


Abstract
Nuclear architecture influences gene regulation and cell identity by controlling the three-dimensional organization of genes and their distal regulatory sequences, which may be far apart in linear space. The genome is functionally and spatially segregated in the eukaryotic nucleus with transcriptionally active regions in the nuclear interior separated from repressive regions, including those at the nuclear periphery. Here, we describe the identification of a novel type of nuclear peripheral chromatin domain that is enriched for tissue-specific transcriptional enhancers. Like other chromatin at the nuclear periphery, these regions are marked by H3K9me2. But unlike the nuclear peripheral Lamina-Associated Domains (LADs), these novel, enhancer-rich domains have limited Lamin B interaction. We therefore refer to them as H3K9me2-Only Domains (KODs). In mouse embryonic stem cells, KODs are found in Hi-C-defined A compartments and feature relatively accessible chromatin. KODs are characterized by low gene expression and enhancers located in these domains bear the histone marks of an inactive or poised state. These results indicate that KODs organize a subset of inactive, tissue-specific enhancers at the nuclear periphery. We hypothesize that KODs may play a role in facilitating and perhaps constraining the enhancer-promoter interactions underlying spatiotemporal regulation of gene expression programs in differentiation and development.
Graphical Abstract
Graphical Abstract.
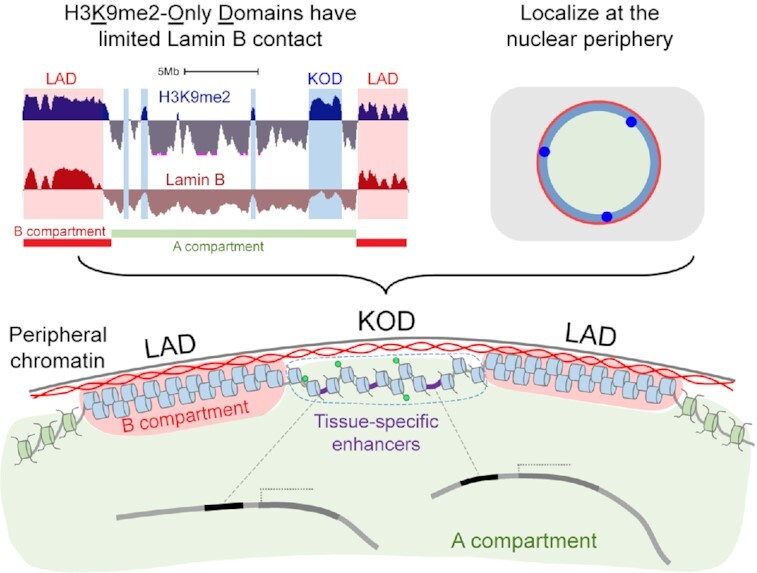
Histone H3K9me2-Only Domains (KODs) represent a subclass of peripheral chromatin with minimal nuclear lamina contact and enrichment for tissue-specific enhancers.
INTRODUCTION
The precise arrangement of the genome in the 3D space of the eukaryotic nucleus is nonrandom and plays a critical role in establishing and maintaining cellular identity (1–5). One of the most prominent aspects of nuclear architecture is the segregation of transcriptionally active euchromatin and repressed heterochromatin into separate compartments of the nucleus. Early microscopy studies of eukaryotic nuclei identified stereotypical characteristics of chromatin compaction and localization, including observations of euchromatin in the nuclear interior and heterochromatin at the nuclear periphery (6,7). Heterochromatin contains densely packed nucleosomes with repressive histone modifications (8,9) and tends to be transcriptionally silent. In contrast, euchromatin features more loosely packed histones marked with modifications associated with active gene transcription, accessibility to the DNA and higher levels of transcriptional activity. Spatial partitioning of these functionally distinct types of chromatin is also evident in interaction maps generated by chromosome conformation capture technologies such as Hi-C (10). Hi-C maps segregate the genome into two major spatial compartments: compartment A has a greater number of accessible regions with active histone modifications that tend to cluster together; compartment B has more compact chromatin with less accessibility and histone modifications associated with transcriptional repression. Regions from each of these different compartments are more likely to interact or cluster with those of the same compartment type. Such chromatin compartmentalization allows genomic regions that may be far apart along the linear genome to interact with each other or with substructures in the nucleus. These interactions have been shown to change during cell differentiation (11–13).
In addition to A/B compartment segregation, the genome is organized through anchoring to substructures of the nucleus such as the nuclear lamina. In metazoan cells, a large fraction of chromatin is found just inside the nuclear envelope, where it interacts with the nuclear lamina and its associated proteins. Here, heterochromatin forms extended regions of hundreds of kilobases called Lamina-Associated Domains (LADs). LADs were originally identified using the Dam-ID method which labels genomic regions that are in contact with nuclear lamina proteins including Lamin B1 (3,14). LADs have also been mapped through chromatin immunoprecipitation (ChIP) of Lamin B-interacting DNA isolated with an anti-Lamin B antibody (1,15,16). Multiple studies have demonstrated that approximately one-third of the genome is in LADs and that they are associated with different regions in different cell types. LADs often overlap B compartment designations and share the repressive histone modifications, nucleosome density, and reduced gene transcription of heterochromatin (17,18). Comparisons of LAD organization among different cell types revealed that some, so-called constitutive LADs, are conserved among cell types and in syntenic regions of different species (19), suggesting a functional role in spatial organization of the genome. However, not all LADs are constitutive. It has been documented that changes in chromatin-lamina interactions accompany differentiation (3,20–22) and that disruption of normal chromatin-lamina interactions during mouse embryonic stem cell (mESC) differentiation can lead to aberrant cell-type specification (1,23). The dynamics of chromatin localization have also been linked to gene expression. Multiple studies report chromatin detachment from the nuclear periphery leading to gene activation as well as increased chromatin-lamina contacts correlating with gene repression (1,3,20,24–26). Localization of chromatin at the nuclear lamina has been associated with strong, but not absolute, transcriptional repression (27). These findings demonstrate the important role of chromatin organization at the nuclear periphery in maintaining and/or regulating gene programs.
Not all chromatin at the nuclear periphery shares the same characteristics. Histone H3 post-translational modifications H3K9me2 and H3K9me3 have been shown to be enriched in LADs and are often associated with transcriptional repression (1,3,14). H3K9me3-marked chromatin is found both at the nuclear periphery and in clusters in the nuclear interior where it labels highly repetitive, pericentric heterochromatin (28,29). H3K9me2, on the other hand, is a histone modification specific for chromatin that localizes to the nuclear periphery in species from C. elegans to humans (1,30–32). Genetic studies in C. elegans have provided evidence that H3K9me1/2 mediates chromatin anchoring at the nuclear periphery whereas H3K9me3 is required for transcriptional repression (33). Studies in mammalian cells have demonstrated that targeted H3K9me2 modification of a chosen chromatin region promotes its localization to the nuclear periphery (34), while disruption of chromatin-nuclear lamina interaction through targeted VP16 activation was shown to reposition the region to the nuclear interior (35) and reportedly reduce H3K9me2 (36). Together, these findings implicate H3K9me2 as a mark of chromatin specified for nuclear peripheral localization. This is consistent with the previous observation that H3K9me2-marked chromatin regions are highly correlated with LADs (1).
Here, we present evidence that a portion of H3K9me2-marked chromatin forms large domains that are localized at the nuclear periphery but are physically separate and epigenetically distinct from LADs. These H3K9me2-Only Domains (KODs) have minimal interaction with Lamin B, are depleted for H3K9me3, and feature accessible chromatin and acetylated histones. Notably, KODs are enriched for distal enhancers of transcription and the histone modifications that define them. We propose a model in which KODs serve as a scaffold to organize enhancers and influence long-range chromatin interactions through H3K9me2-mediated nuclear positioning.
MATERIALS AND METHODS
Cell lines
Murine embryonic multipotent stem cell was obtained from the American Type Culture Collection (ATCC, cat# CRL-1934). All cells tested negative for mycoplasma contamination. Mouse ES cells were maintained at 37°C on a feeder layer of mitotically inactivated MEFs in DMEM with 15% FBS (Fisher Scientific #SH3007003) and ESGRO LIF (EMD Millipore, cat#ESG1106).
Immunofluorescence and DNA oligo FISH
Mouse ESCs were grown on 0.1% porcine gelatin (Sigma, cat#G2500) coated glass coverslips (EMS, cat#3406), mESCs were fixed with 2% PFA for 10 min at RT, washed three times with DPBS (Gibco, cat#14190-136), then permeabilized with 0.5% Triton X-100 (Thermo Scientific, cat#28314) for 10 min. After permeabilization, cells were washed three times with DPBS for 5 min, then blocked in 1% BSA (Sigma, cat#A4503) in PBST (DPBS with 0.05% Tween 20, pH 7.4 (Thermo Scientific, cat#28320)) for 60 min at RT. After block samples were incubated with primary antibodies for 1 h at RT and washed three times with PBST for 5 min. Then incubated with secondary antibodies for 30 min at RT. After incubation with secondary antibodies, samples were washed with DPBS and post-fixed with 2% PFA for 10 min at RT, washed 3 times with DPBS and permeabilized with 0.7% Triton X-100 for 10 min at RT and rinsed with DPBS. Next sample were prepared for hybridization with DNA oligo probes. Coverslips were incubated in 70% ethanol, 90% ethanol and 100% ethanol for 2 min each, then washed in 2× SSC (Corning, cat# 46-020-CM) for 5 min. Coverslips were equilibrated in 2× SSCT (2× SSC with 0.1% Tween) with 50% Formamide for 5 min at RT. DNA denaturation was performed in 2× SSCT with 50% Formamide for 2–3 min at 92°C in water bath, then additional 20 min at 60°C in water bath. After DNA denaturation, coverslips were cooled to RT in humid conditions for 2–3 min. Coverslips were places on a slide with 10–20 μl of hybridization mix (50% Formamide, 10% dextran sulfate, 50–100 pmol primary DNA oligo probes). Coverslips were heated at 92°C for 3 min on a heat block, then incubated overnight at 37°C in a humid chamber. After hybridization with primary DNA oligo probes coverslips were washed in 2× SSCT for 15 min at 60°C, then for 10 min in 2× SSCT for 10 min at RT, and then transferred in 2× SSC for 5 min. Next, coverslips were hybridized with a secondary fluorescent DNA oligo probes (10% Formamide, 10% dextran sulfate, 10 pmol secondary DNA probes) in dark humidified chamber for 3 h at RT. After secondary hybridization coverslips were washed for 5 min in 2× SSCT and 2× SSC buffers at RT. Cells were counterstained with DAPI solution 400 ng/ml (Sigma, cat#D9542) for 10 min at RT, then rinsed with PBS. Coverslips were mounted on slides using 80% glycerol mounting media: 80% glycerol (Invitrogen, cat# 15514-011), 0.1% sodium azide (Sigma, cat#S2002), 0.5% propyl gallate (Sigma, cat# 02370), 20 mM Tris–HCl, pH 8.0 (Invitrogen, cat#15568–025). This protocol was adopted from previously published protocols (30,37). Probes were designed as described previously (30,37).
Antibodies
Antibodies against H3K9me2 (Active Motif, cat# 39239) and Lamin B (Santa Cruz, cat# sc-6217) were previously tested for specificity (1).
Image acquisition and analysis
All confocal immunofluorescent and oligoFISH images were taken with a Leica TCS SP8 3X STED confocal microscopes using 63×/1.40 oil objective. DAPI staining (blue channel) were acquired using a PMT detector with offset –0.1%. All other fluorescent staining (green, red and far red channels) were acquired using HyD detectors in the standard mode with 100% gain. All images were taken with minimal laser power to avoid saturation. 3D images were taken as Z-stacks with 0.05 μm intervals. Confocal 3D images were deconvoluted using Huygens Professional software using the microscope parameters, standard PSF and automatic settings for background estimation. Confocal channel shift alignment was performed using 0.1 μm TetraSpeck fluorescent beads (Invitrogen, cat# T7279).
Confocal images on figures are representative images of a single focal plane. 3D image reconstructions were performed using Imaris 9.0.1 software (Bitplane AG, Switzerland) as described before (1,30). In brief, nuclear lamina, nuclear DNA volume, and H3K9me2-marked chromatin structure were created using Surfaces tool with automatic settings based on the fluorescent signals from the anti-Lamin B, DAPI staining, and anti-H3K9me2 antibodies. DNA oligo probe spots were identified using the Spots tool with a 300 nm diameter, created at the intensity mass center of the fluorescent probe signal. Distance from the center of the DNA oligo FISH spot to the edge of the nuclear lamina surface was quantified using the Distance Transformation tool. In cases when the DNA oligo FISH signal was imbedded into the generated nuclear lamina surface, the measurement returned negative distances. If the distance from the DNA oligo FISH spot to the nuclear lamina was smaller than (or equal to) the average thickness of peripheral chromatin, then the spot was counted as localized to nuclear periphery. The thickness of the H3K9me2-marked peripheral chromatin layer in mESC was measured previously (1) and equals 0.3 μm (distance from the H3K9me2 surface inner edge to the nuclear lamina surface inner edge.
LaminB and H3K9me2 ChIPseq
Previously published LaminB and H3K9me2 ChIPseq datasets (GEO GSE97877) generated and processed as described (1) were used in this study. Briefly, mouse embryonic stem cells (mESCs) derived from 129/Sv mouse strain were cultured in serum-free, feeder-free 2i media (50% DMD-F12 (Invitrogen); 50% Neurobasal medium (Invitrogen); 0.5× N2 supplement (Invitrogen); 0.5× B27 supplement (Invitrogen); 0.05% BSA (Invitrogen); l-glutamine (Invitrogen); penicilin/streptomycin (Invitrogen) and 150 μM β-mercaptoethanol (Sigma); 20 ng/ml LIF (eBioscience), 3 μM CHIR (Stemgent) and 1 μM PD325901 (Stemgent)). Cells were crosslinked, ChIP performed and sequencing libraries prepared as described (1). Primary antibodies used for ChIPseq were Santa Cruz Goat anti-Lamin B (M-20) (cat# sc-6217) and Abcam Mouse anti-H3K9me2 (cat# ab1220). Libraries were sequenced on the Illumina NextSeq500 platform (vII; 75bp single end sequencing; Illumina). Fastq files were aligned to the mm9 reference genome using STAR with the following parameters –alignIntronMax 1 –alignEndsType EndToEnd. Duplicate reads were marked and removed using Picard Tools and Bamtools. Replicates were merged and downsampled using PicardTools. Coverage tracks were created using DeepTools BamCompare (38). For visualization purposes, bam files were converted to bigwig files using bamCoverage (38) and visualized using the UCSC genome browser (39).
H3K9-Only Domain (KOD) Identification
Using previously published ChIPseq datasets (GEO GSE97877), LaminB (LB) and H3K9me2 read counts of two replicates each in 10kb bins were calculated using DeepTools BamCompare. Averages of the replicates were converted to z-scores to account for small differences in dynamic range between experiments. Z-scores were then smoothed using the mean of rolling, centered windows of size 10 to produce final scores in 10 kb bins. LADs in mESCs had LB and H3K9me2 scores above the genome-wide median score for each. For KODs in mESCs, we calculated the difference between the H3K9me2 score and LB score across all bins and set a stringent threshold to select only those bins with differences above the 80th percentile and LB below the genome median. The parameters for LADs were set to maximize the number of regions that overlap with cLADs as assigned in Meuleman et al. (19). The parameters for KODs were set to maximize the number of contiguous regions and minimize overlap with cLADs and our own mESC LAD assignments. For both LADs and KODs, adjacent 10 kb bins that met the respective criteria were merged using bedtools (v2.29.2) merge (maximum distance between features: 0) to assign individual domains for further analyses (40). Those KODs that were at least 50 kb away from the nearest LAD were designated as stand-alone KODs (saKODs). Coordinates (mm9) for all KODs and saKODs are included in Supplemental Table S1. KODs that overlap fLADs, KODs/fLADs, are KODs that share ≥90% of the domain with fLADs; KODs (non-fLADs) are KODs that share ≤10% of the domain with fLADs.
Integration of ChIPseq and gene expression data
Publicly available datasets for various genomic features in mESCs were downloaded from GEO (see below) and included peak calls where available. Each of these features was assessed for overlap with regions of mm9 genome designated as cLADs (GSE17051), nonLADs (GSE17051), KODs or saKODs (this study). In this study, fLADs refer to non-repetitive regions of the genome that were neither constitutive nonLADs nor mESC LADs designated as described above. To determine densities of features with discrete peaks (narrow histone modifications, CpG islands, ATACseq, DNaseHS), bedtools intersect –c flag was used to count events per domain of each type. For broad features (A/B compartments, H3K9me3), bedtools intersect –wo flag was used to calculate coverage as basepairs of overlap for each domain type which was then expressed as a percentage of the total genome coverage of that domain type. Distributions of scores reflect the signal intensity values underlying the peak assignments. Whereas peaks reflect the relative enrichment of signal in a region, scores reflect the signal intensity values underlying peak assignments.
Enrichment across annotated genomic features
Feature enrichment analysis was performed using Genomic Association Tester (GAT) (41). The significance of overlap between sets of genomic intervals was calculated based on simulation using a permutation-based approach and accounting for genome organization regions of low mapability. All enrichment analyses were subjected to 1000 simulations. The fold enrichment is expressed as the ratio of observed / expected. P values reflect an estimate of the probability to obtain an observed (or larger) overlap between two segment sets by chance.
Publicly available datasets used in this study
For analysis of histone features in mESCs, peak coordinates and underlying signal values (when available) for the following datasets (all obtained from undifferentiated mESCs derived from the same strain) were downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/):
H3K27me3: GSM1000089
H3K4me1: GSM1000121
H3K9ac: GSM1000123
H3K4me3: GSM1000124
H3K27ac: GSM1000126
H3K9me3: GSM3123483
H3K14Ac: GSM775314
DNaseHS1: GSM1014154
ATAC: GSM2156964
A/B Compartments: GSE113985
RNA: GSE89211
CpG islands (42) and Gene annotations (43) were downloaded from UCSC Genome Browser (NCBI37/mm9) Assembly.
Gene annotations: GENCODE VM23 release—protein-coding genes only as downloaded from UCSC GENCODE Genes track was used for gene density assessment.
For primary enhancer–promoter analyses (Figure 4), enhancers (Replicated set) and promoters (EPDnew replicated set) (44) were downloaded from UCSC Genome Browser (GRCm38/mm10). For supplementary analyses (Supplementary Figure S4), ENCODE Candidate Cis-Regulatory Elements (cCREs) (45) were downloaded from UCSC Genome Browser (GRCm38/mm10) Assembly and coordinates for promoter-like signatures (cCRE-PLS) were used for promoters, and enhancer-like signatures that are greater than 2 kb from a TSS, denoted distal (cCRE-dELS), were used for enhancers following translation from mm10 to mm9 using UCSC Liftover Tool (39).
Figure 4.

KODs are enriched for transcriptional enhancers. (A) UCSC genome browser view (mm10) of the Sox2 locus with its associated enhancers, many of which are in KODs surrounding the Sox2 gene body (GENCODE Track). Displayed tracks are Lamin B ChIPseq, histone marks as indicated, DNase and ATAC peaks, and CpG islands (all datasets from mESCs). Enhancers and predicted Enhancer-Promoter associations based on ENCODE panel of 72 mouse tissue-stages (44). (B) Domain-wide average gene density per kb of each domain type. (C, D) Domain-wide average density per kb of each domain by type (left panel) and enrichment (fold change observed over expected for randomly permuted genomic regions of equal size) for each domain type (right panel) for promoters (C) and enhancers (D). (E, F) Domain-wide average density per kb of (E) H3K4me3 and (F) H3K4me1 peaks per domain type. (G) Enrichment of mESC-specific enhancers by domain type. (H) Enrichment of all tissue-specific enhancers (not including mESC-specific enhancers) by domain type. (I) Transcription (FPKM) for genes associated with promoters or enhancers located in each domain type. Dotted lines in B–F left panels indicate genome-wide average for each category plotted. Statistical analysis for enrichment was performed using GAT with 1,000 permutations each; all other statistical analysis was performed using ANOVA Kruskal-Wallis test with Dunn's multiple comparisons; see Supplemental Tables S2 and S3.
RESULTS
H3K9me2-Only Domains (KODs) specify broad H3K9-dimethylated regions of the genome with minimal Lamin B contact
H3K9me2 is a specific mark of nuclear peripheral chromatin. H3K9me2-modified chromatin is visible by immunofluorescence as a layer adjacent to the nuclear lamina as shown in undifferentiated mouse embryonic stem cells (mESCs) with the nuclear lamina marker Lamin B (Figure 1A) (1,30). Two genome-wide sequencing methods for identifying chromatin that is in close proximity to the nuclear lamina are Lamin B-ChIPseq and Lamin B-DamID. These assays produce comparable results and have been used to define the 30–40% of the genome that is found in Lamina-Associated Domains (LADs) (Figure 1B) (16). As previously reported, most chromatin in LADs is enriched for H3K9me2 (Figure 1B) (1,14). We used ChIPseq analyses to define LADs in mESCs and found 40.8% of the genome to be Lamin B-associated. The vast majority of the chromatin in mESC LADs (94%) is H3K9me2-modifed (Figure 1B, C). But H3K9me2 also marks a separate 14% of the genome that is not associated with Lamin B (as assayed by Lamin B-ChIPseq from the same cells) (Figure 1B, C). We refer to these large chromatin regions as H3K9me2-Only Domains, or KODs. We identified >2000 KODs in mESCs. By definition, KODs do not overlap LADs, but some are found at the edges of LADs (Figure 1B). More than 75% of KODs are at least 50 kb away from the nearest LAD and we have designated these as ‘stand-alone KODs’ (saKODs) (Figure 1B). In order to assess possible effects of LAD edges, we conducted analyses on all KODs together as well as on the saKODs subset separately.
Figure 1.

H3K9me2-Only Domains (KODs) mark nuclear peripheral chromatin with minimal Lamin B association. (A) Representative immunofluorescent confocal images of a mouse embryonic stem cell (mESC), counterstained with DAPI. Area in dotted box highlighted in bottom panel. Scale bars, 5 and 1 μm. (B) Representative genome tracks from mESCs for H3K9me2 ChIPseq, Lamin B ChIPseq (1), and Lamin B DamID (3). Examples of LADs (red), nonLADs (green), KODs and stand-alone KODs (saKODs, blue) are highlighted. (C) Genome coverage per domain type (expressed as percent of total genome) in mESCs. LADs, nonLADs, H3K9me2-modified chromatin, and KODs as defined by Lamin B and H3K9me2 ChIPseq in mESCs ((1); this study). (D) Genome coverage for constitutive LADs, facultative LADs, and constitutive nonLADs as defined by Lamin B1 DamID across aggregate cell types (19). Percentage of KOD basepairs overlapping nonLADs (52%), fLADs (44%), and cLADs (4%, not shown). (E) Distribution of domain sizes (in kb) for KODs (n = 2603, all KODs), saKODs (n = 1969, a subset of all KODs), and cLADs (n = 3877). (F, G) Distributions of average ChIPseq scores per domain for nonLADs, KODs, saKODs, cLADs, and fLADs in mESCs for Lamin B (F) and H3K9me2 (G). Lines on violin plots show median, 25% and 75% quartiles. Statistical analysis was performed using ANOVA Kruskal-Wallis test with Dunn's multiple comparisons and Mann–Whitney test; see Supplemental Table S2.
In a previous study, Meuleman et al. used genome-wide Lamin B-DamID data from four mouse cell types to define regions of the genome with cell-type-invariant and cell-type-specific LADs (19). Constitutive LADs (cLADs) are regions that were consistently identified as LADs in all cell types examined; facultative LADs (fLADs) are those that varied among the cell types; and constitutive nonLADs (nonLADs) are regions that were not lamina-associated in any cell type. cLADs and nonLADs each encompass 35% of the genome, and the remaining 30% of the genome is variable fLADs (Figure 1D). The group also reported an evolutionary conservation of nuclear lamina interactions shared between mouse and human which was most pronounced in the constitutive domains (19).
Since we defined LADs and KODs in mESCs using ChIPseq data and different criteria from that used by Meuleman et al. (19), we compared our domains with their aggregate DamID-defined cLADs, fLADs, and nonLADs. Consistent with KODs marking regions separate from LADs, the regions we defined as KODs in mESCs overlap <2% of the base pairs defined by Meuleman et al. as cLADs. We also compared regions that were marked by KODs in mESCs with fLADs—regions identified as LADs in at least one but not all cell types. Almost half of all basepairs in KODs overlap fLADs (Figure 1B, D; Supplementary Figure S1), which indicates that some KODs in mESCs will become LADs in other cells or tissues. In order to characterize the epigenetic and genomic features of KODs, we compared them primarily with the cell-type independent sets of cLADs and nonLADs since these constitutive regions likely feature the most representative characteristics of each domain type. We included comparisons to fLADs in some analyses to consider regions that are variable among cell types.
One striking aspect of KODs is the size of these domains. We found that individual KODs, like cLADs, span tens to hundreds of kilobases each (KOD median domain size: 70 kb, average domain size: 159 kb; cLADs median: 76 kb, average: 232 kb; Figure 1E). This is at least one order of magnitude different from so-called ‘broad marks’ such as H3K36me2/3 that cover gene bodies, or ‘narrow marks’ like H3K4me3 that span a small number of nucleosomes. We also assessed each domain type for relative Lamin B and H3K9me2 ChIPseq signal intensities (Z-score; see Materials and Methods). This analysis confirmed that cLADs are enriched for both Lamin B and H3K9me2, and nonLADs are depleted for both Lamin B interaction and H3K9me2-modified chromatin (Figure 1F, G). Lamin B scores for fLADs are broadly distributed, reflecting the variability of this domain type. KODs are the only subset consistently enriched for H3K9me2 and depleted for Lamin B (Figure 1F, G).
KODs are positioned at the nuclear periphery
We previously showed that H3K9me2 specifically marks nuclear peripheral chromatin and verified the proximity of H3K9me2 chromatin to the nuclear lamina through super-resolution microscopy (30). KODs are, by definition, H3K9me2-modifed chromatin domains and are therefore expected to be positioned at the nuclear periphery. ChIPseq data reflect genome-wide H3K9me2 modification and Lamin B interactions from pools of large populations of cells; hence, we sought to examine the location of KODs in individual nuclei of mESCs. We designed libraries of fluorescent DNA oligo probes targeting specific KODs, LADs, and nonLADs from a variety of regions across the genome (Supplementary Figure S2; (30)). Examples of a LAD-targeted probe (Figure 2A, left) and a KOD-targeted probe (Figure 2A, right) are shown in the context of H3K9me2 and Lamin B ChIPseq profiles for each region. We used fluorescence in situ hybridization (FISH)-based imaging to monitor the positions of these individual regions in single cells and found both LAD probes and KOD probes to be at the periphery of most nuclei (Figure 2B). A pool of probes labeling 6 different KODs showed a broader distribution of positions than LADs, with the median distance from the periphery approximately matching the thickness of the H3K9me2 chromatin layer as measured in mESCs (shown as a dotted line in Figure 2C–E). Plots of distances to the nuclear lamina for individual LADs or KODs (25 cells for each probe) illustrate the range of positions possible for a given region and demonstrates the propensity for KODs to be localized at or near the nuclear lamina (Figure 2D, E).
Figure 2.

KODs are localized at the nuclear periphery. (A) Genomic locations of DNA oligo probes targeting a LAD (left panel; red highlight), a KOD (right panel; purple highlight) or a non-LAD (right panel; green highlight) shown with corresponding genome tracks (mm9) for H3K9me2 and Lamin B ChIPseq from mESCs. 250 kb regions targeted with DNA oligo probes shown as black bars above Lamin B tracks. (B) Localization of regions from a LAD and a KOD in interphase mESCs. Representative immuno-FISH images of cells hybridized with fluorescent DNA oligo probes targeting an individual LAD (red; left 3 panels) and an individual KOD (purple; right 3 panels), and immunostained for Lamin B (cyan) and DAPI (blue). Area in dotted boxes highlighted in right panel of each set. Scale bars: 5 and 1 μm. (C) Violin plots show distribution of distances to the nuclear periphery (as defined by Lamin B) of individual LAD, KOD, or nonLAD probes for ≥300 target loci. (D, E) Violin plots show distribution of distances to the nuclear periphery (as defined by Lamin B) for six individual (D) LADs or (E) KODs; n = 50 target loci in 25 cells. All KOD probes target saKODs. For C–E, dotted line indicates average thickness of H3K9me2 peripheral chromatin layer. Lines on violin plots show median, 25% and 75% quartiles. Statistical analysis was performed using ANOVA Kruskal-Wallis test with Dunn's multiple comparisons and Mann Whitney test; see Supplemental Table S2.
KODs feature permissive chromatin accessibility
Since KODs and LADs both contain H3K9me2-modified chromatin and localize mainly to the nuclear periphery, but differ in their Lamin B interaction, we asked what other characteristics distinguish KODs from LADs. Since we identified KODs in undifferentiated mESCs, we took advantage of the abundance of high-throughput data available for mESCs to perform an in-depth epigenetic characterization of H3K9me2-modified chromatin associated with KODs as compared to constitutive LADs and nonLADs. First, we examined the relationship between KODs and Hi-C-defined A and B subnuclear compartments. These higher-order compartments feature accessible chromatin in A, and inaccessible or less accessible chromatin in B (10). cLADs have been shown to be enriched in B compartments (46), but we found this was not the case for KODs. We examined KODs for overlap with A or B compartments using compartment designations from undifferentiated mESCs (47), and observed most KODs were found in A compartments: 76% of all basepairs in KODs overlapped A, while 21% overlapped B (Figure 3A, B; Statistical analysis for enrichment, performed with 1000 permutations each (see Methods, Supplemental Table S3) showed enrichment of KODs in A: 1.74-fold observed over expected; and depletion of KODs in B: 0.44-fold observed over expected; P < 0.001). Stand-alone KODs (saKODs) show a stronger enrichment with 81% in A compartments (saKODs in A: 1.86-fold; saKODs in B: 0.33; P < 0.001). For comparison, only 3.9% of cLADs bases were found to be in A whereas 81.0% were in B compartments (cLADs in A: 0.10-fold; cLADs in B: 1.81; P < 0.001). KODs more closely resembled constitutive nonLADs, 86.6% of which are located in A compartments (nonLADs in A: 2.02-fold; nonLADs in B: 0.18; P < 0.001; Figure 3B).
Figure 3.

KODs mark regions of accessible chromatin. (A) UCSC genome browser view (mm10) showing LADs (red bars), nonLADs (green bars), KODs (blue bars), and saKODs (purple bars) with representative genome tracks for Lamin B ChIPseq, histone marks as indicated, DNase and ATAC peaks, and A/B compartments. All datasets from mESCs. An example LAD (red highlight) and 2 example KODs (blue highlights) are shown. (B) Percent of each domain type associated with A (left) or B (right) compartments. (C) ATAC peak density per kb plotted as a domain-wide average (left panel) or distribution of densities across all domains of each type (right panel). (D) DNase peak density per kb plotted as a domain-wide average (left panel) or distribution of densities across all domains of each type (right panel). (E, F) Density per kb plotted as domain-wide average (left panel) or distribution of scores (signal intensity) per domain for each type (right panel) for (E) H3K14ac and (F) H3K27me3 peaks. (G) Percent of each domain type with H3K9me3 enrichment (left panel) and distribution of H3K9me3 scores per domain for each type (right panel). Dotted lines in left panels of C-G indicate genome-wide average for each category plotted. Lines on violin plots show median, 25% and 75% quartiles. Statistical analysis was performed using ANOVA Kruskal-Wallis test with Dunn's multiple comparisons and Mann–Whitney test; see Supplemental Table S2.
Consistent with established correlations between A compartments and relatively accessible chromatin (10), we also observed intermediate densities of both mESC ATACseq peaks and mESC DNase-hypersensitivity (DNase) peaks in KODs (Figure 3A, C–D, Supplementary Figure S3) (48,49). These markers of chromatin accessibility were found in KODs at approximately the same frequency as the genome-wide average distribution (Figure 3C–D, Supplementary Figure S3). We examined enrichment compared to randomly permuted genomic regions of equal size for each domain type and found ATAC and DNase peaks were enriched in nonLADs (enrichment scores and P values for observed over expected for 1,000 permutations (see Methods): ATAC 2.29-fold enrichment; DNase 2.05; P < 0.001) and depleted from cLADs (ATAC 0.10; DNase 0.22; P < 0.001) (Figure 3C-D, Supplementary Figure S3, Table S3). In KODs, the density per domain as well as the median distributions of both ATAC and DNase peak scores, reflecting the signal underlying the designated peaks, were between those of nonLADs and cLADs (Figure 3C–D, Supplementary Figure S3). Chromatin accessibility has been described as a continuum from closed to permissive to open (50), and these data indicate that KODs fall into the intermediate, permissive category. Consistent with this observation, acetylated histone marks such as H3K14ac were found in greater average densities in KODs than the genome-wide average (Figure 3A, E). Both density of H3K14ac peaks per individual domain and scores reflecting the underlying ChIPseq signal in those regions were higher for KODs than cLADs (Figure 3E). H3K9ac was also more commonly found in KODs than in cLADs, but it is well below the genome average in both types of domains (Figure 3A, Supplementary Figure S3). cLADs showed very low densities for both of these acetylated histone marks (51) (Figure 3A, E; Supplementary Figure S3). We next examined the relationship between KODs and repressive epigenetic modifications.
H3K27me3, a histone modification associated with facultative heterochromatin, was observed at a higher density in KODs than the genome-wide average but was well below average in cLADs (Figure 3A, F). H3K9me3, a hallmark of constitutive heterochromatin, is enriched in cLADs (1.68, P < 0.001), as previously reported (52). Relative to random permutations of equivalent regions, both nonLADs and KODs were depleted for the repressive H3K9me3 modification (nonLADs: 0.42-fold; KODs: 0.60; saKODs: 0.56; P < 0.001; Supplemental Table S3). Epigenetic characterization of KODs revealed that these domains are different from cLADs in terms of H3K9me3 as well as acetylated histone content and chromatin accessibility. These data indicate that, while KODs and cLADs are both enriched for H3K9me2 and positioned at the nuclear periphery, they specify distinct chromatin environments.
KODs are enriched for enhancers
The characteristics of relatively accessible chromatin found in KODs led us to ask what genic features were associated with these domains. The density of protein-coding genes (GENCODE VM23) (43) in KODs is approximately the same as the genome-wide average, while genes were more rare in cLADs, as has been reported (3) (Figure 4A, B). As expected for euchromatic nonLADs, these regions were enriched for both gene bodies and promoters (promoters: 2.27-fold observed over expected; P < 0.001). Promoter density in KODs is below the genome average (KODs 0.80; saKODs 0.83 P < 0.001), but promoters are even more rare in cLADs (0.07; P < 0.001) (Figure 4A, C). Predictably, CpG island densities in KODs, cLADs and nonLADs correlate with the observed promoter densities in each domain category (Supplementary Figure S4).
We next asked whether transcriptional enhancers were more or less likely to be located in nonLADs, KODs, or cLADs. By integrating data obtained from a wide variety of tissues across eight stages of mouse development, the ENCODE project identified tens of thousands of distal transcriptional enhancers (44). Assessment of the density of these enhancers in each domain type revealed that both nonLADs and KODs were notably enriched for distal enhancers (Figure 4A, D). The average density of enhancers in KODs approached that found in nonLADs, and enhancer enrichment in KODs was 1.55-fold (saKODs 1.66; P < 0.001) and 2.13-fold for nonLADs (P < 0.001; Figure 4D). Facultative LADs (fLADs) are domains that were observed to be LADs only in some cell types (19). These variable regions were depleted for enhancers (fLADs: 0.69; P < 0.001), as were cLADs (0.07; P < 0.001; Figure 4D). We performed the same analysis on an even more extensive set of putative promoters and enhancers from the recently published Phase III of the mouse ENCODE project and confirmed these results (45) (Supplementary Figure S4). Across all datasets, KODs were the only domain type that was depleted for genes and promoters while being enriched for distal enhancers.
The modified histones H3K4me3 and H3K4me1 mark promoters and enhancers, respectively (53,54). We used H3K4me3 and H3K4me1 ChIPseq datasets generated from mESCs and assessed how often these two histone modifications were found in KODs, cLADs and nonLADs. H3K4me3 was observed at densities consistent with the numbers of promoters found within each domain type: nonLADs were enriched for the active promoter mark H3K4me3, while the KODs and cLADs were depleted for H3K4me3 (Figures 3A, 4E; Supplementary Figure S4). Although both nonLADs and KODs were enriched for enhancers, only nonLADs showed enrichment for the enhancer mark, H3K4me1 (Figure 4F). H3K4me1 peak density in KODs was below the genome-wide average, and approximately the same as the genome-wide average in saKODs (Figure 4F). This is in contrast to the marked enrichment of enhancers we observed in both KODs and saKODs (Figure 4D). The difference between enhancer enrichment and H3K4me1-modification enrichment likely reflects the fact that the histone modification marks the regulatory element in the single cell type in which it was assayed—in this case mESCs. Enhancers (and promoters), on the other hand, are designated based on cumulative data from many different cell types and represent the broad range of regulatory elements possible in any of the hundreds of cell types and developmental contexts assayed (44). These data suggest that KODs contain putative enhancers, but not necessarily enhancers that are functional in mESCs. In fact, we found that KODs were depleted for both mESC-specific enhancers (55) (KODs: 0.68-fold; saKODs: 0.70; P < 0.001; Figure 4G) and for enhancers linked to housekeeping genes (KODs: 0.40; saKODs: 0.40; P < 0.001; Supplementary Figure S4) (56). When we examined the density of tissue-specific enhancers (excluding mESC-specific elements) (55), we found that they were enriched in both KODs and saKODs (KODs: 1.70; saKODs: 1.73; P < 0.001; Figure 4H). This is the only class of enhancers that was more highly enriched in KODs than nonLADs (nonLADs: 1.50; P < 0.001; Supplemental Table S3). Together these results indicate that KODs organize tissue-specific transcriptional enhancers that may be activated at a future stage of development.
KODs overlapping fLADs share characteristic features of KODs
Since approximately half of all KODs overlap regions defined by Meuleman et al. (19) as facultative LADs (fLADs), we characterized the subtypes of KODs (overlapping and non-overlapping fLADs) separately. Features of KODs that set them apart from LADs, such as being found primarily in A compartments and depleted for H3K9me3, apply to both non-fLAD KODs and KODs that overlap fLADs (KODs/fLADs; Supplementary Figure S5; Table S3). Further, both subtypes of KODs are depleted for promoters but not for enhancers. Most importantly, both non-fLAD KODs and KODs/fLADs are enriched for tissue-specific enhancers but depleted for enhancers of housekeeping genes and mESC-specific enhancers (Supplementary Figure S5; Table S3). All KODs, defined and characterized in mESCs, share several distinguishing features regardless of whether some of these regions may become LADs in another cell type upon differentiation.
KODs mark chromatin with limited protein-coding gene transcription
According to previous studies, genes in LADs tend to be transcriptionally inactive and loss of nuclear lamina interaction can be correlated with transcriptional activation (1,3,14). This prompted us to ask whether genes with regulatory elements located in KODs were transcriptionally active or repressed. Using published gene expression data from mESCs (57), we surveyed the expression levels of genes whose promoters lie in KODs, cLADs or nonLADs and found that transcriptional activity was extremely low for most genes with KOD- or cLAD-associated promoters (Figure 4I). In comparison, many genes with promoters in euchromatic nonLADs were expressed at a much higher level (Figure 4I). We made use of the thousands of enhancer–promoter interaction predictions made by Gorkin et al. (44) to ask whether genes linked to the distal enhancers found in KODs (Figure 4D) were expressed or repressed. We observed a broader range of transcript abundance for genes with an enhancer in a KOD compared to genes with a promoter in a KOD, but the median expression level was well below that of genes with a nonLAD-associated enhancer or promoter and below the genome-wide average of all mESC gene expression (Figure 4I). Expression levels for genes with cLAD-associated regulatory elements were very low for both promoter- and enhancer-associated genes, consistent with the heterochromatin characteristics of cLADs (Figure 4I). While we did observe more genes with higher transcript levels for KOD-associated enhancers than for KOD-associated promoters, this analysis is limited by the incomplete information regarding which enhancer (located in which domain) is responsible for activating the gene in mESCs. The result, therefore, may not reflect the transcriptional activity of a KOD-associated enhancer, but rather any of multiple enhancers linked to the gene including active enhancers located outside of a KOD. Taken together, this analysis suggests that KODs mark a chromatin environment that is generally repressive for protein-coding gene transcription.
KODs are depleted for active enhancers
Our observations that KODs are enriched for putative enhancers, depleted for mESC-specific enhancers, and associated with relatively low levels of gene expression led us to examine the functional state of enhancers by domain type. In this study, we identified KODs in undifferentiated mESCs, so we made use of enhancer state designations of active, primed, or poised from undifferentiated mESCs (57). In any given cell, at any given time, an enhancer may exist in one of these three states (58). Based on combinations of modifications at H3K4 and H3K27 together with binding of the histone acetyltransferase P300, Rada-Iglesias's group designated non-overlapping sets of enhancers as active, primed, or poised (57). Enhancers belonging to each of the three categories are enriched for H3K4me1 and bound by P300. Active enhancers are enriched for H3K27ac and drive high levels of transcription; primed enhancers lack H3K27ac and induce basal levels of transcription; and poised enhancers include the repressive histone mark H3K27me3 and are associated with a transcriptionally silent state (59–61). We asked whether mESC KODs were enriched for enhancers in any particular state. By mapping enhancers that were active, primed, or poised in mESCs to KODs, cLADs, and nonLADs, we found that nonLADs were enriched for all three enhancer categories (nonLADs active enhancers: 2.28-fold; primed: 2.40; poised: 2.35; P < 0.001; Figure 5A, C). KODs were depleted for both active and primed enhancers (KODs active enhancers: 0.47-fold; primed: 0.58; saKODs active: 0.50; primed: 0.63; P < 0.001; Figure 5A, C). Depletion of active enhancers in KODs is consistent with the observation of relatively modest transcriptional activity for protein-coding genes linked to KOD-localized enhancers (Figure 4G). Poised enhancers, however, were found in KODs at approximately the same density as the genome-wide average distribution (KODs: 0.95; saKODs: 1.0; Figure 5A). In comparison, cLADs were depleted for all three classes of enhancers, as would be expected given the scarcity of enhancers as a whole in cLADs, as described above (cLADs active enhancers: 0.09-fold; primed: 0.03; poised: 0.08; P < 0.001; Figures 5A, C, 4D). The densities of active and repressive H3K27 modifications – H3K27ac and H3K27me3, respectively – in each domain type correlate with enrichment of enhancers by designated state: nonLADs have higher than average densities of both H3K27ac and H3K27me3, cLADs have lower than average for both enhancer marks, and KODs have higher than the genome-wide average only for H3K27me3 and below average for H3K27ac (Figure 3F, Supplementary Figure S3 and S6).
Figure 5.

KODs are enriched for poised enhancers that become activated through differentiation. (A) Domain-wide average density per Mb of each domain category for enhancers in undifferentiated mESCs plotted by indicated enhancer state: active, primed, and poised. (B) Domain-wide average density per Mb of each domain category for enhancers that were poised in mESCs and transitioned to active in differentiated AntNPCs. Dotted lines in A–B indicate genome-wide average density for each category plotted. (C) Enrichment (fold change observed over expected for randomly permuted genomic regions of equal size) by domain type for active enhancers (in mESCs; left panel) and poised-to-active enhancers (in AntNPCs; right panel). Statistical analysis for enrichment was performed using GAT with 1000 permutations each; see Supplemental Table S3.
In addition to undifferentiated mESCs, Cruz-Molina et al. also tracked enhancer states following differentiation of mESCs into anterior neural progenitors (AntNPCs). Some of the enhancers that had been poised in undifferentiated mESCs were found to have transitioned to active enhancers in the differentiated AntNPCs (57). When we examined the genome-wide distribution of these ‘poised-to-active’ enhancers, we found that they were enriched in KODs (KODs: 1.73, P = 0.004; saKODs: 1.89, P < 0.001; Figure 5B, C). Enrichment of poised-to-active enhancers was similarly observed in nonLADs (nonLADs poised-to-active: 1.85-fold; P < 0.001; Figure 5B). While KODs are depleted for both active enhancers and the active histone modification H3K27ac, KODs are enriched for tissue-specific enhancers and poised enhancers that can become activated in another cell type. Our findings indicate that KODs contribute to the spatial organization of distal enhancers and we postulate that enhancer activity and promoter interactions are regulated, at least in part, by H3K9 dimethylation and association with the nuclear periphery.
DISCUSSION
H3K9me2 is a prominent modification of chromatin at the nuclear periphery of eukaryotic cells and an organizer of the 3D genome (1). H3K9me2 has been shown to be highly correlated with the heterochromatin of LADs and, in KODs, we find that H3K9me2 also marks a separate subset of nuclear peripheral chromatin that has limited association with Lamin B. KODs are enriched for tissue-specific distal transcriptional enhancers. Our genome-wide epigenetic characterization of KODs reveals several features indicative of possible function. First, KODs contain relatively open chromatin compared with the inaccessible heterochromatin of LADs. Chromatin accessibility in KODs features an intermediate degree of permissiveness on the continuum from inaccessible, ‘closed’ heterochromatin to highly accessible, ‘open’ euchromatin. Given this level of accessibility, KODs represent a portion of the chromatin landscape that is competent for physical interactions between transcription factors, chromatin remodelers, and other non-histone chromatin-binding factors and the underlying DNA regulatory elements that govern gene expression. Consistent with this accessible chromatin, most KODs are found in A compartments and we therefore expect that they are competent to interact with other regions of the transcriptionally active euchromatin of the A compartment (62). The most striking feature of KODs is their enrichment for distal transcriptional enhancers. Like LADs, KODs are gene-poor and depleted for promoters, but the density of enhancers in KODs is well above the genome average and approaches that of nonLADs. In undifferentiated mESCs, KODs are enriched for tissue-specific regulatory elements with histone modifications indicative of an inactive or poised state. Together, these characteristics suggest that KODs mark a functional subdivision of the peripheral genome and likely play a role in 3D chromatin conformation as it relates to gene expression.
Prominent questions in the field of gene regulation involve the relationship between enhancer function and architectural features of chromatin. These include understanding how distal enhancers find their target genes, and how these interactions are regulated in a cell-type-specific manner. Based on our findings, we propose the following speculative model (Figure 6) that can be tested experimentally in the future. We predict that KODs play a role in facilitating and organizing enhancer-promoter interactions that form the basis of spatiotemporal regulation of gene expression programs, especially those regulating development and differentiation.
Figure 6.
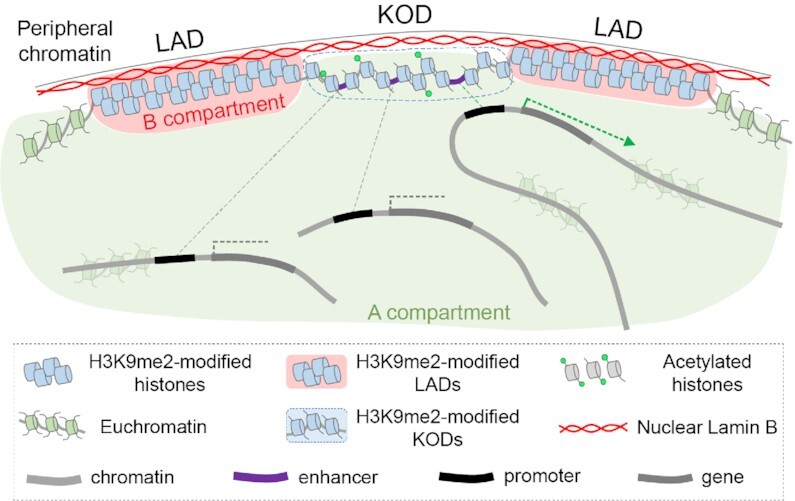
Model: KODs organize the genome at the nuclear periphery and provide a scaffold for distal enhancers.
Just as LADs have been postulated to form a structural backbone to anchor silent chromatin to the nuclear lamina (19), we hypothesize that KODs serve as a platform to position enhancers for timely access to gene expression programs. Enhancers far outnumber genes in mammalian genomes, and it is well appreciated that regulatory elements can be hundreds of kilobases or even megabases away from their target genes (63). It remains unclear how enhancers bridge these distances to find their target genes. Our results suggest that KODs may provide topological constraints to regulatory element organization which may increase the proximity of regulatory elements and reduce the search space required to bring enhancers and promoters together. KODs identified in mESCs are depleted for active mESC-specific enhancers, but they are enriched for inactive or poised enhancers with non-mESC tissue specificity. This suggests that KODs could organize enhancer-promoter interactions according to cell identity. Since roughly half of the KODs in mESCs overlap facultative LADs, we predict that at least some KODs vary by cell type. By maintaining enhancers in an accessible, likely tissue-specific configuration at the nuclear periphery, KODs may contribute to orchestrating cell-type-specific, spatiotemporal gene expression programs. Further experiments will be necessary to explore this hypothesis and confirm or refute this model.
LADs are thought to associate with the nuclear lamina through extended stretches of uninterrupted, multivalent chromatin-lamina contacts mediated by H3K9me2 (30,36). Our FISH data revealed that the median distance from the periphery is greater for KODs than for LADs. This suggests that KODs are less spatially restricted or more dynamic than LADs. B compartment chromatin generally, and LADs specifically, have been shown to exhibit reduced mobility compared with A compartment chromatin (64–67). In KODs, the greater DNA accessibility and presence of acetylated histones likely interrupts some stretches of H3K9me2 and may allow transient movement away from the periphery. This movement could be restricted to a small region around an individual enhancer, or the domain might occupy different positions over time or across different cells as cause or consequence of changes in transcription activity. It will be interesting to examine the biophysical characteristics of KODs over the course of differentiation and as enhancers become activated and engage their cognate promoters. Future studies will determine whether these changing characteristics are necessary for the proper function of KOD-resident enhancers and what impact they have on cell fate specification.
Gene regulation at the nuclear periphery has, to date, been studied in the context of LADs and the nuclear periphery has been regarded as a generally repressive environment for transcription (3,14). Consistent with their peripheral position, KODs also designate a nuclear sub-compartment with low transcriptional activity. Median expression levels are below the genome average for genes with either promoters or enhancers in KODs. KODs are depleted for enhancers that show mESC-specific activity as well as enhancers that regulate transcriptionally active housekeeping genes. Also, while KODs are enriched for enhancer-associated histone marks such as H3K4me1 and H3K27me3, they are depleted for the active enhancer mark, H3K27ac (Figures 3, 4, Supplementary Figure S6). This is reflected in the representation of enhancers in different states: poised enhancers are found in KODs at numbers proportional to their share of the genome while active enhancers are relatively depleted (57) (Figure 5). These results support the current understanding in the field that the nuclear periphery is largely repressive for transcription, but we also provide further evidence that the genomic landscape near the lamina is heterogeneous (27). KODs exemplify regions at the nuclear periphery that are accessible for epigenetic modification and nucleosome remodeling, and permissive for possible enhancer-promoter interactions.
A number of studies have documented a correlation between gene activation and repositioning of loci away from the nuclear periphery (3,21,68); this mechanism may also be relevant for KOD function. In a recent report, disruption of chromatin-lamina interactions occurred in conjunction with the active histone mark H3K27ac, and overexpression of the histone acetyltransferase CBP-1/p300 contributed to the release of heterochromatin from the nuclear periphery (69). Work from the Murre lab has shown that activation of the long non-coding RNA gene ThymoD was required to reposition an enhancer of the nearby Bcl11b gene from the lamina to the nuclear interior and bring the Bcl11b enhancer and promoter into closer proximity (26). Thus, enhancer activation may precede or promote displacement of a locus from the nuclear periphery as part of the process that supports enhancer-promoter engagement. Future research will be aimed at characterizing the epigenetic changes and spatial repositioning of KODs and individual enhancers concordant with transitions in gene activation.
The intermediate accessibility of chromatin in KODs indicates that these regions provide a landscape that is permissive for DNA-binding factors and chromatin remodelers to access regulatory elements as needed. Our results are consistent with a role for H3K9me2 in positioning chromatin at the nuclear periphery in the absence of transcription. Given the scarcity of active enhancers found in KODs, we predict that activation of a poised enhancer involves loss of H3K9me2, locally or more broadly, and that KODs will vary by cell type and reflect the state of enhancers required to maintain the appropriate cell identity. In our model (Figure 6), we propose that KODs provide a platform for tissue-specific enhancers bearing the histone marks of a poised state, in an environment of intermediate chromatin accessibility, with opportunity for interactions with other A compartment chromatin. We postulate that enhancer activity and promoter interactions are regulated, at least in part, by H3K9 dimethylation and association with the nuclear periphery. Going forward, it will be of great interest to explore the spatial and epigenetic response of KODs to development and differentiation, and to determine whether KODs contain ‘facultative’ enhancers that are repositioned and activated in response to developmental morphogens or other inductive signals.
DATA AVAILABILITY
The results published here are based on previously published LaminB and H3K9me2 ChIPseq datasets (GEO GSE97877) generated and processed as described (1).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Casey Brown, Rajan Jain, and Joel Rurik for critical assessment of the manuscript and for helpful suggestions. We thank all members of the Epstein lab for their useful insights and discussion, Andrea Stout from the Penn CDB Microscopy Core for help with imaging, and Apporva Babu for early technical support.
Author contributions: C.L.S., A.P. and J.A.E. conceived of the project and designed experiments. C.L.S. and A.P. performed experiments. C.L.S., A.P. and J.A.E. analyzed data and wrote the manuscript. J.A.E. supervised the project.
Contributor Information
Cheryl L Smith, Department of Cell and Developmental Biology and Penn Epigenetics Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, PA 19104, USA.
Andrey Poleshko, Department of Cell and Developmental Biology and Penn Epigenetics Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, PA 19104, USA.
Jonathan A Epstein, Department of Medicine and Penn Cardiovascular Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, PA 19104, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35 HL140018 to J.A.E.]; Cotswold Foundation (to J.A.E.); WW Smith endowed chair (to J.A.E.). Funding for open access charge: National Institutes of Health [R35 HL140018].
Conflict of interest statement. None declared.
REFERENCES
- 1. Poleshko A., Shah P.P., Gupta M., Babu A., Morley M.P., Manderfield L.J., Ifkovits J.L., Calderon D., Aghajanian H., Sierra-Pagan J.E.et al.. Genome-nuclear lamina interactions regulate cardiac stem cell lineage restriction. Cell. 2017; 171:573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Towbin B.D., Gonzalez-Sandoval A., Gasser S.M.. Mechanisms of heterochromatin subnuclear localization. Trends Biochem. Sci. 2013; 38:356–363. [DOI] [PubMed] [Google Scholar]
- 3. Peric-Hupkes D., Meuleman W., Pagie L., Bruggeman S.W., Solovei I., Brugman W., Graf S., Flicek P., Kerkhoven R.M., van Lohuizen M.et al.. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell. 2010; 38:603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andrey G., Mundlos S.. The three-dimensional genome: regulating gene expression during pluripotency and development. Development. 2017; 144:3646–3658. [DOI] [PubMed] [Google Scholar]
- 5. Buchwalter A., Kaneshiro J.M., Hetzer M.W.. Coaching from the sidelines: the nuclear periphery in genome regulation. Nat. Rev. Genet. 2019; 20:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rae M.M., Franke W.W.. The interphase distribution of satellite DNA-containing heterochromatin in mouse nuclei. Chromosoma. 1972; 39:443–456. [DOI] [PubMed] [Google Scholar]
- 7. Schermelleh L., Carlton P.M., Haase S., Shao L., Winoto L., Kner P., Burke B., Cardoso M.C., Agard D.A., Gustafsson M.G.et al.. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science. 2008; 320:1332–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huisinga K.L., Brower-Toland B., Elgin S.C.. The contradictory definitions of heterochromatin: transcription and silencing. Chromosoma. 2006; 115:110–122. [DOI] [PubMed] [Google Scholar]
- 9. Grewal S.I., Jia S.. Heterochromatin revisited. Nat. Rev. Genet. 2007; 8:35–46. [DOI] [PubMed] [Google Scholar]
- 10. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O.et al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cavalli G., Misteli T.. Functional implications of genome topology. Nat. Struct. Mol. Biol. 2013; 20:290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fortin J.P., Hansen K.D.. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol. 2015; 16:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W.et al.. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015; 518:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guelen L., Pagie L., Brasset E., Meuleman W., Faza M.B., Talhout W., Eussen B.H., de Klein A., Wessels L., de Laat W.et al.. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008; 453:948–951. [DOI] [PubMed] [Google Scholar]
- 15. Shah P.P., Donahue G., Otte G.L., Capell B.C., Nelson D.M., Cao K., Aggarwala V., Cruickshanks H.A., Rai T.S., McBryan T.et al.. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013; 27:1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lund E., Oldenburg A.R., Collas P.. Enriched domain detector: a program for detection of wide genomic enrichment domains robust against local variations. Nucleic Acids Res. 2014; 42:e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kind J., Pagie L., de Vries S.S., Nahidiazar L., Dey S.S., Bienko M., Zhan Y., Lajoie B., de Graaf C.A., Amendola M.et al.. Genome-wide maps of nuclear lamina interactions in single human cells. Cell. 2015; 163:134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Steensel B., Belmont A.S.. Lamina-associated domains: Links with chromosome architecture, heterochromatin, and gene repression. Cell. 2017; 169:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meuleman W., Peric-Hupkes D., Kind J., Beaudry J.B., Pagie L., Kellis M., Reinders M., Wessels L., van Steensel B.. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013; 23:270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robson M.I., de Las Heras J.I., Czapiewski R., Le Thanh P., Booth D.G., Kelly D.A., Webb S., Kerr A.R.W., Schirmer E.C. Tissue-Specific gene repositioning by muscle nuclear membrane proteins enhances repression of critical developmental genes during myogenesis. Mol. Cell. 2016; 62:834–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kohwi M., Lupton J.R., Lai S.L., Miller M.R., Doe C.Q.. Developmentally regulated subnuclear genome reorganization restricts neural progenitor competence in Drosophila. Cell. 2013; 152:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meister P., Gehlen L.R., Varela E., Kalck V., Gasser S.M.. Visualizing yeast chromosomes and nuclear architecture. Methods Enzymol. 2010; 470:535–567. [DOI] [PubMed] [Google Scholar]
- 23. Gonzalez-Sandoval A., Towbin B.D., Kalck V., Cabianca D.S., Gaidatzis D., Hauer M.H., Geng L., Wang L., Yang T., Wang X.et al.. Perinuclear anchoring of H3K9-Methylated chromatin stabilizes induced cell fate in C. elegans embryos. 2015; 163:1333–1347. [DOI] [PubMed] [Google Scholar]
- 24. Pickersgill H., Kalverda B., de Wit E., Talhout W., Fornerod M., van Steensel B.. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat. Genet. 2006; 38:1005–1014. [DOI] [PubMed] [Google Scholar]
- 25. Lund E., Oldenburg A.R., Delbarre E., Freberg C.T., Duband-Goulet I., Eskeland R., Buendia B., Collas P.. Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. 2013; 23:1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Isoda T., Moore A.J., He Z., Chandra V., Aida M., Denholtz M., Piet van Hamburg J., Fisch K.M., Chang A.N., Fahl S.P.et al.. Non-coding transcription instructs chromatin folding and compartmentalization to dictate enhancer-promoter communication and T cell fate. 2017; 171:103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brueckner L., Zhao P.A., van Schaik T., Leemans C., Sima J., Peric-Hupkes D., Gilbert D.M., van Steensel B.. Local rewiring of genome-nuclear lamina interactions by transcription. EMBO J. 2020; 39:e103159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cruz-Tapias P., Robin P., Pontis J., Maestro L.D., Ait-Si-Ali S. The H3K9 methylation writer SETDB1 and its reader MPP8 cooperate to silence satellite DNA repeats in mouse embryonic stem cells. Genes (Basel). 2019; 10:750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoshioka H., McCarrey J.R., Yamazaki Y.. Dynamic nuclear organization of constitutive heterochromatin during fetal male germ cell development in mice. Biol. Reprod. 2009; 80:804–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Poleshko A., Smith C.L., Nguyen S.C., Sivaramakrishnan P., Wong K.G., Murray J.I., Lakadamyali M., Joyce E.F., Jain R., Epstein J.A.. H3K9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis. Elife. 2019; 8:e49278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu R., Terry A.V., Singh P.B., Gilbert D.M.. Differential subnuclear localization and replication timing of histone H3 lysine 9 methylation states. Mol. Biol. Cell. 2005; 16:2872–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yokochi T., Poduch K., Ryba T., Lu J., Hiratani I., Tachibana M., Shinkai Y., Gilbert D.M.. G9a selectively represses a class of late-replicating genes at the nuclear periphery. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:19363–19368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Towbin B.D., Gonzalez-Aguilera C., Sack R., Gaidatzis D., Kalck V., Meister P., Askjaer P., Gasser S.M.. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell. 2012; 150:934–947. [DOI] [PubMed] [Google Scholar]
- 34. See K., Kiseleva A.A., Smith C.L., Liu F., Li J., Poleshko A., Epstein J.A.. Histone methyltransferase activity programs nuclear peripheral genome positioning. Dev. Biol. 2020; 466:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Therizols P., Illingworth R.S., Courilleau C., Boyle S., Wood A.J., Bickmore W.A.. Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science. 2014; 346:1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kind J., Pagie L., Ortabozkoyun H., Boyle S., de Vries S.S., Janssen H., Amendola M., Nolen L.D., Bickmore W.A., van Steensel B.. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 2013; 153:178–192. [DOI] [PubMed] [Google Scholar]
- 37. Rosin L.F., Nguyen S.C., Joyce E.F.. Condensin II drives large-scale folding and spatial partitioning of interphase chromosomes in Drosophila nuclei. PLos Genet. 2018; 14:e1007393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Quinlan A.R. BEDTools: The swiss-army tool for genome feature analysis. Curr. Protoc. Bioinformatics. 2014; 47:doi:10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Benton M.C., Lea R.A., Macartney-Coxson D., Sutherland H.G., White N., Kennedy D., Mengersen K., Haupt L.M., Griffiths L.R.. Genome-wide allele-specific methylation is enriched at gene regulatory regions in a multi-generation pedigree from the Norfolk Island isolate. Epigenetics Chromatin. 2019; 12:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gardiner-Garden M., Frommer M.. CpG islands in vertebrate genomes. J. Mol. Biol. 1987; 196:261–282. [DOI] [PubMed] [Google Scholar]
- 43. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J.et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gorkin D.U., Barozzi I., Zhao Y., Zhang Y., Huang H., Lee A.Y., Li B., Chiou J., Wildberg A., Ding B.et al.. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature. 2020; 583:744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Consortium E.P., Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A.et al.. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020; 583:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zheng X., Hu J., Yue S., Kristiani L., Kim M., Sauria M., Taylor J., Kim Y., Zheng Y.. Lamins organize the global three-dimensional genome from the nuclear periphery. Mol. Cell. 2018; 71:802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miura H., Takahashi S., Poonperm R., Tanigawa A., Takebayashi S.I., Hiratani I.. Single-cell DNA replication profiling identifies spatiotemporal developmental dynamics of chromosome organization. Nat. Genet. 2019; 51:1356–1368. [DOI] [PubMed] [Google Scholar]
- 48. Vierstra J., Rynes E., Sandstrom R., Zhang M., Canfield T., Hansen R.S., Stehling-Sun S., Sabo P.J., Byron R., Humbert R.et al.. Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science. 2014; 346:1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu J., Huang B., Chen H., Yin Q., Liu Y., Xiang Y., Zhang B., Liu B., Wang Q., Xia W.et al.. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature. 2016; 534:652–657. [DOI] [PubMed] [Google Scholar]
- 50. Klemm S.L., Shipony Z., Greenleaf W.J.. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019; 20:207–220. [DOI] [PubMed] [Google Scholar]
- 51. Eberharter A., Becker P.B.. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002; 3:224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gesson K., Rescheneder P., Skoruppa M.P., von Haeseler A., Dechat T., Foisner R.. A-type lamins bind both hetero- and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 2016; 26:462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S.A., Flynn R.A., Wysocka J.. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011; 470:279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W.et al.. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009; 459:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shen Y., Yue F., McCleary D.F., Ye Z., Edsall L., Kuan S., Wagner U., Dixon J., Lee L., Lobanenkov V.V.et al.. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012; 488:116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Osterwalder M., Barozzi I., Tissieres V., Fukuda-Yuzawa Y., Mannion B.J., Afzal S.Y., Lee E.A., Zhu Y., Plajzer-Frick I., Pickle C.S.et al.. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature. 2018; 554:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cruz-Molina S., Respuela P., Tebartz C., Kolovos P., Nikolic M., Fueyo R., van Ijcken W.F.J., Grosveld F., Frommolt P., Bazzi H.et al.. PRC2 facilitates the regulatory topology required for poised enhancer function during pluripotent stem cell differentiation. Cell Stem Cell. 2017; 20:689–705. [DOI] [PubMed] [Google Scholar]
- 58. Calo E., Wysocka J.. Modification of enhancer chromatin: what, how, and why?. Mol. Cell. 2013; 49:825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A.et al.. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rada-Iglesias A., Grosveld F.G., Papantonis A.. Forces driving the three-dimensional folding of eukaryotic genomes. Mol. Syst. Biol. 2018; 14:e8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zentner G.E., Tesar P.J., Scacheri P.C.. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011; 21:1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eagen K.P. Principles of chromosome architecture revealed by Hi-C. Trends Biochem. Sci. 2018; 43:469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bulger M., Groudine M.. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011; 144:327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Robson M.I., Ringel A.R., Mundlos S.. Regulatory landscaping: how enhancer-promoter communication is sculpted in 3D. Mol. Cell. 2019; 74:1110–1122. [DOI] [PubMed] [Google Scholar]
- 65. Pope B.D., Ryba T., Dileep V., Yue F., Wu W., Denas O., Vera D.L., Wang Y., Hansen R.S., Canfield T.K.et al.. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014; 515:402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chubb J.R., Boyle S., Perry P., Bickmore W.A.. Chromatin motion is constrained by association with nuclear compartments in human cells. Curr. Biol. 2002; 12:439–445. [DOI] [PubMed] [Google Scholar]
- 67. Ou H.D., Phan S., Deerinck T.J., Thor A., Ellisman M.H., O'Shea C.C. ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017; 357:eaag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shachar S., Misteli T.. Causes and consequences of nuclear gene positioning. J. Cell Sci. 2017; 130:1501–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cabianca D.S., Munoz-Jimenez C., Kalck V., Gaidatzis D., Padeken J., Seeber A., Askjaer P., Gasser S.M.. Active chromatin marks drive spatial sequestration of heterochromatin in C. elegans nuclei. Nature. 2019; 569:734–739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The results published here are based on previously published LaminB and H3K9me2 ChIPseq datasets (GEO GSE97877) generated and processed as described (1).