


Abstract
Aptamers can control the biological functions of enzymes, thereby facilitating the development of novel biosensors. While aptamers that inhibit catalytic reactions of enzymes were found and used as signal transducers to sense target molecules in biosensors, no aptamers that amplify enzymatic activity have been identified. In this study, we report G-quadruplex (G4)-forming DNA aptamers that upregulate the peroxidase activity in myoglobin specifically for luminol. Using in vitro selection, one G4-forming aptamer that enhanced chemiluminescence from luminol by myoglobin's peroxidase activity was discovered. Through our strategy—in silico maturation, which is a genetic algorithm-aided sequence manipulation method, the enhancing activity of the aptamer was improved by introducing mutations to the aptamer sequences. The best aptamer conserved the parallel G4 property with over 300-times higher luminol chemiluminescence from peroxidase activity more than myoglobin alone at an optimal pH of 5.0. Furthermore, using hemin and hemin-binding aptamers, we demonstrated that the binding property of the G4 aptamers to heme in myoglobin might be necessary to exert the enhancing effect. Structure determination for one of the aptamers revealed a parallel-type G4 structure with propeller-like loops, which might be useful for a rational design of aptasensors utilizing the G4 aptamer-myoglobin pair.
INTRODUCTION
Aptamers (1,2) are nucleic acid-based synthetic ligands that can be obtained against many target molecules with high affinity and specificity. Some aptamers that bind to proteins are reported as not only a specific ligand but also regulators of biological functions. For example, aptamers that specifically bind to enzymes have been reported to directly inhibit their enzymatic activity (3–12). The revealed mechanism for inhibiting the enzymatic activity by the aptamers is their binding activity around the substrate binding site of the enzyme and competition with the substrate (7–10,12,13). These mechanisms are expected to contribute to the pharmacological action of the enzyme-binding aptamers: several thrombin-binding aptamers, which are the most frequently studied enzyme-inhibiting aptamers that work as competitive inhibitors of the substrate, are or are being tested in clinical trials (14).
Additionally, application of the enzyme-inhibiting aptamers to biosensor development can be anticipated because enzyme acts as a remarkable signal amplifier. We developed the first aptameric enzyme modulator—aptameric enzyme subunit (AES) by utilizing a thrombin-inhibiting aptamer, it changes the enzyme activity depending on the binding activity of the target molecule (15). In the previous work, AES was constructed by insertion of target-binding oligonucleotides such as the aptamer or complementary oligonucleotides of targeted DNA into the thrombin-inhibiting aptamer to allosterically control the enzymatic activity with the existence of target molecules (6). The AES can bind to the thrombin's substrate-binding site and inhibit thrombin activity depending on the concentration of targeted molecules, so that adenosine triphosphate (ATP) (15), oligonucleotides harboring a part of the SARS coronavirus (SARS-CoV) gene (16,17), immunoglobulin E (18), and insulin (19) were detected by monitoring an increase or decrease in the thrombin activity. By using a DNA aptamer that inhibits DNA polymerase (4), sensing systems to detect target oligonucleotides of disease-related bacteria (20) and lysozyme (21) were also created. Consequently, utilizing enzymatic activity-inhibiting aptamers could apply to the development of a universal target detection method. However, there is a limitation of enzyme-inhibiting aptamers suitable for signal amplification.
Alternatively, after Sen's group discovered hemin-binding DNA aptamers that enhance the peroxidase activity of hemin when it binds to the DNA, thereby forming a G-quadruplex (G4) (22), a large number of reports on the development of biosensors utilizing the G4-hemin complex as a DNAzyme has been published (23,24). This rapid progress in the usage of G4-hemin complex for biosensor development is because G4 is a four-stranded tertiary structure formed in guanine-rich sequences and the interaction between hemin and G4 that causes the enhancement of peroxidase activity has frequently been observed as not only the first-reported hemin-binding aptamer (25–27). Moreover, this peroxidase reaction can catalyze the oxidation of luminol with hydrogen peroxide producing chemiluminescent signals, thereby allowing for highly sensitive analysis. However, since hemin would likely react with G4-forming aptamers that recognize target molecules, it is difficult to create a simple and universal design of aptasensors that includes the G4-hemin complex compared with the detection principle like AES; especially, the amplification of G4 or bound-free separation or both is required and often combined (28,29). Therefore, to open a new field of simple, universal, and sensitive aptasensor, novel aptamers that upregulate catalytic activity of proteinaceous enzymes would be highly demanded. Although aptamers with a high affinity with Kd (dissociation constant) <10−7 M can play some roles in the catalytic reaction when they bind to the reactive center of the enzyme, to date, only one study that describes the small enhancement of enzymatic activity of matrix metalloproteinase-9 by the nonspecific binding of nucleic acids such as ssDNA, ssRNA and dsDNA (30).
Here, we report a novel AES, which can enhance the peroxidase activity in myoglobin (Figure 1A). Myoglobin contains heme as a cofactor and displays weak peroxidase activity. The importance of the reaction center around heme for enzymatic reactions has been intensively investigated by comparing it with the case of horseradish peroxidase (HRP) (31,32). The histidine and arginine located near the sixth coordination site of the Fe ion in the heme center are expected to play important roles. Histidine serves as a proton donor or acceptor and arginine, which might have positive charges, serves to stabilize the hydrogen peroxide located at the sixth coordination site of the Fe ion in the heme. Myoglobin has much lower peroxidase activity than HRP, and is attributable to differences in the constituent of histidine and arginine (33). By comparing the three-dimensional structures of myoglobin and HRP, the histidine and arginine near the sixth coordination site of the Fe ion in the heme of the myoglobin were located distantly when compared with those of HRP (Supplementary Figures S1a and S1b). Indeed, their configuration is different and might be the cause of the low peroxidase activity of myoglobin (34).
Figure 1.
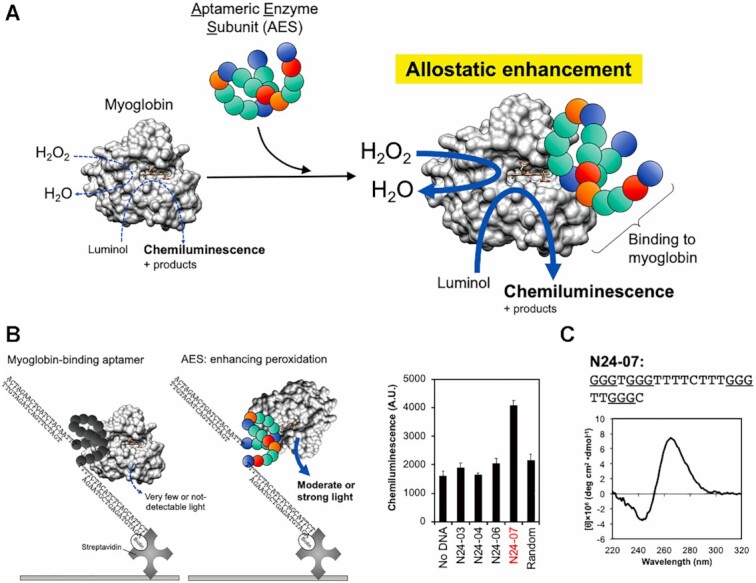
(A) Schematic illustration of the concept that aptameric enzyme subunit enhances myoglobin-derived peroxidase reaction. The aptameric enzyme subunit binds to myoglobin directly and enhances the performance of peroxidase activity, which catalyzes H2O2 and luminol. (B) Selection of aptameric enzyme subunit from the myoglobin-binding aptamers obtained by the systematic evolution of ligands by exponential enrichment. The aptameric enzyme subunit was selected by the investigation of aptamer-captured myoglobin. After the removal of free-myoglobin, chemiluminescent signal derived from the catalytic reaction of myoglobin trapped by the aptamer was analyzed with luminol. The mean ± SD is shown in the graph (N = 3). (C) Sequence of N24-07 except for the primer-binding site and circular dichroism spectrum of N24-07. Three consecutive guanines are underlined.
Based on the observation of the amino acid distribution of myoglobin, the proximal region of the heme binding site can be positively charged (Supplementary Figure S1c), and thus this region is likely to interact with the DNA aptamer harboring intensive negative charges due to electrostatic interaction derived from its sugar-phosphate backbone. Therefore, if the DNA aptamer interacts with the heme-surrounding positively-charged surface of myoglobin, the aptamer could affect myoglobin itself, upregulating peroxidation, as AES. There are reports on the improvement of peroxidase activity of myoglobin by the introduction of a few histidine residues near the sixth coordination site of Fe in the heme (33,35,36), which corresponds to our hypothesis. Additionally, ribosome, a complex of ribozyme and the ribosomal protein, might coordinately withdraw a proton from the surrounding nucleic and amino acids (37). A proton donor or acceptor is required to reduce H2O2 and to oxidize the secondary substrate, respectively, so that bases of DNA close to the heme may serve as electron mediators in the peroxidase reaction.
First, we screened for DNA aptamers against myoglobin with conventional SELEX (Systematic Evolution of Ligands by Exponential Enrichment) (1,2), and we investigated the enhancement activity of the aptamers for peroxidation. As a result, one G4-forming DNA aptamer that enhanced myoglobin peroxidase reaction was identified. We thus attempted to improve and investigate its enhancement effect using our strategy named—the in silico maturation (ISM) (6,38,39) that combines G4 structural analysis with the circular dichroism (CD) spectrum and NMR study. The NMR structure of the AES was solved and a parallel-stranded G4 structure with a flexible loop was determined. Finally, the best AES selected through the ISM study exhibited up to a 300-fold enhanced luminol chemiluminescence from the peroxidase activity of myoglobin.
MATERIALS AND METHODS
Materials
Myoglobin was purchased from Life Diagnostics (West Chester, PA, USA). DNA oligonucleotides (Supplementary Data) were purchased from Eurofins Genomics (Tokyo, Japan) or FASMAC (Kanagawa, Japan). Streptavidin-alkaline phosphatase conjugate was purchased from Sigma-Aldrich (St. Louis, MO, USA). BM Chemiluminescence ELISA Substrate (POD) was purchased from Roche (Basel, Switzerland). Luminol and 4-iodophenol were purchased from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan). A hydrogen peroxide-containing stabilizer was purchased from Sigma-Aldrich (St. Louis, MO).
Aptamer blotting assay and measurement of the peroxidase reaction of aptamer-myoglobin complex
Myoglobin was prepared in TBS and immobilized on nitrocellulose membranes (3.5 μg per spot). To prevent nonspecific binding, membranes were incubated with 5% (w/v) bovine serum albumin in TBS-T (0.05% (v/v) Tween 20 in TBS) for 1 h. Following washing with TBS-T, the membranes were incubated with 200 nM of 5′-biotin-labeled DNA oligonucleotide prepared in TBS-T for 1 h. After washing, the membranes were incubated with 0.1 μg/ml of streptavidin-alkaline phosphatase for 1 h. Following washing, ALP chemiluminescence was detected by LAS 4000mini (GE Healthcare) using CDP-Star, ready-to-use (Roche). To explore the enhancement activity of the myoglobin-binding aptamers on the peroxidase reaction, we immobilized aptamer-myoglobin complex on the 96-well plate via the avidin-biotin interaction and measured the chemiluminescent signal from catalysis of luminol. The aptamer was labeled with biotin by the hybridization of 5′-biotinylated oligonucleotide complement to the primer-binding site and immobilized streptavidin-coated 96-well plate. The plate surface was blocked with 4% skim milk solution containing 1 mM biotin. We added 200 nM of myoglobin to the well and incubated for 1 h, which followed by removal of free-myoglobin. By adding 100 μl of BM Chemiluminescence ELISA Substrate (POD) and performing a 5 min incubation, chemiluminescent signal was detected with a plate reader (PerkinElmer Co., Ltd.).
Circular dichroism (CD) spectroscopy analysis
CD spectroscopic measurements were obtained using J-820 (JASCO Co., Ltd., Tokyo, Japan). CD spectra of the non-labeled aptamers (5 μM) were recorded from 320 to 220 nm at 20°C. The scanning rate was 100 nm/min, and cumulative number was 5. The results were presented as a different spectrum using the spectrum of each buffer without aptamer as the background.
In silico maturation: myoglobin-derived peroxidase activity measurement
50 μl of myoglobin (f.c. 100 nM) and 50 μl of AES candidate oligonucleotide (f.c. 100 nM) were diluted in 10 mM potassium phosphate buffer (pH 6.5) were incubated in a 96-well plate for 1 h at room temperature. 50 μl of BM Chemiluminescence ELISA Substrate (POD) was then added to the aptamer-myoglobin mixture. After incubation for 5 min, chemiluminescent signals resulting from peroxidase activity was detected using a plate reader.
In silico maturation: sequence generation
For the first cycle of ISM, five sequences (N24-07, Annexin II-PQS, PSA-PQS2, HER2-PQS3 and aFGF-PQS20) were selected as parent AESs (Supplementary Figure S2). We ignored several consecutive guanines to make the variable G4 structure of these oligonucleotides simple; that is, we limited just four blocks of consecutive guanines to investigate oligonucleotides using ISM. The binding affinity of the predictive G4-forming sequences to myoglobin was evaluated by an aptamer-blotting assay, as stated above.
From five sequences, we fixed four blocks of three consecutive guanines and selected the sequences of loop regions: five sequences for the first loop (5′-CC-3′, 5′-AT-3′, 5′-A-3′, 5′-T-3′, 5′-AA-3′), seven sequences for the second loop (5′-A-3′, 5′-TT-3′, 5′-TG-3′, 5′-AA-3′, 5′-TTTCTTT-3′, 5′-CACTCACAGAAT-3′, 5′-TTTCTT-3′), and five sequences for the third loop (5′-CA-3′, 5′-TGTAAAGT-3′, 5′-A-3′, 5′-TT-3′). We then shuffled the sequences at each loop with the same appearance rate to create the next 20 sequences. Single base mutation was introduced into each of the 20 sequences with a mutation rate of a single base per sequence. Subsequently, we chemically synthesized the 20 generated sequences for the next round.
From the second to the fourth round, single base deletion was introduced at the same rate as the single base mutation in addition to the same sequence production as the first round. At the fourth round, the introduction of a single base at 5′ or 3′ end of the aptamers was also performed.
NMR spectroscopy
All NMR spectra were recorded on Bruker Avance III 700 and 800 MHz spectrometers equipped with a triple-resonance CryoProbe at 25°C and 15°C. DNA samples (concentration, 0.3–1.0 mM) were dissolved in a buffer containing 10 mM potassium phosphate buffer (pH 6.5). The nuclear overhauser effect spectroscopy in D2O and H2O/D2O (9.5:0.5) were acquired at mixing times of 50, 80, 250, 400 and 500 ms. Total correlation spectroscopy (TOCSY) spectra were recorded with the standard DIPSI-2 spin-lock sequence and a mixing time of 60 and 120 ms. In most of the experiments in H2O, water suppression was achieved by including a WATERGATE module in the pulse sequence before the acquisition. Spectral assignments were completed by NOESY, TOCSY, double-quantum filtered correlation spectroscopy (DQF-COSY) and (13C–1H)-HSQC using standard methods (40). Inter-proton distances were deduced from the NOESY experiments at various mixing times. All spectral analyses were performed using TopSpin (Bruker), NMRPipe (41) and SPARKY (42).
Distance and dihedral angle constraints
Interproton distances were obtained from the NOESY spectrum with a mixing time of 50 ms in 2H2O and H2O, supplemented by those with a mixing time of 500 ms in 2H2O and 250 ms in H2O, as described previously (43,44). In total, 480 distance constraints were obtained. The dihedral angle constraints for the δ, and endocyclic ν0, ν1, ν2, ν3 and ν4 torsion angles were derived from the 3JH1′-H2′, 3JH2″-H3′ and 3JH3–H4′ couplings, as described previously (43,44). The sugar pucker was determined as the C1′-exo conformation for all residues. The δ and endocyclic ν0 –ν4 torsion angles of the residues assuming the C1′-exo conformation was moderately constrained, leaving the sugar free to assume any conformation without an energy penalty between O4′-endo and C2′-endo, including C1′-exo in the pseudorotation cycle.
Structure calculations
Structure calculations were performed using distance and dihedral angle constraints with a simulated annealing protocol supplied by Xplor-NIH Version 2.48 (45). The hydrogen‐bonding constraints for each G:G base pair of the three quartets were also included. Ten final structures were selected from 100 calculations based on the criteria of the smallest residual energy values. None violated the distance constraints by >0.5 Å or the dihedral angle constraints by >5°.
Absorption spectroscopy
Myoglobin was mixed (f.c. 10 μM) and incubated with PEA3-01, PEA1-20, N24-07, and 17-mer poly T (T17) (f.c. 10 μM) in potassium phosphate buffer (pH 5.0) for 30–60 min. The absorption spectra were recorded using a V630 UV-VIS spectrophotometer at 25°C (JASCO Co., Ltd., Tokyo, Japan) between 300–700 nm.
Hemin-derived peroxidase activity measurement for luminol
Two hemin-binding DNAzymes, CatG4 (5′-TGGGTAGGGCGGGTTGGGAAA-3′) (46) and PS2.M (5′-GTGGGTAGGGCGGGTTGG-3′) (22), were purchased from Eurofins Genomics (Tokyo, Japan). Chlorohemin (Tokyo Chemical Industry Co., Ltd.) was dissolved in 10 mM dimethylformamide (DMF) and then diluted in hemin reaction buffer (10 mM potassium acetate buffer, pH 5.0, 1% DMF and 0.05% Triton X-100). Fifty microliters of hemin (f.c. 100 nM) was incubated with 50 μl of PEA3-01, PEA1-20, N24-07, T17, CatG4 and PS2M (f.c. 200 nM). After incubation for 1 h, the peroxidase activity of the DNA–hemin complex was measured by adding 50 μl of BM Chemiluminescence ELISA Substrate (POD) and compared the chemiluminescent signal from DNA–hemin complex to that from hemin alone.
Competition assay using G4-specific ligand
Small ligands that specifically bind to the G-quartet were used in this study. 6OTD (47), Phen-DC3 (48) and TmPyP4 (49) were dissolved in DMSO and then diluted in potassium phosphate buffer (pH 7.4). Before heat-treating PEA3-01 (f.c. 100 nM), the G4 ligand (f.c. 500 nM) was added. The ligand–DNA complex was mixed with myoglobin (f.c. 100 nM) and incubated for 1 h. Peroxidase activity using BM Chemiluminescence ELISA Substrate (POD) was carried out as described in the section ‘In silico maturation: myoglobin-derived peroxidase activity measurement,’ and we compared chemiluminescent signal intensity to intact PEA3-01-myoglobin complex.
Effect of luminol-concentration on the activity of PEA3-01-myoglobin complex
Luminol was dissolved in DMSO at a concentration of 20 mM, and p-iodophenol was dissolved at 100 mM in DMF. The luminol, p-iodophenol, and hydrogen peroxide solutions were diluted in 150 mM Tris–HCl (pH 8.8). Then, 2 μM myoglobin and 2 μM PEA3-01 were mixed in 10 mM potassium acetate buffer (pH 5.0) for 1 h at room temperature (approximately 1 μM each). After the formation of the PEA3-01-myoglobin complex, 10 μl of the solution was added to the 96-well plate well, followed by the addition of 90 μl luminol substrate containing luminol (f.c. 6.3, 12.5, 25, 50, 100 μM), p-iodophenol (f.c. 100 μM), and hydrogen peroxide (f.c. 1 mM). The solution was immediately mixed, the mixture was incubated for 4 min, and chemiluminescence was measured using a plate reader.
RESULTS
Effect of the myoglobin-binding aptamer screened by SELEX on the peroxidase activity of myoglobin
After seven rounds of SELEX, we obtained 20 aptamer-candidates (Supplementary Table S1) and found that N24-01, 03, 04, 06 and 07 had higher binding ability than the initial library in the aptamer-blotting assay (Supplementary Figure S3). We thus investigated the peroxidase activity of myoglobin using the chemiluminescent reaction following the formation of complexes with the myoglobin-binding aptamers. We found significantly higher chemiluminescence (f.c. >2-fold) derived from the peroxidase activity in myoglobin captured by N24-07 compared with other DNA aptamers (Figure 1B) although all aptamers can bind to myoglobin (Supplementary Figure S3). This result suggested that, while N24-01, 03, 04, 06 did not enhance the peroxidase reaction of myoglobin as AES, N24-07 did. N24-07 (5′-GGGTGGGTTTTCTTTGGGTTGGGC-3′) has four blocks of consecutive guanines, giving it the tendency to form the G4 structure. We checked the CD spectrum of N24-07 and found a negative peak around 240 nm and a positive peak ∼265 nm, which corresponded to the typical parallel-type G4 structure (Figure 1C).
Improvement of AES in the enhancement of peroxidase activity of myoglobin by in silico maturation
To investigate the key motif of AES for increasing the peroxidase activity of myoglobin with the goal of obtaining advanced AESs with a stronger activity than that of the original N24-07, we conducted ‘in silico maturation (ISM)’ (6,38,39) (Figure 2A). ISM is our original strategy for the functional evolution of aptamers and has also recently been applied to improve the function of aptamers by other groups (50,51). To start the ISM, pre-selected aptamers, called ‘parent aptamers,’ are necessary. The parent aptamer sequences are replicated, crossed over, and mutation(s) are introduced in silico to produce new sequences of aptamer candidates. These aptamer candidates generated from parent aptamers belong to the first generation of the ISM study. The target function to be improved, such as affinity or specificity, was then evaluated for each altered sequence in vitro. The aptamer candidates in the first generation are ranked by the level of their abilities for the function of interest, and sequences with good ability are used to produce the next generation as parent aptamer sequences. By repeating these steps, sequences of aptamers were optimized and aptamers with a greater ability were obtained.
Figure 2.
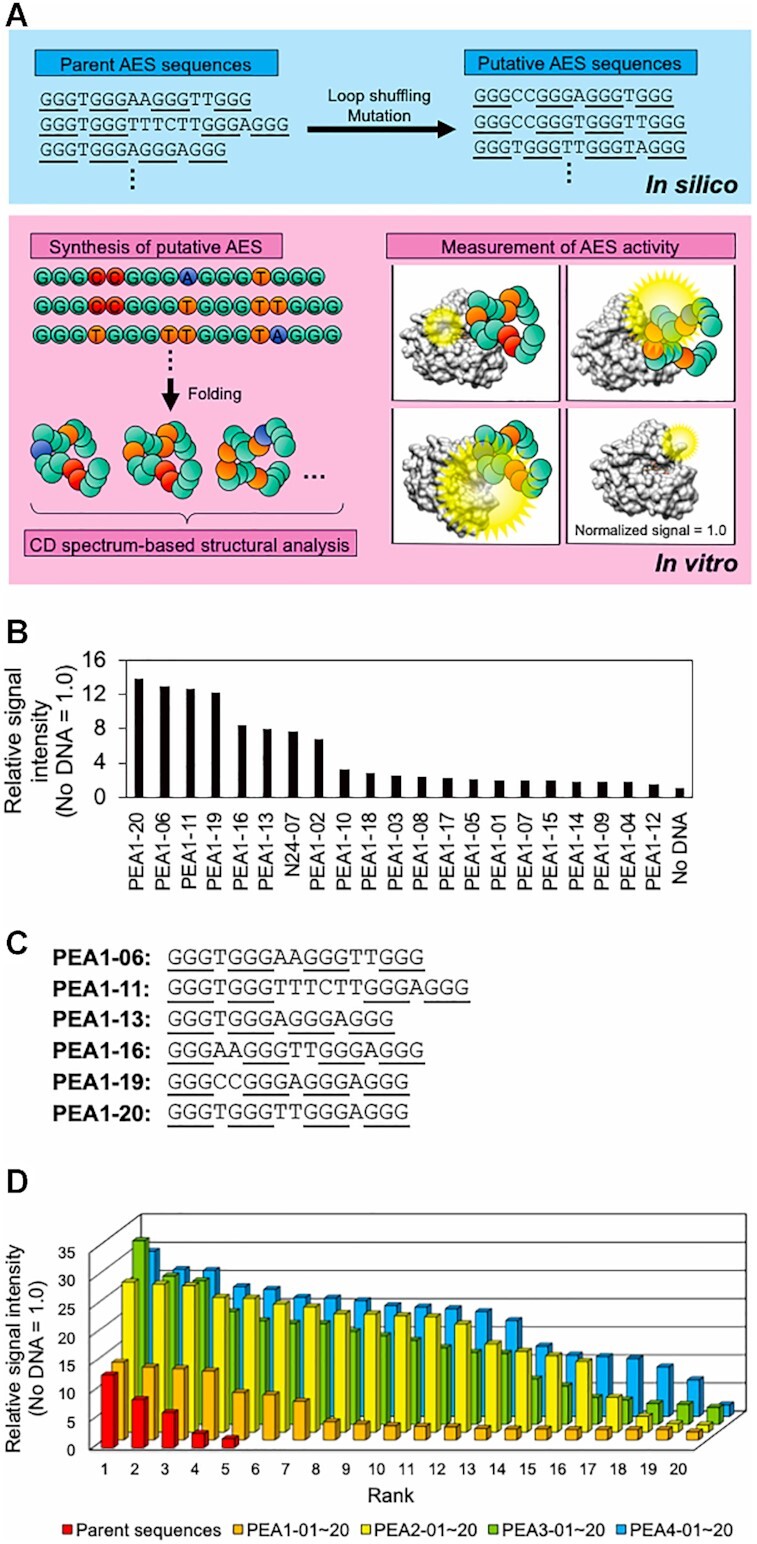
ISM to improve the AES activity and to investigate the structural property of the AES. (A) Schematic illustration of the ISM process. By performing loop shuffling between the conserved guanines (underlined) and introducing a single mutation into oligonucleotide sequence of selected AESs except for the guanine blocks, putative AES sequences were generated. CD spectrum analysis of chemically-synthesized putative AESs in vitro was performed and AES activity, which enhances peroxidization of myoglobin was measured. (B) Ranking of the enhanced level of the chemiluminescence signal in the AES candidates obtained after the first round of ISM and (C) sequences of the AES accompanied by highly increasing peroxidase reaction relative to N24-07. Three consecutive guanines are underlined. (D) Evolution of AES activity to myoglobin by four rounds of ISM. Each bar indicates an increased signal intensity by the addition of AES candidate relative to the no DNA condition. Sequence, enhanced level, and rank in each ISM procedure of all oligonucleotides are shown in Supplementary Data.
Because we assumed that the G4 structure might be a good scaffold for binding to myoglobin, we chemically-synthesized DNA oligonucleotides harboring 22 putative G4 forming sequences (PQSs) existing in the promoter region of genes (52) and and seven aptamers previously reported to obtain the first round of parent AES for ISM. Thereafter, we found that, among the 29 evaluated sequences, Annexin II-PQS, PSA-PQS2, HER2-PQS3 and aFGF-PQS20 (Supplementary Table S2) bound to myoglobin (data not shown). We thus investigated the peroxidase activity of myoglobin with chemiluminescent reaction following the addition of aptamers, and we also found upregulated chemiluminescent signals when these G4 aptamers were mixed with myoglobin (Supplementary Table S2). Therefore, these sequences with the G4-forming motifs were selected in addition to the existing AESs and N24-07, as the parent sequence for the ISM to improve the enhancement activity (Supplementary Figure S2). Here, sequence alterations such as shuffling and mutations were introduced into the parent AES. Importantly, to produce the G4-forming sequence of the first generation, we fixed the core sequence for the G4 structure, which was four blocks of three consecutive guanines, and shuffled the other G4-loop regions between the three consecutive guanines (Figure 2A, blue box). Point mutation was also introduced into loop regions with a mutation rate of 10%. In this ISM study, we evaluated the level of enhancement of chemiluminescent signals derived from AES-myoglobin complex activity (Figure 2A, pink box).
When the twenty peroxidase-activity enhancing AES (PEA) candidates obtained from the first generation of ISM by aptamer-blotting analysis were evaluated for their binding ability, PEA1-02, PEA1-06, PEA1-11, PEA1-13, PEA1-16, PEA1-19 and PEA1-20 showed binding signals equal to or higher than the original N24-07 (Supplementary Figure S4). Additionally, PEA1-06, PEA1-11, PEA1-13, PEA1-16, PEA1-19 and PEA1-20 showed an equal or higher enhancement effect on peroxidase activity of myoglobin than N24-07 (Figure 2B). Compared to N24-07, a long oligonucleotide loop between the second and third guanine block was substituted with the 1–6-mer of a short oligonucleotide loop (Figure 2C). CD spectrum analysis of the twenty oligonucleotides demonstrated that not all oligonucleotides folded into the G4 structure. We found that six AESs could enhance the peroxidase activity of myoglobin, which had a similar CD spectrum to N24-07, suggesting that they commonly folded into a parallel G4 structure (Supplementary Figure S5). Notably, the rank orders of aptamers based on their ability to bind to myoglobin do not completely correspond with the rank orders based on the enhancement activity. This difference could indicate that AES activity requires not only binding ability but also specific binding to the proper site around the heme in myoglobin, supporting our hypothesis.
To further improve the enhancement activity and explore the relationship of the structural activity of a series of PEA, we continued with three cycles of ISM (Figure 2D). For the second cycle, PEA1-02, PEA1-06, PEA1-11, PEA1-19 and PEA1-20 were selected to produce 20 new sequences (the second generation, PEA2-01–20, Supplementary Data). PEA2-11, PEA2-13 and PEA2-14 had a higher enhancement effect on peroxidase activity of myoglobin than other oligonucleotides. We selected (PEA2-11, PEA2-13 and PEA2-14) and (PEA1-20, PEA1-06 and PEA1-11) for use as parent sequences for the production of the third-generation oligonucleotides (the third generation, PEA3-01-20, Supplementary Data). In the third generation, six oligonucleotides (PEA3-01, PEA3-02, PEA3-05, PEA3-06, PEA3-12 and PEA3-17) indicated an enhancement in peroxidase activity of myoglobin. PEA3-01 exerted a 33-fold enhancement in myoglobin peroxidase activity. To obtain various sequences, four rounds of ISM were performed using the six sequences from the third round and PEA2-11 (fourth generation, PEA4-01–20, Supplementary Data). All mutated sequences in the fourth generation, except PEA4-16 and PEA4-17 had a higher level of enhanced chemiluminescence than N24-07. However, no oligonucleotide caused a higher chemiluminescence than PEA3-01. We summarized the sequences, signal enhancement levels, and predicted topology analyzed by CD spectrum measurement for the top ten AESs in the 85 oligonucleotides obtained by SELEX, G4 screening, and ISM (Table 1). The ten AESs with high enhancement effects by 20–30-fold had short loops between the three consecutive guanine repeats and seemed to have parallel G4-fold in CD spectrum analysis (Supplementary Figures S5–S8).
Table 1.
The top-ranking of AESs by enhancement level of peroxidase activity derived from myoglobin
| Rank | Name | Sequence (5′ to 3′) | Length (mer) | Topology |
|---|---|---|---|---|
| 1 | PEA3-01 | GGGCGGGTTGGGCTGGG | 17 | parallel |
| 2 | PEA4-03 | AGGGTGGGAAGGGAGGG | 17 | parallel |
| 3 | PEA2-03 | GGGCCGGGTGGGTTGGG | 17 | parallel |
| 4 | PEA3-02 | GGGTGGGAGGGCTGGG | 16 | parallel |
| 5 | PEA2-02 | GGGCCGGGAGGGTGGG | 16 | parallel |
| 6 | PEA4-19 | GGGTGGGCTGGGTTGGG | 17 | parallel |
| 7 | PEA2-01 | GGGTGGGCGGGTGTAGAGTGGG | 22 | parallel |
| 8 | PEA4-18 | GGGCGGGTTGGGTCGGG | 17 | parallel |
| 9 | PEA3-05 | GGGCGGGTTGGGTTGGG | 17 | parallel |
| 10 | PEA2-13 | GGGTGGGTTGGGCTGGG | 17 | parallel |
For higher peroxidase activity, strong interaction of AES with the protein backbone of myoglobin around heme was expected to be crucial. Since electrostatic interaction between the AES and myoglobin appeared to be important for the AES-binding mode, we evaluated the effect of pH change in the incubation buffer on enhancing peroxidase activity derived from the formation of a complex of myoglobin with N24-07 (5′-GGGTGGGTTTTCTTTGGGTTGGGC-3′) and PEA3-01 (5′-GGGCGGGTTGGGCTGGG-3′), respectively (Supplementary Figure S9). By adding PEA3-01, the highest peroxidase activity was observed when PEA3-01 was mixed with myoglobin in potassium acetate buffer at pH 5.0, and the signal was 47-fold greater than that in the absence of PEA3-01. N24-07 also showed the highest enhancement activity at a pH of 5.0. As polyT was added to the myoglobin-containing buffer, there was no enhanced peroxidase activity even at pH 5.0. Therefore, incubation with PEA and myoglobin at pH 5.0 would be optimal for enhancing the peroxidase reaction on the luminol–H2O2 system.
Using SPR analysis, we determined that the dissociation constant (Kd) of PEA3-01 against myoglobin at pH 4.7, was 5.8 nM (Supplementary Figure S10). This result indicated that at least 80% of added myoglobin complexed with PEA3-01 when 100 nM myoglobin was mixed with 100 nM of PEA3-01. In accordance with the pH dependency of the enhancing level, we did not determine the Kd of PEA3-01 against myoglobin at pH 7.4, due to low binding SPR signals (data not shown). Therefore, the binding of PEA to myoglobin at pH 5.0 may be appropriate for the formation of an active DNA-myoglobin complex.
Structural investigation of myoglobin-derived peroxidase activity enhancing AESs in the NMR study
The best AES, PEA3-01 (5′-GGGCGGGTTGGGCTGGG-3′), was 17-mer with a short loop region consisting of cytosine or thymine between the guanine blocks. Unfortunately, PEA3-01 aggregated into a multimer at a high concentration of oligonucleotide above 0.5 mM for the NMR study, which indicated the difficulty of performing NMR structure determination. Since we found that PEA1-20 (5′-GGGTGGGTTGGGAGGG-3′), the first generation's top aptamer (Figure 2B), can fold into a monomeric G4 by using polyacrylamide gel electrophoresis (PAGE) analysis (data not shown), we solved the structure of PEA1-20 and compared its structural features with PEA3-01.
CD spectrum of PEA3-01 showed a positive peak at 260 nm and a negative peak at 240 nm, suggesting that its folding yielded a parallel G4, similar to that of PEA1-20 (Figure 3A). We then proceeded to examine the G4 structure of PEA3-01 by comparing the NMR spectra of PEA3-01 to that of PEA1-20. It was difficult to assign all guanines because some peaks around 11.2 ppm were not clearly separated under this experimental condition. However, 1D 1H-NMR spectra of PEA3-01 (Figure 3B) showed several major sharp imino proton peaks, similar to PEA1-20, ∼10.8–12.0 ppm, suggesting that both aptamers form similar G4 containing three G-quartets. Structural similarity was also demonstrated in the 2D 1H–15N-HSQC (heteronuclear single quantum coherence) spectra displaying the imino proton from the G-quartet of PEA3-01 neighboring the PEA1-20 spectrum (Figure 3C).
Figure 3.
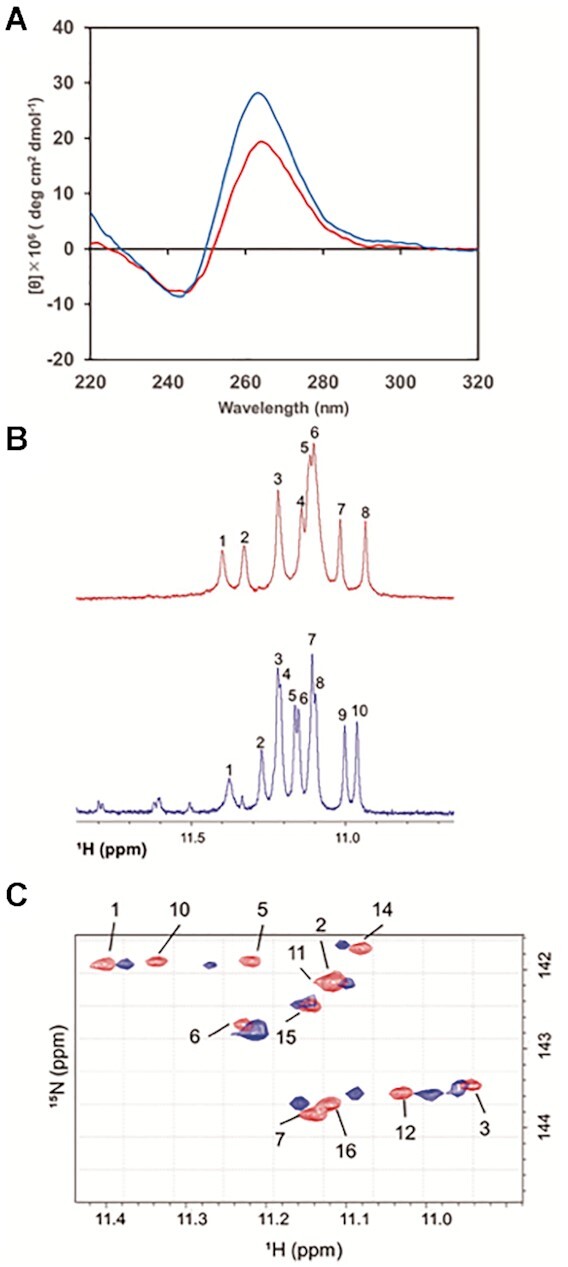
(A) CD spectra of PEA1-20 (red) and PEA3-01 (blue). (B) 1D 1H NMR spectra displaying the imino proton resonances of PEA1-20 (red) and PEA3-01 (blue). Imino proton peaks from 10.9–11.4 ppm indicate the formation of the G-quartet. (C) 2D 1H–15N-HSQC spectra displaying the imino group of PEA1-20 (red) and PEA3-01 (blue). The spectrum of PEA1-20 is displayed with the number of assigned guanines.
Here, to determine the structural feature of the parallel G4, which increases myoglobin-derived peroxidase activity, we solved the NMR structure of PEA1-20, which is a 16-mer oligonucleotide (5′-GGGTGGGTTGGGAGGG-3′). Based on the solved structure, we discovered that PEA1-20 is a monomeric parallel-stranded G4 structure that has three G-quartet planes, consisting of G1-3, G5-7, G10-12 and G14-16 (Figure 4). The other nucleotides, T4, T8, T9 and A13, are located at the loop structures while T4, T8, and A13 are exposed to the exterior of the G4 core (Figure 4A). The most stable NMR structure of PEA1-20 was calculated and the T9 faces the G7 belonging to the G-quartet in the structure (Figure 4B, C). According to insights from the 10 superimposed calculated structure, three loops consisting of one or two residues were expected to be very flexible, as opposed to the three G-quartet cores (Figure 4D–H). As indicated by the PAGE analysis, we considered the monomer state of PEA3-01 at a low concentration (under 5 μM) (data not shown), we presumed that PEA3-01 folded into the same parallel G4 structure with short and flexible loops as did PEA1-20.
Figure 4.
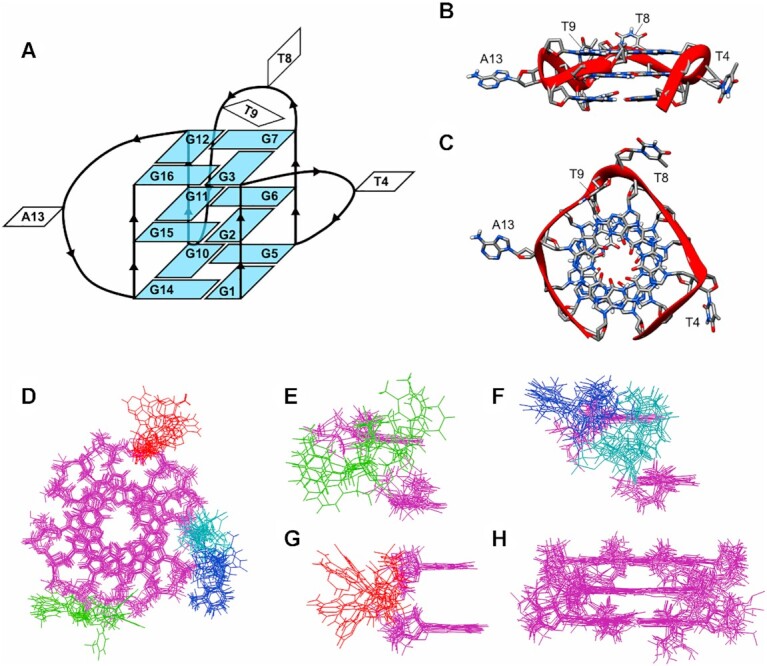
Three-dimensional structure of the aptamer, PEA1-20 (PDB ID: 7E5P). (A) G4 topology of PEA1-20. Residues forming the G-quartet are shown in light blue. (B) Side view and (C) top view of the best quadruplex structure. Phosphate sugar backbone is represented by the red ribbon. (D) Ten lowest energy structures were superimposed using PyMOL Version 2.3.1 (https://pymol.org/2/) and top view (D), side view focusing on T4 (E), T8 and T9 (F), A13 (G) and quadruplex core (H) of the superimposition is shown. In the superimposed figures, guanines, T4, T8, T9 and A13 are highlighted in magenta, green, blue, cyan and red, respectively.
Absorption spectrum analysis for the PEA–myoglobin complex
It is known that the spectral peaks of myoglobin in the Soret band (∼400 nm) and visible spectral region (500–700 nm) are affected by environmental changes around heme with the protein folding property of myoglobin (53,54). By examining the absorption spectrum of myoglobin in the presence of the PEA series, we investigated the binding of PEA to the periphery of heme in myoglobin, which is important for enhancing the activity of myoglobin. In Figure 5A, a 1:1 mixture of PEA3-01, PEA1-20 and N24-07 with myoglobin showed hyperchromicity of the Soret band at pH 5.0. T17 showed increased absorption; however, there was no significant peak in the Soret band. Therefore, the increased absorption at the Soret band of myoglobin in the presence of PEA was attributed to the location of PEA in position to interact with heme in myoglobin. In the 500–700 nm range, the peak position of the absorption spectrum in the presence of PEA was also different from that of myoglobin in the absence of oligonucleotides (Figure 5B). Myoglobin alone showed two peaks at 538 nm and 575 nm, indicating the presence of oxymyoglobin (Figure 5B, black solid line) (54). We also observed an unknown strong absorption spectrum in the visible region (400–700 nm range) with the addition of PEA3-01 and T17 (Figure 5B, red and gray solid line). An aptamer blotting assay (Supplementary Figure S11a) indicated that T17 was non-specifically bound to myoglobin at pH 5.0. Meanwhile, when T17 was dissolved in PBS (pH 7.4) and incubated with myoglobin, the non-specific binding of T17 to myoglobin was prevented (Supplementary Figure S11a) and the unknown absorption disappeared at pH 7.4 (Supplementary Figure S11b). Therefore, the unknown strong absorbance in the 400–700 nm range might be caused by non-specific binding of the oligonucleotide to myoglobin at pH 5.0. By adding PEA1-20 and N24-07 to myoglobin, we observed new peaks at 500 and 625 nm (Figure 5B, yellow and green solid line), which were likely metmyoglobin-derived peaks in the visible region (54). It is unclear whether oxygen in oxymyoglobin is released by the binding of PEA; however, the results suggested that the interaction of PEA1-20 and N24-07 with myoglobin might alter the environment around heme in myoglobin. As is the case with the hyperchromicity of the Soret band, these results indicated that PEA formed a complex with myoglobin and the bound PEA was located around heme in myoglobin.
Figure 5.

Absorption spectra of myoglobin (Mb) alone, DNA alone, and myoglobin-oligonucleotide mixtures in potassium acetate buffer (pH 5.0). T17 was used as a negative control. (A) Spectra in the 300–700 nm range to observe a Soret peak. (B) Graph showing enlarged spectra in a visible region (400–700 nm) from (a). DNA and myoglobin concentrations were each set at 10 μM. The arrows indicate generated peaks distinct from myoglobin only when PEA-myoglobin complex formation occurred.
Investigation of G4–heme interaction associated with enhancing the activity of myoglobin
Given the large number of hemin-binding G4 and PEA folded into a parallel G4 structure, a mechanism that enhanced peroxidase activity of myoglobin was expected to include direct binding of PEA to heme in myoglobin. Indeed, we observed that PEA1-20 folds into a parallel G4 structure (Figure 4) and hemin prefers stacking to the planar surface of the G-quartet. Therefore, to investigate whether PEA can bind to heme directly and whether the heme-binding property can be associated with the enhanced activity of PEA, we investigated the binding property of PEA to hemin (heme). The addition of PEA3-01, PEA1-20, and N24-07 to hemin dissolved in hemin reaction buffer containing 1% DMF and 0.05% Triton-X 100 significantly increased the peroxidase activity of hemin (Figure 6A, B). We also used two hemin-binding aptamers, CatG4 (5′-TGGGTAGGGCGGGTTGGGAAA-3′) and PS2.M (5′-GTGGGTAGGGCGGGTTGG-3′), as positive controls. The enhancement levels of the three PEAs in hemin peroxidase activity were equal to or higher than those of the DNAzymes (Figure 6A, B). Taken together, these data indicate that the PEA series can also enhance hemin-derived peroxidase activity. We analyzed the absorption spectrum of hemin to validate the interaction between PEA and hemin. Similar to CatG4 and PS2.M, hyperchromicity of the Soret band in the presence of PEA was also observed (Supplementary Figure S12), indicating the binding of PEA to hemin.
Figure 6.
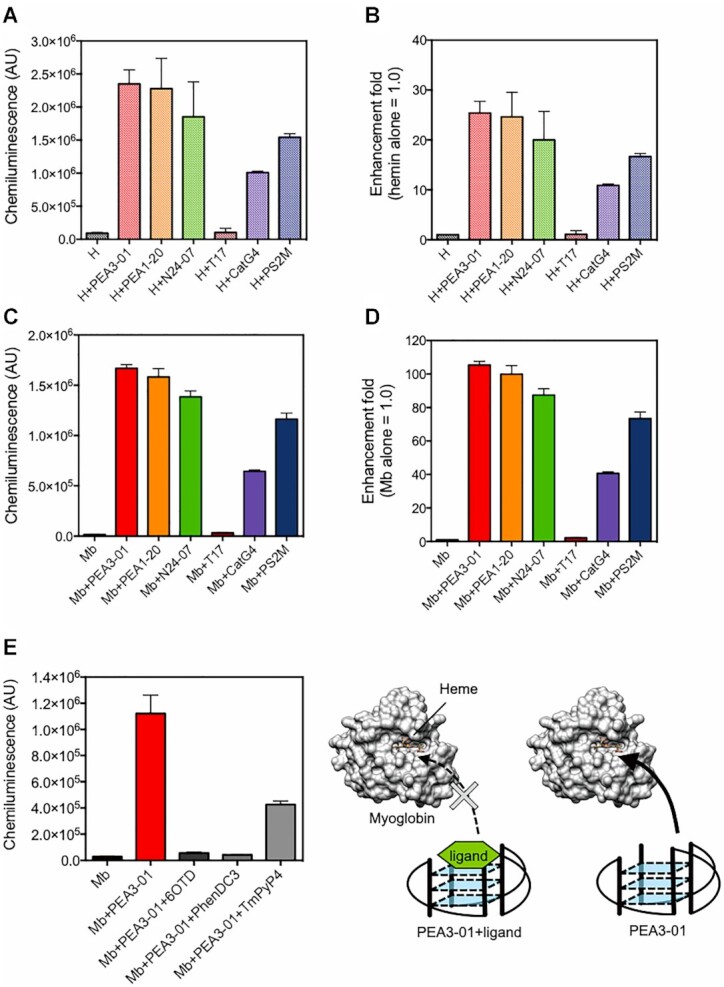
PEA series upregulated peroxidase activity of hemin, and hemin-binding aptamers enhanced chemiluminescent signal from luminol reaction catalyzed by myoglobin. T17 was used as a negative control. All measurements were performed in triplicate except for the data of hemin-T17 and hemin-CatG4 (one experimental result was lost for each) and the data are shown as mean ± SD in the graph. (A–B) 100 nM Hemin (H) was incubated with 200 nM PEA3-01, PEA1-20, N24-07, T17, CatG4 or PS2M in potassium acetate buffer (pH 5.0). Enhancement of the chemiluminescent signal corresponding to luminol substrate oxidation was evaluated (A). Relative enhancement fold (hemin alone = 1.0) was shown in (B). (C–D) Enhanced chemiluminescent signals with hemin-binding G4 aptamers, CatG4 and PS2.M. 100 nM myoglobin (Mb) was incubated with 200 nM PEA3-01, PEA1-20, N24-07, T17, CatG4 or PS2M in potassium acetate buffer (pH 5.0). Chemiluminescent signal (C) and relative enhancement (myoglobin alone = 1.0) (D) are shown. (E) Competitive assay using G4 ligands. Activity measurement was performed using 100 nM PEA3-01, 0 or 500 nM G4 specific ligands, and 100 nM myoglobin.
If the hemin-binding property of PEA is necessary for the activation of myoglobin, we hypothesized that hemin-binding aptamers, such as CatG4 and PS2.M, also work as an AES that enhances the peroxidase activity of myoglobin. Therefore, we examined the enhancement of CatG4 and PS2.M for chemiluminescence produced by the catalytic activity of myoglobin. In the presence of CatG4 and PS2.M, the chemiluminescence from myoglobin activity was significantly increased (Figure 6C, D), as expected. The PS2.M-myoglobin complex showed an almost equal signal to the N24-07-myoglobin complex. As for the comparison with PEA1-20 and PEA3-01, these two PEAs showed stronger enhancement in chemiluminescence than CatG4 and PS2.M. The higher signal from the PEAs may result from sequence/structure optimization by using ISM.
Furthermore, to investigate whether G4 in PEA plays an important role in activating myoglobin activity, a competitive assay using G4 ligands that specifically bind to the plane of G-quartet was carried out. When G4 ligands [6OTD (47), Phen-DC3 (48) and TmPyP4 (49)] were preincubated with PEA3-01 before addition to myoglobin, the enhancement activity of PEA3-01 was dramatically decreased (Figure 6E). Therefore, we concluded that PEA binds to heme, and the direct binding of PEA to heme in myoglobin may be involved in the mechanism of the enhanced activity.
Substrate specificity of enhanced peroxidase activity
To examine whether a chromogenic substrate can be catalyzed by the PEA-myoglobin complex, we attempted to determine the catalytic activity of the PEA-myoglobin complex for 3,3′,5,5′-tetramethylbenzidine (TMB). Notably, PEA3-01 and PEA1-20 did not enhance the catalytic activity of myoglobin for TMB (Figure 7A). Only N24-07, the AES selected using SELEX, showed twice the signal increase. In contrast, when PEA formed a complex with hemin, PEA3-01 and PEA1-20 enhanced hemin-derived peroxidase activity for TMB by 4- and 5-fold, respectively. Previously reported DNAzyme, a CatG4-hemin complex, oxidized TMB to a greater extent (Figure 7B), indicating that the experimental protocol was correct. This result indicates that the PEA developed by using ISM did not enhance myoglobin-derived activity for TMB. We also checked the colorimetric response of ABTS [2,2-azino-di-(3-ethylbenzthiazoline sulfonic acid)] to the PEA3-01-myoglobin complex; however, no enhancement was observed (data not shown). For luminol with enhancer (p-iodophenol), the PEA–myoglobin complex showed a luminol-concentration dependent signal (Figure 7C), which suggested that PEA selectively enhanced the catalytic activity of myoglobin against luminol. Finally, the PEA3-01-myoglobin complex showed a 300-fold higher chemiluminescent signal (3.1 × 106 ± 1.3 × 105 AU) than myoglobin alone (1.0 × 104 ± 6.6 × 102 AU) when using 100 μM of luminol and enhancer (Figure 7C).
Figure 7.
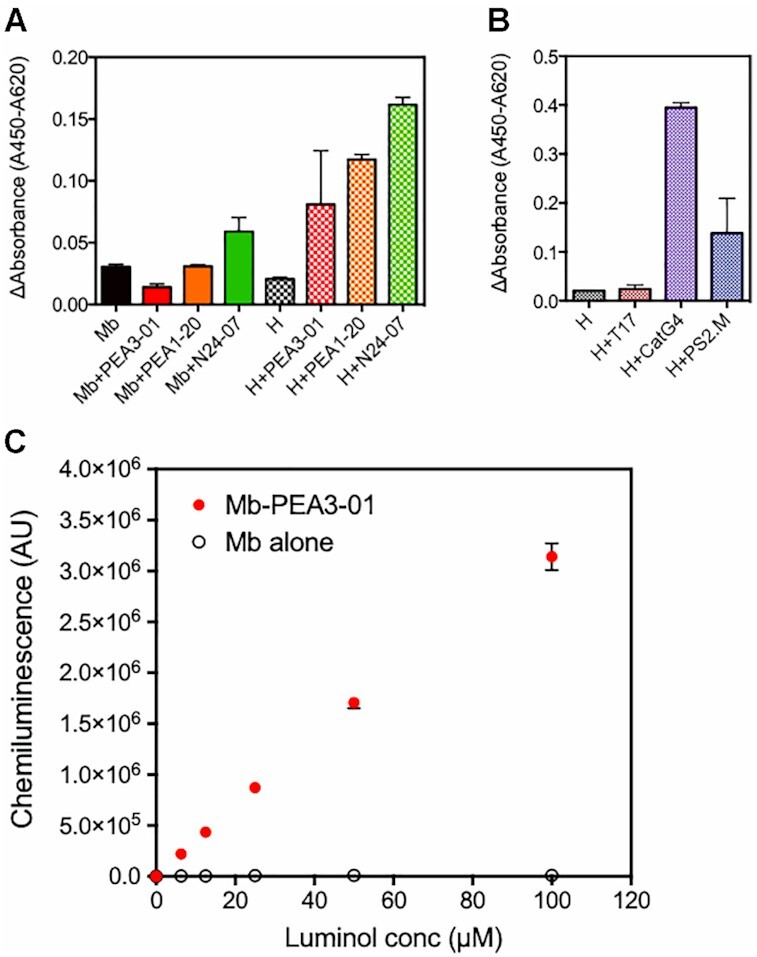
Substrate selectivity of PEA-myoglobin complex for TMB and luminol. All measurements were performed in triplicate and the data are shown as mean ± SD in the graph. (A) Catalytic activity of myoglobin (Mb)–PEA complex and hemin (H)-PEA complex for TMB. Change in absorption of TMB after addition of the complex (f.c. 100 nM) was measured. Myoglobin and hemin alone were also evaluated. (B) Hemin-hemin binding aptamer complex was used as a positive control of this activity measurement for TMB. T17 was used as a negative control. (C) Luminol concentration-dependency in the chemiluminescent signal from myoglobin and PEA3-01-myoglobin complex. Activity of 100 nM PEA3-01-myoglobin complex (red circle) and intact myoglobin (white circle) was measured under a series of luminol concentrations and 100 μM p-iodophenol.
DISCUSSION
Here, we discovered novel AESs that enabled the enhancement of the peroxidase activity of myoglobin through the formation of a complex between the aptamer and myoglobin. This is the first study to report the enhancement of the enzymatic activity of a protein by specific binding of DNA aptamer. We improved the enhancement activity of the peroxidase-activity-enhancing AES, named PEA, against the peroxidase activity of myoglobin by using ISM based on a set of G4-folding oligonucleotides. Finally, PEA3-01, generated using ISM, enhanced myoglobin activity significantly, resulting in a 300-fold increase in chemiluminescent signal from luminol.
We performed aptamer blotting experiments (Supplementary Figures S3, S4, S11) and succeeded in identifying the Kd value of PEA3-01 to myoglobin using SPR analysis (Supplementary Figure S10), ensuring binding of PEA to myoglobin. At its optimal pH, due to the high binding ability of PEA3-01 (Kd of 5.8 nM), 80% of PEA3-01 and myoglobin could be complexed at 100 nM in the solution. The molecular mechanism underlying the ability of AES to enhance the catalysis of luminol by myoglobin has not yet been identified, but we considered that the enhancement of chemiluminescence by the peroxidase reaction for luminol might be caused by the interaction of the aptamer with myoglobin. Meanwhile, we revealed that PEA3-01, PEA1-20 and N24-07 bound to hemin directly and activated hemin peroxidase activity, so that G4 DNAzyme-hemin-type interaction may be involved in the enhancement mechanism of PEA. Previous studies that investigated the role of G4 DNAzyme in enhancing hemin-derived peroxidase activity suggested that a G-quartet (27,55) or adenine (56) of the G4 DNAzyme may serve as the proximal ligand, similar to the role of the histidine residue in HRP, resulting in facilitation of the peroxidase reaction. According to the solved structure of PEA1-20 (Figure 4), PEA1-20 has a flat G-quartet and extended loops, which can serve as the proximal ligand likely G4 DNAzymes. CD spectrum analysis of 80 AES candidates (Supplementary Figures S5–S8) also indicated that PEA might fold into the G4 structure like PEA1-20. Additionally, blocking the G-quartet plane of PEA with small molecular ligands significantly reduced the enhancing activity of PEA (Figure 6E). Supplementary Figure S13 shows the structure of myoglobin and PEA1-20 on the same scale, providing insight into the potential of PEA1-20 to interact with surface-exposed heme in myoglobin via the loops or G-quartet plane of PEA. Therefore, the enhancement of luminol chemiluminescence by myoglobin may be attributed to the presence of functional groups that function as proton acceptors or donors in myoglobin-binding G4 aptamers. Hemin-binding aptamers, CatG4 and PS2.M, also significantly increased myoglobin activity for luminol (Figure 6C and D), supporting this hypothesis. The enhancement level of the hemin-binding aptamer was lower than that of the PEAs developed using ISM and also than that of the original AES N24-07 (Figure 6D). This result also suggested that interaction with heme may be essential for enhancing activity, but binding ability to myoglobin itself would be very important to exert a marked enhancing effect in the catalysis of luminol.
Our results indicate the importance of G4–heme–myoglobin interaction for AES properties. Regarding the mechanism of PEA-enhancing luminol oxidation, we assumed a possibility that contributed to the repositioning of amino acid residues around the myoglobin-pseudoperoxidase active center by the binding of PEA, too. As described in introduction and as shown in Supplementary Figure S1, myoglobin has two histidine residues and an arginine residue near the sixth coordination site of the Fe ion in the heme, which are required by amino acids to catalyze the oxidation of substrates. Binding of the PEA to myoglobin may allosterically alter the location of parts of myoglobin, including those histidine and arginine residues.
When we compared the enzymatic properties of the myoglobin–PEA complex to that of hemin-G4 DNAzyme, the myoglobin–PEA complex selectively showed marked signal enhancement selectively for luminol, not TMB or ABTS. Because N24-07 showed a weak enhancing effect on the myoglobin peroxidase activity for TMB (Figure 7A), we assumed that this distinctive selectivity for luminol was potentially caused by the ISM study performed with luminol as a substrate. The reason for selective enhancement of the peroxidase activity of luminol by PEA has not yet been elucidated. However, we hypothesize that because the heme in the myoglobin–PEA complex is surrounded by not only DNA but also the protein framework, accessibility of the secondary substrate to heme in the myoglobin–PEA complex may be different from that in hemin-DNAzyme. In addition, the AES for myoglobin has a remarkable enhancement of the peroxidase activity of myoglobin for luminol rather than the hemin-binding aptamer for hemin. PEA3-01 upregulated hemin-derived chemiluminescence by up to 25-fold (Figure 6B). On the other hand, PEA3-01 showed up to a 105-fold increase in myoglobin under the same experimental conditions (Figure 6B). When we used 100 μM of luminol and enhancer, PEA3-01 enhanced chemiluminescence up to 300-fold (Figure 7C). Although additional experiments are still required, the peculiar environment around the heme, which is formed by binding of DNA to myoglobin, may strongly relate to this large enhancement of the PEA and the substrate specificity of the PEA–myoglobin complex for luminol. Therefore, a study combining aptamers and enzymes would be promising for the production of advanced enzyme engineering technologies with allosteric regulation of enzymatic activity.
Solving the 3D structure of the AES is crucial for a rational design of the aptameric biosensing system employing the myoglobin-activating AES likely hemin-binding DNA aptamers. We, therefore, solved the NMR structure of PEA and conducted structural investigation by CD spectrum analysis along with the ISM. The solved NMR structure of PEA1-20 (the best PEA in the first round of ISM) had a monomeric, parallel, and square planar arrangement with three overhanging loops (Figure 4). Comparison of NMR spectrum also revealed that the structural property of PEA3-01 is close to that of PEA1-20 and CD spectrum analysis strongly suggested that PEA3-01 folded into the parallel G4 structure, like PEA1-20. In addition to the NMR study, we explored the relationship in the structural-activity of 80 sequences produced via four cycles of the ISM and discovered that the parallel type of G4-forming aptamer could enhance the peroxidase activity of myoglobin. This topological feature of the G4 was highly conserved in the top ten aptamers from ISM. Therefore, the active PEA adopts a conformation of the parallel G4 structure.
In general, sequence similarity is important for estimating the function of an aptamer during aptamer development. However, in this study, we found that the sequence similarity of AES may not be important in the expression of the enhancement ability. For example, an PEA candidate, PEA4-17 (5′-GGGCGGGAAGGGCTGGG-3′), a loop mutant of PEA3-01 (5′-GGGCGGGTTGGGCTGGG-3′) that has its second ‘TT’ loop replaced with AA, which has a small difference in sequence, considerably lost its activation property and parallel-type G4 folding when compared to PEA3-01 (Supplementary Figure S8 and Supplementary Data). In addition, the sequence similarity of the top ten PEA was not observed. Taken together, our findings indicate that the formation of parallel G4 is the most crucial contributor to the complementation of the myoglobin peroxidase activity. This observation may indicate the possibility that parallel G4 DNAs such as telomere G4 and promoter G4 are involved in physiological processes that enhance the peroxidase reaction. Thus, from this perspective and the standpoint of biosensor development, contamination of unknown parallel G4 oligonucleotides to the analyte may interfere with enhancement of PEA activity by competition. Removal of DNA from samples might be required to reduce the interference to the AES-myoglobin signal system. Since PEA3-01 showed a Kd of 5.8 nM for myoglobin, the use of PEA3-01 would avoid competition with other interfering G4. Importantly, PEA itself did not show any peroxidase activity. Therefore, a larger number of PEAs can be used as a more sensitive signal amplifier without increasing the background signal.
In conclusion, we developed novel AESs, oligonucleotide-based enzyme regulators to control the peroxidase activity by binding to myoglobin. This AES would offer a new strategy for future studies on biosensor applications. When the parallel G4 formation of AES is controlled by combining target-responding modification such as the insertion of another aptamer, myoglobin's activity-based chemiluminescent aptasensors could be developed. Our finding of a DNA aptamer that enhances the enzymatic activity of a protein by its specific binding activity also adds important knowledge to understanding the control of the catalytic reaction by protein–DNA complexes.
DATA AVAILABILITY
Atomic coordinates and structural factors have been deposited with the Protein Data bank under accession number 7E5P.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Professor Kazuo Nagasawa (Tokyo University of Agriculture and Technology) for providing 6OTD. We are grateful to Professor Stefano Ferri (Shizuoka University) for his helpful comments on the title of the paper.
Contributor Information
Kaori Tsukakoshi, Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo 184-8588, Japan.
Yasuko Yamagishi, Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo 184-8588, Japan.
Mana Kanazashi, DENSO CORPORATION, 1-1 Showa-cho, Kariya, Aichi 448-8661, Japan.
Kenta Nakama, Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo 184-8588, Japan.
Daiki Oshikawa, Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo 184-8588, Japan.
Nasa Savory, Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo 184-8588, Japan.
Akimasa Matsugami, Advanced NMR Application and Platform Team, NMR Research and Collaboration Group, NMR Science and Development Division, RIKEN SPring-8 Center, 1-7-22 Suehiro-cho, Tsurumi, Yokohama, Kanagawa 230-0045, Japan.
Fumiaki Hayashi, Advanced NMR Application and Platform Team, NMR Research and Collaboration Group, NMR Science and Development Division, RIKEN SPring-8 Center, 1-7-22 Suehiro-cho, Tsurumi, Yokohama, Kanagawa 230-0045, Japan.
Jinhee Lee, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Chapel Hill, NC 27599, USA.
Taiki Saito, Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo 184-8588, Japan.
Koji Sode, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Chapel Hill, NC 27599, USA.
Kanjana Khunathai, DENSO CORPORATION, 1-1 Showa-cho, Kariya, Aichi 448-8661, Japan.
Hitoshi Kuno, DENSO CORPORATION, 1-1 Showa-cho, Kariya, Aichi 448-8661, Japan.
Kazunori Ikebukuro, Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo 184-8588, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Japan Agency for Medical Research and Development (AMED)-SENTAN program. Funding for open access charge: Tokyo University of Agriculture and Technology.
Conflict of interest statement. M.K., K.K. and H.K. are employees of DENSO CORPORATION. The remaining authors declare no conflict of interests.
REFERENCES
- 1. Ellington A.D., Szostak J.W.. Invitro selection of Rna molecules that bind specific ligands. Nature. 1990; 346:818–822. [DOI] [PubMed] [Google Scholar]
- 2. Tuerk C., Gold L.. Systematic evolution of ligands by exponential enrichment - RNA ligands to bacteriophage-T4 DNA-polymerase. Science. 1990; 249:505–510. [DOI] [PubMed] [Google Scholar]
- 3. Bock L.C., Griffin L.C., Latham J.A., Vermaas E.H., Toole J.J.. Selection of single-stranded-DNA molecules that bind and inhibit human thrombin. Nature. 1992; 355:564–566. [DOI] [PubMed] [Google Scholar]
- 4. Dang C., Jayasena S.D.. Oligonucleotide inhibitors of Taq DNA polymerase facilitate detection of low copy number targets by PCR. J. Mol. Biol. 1996; 264:268–278. [DOI] [PubMed] [Google Scholar]
- 5. Urvil P.T., Kakiuchi N., Zhou D.M., Shimotohno K., Kumar P.K.R., Nishikawa S.. Selection of RNA aptamers that bind specifically to the NS3 protease of hepatitis C virus. Eur. J. Biochem. 1997; 248:130–138. [DOI] [PubMed] [Google Scholar]
- 6. Ikebukuro K., Okumura Y., Sumikura K., Karube I.. A novel method of screening thrombin-inhibiting DNA aptamers using an evolution-mimicking algorithm. Nucleic Acids Res. 2005; 33:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kato K., Ikeda H., Miyakawa S., Futakawa S., Nonaka Y., Fujiwara M., Okudaira S., Kano K., Aoki J., Morita J.et al.. Structural basis for specific inhibition of Autotaxin by a DNA aptamer. Nat. Struct. Mol. Biol. 2016; 23:395–401. [DOI] [PubMed] [Google Scholar]
- 8. Donkor D.A., Bhakta V., Eltringham-Smith L.J., Stafford A.R., Weitz J.I., Sheffield W.P.. Selection and characterization of a DNA aptamer inhibiting coagulation factor XIa. Sci. Rep. 2017; 7:2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miropolskaya N., Feklistov A., Nikiforov V., Kulbachinskiy A.. Site-specific aptamer inhibitors of Thermus RNA polymerase. Biochem. Biophys. Res. Commun. 2018; 495:110–115. [DOI] [PubMed] [Google Scholar]
- 10. Jung J.I., Han S.R., Lee S.W.. Development of RNA aptamer that inhibits methyltransferase activity of dengue virus. Biotechnol. Lett. 2018; 40:315–324. [DOI] [PubMed] [Google Scholar]
- 11. Malicki S., Ksiazek M., Majewski P., Pecak A., Mydel P., Grudnik P., Dubin G.. Development of a novel, high-affinity ssDNA trypsin inhibitor. J. Enzyme Inhib. Med. Chem. 2019; 34:638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang L., Lee J.Y., Gao L., Yin J., Duan Y., Jimenez L.A., Adkins G.B., Ren W., Li L., Fang J.et al.. A DNA aptamer for binding and inhibition of DNA methyltransferase 1. Nucleic Acids Res. 2019; 47:11527–11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Padmanabhan K., Padmanabhan K.P., Ferrara J.D., Sadler J.E., Tulinsky A.. The structure of alpha-thrombin inhibited by a 15-mer single-stranded DNA aptamer. J. Biol. Chem. 1993; 268:17651–17654. [DOI] [PubMed] [Google Scholar]
- 14. Ponce A.T., Hong K.L.. A mini-review: clinical development and potential of aptamers for thrombotic events treatment and monitoring. Biomedicines. 2019; 7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshida W., Sode K., Ikebukuro K.. Aptameric enzyme subunit for biosensing based on enzymatic activity measurement. Anal. Chem. 2006; 78:3296–3303. [DOI] [PubMed] [Google Scholar]
- 16. Yoshida W., Sode K., Ikebukuro K.. Homogeneous DNA sensing using enzyme-inhibiting DNA aptamers. Biochem. Biophys. Res. Commun. 2006; 348:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ikebukuro K., Yoshida W., Sode K.. Aptameric enzyme subunit for homogeneous DNA sensing. Biotechnol. Lett. 2008; 30:243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoshida W., Sode K., Ikebukuro K.. Label-free homogeneous detection of immunoglobulin E by an aptameric enzyme subunit. Biotechnol. Lett. 2008; 30:421–425. [DOI] [PubMed] [Google Scholar]
- 19. Yoshida W., Mochizuki E., Takase M., Hasegawa H., Morita Y., Yamazaki H., Sode K., Ikebukuro K.. Selection of DNA aptamers against insulin and construction of an aptameric enzyme subunit for insulin sensing. Biosens. Bioelectron. 2009; 24:1116–1120. [DOI] [PubMed] [Google Scholar]
- 20. Park K.S., Lee C.Y., Park H.G.. Target DNA induced switches of DNA polymerase activity. Chem. Commun. (Camb.). 2015; 51:9942–9945. [DOI] [PubMed] [Google Scholar]
- 21. Jung Y., Lee C.Y., Park K.S., Park H.G.. Sensitive and specific detection of proteins based on target-responsive DNA polymerase activity. Anal. Chim. Acta. 2019; 1059:80–85. [DOI] [PubMed] [Google Scholar]
- 22. Travascio P., Li Y.F., Sen D.. DNA-enhanced peroxidase activity of a DNA aptamer-hemin complex. Chem. Biol. 1998; 5:505–517. [DOI] [PubMed] [Google Scholar]
- 23. Ruttkay-Nedecky B., Kudr J., Nejdl L., Maskova D., Kizek R., Adam V.. G-quadruplexes as sensing probes. Molecules. 2013; 18:14760–14779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kosman J., Juskowiak B.. Seitz H., Stahl F., Walter J-.G.. Catalytically Active Nucleic Acids. 2020; Cham: Springer International Publishing; 59–84. [Google Scholar]
- 25. Cheng M.P., Zhou J., Jia G.Q., Ai X.J., Mergny J.L., Li C.. Relations between the loop transposition of DNA G-quadruplex and the catalytic function of DNAzyme. BBA-Gen. Subj. 2017; 1861:1913–1920. [DOI] [PubMed] [Google Scholar]
- 26. Shibata T., Nakayama Y., Katahira Y., Tai H., Moritaka Y., Nakano Y., Yamamoto Y.. Characterization of the interaction between heme and a parallel G-quadruplex DNA formed from d(TTGAGG). BBA-Gen. Subj. 2017; 1861:1264–1270. [DOI] [PubMed] [Google Scholar]
- 27. Shinomiya R., Katahira Y., Araki H., Shibata T., Momotake A., Yanagisawa S., Ogura T., Suzuki A., Neya S., Yamamoto Y.. Characterization of catalytic activities and heme coordination structures of heme-DNA complexes composed of some chemically modified hemes and an all parallel-stranded tetrameric G-quadruplex DNA formed from d(TTAGGG). Biochemistry-US. 2018; 57:5930–5937. [DOI] [PubMed] [Google Scholar]
- 28. Tian Y., He Y., Mao C.. Cascade signal amplification for DNA detection. ChemBioChem. 2006; 7:1862–1864. [DOI] [PubMed] [Google Scholar]
- 29. Kosman J., Juskowiak B.. Peroxidase-mimicking DNAzymes for biosensing applications: a review. Anal. Chim. Acta. 2011; 707:7–17. [DOI] [PubMed] [Google Scholar]
- 30. Duellman T., Chen X., Wakamiya R., Yang J.. Nucleic acid-induced potentiation of matrix metalloproteinase-9 enzymatic activity. Biochem. J. 2018; 475:1597–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berglund G.I., Carlsson G.H., Smith A.T., Szoke H., Henriksen A., Hajdu J.. The catalytic pathway of horseradish peroxidase at high resolution. Nature. 2002; 417:463–468. [DOI] [PubMed] [Google Scholar]
- 32. Ozaki S., Matsui T., Roach M.P., Watanabe Y.. Rational molecular design of a catalytic site: engineering of catalytic functions to the myoglobin active site framework. Coordin Chem Rev. 2000; 198:39–59. [Google Scholar]
- 33. Matsui T., Ozaki S., Liong E., Phillips G.N. Jr, Watanabe Y.. Effects of the location of distal histidine in the reaction of myoglobin with hydrogen peroxide. J. Biol. Chem. 1999; 274:2838–2844. [DOI] [PubMed] [Google Scholar]
- 34. Du J., Huang X., Sun S., Wang C., Lebioda L., Dawson J.H.. Amphitrite ornata dehaloperoxidase (DHP): investigations of structural factors that influence the mechanism of halophenol dehalogenation using “peroxidase-like” myoglobin mutants and “myoglobin-like” DHP mutants. Biochemistry-Us. 2011; 50:8172–8180. [DOI] [PubMed] [Google Scholar]
- 35. Cai Y.B., Li X.H., Jing J., Zhang J.L.. Effect of distal histidines on hydrogen peroxide activation by manganese reconstituted myoglobin. Metallomics. 2013; 5:828–835. [DOI] [PubMed] [Google Scholar]
- 36. Wu L.B., Du K.J., Nie C.M., Gao S.Q., Wen G.B., Tan X.S., Lin Y.W.. Peroxidase activity enhancement of myoglobin by two cooperative distal histidines and a channel to the heme pocket. J. Mol. Catal. B-Enzym. 2016; 134:367–371. [Google Scholar]
- 37. Hountondji C., Crechet J.B., Tanaka M., Suzuki M., Nakayama J.I., Aguida B., Bulygin K., Cognet J., Karpova G., Baouz S.. Ribosomal protein eL42 contributes to the catalytic activity of the yeast ribosome at the elongation step of translation. Biochimie. 2019; 158:20–33. [DOI] [PubMed] [Google Scholar]
- 38. Savory N., Abe K., Sode K., Ikebukuro K.. Selection of DNA aptamer against prostate specific antigen using a genetic algorithm and application to sensing. Biosens. Bioelectron. 2010; 26:1386–1391. [DOI] [PubMed] [Google Scholar]
- 39. Ikebukuro K., Yoshida W., Noma T., Sode K.. Analysis of the evolution of the thrombin-inhibiting DNA aptamers using a genetic algorithm. Biotechnol. Lett. 2006; 28:1933–1937. [DOI] [PubMed] [Google Scholar]
- 40. Varani G., Aboulela F., Allain F.H.T.. NMR investigation of RNA structure. Prog. Nucl. Mag. Res. Sp. 1996; 29:51–127. [Google Scholar]
- 41. Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A.. Nmrpipe - a multidimensional spectral processing system based on unix pipes. J. Biomol. NMR. 1995; 6:277–293. [DOI] [PubMed] [Google Scholar]
- 42. Goddard T.D., Kneller D.G.. 3.115 ed. 2007; San Fransico: University of California. [Google Scholar]
- 43. Matsugami A., Ouhashi K., Kanagawa M., Liu H., Kanagawa S., Uesugi S., Katahira M.. An intramolecular quadruplex of (GGA)(4) triplet repeat DNA with a G:G:G:G tetrad and a G(:A):G(:A):G(:A):G heptad, and its dimeric interaction. J. Mol. Biol. 2001; 313:255–269. [DOI] [PubMed] [Google Scholar]
- 44. Matsugami A., Okuizumi T., Uesugi S., Katahira M.. Intramolecular higher order packing of parallel quadruplexes comprising a G:G:G:G tetrad and a G(:A):G(:A):G(:A):G heptad of GGA triplet repeat DNA. J. Biol. Chem. 2003; 278:28147–28153. [DOI] [PubMed] [Google Scholar]
- 45. Schwieters C.D., Kuszewski J.J., Tjandra N., Clore G.M.. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003; 160:65–73. [DOI] [PubMed] [Google Scholar]
- 46. Shumayrikh N., Sen D.. Heme*G-quadruplex DNAzymes: conditions for maximizing their peroxidase activity. Methods Mol. Biol. 2019; 2035:357–368. [DOI] [PubMed] [Google Scholar]
- 47. Tera M., Ishizuka H., Takagi M., Suganuma M., Shin-ya K., Nagasawa K.. Macrocyclic hexaoxazoles as sequence- and mode-selective G-quadruplex binders. Angew. Chem. Int. Ed. Engl. 2008; 47:5557–5560. [DOI] [PubMed] [Google Scholar]
- 48. De Cian A., Delemos E., Mergny J.L., Teulade-Fichou M.P., Monchaud D.. Highly efficient G-quadruplex recognition by bisquinolinium compounds. J. Am. Chem. Soc. 2007; 129:1856–1857. [DOI] [PubMed] [Google Scholar]
- 49. Phan A.T., Kuryavyi V., Gaw H.Y., Patel D.J.. Small-molecule interaction with a five-guanine-tract G-quadruplex structure from the human MYC promoter. Nat. Chem. Biol. 2005; 1:167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mousivand M., Anfossi L., Bagherzadeh K., Barbero N., Mirzadi-Gohari A., Javan-Nikkhah M.. In silico maturation of affinity and selectivity of DNA aptamers against aflatoxin B1 for biosensor development. Anal. Chim. Acta. 2020; 1105:178–186. [DOI] [PubMed] [Google Scholar]
- 51. Navien T.N., Thevendran R., Hamdani H.Y., Tang T.H., Citartan M.. In silico molecular docking in DNA aptamer development. Biochimie. 2021; 180:54–67. [DOI] [PubMed] [Google Scholar]
- 52. Yokoyama T., Tsukakoshi K., Yoshida W., Saito T., Teramoto K., Savory N., Abe K., Ikebukuro K.. Development of HGF-binding aptamers with the combination of G4 promoter-derived aptamer selection and in silico maturation. Biotechnol. Bioeng. 2017; 114:2196–2203. [DOI] [PubMed] [Google Scholar]
- 53. Puett D. The equilibrium unfolding parameters of horse and sperm whale myoglobin. Effects of guanidine hydrochloride, urea, and acid. J. Biol. Chem. 1973; 248:4623–4634. [PubMed] [Google Scholar]
- 54. Schenkman K.A., Marble D.R., Burns D.H., Feigl E.O.. Myoglobin oxygen dissociation by multiwavelength spectroscopy. J. Appl. Physiol. 1997; 82:86–92. [DOI] [PubMed] [Google Scholar]
- 55. Shumayrikh N., Huang Y.C., Sen D.. Heme activation by DNA: isoguanine pentaplexes, but not quadruplexes, bind heme and enhance its oxidative activity. Nucleic Acids Res. 2015; 43:4191–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li W., Li Y., Liu Z., Lin B., Yi H., Xu F., Nie Z., Yao S.. Insight into G-quadruplex-hemin DNAzyme/RNAzyme: adjacent adenine as the intramolecular species for remarkable enhancement of enzymatic activity. Nucleic Acids Res. 2016; 44:7373–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structural factors have been deposited with the Protein Data bank under accession number 7E5P.