transferred to 1.5 ml tubes, then incubated for 15 min on ice. Purification of FLAG-tagged protein was performed using DDDDK-tagged Protein Magnetic Purification Kit (MBL) according to the protocol provided by the manufacturer. The resulting elutes were analyzed by SDS-PAGE and western blotting using following antibodies: anti-His-tag mAb (1:10 000) (MBL), anti-DDDDK-tag mAb (1:10 000) (MBL), anti-β-actin mAb (1:10 000) (MBL), anti-Mouse IgG (H+L) and HRP Conjugate (1:10 000) (Promega). The experiments were repeated three times independently with similar results.
DNA deletion analysis by long-range PCR
To detect DNA deletion in HEK293T cells, long-range PCR and cloning of a pool of long PCR products was performed. Genomic DNA was first extracted from HEK293T cells using the Geno Plus™ Genomic DNA Extraction Miniprep System (Viogene-BioTek). Nested PCR was then performed to amplify long-range DNA region specifically. At first, the extracted genome DNA was used as a template for amplification of the target DNA region using several specific kinds of primer sets for long-range PCR (Supplementary Table S5), which were designed to amplify target DNA region of various lengths (3.5–25 kb). The first PCR reactions were performed using KOD ONE Master Mix (TOYOBO) under the following conditions: 35 cycles of 10 s at 98°C, 5 s at 60°C, 50 s (amplicon: <10 kb) or 150 s (amplicon: 10–15 kb) or 200 s (amplicon: 15–20 kb) at 68°C. These PCR products were then diluted 100–10 000 times and used as a template for nested PCR. The nested PCR was performed under same conditions mentioned above. The PCR products were separated by electrophoresis on 1% agarose gel and visualized by staining with GelRed™ Nucleic Acid Gel Stain (Biotium). Nested PCR products were pooled and purified using Monofas® DNA purification Kit I (GL Sciences). The purified mixture of PCR products was cloned into pMD20-T vector using a Mighty TA-cloning Kit (TaKaRa). The 119 clones for AAVS- GTC_70–107 (+) and 20 clones for hEMX1- GTT_998–1036 (–) were picked up and Sanger sequenced using M13 Uni and M13 RV primers. Sanger sequencing results were analyzed using BLASTN search and ClustalW program to identify DNA deletions.
Mutation analyses in short-range PCR products
To evaluate mutations introduced in transfected human cells, a region of about 400 bp surrounding the target locus of gRNA was amplified by short-range PCR using a PCR kit as described above. In heteroduplex mobility assay (HMA), the PCR fragments were analyzed directly using a microchip electrophoresis system with MCE202 MultiNA (Shimazu). PCR amplicons were also cloned into the TA cloning vector (TaKaRa) to determine their sequences by the Sanger method. All primers used for short-range PCRs used in the mutation analyses are listed in Supplementary Table S4.
Multiple alignment
A multiple alignment of the HD domain of Cas3 and Cas10d protein sequences was constructed by using ClustalX (http://www.clustal.org/clustal2/), with pairwise parameters as follows: gap opening = 10, gap extend = 0.1 and protein weight matrix = identity matrix; multiple parameters as gap opening = 10, gap extension = 0.2, delay divergent sequence (%) = 15, protein weight matrix = identity matrix. Gene accession numbers of protein sequences used for the alignment were MaCas10d: M. aeruginosa (WP_002791883.1), AcCas3a: Anabaena cylindrica (WP_081593764.1), BtCas3a: Bacillus thuringiensis (WP_000506550.1), HhCas3a Halobacteroides halobius (WP_015326643.1), BcCas3b: Bacillus cytotoxicus (NC_009674), LrCas3b: Lactobacillus ruminis (NC_015975), SaCas3b: Sulfobacillus acidophilus (NC_016884), ErCas3c: Eubacterium rectale (NC_021044), LfCas3c: Lactobacillus fermentum (NC_017465), XoCas3c: Xanthomonas oryzae (NC_007705), EcCas3e: Escherichia coli (EIG78940.1), PaCas3e: Pseudomonas aeruginosa (NC_021577), TfCas3e: Thermobifida fusca (AAZ55629.1), EcCas3f: Escherichia coli (NC_008563), LpCas3f: Legionella pneumophila (NC_006366), YpCas3f: Yersinia pestis (NC_017168).
Molecular dynamics simulations of the in silico model of cascade complexes of type I-D CRISPR–Cas, type I-E CRISPR–Cas and type I-F CRISPR–Cas
The Cas7d structure in the type I-D CRISPR–Cas Cascade complex was predicted using a homology modeling method, MODELLER (41), and an in silico structure prediction method, ROSETTA (42). To check the stability of the in silico model structure of Cas7d, 100 ns molecular dynamics (MD) simulations were performed. The type I-D Cascade complex structure was constructed using five in silico models for Cas7d, and a crRNA. The structure of the Type I-E CRISPR Cascade complex (14) was used as the reference conformation for the type I-D Cascade.
The MD simulations of the in silico model structure of the type I-D Cascade complex, the Type I-E CRISPR Cascade complex (PDB ID:5H9F), and the Type I-F CRISPR Cascade complex (PDB ID: 5UZ9) (43) were performed using GROMACS software (44) with Amber14sb-permbsc1 force field (45). In the case of the Type I-F structure, we removed the anti-CRISPR molecules, before simulation. Concentrations of K+ and Cl– were set to 150 mM, and the temperature and pressure in the system were set to 300 K and 1 bar, respectively. The MD simulation times were 1.5 μs for Type I-E, and 1.0 μs for Type I-F CRISPR systems. The MD simulation of the in silico model system of type I-D was performed for 100 ns. VMD software (46) was used to display the structures of proteins, DNA, and crRNA.
RESULTS AND DISCUSSION
The type I-D CRISPR–Cas system is composed of Cas effector proteins with a nuclease module
Components of Cas effector proteins and the crRNA sequence from Microcystis aeruginosa were evaluated using BLAST. The 7.6 kb type I-D CRISPR–Cas locus of M. aeruginosa strain PCC9808 consists of eight cas genes (cas1d–cas7d, cas10d) followed by an array of 36 repeat-spacer units (Figure 1A and B). The HD domain (47) that functions in DNA cleavage in CRISPR type I-A, B, C, E and F (13,21,26,48–50) is lacking in Cas3d. Instead, Cas10d, which belongs to the Cas10 superfamily protein, harbors an HD-like nuclease domain in its N-terminal region (Figure 1C and Supplementary Figure S1). Cas10—a signature protein of CRISPR type III systems (51,52)—functions in DNA and RNA degradation (53–55). Recently, an evolutional model of CRISPR systems proposed that the origin of cas10 is as an ancestral factor in the prokaryotic immune system (52). In this model, the ancestral cas10 gene locus has evolved into an ancestral Class1 system, with this ancestral Class1 system further divided into type I and type III systems. During this period, incorporation of cas3 and loss of cas10 occurred in the typical type I system (52). Therefore, CRISPR type I-D is a unique system that possesses both type I and III signature genes, although this sub-type was assigned as type I.
Figure 1.

DNA cleavage activity of CRISPR type I-D. (A) CRISPR type I-D structure. Top: CRISPR type I-D locus in Microcystis aerginosa. Bottom (left side): schematic organization of type I-D CRISPR–Cas subunits and mature crRNA (black line), Bottom (right side): schematic organization of type I-E CRISPR–Cas subunits and mature crRNA (black line). PAM for type I-D or type I-E is indicated as a red box. (B) Structure of pre-mature and mature crRNAs including repeat and spacer. Top: pre-mature crRNA sequence. Bottom: predicted mature crRNA sequence. The potential stem loop sequence recognized by Cas6d was underlined. (C) Amino acid sequence alignment of the HD domain in Cas3 and Cas10d. Multiple alignments were generated by ClustalX using the Cas3 HD domains from various bacteria and a Cas10d HD domain from M. aeruginosa. Amino acids were colored as default coloring setting of the CLUSTAL program as blue: hydrophobic amino acids (A, I, L, M, F, W, V, C); red: positively charged amino acids (K, R); magenta: negatively charged amino acids (E, D); green: polar amino acids (N, Q, S, T); orange: glycine; yellow: proline; cyan: aromatic amino acids (H, Y). Asterisks indicate amino acids found in all protein sequences. The multiple alignment of full Cas10d protein sequences is shown in Supplementary Figure S1. (D–F) Detection of ssDNA nuclease activity by in vitro assay. M13mp18 single-stranded DNA was used as a substrate. (D) Time course study of ssDNA nuclease activity of Cas10d. Ctrl: control reaction without Cas proteins, Cas10d: reaction with Cas10d protein, Cas3d: reaction with Cas3d protein, red arrows; undigested DNA. (E) Dependence of ssDNA nuclease activity on Cas10d concentrations. (F) Effect of dications on Cas10d ssDNA nuclease activity. ssDNA nuclease assay was performed with 25 mM EDTA or various concentrations of dications (Mg2+, Ni 2+, Co 2+). (G) Detection of dsDNA nuclease activity by in vitro assay; linearized DNA was used as a substrate. Ctrl: control reaction without Cas proteins, Cas10d: reaction with Cas10d protein, Cas3d: reaction with Cas3d protein, red arrows; undigested DNA.
It has been reported that Cas3 itself has the ability to degrade single-stranded DNA non-specifically (20,39). Therefore, to confirm the nuclease function of Cas10d, we performed in vitro nuclease assays. To this test, Cas10d and Cas3d proteins were first expressed and purified from human HEK293T cells (Supplementary Figure S2A and B) and identified by LC-MS/MS analysis (Supplementary Figure S2C) for use in further assays. The Cas10d protein, but not Cas3d protein, was able to cleave M13mp18 ssDNAs in the presence of Mg2+, Ni2+ and Co2+ ions (Figure 1D), suggesting that Cas10d acts as a nuclease in the type I-D system.
Cas10d concentration-dependent nuclease activity was also detected (Figure 1E). Reactions without metal ions or with EDTA resulted in no cleavage of ssDNAs, suggesting an important role of divalent cations in Cas10d-mediated ssDNA cleavage (Figure 1F). We next investigated the effect of different divalent cations on ssDNA cleavage activity in the range of concentrations used by Mulepati and Bailey (39) and found that the presence of Mg2+ or Co2+ or Ni2+ ions stimulated the ssDNA cleavage activity of Cas10d (Figure 1F). Looking at double strand DNA (dsDNA) nuclease activity, neither Cas10d nor Cas3d cleaved dsDNA in the presence of ATP, Mg2+, Co2+ and Ni2+ ions (Figure 1G). These results indicated that Cas10d has a nuclease activity specific for ssDNA, not dsDNA. Although E. coli and Streptococcus thermophilus type I-E Cas3 proteins have also been reported to have ssDNA nuclease activity, the requirements for divalent cations differed from those of type I-D Cas10d (EcCas3; Cu2+ or Ni2+, StCas3; Mg2+) (20,39). These differences may influence genome editing activity in human cells, according to the physiological concentration of each divalent cation inside human nuclei. Our results suggest that Cas10d in the type I-D system has unique characteristics as a nuclease that is distinct from other known Cas3 proteins.
We next investigated whether Cas10d protein has ATPase activity by incubating Cas10d with ATP because we found that Cas10d has possible ATP binding motifs (Supplementary Figure S1). The amount of phosphate liberated from the ATP increased upon incubation with Cas10d and also in the presence of ssDNA, but not dsDNA (Figure 2A), indicating that Cas10d has a ssDNA-stimulated ATPase activity. A time course study indicated that the liberated phosphate accumulated with reaction time in the presence of Cas10d (Figure 2B). The reaction rate was calculated based on the slope of the approximate straight line, resulting in 1.22 min–1 at 2 mM of ATP, which was lower than the reaction rate of StCas3 (38 min–1 at 0.5 mM of ATP) (20), suggesting that the ATPase activity of Cas10d is weaker than that of StCas3. As Cas3d protein contains ATP-binding domain motifs, we investigated whether Cas3d also has ATPase activity. The results indicated that Cas3d protein exhibit weak ATPase activity (Figure 2A). The results combined together with our previous study (37) suggest that both Cas10d and Cas3d cooperate for target DNA cleavage in the type I-D system.
Figure 2.

ATPase and helicase activity of Cas10d and Cas3d. (A) Dependence of ATPase activity of Cas10d (left) and Cas3d (right) on concentration of ssDNA or dsDNA. Various amounts of ssDNA (M13mp18) or dsDNA (pNEB193) were mixed with 700 nM of Cas10d in the presence of Co2+, Ni2+, and ATP, and reacted at 37 °C for 2 hours before measuring the free phosphate liberated from ATP. Data are means ± S.E. of independent experiments (n = 3). (B) Time course study of ATPase activity of Cas10d. The reaction was performed with 700 nM of Cas10d and 3 nM ssDNA at 37°C in the same buffer used in (A). The reaction rate calculated from the slope of the time course is 1.22 min–1. The experiments were repeated three times independently. Data are means ± S.E. of independent experiments (n = 3). (C) Helicase assay for Cas3d and Cas10d. Partial duplex DNAs labeled with ATTO532 were used as a dsDNA in this experiment. ATTO532-labeled oligo (100 nt) was used as a ssDNA. Cas10d or Cas3d or both were incubated with DNAs with or without ATP at 37°C for 2 h. The products were separated by non-denaturing gel electrophoresis and visualized by Typhoon FLA9500 (Cytiva). The experiments were repeated three times independently.
To test whether the Cas10 protein has helicase activity, we performed the helicase assay using partial duplex DNA as a substrate. The reaction was performed in the same buffer used in the StCas3 helicase assay reported in Sinkunas et al. (20); however, DNA strand separation by Cas10d helicase activity was not detected (Figure 2C). The Cas3d protein and the combination of Cas10d and Cas3d were also examined for helicase activity; however, no helicase activity was detected, irrespective of the presence or absence of ATP (Figure 2C). These results suggest that Cas10d and Cas3d in the type I-D system do not have helicase activity under these conditions.
The results above suggest unique features of Cas10d in the type I-D system in DNA cleavage compared with another type I sub-types. The typical cas8 gene—the common effector in CRISPR type I-A, B, C, E and F (36,51,52)—is missing from the CRISPR–Cas type I-D locus of M. aeruginosa, also predicting different mechanisms of Cascade complex stability and DNA cleavage activity of type I-D in cells compared with other type I sub-types (Figure 1A).
Type I-D CRISPR–Cas is biologically active in human cells as a genome editing tool
To confirm protein expression in heterogenous host cells and complex formation of type I-D Cas proteins, Cas expression vectors (Figure 3A and Supplementary Figure S4B) for mammalian cells were constructed and transfected into HEK293T cells. Complex formation of Cas3d, Cas5d, Cas6d, Cas7d and Cas10d with crRNA in human cells was shown by pull-down assay (Figure 3B and Supplementary Figure S4A). The results also show that the absence of Cas10d destabilized binding of Cas3d to the Cascade complex. Direct interaction of Cas3d and other Cas proteins was also investigated (Supplementary Figures S4 and S5). Cas3d interacted directly with Cas10d but not with other Cas proteins. The interactions were independent of the expression of crRNA. The results suggest that Cas10d is necessary to recruit Cas3d into the Cascade complex in the type I-D system. In another type I system, Cas8 has been reported to interact with Cas5/Cas7/crRNA and recruits Cas3 protein in the Cascade complex. On the other hand, the type I-D system does not contain cas8. Our results suggest that Cas10d plays a similar role to Cas8 in type I-D system, i.e., recruitment of Cas3d protein and formation of Cascade complex.
Figure 3.
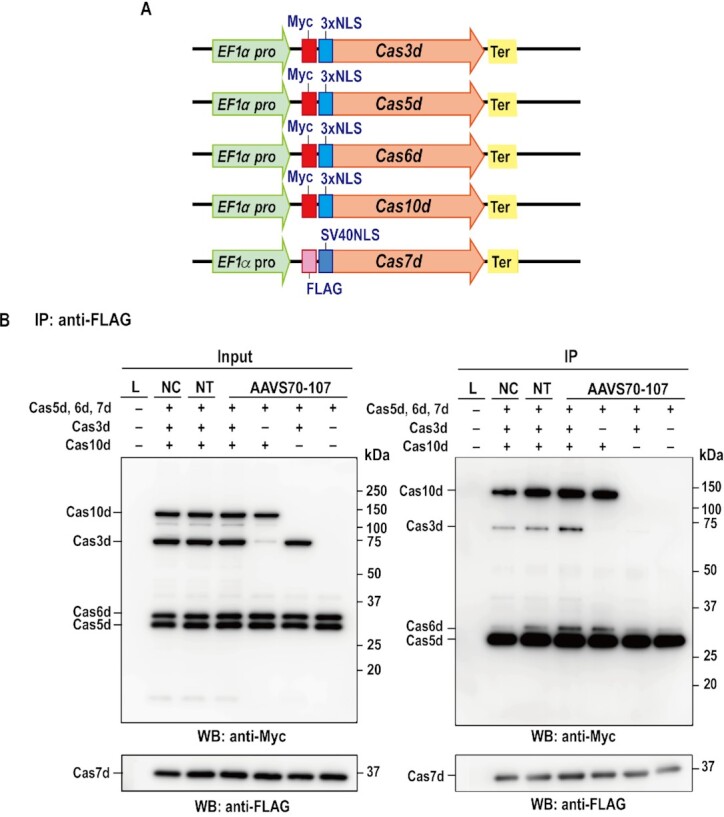
Co-immunoprecipitation assay of type I-D Cas proteins. (A) Schematic illustration of vectors used for the expression of each Cas protein. The five types of vectors (Flag-NLS-Cas7d, Myc-bpNLS-Cas5d-bpNLS-6xHis, Myc-bpNLS-Cas6d-bpNLS-6xHis, Myc-bpNLS-Cas3d-bpNLS-6xHis, Myc-bpNLS-Cas10d-bpNLS-6xHis) and gRNA (non-target gRNA or AAVS 70–107, Supplementary Table S2) were transfected into human HEK293T cells. (B) Co-immunoprecipitation assay using cell lysates was performed with the antibodies described in the figures. Experiments were repeated three times independently. L, lysate from non-transfected sample, NC, five kinds of vectors were introduced without gRNA, NT, five kinds of vectors were introduced with non-target gRNA. AAVS70-07; five kinds of vectors were introduced with AAVS GTC70–107 gRNA.
Genome editing activity of the type I-D CRISPR–Cas complex was then evaluated using the luciferase single-strand annealing (SSA) recombination system using NanoLuc luciferase (56). We developed a split type NanoLUxxUC system that consists of two vectors: pCAG-nLuxxUC_Block1 and pCAG-nLUxxUC Block2 (Supplementary Figures S10 and S13). Each vector contains a part of the NanoLuc genes containing a 300 bp homologous arm and the human AAVS1 or EMX1 gene fragments containing the type I-D target site following, or followed by, the homologous arm, respectively (Supplementary Figure S13; target DNA fragments listed in Supplementary Table S3). In this system, DSBs at target sites induced by the type I-D system can be repaired via homology-dependent repair (HDR) or the single-strand annealing (SSA) pathway, which induces recombination between homology arms. This recombination recovers a functional NanoLuc gene, resulting in the emission of luminescence. We compared the Nanoluc/Fluc ratio of the tested crRNA with that of non-target crRNA and evaluated genome editing activity. A schematic illustration of the crRNA selection process is shown in Supplementary Figure S10.
It has been reported that the sequence 5′-GTT-3′ is used for CRISPR type I-D PAM in Synechocystis sp. PCC 6803 (32). We performed E. coli negative screening using a PAM library with the ccdB expression system, and identified that the type I-D from M. aeruginosa recognized only 5′-GTH-3′ (H = A, C or T) PAM in E. coli cells (37). We then examined whether GTT is active as the PAM in mammalian cells, and also analyzed the activity of GTC and GTA PAMs (Figure 4A, Supplementary Table S6; crRNAs listed in Supplementary Tables S2 and S6). The type I-D crRNAs were screened by luc reporter assay, in which HEK293T cells were transfected simultaneously with type I-D cas genes and crRNA and activities detected 48 hr after transfection (Figure 4A and Supplementary Table S6). The data indicated that expression of type I-D Cas proteins led to notably higher genome editing activity, and that type I-D CRISPR–Cas prefers the GTT and GTC PAM over GTA PAM in mammalian cells.
Figure 4.

Detection of type I-D CRISPR–Cas-mediated genome editing by luc reporter assay. (A) The Cas expression vectors and luc reporter vectors harboring the target sequence were transfected into HEK293T cells; an endonuclease cleavage assay produced luminescence. The charts show the selection of gRNAs for AAVS and hEMX1 genes in the type I-D system using the luc reporter assay. Data are means ± S.E. of independent experiments (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.005 are determined by Student's t tests. (B) Effect on the HD domain of Cas10d. The luc reporter assay was performed using the AAVS GTC_70–107 (+), AAVS GTC_159–196 (+), and non-target gRNAs. White bars: luc reporter assay of the type I-D system with the wild-type Cas10d, black bars: luc reporter assay of the type I-D system with the HD-mutated dCas10d (H177A). Data are means ± S.E. of independent experiments (n = 3). Asterisks indicate statistically significant differences determined by Student's t tests. *P < 0.05. (Lower panel) The expression levels of Cas10 and dCas10d (H177A) detected by the antibodies for the tags fused to the N- (Myc) or C- (His) terminus. For this experiment, pEFs-Myc-bpNLS-Cas10d or dCas10d (H177A)-bpNLS-6xHis and pEFs-SV40NLS HA-Cas3d, Strept-Cas5d, Myc-Cas6d, or FLAG-Cas7d were used for transfection. (C) Effect of gRNA form on genome editing activity of the type I-D system. White bars: luc reporter assay of the type I-D system with crRNA with a repeat-spacer-repeat structure (pre-mature crRNA); black bars: luc reporter assay of the type I-D system with crRNA with a spacer-repeat structure (mature crRNA). Data are means ± S.E. of independent experiments (n = 3). *P < 0.05 is determined by Student's t tests.
Wild-type Cas10d and Cas10d carrying a mutation in the HD-like domain [Cas10d (H177A)] were then tested in the luciferase reporter assay using gRNAs AAVS GTC_70–107(+) and AAVS GTC_159–196(+) (Figure 4B and Supplementary Table S2). The results indicated that Cas10d indeed possesses a nuclease activity in the HD-like domain that can be utilized for genome editing in human cells. However, in vitro ssDNA nuclease assay indicated that purified Cas10d (H177A) still maintains ssDNA nuclease activity (Supplementary Figure S3). This result suggests that the H177 residue in Cas10d is important for dsDNA nuclease activity in human cells but not necessary for ssDNA nuclease activity in vitro. Recently, Lin et al. (34) have also reported the effects of HD domain mutations on the nuclease activity of the Sulfolobus islandicus type I-D system in vitro. They showed that the reconstructed type I-D Cas complex [SiCas5, SiCas7, processed crRNA from S. islandicus and small subunits (SS), SiCas10 carrying HD domain mutations purified from E. coli] failed to cleave dsDNA but could cleave complementary ssDNA in vitro. Although the type I-D system from M. aeruginosa and that from S. islandicus shows different characteristics, for example, SiCas10/SS alone does not bind to ssDNA, and SiCas7 in a backbone complex catalyzes ssDNA cleavage, Cas10d plays the major role in the DNA cleavage function on the target DNA. Further studies will be needed to understand the mechanisms of DNA cleavage by type I-D systems in more detail.
Cas6 is known as an endoribonuclease, and cleaves repeat sequences of the pre-mature crRNA at specific positions that hold the repeat-derived 3′ handle of the mature crRNA (5–7,32). In type I-E, processing of the crRNA sequence by Cas6e was required for genome editing activity in cells (24). In contrast, the processed mature crRNA in type I-B has activity in cells (57). To test whether the processed crRNA in type I-D system has genome editing activity, we performed the luc reporter assay using pre-mature crRNA and the predicted mature crRNA sequences (Figure 4C). We predicted mature crRNA sequences based on Shao et al. (6) and stability of the secondary structure was analyzed using the RNAfold web server. The most stable structure was predicted as a mature crRNA. The luc reporter assay indicated that genome editing activity of type I-D required the expression of pre-mature crRNA rather than processed mature crRNA (Figure 4C). Processing of pre-crRNA into its mature form might be important for the crRNA interaction with Cascade and functional genome editing activity of type I-D in cells, as in the type I-E system.
We also evaluated the effect of mutations in the ATP-binding domain motifs of Cas3d and Cas10d on genome editing activity using the luc reporter assay. The mutations abolished the genome editing activity of the type I-D system (Supplementary Figure S6). These results indicate that ATPase activity of both Cas3d and Cas10d proteins are necessary for genome editing in human cells. These results also suggest that the type I-D system has a unique mechanism compared to other known CRISPR–Cas systems in which only Cas3 protein has ATPase activity. Cas3d and Cas10d mutant proteins formed the complex with other Cas proteins (Supplementary Figure S7), indicating that the abolished genome editing activity was not due to the failure of complex formation.
We also evaluated the effects of the position and number of nuclear localization signals (NLS) by using two types of NLS: monopartite-NLS (SV40NLS) and bipartite-NLS (bpNLS). bpNLS functioned as effectively at both N-terminal and C-terminal of Cas3d, Cas5d, Cas6d and Cas10d as SV40NLS did at N-terminus (Supplementary Figure S8A–C). On the contrary, bpNLS-6xHis attached to the C-terminus of Cas7d disrupted the genome editing activity of type I-D (Supplementary Figure S9), although it was expressed strongly in the nucleus (Supplementary Figure S8C). We hypothesized that the function of Cas7d was affected by fusing 6xHis. To test this hypothesis, we performed the luc reporter assay with Cas7d fused to 6xHis at either N-terminus or C-terminus. The result indicated that the genome editing activity of type I-D was decreased when Cas7d fused with 6xHis-tag (Supplementary Figure S9). Positively charged 6xHis might affect the Cas7d function of RNA binding or Cascade formation.
Effect of number and positions of mismatches between spacers and target DNA, and the Cas7d mutations on genome editing activity of type I-D
We next explored the mismatch effect of spacer sequence on genome editing activity using the luc reporter assay (Figure 5 and Supplementary Figure S11). The 23 nucleotides just adjacent to the PAM, with the exception of nucleotides at 1-, 6-, 12- and 18-nt downstream of PAM, were identified as important for genome editing activity; however, a single mismatch at 24-, 25-, 30- or 35-nt showed no effect on genome editing activity (Figure 5A and B). Previous studies have shown that every 6th nucleotide of the protospacer is not engaged in base pairing with the target DNA strand in the Cascade complex in the type I-E system (17,58), because every 6th nucleotide binds with the thumb domain of Cas7 subunits in the Cascade complex (18,59–61). The obvious difference between type I-D and type I-E was the effect of a mismatch at 1-nt, where the DNA cleavage activity in the type I-E system is markedly reduced (24), suggesting that the mode and/or position of RNA-Cas7 binding might differ between the two systems. In the type I-E system, the large subunit Cas8 (Cse1 in the type I-E) and two small Cas11 subunits function to stabilize the entire R-loop (14). The structural difference in Cascade between type I-D and type I-E might influence the mismatch effect of each system. We further analyzed the effect of mismatch number and position in the gRNA sequence. Introduction of two mismatches within the region covered by nucleotides 1–28 markedly reduced genome editing activity except for the combination of mismatches at position 24-nt and 25-nt; however, within nucleotide positions 29-nt to 35-nt, two mismatches had less of a reducing effect on genome editing activity (Figure 5C). On the other hand, the introduction of three mismatches completely abolished genome editing activity (Figure 5D). These findings highlight important aspects of the specific interaction between protospacer and target DNAs, and the subsequent DNA cleavage mechanism of type I-D CRISPR–Cas system.
Figure 5.

Detection of off-target effects of type I-D CRISPR–Cas-mediated genome editing. (A) Schematic illustration of gRNA and target DNA heteroduplex. (B–D) Evaluation of off-target effects in the type I-D gRNA. The critical nucleotides in AAVS GTC_70–107(+) gRNA sequence for genome editing activity of type I-D system were evaluated using the luc reporter assay in human HEK293T cells. The gRNAs were designed to contain a single mismatch (B) or two (C) or three (D) mismatch nucleotides. The nucleotides indicated with blue arrows in (A), the nucleotides at 6-, 12-, 18- and 24-nt positions, weakly affect the genome editing activity of type I-D. The first nucleotide (red arrow) directly PAM-adjacent also has a weak effect on the genome editing activity of the type I-D system. NT: non-target, PC: positive control with non-mutated gRNA. Data are means ± S.E. of independent experiments (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.005 are determined by Student's t tests and statistically significant differences compared with NT.
We next focused on interactions between crRNA, target DNA and the type I-D Cas effector protein, especially the Cas7d protein that binds to crRNA and forms a helical backbone in the Type I-E Cascade complex (14,58). MD simulations of type I-E Cas7e, type I-F Cas7f, and type I-D Cas7d models including cRNA and DNA were first performed (Figure 6A–C and Supplementary Movies S1–S3). Examination of the crystal structure together with biochemical analyses suggested that K45 and R46 in Cas7e, and Q326 and K327 in Cas7f are important in forming the complex with Cascade and the target DNA (14,43). Comparison of these models suggested that the three amino acid residues (K67, R68 and K69) in α-helical regions are important for the interaction between Cas7d, crRNA, and the target DNA (Figure 6A-C). To evaluate the role of these amino acids in complex formation, we designed 12 types of mutant Cas7d [Cas7d (K58A), Cas7d (R62A), Cas7d (K67A), Cas7d (R68A), Cas7d (K69A), Cas7d (R75A), Cas7d (E148Q), Cas7d (K67S/R68Q), Cas7d (K67A/R68A), Cas7d (F66K/K67S/R68Q), Cas7d(F66R/K67A/R68M/K69R), Cas7d (K67S/R68Q/K69Q)] (Figure 6D) and then investigated the effects of these mutations on genome editing activity of type I-D. The luc reporter assay indicated that Cas7d (K58A) did not affect genome editing activity but other types of mutations abolished activity, suggesting that these amino acid residues in Cas7d are important for genome editing using the type I-D CRISPR–Cas system (Figure 6E and F). To investigate the mechanism underlying the loss of activity in Cas7d mutants, we performed co-immunoprecipitation assays using Cas7d mutants and other type I-D CRISPR–Cas proteins with or without crRNA. The results indicated that the mechanism underlying loss of activity differed depending on the Cas7d mutant used (Supplementary Figure S12). Cas7d (K67A), Cas7d (E148Q), Cas7d (F66K/K67S/R68Q) did not affect binding of Cas7d to the complex. This suggests that loss of genome editing activity was caused by factors other than complex formation. On the other hand, other Cas7d mutants caused the decrease of Cas7d protein bound to the complex, suggesting that complex formation was impaired in these mutants. Xue et al. (62) also indicated some Cas7d mutations caused the loss of binding to dsDNA in vitro, and that this was not due to defects in complex formation. Further studies will be required to elucidate the mechanisms underlying loss of activity in these Cas7d mutants.
Figure 6.

Effect of Cas7d mutations on type I-D CRISPR–Cas-mediated genome editing. (A–C) Snapshot of the MD simulation trajectory of the cascade of Type I-E (A), Type I-F (B), and Type I-D (C) system. An opaque ribbon, translucent red ribbon with ball-and-stick, and translucent blue ribbon show one of the Cas7e (A), Cas7f (B), or Cas7d (C) molecules, crRNA and DNA, respectively. The ball-and-stick indicates the crRNA backbone. The essential residues that interact with crRNA, i.e. K45, R46 and R49 of Cas7e (A), Q326 and K327 of Cas7f (B), and K67, R68 and K69 of Cas7d (C), are depicted by space-filling models. (D) Cas7d amino acid sequences and the mutations introduced in this experiment. (E, F) Luc reporter assay of single mutants (E) and double, triple and quadruple mutants (F). Data are means ± S.E. of independent experiments (n = 3). *P < 0.05 is determined by Student's t tests.
These results highlight important aspects of the interaction in type I-D Cas7d, crRNA, and the target DNA; however, further studies, including structural analysis of the type I-D Cas complex, are needed to understand the finer details of the system.
Targeted-mutagenesis by type I-D CRISPR–Cas system in HEK293 cells
We next targeted an internal genomic DNA region corresponding to the type I-D crRNA target to detect the type I-D-induced mutagenesis in mammals. We constructed single expression vectors for each cas gene, a vector containing a triple-gene expression cassette connected via a 2A self-cleavage peptide sequence (2A), and a vector containing a quintuple gene expression cassette connected via 2A (Supplementary Figures S8A and S12). Of all these configurations, transfection with the single cas gene expression vectors yielded the highest Cas protein expression levels in human cells (Supplementary Figure S8D). The expression of Cas5 was rather poor but this could be resolved by expressing from the pCAG plasmid using the CAG promoter, which is a strong synthetic promoter (63) (Supplementary Figure S8E). A second round of codon optimization would also be an option to improve Cas5 expression, as reported by Pickar-Oliver et al. (27).
Following a recent genome editing study using CRISPR–Cas type I-E (23,24), we speculated that long-range deletion might be introduced near the type I-D target site. To investigate this, we selected targets hEMX1 GTT_998–1036 (–) for the human EMX1 gene and AAVS GTC_70–107(+) for the AAVS gene based on the luciferase SSA assay (Figure 4A and B). Using several primer sets flanking 3–15 kbp around the AAVS and hEMX1 target sites and also over 19 kbp around the AAVS target site (Figure 7A and B), DNA fragments PCR-amplified from total DNA from HEK293T cells transfected with the type I-D CRISPR–Cas components were cloned. Sequencing revealed that type I-D CRISPR–Cas introduced long-range deletions ranging from 2.5 kb to over 19 kb at target sites (Figure 7A-C; Supplementary Tables S7 and S8). AAVS GTC_70–107(+) mutated sequences shared some specific features: the size of major long-range deletions was not random but showed some restrictions in the case of the target AAVS GTC_70–107(+) as 5.2–5.5 kb and 17–18 kb deletions; with some minor exceptions, mostly bi-directional deletions were detected (Figure 7A-C; Supplementary Tables S7 and S8). These features differed from those following mutation by type I-E (23,24), and type II effectors such as Cas9 (1,2) and Cas12a (3). Microhomology and insertions were also observed in mutation sites (Figure 7A-C; Supplementary Tables S7 and S8). The mutation rates for long-range deletion in the cloned DNA fragments were 55.0% and 57.1% with hEMX1 GTT_998–1036 (–) and AAVS GTC_70–107(+), respectively. Furthermore, the mutation distribution in cloned PCR products varied among targets; mutations by hEMX1 998–1036 (–) showed low mosaic, whereas mutations by AAVS GTC_70–107(+) showed more varied mutated sequences (Figure 7A-C and Supplementary Table S7). The detailed sequences of PCR fragments derived from long deletion mutations are listed in Supplementary Table S9.
Figure 7.
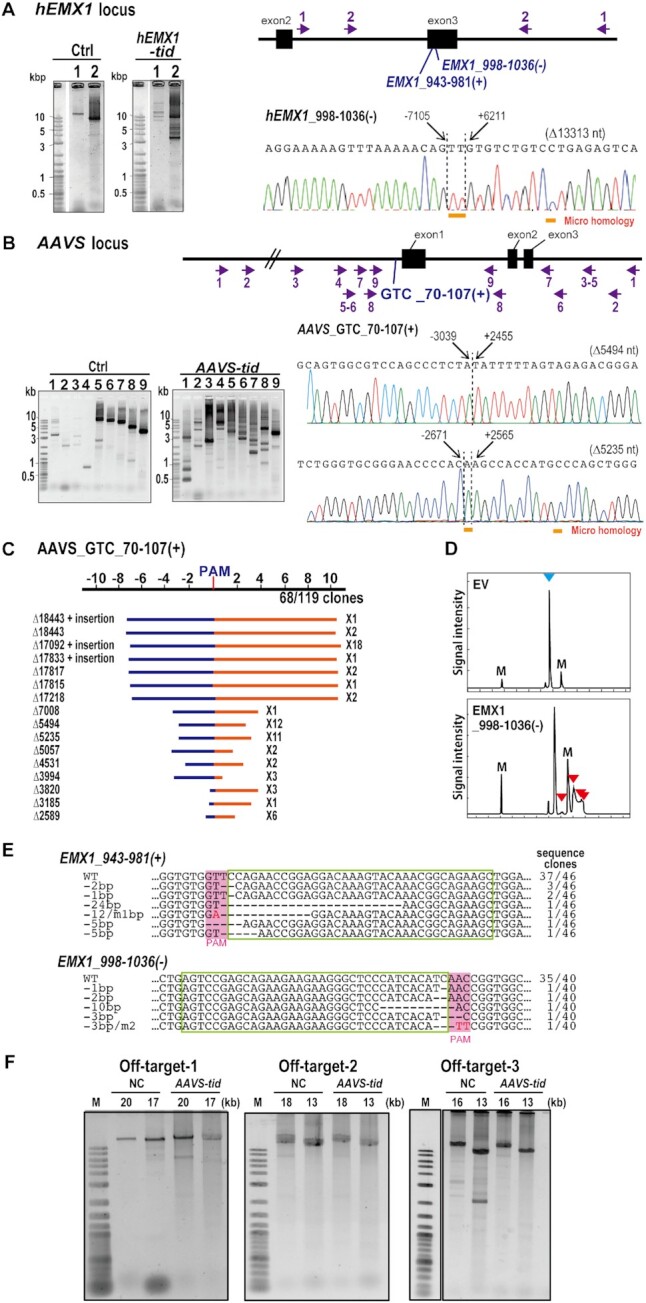
Type I-D CRISPR–Cas mediates genome editing in human cells. (A, B) Detection of long-range deletion mutations in the EMX1 and AAVS genes induced by the type I-D CRISPR–Cas. Upper: gene structure and various primer sets to amplify the mutation (arrows). The primer sets for hEMX1 gene amplify 14,915 bp (Primer set 1) and 9,109 bp (Primer set 2), respectively. The primer sets for AAVS locus amplify 19031 bp (Primer set 1), 16624 bp(Primer set 2), 12659 bp (Primer set 3), 10482 bp (Primer set 4), 9246 bp (Primer set 5), 8359 bp (Primer set 6), 7077 bp (Primer set 7), 4764 bp (Primer set 8), 3594 bp (Primer set 9), respectively. Lower (left): The PCR-amplified fragments were separated on agarose gels. Ctrl indicates the samples co-transfected with type I-D Cas expression vectors with non-target gRNA vector. Numbers correspond to the primer sets shown with the gene structures. Lower (right); The large deletion mutations analyzed by the Sanger sequencing of the cloned DNA from the type I-D Cas infected cells. Dashed lines indicate the deletion/repair junctions. The nucleotide distances from the PAM were indicated on the sequence. Sizes of deletions are shown in parentheses. The sequences underlined with orange bar indicates the microhomology found in junctions. (C) Long-range deletion pattern induced by AAVS GTC_70–107 (+). Blue bar: deletion in the 5′ upstream from the PAM, red bar: deletion in the 3′ downstream from the PAM. The x-axis indicates the distance from PAM site (kbp). The upstream region was referred to as (–), downstream region is referred to as (+). The distribution of longer deletion mutation is shown in Supplementary Figure S4C. (D) Heteroduplex mobility assay of the mutation in the hEMX1 gene induced by type I-D CRISPR–Cas. Multiple heteroduplex peaks (red arrows) were detected in PCR amplicons from the HEK293T cells infected with type I-D CRISPR–Cas that targets to hEMX1 gene [EMX1 998–1036 (–)]. blue arrow; Wild-type peak. EV: the cells infected with empty vector. M: internal markers. (E) Mutation sequences in HEK293T cells transfected with type I-D CRISPR–Cas that target to hEMX gene. WT: wild-type sequences. Two gRNAs for CRISPR type I-D [EMX1_943–981(+) and 998–1036(–)] were infected separately to HEK293T cells. gRNA target sequences are indicated in green boxes and PAM is indicated in pink boxes. The sequence frequencies in the cloned PCR products are indicated to the right of the sequence. (F) Analysis of off-target effect of type I-D in human cells. Off target sites were selected based on the mismatch number (off-target-1, -2 and -3 for AAVS GTC_70–107) and long deletions were investigated by long PCR analysis. The expected sizes of amplified fragment were indicated at the top of the gels.
The luciferase SSA assay, with a short (300–500 bp) fragment spanning two homologous regions of the luciferase gene, clearly showed the genome editing activity of type I-D CRISPR–Cas, thus we next asked whether, like Cas9 and Cas12a, the type I-D system could also introduce small changes at the target site. To test this, the 300–500 bp target region was PCR-amplified and analyzed by HMA. Typical multiple heteroduplex HMA peaks were detected in PCR amplicons from HEK293FT cells transfected with type I-D CRISPR–Cas with hEMX1 GTT_998–1036 (–) gRNA (Figure 7D). Sequence analysis of cloned PCR products from HEK293FT cells transfected with gRNAs hEMX1 GTT_943–981 (+) and hEMX1 GTT_998–1036 (–) showed that gRNA coupled with type I-D CRISPR–Cas proteins could introduce double-strand breaks (DSBs) and small indels at the target site at mutation rates of 19.6% [hEMX1 GTT_943–981 (+)] and 12.5% [hEMX1 GTT_998–1036 (–)] in the cloned DNAs (Figure 7E).
Recently, Dolan et al. (23) and Morisaka et al. (24) reported that the type I-E CRISPR–Cas3 system induced large deletions. Morisaka et al. (24) investigated the mutations with an efficiency of 16.1% for the EMX1 gene and 4.6% for the CCR5 gene in HET293T cells, whereas SpyCas9-mediated indels were 23.4% for EMX1 and 23.7% for CCR5. Dolan et al. (23) showed the mutations efficiency of 13% for the GFP gene in hESC cells. Both studies indicated that type I-E CRISPR–Cas3 induced long-range deletions through a long stretch of the target region, mostly upstream of the PAM/target site, with no defined end spots. Morisaka et al. (24) reported that 86.3% and 81.6% of deletions were over 10 kb for the EMX1 and CCR5 genes, respectively, and deletion size was up to 78 kb. In the study by Dolan et al. (23), most deletions were concentrated within 30 kb and the deletion sizes ranged from a few hundred bp to 100 kb. On the other hand, the type I-D produced different editing outcomes compared to the type I-E system; the type I-D can induce both small indels and bi-directional long-range deletions.
Some studies have reported that out-of-frame mutations induced by CRISPR–Cas9 result in the production of aberrant mRNA or protein (64,65). The ability to induce long-range deletions is useful to knockout a gene-of-interest by deleting the entire region harboring the gene. It could also be useful to induce deletions in non-coding regions such as promoters, UTRs, introns and repetitive sequences. Morisaka et al. (24) indicated that the type I-E system can induce exon skipping more efficiently than the CRISPR–Cas9 system. It can be expected that the type I-D system is also used for this purpose. Further development to control the mutations and increase efficiency should expand the applicability of this technology. The engineering of Cas proteins or fusing functional proteins, and the application of anti-Cas protein will contribute to the development of this technology.
The significant difference between the type I-E CRISPR–Cas3 system and our type I-D system is that CRISPR–Cas3 induces only unidirectional long deletion, but the type I-D system induces bi-directional long-range deletion and also small indels. Small indels might be the result of low helicase activity in the type I-D system. The ability to induce small indels can be applied for mutagenesis, such as that performed using CRISPR–Cas9 or Cas12a. While this study has shown that the type I-D system can introduce a wide variety of mutations into the target, it is difficult to accurately calculate the mutation efficiency of type I-D, which exhibits diverse and complex mutation profiles with bi-directional long-range deletions and short indels. To characterize the mutation efficiency mediated by the type I-D system in depth, and compare it with those of other tools, a novel next-generation sequencing-based approach should be established to detect and analyze the variety of both bi-directional long-range deletions and short indels.
We also analyzed whether off-target mutations occurred in genome-edited samples by long-range PCR analysis. As potential off-target sites with less than 8 mismatches were not found for AAVS GTC_70–107, we evaluated three potential sites with 9 and 10 mismatches (Supplementary Table S10) by long-range PCR. The result showed that off-target mutations were not induced by the type I-D system (Figure 7F). This could be attributed to the recognition of longer target sequences by type I-D than Cas9. Although further off-target mutation analysis is needed, results suggest that the type I-D system is a useful genome editing system with high specificity, and further engineering of crRNA could increase the specificity of the type I-D system.
CONCLUSIONS
Detailed studies on prokaryote genomes have identified nine subtypes of CRISPR type I families to date (4,51), some of which, such as the type I-D CRISPR–Cas, remain less well characterized. A unique member of the type I subtype, the type I-D CRISPR–Cas does not possess a Cas3 nuclease but instead has a Cas10d nuclease as its functional component. In this study, we developed the type I-D CRISPR–Cas system for use as a genome editing tool, and showed efficient site-directed mutagenesis with high specificity in human cells. Notably, in the type I-D CRISPR–Cas system, Cas10d—a unique Cas effector protein of the type I-D system—is a multifunctional effector for PAM recognition, stabilization and target DNA degradation. The DNA cleavage mechanism of the type I-D CRISPR–Cas and the subsequent DNA repair pathway may differ from those of Cas3, Cas9 and Cas12a. The ability of type I CRISPR to generate such a diverse range of large deletions from a single targeted site could potentially enable long-range chromosome engineering that would allow genetic analysis by deleting the full length gene-of-interest, gene clusters and non-coding regions and also simple and effective multi-gene function screening. Other promising applications, such as base editing, transcriptional control, epigenome editing, imaging of genomic loci, and virus detection would also be amenable to the application of the type I-D system. As a novel technology in the CRISPR toolbox, the type I-D CRISPR–Cas opens new possibilities in genome engineering.
Supplementary Material
ACKNOWLEDGEMENTS
We thank M. Fukuhara, and E. Niimi for their technical assistance. Simulations used computational resources of MOMO provided by BOST Computer Center KINDAI University, Oakforest-PACS provided by CCS Tsukuba University (Project ID: xg18i072), and Reedbush-L provided by The University of Tokyo.
Contributor Information
Keishi Osakabe, Graduate School of Technology, Industrial and Social Sciences, Tokushima University, Tokushima, Tokushima 770-8503, Japan.
Naoki Wada, Graduate School of Technology, Industrial and Social Sciences, Tokushima University, Tokushima, Tokushima 770-8503, Japan.
Emi Murakami, Graduate School of Technology, Industrial and Social Sciences, Tokushima University, Tokushima, Tokushima 770-8503, Japan.
Naoyuki Miyashita, Department of Computational Systems Biology, Faculty of Biology-Oriented Science and Technology, Kindai University, Kinokawa, Wakayama 649-6493, Japan.
Yuriko Osakabe, Graduate School of Technology, Industrial and Social Sciences, Tokushima University, Tokushima, Tokushima 770-8503, Japan; School of Life Science and Technology, Tokyo Institute of Technology, Yokohama, Kanagawa 226-8502, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
New Energy and Industrial Technology Development Organization (NEDO; to K.O. and N.M.); Program on Open Innovation Platform with Enterprises, Research Institute and Academia (OPERA); Japan Science and Technology Agency (to K.O.); Japan Science and Technology Agency (JST), Adaptable and Seamless Technology transfer Program through Target-driven R&D (A-STEP) (to K.O.); Funding for open access charge: Japan Science and Technology Agency, Adaptable and Seamless Technology transfer Program through Target-driven R&D (to K.O.).
Conflict of interest statement. Two patent applications have been filed relating to the data presented. The authors have no conflict interest to declare.
REFERENCES
- 1. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F.. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M.. RNA-guided human genome engineering via Cas9. Science. 2013; 339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A.et al.. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR–Cas system. Cell. 2015; 163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Makarova K.S., Wolf Y.I., Iranzo J., Shmakov S.A., Alkhnbashi O.S., Brouns S.J.J., Charpentier E., Cheng D., Haft D.H., Horvath P.et al.. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020; 18:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang R., Preamplume G., Terns M.P., Terns R.M., Li H.. Interaction of the Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure. 2011; 19:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shao Y., Li H.. Recognition and cleavage of a nonstructured CRISPR RNA by its processing endoribonuclease Cas6. Structure. 2013; 21:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Niewoehner O., Jinek M., Doudna J.A.. Evolution of CRISPR RNA recognition and processing by Cas6 endonucleases. Nucleic Acids Res. 2014; 42:1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brouns S.J.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J.H., Snijders A.P.L., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J.. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carte J., Wang R., Li H., Terns R.M., Terns M.P.. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008; 22:3489–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mojica F.J., Díez-Villaseñor C., García-Martínez J., Almendros C.. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009; 155:733–740. [DOI] [PubMed] [Google Scholar]
- 11. Shah S.A., Erdmann S., Mojica F.J., Garrett R.A.. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol. 2013; 10:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leenay R.T., Maksimchuk K.R., Slotkowski R.A., Agrawal R.N., Gomaa A.A., Briner A.E., Barrangou R., Beisel C.L.. Identifying and visualizing functional PAM diversity across CRISPR–Cas Systems. Mol. Cell. 2016; 62:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hochstrasser M.L., Taylor D.W., Kornfeld J.E., Nogales E., Doudna J.A.. DNA targeting by a minimal CRISPR RNA-guided cascade. Mol. Cell. 2016; 63:840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hayes R.P., Xiao Y., Ding F., van Erp P.B., Rajashankar K., Bailey S., Wiedenheft B., Ke A.. Structural basis for promiscuous PAM recognition in type I-E cascade from E. coli. Nature. 2016; 530:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sashital D.G., Wiedenheft B., Doudna J.A.. Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol. Cell. 2012; 46:606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anders C., Niewoehner O., Duerst A., Jinek M.. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014; 513:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hochstrasser M.L., Taylor D.W., Bhat P., Guegler C.K., Sternberg S.H., Nogales E., Doudna J.A.. CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:6618–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xiao Y., Luo M., Dolan A.E., Liao M., Ke A.. Structure basis for RNA-guided DNA degradation by cascade and Cas3. Science. 2018; 361:eaat0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sapranauskas R., Gasiunas G., Fremaux C., Barrangou R., Horvath P., Siksnys V.. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011; 39:9275–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sinkunas T., Gasiunas G., Fremaux C., Barrangou R., Horvath P., Siksnys V.. Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J. 2011; 30:1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sinkunas T., Gasiunas G., Siksnys. Cas3 nuclease-helicase activity assays. Methods Mol. Biol. 2015; 1311:277–291. [DOI] [PubMed] [Google Scholar]
- 22. Huo Y., Nam K.H., Ding F., Lee H., Wu L., Xiao Y., Farchione M.D. Jr, Zhou S., Rajashankar K., Kurinov I.et al.. Structures of CRISPR Cas3 offer mechanistic insights into cascade-activated DNA unwinding and degradation. Nat. Struct. Mol. Biol. 2014; 21:771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dolan A.E., Hou Z., Xiao Y., Gramelspacher M.J., Heo J., Howden S.E., Freddolino P.L., Ke A., Zhang Y.. Introducing a spectrum of long-range genomic deletions in human embryonic stem cells using type i CRISPR–Cas. Mol. Cell. 2019; 74:936–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morisaka H., Yoshimi K., Okuzaki Y., Gee P., Kunihiro Y., Sonpho E., Xu H., Sasakawa N., Naito Y., Nakada S.et al.. CRISPR–Cas3 induces broad and unidirectional genome editing in human cells. Nat. Commun. 2019; 10:5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Luo M.L., Mullis A.S., Leenay R.T., Beisel C.L.. Repurposing endogenous type I CRISPR–Cas systems for programmable gene repression. Nucleic Acids Res. 2015; 43:674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Y., Pan S., Zhang Y., Ren M., Feng M., Peng N., Chen L., Liang Y.X., She Q.. Harnessing Type I and Type III CRISPR–Cas systems for genome editing. Nucleic Acids Res. 2016; 44:e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pickar-Oliver A., Black J.B., Lewis M.M., Mutchnick K.J., Klann T.S., Gilcrest K.A., Sitton M.J., Nelson C.E., Barrera A., Bartelt L.C.et al.. Targeted transcriptional modulation with type I CRISPR–Cas systems in human cells. Nat. Biotechnol. 2019; 37:1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cameron P., Coons M.M., Klompe S.E., Lied A.M., Smith S.C., Vidal B., Donohoue P.D., Rotstein T., Kohrs B.W., Nyer D.B.et al.. Harnessing type I CRISPR–Cas systems for genome engineering in human cells. Nat. Biotechnol. 2019; 37:1471–1477. [DOI] [PubMed] [Google Scholar]
- 29. Young J.K., Gasior S.L., Jones S., Wang L., Navarro P., Vickroy B., Barrangou R.. The repurposing of type I-E CRISPR–Cascade for gene activation in plants. Commun Biol. 2019; 2:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu Z., Li Y., Li M., Xiang H., Yan A.. Harnessing the type I CRISPR–Cas systems for genome editing in prokaryotes. Environ. Microbiol. 2020; 23:542–558. [DOI] [PubMed] [Google Scholar]
- 31. Chen Y, Liu J, Zhi S, Zheng Q, Ma W, Huang J, Liu Y, Liu D, Liang P, Songyang Z.. Repurposing type I-F CRISPR–Cas system as a transcriptional activation tool in human cells. Nat. Commun. 2020; 11:3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jesser R., Behler J., Benda C., Reimann V., Hess W.R.. Biochemical analysis of the Cas6-1 RNA endonuclease associated with the subtype I-D CRISPR–Cas system in Synechocystis sp. PCC 6803. RNA Biol. 2019; 16:481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hrle A., Maier L.K., Sharma K., Ebert J., Basquin C., Urlaub H., Marchfelder A., Conti E.. Structural analyses of the CRISPR protein Csc2 reveal the RNA-binding interface of the type I-D Cas7 family. RNA Biol. 2014; 11:1072–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin J., Fuglsang A., Kjeldsen A.L., Sun K., Bhoobalan-Chitty Y., Peng X.. DNA targeting by subtype I-D CRISPR–Cas shows type I and type III features. Nucleic Acids Res. 2020; 48:10470–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McBride T.M., Schwartz E.A., Kumar A., Taylor D.W., Fineran P.C., Fagerlund R.D.. Diverse CRISPR–Cas complexes require independent translation of small and large subunits from a single gene. Mol. Cell. 2020; 80:971–979. [DOI] [PubMed] [Google Scholar]
- 36. Cass S.D., Haas K.A., Stoll B., Alkhnbashi O.S., Sharma K., Urlaub H., Backofen R., Marchfelder A., Bolt E.L.. The role of Cas8 in type I CRISPR interference. Biosci. Rep. 2015; 35:pii: e00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Osakabe K., Wada N., Miyaji T., Murakami E., Marui K., Ueta R., Hashimoto R., Abe-Hara C., Kong B., Yano K., Osakabe Y.. Genome editing in plants using CRISPR type I-D nuclease. Comm. Biol. 2020; 3:648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. López-Perrote A., Castaño R., Melero R., Zamarro T., Kurosawa H., Ohnishi T., Uchiyama A., Aoyagi K., Buchwald G., Kataoka N.et al.. Human nonsense-mediated mRNA decay factor UPF2 interacts directly with eRF3 and the SURF complex. Nucleic Acids Res. 2016; 44:1909–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mulepati S., Bailey S.. In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. J. Biol. Chem. 2013; 288:22184–22192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Katoh Y., Nozaki S., Hartanto D., Miyano R., Nakayama K.. Architectures of multisubunit complexes revealed by a visible immunoprecipitation assay using fluorescent fusion proteins. J. Cell Sci. 2015; 128:2351–23620. [DOI] [PubMed] [Google Scholar]
- 41. Webb B., Sali A.. Comparative Protein Structure Modeling Using Modeller. Current Protocols in Bioinformatics 54. 2016; John Wiley & Sons, Inc; 5.6.1–5.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Simons K., Kooperberg C., Huang E., Baker D.. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 1997; 268:209–225. [DOI] [PubMed] [Google Scholar]
- 43. Chowdhury S., Carter J., Rollins M.F., Golden S.M., Jackson R.N., Hoffmann C., Nosaka L., Bondy-Denomy J., Maxwell K.L., Davidson A.R.et al.. Structure reveals mechanisms of viral suppressors that intercept a crispr rna-guided surveillance complex. Cell. 2017; 169:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Berendsen H.J.C., van der Spoel D., van Drunen R.. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995; 91:43–56. [Google Scholar]
- 45. Galindo-Murillo R., Robertson J.C., Zgarbová M., Šponer J., Otyepka M., Jurečka P., Cheatham T.E. III. Assessing the current state of amber force field modifications for DNA. J. Chem. Theory Comput. 2016; 12:4114–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Humphrey W., Dalke A., Schulten K.. VMD: visual molecular dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 47. Aravind L., Koonin E.V.. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. 1998; 23:469–472. [DOI] [PubMed] [Google Scholar]
- 48. Gong B., Shin M., Sun J., Jung C.H., Bolt E.L., van der Oost J., Kim J.S.. Molecular insights into DNA interference by CRISPR-associated nuclease-helicase Cas3. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:16359–16364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pyne M.E., Bruder M.R., Moo-Young M., Chung D.A., Chou C.P.. Harnessing heterologous and endogenous CRISPR–Cas machineries for efficient markerless genome editing in Clostridium. Sci. Rep. 2016; 6:25666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Guo T.W., Bartesaghi A., Yang H., Falconieri V., Rao P., Merk A., Eng E.T., Raczkowski A.M., Fox T., Earl L.A.et al.. Cryo-EM structures reveal mechanism and inhibition of dna targeting by a crispr-cas surveillance complex. Cell. 2017; 171:414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J., Charpentier E., Haft D.H.et al.. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Makarova K.S., Zhang F., Koonin E.V.. SnapShot: Class 1 CRISPR–Cas systems. Cell. 2017; 168:946–946. [DOI] [PubMed] [Google Scholar]
- 53. Elmore J.R., Sheppard N.F., Ramia N., Deighan T., Li H., Terns R.M., Terns M.P.. Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR–Cas system. Genes Dev. 2016; 30:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Estrella M.A., Kuo F.-T., Bailey S.. RNA-activated DNA cleavage by the Type III-B CRISPR–Cas effector complex. Genes Dev. 2016; 30:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kazlauskiene M., Tamulaitis G., Kostiuk G., Venclovas Č., Siksnys V.. Spatiotemporal control of type III-A CRISPR–Cas immunity: coupling DNA degradation with the target RNA recognition. Mol. Cell. 2016; 62:295–306. [DOI] [PubMed] [Google Scholar]
- 56. Hall M.P., Unch J., Binkowski B.F., Valley M.P., Butler B.L., Wood M.G., Otto P., Zimmerman K., Vidugiris G., Machleidt T.et al.. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012; 7:1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maier L.K., Stachler A.E., Saunders S.J., Backofen R., Marchfelder A.. An active immune defense with a minimal CRISPR (clustered regularly interspaced short palindromic repeats) RNA and without the Cas6 protein. J. Biol. Chem. 2015; 290:4192–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jung C., Hawkins J.A., Jones S.K. Jr, Xiao Y., Rybarski J.R., Dillard K.E., Hussmann J., Saifuddin F.A., Savran C.A., Ellington A.D.et al.. Massively parallel biophysical analysis of CRISPR–Cas complexes on next generation sequencing chips. Cell. 2017; 170:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jackson R.N., Golden S.M., van Erp P.B., Carter J., Westra E.R., Brouns S.J., van der Oost J., Terwilliger T.C., Read R.J., Wiedenheft B.. Structural biology. Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science. 2014; 345:1473–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mulepati S., Héroux A., Bailey S.. Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science. 2014; 345:1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhao H., Sheng G., Wang J., Wang M., Bunkoczi G., Gong W., Wei Z., Wang Y.. Crystal structure of the RNA-guided immune surveillance Cascade complex in Escherichia coli. Nature. 2014; 515:147–150. [DOI] [PubMed] [Google Scholar]
- 62. Xue C., Zhu Y., Zhang X., Shin Y.K., Sashital D.G.. Real-time observation of target search by the CRISPR surveillance complex cascade. Cell Rep. 2017; 21:3717–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Niwa H., Yamamura K., Miyazaki J.. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991; 108:193–199. [DOI] [PubMed] [Google Scholar]
- 64. Makino S., Fukumura R., Gondo Y.. Illegitimate translation causes unexpected gene expression from on-target out-of-frame alleles created by CRISPR–Cas9. Sci. Rep. 2016; 6:39608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tuladhar R., Yeu Y., Tyler Piazza, J., Tan Z., Rene Clemenceau, J., Wu X., Barrett Q., Herbert J., Mathews D.H., Kim J.et al.. CRISPR–Cas9-based mutagenesis frequently provokes on-target mRNA misregulation. Nat. Commun. 2019; 10:4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.