

Abstract
Formic acid (HCOOH) is an important component of atmospheric acidity but its budget is poorly understood, with prior observations implying substantial missing sources. Here we combine pole-to-pole airborne observations from the Atmospheric Tomography Mission (ATom) with chemical transport model (GEOS-Chem CTM) and back trajectory analyses to provide the first global in-situ characterization of HCOOH in the remote atmosphere. ATom reveals sub-100 ppt HCOOH concentrations over most of the remote oceans, punctuated by large enhancements associated with continental outflow. Enhancements correlate with known combustion tracers and trajectory-based fire influences. The GEOS-Chem model underpredicts these in-plume HCOOH enhancements, but elsewhere we find no broad indication of a missing HCOOH source in the background free troposphere. We conclude that missing non-fire HCOOH precursors inferred previously are predominantly short-lived. We find indications of a wet scavenging underestimate in the model consistent with a positive HCOOH bias in the tropical upper troposphere. Observations reveal episodic evidence of ocean HCOOH uptake, which is well-captured by GEOS-Chem; however, despite its strong seawater undersaturation HCOOH is not consistently depleted in the remote marine boundary layer. Over fifty fire and mixed plumes were intercepted during ATom with widely varying transit times and source regions. HCOOH:CO normalized excess mixing ratios in these plumes range from 3.4 to >50 ppt/ppb CO and are often over an order of magnitude higher than expected primary emission ratios. HCOOH is thus a major reactive organic carbon reservoir in the aged plumes sampled during ATom, implying important missing pathways for in-plume HCOOH production.
Keywords: formic acid, remote atmosphere, fire, deposition, Atmospheric Tomography Mission, iodide CIMS, chemical transport model, back trajectory
TOC Graphic:
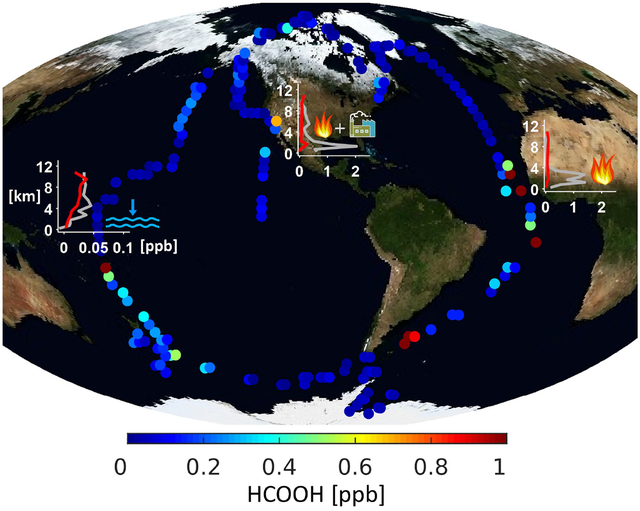
Observed formic acid (HCOOH) disribution below 5 km during ATom-3. Each data point is colored by the mean HCOOH mixing ratio for the surrounding 1.5° latitude ×1.5° longitude bin along the flight track. The 3 panels inset show the modeled (red) vs. observed (grey) median vertical profiles for example flights demonstrating the influence of fire emissions (right, flight on 05/14/2018 off the west African coast), combined fire and urban sources (middle, flight on 05/21/2018 over California), and ocean uptake (left, a tropical Pacific flight on 10/06/2017) on atmospheric HCOOH.
1. Introduction
Formic acid (HCOOH) is ubiquitous in the atmosphere, one of the largest drivers of precipitation acidity in remote regions1–3, and a major OH sink in the aqueous phase4. HCOOH therefore acts as a pH buffer and affects aqueous phase chemistry through its effects on oxidant levels, pH-dependent reaction rates, and solubilities. Prior work has shown HCOOH to be present in the atmosphere at levels far greater than can be explained based on its known sources and sinks5–8; however, such studies have mainly focused on terrestrial and near-source regions. Here we combine observations from the NASA DC-8 flying laboratory during the Atmospheric Tomography Mission (ATom-3 and 4) with a global chemical transport model (GEOS-Chem CTM) to provide the first global in-situ characterization of HCOOH in the remote atmosphere.
Recent evaluations of the global HCOOH budget have estimated its total known source at ~60 Tg/yr with subsequent removal dominated by deposition5, 6. Photochemical production from volatile organic compounds (VOCs) is believed to be the dominant HCOOH source over forested regions9, 10, urban areas11, and on a global scale5, 6, with direct emissions from the terrestrial biosphere12, soils13, ants14, and urban/industrial activities15 playing more modest roles. Secondary formation pathways that have been identified include hydroxyl radical (OH)- and ozone (O3)-initiated oxidation of isoprene, monoterpenes, and their oxidation products16, ozonolysis of other alkenes17, and alkyne oxidation18. Acetaldehyde has also been shown to be a precursor of HCOOH via its photo-tautomerization to vinyl alcohol19; subsequent work using the GEOS-Chem CTM found that this pathway can account up to 60% of the modeled HCOOH production over remote oceans20. Aerosol aging has been implicated as another HCOOH source through heterogeneous (or condensed-phase) oxidation by OH21, ozonolysis22, and photolysis23–26.
HCOOH is also produced from biomass burning27–30, and recent work indicates that this source may be larger than previously thought. Yokelson et al. (2009)31 find that in many biomass burning plumes organic acids are the third most important carbon reservoir—behind CO2 and CO and ahead of CH4. The same authors report organic acid:CO enhancement ratios of up to 18% (mol/mol) in aged fire plumes—with secondary acid production exceeding the abundance of known precursors—and invoke unmeasured pyrogenic VOCs as explanation. Chaliyakunnel et al (2016)32, Pommier et al. (2017)33, and Yamanouchi et al. (2020)34 likewise observed fire HCOOH:CO enhancement ratios much higher than primary emission ratios could explain, implying substantial HCOOH production during fire plume aging.
High levels of HCOOH have also been measured in remote areas, with a variety of formation mechanisms proposed. For example, Baboukas et al. (2000)35 argued that an indirect marine HCOOH source of ~50 Tg/yr globally was required to explain measured mixing ratios (averaging ~450 ppt) in the Atlantic marine boundary layer (MBL). Dibb and Arsenault (2002)36 measured higher HCOOH in firn air (up to 6 ppb) than in the overlying atmosphere and invoked a source from the oxidation of organics in snow. Dibb et al. (1996)37 presented strong evidence of enhanced formic acid at Summit, Greenland from the long-range transport of wildfire smoke. Jones et al. (2014)38 measured HCOOH concentrations in the European Arctic MBL that were 50% higher than over the surrounding land areas, and proposed a source from stabilized Criegee intermediates following photolysis of ocean-released diiodomethane (CH2I2). Mungall et al. (2017)39 reported HCOOH levels of up to 4 ppb in the Canadian Arctic MBL, with evidence pointing to a photochemical or heterogeneous source from the sea surface microlayer.
The above findings highlight the need for a better understanding of atmospheric HCOOH and its sources and sinks. A key open question is whether the inferred missing HCOOH source persists throughout the global atmosphere (for example, as inferred for acetaldehyde in Wang et al. (2019)40), which would imply a ubiquitous long-lived precursor pool, or if it primarily manifests near and downwind of continental VOC source regions. The ATom campaign41 collected observations of HCOOH and a suite of related tracers from pole-to-pole over the remote Pacific and Atlantic Oceans with continuous vertical profiling and across multiple seasons (Fig. 1). The dataset thus provides a new opportunity to address the above science issues on a global scale. In this paper we apply the GEOS-Chem CTM to interpret the ATom measurements in terms of their implications for the HCOOH budget in the remote atmosphere.
Figure 1.
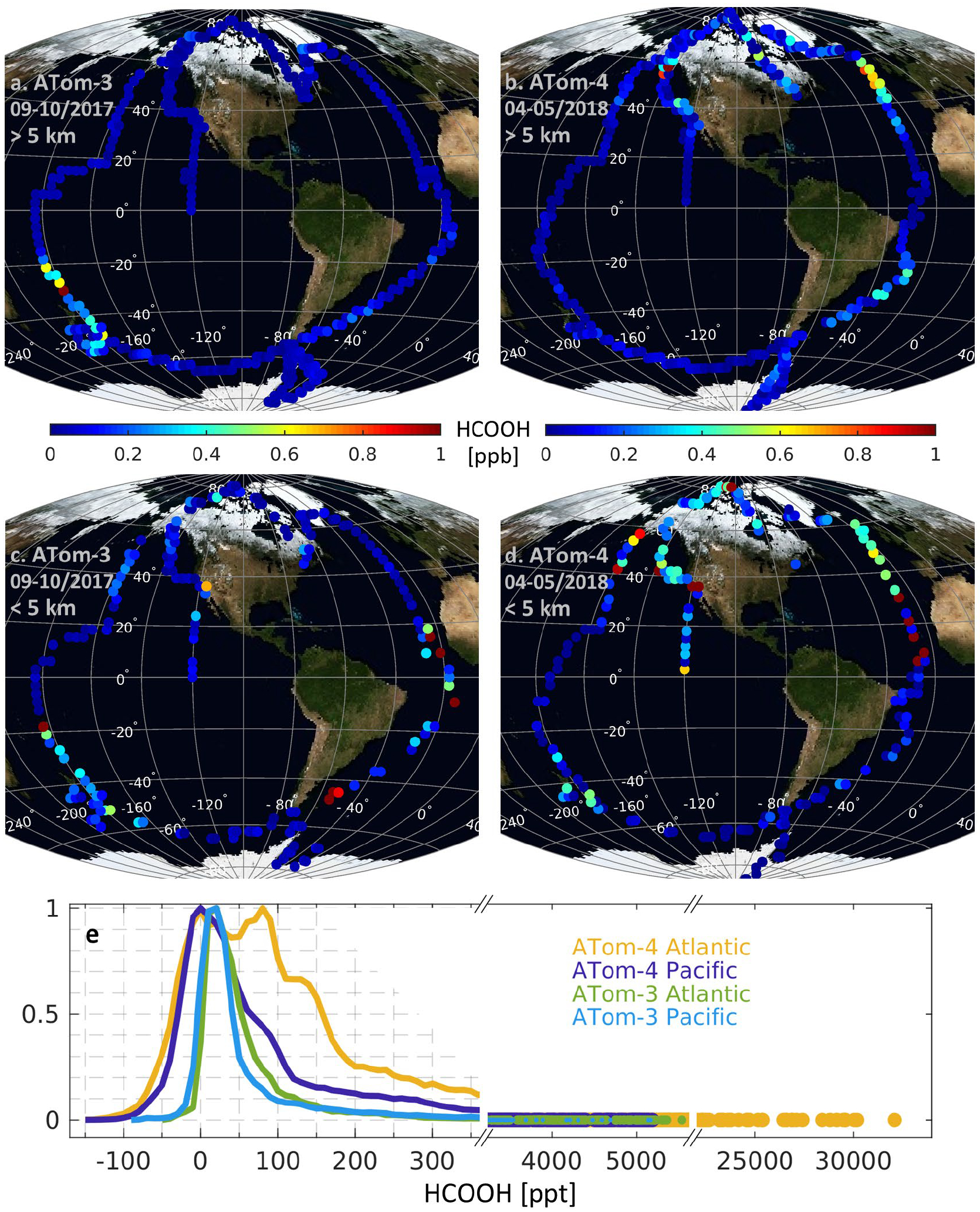
Observed HCOOH distribution during ATom-3 and 4. Panels a and c map the HCOOH mixing ratios measured during ATom-3 above (a) and below (c) 5 km altitude. Panels (b) and (d) show the corresponding plots for ATom-4. Each datapoint in panels a–d is colored by the mean HCOOH mixing ratio for the surrounding 1.5° latitude × 1.5° longitude bin. Panel e shows normalized frequency distributions for the ATom HCOOH observations with different linear scales at the upper range of the distribution. Some negative values occur reflecting uncertainty and variability in the instrument background signal.
2. Methods
2.1. Atmospheric Tomography (ATom) 3 and 4 aircraft campaigns
HCOOH measurements were conducted onboard the NASA DC-8 aircraft as part of the two final ATom deployments41 during September-October 2017 (ATom-3) and April-May 2018 (ATom-4). ATom was designed to (i) perform detailed chemical characterization of the global remote atmosphere, (ii) establish a comprehensive dataset of climatically and chemically important atmospheric constituents, and (iii) provide new observations to improve models42. Each ATom deployment spans nearly pole-to-pole over the Pacific and Atlantic Oceans with continuous vertical profiling from within the MBL to above the tropopause.
2.1.1. HCOOH measurements by Iodide Ion Time-of-Flight Chemical Ionization Mass Spectrometry (I-CIMS) during ATom-3 and 4
HCOOH was measured once per second using an iodide-ion time of flight chemical ionization mass spectrometer (I-CIMS), as described in Veres et al. (2020)43. Pressure and temperature control of the sampling inlet and humidity control of the ion molecule reaction region guaranteed that the instrument response was independent of aircraft altitude and ambient conditions. The 70 cm long Teflon inlet tube was controlled to 40 °C and at ambient pressure. The pressure upstream of the ion-molecule reaction region was controlled to 140 mb, and the ion-molecule reaction region itself was controlled to 40 mb (ATom-4) or 50 mb (ATom-3) to maintain constant ion-molecule reaction times. The measurement sensitivity to HCOOH depends on water concentration in the ion molecule reaction region, and water vapor was added to this region to compensate for changing ambient humidity and maintain constant detection sensitivity. The ratio of the two reagent ions (iodide and the iodide-water cluster) served as a proxy for humidity in the instrument, and water was controlled to maintain a constant reagent-ion cluster ratio. Effects of any residual humidity changes were then corrected based on a laboratory quantification of the water-dependent HCOOH sensitivity as a linear function of the iodide:iodide-water ion ratio.
HCOOH was detected as a cluster with iodide at m/z 172.9105, with a resolution of m/Δm=5000. The instrument sensitivity to HCOOH was determined before and after the campaign by laboratory-based standard addition of HCOOH from a permeation tube source with the instrument in its flight configuration. The permeation tube was held in an oven controlled to 40 °C with a 50 sccm continuous flow of N2. The HCOOH emission from the permeation tube was determined by catalytically converting HCOOH to CO2 followed by CO2 detection.44 The 47 ng/min HCOOH permeation tube emission was mixed with the 6 slm inlet flow to give a 3.8 ppb calibration mixing ratio. The uncertainty in this calibration process is approximately 15%. Under typical operating conditions, the instrument sensitivity was approximately 8 ion counts/second/ppt HCOOH, with reagent ion signals of 3 MHz for iodide and 1.5 MHz for the iodide-water cluster. In-flight calibrations of chlorine and nitric acid demonstrated that the reagent-ion-normalized instrument sensitivity did not change over the course of each campaign.
The instrument zeros were determined in-flight for 30 s every 8 min by overfilling the inlet with ambient air drawn through a scrubber. After ATom-4, an additional background measurement capability was added that involved the periodic addition of dry N2 at the inlet tip without going through a scrubber. These two zeroing methods were found to agree to within the variability of the background determinations. For ATom, average background signals were typically 300 ppt; these were interpolated between consecutive determinations and subtracted from the total signal to determine the ambient HCOOH contribution. Background signals varied by 50 ppt to 100 ppt between successive quantifications, imposing an additional 50–100 ppt measurement uncertainty (and a lower limit of detection) that is not reduced by averaging over times less than background measurement interval. The measurement precision was better than the instrument background uncertainty, and was typically below 10 ppt for 1 s measurements, ranging from 3–15 ppt.
2.1.2. Other measurements
The DC-8 aircraft featured a large instrument payload during ATom, and we employ the following measurements to aid interpretation of the HCOOH dataset. For species measured by more than one instrument, dataset selection was based on time response, data availability, and nominal accuracy. Carbon monoxide (CO) was measured with the NOAA Picarro cavity ring-down spectrometer (G2401-m)45 and with the Harvard Quantum Cascade Laser System (QCLS)46. We use here the merged CO.X product41, which is based primarily on the QCLS observations with gaps filled using the NOAA Picarro observations. We use hydrogen cyanide (HCN) observations from the Caltech chemical ionization mass spectrometer using CF3O− reagent ions (CIT-CIMS)47, 48, and formaldehyde (HCHO) observations from the NASA In Situ Airborne Formaldehyde (ISAF) instrument49, 50. VOC measurements used here were obtained with the NCAR Trace Organic Gas Analyzer (TOGA)51 and the UC Irvine Whole Air Sampling System (WAS)52. NOx (NO+NO2) was measured by the NOAA Nitrogen Oxides and Ozone (NOyO3) instrument53. Black carbon (BC) was measured with the NOAA Single Particle Soot Photometer (SP2)54. Submicron organic aerosol (OA) was measured with the University of Colorado High-Resolution Time-of-Flight Aerosol Mass Spectrometer (HR-AMS)55, 56. BC and OA are both reported for T = 273 K and P = 1013 hPa; we perform all unit conversions accordingly. Submicron organic carbon aerosol (OC) is derived from OA based on the OA:OC ratio reported by the instrument team. We use the Particle Analysis by Laser Mass Spectrometry (PALMS) biomass burning particle number fraction57 in our analysis to aid in plume source attribution. Finally, we use the cloud indicator values from the Cloud, Aerosol, and Precipitation Spectrometer (CAPS)58 to identify within-cloud sampling periods.
2.2. GEOS-Chem simulation
We use the GEOS-Chem CTM in its Classic mode59, with detailed HOx-NOx-VOC-ozone-halogen-aerosol chemistry following JPL/IUPAC recommendations and photolysis frequencies calculated using Fast-JX algorithms60–62. The simulation is driven by assimilated meteorological data (Goddard Earth Observation System Forward Processing, GEOS-FP) from the NASA Global Modeling and Assimilation Office (GMAO) and conducted at 2°×2.5° horizontal resolution with time steps of 15 min (transport/convection) and 30 min (chemistry/emission). We use the TPCORE advection algorithm63, convective mass fluxes from the meteorological archive64, and non-local boundary layer mixing65.
We employ a customized GEOS-Chem simulation for HCOOH building on prior work by Shaw et al. (2018)20, Millet et al. (2015)5, and Paulot et al. (2011)6. Photochemical HCOOH production proceeds as described by Millet et al. (2015), with photo-tautomerization of acetaldehyde updated according to Shaw et al. (2018)20. Global anthropogenic emissions for HCOOH and other species are from the Community Emissions Data System inventory66, overwritten for Africa by the Diffuse and Inefficient Combustion Emissions in Africa dataset (DICE-Africa)67. Global HCOOH emissions from agriculture and natural soils are scaled to the corresponding ammonia and NOx fluxes following prior work6. Direct HCOOH biogenic emissions are calculated using the Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN v2.1)68, implemented in GEOS-Chem as described by Hu et al. (2015)69. Biomass burning emissions are based on the Global Fire Emissions Database with small fires (GFED4s)70 with recommended species-specific emission factors (http://www.globalfiredata.org)71. Ocean emissions of HCOOH precursors (alkenes and isoprene) are estimated following Millet et al. (2015)5 and Paulot et al. (2011)6. Dry deposition of HCOOH and other species is computed using a modified Wesely scheme72, 73; over oceans this is equivalent to an infinite liquid-phase sink and hence unidirectional air-to-sea flux. The resulting monthly-mean HCOOH dry deposition velocities during the timeframes of ATom-3 and ATom-4 typically range between near-zero and 0.8 cm/s over land and reach 2.2 cm/s over ocean. Wet deposition is as described by Amos et al. (2012)74 and Mari et al. (2000)75. Uptake of HCOOH on ice and dust follows Paulot et al. (2011)6.
2.3. Back trajectory analysis
We use atmospheric back trajectories computed along the ATom flight tracks with the Traj3D model76, 77 to interpret the observations in terms of their source footprint and source/sink influences. The model is driven by National Centers for Environmental Prediction (NCEP) Global Forecast System (GFS) 0.5°×0.5° resolution meteorology. For each minute along the flight track, a cluster of 245 trajectories is initialized in a 0.3°latitude × 0.3° longitude × 20 hPa cube centered on the aircraft position. Each trajectory is then run 30 days backwards with a 3-hour time-step. Traj3D model results used here include individual trajectories, cluster ensemble distributions, and cluster-average results.
Two influence processes are employed in this work, as follows. (1) Fire influence. A trajectory is considered fire-influenced if it encountered a fire hotspot at a time and location indicated by one of the following three products: Moderate Resolution Imaging Spectroradiometer (MODIS) Fire Radiative Power (FRP) (Collection 6)78; Visible Infrared Imaging Radiometer Suite (VIIRS) 375-m FRP79; or Global Fire Emissions Database (GFED) v2. Fire plume heights were calculated based on FRP for MODIS and VIIRS, and assumed to be within the boundary layer for GFED. (2) Land boundary layer (BL). A trajectory is considered to be influenced by the land boundary layer if it entered the BL over any continent.
3. Results and Discussion
3.1. HCOOH distribution over remote oceans and role of fire plumes in driving variability
Figure 1 shows horizontal and frequency distributions of HCOOH mixing ratios observed during ATom-3 and 4. We see that the ATom flights sampled a broad range of HCOOH concentrations from below the instrumental LOD to above 30 ppb (Fig. 1f). However, the majority of the distribution is characterized by very low concentrations, with 80% (ATom-3) and 61% (ATom-4) of the data falling below 100 ppt (Fig. 1). While the instrumental LOD prevents accurate quantification of the lowest observed concentrations, the data clearly reveal sub-100 ppt HCOOH background concentrations over most of the remote oceans. More frequent HCOOH enhancements are apparent during ATom-4 than ATom-3 (Fig. 1e–f); as will be seen this reflects strong fire influence during this campaign. In general, lower HCOOH mixing ratios are seen aloft (Figs. 1a/1b vs. 1c/1d), but with clear enhancements in certain regions: east of Australia, east of South America, and over the springtime northern oceans.
Figure 2a compares the vertical HCOOH distribution observed during ATom to that seen in prior aircraft campaigns (Table S1) over the ocean. We see that the median ATom HCOOH profile is very similar to that observed during SONEX (10–11/1997)80 and PEM-Tropics A (08–09/1996)81, particularly in the free troposphere. PEM-West A (09–10/1991)82, PEM-West B (02–03/1994)83, and INTEX-B (04–05/2006)84, 85 sampled closer to land, with the associated Asian/North American outflow leading to higher median HCOOH concentrations. TRACE-A (09–10/1992)86 encountered extremely high HCOOH levels in the south Atlantic due to biomass burning in South America and southern Africa. Together, these campaigns are consistent in revealing HCOOH concentrations on the order of <100 ppt over the remote oceans, rising to >100 ppt closer to continental regions, and much higher under strong fire influence.
Figure 2.
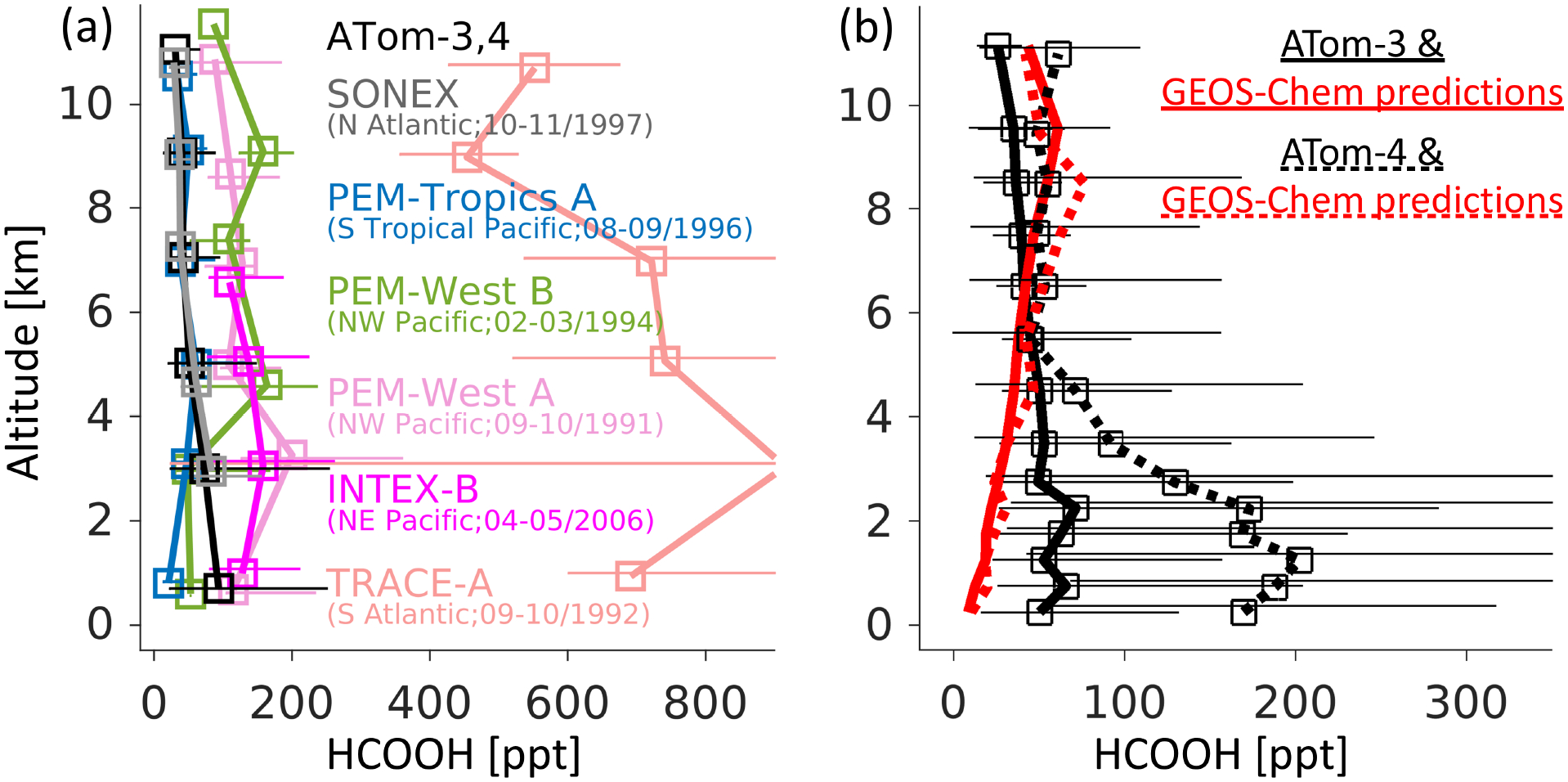
Median HCOOH vertical profiles measured during ATom-3 and 4, and comparison with (a) prior aircraft observations over ocean and (b) GEOS-Chem model predictions. The vertical bin resolution differs between panels (a) and (b) (a: 2 km; b: 0.5 km below 3 km altitude, 1 km above 3 km altitude) to reflect differing sampling densities between the campaigns. Horizontal bars indicate 25th-75th percentiles for each vertical bin. For panel (a) observations over land have been filtered out, and the SONEX dataset further employs ozone and latitude filters following Singh et al. (2000)134. See Table S1 for details on the aircraft campaigns plotted in panel (a).
Figure 2b compares the ATom-3 and 4 vertical HCOOH profiles with those simulated by GEOS-Chem. For both campaigns the model significantly underestimates the median observed concentrations in the lower atmosphere, with the discrepancy more severe during ATom-4. On the other hand, the model captures the median HCOOH concentrations seen in the mid-to-upper free troposphere with no appreciable bias. This contrasting behavior will be further discussed in Sect. 3.2.
Figure 3 compares interpolated (0.05° latitude × 250 m) latitude curtains for HCOOH, CO, BC, and HCN based on the ATom-3 and 4 observations. While CO exhibits a clear latitudinal gradient with elevated concentrations throughout the northern high latitudes, no consistent gradient is detectable for HCOOH. Instead, we observe persistently low (< 100 ppt) background HCOOH concentrations throughout most of the remote atmosphere with discrete enhancements at specific locations associated with terrestrial outflow. The strongest such enhancements occur during spring (Southern Hemisphere for ATom-3; Northern Hemisphere for ATom-4) and consistently over the tropical Atlantic (Fig. 3). This pattern is very similar to that seen for BC, a known combustion tracer. In fact, the HCOOH correlation with BC is higher than with any other ATom-measured species (R = 0.72 and 0.74 for the entire ATom-3 and 4 datasets, respectively; flight-specific values reach 0.86; Fig. S1). The localized nature of the strongest enhancements, their locations and their distance from population centers, together point to a predominant role for biomass burning in driving remote HCOOH variability.
Figure 3.
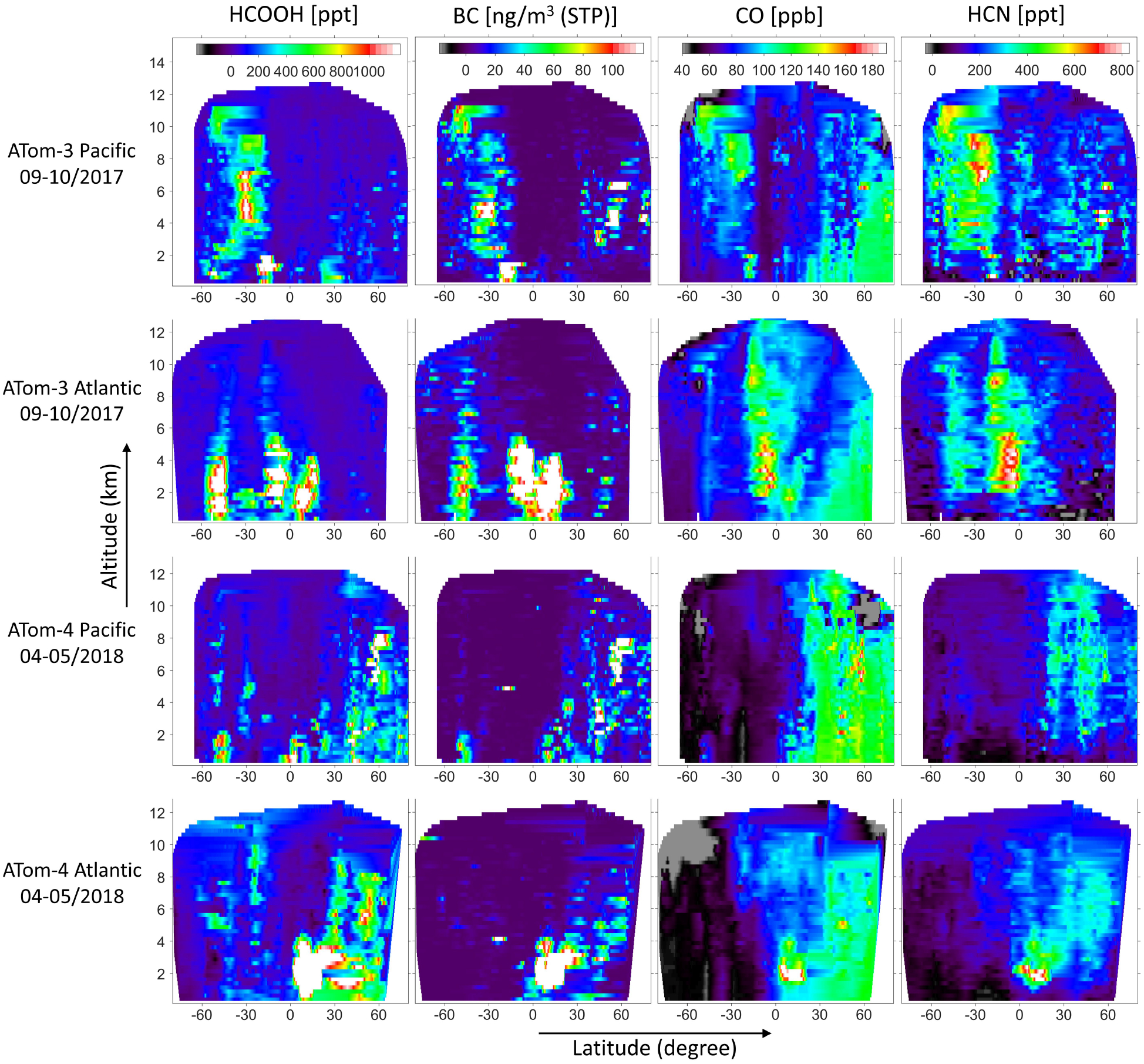
Interpolated tracer latitude-altitude cross-sections based on ATom-3 (Northern Hemisphere fall) and 4 (Northern Hemisphere spring) in-situ observations. 1 Hz data are gridded and interpolated into 0.05° latitude × 250m altitude bins.
BC is also emitted from other form of combustion, and the Figure 3 enhancements presumably also include some contributions from non-fire sources such as urban pollution. Notably, the HCOOH correlation with HCN (a biomass burning tracer) is not as high as with BC (Fig. S1); this may reflect the large variability in fire HCN emissions87 as well as the differing removal pathways for HCN and HCOOH. Overall, the strong importance of fires for HCOOH is confirmed by its tight correlation with the trajectory-derived fire influence (Fig. S2)—HCOOH enhancements under recent fire influence are an order of magnitude higher than under general land boundary layer influence. Later (Sect. 3.4.2) we assess the importance of primary emissions versus in-plume production in driving these HCOOH enhancements, and explore the role of HCOOH as a reactive carbon reservoir in highly aged fire plumes.
Despite the high overall HCOOH-BC correlation, we see in Figure 3 instances where their spatial patterns differ. For example, elevated HCOOH is observed in the MBL at ~30° N over the Pacific during ATom-3 with no accompanying BC enhancement. Also during ATom-3, comparable HCOOH enhancements are seen over the tropical and southern Atlantic, whereas for BC the tropical enhancements are far more pronounced. Such differences can arise from factors such as secondary production of HCOOH, non-pyrogenic sources, and differing removal rates.
The following sections combine the ATom observations with GEOS-Chem model predictions to i) evaluate current model treatment of HCOOH in the remote marine atmosphere (outside of fire plumes) in terms of evidence for broad-scale missing sources, and ii) diagnose signatures of HCOOH removal and the degree to which these are captured in the GEOS-Chem simulation. We return to examine the role of fires for remote HCOOH in Section 3.4.
3.2. Is there a ubiquitous missing HCOOH source in the background free troposphere?
Several prior studies have demonstrated the existence of large missing HCOOH sources5–8, 16, 39, which is consistent with the model-measurement discrepancies seen during ATom in the lower troposphere (Fig. 2b). On the other hand, Figure 2b shows that for both ATom-3 and ATom-4 GEOS-Chem is able to capture the median observed HCOOH abundance in the free troposphere with no detectable low bias. Median model levels in the free troposphere are generally in the 50–100 ppt range; if there were persistent missing sources leading to concentrations significantly >100 ppt they would be detectable given the instrumental LOD. Strong model HCOOH underestimates are seen in the vicinity of plumes and in the springtime northern latitudes, but do not occur ubiquitously across the remote free troposphere (Fig. 4) or at any altitude when >10 days removed from any land boundary layer or fire influence (Fig. S2). In fact, there is some indication of a positive model bias in the free troposphere (e.g., at high altitudes over the tropical Atlantic; Fig. 4) which we discuss further in Section 3.3.
Figure 4.
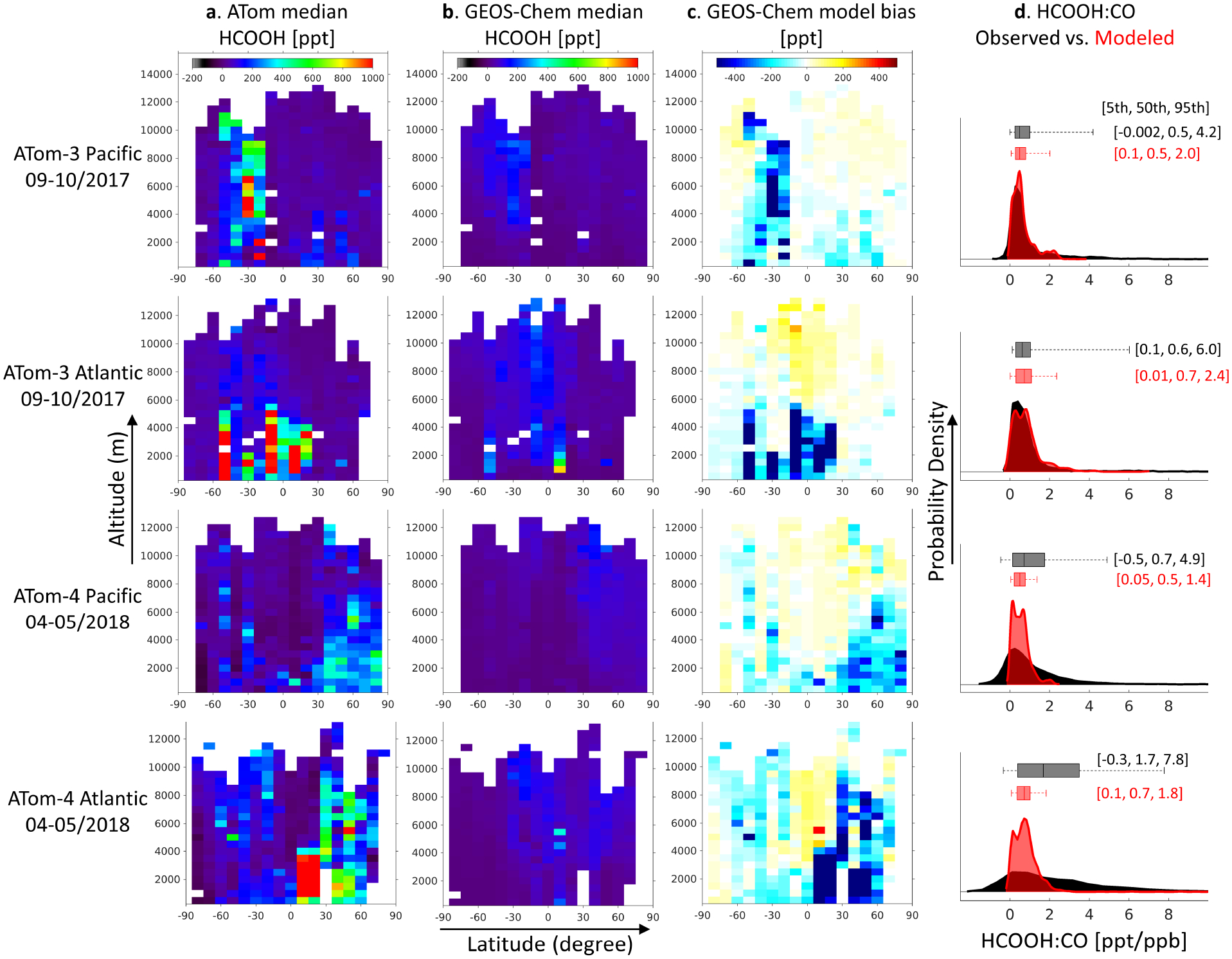
(a–c): Median gridded (10° latitude × 500m altitude) HCOOH cross-sections. Plotted are the (a) ATom observations, (b) GEOS-Chem model predictions, and (c) absolute model bias. (d): Kernel-smoothed probability density distributions and box-whisker plots (5th-25th-50th-75th-95th percentiles) for the HCOOH:CO ratio as measured during ATom (black) and modeled by GEOS-Chem (red) with numbers inset representing (left to right) the 5th, 50th, and 95th percentiles of the distribution.
The comparisons above imply that the large missing sources of HCOOH inferred in prior work are mainly terrestrial and sufficiently short-lived that their effects do not manifest as a consistent detectable model bias in the ATom dataset. Figure S3 shows that median levels for key measured HCOOH precursors are indeed very low during ATom, and this is generally captured by the model (though with underestimates for ethyne and acetaldehyde). We likewise conclude that the fire plume enhancements discussed earlier do not reflect a source of sufficient global magnitude to yield a detectable model HCOOH bias throughout the remote atmosphere.
Recent ATom-based analyses by Wang et al. (2019)40 and Travis et al. (2020)88 point to a widespread missing acetaldehyde source in the free troposphere over remote oceans, which is also seen here (Fig. S3). Unidentified precursor VOCs are one possible explanation; if so, it appears that those VOCs do not give rise to large amounts of HCOOH. In addition, while acetaldehyde itself is a HCOOH precursor via tautomerization to vinyl alcohol19, 89–92, we find that the model bias for this species is insufficient to have a large broad-scale impact on HCOOH. Specifically, a sensitivity simulation doubling vinyl alcohol source from acetaldehyde yields a modeled HCOOH increase of <1 ppt over most remote areas. Thus, while acetaldehyde tautomerization can be a large fraction of the total modeled HCOOH production rate in remote areas20, the absolute magnitude is then relatively small.
3.3. Air-sea exchange and wet scavenging of HCOOH
HCOOH has a high gas-to-aqueous partitioning tendency with an intrinsic Henry’s law constant of kH = 8917 M/atm at 298K93–95. Its aqueous uptake is further enhanced by dissociation (pKa = 3.75) in natural solvents such as rainwater (pH ~ 5.6; M/atm at T = 298 K) and ocean water (pH ~ 8.1; M/atm at T = 298 K). As a result, wet scavenging is thought to be the dominant global sink of atmospheric HCOOH6, and the signature of this process is indeed evident in the ATom dataset. For example, HCN and CO co-enhancements in the tropical Atlantic upper troposphere (Fig. 3) indicate the presence of transported fire plumes. However, no comparably strong HCOOH or BC enhancements are apparent in the same locations, implying wet removal of these species by convective updrafts, clouds, or precipitation during plume transport. The differing behavior for HCOOH versus HCN can be explained by the much weaker solubility (kH = 12 M/atm at 298K93) and acid dissociation (pKa = 9.2) for HCN. In the GEOS-Chem simulation we thus derive a global HCOOH lifetime of ~8 days against wet deposition, whereas this is a negligible removal pathway for HCN96, 97. The fact that the model frequently overestimates HCOOH at high altitude, particularly over the tropical Atlantic (Fig. 4; Section 3.2), may reflect an underestimate of convective HCOOH removal–as has been identified previously in the model for black carbon and sea salt aerosols98.
The ATom data also include segments with clear evidence of oceanic HCOOH uptake. Figure 5 shows measurements of HCOOH and related tracers during two flights with minimal continental influence (based on CO, BC, and HCN) over the Tropical Pacific and Antarctic Oceans. We see strong HCOOH depletion in the MBL for nearly all of the vertical profiling maneuvers (others are apparently influenced by fire or pollution plumes). HCN is likewise depleted in the MBL, whereas CO is enhanced. MBL depletion of HCOOH could in theory be explained either by cloud scavenging or by ocean uptake. However, the timeseries in Figure 5 show that MBL depletion of HCOOH is not dependent on cloudy conditions—arguing against cloud scavenging as the major driver. Similar HCOOH depletion in the MBL was previously observed during an airborne ascent/descent over the European Arctic ocean38.
Figure 5.
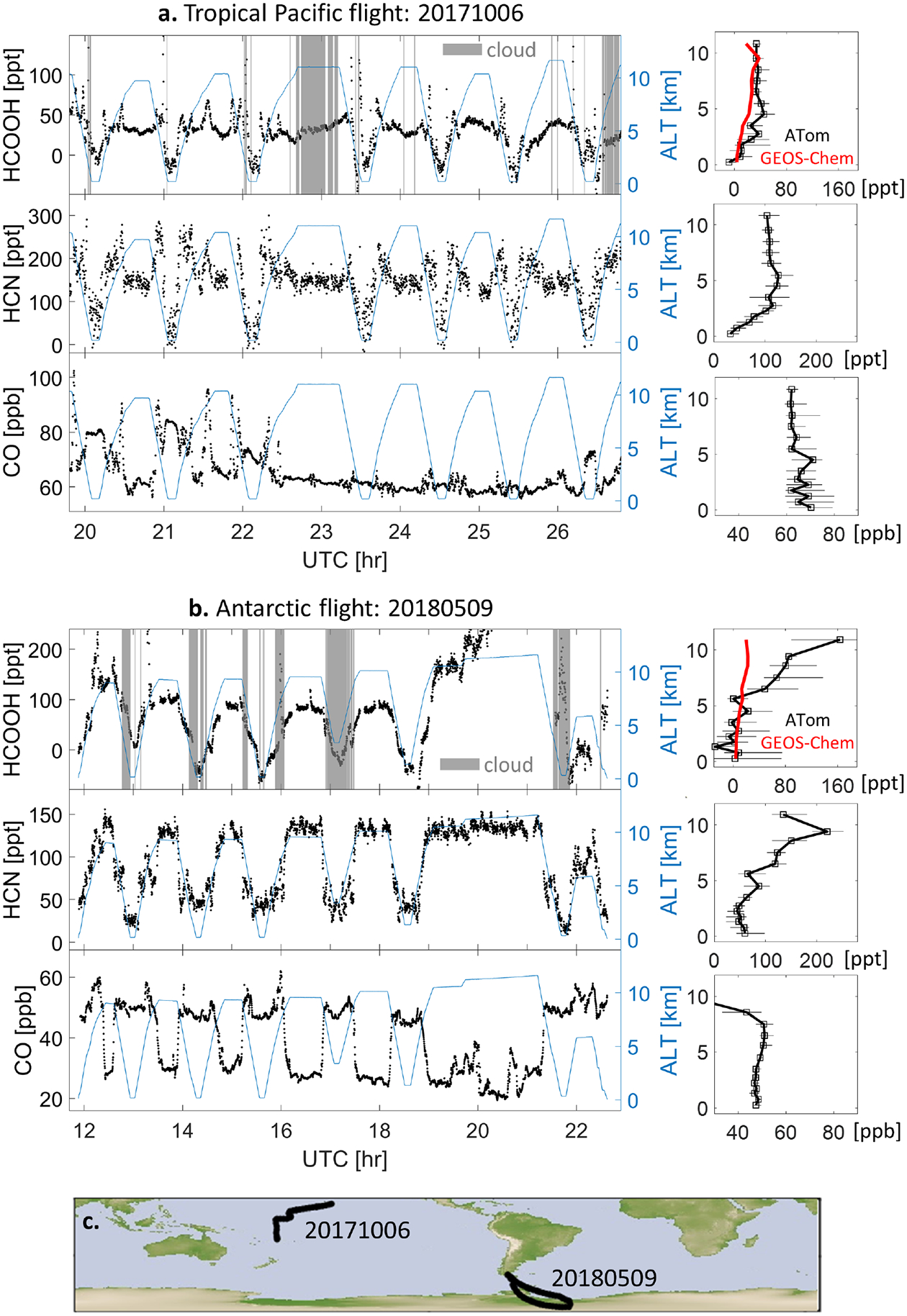
Ocean uptake of HCOOH as evident during two background flights: (a) a tropical Pacific flight on 10/06/2017, and (b) an Antarctic flight on 05/09/2018. Left panels show timeseries for three trace gases (black) and altitude (blue). Shaded regions (grey) in the HCOOH panel indicate cloudy conditions (liquid-phase, mixed-phase, or cirrus clouds) based on CAPS observations. Right panels show the corresponding median observed vertical profiles for the entire flight (in black) with horizontal bars denoting the 25th–75th percentiles. The observed HCOOH profile is compared to GEOS-Chem model predictions (in red). Panel (c) shows the two flight tracks (black).
Figure 5 also compares the HCOOH vertical profiles from the above remote flights with model predictions, showing that GEOS-Chem generally reproduces the observed 0–5 km vertical gradient. While the observations have high relative uncertainty at these low concentrations, this agreement supports the idea that an air-side resistance-in-series deposition mechanism can adequately represent ocean HCOOH uptake without considering its degree of saturation or liquid-side resistance. For example, based on the effective Henry’s constant above, 1 ppb and 1 ppt of atmospheric HCOOH correspond at equilibrium to surface ocean [HCOOH(aq) + HCOO−] concentrations of 0.2 M and 0.2 mM, respectively (and higher at lower temperatures), with formate as the predominant dissolved form ([HCOOH(aq)]:[HCOO−] = 4.5×10−5 at pH = 8.1). With very few exceptions99, literature-reported seawater formate concentrations are in the μM or nM range (Table S2100–104)—orders of magnitude below the saturation concentration. It is clear therefore that the oceans are strongly undersaturated for HCOOH and that the bulk ocean-atmosphere flux should be effectively unidirectional. In this way we find in GEOS-Chem that ocean uptake accounts for 14% of global annual HCOOH removal through dry deposition, corresponding to 5% of the total sink (Table S3).
However, the MBL depletion of HCOOH discussed above is not consistently present across all of the ATom flights and profiles. In particular, the mean ATom-3 and ATom-4 latitude-altitude cross-sections (Fig. 3) show consistent MBL depletion for HCN but not for HCOOH. Based on solubility alone we would conversely expect a stronger uptake signal for HCOOH than for HCN; the lack of a consistent HCOOH ocean uptake signature in the ATom observations implies that other processes are obscuring that signal. For example, Figure S4 shows a series of ATom vertical profiles in which HCN is characteristically depleted in the MBL but HCOOH is strongly enhanced. BC and CO co-enhancements for these same profiles suggest fire and/or pollution influence. However, given their expected co-emission and solubility difference it is then surprising that HCN would be scrubbed from the plume while the HCOOH enhancement persists. This may reflect ongoing secondary production during plume transport, or a marine boundary layer source of uncertain mechanism38, 39.
3.4. A large and variable HCOOH sources from fires
We have seen above that fire plumes are key drivers of HCOOH variability in the remote atmosphere sampled by ATom. This influence is strongly underestimated by GEOS-Chem. Figure 4 plots the latitude-altitude HCOOH curtains from Figure 3 as median values over 10° latitude × 500 m altitude bins for comparison with the model. The enhancement patterns visible in Figure 3 still emerge in the median values and at the coarser spatial resolution of Figure 4. While the model exhibits some fidelity in the simulated enhancement locations, predicted magnitudes are far weaker than observed. This tendency is also seen in the HCOOH:CO concentration ratios: Figure 4 shows that during ATom-3 and the Pacific leg of ATom-4 the model captures the central part of the HCOOH:CO distribution but misses the upper tail. A larger model bias is evident over the Atlantic during ATom-4, where the model underestimates the median HCOOH:CO ratio by a factor of >2 and the 95th percentile by a factor of >4.
We proceed to examine these HCOOH enhancements in terms of potential reasons for the model underpredictions, employing as comparison metric the normalized excess mixing ratios (NEMR) relative to CO. The NEMR is commonly used to quantify dilution-corrected above-background plume enhancements: at-source the NEMR corresponds to a molar emission ratio (ER), while downwind values also reflect ensuing production and loss. We performed a case-by-case identification of plumes intercepted during ATom meeting any two of the following criteria: (a) peak HCOOH > 1 ppb, (b) peak BC > 100 ng/m3, (c) peak CO > 175 ppb (using one-second data in all cases). In this way, 53 plume intercepts were identified during ATom-3 and 4 ranging from <1 min to >20 min duration. Plume identification, NEMR calculation, and source attribution are described for each intercept in the Supporting Information (Table S4, Sect. S2). In particular, we find that 38 of the 53 identified plume intercepts exhibit clear biomass burning signatures and are thus treated as fire plumes in the analyses that follow. The remaining 15 are likely to be partly or mainly anthropogenic.
HCOOH enhancements across all 53 plume intercepts vary from ~1 to >30 ppb, with corresponding CO enhancements of 9–270 ppb. The resulting HCOOH NEMRs (calculated for each plume intercept as the reduced major axis HCOOH:CO slope; Table S4 and Fig. 6) range from 3.4 ppt/ppb CO in the northern Pacific (plume intercept #19, Sect. S2) to >200 ppt/ppb CO in plumes with potential urban/anthropogenic influence (plume intercepts #50–53, Sect. S2).
Figure 6.
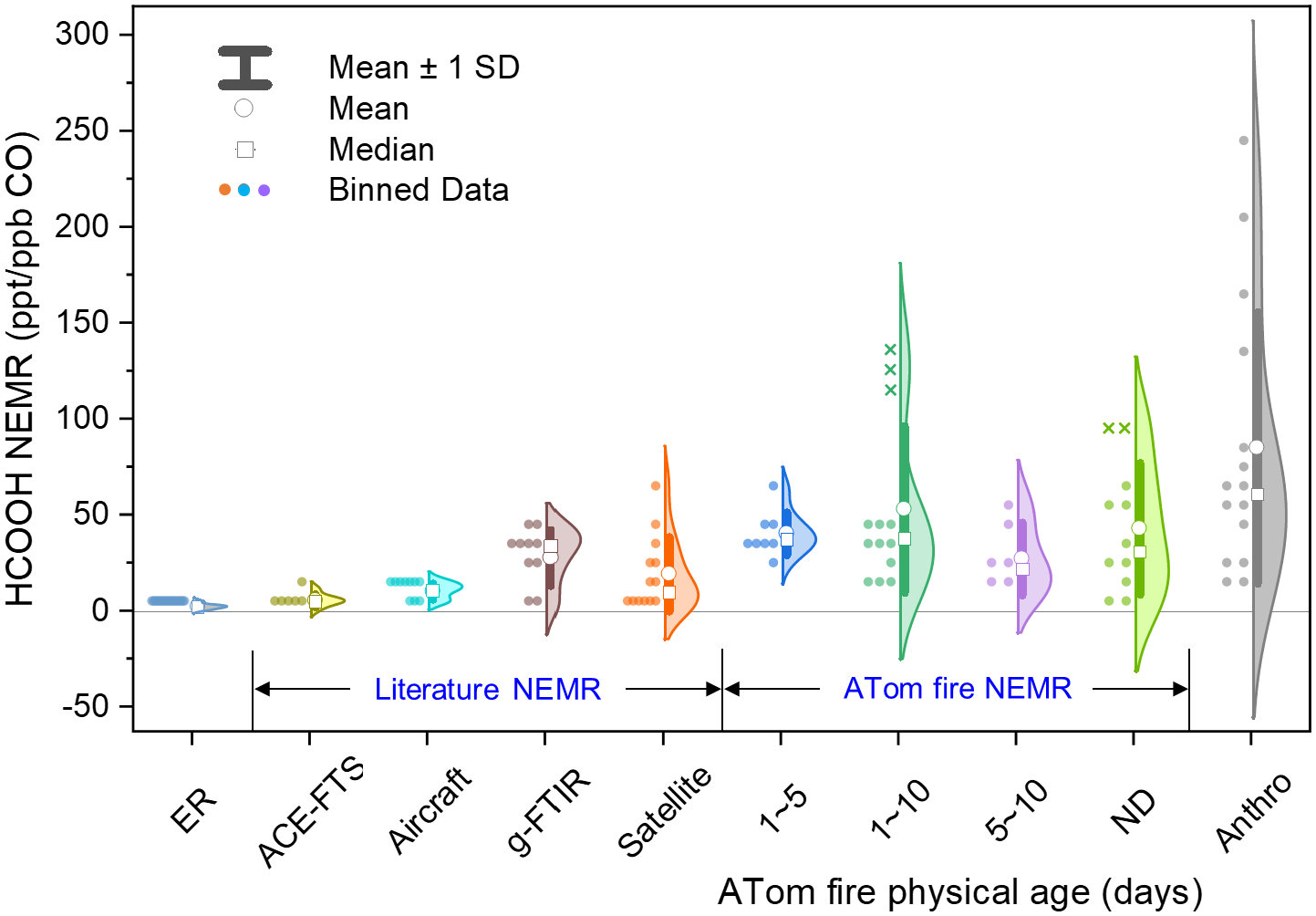
HCOOH normalized excess mixing ratios (NEMRs) relative to CO as a function of transport time for ATom plumes, and comparison to prior studies. Transport times (ND: not determined) are estimated based on back trajectory analysis as described in-text. Colored dots indicate fire plumes and plumes with anthropogenic influence (grey). Plumes marked with an “×” denote encounters after a mid-flight instrumental change on 05/14/2018 (see text). Also shown are primary HCOOH emission ratios (ER) as compiled by Andreae (2019)125 and Akagi et al. (2011)71, and HCOOH NEMR estimates inferred from prior atmospheric observations. These include: aircraft studies featuring downwind plume sampling (up to 4.5 hours aging)27, 31, 105, 121, ground-based remote sensing by Fourier transform infrared spectrometry (g-FTIR)34, 135–137, and space-based observations from the ACE-FTS29, 30, 123, 124, 138, 139, and TES/IASI satellite instruments32, 33, 140.
3.4.1. Case study: Large HCOOH enhancements in African plumes
We find that 19 of the 53 identified plume intercepts are due to African fires (with 15 from northwest Africa and 4 from southern Africa; Sect. S2.1). Of these, plume intercept #40 off the African coast (~16 UTC on 05/14/2018; Fig. 7a) featured HCOOH mixing ratios exceeding 15–30 ppb—the highest values seen across all of ATom. Highly enhanced levels of CO (~350 ppb), HCN (~1.5 ppb), and BC (~2 μg/m3) were also observed. Back-trajectory analysis indicates that the above plume underwent >2 days transport after emission in northwest Africa before being intercepted by the aircraft (Fig. 7c and Sect. S2.1). Explaining the observed in-plume HCOOH mixing ratios requires i) sufficiently large primary HCOOH emissions that 15–30 ppb still remained after >2 days dilution, ii) strong secondary HCOOH production during plume aging, or iii) some combination of the two. HCOOH enhancements this large have only been reported once previously: for a fresh grass fire plume in Southern Africa105. In that case the HCOOH enhancement dropped to 5 ppb after 30 min and to below the detection limit after 40 min. Most other in-situ studies (even those conducted near-source) have reported HCOOH mixing ratios below 12 ppb5, 9, 106–111.
Figure 7.
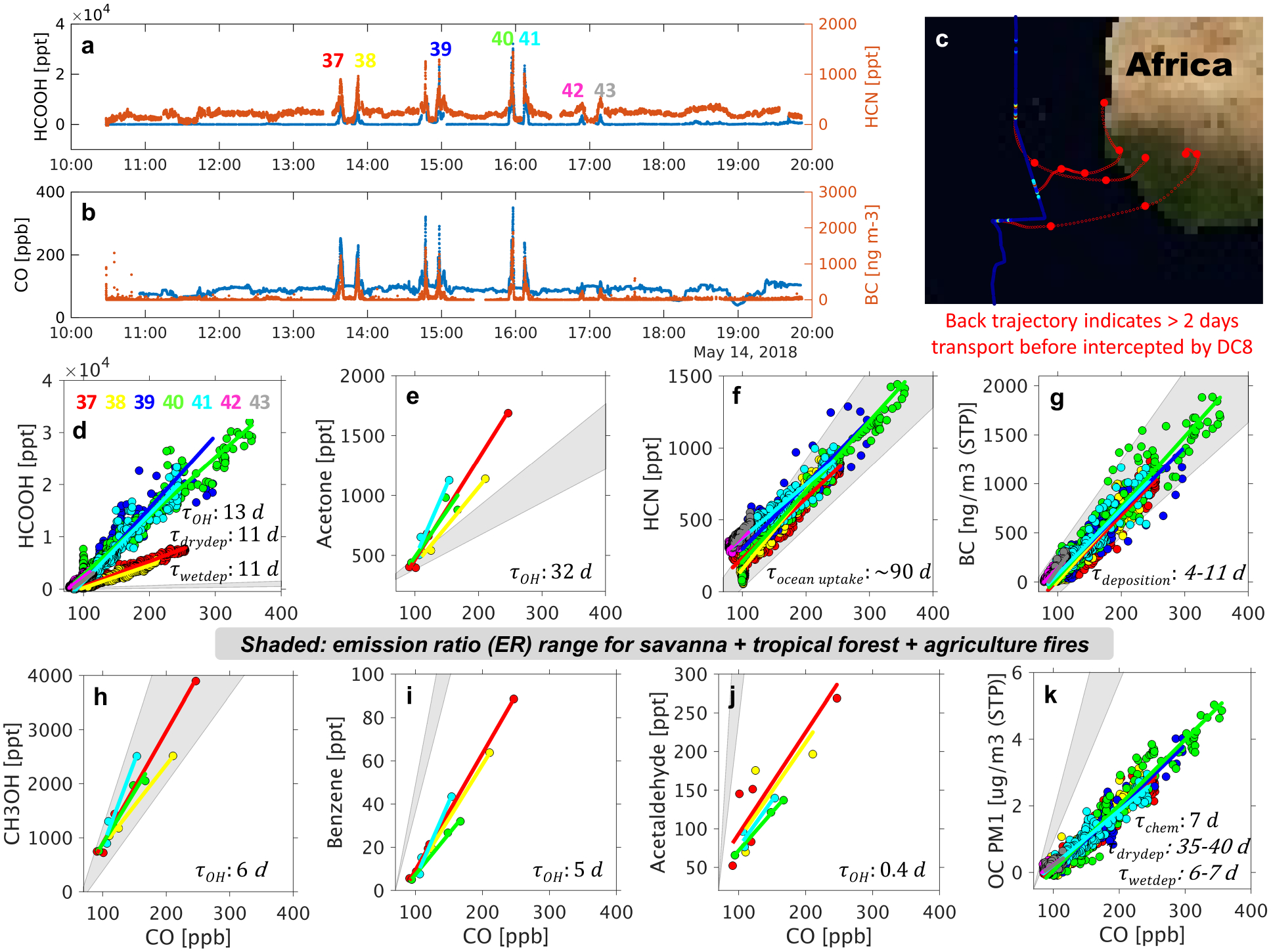
Aged west African fire plumes sampled on 05/14/2018. Panels (a) and (b) show HCOOH, HCN, CO, and BC time series for the entire flight, with 7 plume intercepts (#37–43) marked inset. An additional plume (between #38 and #39) was encountered during an HCOOH instrumental zero and is therefore omitted. Panel (c) shows HYSPLIT back trajectories (red, 1-day interval marked) for 3 example plume intercepts. Panels (d)–(k) show correlations between select pyrogenic species and CO for all plume intercepts. The resulting NEMR range (obtained from the regression slopes for the 7 plume intercepts) is compared to expected primary emission ratios (ER, shaded in grey) for that species based on compiled data ranges for relevant fuel types (forests, savanna and grassland, and agricultural residue125). For TOGA-measured species only plume intercepts including 3 or more data points (#37, #38, #40, #41) are retained for NEMR analysis. Convective influence is negligible for these plumes based on back trajectory analysis. The lifetime (unit: days) is shown for each species with respect to its dominant loss pathway5, 141–144. An OH concentration of 2×106 molecule/cm3 is used to calculate nominal lifetimes to OH oxidation145, 146.
The same 05/14/2018 ATom-4 flight included a total of 8 plume intercepts identified as fire outflow from west Africa, and HCOOH:CO NEMR values were quantified for 7 of these (the 8th occurred during an instrument zeroing procedure). Figure 7 shows that HCOOH exhibits quite different behavior compared to other species sampled across these plumes. HCN and BC, primary fire tracers that are unreactive towards OH, exhibit similar NEMRs across the 7 plume intercepts that are within the range of literature-reported ERs for relevant fire types (savanna, tropical forest, agriculture). Benzene, a primary combustion tracer that is more photochemically reactive (τ=4.8 days at OH=2×106 molecules/cm3 and kbenzene+OH=1.2×10−12 cm3/molecule/s112) exhibits NEMRs 44–78% lower than the expected primary ERs—consistent with a few days of aging. Acetaldehyde likewise exhibits NEMR values lower than its primary ERs, reflecting the net effect of direct emissions, secondary production, and a short photochemical lifetime. NEMR values for acetone, on the other hand, are 1.4–4.2× higher than the corresponding ERs and indicate net secondary production during plume aging113, 114. Methanol, despite having a photochemical lifetime similar to that of benzene, exhibits NEMR values consistent with its primary ERs, likely reflecting in-plume secondary production offsetting chemical loss115–117—as also inferred from the ATom observations by Bates et al. (2021)118. The I-CIMS instrument underwent an operational change at ~14:30 UTC during this flight which may explain the HCOOH NEMR bifurcation seen in Figure 7. However, even if we take the conservative assumption that the post-14:30 HCOOH NEMRs on this flight should be reduced by ~2× to match the earlier values, Figure 7 shows that HCOOH is unique among the plotted tracers in exhibiting in-plume NEMRs 1–2 orders of magnitude higher than expected primary ERs—suggesting substantial secondary production en-route.
3.4.2. HCOOH NEMR variability and relationship with plume age
HCOOH NEMRs vary significantly across the ATom plumes investigated here. Values range from a low of 3.4 ppt/ppb CO in a high-altitude fire plume over the North Pacific (plume intercept #19, flight 20171027; the same plume featured the highest HCN mixing ratios encountered during ATom) to >50 ppt/ppb CO. Particularly high NEMR values were observed in a number of plumes that appear to contain a mixture of biomass burning and urban/anthropogenic contributions (Sect. S2.2 and Fig. 6). These large HCOOH enhancements (e.g., >200 ppt/ppb CO for California plumes encountered near the end of ATom-4) may reflect accelerated oxidation of VOC-rich biomass burning emissions in the presence of ample anthropogenic NOx119, 120.
The highest HCOOH NEMRs seen during ATom and highlighted above exceed any values reported previously in fire emissions or urban plumes. However, HCOOH has not been routinely measured in airborne fire campaigns, and prior studies including HCOOH measurements have focused on relatively fresh plumes (< 4.5 hrs) without characterizing their longer-term evolution. For example, Goode et al. (2000)121 studied the evolution of two Alaskan forest fire plumes and found that the HCOOH NEMR doubled (from 6–8 to 12–16 ppt/ppb CO) after 2–3 hrs evolution. An immediate increase of the HCOOH NEMR (to 10 ppt/ppb CO) was observed in the first 20 min after emission from a savanna fire in southern Africa, but this was soon followed by a decrease to below the instrumental detection limit105. During MILAGRO, the HCOOH NEMR in a tropical fire plume was observed to increase at a rate of 0.0033/hr (from 2.5 to ~5 ppt/ppb CO) over the first 1.5 hr of aging31. Akagi et al. (2012)27 report for a prescribed chaparral fire plume in California that the HCOOH NEMR increased by a factor of 7 (from 0.7 to 4.8 ppt/ppb CO) over 4–4.5 hrs aging. Finally, Yokelson et al. (2009)31 report an even higher NEMR for total organic acids of up to 180 ppt C/ppb CO in aged smoke, but do not specify the fraction due to HCOOH. In the case of urban/anthropogenic sources11, direct HCOOH emissions have been found to be relatively small (~ 1 ppt/ppb CO106, 109, 110) with subsequent secondary production yielding enhancements of up to ~20 ppt/ppb CO122.
Enhanced HCOOH levels in fire plumes have also been quantified using remote sensing observations. Tereszchuk et al. (2011, 2013)123, 124 combined ACE-FTS data with trajectory modeling to examine the evolution of pyrogenic trace species in biomass burning plumes from a variety of geographic regions and found a net decrease of the HCOOH NEMR over 7 days aging. More recently, satellite data from IASI33 and TES32 have been used to study the HCOOH source from key biomass burning regions, with the latter study inferring substantially higher enhancement ratios than expected solely from direct emissions—indicating significant secondary HCOOH production in fire plumes.
One hypothesis to explain the variable HCOOH enhancements observed during ATom is that longer plume transport times and hence aging lead to additional net HCOOH production. We test this idea by estimating physical plume ages for the identified fire intercepts. Ages are computed for each minute of the intercept window as the time since most recent fire influence based on back-trajectory analysis (Methods; Sect. S2). We find in this way that the 53 identified plume intercepts can be categorized as follows: (i) one single unambiguous transport time; (ii) a unimodal transport time distribution; (iii) a multimodal transport time distribution; (iv) variable trajectory cluster results across the plume intercept window; (v) low fire influence probability based on the back trajectories and therefore an undetermined age. We then obtain a best-estimate transport time for each plume intercept from the ensemble of trajectory results across the intercept. Finally, we group the results into plume age categories of (a) <1 day, (b) 1–5 days, (c) 5–10 days, (d) >10 days, or (e) a mixture of two neighboring classes within a-d. More details on this classification process are provided in Section S2.
Figure 6 shows the resulting HCOOH:CO plume-age dependence for the ATom dataset in relation to prior NEMR observations and direct pyrogenic emission ratios. We see that the strongest enhancements (>100 ppt/ppb CO) manifest for aged and anthropogenically-influenced plumes (with their likely faster chemistry). Conceptually, we expect increasing NEMRs during early plume aging if the rate of secondary HCOOH production exceeds that of CO, with a subsequent decrease as the plume age approaches the HCOOH photochemical lifetime and precursor VOCs become depleted. The mean trend in Figure 6 does not contradict this expectation, but the main emergent feature is that of highly variable HCOOH enhancements across plumes, with NEMRs spanning a factor of 50 or more within individual age classes. While our trajectory-based plume age estimation and the instrumental calibration for HCOOH (Sect. 2.1.1) both have some inherent uncertainty, it appears that this remaining variability must mainly reflect other factors such as source type and burn conditions that affect the HCOOH ER and subsequent plume chemistry.
3.4.3. HCOOH as a major C reservoir in aged plumes
Figure 8 presents a global view of the gas-phase reactive organic carbon (ROC) composition of the plumes encountered during ATom-3 and 4. We define ROC here to include all VOCs measured during ATom with sufficient in-plume sampling coverage to determine regression-based NEMRs, plus submicron aerosol OC (Sect. S3). We see in the map plumes spanning a wide range of intercept locations, source regions, and transit times, including: transported Eurasian fire plumes sampled over the Arctic, Alaska, and the north Pacific; plumes from the northwestern US/southwestern Canada sampled over California; Central American fire plumes sampled over the equatorial Pacific; plumes downwind of Australia, South America, and Africa; and near-source plumes sampled over California and Fiji.
Figure 8.
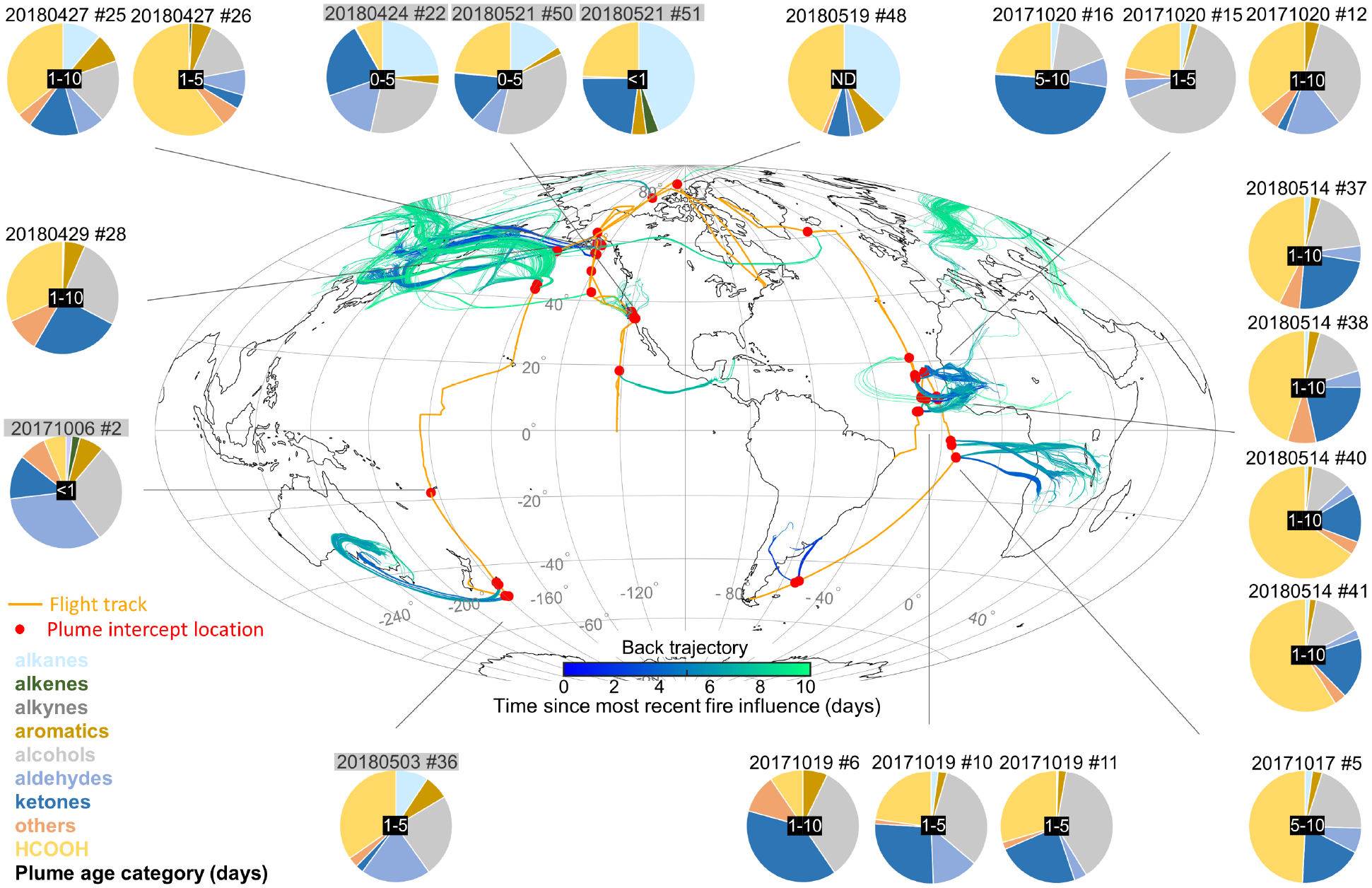
Gas-phase reactive organic carbon (gROC) composition in ATom-3 and ATom-4 fire plumes. Grey shaded plume intercept numbers are plumes with anthropogenic influence. Identified plumes (filled red circles) are denoted along the flight tracks (orange lines) with associated back trajectory clusters colored by the time since most recent fire influence. Plumes and trajectories shown are restricted to those with identified fire influence within the previous 10 days. Pie charts show in-plume gROC speciation on an NEMR basis (ppt C/ppb CO). VOCs that are measured only with the WAS system are omitted here for consistency across plumes; Figure S5 includes such species when available. See Figure S6 for a version of this figure that includes submicron aerosol OC.
The pie charts in Figure 8 quantify the gas-phase ROC (gROC) composition for selected sampled plumes on an NEMR basis (ppt C/ppb CO). The 21 plume intercepts shown (of 53 total) reflect those with sampling coverage for the I-CIMS, CF3O− CIMS, ISAF, and TOGA instruments (see Methods). VOCs measured only by WAS (with its lower temporal resolution) are excluded to prevent species coverage differences between plumes. Figure S5 shows that including these species (when available) changes the HCOOH contribution by 1–26% on an NEMR basis.
We see for the plumes plotted in Figure 8 that HCOOH, with NEMRs ranging from 11–129 ppt/ppb CO, accounts for between 6 and 60% of the total detected gROC enhancements. This fractional contribution depends on measurement coverage, which is incomplete as ATom was not explicitly a biomass-burning focused mission. Based on prior estimates for the importance of unmeasured/unidentified pyrogenic VOCs71, the HCOOH fractions in Fig. 8 (and S5) could be 2–3× lower if all species were accounted for.
The lowest HCOOH contributions (6–23%) are seen for relatively near-source plumes over California and Fiji, contrasting with >30% contributions for several transported Asian and African plumes. Figure S6 shows that when we expand the analysis to include both gas and particle-phase OC, OC is frequently the largest component of the in-plume ROC enhancement. However, HCOOH still typically contributes between 4 and 33% of the total gas + particle ROC enhancements shown in Figure S6. HCOOH therefore represents a surprisingly large fraction of the detected ROC pool in these plumes, despite being only a small component of primary biomass burning and anthropogenic ROC emissions. As a result, when not subject to wet deposition, HCOOH can be an important pathway for transporting combustion-emitted reactive carbon to the remote atmosphere.
3.4.4. Implied in-plume HCOOH production greater than predicted from known precursors
The fact that the HCOOH enhancements observed during ATom are, in most cases, far larger than can be explained solely based on its direct pyrogenic emissions (Fig. 6) points to secondary production as the dominant HCOOH source in these plumes. The total summed direct ER for all identified gas+particle-phase ROC components is estimated to range from ~300–500 ppt C/ppb CO across forest, savanna/grassland/shrubland, and cropland fires, which are the predominant global fire types (Table S6)71, 125, 126. This increases to ~500–1200 ppt C/ppb CO when also including estimated emissions of unidentified/unmeasured ROC components71. The median HCOOH enhancement observed across the ATom fire plumes of 36 ppt/ppb CO therefore corresponds to approximately 3–7% of the total expected ROC emission from fires on an NEMR basis.
However, only a fraction of fire-emitted ROC species have been identified as HCOOH precursors, including alkenes (ethene, propene, butene, isoprene, monoterpenes), ethyne, several OVOCs (MVK, MACR, acetaldehyde), hydroxyacetone, hydroxyacetaldehyde, and submicron aerosol OC (Table S6)17, 18, 21, 23, 127–130. The summed direct ER for these known gas-phase HCOOH precursors is estimated to range from 91 to 189 ppt C/ppb CO for forest, savanna/grassland/shrubland, and cropland fires (Table S6). Accounting for the median in-plume ATom HCOOH enhancement of 36 ppt/ppb CO based on these precursor emissions would thus require an average carbon-based yield of 19–40%—much higher than has been reported5, 16, 130, 131. Primary ERs for submicron aerosol OC are estimated at 98–226 ppt C/ppb CO (Table S6); in this case an average per-carbon HCOOH yield of 16–37% from aerosol OC would be needed to explain a HCOOH enhancement of 36 ppt/ppb CO. Malecha and Nizkorodov (2016)23 report based on chamber measurements that OA mass is photochemically converted to HCOOH at a rate of 1.9/yr. Assuming that photodegradation is the major loss pathway for biomass burning OA (at a similar loss rate as derived in chamber experiments for biogenic/anthropogenic secondary), the 1.9/yr corresponds to a carbon-based HCOOH yield of ~0.3–3% over 1–10 days of aging, which again is far lower (>10–100×) than required to account for the ATom observations. Together, the above calculations indicate that additional, unknown production pathways are needed to account for the HCOOH enhancements observed in many of the aged fired fire plumes sampled by ATom.
4. Summary and implications
The ATom campaign sampled from pole-to-pole over the remote Pacific and Atlantic Oceans. Outside of continental plumes, HCOOH concentrations were generally less than 100 ppt with no consistent latitudinal gradient that was detectable. Observed background concentrations in the free troposphere are consistent with prior aircraft profiling of the remote atmosphere and with GEOS-Chem model predictions. We conclude that the missing HCOOH precursors identified in earlier work must be mainly terrestrial in nature and short-lived so that their effects do not manifest throughout the remote atmosphere. Similarly, the missing acetaldehyde source in the remote background atmosphere inferred by Wang et al. (2019)40 apparently does not lead to substantial background HCOOH production.
ATom observations provide signatures of HCOOH removal via wet scavenging and ocean uptake. The contrasting behavior between HCOOH and HCN observed in these situations qualitatively matches expectations in the case of wet scavenging, with stronger removal for the more-soluble HCOOH. The GEOS-Chem model appears to be underestimating HCOOH removal via wet scavenging, and this is consistent with a small but positive model bias in many parts of the clean mid-to-upper troposphere. In the case of ocean-atmosphere exchange, we find that while HCN is consistently depleted in the marine boundary layer, this is only sporadically the case for HCOOH—despite its strong air-to-sea partitioning tendency and seawater undersaturation. We attribute this to that ongoing secondary production in continental outflow that often exceeds the rate of HCOOH dry deposition to the ocean; other processes related to the sea surface38, 39 may also be playing a role.
HCOOH enhancements observed during ATom correlate most strongly with BC and are also closely related to the trajectory-derived time since fire influence. These findings reveal a significant role for fires in driving remote HCOOH variability and highlight the general influence of biomass burning in the remote atmosphere57. Normalized excess mixing ratios (NEMRs) for HCOOH in the fire plumes sampled during ATom are frequently 1–2 orders of magnitude higher than expected solely from its primary emissions, implying substantial secondary production in fire plumes (as found in prior near-source and satellite-based studies). We find that HCOOH is a major reactive carbon reservoir in aged fire plumes (up to 33% of the total gas+particle-phase reactive organic carbon, ROC). Such enhancements are larger than can be explained based on its known pyrogenic precursors and current understanding of its formation pathways.
The range of HCOOH NEMR values across the 38 mainly-pyrogenic ATom plumes identified here is consistent with findings from prior satellite-based studies, highlighting the potential for using satellite data in future investigations of fire plume aging. Unusually high HCOOH NEMRs observed in several mixed anthropogenic+fire plumes sampled during ATom may reflect the vigorous oxidation that can occur when biomass burning VOCs mix with elevated urban-derived NOx. Further research is needed to better understand the chemistry and associated air quality impacts during such urban-fire interactions, which can lead to extreme air pollution events for cities in wildfire-prone areas such as the western US.
Fire plumes sampled during ATom range in age up to ~10 days. While the largest HCOOH enhancements occur in the aged plumes, observed HCOOH NEMRs are highly variable and do not correlate consistently with plume age. What drives this variability? Answering this question requires dedicated in-situ plume Lagrangian sampling that is chemically comprehensive and spans a wider age range than typically covered in field studies (<10 hrs). Such extended sampling is especially important for relatively long-lived species such as HCOOH (lifetime of ~weeks). Satellite observations132, 133, while more limited in species coverage, also have potential for sampling across the entire plume evolution spectrum.
Supplementary Material
Acknowledgments
This research was primarily supported by the National Aeronautics and Space Administration (grant NNX14AP89G). We thank Armin Wisthaler and Hanwant Singh for their support of this project. Computing resources were provided by the Minnesota Supercomputing Institute (https://www.msi.umn.edu) at the University of Minnesota.
We acknowledge the contributions of Steven Wofsy (Harvard QCLS and ATom CO.X); Daniel Murphy (NOAA CSL PALMS); Paul Wennberg (CIT-CIMS); Bernadett Weinzierl (University of Vienna CAPS); Glenn Wolfe and Thomas Hanisco (NASA GSFC ISAF); Thomas Ryerson (NOAA CSL NOyO3); and Robert Talbot (NASA GTE MC/IC). We thank Gordon Novak, Jeffery Pierce, and Anna Hodshire for helpful discussions.
ATom was funded by the NASA Earth Venture program through grant NNX15AJ23G. NCAR researchers acknowledge support from the National Center for Atmospheric Research, which is a major facility sponsored by the National Science Foundation under Cooperative Agreement No. 1852977. The CU Boulder HR-AMS team (BAN, PCJ and JLJ) were supported by NASA grants NNX15AH33A, 80NSSC19K0124 and 80NSSC18K0630.
Footnotes
Supporting Information (SI) Available:
S1. Tables S1–S3; Figures S1–S3
Table S1. Airborne HCOOH measurements in the remote atmosphere; Table S2. Reported surface seawater + lakewater concentrations of HCOOH and formate; Table S3. Global HCOOH sinks; Figure S1. Correlation between HCOOH and other observed species; Figure S2. Back trajectory-derived influence from fires, convection, boundary layer on HCOOH; Figure S3. Modeled vs. observed key HCOOH precursors. Figure S4. Example timeseries showing MBL enhancements of HCOOH
S2. Plume attribution
Table S4. Summary of the 53 identified plume intercepts; S2.1 African fire plume intercepts; S2.2 Plumes with unusually high HCOOH:CO NEMRs; S2.3 Other fire plume intercepts; S2.4 Other fire plume intercepts with anthropogenic influence
S3. ROC related tables and figures
Figure S5. Same as Figure 8, but including those species measured only by WAS; Figure S6. Same as Figure 8, but including submicron aerosol OC; Table S5. ROC speciation in Figures 8, S3 and S4; Table S6. Primary emission ratios (ERs, ppt C/ppb CO) for fire-emitted ROC species
Reference:
- (1).Keene WC, Galloway JN, and Holden JD, Measurement of weak organic acidity in precipitation from remote areas of the world. J. Geophys. Res.: Oceans 1983, 88 (C9), 5122–5130, 10.1029/JC088iC09p05122. [DOI] [Google Scholar]
- (2).Andreae MO, Talbot RW, Andreae TW, and Harriss RC, Formic and acetic acid over the central Amazon region, Brazil: 1. Dry season. J. Geophys. Res.: Atmos 1988, 93 (D2), 10.1029/JD093iD02p01616. [DOI] [Google Scholar]
- (3).Galloway JN, Likens GE, Keene WC, and Miller JM, The composition of precipitation in remote areas of the world. J. Geophys. Res.: Oceans 1982, 87 (C11), 10.1029/JC087iC11p08771. [DOI] [Google Scholar]
- (4).Jacob DJ, Chemistry of OH in remote clouds and its role in the production of formic acid and peroxymonosulfate. J. Geophys. Res.: Atmos 1986, 91 (D9), 10.1029/JD091iD09p09807. [DOI] [Google Scholar]
- (5).Millet DB, Baasandorj M, Farmer DK, Thornton JA, Baumann K, Brophy P, Chaliyakunnel S, de Gouw JA, Graus M, Hu L, Koss A, Lee BH, Lopez-Hilfiker FD, Neuman JA, Paulot F, Peischl J, Pollack IB, Ryerson TB, Warneke C, Williams BJ, and Xu J, A large and ubiquitous source of atmospheric formic acid. Atmos. Chem. Phys 2015, 15 (11), 6283–6304, 10.5194/acp-15-6283-2015. [DOI] [Google Scholar]
- (6).Paulot F, Wunch D, Crounse JD, Toon GC, Millet DB, DeCarlo PF, Vigouroux C, Deutscher NM, González Abad G, Notholt J, Warneke T, Hannigan JW, Warneke C, de Gouw JA, Dunlea EJ, De Mazière M, Griffith DWT, Bernath P, Jimenez JL, and Wennberg PO, Importance of secondary sources in the atmospheric budgets of formic and acetic acids. Atmos. Chem. Phys 2011, 11 (5), 1989–2013, 10.5194/acp-11-1989-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cady-Pereira KE, Chaliyakunnel S, Shephard MW, Millet DB, Luo M, and Wells KC, HCOOH measurements from space: TES retrieval algorithm and observed global distribution. Atmos. Meas. Tech 2014, 7 (7), 2297–2311, 10.5194/amt-7-2297-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Stavrakou T, Müller JF, Peeters J, Razavi A, Clarisse L, Clerbaux C, Coheur PF, Hurtmans D, De Mazière M, Vigouroux C, Deutscher NM, Griffith DWT, Jones N, and Paton-Walsh C, Satellite evidence for a large source of formic acid from boreal and tropical forests. Nat. Geosci 2012, 5 (1), 26–30, 10.1038/ngeo1354. [DOI] [Google Scholar]
- (9).Alwe HD, Millet DB, Chen X, Raff JD, Payne ZC, and Fledderman K, Oxidation of Volatile Organic Compounds as the Major Source of Formic Acid in a Mixed Forest Canopy. Geophys. Res. Lett 2019, 46 (5), 2940–2948, 10.1029/2018GL081526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schobesberger S, Lopez-Hilfiker FD, Taipale D, Millet DB, D’Ambro EL, Rantala P, Mammarella I, Zhou P, Wolfe GM, Lee BH, Boy M, and Thornton JA, High upward fluxes of formic acid from a boreal forest canopy. Geophys. Res. Lett 2016, 43 (17), 9342–9351, 10.1002/2016gl069599. [DOI] [Google Scholar]
- (11).Veres PR, Roberts JM, Cochran AK, Gilman JB, Kuster WC, Holloway JS, Graus M, Flynn J, Lefer B, Warneke C, and de Gouw J, Evidence of rapid production of organic acids in an urban air mass. Geophys. Res. Lett 2011, 38 (17), 10.1029/2011gl048420. [DOI] [Google Scholar]
- (12).Kesselmeier J, Bode K, Gerlach C, and Jork EM, Exchange of atmospheric formic and acetic acids with trees and crop plants under controlled chamber and purified air conditions. Atmos. Environ 1998, 32 (10), 1765–1775, 10.1016/s1352-2310(97)00465-2. [DOI] [Google Scholar]
- (13).Mielnik A, Link M, Mattila J, Fulgham SR, and Farmer DK, Emission of formic and acetic acids from two Colorado soils. Environ. Sci. Processes Impacts 2018, 20 (11), 1537–1545, 10.1039/c8em00356d. [DOI] [PubMed] [Google Scholar]
- (14).Fulgham SR, Brophy P, Link M, Ortega J, Pollack I, and Farmer DK, Seasonal Flux Measurements over a Colorado Pine Forest Demonstrate a Persistent Source of Organic Acids. ACS Earth Space Chem. 2019, 3 (9), 2017–2032, 10.1021/acsearthspacechem.9b00182. [DOI] [Google Scholar]
- (15).Friedman B, Link MF, Fulgham SR, Brophy P, Galang A, Brune WH, Jathar SH, and Farmer DK, Primary and Secondary Sources of Gas-Phase Organic Acids from Diesel Exhaust. Environ. Sci. Technol 2017, 51 (18), 10872–10880, 10.1021/acs.est.7b01169. [DOI] [PubMed] [Google Scholar]
- (16).Link MF, Nguyen TB, Bates K, Müller J, and Farmer DK, Can Isoprene Oxidation Explain High Concentrations of Atmospheric Formic and Acetic Acid over Forests? ACS Earth Space Chem. 2020, 4 (5), 730–740, 10.1021/acsearthspacechem.0c00010. [DOI] [Google Scholar]
- (17).Neeb P, Sauer F, Horie O, and Moortgat GK, Formation of hydroxymethyl hydroperoxide and formic acid in alkene ozonolysis in the presence of water vapour. Atmos. Environ 1997, 31 (10), 1417–1423, 10.1016/s1352-2310(96)00322-6. [DOI] [Google Scholar]
- (18).Hatakeyama S, Washida N, and Akimoto H, Rate constants and mechanisms for the reaction of hydroxyl (OD) radicals with acetylene, propyne, and 2-butyne in air at 297 ± 2 K. J. Phys. Chem 1986, 90 (1), 173–178, 10.1021/j100273a039. [DOI] [Google Scholar]
- (19).Andrews DU, Heazlewood BR, Maccarone AT, Conroy T, Payne RJ, Jordan MJ, and Kable SH, Photo-tautomerization of acetaldehyde to vinyl alcohol: a potential route to tropospheric acids. Science 2012, 337 (6099), 1203–6, 10.1126/science.1220712. [DOI] [PubMed] [Google Scholar]
- (20).Shaw MF, Sztaray B, Whalley LK, Heard DE, Millet DB, Jordan MJT, Osborn DL, and Kable SH, Photo-tautomerization of acetaldehyde as a photochemical source of formic acid in the troposphere. Nat. Commun 2018, 9 (1), 2584, 10.1038/s41467-018-04824-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Molina MJ, Ivanov AV, Trakhtenberg S, and Molina LT, Atmospheric evolution of organic aerosol. Geophys. Res. Lett 2004, 31 (22), 10.1029/2004gl020910. [DOI] [Google Scholar]
- (22).Park J, Gomez AL, Walser ML, Lin A, and Nizkorodov SA, Ozonolysis and photolysis of alkene-terminated self-assembled monolayers on quartz nanoparticles: implications for photochemical aging of organic aerosol particles. Phys. Chem. Chem. Phys 2006, 8 (21), 2506–12, 10.1039/b602704k. [DOI] [PubMed] [Google Scholar]
- (23).Malecha KT, and Nizkorodov SA, Photodegradation of Secondary Organic Aerosol Particles as a Source of Small, Oxygenated Volatile Organic Compounds. Environ. Sci. Technol 2016, 50 (18), 9990–7, 10.1021/acs.est.6b02313. [DOI] [PubMed] [Google Scholar]
- (24).Malecha KT, Cai Z, and Nizkorodov SA, Photodegradation of Secondary Organic Aerosol Material Quantified with a Quartz Crystal Microbalance. Environ. Sci. Technol. Lett 2018, 5 (6), 366–371, 10.1021/acs.estlett.8b00231. [DOI] [Google Scholar]
- (25).Pan X, Underwood JS, Xing JH, Mang SA, and Nizkorodov SA, Photodegradation of secondary organic aerosol generated from limonene oxidation by ozone studied with chemical ionization mass spectrometry. Atmos. Chem. Phys 2009, 9 (12), 3851–3865, 10.5194/acp-9-3851-2009. [DOI] [Google Scholar]
- (26).Walser ML, Park J, Gomez AL, Russell AR, and Nizkorodov SA, Photochemical aging of secondary organic aerosol particles generated from the oxidation of d-limonene. J. Phys. Chem. A 2007, 111 (10), 1907–13, 10.1021/jp066293l. [DOI] [PubMed] [Google Scholar]
- (27).Akagi SK, Craven JS, Taylor JW, McMeeking GR, Yokelson RJ, Burling IR, Urbanski SP, Wold CE, Seinfeld JH, Coe H, Alvarado MJ, and Weise DR, Evolution of trace gases and particles emitted by a chaparral fire in California. Atmos. Chem. Phys 2012, 12 (3), 1397–1421, 10.5194/acp-12-1397-2012. [DOI] [Google Scholar]
- (28).Franco B, Clarisse L, Stavrakou T, Müller JF, Taraborrelli D, Hadji‐Lazaro J, Hannigan JW, Hase F, Hurtmans D, Jones N, Lutsch E, Mahieu E, Ortega I, Schneider M, Strong K, Vigouroux C, Clerbaux C, and Coheur PF, Spaceborne Measurements of Formic and Acetic Acids: A Global View of the Regional Sources. Geophys. Res. Lett 2020, 47 (4), 10.1029/2019gl086239. [DOI] [Google Scholar]
- (29).González Abad G, Bernath PF, Boone CD, McLeod SD, Manney GL, and Toon GC, Global distribution of upper tropospheric formic acid from the ACE-FTS. Atmos. Chem. Phys 2009, 9 (20), 8039–8047, 10.5194/acp-9-8039-2009. [DOI] [Google Scholar]
- (30).Rinsland CP, Boone CD, Bernath PF, Mahieu E, Zander R, Dufour G, Clerbaux C, Turquety S, Chiou L, McConnell JC, Neary L, and Kaminski JW, First space-based observations of formic acid (HCOOH): Atmospheric Chemistry Experiment austral spring 2004 and 2005 Southern Hemisphere tropical-mid-latitude upper tropospheric measurements. Geophys. Res. Lett 2006, 33 (23), 10.1029/2006gl027128. [DOI] [Google Scholar]
- (31).Yokelson RJ, Crounse JD, DeCarlo PF, Karl T, Urbanski S, Atlas E, Campos T, Shinozuka Y, Kapustin V, Clarke AD, Weinheimer A, Knapp DJ, Montzka DD, Holloway J, Weibring P, Flocke F, Zheng W, Toohey D, Wennberg PO, Wiedinmyer C, Mauldin L, Fried A, Richter D, Walega J, Jimenez JL, Adachi K, Buseck PR, Hall SR, and Shetter R, Emissions from biomass burning in the Yucatan. Atmos. Chem. Phys 2009, 9 (15), 5785–5812, 10.5194/acp-9-5785-2009. [DOI] [Google Scholar]
- (32).Chaliyakunnel S, Millet DB, Wells KC, Cady-Pereira KE, and Shephard MW, A Large Underestimate of Formic Acid from Tropical Fires: Constraints from Space-Borne Measurements. Environ. Sci. Technol 2016, 50 (11), 5631–40, 10.1021/acs.est.5b06385. [DOI] [PubMed] [Google Scholar]
- (33).Pommier M, Clerbaux C, and Coheur P, Determination of enhancement ratios of HCOOH relative to CO in biomass burning plumes by the Infrared Atmospheric Sounding Interferometer (IASI). Atmos. Chem. Phys 2017, 17 (18), 11089–11105, 10.5194/acp-17-11089-2017. [DOI] [Google Scholar]
- (34).Yamanouchi S, Strong K, Lutsch E, and Jones DBA, Detection of HCOOH, CH3OH, CO, HCN, and C2H6 in Wildfire Plumes Transported Over Toronto Using Ground‐Based FTIR Measurements From 2002–2018. J. Geophys. Res.: Atmos 2020, 125 (16), 10.1029/2019jd031924. [DOI] [Google Scholar]
- (35).Baboukas ED, Kanakidou M, and Mihalopoulos N, Carboxylic acids in gas and particulate phase above the Atlantic Ocean. J. Geophys. Res.: Atmos 2000, 105 (D11), 14459–14471, 10.1029/1999jd900977. [DOI] [Google Scholar]
- (36).Dibb JE, and Arsenault M, Shouldn’t snowpacks be sources of monocarboxylic acids? Atmos. Environ 2002, 36 (15–16), 2513–2522, 10.1016/s1352-2310(02)00131-0. [DOI] [Google Scholar]
- (37).Dibb JE, Talbot RW, Whitlow SI, Shipham MC, Winterle J, McConnell J, and Bales R, Biomass burning signatures in the atmosphere and snow at Summit, Greenland: An event on 5 August 1994. Atmos. Environ 1996, 30 (4), 553–561, 10.1016/1352-2310(95)00328-2. [DOI] [Google Scholar]
- (38).Jones BT, Muller JBA, O’Shea SJ, Bacak A, Le Breton M, Bannan TJ, Leather KE, Booth AM, Illingworth S, Bower K, Gallagher MW, Allen G, Shallcross DE, Bauguitte SJB, Pyle JA, and Percival CJ, Airborne measurements of HC(O)OH in the European Arctic: A winter – summer comparison. Atmos. Environ 2014, 99, 556–567, 10.1016/j.atmosenv.2014.10.030. [DOI] [Google Scholar]
- (39).Mungall EL, Abbatt JPD, Wentzell JJB, Lee AKY, Thomas JL, Blais M, Gosselin M, Miller LA, Papakyriakou T, Willis MD, and Liggio J, Microlayer source of oxygenated volatile organic compounds in the summertime marine Arctic boundary layer. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (24), 6203–6208, 10.1073/pnas.1620571114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wang S, Apel EC, Hornbrook RS, Hills A, Emmons LK, Tilmes S, Lamarque JF, Jimenez JL, Campuzano-Jost P, Nault BA, Crounse JD, Wennberg PO, Ryerson TB, Thompson CR, Peischl J, Moore F, Nance D, Hall B, Elkins J, Tanner D, Gregory Huey L, Hall SR, Ullmann K, Orlando JJ, Tyndall GS, Flocke FM, Ray E, Hanisco TF, Wolfe GM, St Clair J, Commane R, Daube B, Barletta B, Blake DR, Weinzierl B, Dollner M, Conley A, Vitt F, Wofsy SC, and Riemer DD, Atmospheric Acetaldehyde: Importance of Air-Sea Exchange and a Missing Source in the Remote Troposphere. Geophys. Res. Lett 2019, 46 (10), 5601–5613, 10.1029/2019GL082034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wofsy SC, Afshar S, Allen HM, Apel E, Asher EC, Barletta B, Bent J, Bian H, Biggs BC, Blake DR, Blake N, Bourgeois I, Brock CA, Brune WH, Budney JW, Bui TP, Butler A, Campuzano-Jost P, Chang CS, Chin M, Commane R, Correa G, Crounse JD, Cullis PD, Daube BC, Day DA, Dean-Day JM, Dibb JE, DiGangi JP, Diskin GS, Dollner M, Elkins JW, Erdesz F, Fiore AM, Flynn CM, Froyd K, Gesler DW, Hall SR, Hanisco TF, Hannun RA, Hills AJ, Hintsa EJ, Hoffman A, Hornbrook RS, Huey LG, Hughes S, Jimenez JL, Johnson BJ, Katich JM, Keeling RF, Kim MJ, Kupc A, Lait LR, Lamarque J-F, Liu J, McKain K, Mclaughlin RJ, Meinardi S, Miller DO, Montzka SA, Moore FL, Morgan EJ, Murphy DM, Murray LT, Nault BA, Neuman JA, Newman PA, Nicely JM, Pan X, Paplawsky W, Peischl J, Prather MJ, Price DJ, Ray E, Reeves JM, Richardson M, Rollins AW, Rosenlof KH, Ryerson TB, Scheuer E, Schill GP, Schroder JC, Schwarz JP, St.Clair JM, Steenrod SD, Stephens BB, Strode SA, Sweeney C, Tanner D, Teng AP, Thames AB, Thompson CR, Ullmann K, Veres PR, Vieznor N, Wagner NL, Watt A, Weber R, Weinzierl B, Wennberg P, Williamson CJ, Wilson JC, Wolfe GM, Woods CT, and Zeng LH, ATom: Merged Atmospheric Chemistry, Trace Gases, and Aerosols. ORNL DAAC, Oak Ridge, Tennessee, USA. 2018, 10.3334/ornldaac/1581. [DOI] [Google Scholar]
- (42).Prather MJ, Zhu X, Flynn CM, Strode SA, Rodriguez JM, Steenrod SD, Liu J, Lamarque J-F, Fiore AM, Horowitz LW, Mao J, Murray LT, Shindell DT, and Wofsy SC, Global atmospheric chemistry – which air matters. Atmos. Chem. Phys 2017, 17 (14), 9081–9102, 10.5194/acp-17-9081-2017. [DOI] [Google Scholar]
- (43).Veres PR, Neuman JA, Bertram TH, Assaf E, Wolfe GM, Williamson CJ, Weinzierl B, Tilmes S, Thompson CR, Thames AB, Schroder JC, Saiz-Lopez A, Rollins AW, Roberts JM, Price D, Peischl J, Nault BA, Moller KH, Miller DO, Meinardi S, Li Q, Lamarque JF, Kupc A, Kjaergaard HG, Kinnison D, Jimenez JL, Jernigan CM, Hornbrook RS, Hills A, Dollner M, Day DA, Cuevas CA, Campuzano-Jost P, Burkholder J, Bui TP, Brune WH, Brown SS, Brock CA, Bourgeois I, Blake DR, Apel EC, and Ryerson TB, Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (9), 4505–4510, 10.1073/pnas.1919344117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Veres P, Gilman JB, Roberts JM, Kuster WC, Warneke C, Burling IR, and de Gouw J, Development and validation of a portable gas phase standard generation and calibration system for volatile organic compounds. Atmos. Meas. Tech 2010, 3 (3), 683–691, 10.5194/amt-3-683-2010. [DOI] [Google Scholar]
- (45).McKain K, and Sweeney C, Readme for NOAA-Picarro on ATom-1,2,3,4. 2018, https://espo.nasa.gov/sites/default/files/archive_docs/NOAA-Picarro_ATom1234_readme.pdf.
- (46).Commane R, Budney JW, Gonzalez ramos Y, Sargent M, Wofsy SC, and Daube BC, ATom: Measurements from the Quantum Cascade Laser System (QCLS). 2020, 10.3334/ORNLDAAC/1747. [DOI]
- (47).Allen HM, Crounse JD, Kim MJ, Teng AP, and Wennberg PO, ATom: L2 In Situ Data from Caltech Chemical Ionization Mass Spectrometer (CIT-CIMS). 2019, 10.3334/ORNLDAAC/1713. [DOI]
- (48).Crounse JD, McKinney KA, Kwan AJ, and Wennberg PO, Measurement of gas-phase hydroperoxides by chemical ionization mass spectrometry. Anal. Chem 2006, 78 (19), 6726–32, 10.1021/ac0604235. [DOI] [PubMed] [Google Scholar]
- (49).Hanisco TF, Bian H, Nicely JM, Pan X, Hannun RA, St.Clair JM, and Wolfe GM, ATom: L2 Measurements of In Situ Airborne Formaldehyde (ISAF). 2019, 10.3334/ORNLDAAC/1730. [DOI]
- (50).Cazorla M, Wolfe GM, Bailey SA, Swanson AK, Arkinson HL, and Hanisco TF, A new airborne laser-induced fluorescence instrument for in situ detection of formaldehyde throughout the troposphere and lower stratosphere. Atmos. Meas. Tech 2015, 8 (2), 541–552, 10.5194/amt-8-541-2015. [DOI] [Google Scholar]
- (51).Apel EC, Asher EC, Hills AJ, and Hornbrook RS, ATom: L2 Volatile Organic Compounds (VOCs) from the Trace Organic Gas Analyzer (TOGA). 2019, 10.3334/ORNLDAAC/1749. [DOI]
- (52).Barletta B, Biggs BC, Blake DR, Blake N, Hoffman A, Hughes S, Meinardi S, Vieznor N, and Woods CT, ATom: L2 Halocarbons and Hydrocarbons from the UC-Irvine Whole Air Sampler (WAS). 2019, 10.3334/ORNLDAAC/1751. [DOI]
- (53).Ryerson TB, Thompson CR, Peischl J, and Bourgeois I, ATom: L2 In Situ Measurements from NOAA Nitrogen Oxides and Ozone (NOyO3) Instrument. 2019, 10.3334/ORNLDAAC/1734. [DOI]
- (54).Schwarz JP, and Katich JM, ATom: L2 In Situ Measurements from Single Particle Soot Photometer (SP2). 2019, 10.3334/ORNLDAAC/1672. [DOI]
- (55).Jimenez JL, Campuzano-Jost P, Day DA, Nault BA, Price DJ, and Schroder JC, ATom: L2 Measurements from CU High-Resolution Aerosol Mass Spectrometer (HR-AMS). 2019, 10.3334/ORNLDAAC/1716. [DOI]
- (56).Hodzic A, Campuzano-Jost P, Bian H, Chin M, Colarco PR, Day DA, Froyd KD, Heinold B, Jo DS, Katich JM, Kodros JK, Nault BA, Pierce JR, Ray E, Schacht J, Schill GP, Schroder JC, Schwarz JP, Sueper DT, Tegen I, Tilmes S, Tsigaridis K, Yu P, and Jimenez JL, Characterization of organic aerosol across the global remote troposphere: a comparison of ATom measurements and global chemistry models. Atmos. Chem. Phys 2020, 20 (8), 4607–4635, 10.5194/acp-20-4607-2020. [DOI] [Google Scholar]
- (57).Schill GP, Froyd KD, Bian H, Kupc A, Williamson C, Brock CA, Ray E, Hornbrook RS, Hills AJ, Apel EC, Chin M, Colarco PR, and Murphy DM, Widespread biomass burning smoke throughout the remote troposphere. Nat. Geosci 2020, 13 (6), 422–427, 10.1038/s41561-020-0586-1. [DOI] [Google Scholar]
- (58).Spanu A, Dollner M, Gasteiger J, Bui TP, and Weinzierl B, Flow-induced errors in airborne in situ measurements of aerosols and clouds. Atmos. Meas. Tech 2020, 13 (4), 1963–1987, 10.5194/amt-13-1963-2020. [DOI] [Google Scholar]
- (59).GEOS-Chem 12.1.1, 2018, 10.5281/zenodo.2249246. [DOI]
- (60).Bian H, and Prather MJ, Fast-J2: Accurate Simulation of Stratospheric Photolysis in Global Chemical Models. J. Atmos. Chem 2002, 41 (3), 281–296, 10.1023/a:1014980619462. [DOI] [Google Scholar]
- (61).Mao J, Jacob DJ, Evans MJ, Olson JR, Ren X, Brune WH, Clair JMS, Crounse JD, Spencer KM, Beaver MR, Wennberg PO, Cubison MJ, Jimenez JL, Fried A, Weibring P, Walega JG, Hall SR, Weinheimer AJ, Cohen RC, Chen G, Crawford JH, McNaughton C, Clarke AD, Jaeglé L, Fisher JA, Yantosca RM, Le Sager P, and Carouge C, Chemistry of hydrogen oxide radicals (HOx) in the Arctic troposphere in spring. Atmos. Chem. Phys 2010, 10 (13), 5823–5838, 10.5194/acp-10-5823-2010. [DOI] [Google Scholar]
- (62).Eastham SD, Weisenstein DK, and Barrett SRH, Development and evaluation of the unified tropospheric–stratospheric chemistry extension (UCX) for the global chemistry-transport model GEOS-Chem. Atmos. Environ 2014, 89, 52–63, 10.1016/j.atmosenv.2014.02.001. [DOI] [Google Scholar]
- (63).Lin S, and Rood RB, Multidimensional Flux-Form Semi-Lagrangian Transport Schemes. Mon. Weather Rev 1996, 124 (9), 2046–2070, . [DOI] [Google Scholar]
- (64).Wu S, Mickley LJ, Jacob DJ, Logan JA, Yantosca RM, and Rind D, Why are there large differences between models in global budgets of tropospheric ozone? J. Geophys. Res.: Atmos 2007, 112 (D5), 10.1029/2006jd007801. [DOI] [Google Scholar]
- (65).Lin J, and McElroy MB, Impacts of boundary layer mixing on pollutant vertical profiles in the lower troposphere: Implications to satellite remote sensing. Atmos. Environ 2010, 44 (14), 1726–1739, 10.1016/j.atmosenv.2010.02.009. [DOI] [Google Scholar]
- (66).Hoesly RM, Smith SJ, Feng L, Klimont Z, Janssens-Maenhout G, Pitkanen T, Seibert JJ, Vu L, Andres RJ, Bolt RM, Bond TC, Dawidowski L, Kholod N, Kurokawa J, Li M, Liu L, Lu Z, Moura MCP, O’Rourke PR, and Zhang Q, Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS). Geosci. Model Dev 2018, 11 (1), 369–408, 10.5194/gmd-11-369-2018. [DOI] [Google Scholar]
- (67).Marais EA, and Wiedinmyer C, Air Quality Impact of Diffuse and Inefficient Combustion Emissions in Africa (DICE-Africa). Environ. Sci. Technol 2016, 50 (19), 10739–10745, 10.1021/acs.est.6b02602. [DOI] [PubMed] [Google Scholar]
- (68).Guenther AB, Jiang X, Heald CL, Sakulyanontvittaya T, Duhl T, Emmons LK, and Wang X, The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions. Geosci. Model Dev 2012, 5 (6), 1471–1492, 10.5194/gmd-5-1471-2012. [DOI] [Google Scholar]
- (69).Hu L, Millet DB, Baasandorj M, Griffis TJ, Turner P, Helmig D, Curtis AJ, and Hueber J, Isoprene emissions and impacts over an ecological transition region in the U.S. Upper Midwest inferred from tall tower measurements. J. Geophys. Res.: Atmos 2015, 120 (8), 3553–3571, 10.1002/2014jd022732. [DOI] [Google Scholar]
- (70).van der Werf GR, Randerson JT, Giglio L, van Leeuwen TT, Chen Y, Rogers BM, Mu M, van Marle MJE, Morton DC, Collatz GJ, Yokelson RJ, and Kasibhatla PS, Global fire emissions estimates during 1997–2016. Earth Syst. Sci. Data 2017, 9 (2), 697–720, 10.5194/essd-9-697-2017. [DOI] [Google Scholar]
- (71).Akagi SK, Yokelson RJ, Wiedinmyer C, Alvarado MJ, Reid JS, Karl T, Crounse JD, and Wennberg PO, Emission factors for open and domestic biomass burning for use in atmospheric models. Atmos. Chem. Phys 2011, 11 (9), 4039–4072, 10.5194/acp-11-4039-2011. [DOI] [Google Scholar]
- (72).Wesely ML, Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models. Atmos. Environ 1989, 23 (6), 1293–1304, 10.1016/0004-6981(89)90153-4. [DOI] [Google Scholar]
- (73).Wang Y, Jacob DJ, and Logan JA, Global simulation of tropospheric O3-NOx-hydrocarbon chemistry: 1. Model formulation. J. Geophys. Res.: Atmos 1998, 103 (D9), 10713–10725, 10.1029/98jd00158. [DOI] [Google Scholar]
- (74).Amos HM, Jacob DJ, Holmes CD, Fisher JA, Wang Q, Yantosca RM, Corbitt ES, Galarneau E, Rutter AP, Gustin MS, Steffen A, Schauer JJ, Graydon JA, Louis VLS, Talbot RW, Edgerton ES, Zhang Y, and Sunderland EM, Gas-particle partitioning of atmospheric Hg(II) and its effect on global mercury deposition. Atmos. Chem. Phys 2012, 12 (1), 591–603, 10.5194/acp-12-591-2012. [DOI] [Google Scholar]
- (75).Mari C, Jacob DJ, and Bechtold P, Transport and scavenging of soluble gases in a deep convective cloud. J. Geophys. Res.: Atmos 2000, 105 (D17), 22255–22267, 10.1029/2000jd900211. [DOI] [Google Scholar]
- (76).Bowman KP, Large-scale isentropic mixing properties of the Antarctic polar vortex from analyzed winds. J. Geophys. Res.: Atmos 1993, 98 (D12), 10.1029/93JD02599. [DOI] [Google Scholar]
- (77).Bowman KP, and Carrie GD, The Mean-Meridional Transport Circulation of the Troposphere in an Idealized GCM. J. Atmos. Sci 2002, 59 (9), 1502–1514, . [DOI] [Google Scholar]
- (78).Freeborn PH, Wooster MJ, Roy DP, and Cochrane MA, Quantification of MODIS fire radiative power (FRP) measurement uncertainty for use in satellite-based active fire characterization and biomass burning estimation. Geophys. Res. Lett 2014, 41 (6), 1988–1994, 10.1002/2013GL059086. [DOI] [Google Scholar]
- (79).Schroeder W, Oliva P, Giglio L, and Csiszar IA, The New VIIRS 375m active fire detection data product: Algorithm description and initial assessment. Remote Sens. Environ 2014, 143, 85–96, 10.1016/j.rse.2013.12.008. [DOI] [Google Scholar]
- (80).Singh HB, Thompson AM, and Schlager H, SONEX airborne mission and coordinated POLINAT-2 activity: Overview and accomplishments. Geophys. Res. Lett 1999, 26 (20), 3053–3056, 10.1029/1999gl900588. [DOI] [Google Scholar]
- (81).Hoell JM, Davis DD, Jacob DJ, Rodgers MO, Newell RE, Fuelberg HE, McNeal RJ, Raper JL, and Bendura RJ, Pacific Exploratory Mission in the tropical Pacific: PEM-Tropics A, August-September 1996. J. Geophys. Res.: Atmos 1999, 104 (D5), 5567–5583, 10.1029/1998jd100074. [DOI] [Google Scholar]
- (82).Hoell JM, Davis DD, Liu SC, Newell R, Shipham M, Akimoto H, McNeal RJ, Bendura RJ, and Drewry JW, Pacific Exploratory Mission-West A (PEM-West A): September-October 1991. J. Geophys. Res.: Atmos 1996, 101 (D1), 1641–1653, 10.1029/95jd00622. [DOI] [Google Scholar]
- (83).Hoell JM, Davis DD, Liu SC, Newell RE, Akimoto H, McNeal RJ, and Bendura RJ, The Pacific Exploratory Mission-West Phase B: February-March, 1994. J. Geophys. Res.: Atmos 1997, 102 (D23), 28223–28239, 10.1029/97jd02581. [DOI] [Google Scholar]
- (84).Kleb MM, Chen G, Crawford JH, Flocke FM, and Brown CC, An overview of measurement comparisons from the INTEX-B/MILAGRO airborne field campaign. Atmos. Meas. Tech 2011, 4 (1), 9–27, 10.5194/amt-4-9-2011. [DOI] [Google Scholar]
- (85).Singh HB, Brune WH, Crawford JH, Flocke F, and Jacob DJ, Chemistry and transport of pollution over the Gulf of Mexico and the Pacific: spring 2006 INTEX-B campaign overview and first results. Atmos. Chem. Phys 2009, 9 (7), 2301–2318, 10.5194/acp-9-2301-2009. [DOI] [Google Scholar]
- (86).Fishman J, Hoell JM, Bendura RD, McNeal RJ, and Kirchhoff VWJH, NASA GTE TRACE A experiment (September-October 1992): Overview. J. Geophys. Res.: Atmos 1996, 101 (D19), 23865–23879, 10.1029/96jd00123. [DOI] [Google Scholar]
- (87).Coggon MM, Veres PR, Yuan B, Koss A, Warneke C, Gilman JB, Lerner BM, Peischl J, Aikin KC, Stockwell CE, Hatch LE, Ryerson TB, Roberts JM, Yokelson RJ, and de Gouw JA, Emissions of nitrogen-containing organic compounds from the burning of herbaceous and arboraceous biomass: Fuel composition dependence and the variability of commonly used nitrile tracers. Geophys. Res. Lett 2016, 43 (18), 9903–9912, 10.1002/2016GL070562. [DOI] [Google Scholar]
- (88).Travis KR, Heald CL, Allen HM, Apel EC, Arnold SR, Blake DR, Brune WH, Chen X, Commane R, Crounse JD, Daube BC, Diskin GS, Elkins JW, Evans MJ, Hall SR, Hintsa EJ, Hornbrook RS, Kasibhatla PS, Kim MJ, Luo G, McKain K, Millet DB, Moore FL, Peischl J, Ryerson TB, Sherwen T, Thames AB, Ullmann K, Wang X, Wennberg PO, Wolfe GM, and Yu F, Constraining remote oxidation capacity with ATom observations. Atmos. Chem. Phys 2020, 20 (13), 7753–7781, 10.5194/acp-20-7753-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Clubb AE, Jordan MJ, Kable SH, and Osborn DL, Phototautomerization of Acetaldehyde to Vinyl Alcohol: A Primary Process in UV-Irradiated Acetaldehyde from 295 to 335 nm. J. Phys. Chem. Lett 2012, 3 (23), 3522–6, 10.1021/jz301701x. [DOI] [PubMed] [Google Scholar]
- (90).So S, Wille U, and da Silva G, Atmospheric chemistry of enols: a theoretical study of the vinyl alcohol + OH + O2 reaction mechanism. Environ. Sci. Technol 2014, 48 (12), 6694–701, 10.1021/es500319q. [DOI] [PubMed] [Google Scholar]
- (91).da Silva G, Carboxylic acid catalyzed keto-enol tautomerizations in the gas phase. Angew. Chem., Int. Ed. Engl 2010, 49 (41), 7523–5, 10.1002/anie.201003530. [DOI] [PubMed] [Google Scholar]
- (92).Archibald AT, McGillen MR, Taatjes CA, Percival CJ, and Shallcross DE, Atmospheric transformation of enols: A potential secondary source of carboxylic acids in the urban troposphere. Geophys. Res. Lett 2007, 34 (21), 10.1029/2007GL031032. [DOI] [Google Scholar]
- (93).Sander R, Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys 2015, 15 (8), 4399–4981, 10.5194/acp-15-4399-2015. [DOI] [Google Scholar]
- (94).Sander SP, Abbatt J, Barker JR, Burkholder JB, Friedl RR, Golden DM, Huie RE, Kolb CE, Kurylo MJ, Moortgat GK, Orkin VL, and Wine PH, Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 17, JPL Publication 10–6, Jet Propulsion Laboratory, Pasadena, available at: https://jpldataeval.jpl.nasa.gov/pdf/JPL%2010-6%20Final%2015June2011.pdf (last access: 29 May 2018), 2011. 2011. [Google Scholar]
- (95).Johnson BJ, Betterton EA, and Craig D, Henry’s law coefficients of formic and acetic acids. J. Atmos. Chem 1996, 24 (2), 113–119, 10.1007/bf00162406. [DOI] [Google Scholar]
- (96).Singh HB, Salas L, Herlth D, Kolyer R, Czech E, Viezee W, Li Q, Jacob DJ, Blake D, Sachse G, Harward CN, Fuelberg H, Kiley CM, Zhao Y, and Kondo Y, In situ measurements of HCN and CH3CN over the Pacific Ocean: Sources, sinks, and budgets. J. Geophys. Res.: Atmos 2003, 108 (D20), 10.1029/2002jd003006. [DOI] [Google Scholar]
- (97).Crutzen PJ, and Lawrence MG, The Impact of Precipitation Scavenging on the Transport of Trace Gases: A 3-Dimensional Model Sensitivity Study. J. Atmos. Chem 2000, 37 (1), 81–112, 10.1023/a:1006322926426. [DOI] [Google Scholar]
- (98).Yu P, Froyd KD, Portmann RW, Toon OB, Freitas SR, Bardeen CG, Brock C, Fan T, Gao RS, Katich JM, Kupc A, Liu S, Maloney C, Murphy DM, Rosenlof KH, Schill G, Schwarz JP, and Williamson C, Efficient In‐Cloud Removal of Aerosols by Deep Convection. Geophys. Res. Lett 2019, 46 (2), 1061–1069, 10.1029/2018GL080544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Koyama T, and Thoivipson TG, Identification and Determination of Organic Acids in Sea Water by Partition Chromatography. J. Oceanogr. Soc. Jpn 1964, 20 (5), 209–220, 10.5928/kaiyou1942.20.209. [DOI] [Google Scholar]
- (100).Hama T, and Handa N, Volatile Organic Acids in Lake Water;Their Gas Chromatographic Determination and Ecological Significance. Japanese Journal of Limnology (Rikusuigaku Zasshi) 1981, 42 (1), 8–19, 10.3739/rikusui.42.8. [DOI] [Google Scholar]
- (101).Kieber DJ, Vaughan GM, and Mopper K, Determination of formate in natural waters by a coupled enzymatic/high-performance liquid chromatographic technique. Anal. Chem 2002, 60 (17), 1654–1659, 10.1021/ac00168a004. [DOI] [Google Scholar]
- (102).Vaughan GM, and Mopper K, Determination of nanomolar levels of formate in natural waters based on a luminescence enzymatic assay. Anal. Chim. Acta 1990, 231, 299–303, 10.1016/s0003-2670(00)86430-2. [DOI] [Google Scholar]
- (103).Mopper K, and Kieber DJ, Distribution and biological turnover of dissolved organic compounds in the water column of the Black Sea. Deep Sea Res. Part A 1991, 38, S1021–S1047, 10.1016/s0198-0149(10)80022-6. [DOI] [Google Scholar]
- (104).Talaie A, Boger Z, Romagnoli JA, Adeloju SB, and Yuan YJ, Data acquisition, signal processing and modelling: a study of a conducting polypyrrole formate biosensor Part 1: Batch experiment. Synth. Met 1996, 83 (1), 21–26, 10.1016/s0379-6779(97)80048-3. [DOI] [Google Scholar]
- (105).Hobbs PV, Sinha P, Yokelson RJ, Christian TJ, Blake DR, Gao S, Kirchstetter TW, Novakov T, and Pilewskie P, Evolution of gases and particles from a savanna fire in South Africa. J. Geophys. Res.: Atmos 2003, 108 (D13), 10.1029/2002jd002352. [DOI] [Google Scholar]
- (106).Yuan B, Veres PR, Warneke C, Roberts JM, Gilman JB, Koss A, Edwards PM, Graus M, Kuster WC, Li SM, Wild RJ, Brown SS, Dubé WP, Lerner BM, Williams EJ, Johnson JE, Quinn PK, Bates TS, Lefer B, Hayes PL, Jimenez JL, Weber RJ, Zamora R, Ervens B, Millet DB, Rappenglück B, and de Gouw JA, Investigation of secondary formation of formic acid: urban environment vs. oil and gas producing region. Atmos. Chem. Phys 2015, 15 (4), 1975–1993, 10.5194/acp-15-1975-2015. [DOI] [Google Scholar]
- (107).Nah T, Ji Y, Tanner DJ, Guo H, Sullivan AP, Ng NL, Weber RJ, and Huey LG, Real-time measurements of gas-phase organic acids using chemical ionization mass spectrometry. Atmos. Meas. Tech 2018, 11 (9), 5087–5104, 10.5194/amt-11-5087-2018. [DOI] [Google Scholar]
- (108).Mungall EL, Abbatt JPD, Wentzell JJB, Wentworth GR, Murphy JG, Kunkel D, Gute E, Tarasick DW, Sharma S, Cox CJ, Uttal T, and Liggio J, High gas-phase mixing ratios of formic and acetic acid in the High Arctic. Atmos. Chem. Phys 2018, 18 (14), 10237–10254, 10.5194/acp-18-10237-2018. [DOI] [Google Scholar]
- (109).Bannan TJ, Murray Booth A, Le Breton M, Bacak A, Muller JBA, Leather KE, Khan MAH, Lee JD, Dunmore RE, Hopkins JR, Fleming ZL, Sheps L, Taatjes CA, Shallcross DE, and Percival CJ, Seasonality of Formic Acid (HCOOH) in London during the ClearfLo Campaign. J. Geophys. Res.: Atmos 2017, 122 (22), 10.1002/2017jd027064. [DOI] [Google Scholar]
- (110).Li K, Li J, Tong S, Wang W, Huang R, and Ge M, Characteristics of wintertime VOCs in suburban and urban Beijing: concentrations, emission ratios, and festival effects. Atmos. Chem. Phys 2019, 19 (12), 8021–8036, 10.5194/acp-19-8021-2019. [DOI] [Google Scholar]
- (111).Brophy P, and Farmer DK, A switchable reagent ion high resolution time-of-flight chemical ionization mass spectrometer for real-time measurement of gas phase oxidized species: characterization from the 2013 southern oxidant and aerosol study. Atmos. Meas. Tech 2015, 8 (7), 2945–2959, 10.5194/amt-8-2945-2015. [DOI] [Google Scholar]
- (112).IUPAC, http://iupac.pole-ether.fr. 2008.
- (113).Jost C, Trentmann J, Sprung D, Andreae MO, McQuaid JB, and Barjat H, Trace gas chemistry in a young biomass burning plume over Namibia: Observations and model simulations. J. Geophys. Res.: Atmos 2003, 108 (D13), 10.1029/2002jd002431. [DOI] [Google Scholar]
- (114).Hornbrook RS, Blake DR, Diskin GS, Fried A, Fuelberg HE, Meinardi S, Mikoviny T, Richter D, Sachse GW, Vay SA, Walega J, Weibring P, Weinheimer AJ, Wiedinmyer C, Wisthaler A, Hills A, Riemer DD, and Apel EC, Observations of nonmethane organic compounds during ARCTAS − Part 1: Biomass burning emissions and plume enhancements. Atmos. Chem. Phys 2011, 11 (21), 11103–11130, 10.5194/acp-11-11103-2011. [DOI] [Google Scholar]
- (115).de Gouw JA, Warneke C, Stohl A, Wollny AG, Brock CA, Cooper OR, Holloway JS, Trainer M, Fehsenfeld FC, Atlas EL, Donnelly SG, Stroud V, and Lueb A, Volatile organic compounds composition of merged and aged forest fire plumes from Alaska and western Canada. J. Geophys. Res.: Atmos 2006, 111 (D10), 10.1029/2005jd006175. [DOI] [Google Scholar]
- (116).Holzinger R, Williams J, Salisbury G, Klüpfel T, de Reus M, Traub M, Crutzen PJ, and Lelieveld J, Oxygenated compounds in aged biomass burning plumes over the Eastern Mediterranean: evidence for strong secondary production of methanol and acetone. Atmos. Chem. Phys 2005, 5 (1), 39–46, 10.5194/acp-5-39-2005. [DOI] [Google Scholar]
- (117).Yokelson RJ, Bertschi IT, Christian TJ, Hobbs PV, Ward DE, and Hao WM, Trace gas measurements in nascent, aged, and cloud-processed smoke from African savanna fires by airborne Fourier transform infrared spectroscopy (AFTIR). J. Geophys. Res.: Atmos 2003, 108 (D13), 10.1029/2002jd002322. [DOI] [Google Scholar]
- (118).Bates KH, Jacob DJ, Wang S, Hornbrook RS, Apel EC, Kim MJ, Millet DB, Wells KC, Chen X, Brewer JF, Ray E, Commane R, Diskin GS, and Wofsy SC, The global budget of atmospheric methanol: new constraints on secondary, oceanic, and terrestrial sources. J. Geophys. Res.: Atmos 2021, 10.1029/2020JD033439. [DOI] [Google Scholar]
- (119).Hecobian A, Liu Z, Hennigan CJ, Huey LG, Jimenez JL, Cubison MJ, Vay S, Diskin GS, Sachse GW, Wisthaler A, Mikoviny T, Weinheimer AJ, Liao J, Knapp DJ, Wennberg PO, Kürten A, Crounse JD, Clair JS, Wang Y, and Weber RJ, Comparison of chemical characteristics of 495 biomass burning plumes intercepted by the NASA DC-8 aircraft during the ARCTAS/CARB-2008 field campaign. Atmos. Chem. Phys 2011, 11 (24), 13325–13337, 10.5194/acp-11-13325-2011. [DOI] [Google Scholar]
- (120).Singh HB, Cai C, Kaduwela A, Weinheimer A, and Wisthaler A, Interactions of fire emissions and urban pollution over California: Ozone formation and air quality simulations. Atmos. Environ 2012, 56, 45–51, 10.1016/j.atmosenv.2012.03.046. [DOI] [Google Scholar]
- (121).Goode JG, Yokelson RJ, Ward DE, Susott RA, Babbitt RE, Davies MA, and Hao WM, Measurements of excess O3, CO2, CO, CH4, C2H4, C2H2, HCN, NO, NH3, HCOOH, CH3COOH, HCHO, and CH3OH in 1997 Alaskan biomass burning plumes by airborne Fourier transform infrared spectroscopy (AFTIR). J. Geophys. Res.: Atmos 2000, 105 (D17), 22147–22166, 10.1029/2000jd900287. [DOI] [Google Scholar]
- (122).de Gouw JA, Gilman JB, Kim SW, Alvarez SL, Dusanter S, Graus M, Griffith SM, Isaacman-VanWertz G, Kuster WC, Lefer BL, Lerner BM, McDonald BC, Rappenglück B, Roberts JM, Stevens PS, Stutz J, Thalman R, Veres PR, Volkamer R, Warneke C, Washenfelder RA, and Young CJ, Chemistry of Volatile Organic Compounds in the Los Angeles Basin: Formation of Oxygenated Compounds and Determination of Emission Ratios. J. Geophys. Res.: Atmos 2018, 123 (4), 2298–2319, 10.1002/2017jd027976. [DOI] [Google Scholar]
- (123).Tereszchuk KA, González Abad G, Clerbaux C, Hadji-Lazaro J, Hurtmans D, Coheur PF, and Bernath PF, ACE-FTS observations of pyrogenic trace species in boreal biomass burning plumes during BORTAS. Atmos. Chem. Phys 2013, 13 (9), 4529–4541, 10.5194/acp-13-4529-2013. [DOI] [Google Scholar]
- (124).Tereszchuk KA, González Abad G, Clerbaux C, Hurtmans D, Coheur PF, and Bernath PF, ACE-FTS measurements of trace species in the characterization of biomass burning plumes. Atmos. Chem. Phys 2011, 11 (23), 12169–12179, 10.5194/acp-11-12169-2011. [DOI] [Google Scholar]
- (125).Andreae MO, Emission of trace gases and aerosols from biomass burning – an updated assessment. Atmos. Chem. Phys 2019, 19 (13), 8523–8546, 10.5194/acp-19-8523-2019. [DOI] [Google Scholar]
- (126).Giglio L, Randerson JT, and van der Werf GR, Analysis of daily, monthly, and annual burned area using the fourth-generation global fire emissions database (GFED4). J. Geophys. Res.: Biogeosci 2013, 118 (1), 317–328, 10.1002/jgrg.20042. [DOI] [Google Scholar]
- (127).Butkovskaya NI, Pouvesle N, Kukui A, and Bras GL, Mechanism of the OH-initiated oxidation of glycolaldehyde over the temperature range 233–296 K. J. Phys. Chem. A 2006, 110 (50), 13492–9, 10.1021/jp064993k. [DOI] [PubMed] [Google Scholar]
- (128).Butkovskaya NI, Pouvesle N, Kukui A, Mu Y, and Le Bras G, Mechanism of the OH-initiated oxidation of hydroxyacetone over the temperature range 236–298 K. J. Phys. Chem. A 2006, 110 (21), 6833–43, 10.1021/jp056345r. [DOI] [PubMed] [Google Scholar]
- (129).Leather KE, McGillen MR, Cooke MC, Utembe SR, Archibald AT, Jenkin ME, Derwent RG, Shallcross DE, and Percival CJ, Acid-yield measurements of the gas-phase ozonolysis of ethene as a function of humidity using Chemical Ionisation Mass Spectrometry (CIMS). Atmos. Chem. Phys 2012, 12 (1), 469–479, 10.5194/acp-12-469-2012. [DOI] [Google Scholar]
- (130).Lee A, Goldstein AH, Kroll JH, Ng NL, Varutbangkul V, Flagan RC, and Seinfeld JH, Gas-phase products and secondary aerosol yields from the photooxidation of 16 different terpenes. J. Geophys. Res.: Atmos 2006, 111 (D17), 10.1029/2006jd007050. [DOI] [Google Scholar]
- (131).Friedman B, and Farmer DK, SOA and gas phase organic acid yields from the sequential photooxidation of seven monoterpenes. Atmos. Environ 2018, 187, 335–345, 10.1016/j.atmosenv.2018.06.003. [DOI] [Google Scholar]
- (132).Theys N, Volkamer R, Müller JF, Zarzana KJ, Kille N, Clarisse L, De Smedt I, Lerot C, Finkenzeller H, Hendrick F, Koenig TK, Lee CF, Knote C, Yu H, and Van Roozendael M, Global nitrous acid emissions and levels of regional oxidants enhanced by wildfires. Nat. Geosci 2020, 13 (10), 681–686, 10.1038/s41561-020-0637-7. [DOI] [Google Scholar]
- (133).Alvarado LMA, Richter A, Vrekoussis M, Hilboll A, Kalisz Hedegaard AB, Schneising O, and Burrows JP, Unexpected long-range transport of glyoxal and formaldehyde observed from the Copernicus Sentinel-5 Precursor satellite during the 2018 Canadian wildfires. Atmos. Chem. Phys 2020, 20 (4), 2057–2072, 10.5194/acp-20-2057-2020. [DOI] [Google Scholar]
- (134).Singh H, Chen Y, Tabazadeh A, Fukui Y, Bey I, Yantosca R, Jacob D, Arnold F, Wohlfrom K, Atlas E, Flocke F, Blake D, Blake N, Heikes B, Snow J, Talbot R, Gregory G, Sachse G, Vay S, and Kondo Y, Distribution and fate of selected oxygenated organic species in the troposphere and lower stratosphere over the Atlantic. J. Geophys. Res.: Atmos 2000, 105 (D3), 3795–3805, 10.1029/1999jd900779. [DOI] [Google Scholar]
- (135).Viatte C, Strong K, Hannigan J, Nussbaumer E, Emmons LK, Conway S, Paton-Walsh C, Hartley J, Benmergui J, and Lin J, Identifying fire plumes in the Arctic with tropospheric FTIR measurements and transport models. Atmos. Chem. Phys 2015, 15 (5), 2227–2246, 10.5194/acp-15-2227-2015. [DOI] [Google Scholar]
- (136).Vigouroux C, Stavrakou T, Whaley C, Dils B, Duflot V, Hermans C, Kumps N, Metzger JM, Scolas F, Vanhaelewyn G, Müller JF, Jones DBA, Li Q, and De Mazière M, FTIR time-series of biomass burning products (HCN, C2H6, C2H2, CH3OH, and HCOOH) at Reunion Island (21° S, 55° E) and comparisons with model data. Atmos. Chem. Phys 2012, 12 (21), 10367–10385, 10.5194/acp-12-10367-2012. [DOI] [Google Scholar]
- (137).Paton-Walsh C, Jones NB, Wilson SR, Haverd V, Meier A, Griffith DWT, and Rinsland CP, Measurements of trace gas emissions from Australian forest fires and correlations with coincident measurements of aerosol optical depth. J. Geophys. Res.: Atmos 2005, 110 (D24), 10.1029/2005jd006202. [DOI] [Google Scholar]
- (138).Rinsland CP, Dufour G, Boone CD, Bernath PF, Chiou L, Coheur P, Turquety S, and Clerbaux C, Satellite boreal measurements over Alaska and Canada during June-July 2004: Simultaneous measurements of upper tropospheric CO, C 2H6, HCN, CH3Cl, CH4, C2H2, CH3OH, HCOOH, OCS, and SF6 mixing ratios. Global Biogeochem. Cycles 2007, 21 (3), 10.1029/2006gb002795. [DOI] [Google Scholar]
- (139).Coheur PF, Herbin H, Clerbaux C, Hurtmans D, Wespes C, Carleer M, Turquety S, Rinsland CP, Remedios J, Hauglustaine D, Boone CD, and Bernath PF, ACE-FTS observation of a young biomass burning plume: first reported measurements of C2H4, C3H6O, H2CO and PAN by infrared occultation from space. Atmos. Chem. Phys 2007, 7 (20), 5437–5446, 10.5194/acp-7-5437-2007. [DOI] [Google Scholar]
- (140).R’Honi Y, Clarisse L, Clerbaux C, Hurtmans D, Duflot V, Turquety S, Ngadi Y, and Coheur PF, Exceptional emissions of NH3 and HCOOH in the 2010 Russian wildfires. Atmos. Chem. Phys 2013, 13 (8), 4171–4181, 10.5194/acp-13-4171-2013. [DOI] [Google Scholar]
- (141).Li Q, Jacob DJ, Bey I, Yantosca RM, Zhao Y, Kondo Y, and Notholt J, Atmospheric hydrogen cyanide (HCN): Biomass burning source, ocean sink? Geophys. Res. Lett 2000, 27 (3), 357–360, 10.1029/1999gl010935. [DOI] [Google Scholar]
- (142).Hodzic A, Kasibhatla PS, Jo DS, Cappa CD, Jimenez JL, Madronich S, and Park RJ, Rethinking the global secondary organic aerosol (SOA) budget: stronger production, faster removal, shorter lifetime. Atmos. Chem. Phys 2016, 16 (12), 7917–7941, 10.5194/acp-16-7917-2016. [DOI] [Google Scholar]
- (143).Tsigaridis K, Daskalakis N, Kanakidou M, Adams PJ, Artaxo P, Bahadur R, Balkanski Y, Bauer SE, Bellouin N, Benedetti A, Bergman T, Berntsen TK, Beukes JP, Bian H, Carslaw KS, Chin M, Curci G, Diehl T, Easter RC, Ghan SJ, Gong SL, Hodzic A, Hoyle CR, Iversen T, Jathar S, Jimenez JL, Kaiser JW, Kirkevåg A, Koch D, Kokkola H, Lee YH, Lin G, Liu X, Luo G, Ma X, Mann GW, Mihalopoulos N, Morcrette JJ, Müller JF, Myhre G, Myriokefalitakis S, Ng NL, O’Donnell D, Penner JE, Pozzoli L, Pringle KJ, Russell LM, Schulz M, Sciare J, Seland Ø, Shindell DT, Sillman S, Skeie RB, Spracklen D, Stavrakou T, Steenrod SD, Takemura T, Tiitta P, Tilmes S, Tost H, van Noije T, van Zyl PG, von Salzen K, Yu F, Wang Z, Wang Z, Zaveri RA, Zhang H, Zhang K, Zhang Q, and Zhang X, The AeroCom evaluation and intercomparison of organic aerosol in global models. Atmos. Chem. Phys 2014, 14 (19), 10845–10895, 10.5194/acp-14-10845-2014. [DOI] [Google Scholar]
- (144).Bond TC, Doherty SJ, Fahey DW, Forster PM, Berntsen T, DeAngelo BJ, Flanner MG, Ghan S, Kärcher B, Koch D, Kinne S, Kondo Y, Quinn PK, Sarofim MC, Schultz MG, Schulz M, Venkataraman C, Zhang H, Zhang S, Bellouin N, Guttikunda SK, Hopke PK, Jacobson MZ, Kaiser JW, Klimont Z, Lohmann U, Schwarz JP, Shindell D, Storelvmo T, Warren SG, and Zender CS, Bounding the role of black carbon in the climate system: A scientific assessment. J. Geophys. Res.: Atmos 2013, 118 (11), 5380–5552, 10.1002/jgrd.50171. [DOI] [Google Scholar]
- (145).Brune WH, Miller DO, Thames AB, Allen HM, Apel EC, Blake DR, Bui TP, Commane R, Crounse JD, Daube BC, Diskin GS, DiGangi JP, Elkins JW, Hall SR, Hanisco TF, Hannun RA, Hintsa EJ, Hornbrook RS, Kim MJ, McKain K, Moore FL, Neuman JA, Nicely JM, Peischl J, Ryerson TB, St. Clair JM, Sweeney C, Teng AP, Thompson C, Ullmann K, Veres PR, Wennberg PO, and Wolfe GM, Exploring Oxidation in the Remote Free Troposphere: Insights From Atmospheric Tomography (ATom). J. Geophys. Res.: Atmos 2020, 125 (1), 10.1029/2019jd031685. [DOI] [Google Scholar]
- (146).Wolfe GM, Nicely JM, St Clair JM, Hanisco TF, Liao J, Oman LD, Brune WB, Miller D, Thames A, Gonzalez Abad G, Ryerson TB, Thompson CR, Peischl J, McCain K, Sweeney C, Wennberg PO, Kim M, Crounse JD, Hall SR, Ullmann K, Diskin G, Bui P, Chang C, and Dean-Day J, Mapping hydroxyl variability throughout the global remote troposphere via synthesis of airborne and satellite formaldehyde observations. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (23), 11171–11180, 10.1073/pnas.1821661116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.