


Abstract
Polycomb repressive complex 2 (PRC2) is an essential protein complex that silences gene expression via post-translational modifications of chromatin. This paper combined homology modeling, atomistic and coarse-grained molecular dynamics simulations, and single-molecule force spectroscopy experiments to characterize both its full-length structure and PRC2-DNA interactions. Using free energy calculations with a newly parameterized protein-DNA force field, we studied a total of three potential PRC2 conformations and their impact on DNA binding and bending. Consistent with cryo-EM studies, we found that EZH2, a core subunit of PRC2, provides the primary interface for DNA binding, and its curved surface can induce DNA bending. Our simulations also predicted the C2 domain of the SUZ12 subunit to contact DNA. Multiple PRC2 complexes bind with DNA cooperatively via allosteric communication through the DNA, leading to a hairpin-like looped configuration. Single-molecule experiments support PRC2-mediated DNA looping and the role of AEBP2 in regulating such loop formation. The impact of AEBP2 can be partly understood from its association with the C2 domain, blocking C2 from DNA binding. Our study suggests that accessory proteins may regulate the genomic location of PRC2 by interfering with its DNA interactions.
Graphical Abstract
Graphical Abstract.
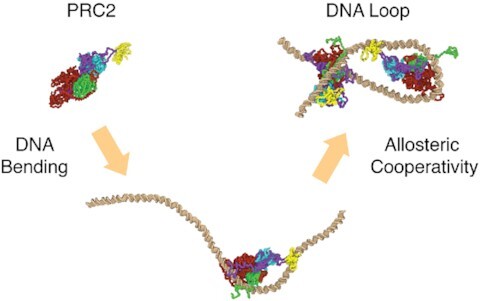
Multiple PRC2 complexes bind with DNA cooperatively via allosteric communication through the DNA, leading to the formation of hairpin-like looped configurations.
INTRODUCTION
Polycomb Group (PcG) proteins are a key class of chromatin regulators that repress gene expression via epigenetic modifications (1–4). A crucial member of PcG proteins is the Polycomb Repressive Complex 2 (PRC2), a methyltransferase for mono-, di- and tri-methylation of the lysine residue 27 of histone H3 (H3K27me1/2/3) (3,5–7). The core complex of PRC2 consists of four subunits: suppressor of zeste 12 (SUZ12), enhancer of zeste homologs 1/2 (EZH1/2), embryonic ectoderm development (EED), and Retinoblastoma-binding protein 4 (RBBP4) (Figure 1 A). Biochemical studies have provided insight for the function of those subunits (8–12). In particular, EZH2, EED, and the C-terminal part of SUZ12, are responsible for chromatin binding and methylation mark propagation across nucleosomes (13–20). Other subunits were found to maintain the complex’s integrity and implicated in the binding to long non-coding RNAs (19,21,22). Structural characterization of the entire complex and its interaction with chromatin could provide further insight into the cooperation among subunits to establish and maintain histone modifications.
Figure 1.

Illustration of the three conformations constructed for the PRC2 core complex via homology modeling. (A) Schematic diagram highlighting the ordered (box) and disordered (line) domains in each subunit. (B–D) corresponds to the conformation that adopts a free C2 domain (B), a bound C2 domain (C) and a dimeric structure (D), respectively. The coloring scheme is the same as in part A. Disordered regions are not shown for visual clarity.
Significant progress has been made in determining high-resolution PRC2 structures using cryogenic electron microscopy (cryo-EM) (19,20,23–25) and X-ray crystallography (14–17,26–30). These studies have greatly improved our understanding of the complex’s organization. They also revealed PRC2’s structural plasticity in response to the binding of accessory proteins. In particular, whether AEBP2 is present or not can alter the conformation of the subunit SUZ12 dramatically (30). Accessory proteins also appear to regulate the protein stoichiometry, and PHF19 was found to stabilize a dimeric conformation with protein-protein interface involving SUZ12 and RBBP4 subunits (30,31).
The impact of various conformations on PRC2’s ability to bind chromatin and induce changes in chromatin organization is less clear. A recent cryo-EM study captured PRC2 with a di-nucleosome, resolving a structure in which the protein complex simultaneously contacts two nucleosomes (20). We further demonstrated that such contacts could extend beyond nearest neighbors, using a combination of single-molecular force spectroscopy and computational modeling (32). Importantly, we found that PRC2 chromatin interactions are significantly weakened with the presence of AEBP2, supporting the role of accessory proteins and PRC2 conformation on chromatin binding. The non-neighboring, long-range contacts between PRC2 and nucleosomes have significant implications. They suggest that multiple PRC2 may act as cross-linkers to compact and condense chromatin. Notably, PRC2 was indeed found to bind with nucleosome arrays cooperatively (10). Recent atomic force microscopy (AFM) experiments (33) provided direct observations of PRC2-induced bending and looping of kilobase-long DNAs. A more systematic study on PRC2–DNA interactions could help uncover molecular details into the collective impact of multiple PRC2s on DNA conformation.
In this work, we characterized different conformational states of the PRC2 core complex and its interaction with the DNA. We constructed three PRC2 structures that differ in their oligomeric states and the conformation of the C2 domain from the SUZ12 subunit. Explicit solvent all-atom molecular dynamics simulations support the stability of the modeled structures. Coarse-grained simulations further revealed that PRC2 binds DNA via the EZH2 subunit, which induces DNA bending due to the protein surface’s intrinsic curvature. The PRC2 structure with a bound C2, a conformation that is often observed in the presence of AEBP2, bends DNA less significantly than the one with a free C2. Conformation-dependent DNA interactions become more evident in the presence of two PRC2s, and complexes with a free C2 conformation are highly cooperative in looping the DNA molecule. Cooperativity between PRC2 complexes does not require direct protein–protein interactions but arise from allostery through the DNA. These simulations suggest a weakening of DNA binding and bending upon AEBP2 association, an observation that is supported with single-molecule force spectroscopy experiments. Our study indicates that PRC2 accessory proteins may modulate the core complex’s DNA binding affinity to relocate it towards specific chromatin regions for targeted histone modification.
MATERIALS AND METHODS
Homology modeling of full-length PRC2
We built the structures for PRC2 in its entirety via homology modeling from multiple templates. The software Modeller was used to add missing regions as random loops and resolve steric clashes (34). The cryo-EM structure of the monomeric PRC2 core complex (PDB ID: 6c23) (19) was used together with two earlier X-ray crystal structures (PDB ID: 5wai and 5hyn) (16,29) for building PRC2b. For PRC2d, in addition to these three structures, we incorporated a dimeric PRC2 structure (PDB ID: 6nq3) that contains the N-terminal part of SUZ12 and RBBP4 (30). To avoid conflicts among the templates, we did not include the N-terminal part of SUZ12 in the monomeric structures for structural modeling. Finally, PRC2f was modeled by using one of the monomeric structures built in PRC2d.
All-atom molecular dynamics simulations
To evaluate the stability of PRC2 structures from homology modeling, we carried out explicit solvent all-atom simulations with the CHARMM36m force field (35). Parameters in the force field have been tuned to model both ordered and disordered proteins. We solvated PRC2b and PRC2f with TIP3P water molecules (36) in dodecahedron boxes of size 23,500 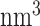 and 21,145
and 21,145 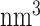 . Na+ and Cl− ions were then added to neutralize the system at a physiological concentration of 0.15 M. Both systems were simulated at constant temperature and constant pressure (NPT) using the V-rescale thermostat (37) at 300 K and the Parrinello-Rahman barostat (38) at 1.0 bar. Gromacs 2019 was used to carry out the simulations with a time step of 2.0 fs (39).
. Na+ and Cl− ions were then added to neutralize the system at a physiological concentration of 0.15 M. Both systems were simulated at constant temperature and constant pressure (NPT) using the V-rescale thermostat (37) at 300 K and the Parrinello-Rahman barostat (38) at 1.0 bar. Gromacs 2019 was used to carry out the simulations with a time step of 2.0 fs (39).
Coarse-grained protein–DNA model
We combined the 3SPN.2C DNA model (40,41) and the structure-based Cα model (42) for coarse-grained simulations of PRC2–DNA interactions. Parameters from 3SPN.2C (40) were directly applied to model intra-DNA interactions. Intra-protein interactions were modeled using the SMOG web server based on the structures obtained from homology modeling (43,44). Since the protein parameters were not tuned to perform simulations at the room temperature, we scaled them by a factor of 2.5 to avoid protein unfolding at 300 K.
In addition to the electrostatic potential, we included a residue and base pair specific contact potential to model protein–DNA interactions. The contact potential is a function of the distance rij between amino acid i and DNA bead j with the following form
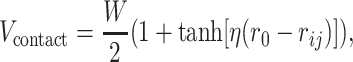 |
(1) |
where r0 = 8 Å and η = 0.7 Å−1. W is a 20 × 12 matrix that depends both on the amino acid and the DNA bead type. We modified W from the knowledge-based statistical potential reported by Skoinick and coworkers (45) to avoid double counting of electrostatic interactions and best fit experimental values of protein–DNA binding free energy (see Supplementary Table S1 and Figure S2).
Coarse-grained molecular dynamics simulations
Coarse grained simulations were carried out using the LAMMPS software package (46) with a timestep of 5 fs. We note that the time scale in coarse grained models is not well defined, and one simulation step in similar models has been mapped to 1 ps by matching the diffusion coefficient of protein domains between coarse grained and all atom simulations (47). For efficient conformational sampling, we rigidified the folded regions of PRC2 to remove fast degree of freedom. The Nos –Hoover thermostat (48,49) was applied separately to the rigid and flexible parts of the system to maintain the simulations at a temperature of 300 K with a damping coefficient of 1 ps. All simulations were carried out inside a cubic box with size of 1000 nm. More details on free energy calculations and in silico pulling can be found in the SI.
–Hoover thermostat (48,49) was applied separately to the rigid and flexible parts of the system to maintain the simulations at a temperature of 300 K with a damping coefficient of 1 ps. All simulations were carried out inside a cubic box with size of 1000 nm. More details on free energy calculations and in silico pulling can be found in the SI.
Single-molecule force spectroscopy experiments
Single-molecule pulling experiments were performed at room temperature on a LUMICKS C-Trap instrument equipped with dual-trap optical tweezers as previously described (32). Briefly, a piece of 10-kb biotinylated-DNA was tethered between two 3.23-micron streptavidin-coated and optically trapped polystyrene beads in PBS buffer. The tether was then transferred to a separate channel containing 500 nM PRC2 4-mer core complex or PRC2-AEBP2 5-mer complex in 10 mM Tris–HCl pH 8.0, 0.1mM EDTA, 200 mM KCl, 0.5 mM MgCl2, 0.1% Tween and 1 mM DTT. PRC2 complexes were purified as previously described (50). The DNA tether was incubated with PRC2 at zero force for 5–10 s and then subjected to mechanical pulling by moving one trap relative to the other at a constant velocity (0.1μm/s)
RESULTS
Structural modeling of the PRC2 core complex
Significant progress has been made in the structural characterization of the PRC2 core complex (24). High-resolution structures have been determined for the four PRC2 subunits, SUZ12, EZH2, EED and RBBP4, via X-ray crystallography (16,29). Cryo-EM structures have also been reported for the assembled complex with the presence of accessory proteins (19,20,25). However, a complete structural model for the full PRC2 core complex remains lacking since many of the subunits contain disordered regions that cannot be resolved experimentally. In addition, the C2 domain of SUZ12 may undergo a large-scale conformational rearrangement depending on whether the core complex is bound with accessory proteins and which protein it is bound to (29,30). We combined homology modeling with all-atom molecular dynamics simulations to build full-length structures for the PRC2 core complex.
As shown in Figure 1, a total of three conformations were constructed via homology modeling (see Methods for details). All three structures adopt essentially the same conformation for the central part of the core complex and only differ in the location of the C2 domain of SUZ12. In the C2-bound form (PRC2b), the C2 domain is close to the subunit RBBP4 (Figure 1 C). This conformation is frequently observed when the core complex is bound with AEBP2 (19,29). In the C2-free structure (PRC2f), the C2 domain extends out with minimal contact with the main part of the core complex. Removing the contacts would lead to a flexible C2 with ill-defined 3D positions, an observation consistent with the difficulty for resolving its electron density map when studying the core complex without AEBP2 (29). Finally, we consider a dimeric structure (PRC2d), in which the two PRC2 core complexes interact via C2 mediated interactions. The hypothesized PRC2f structure was motivated by the recent observation that PRC2 exists in a dynamic equilibrium between the monomeric and dimeric state (30). This equilibrium can be regulated by the binding of the accessory protein PHF19 and AEBP2.
We solvated PRC2b and PRC2f with water molecules and monovalent ions to perform two 100-ns long all-atom molecular dynamics simulations. Our simulations support the stability of both structures, with the root mean squared deviations (RMSDs) from the initial configurations fluctuating around 8 Å near the end of the trajectories (Supplementary Figure S1A). Disordered regions of the complex and the flexible C2 domain were not included when calculating the RMSD. Considering the large size of the protein (2351 aa), the deviation is rather small per amino acid and is localized around flexible loop regions (Supplementary Figure S1B). In addition to the loops, we found that, consistent with its reported flexibility (29), the C2 domain fluctuates significantly in both structures, especially for PRC2f.
Coarse-grained modeling of PRC2–DNA interactions
Using the constructed full-length structures, we characterized PRC2-DNA interactions with free energy calculations. These calculations involve large scale conformational rearrangements that occur on the timescale of micro to milliseconds and are computationally costly for all-atom simulations. We, therefore, resorted to a coarse-grained force field that combines the 3SPN.2C DNA model (41) and the structure-based protein model (43,44,51) (see Materials and Methods). Each amino acid and DNA base was modeled with one and three beads, respectively. Electrostatic interactions between protein and DNA were treated with the Debye–Hückel theory. These near-atomistic models have been widely used to study a wide range of protein-DNA complexes with great success (32,52–57).
To improve force field accuracy, we modeled the specific interactions between amino acids and nucleotides with a knowledge-based statistical potential (45). This potential was derived from curated PDB structures using frequencies of interacting residue-nucleotide pairs. We assessed the force field’s performance in predicting the binding free energy of nine protein-DNA complexes. These complexes constitute a diverse set of binding interfaces, and their dissociation constants have been determined experimentally. As summarized in Figure 2, the computed binding free energy using the potential of mean force method (58) agrees well with experimental values. The Pearson Correlation Coefficient between the two datasets is 0.77. These results improve from a model with only electrostatic interactions and uniform non-specific attraction between protein and DNA molecules, which cannot fully discriminate between strong and weak DNA-binding proteins (Supplementary Figures S3 and S4).
Figure 2.

Coarse-grained force field reproduces protein–DNA binding free energy. (A) A typical free energy profile as a function of the center of mass (COM) distance between protein and DNA molecules used to compute the binding free energy. The shaded region represents standard deviation of the mean. Representative configurations of the protein–DNA complex (PDB ID: 1APL) in the bound and dissociated state are shown on top, with the protein colored in red and DNA in yellow. (B) Comparison between simulated and experimental binding free energy for nine protein–DNA complexes. The PDB ID for each complex is shown in red and the best linear fit line is drawn in blue.
We next applied the coarse-grained model to assess the binding affinity of the PRC2 core complex with a 147 bp long 601 DNA sequence (59,60). For the conformation with a bound C2 domain (PRC2b), we obtained binding free energy of 19.1 ± 0.9 kBT (Supplementary Figure S5), which is in good agreement with the experimental value obtained by the electrophoretic mobility shift assay (10), 17.2–17.8 kBT. We note that a PRC2 5mer with AEBP2 association was used in the experimental study, while our simulations did not explicitly consider AEBP2. Although AEBP2 harbors three zinc-finger motifs, recent experiments indicate that these motifs cannot bind dsDNA (61) and contributions from AEBP2 for DNA binding are expected to be minimal (see Supplementary Figure S5).
We further computed the binding free energy using PRC2f with a free C2 domain and obtained a value of 24.7 ± 1.1kBT. An increase in DNA binding affinity from PRC2b is indeed consistent with prior experimental measurements. Specifically, Wang et al. showed that the PRC2 core complex (a 4mer without AEBP2) binds a CpG repeat sequence 20-times stronger than the 5mer with AEBP2 (10). As aforementioned, PRC2f is expected to be the stable conformation for the 4mer, while the 5mer will adopt a bound C2 conformation (PRC2b) stabilized by AEBP2. Closely examining the simulated protein-DNA contacts revealed that the additional affinity comes mainly from the charged residues on the C2 domain. C2 domains have been widely known for targeting negatively charged membranes (62), and their ability to interact with the DNA molecule is perhaps unsurprising.
PRC2 bends DNA with sequence specificity
Using Atomic Force Microscopy (AFM), Heenan et al. showed that PRC2 binding can induce DNA bending and the formation of kilobase-long loops (33). To provide more structural insight into PRC2-mediated looping, we computed the free energy profile as a function of DNA end-to-end distance with and without PRC2. A 294-bp-long DNA obtained from concatenating two nucleosomal 601 sequences (60) was used in these simulations. Additional simulation details can be found in the supporting information (SI).
As shown in Figure 3A, both PRC2b and PRC2f can induce DNA bending and reduce free energy at small distances. The conformation with the free C2 domain (PRC2f) is slightly more favorable in stabilizing the curved DNA. Binding induced bending is also evident in Figure 3B, which supports a strong correlation between the number of PRC2–DNA contacts and DNA curvature. We quantified the curvature using the bending angle of the most distorted 50-bp long segment across the entire DNA molecule (see Supplementary Figure S6). A straight conformation has an angle of zero degree. As the DNA curves, the angle increases.
Figure 3.

PRC2 binding induces DNA bending. (A) Free energy profiles as a function of the DNA end-to-end distance for the pure DNA (black) and the DNA bound with PRC2b (red) or PRC2f (cyan). (B) The average DNA bending angle as a function of the number of contacts between PRC2 and the DNA. In both A and B, lines represent mean values, and shaded regions correspond to standard deviations of the mean. (C) Residue specific probability map for PRC2b (left) and PRC2f (right) contacting the DNA.
We further colored the amino acids in the two PRC2 conformations using the average number of DNA beads in direct contact with them. Contacts were defined for beads that are within 8 Å estimated from umbrella-sampling simulations restrained at the DNA end-to-end distance of 700 Å (Figure 3 A). As shown in Figure 3C, both PRC2b and PRC2f support the dominance of EZH2 and EED in DNA binding. Similar contacts between EZH2 and DNA were seen in cryo-EM structures of PRC2 bound to nucleosomes (20,25) (see Supplementary Figure S7). The intrinsic curvature of the protein interface drives the observed bending of the DNA molecule. The C2 domain in PRC2f also binds DNA with high probability. These additional interactions, which are absent in PRC2b, lead to a more significantly bent DNA.
To examine the sequence dependence of PRC2–DNA interactions, we computed the free energy profiles of PRC2f with a poly-dG:dC, poly-dA:dT, and 601 sequence. The poly-dG:dC DNA favors more bent configurations than the poly-dA:dT or the 601 sequence (Supplementary Figure S8A), potentially due to its suppression of non-native hybridization and kinked configurations that increase DNA stiffness (63). PRC2f also makes more contacts with the poly-dG:dC DNA (Supplementary Figure S8B), leading to stronger binding interactions. A recent experiment with fluorescence polarization (10) indeed supports a much stronger binding affinity of PRC2 with a GC-rich sequence than with a AT-rich sequence.
Cooperative PRC2 binding loops DNA
While our simulations suggest that PRC2 monomers can bend the DNA, they do not stabilize looped DNA configurations (Figure 3A). For example, DNA configurations with smaller end-to-end distances (∼300 Å) remain high in free energy and are unfavorable compared to less curved structures. The AFM images suggest that multiple copies of PRC2 can simultaneously engage in DNA binding (33). Therefore, we examined whether a pair of PRC2 core complexes can cooperatively bind and bend the DNA.
Towards that end, we computed the DNA bending profile with the presence of two copies of PRC2 monomers in the three forms mentioned above: two PRC2b, two PRC2f and one PRC2d. Introducing an additional copy of PRC2 stabilizes looped DNA configurations, as is evidenced by the shift of the free energy minima to smaller end-to-end distances (Figure 4A). On the other hand, restricting the two complexes in a tightly bound dimer configuration (PRC2d) fails to stabilize the looped DNA. It is plausible, however, that other dimeric structures that place less restriction on the movement of individual complexes could facilitate DNA loop formation (64).
Figure 4.

Two PRC2 complexes can bend the DNA cooperatively to drive loop formation. (A) Free energy profiles as a function of the end-to-end distance for the DNA bound with two copies of PRC2bs (red), two copies of PRC2fs (cyan), and with the dimeric structure PRC2d (orange). The black line is for the pure DNA and provided as a reference. It is identical to the one shown in Figure 3. The shaded regions represent standard deviations of the mean. (B) Example structures from the two most populated clusters for simulations carried with two PRC2f bound to the DNA. DNA is shown in golden and the coloring scheme for the protein is identical to that in Figure 1.
We note that the difference between the curves for PRC2b and PRC2f is much more pronounced than that for the monomer results. The presence of one PRC2f only changes the free energy difference between the open (end-to-end distance 800 Å) and looped DNA (end-to-end distance 200 Å), ΔF, from 4.7 to 0.6 kBT (see Supplementary Figure S9). The introduction of an additional PRC2f shifts the free energy difference to −10.8 kBT. Similarly, ΔF for one and two copies of PRC2b is 2.7 and −2.2 kBT, respectively. Therefore, in both cases, the DNA bending effect is more substantial than a simple sum of two individual monomer results. The non-additive change of ΔF indicates the presence of a cooperative DNA bending for multiple PRC2 complexes.
To provide more insight into the observed cooperativity, we performed a structural clustering of the simulated protein-DNA complexes. The most populated conformation (60%) from PRC2f simulations correspond to a hairpin-like DNA structure (Figure 4B). This structure suggests that bending induced by the first PRC2f, which sits in the middle of the DNA, can impact the binding of additional proteins. Therefore, much like the allosteric effect seen between different parts of a protein, the two complexes can ‘communicate’ without direct contact. Such allosteric communications between distal DNA-binding proteins have been observed in a single molecule study from the Xie group (65). The second PRC2f now bridges the two ends of the DNA to stabilize looped conformations. This new binding mode differs significantly from the conformations observed in single PRC2f simulations. It leads to enhanced binding of both PRC2fs in the hairpin structure and gives rise to the non-additive DNA bending. The second most populated cluster (21%) corresponds to a less or non-cooperative configuration in which two PRC2fs interact with the DNA relatively independently. For PRC2b, we found that the order in the two clusters’ population is reversed (Supplementary Figure S10), explaining its less significant cooperativity. Since the cooperativity arises from the allosteric communication between two independent complexes, it disappeared when they were forced into the dimeric conformation constructed in PRC2d.
To directly visualize the allosteric communication between the two PRC2s via DNA, we introduced a new collective variable to quantify the loop size (Figure 5A). This variable compares the sequence distance between the two longest binding sites for each PRC2 core complex, ΔL1 and ΔL2, and selects the larger one. Detailed definition of the loop length is included in the SI. We then determined the free energy surface as a function of the loop length and DNA end-to-end distance. Two basins at small end-to-end distances that correspond to the two clusters shown in Figure 4B are evident on this surface (Figure 5B). The free energy projected onto the loop length is provided in Supplementary Figure S11. We further computed the average length of the loop formed by the second PRC2f as a function of the number of contacts between DNA and the first PRC2f. As shown in Figure 5C, there is a strong correlation between the two proteins. The enhanced DNA contact of the first PRC2f drives the formation of longer loops by the second complex, supporting the allosteric communication between the two. The corresponding analyses for PRC2b simulations are provided in Supplementary Figure S12, supporting similar conclusions.
Figure 5.

Cooperativity between the two PRC2f complexes arises from an allosteric communication mediated by the DNA. (A) Illustration of the DNA loops formed by the two PRC2f complexes. The collective variable, loop length, is defined as the contour length of the larger loop. (B) Two-dimensional free energy profile as a function of the DNA end-to-end distance and the loop length. (C) The average length of the dominant loop (red in part A) as a function of the number of contacts between the DNA and the PRC2f that forms the smaller loop (blue in part A). The shaded region represents standard deviation of the mean.
Single-molecule experiments support conformation-dependent DNA binding of PRC2
To complement computer simulations, we performed single-molecule force spectroscopy experiments to characterize PRC2–DNA interactions as well. Specifically, we incubated a 10 kbp-long bare DNA with 500 nM PRC2 core complex under a physiological salt condition (see Materials and Methods). The DNA ends were then pulled by optical tweezers at a constant velocity of 0.1 μm/s. Force extension curves were recorded to measure the pulling force as a function of the distance between the two ends. As shown in Figure 6 and Supplementary Figure S13, multiple rupture events with a sudden drop in the recorded force can be seen in the force-extension curve. Notably, when the DNA was incubated with PRC2 5mer with AEBP2, rupture events can no longer be detected.
Figure 6.
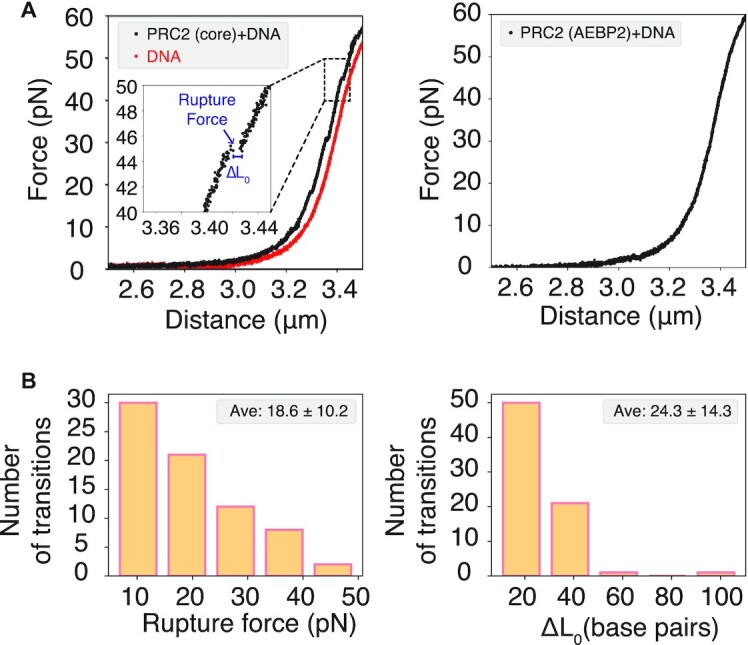
Single molecule experiments revealed distinct behavior of PRC2(core) 4mer and PRC2(AEBP2) 5mer in DNA binding. (A) Example force-extension curves measured for DNA incubated with PRC2 4mer (left) and 5mer (right). The inset highlights the presence of rupture events for DNA bound with 4mer. The trace for bare DNA (red) is provided as a reference. (B) Statistics of the rupture force (left) and DNA contour length change (ΔL0, right) estimated from 73 rupture events detected for DNA bound with PRC2 4mer.
Results from the force spectroscopy experiments are consistent with the free energy profiles shown in Figure 4A. At small forces, PRC2 mediated loops are stable and can compact the DNA. Higher forces will overcome the energetic cost to disrupt PRC2-DNA interactions and release the DNA segment enclosed by the loop. Since the C2-bound configuration (PRC2b), the one adopted by PRC2 5mer with AEBP2, stabilizes the loop to a much lesser extent than PRC2f, the corresponding rupture force is expected to be smaller and may evade detection in pulling experiments. We note that a quantitative interpretation of the experiments with the computed free energy profiles is nontrivial. However, in silico pulling simulations, as shown in Supplementary Figure S14, suggest that unfolding the loop structure presented in Figure 4 can indeed give rise to rupture forces and DNA extension lengths comparable to experimental values.
DISCUSSION
In this work, we characterized conformation-dependent PRC2-DNA interactions with computer simulations and single-molecule experiments. We built atomic structures for the full-length PRC2 core complex via homology modeling and validated their stability with atomistic molecular dynamics simulations. Three conformations that differ in the conformational flexibility of the C2 domain from the SUZ12 subunit were constructed. Via a newly parameterized coarse-grained force field, we were able to reproduce the experimental value for the DNA binding affinity of PRC2. Our simulations support that the C2-domain can engage in direct DNA contacts via its positively charged residues, and the PRC2 structure with a free C2 domain binds more tightly with DNA than the one with a bound C2. Therefore, restricting the exposure of the C2 domain, as is the case upon AEBP2 binding, could reduce the binding affinity of PRC2 to the linker DNA and help recruit the protein to more specific regions for histone methylation (66,67).
We further studied the impact of PRC2 binding on DNA conformation by computing the free energy profile as a function of the DNA end-to-end distance. While binding with DNA tightly, a single copy of PRC2 does not stabilize looped DNA configurations, regardless of the conformation of the C2 domain. PRC2 binding does induce DNA bending due to the curved protein interface. Two copies of PRC2, however, are sufficient for stabilizing DNA looping. We observed a strong cooperative effect of the two proteins in bending the DNA due to a hairpin structure formation. In contrast to other proteins such as HP1 (68,69), multivalent protein-protein interactions are not the driver of the cooperativity. Instead, the cooperativity mainly arises from an allosteric communication between the two proteins due to the ability of PRC2 in DNA bending and the presence of multiple DNA binding domains.
We note that while the computed binding free energy depends on the force field parameters, the presented mechanisms on PRC2-DNA binding are robust. For example, even in simulations carried out without the statistical potential, the same protein-DNA interface shown in Figure 3 was observed (Supplementary Figure S15A). The cooperative role of two PRC2 complexes in DNA looping was preserved as well (Supplementary Figure S15B). The apparent insensitivity of mechanistic insights to model parameters is possibly due to the dominance of electrostatic interactions for PRC2-DNA binding.
Cooperative DNA binding among PRC2 complexes is consistent with results from prior experimental studies (10,33). In particular, Heenan et al. applied AFM imaging to directly visualize the conformation of individual DNA molecules (33). They determined the fraction of DNA adopting compact conformations and looped configurations upon binding with PRC2 at different PRC2 concentrations. Fitting the data with the Hill equation yielded a coefficient of 1.4, which provides direct evidence for a cooperative role of multiple PRC2 in DNA looping. Notably, consistent with the Hill coefficient, two or more PRC2s were observed in looped DNA configurations.
The detection of rupture events in single-molecule force spectroscopy experiments agrees with PRC2-mediated DNA looping. However, the absence of rupture with PRC2 5mers does not rule out looping due to the limit of experimental resolution. Our simulations indeed support the formation of loops by 5mers, though to a lesser extent than 4mers. PRC2 5mer mediated DNA looping has been directly observed in AFM imaging (33). It’s worth noting that the AFM experiments were performed at a different salt concentration from our force spectroscopy experiments. The salt concentration can significantly impact the DNA binding affinity of PRC2 5mer (see Supplementary Figure S16) and the probability for loop formation and detection.
PRC2 mediated looping observed here could play important roles for histone mark spreading inside the nucleus (32,70). As shown by Reinberg and coworkers (71), chromatin loops may serve as nucleation centers from which PRC2 can spread histone marks to both neighboring and distal nucleosomes via 3D diffusion. This nucleation-spreading mechanism could make more efficient use of protein molecules with limited copy numbers (72). Notably, using chromosome conformation capture (Hi-C) experiments to probe contacts between DNA segments inside the nucleus, PRC proteins mediated chromatin loops have been detected (73–78). However, it is worth noting that the current experiments do not distinguish whether PRC2 plays a structural role in stabilizing these loops or merely prepares the chromatin for PRC1 binding (73,79,80). Additional experiments are needed to disentangle the specific role of the two complexes and verify whether PRC2 loops chromatin in vivo via the mechanism uncovered in this study.
Supplementary Material
Contributor Information
Xingcheng Lin, Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA, USA.
Rachel Leicher, Laboratory of Nanoscale Biophysics and Biochemistry, The Rockefeller University, New York, NY 10065, USA; Tri-Institutional PhD Program in Chemical Biology, New York, NY 10065, USA.
Shixin Liu, Laboratory of Nanoscale Biophysics and Biochemistry, The Rockefeller University, New York, NY 10065, USA.
Bin Zhang, Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35GM133580 to B.Z.]; S.L. acknowledges support from the Pershing Square Sohn Cancer Research Alliance; Starr Cancer Consortium; NIH [DP2HG010510]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Margueron R., Reinberg D.. The polycomb complex PRC2 and its mark in life. Nature. 2011; 469:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wiles E.T., Selker E.U.. H3K27 methylation: a promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev. 2017; 43:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schuettengruber B., Bourbon H.-M., Di Croce L., Cavalli G.. Genome regulation by polycomb and trithorax: 70 years and counting. Cell. 2017; 171:34–57. [DOI] [PubMed] [Google Scholar]
- 4. Reinberg D., Vales L.D.. Chromatin domains rich in inheritance. Science. 2018; 361:33–34. [DOI] [PubMed] [Google Scholar]
- 5. Højfeldt J.W., Laugesen A., Willumsen B.M., Damhofer H., Hedehus L., Tvardovskiy A., Mohammad F., Jensen O.N., Helin K.. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol. 2018; 25:225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Laugesen A., Højfeldt J.W., Helin K.. Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol Cell. 2019; 74:8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yu J.-R., Lee C.-H., Oksuz O., Stafford J.M., Reinberg D.. PRC2 is high maintenance. Genes Dev. 2019; 33:903–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davidovich C., Zheng L., Goodrich K.J., Cech T.R.. Promiscuous RNA binding by polycomb repressive complex 2. Nat. Struct. Mol. Biol. 2013; 20:1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davidovich C., Cech T.R.. The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. RNA. 2015; 21:2007–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang X., Paucek R.D., Gooding A.R., Brown Z.Z., Ge E.J., Muir T.W., Cech T.R.. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat. Struct. Mol. Biol. 2017; 24:1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Choi J., Bachmann A.L., Tauscher K., Benda C., Fierz B., Müller J.. DNA binding by PHF1 prolongs PRC2 residence time on chromatin and thereby promotes H3K27 methylation. Nat. Struct. Mol. Biol. 2017; 24:1039–1047. [DOI] [PubMed] [Google Scholar]
- 12. Beltran M., Tavares M., Justin N., Khandelwal G., Ambrose J., Foster B.M., Worlock K.B., Tvardovskiy A., Kunzelmann S., Herrero J.et al.. G-Tract RNA removes polycomb repressive complex 2 from genes. Nat. Struct. Mol. Biol. 2019; 26:899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cao R., Zhang Y.. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell. 2004; 15:57–67. [DOI] [PubMed] [Google Scholar]
- 14. Margueron R., Justin N., Ohno K., Sharpe M.L., Son J., Drury III W.J., Voigt P., Martin S.R., Taylor W.R., De Marco V.et al.. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009; 461:762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiao L., Liu X.. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science. 2015; 350:aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Justin N., Zhang Y., Tarricone C., Martin S.R., Chen S., Underwood E., De Marco V., Haire L.F., Walker P.A., Reinberg D.et al.. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016; 7:11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brooun A., Gajiwala K.S., Deng Y.-L., Liu W., Bolaños B., Bingham P., He Y.-A., Diehl W., Grable N., Kung P.-P.et al.. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat. Commun. 2016; 7:11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Comet I., Riising E.M., Leblanc B., Helin K.. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer. 2016; 16:803–810. [DOI] [PubMed] [Google Scholar]
- 19. Kasinath V., Faini M., Poepsel S., Reif D., Feng X.A., Stjepanovic G., Aebersold R., Nogales E.. Structures of human PRC2 with its cofactors AEBP2 and JARID2. Science. 2018; 359:940–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poepsel S., Kasinath V., Nogales E.. Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat. Struct. Mol. Biol. 2018; 25:154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaneko S., Li G., Son J., Xu C.-F., Margueron R., Neubert T.A., Reinberg D.. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010; 24:2615–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kanhere A., Viiri K., Araújo C.C., Rasaiyaah J., Bouwman R.D., Whyte W.A., Pereira C.F., Brookes E., Walker K., Bell G.W., et al.. Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol. Cell. 2010; 38:675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ciferri C., Lander G.C., Maiolica A., Herzog F., Aebersold R., Nogales E.. Molecular architecture of human polycomb repressive complex 2. Elife. 2012; 1:e00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kasinath V., Poepsel S., Nogales E.. Recent structural insights into polycomb repressive complex 2 regulation and substrate binding. Biochemistry. 2019; 58:346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kasinath V., Beck C., Sauer P., Poepsel S., Kosmatka J., Faini M., Toso D., Aebersold R., Nogales E.. JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science. 2021; 371:eabc3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Han Z., Xing X., Hu M., Zhang Y., Liu P., Chai J.. Structural basis of EZH2 recognition by EED. Structure. 2007; 15:1306–1315. [DOI] [PubMed] [Google Scholar]
- 27. Schmitges F.W., Prusty A.B., Faty M., Stützer A., Lingaraju G.M., Aiwazian J., Sack R., Hess D., Li L., Zhou S.et al.. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell. 2011; 42:330–341. [DOI] [PubMed] [Google Scholar]
- 28. Antonysamy S., Condon B., Druzina Z., Bonanno J.B., Gheyi T., Zhang F., MacEwan I., Zhang A., Ashok S., Rodgers L.et al.. Structural context of disease-associated mutations and putative mechanism of autoinhibition revealed by X-ray crystallographic analysis of the EZH2-SET domain. PLoS ONE. 2013; 8:e84147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen S., Jiao L., Shubbar M., Yang X., Liu X.. Unique structural platforms of Suz12 dictate distinct classes of PRC2 for chromatin binding. Mol. Cell. 2018; 69:840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen S., Jiao L., Liu X., Yang X., Liu X.. A dimeric structural scaffold for PRC2-PCL targeting to CpG island chromatin. Mol. Cell. 2020; 77:1265–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davidovich C., Goodrich K.J., Gooding A.R., Cech T.R.. A dimeric state for PRC2. Nucleic Acids Res. 2014; 42:9236–9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leicher R., Ge E.J., Lin X., Reynolds M.J., Xie W., Walz T., Zhang B., Muir T.W., Liu S.. Single-molecule and in silico dissection of the interaction between polycomb repressive complex 2 and chromatin. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:30465–30475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heenan P.R., Wang X., Gooding A.R., Cech T.R., Perkins T.T.. Bending and looping of long DNA by polycomb repressive complex 2 revealed by AFM imaging in liquid. Nucleic Acids Res. 2020; 48:2969–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.-Y., Pieper U., Sali A.. Comparative protein structure modeling using modeller. Curr. Protoc. Bioinforma. 2006; 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B.L., Grubmüller H., MacKerell A.D.. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017; 14:71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926–935. [Google Scholar]
- 37. Bussi G., Donadio D., Parrinello M.. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007; 126:014101. [DOI] [PubMed] [Google Scholar]
- 38. Parrinello M., Rahman A.. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981; 52:7182–7190. [Google Scholar]
- 39. Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E.. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015; 1-2:19–25. [Google Scholar]
- 40. Hinckley D.M., Lequieu J.P., de Pablo J.J.. Coarse-grained modeling of DNA oligomer hybridization: length, sequence, and salt effects. J. Chem. Phys. 2014; 141:035102. [DOI] [PubMed] [Google Scholar]
- 41. Freeman G.S., Hinckley D.M., Lequieu J.P., Whitmer J.K., de Pablo J.J.. Coarse-grained modeling of DNA curvature. J Chem Phys. 2014; 141:165103. [DOI] [PubMed] [Google Scholar]
- 42. Clementi C., Nymeyer H., Onuchic J.N.. Topological and energetic factors: what determines the structural details of the transition state ensemble and ‘En-Route’ intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol. 2000; 298:937–953. [DOI] [PubMed] [Google Scholar]
- 43. Noel J.K., Whitford P.C., Sanbonmatsu K.Y., Onuchic J.N.. SMOG@ctbp: simplified deployment of structure-based models in GROMACS. Nucleic Acids Res. 2010; 38:W657–W661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Noel J.K., Levi M., Raghunathan M., Lammert H., Hayes R.L., Onuchic J.N., Whitford P.C.. SMOG 2: a versatile software package for generating structure-based models. PLoS Comput. Biol. 2016; 12:e1004794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gao M., Skolnick J.. DBD-Hunter: a knowledge-based method for ithe prediction of DNA– protein Interactions. Nucleic Acids Res. 2008; 36:3978–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Plimpton S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995; 117:1–19. [Google Scholar]
- 47. Takada S., Kanada R., Tan C., Terakawa T., Li W., Kenzaki H.. Modeling structural dynamics of biomolecular complexes by coarse-grained molecular simulations. Acc. Chem. Res. 2015; 48:3026–3035. [DOI] [PubMed] [Google Scholar]
- 48. Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984; 81:511–519. [Google Scholar]
- 49. Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985; 31:1695–1697. [DOI] [PubMed] [Google Scholar]
- 50. Ge E.J., Jani K.S., Diehl K.L., Müller M.M., Muir T.W.. Nucleation and propagation of heterochromatin by the histone methyltransferase PRC2: geometric constraints and impact of the regulatory subunit JARID2. J. Am. Chem. Soc. 2019; 141:15029–15039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Latham A.P., Zhang B.. Maximum entropy optimized force field for intrinsically disordered proteins. J. Chem. Theory Comput. 2020; 16:773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bhattacherjee A., Krepel D., Levy Y.. Coarse-grained models for studying protein diffusion along DNA: coarse-grained models for protein-DNA interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016; 6:515–531. [Google Scholar]
- 53. Niina T., Brandani G.B., Tan C., Takada S.. Sequence-dependent nucleosome sliding in rotation-coupled and uncoupled modes revealed by molecular simulations. PLoS Comput. Biol. 2017; 13:e1005880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang B., Zheng W., Papoian G.A., Wolynes P.G.. Exploring the free energy landscape of nucleosomes. J. Am. Chem. Soc. 2016; 138:8126–8133. [DOI] [PubMed] [Google Scholar]
- 55. Lequieu J., Schwartz D.C., de Pablo J.J.. In silico evidence for sequence-dependent nucleosome sliding. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E9197–E9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Parsons T., Zhang B.. Critical role of histone tail entropy in nucleosome unwinding. J Chem Phys. 2019; 150:185103. [DOI] [PubMed] [Google Scholar]
- 57. Lequieu J., Córdoba A., Schwartz D.C., de Pablo J.J.. Tension-dependent free energies of nucleosome unwrapping. ACS Cent Sci. 2016; 2:660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Trzesniak D., Kunz A.-P.E., van Gunsteren W.F.. A comparison of methods to compute the potential of mean force. ChemPhysChem. 2007; 8:162–169. [DOI] [PubMed] [Google Scholar]
- 59. Lowary P., Widom J.. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998; 276:19–42. [DOI] [PubMed] [Google Scholar]
- 60. Vasudevan D., Chua E.Y., Davey C.A.. Crystal structures of nucleosome core particles containing the ‘601’ strong positioning sequence. J. Mol. Biol. 2010; 403:1–10. [DOI] [PubMed] [Google Scholar]
- 61. Sun A., Li F., Liu Z., Jiang Y., Zhang J., Wu J., Shi Y.. Structural and biochemical insights into human zinc finger protein AEBP2 reveals Interactions with RBBP4. Protein Cell. 2018; 9:738–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ridgway N., McLeod R.. Biochemistry of Lipids, Lipoproteins and Membranes. 2015; 6th ednElsevier Science. [Google Scholar]
- 63. Ortiz V., de Pablo J.J.. Molecular Origins of DNA Flexibility: Sequence Effects on Conformational and Mechanical Properties. Phys. Rev. Lett. 2011; 106:238107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Grau D., Zhang Y., Lee C.H., Valencia-Sánchez M., Zhang J., Wang M., Holder M., Svetlov V., Tan D., Nudler E.et al.. Structures of Monomeric and Dimeric PRC2:EZH1 Reveal Flexible Modules Involved in Chromatin Compaction. Nat. Commun. 2021; 12:714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kim S., Broströmer E., Xing D., Jin J., Chong S., Ge H., Wang S., Gu C., Yang L., Gao Y.Q.et al.. Probing allostery through DNA. Science. 2013; 339:816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Healy E., Mucha M., Glancy E., Fitzpatrick D.J., Conway E., Neikes H.K., Monger C., Van Mierlo G., Baltissen M.P., Koseki Y.et al.. PRC2.1 and PRC2.2 synergize to coordinate H3K27 trimethylation. Mol. Cell. 2019; 76:437–452. [DOI] [PubMed] [Google Scholar]
- 67. Højfeldt J.W., Hedehus L., Laugesen A., Tatar T., Wiehle L., Helin K.. Non-core subunits of the PRC2 complex are collectively required for its target-site specificity. Mol Cell. 2019; 76:423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Larson A.G., Elnatan D., Keenen M.M., Trnka M.J., Johnston J.B., Burlingame A.L., Agard D.A., Redding S., Narlikar G.J.. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature. 2017; 547:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Strom A.R., Emelyanov A.V., Mir M., Fyodorov D.V., Darzacq X., Karpen G.H.. Phase separation drives heterochromatin domain formation. Nature. 2017; 547:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xie W.J., Zhang B.. Learning the formation mechanism of domain-level chromatin states with epigenomics data. Biophys. J. 2019; 116:2047–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Oksuz O., Narendra V., Lee C.H., Descostes N., LeRoy G., Raviram R., Blumenberg L., Karch K., Rocha P.P., Garcia B.A.et al.. Capturing the onset of PRC2-mediated repressive domain formation. Mol. Cell. 2018; 70:1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bonnet J., Lindeboom R.G., Pokrovsky D., Stricker G., Çelik M.H., Rupp R.A., Gagneur J., Vermeulen M., Imhof A., Müller J.. Quantification of proteins and histone marks in Drosophila embryos reveals stoichiometric relationships impacting chromatin regulation. Dev. Cell. 2019; 51:632–644. [DOI] [PubMed] [Google Scholar]
- 73. Joshi O., Wang S.Y., Kuznetsova T., Atlasi Y., Peng T., Fabre P.J., Habibi E., Shaik J., Saeed S., Handoko L.et al.. Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell. 2015; 17:748–757. [DOI] [PubMed] [Google Scholar]
- 74. Schoenfelder S., Sugar R., Dimond A., Javierre B.-M., Armstrong H., Mifsud B., Dimitrova E., Matheson L., Tavares-Cadete F., Furlan-Magaril M.et al.. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat. Genet. 2015; 47:1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vieux-Rochas M., Fabre P.J., Leleu M., Duboule D., Noordermeer D.. Clustering of mammalian Hox genes with other H3K27me3 targets within an active nuclear domain. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:4672–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Eagen K.P., Aiden E.L., Kornberg R.D.. Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:8764–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ogiyama Y., Schuettengruber B., Papadopoulos G.L., Chang J.M., Cavalli G.. Polycomb-dependent chromatin looping contributes to gene silencing during Drosophila development. Mol. Cell. 2018; 71:73–88. [DOI] [PubMed] [Google Scholar]
- 78. Hsieh T. H.S., Cattoglio C., Slobodyanyuk E., Hansen A.S., Rando O.J., Tjian R., Darzacq X.. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Mol. Cell. 2020; 78:539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Plys A.J., Davis C.P., Kim J., Rizki G., Keenen M.M., Marr S.K., Kingston R.E.. Phase separation of polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 2019; 33:799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tatavosian R., Kent S., Brown K., Yao T., Duc H.N., Huynh T.N., Zhen C.Y., Ma B., Wang H., Ren X.. Nuclear condensates of the polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem. 2019; 294:1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.