

Abstract
Voltage-sensitive calcium channels (VSCCs) are ubiquitous multimeric protein complexes that are necessary for the regulation of numerous physiological processes. VSCCs regulate calcium influx and various intracellular processes including muscle contraction, neurotransmission, hormone secretion, and gene transcription, with function specificity defined by the channel‟s subunits and tissue location. The functions of VSCCs in bone are often overlooked since bone is not considered an electrically excitable tissue. However, skeletal homeostasis and adaptation relies heavily on VSCCs. Inhibition or deletion of VSCCs decreases osteogenesis, impairs skeletal structure, and impedes anabolic responses to mechanical loading. While the functions of VSCCs in osteoclasts is less clear, VSCCs have distinct but complementary functions in osteoblasts and osteocytes. This review details the structure, function, and nomenclature of VSCCs, followed by a comprehensive description of the known functions of VSCCs in bone cells and their regulation of bone development, bone formation, and mechanotransduction.
Keywords: Calcium channels, bone, mechanical loading, osteoblast, osteocyte, osteoclast
Components and Physiological Roles of Voltage-Sensitive Calcium Channels
Voltage-sensitive calcium channels (VSCCs) are widely distributed protein complexes serving a variety of functions by transducing electrical potentials into intracellular signals. Acting as a secondary messenger, intracellular calcium (Ca2+) influences physiological processes including muscle contraction, hormone secretion, neurotransmission, and transcriptional regulation [1]. VSCCs are members of a transmembrane cation channel complex superfamily, which includes voltage-gated sodium and potassium channels [2]. This review provides an overview of VSCC structure and function, and how VSCCs control skeletal function.
Structure & Function of VSCCs
VSCCs are multimeric structures consisting of a pore-forming subunit and 2–3 auxiliary subunits that influence channel activity. The α1 subunit forms the channel pore and consists of four homologous voltage-sensing domains (VSD1–4) each with six transmembrane segments (S1-S6) (Fig. 1). While the α1 subunit enables Ca2+ selectivity, the auxiliary subunits (α2δ, β, and γ) contribute unique structural and regulatory properties by modulating Ca2+ influx, altering trafficking of the α1 pore, and regulating interactions with the extracellular matrix (ECM) [3, 4] (Table 1).
Fig 1. VSCC Structure.
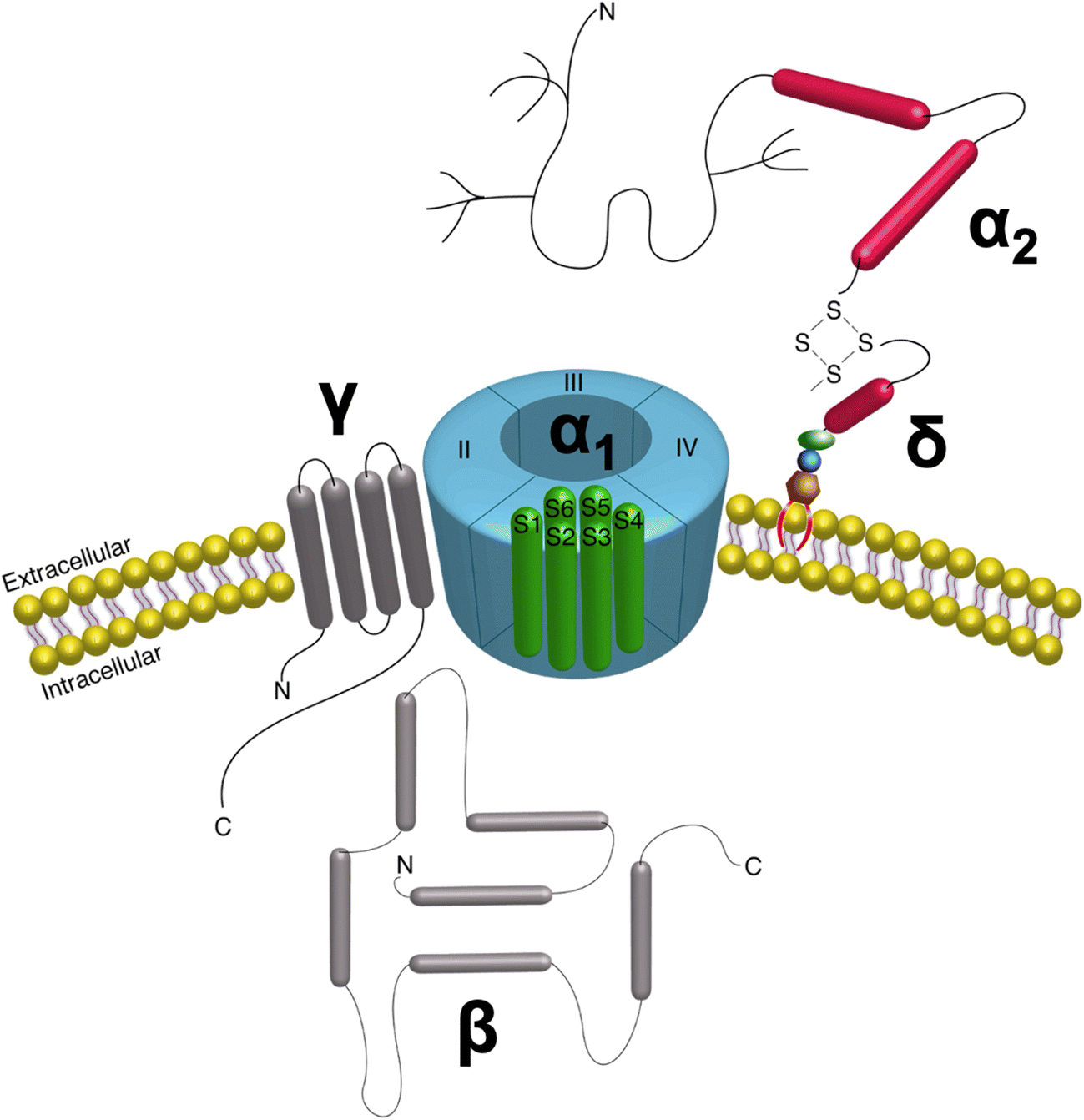
The VSCC complex is composed of the α1 pore-forming subunit with auxiliary β, γ, and α2δ subunits bound to the pore, positioned to alter gating kinetics of the channel. The entirely extracellular α2δ subunit is anchored to the membrane via a GPI-anchor.
Table 1.
Functional effects and tissue distribution of voltage-sensitive calcium channel auxilliary subunits
| Subunit | Functional effects | Tissue Distribution |
|---|---|---|
| α2δ1 | Trafficking α1 to membrane Increases current amplitude Activating/Inactivating calcium gating-kinetetics Site of extracellular ligand interaction Voltage dependence |
Brain, heart, skeletal muscle, bone |
| α2δ2 | Increases current amplitude | lung, testis, brain, heart, pancreas, prostate, skeletal muscle, bone, spinal cord |
| α2δ3 | Amplifying current density | Brain, heart, skeletal muscle |
| α2δ4 | Increases current amplitude | Heart, skeletal muscle, intestine, fetal liver, erythroblasts, adrenal gland, pitutary |
| β1 | Skeletal excitation-contraction coupling Trafficking α1 to membrane Activating/Inactivating calcium gating-kinetetics Voltage dependence Targeting of α11.1 to triads |
Skeletal muscle, brain |
| β2 | Trafficking α1 to membrane Activating/Inactivating calcium gating-kinetetics Voltage dependence Targeting of α11.4 to retina |
Heart, lung, trachea, aorta, brain |
| β3 | Trafficking α1 to membrane Activating/Inactivating calcium gating-kinetetics Voltage dependence |
Smooth muscle, trachea, aorta, lung, brain |
| β4 | Trafficking α1 to membrane Regulating calcium gating-kinetetics Activating/Inactivating calcium gating-kinetetics |
Brain |
| γ1 | Activating/Inactivating calcium gating-kinetetics Inhibiting channel function |
Skeletal muscle |
| γ2 | Activating/Inactivating calcium gating-kinetetics Inhibiting channel function |
Brain |
| γ3 | Activating/Inactivating calcium gating-kinetetics | Brain |
| γ4 | Inactivating calcium gating-kinetetics | Heart, lunc, brain, prostate, spinal cord |
| γ5 | ? | Brain |
| γ6 | Reduces current aplitude | Heart, skeletal muscle, brain |
| γ7 | Reduces current aplitude | Brain, heart, lung, testis |
| γ8 | ? | Brain, testis, spinal cord |
Adapted from Arikkath J & Campbell KP (2003) [3]
The cytosolic β subunit was discovered in purified skeletal muscle dihydropyridine (DHP) receptors [5] and has four isoforms (β1–4) [6–8]. Binding of β to the α1 pore is mediated via the β interacting domain (BID) [9]. This interaction regulates current amplitude [10, 11], voltage dependence [12], activation kinetics [13], and trafficking of α1 to the plasma membrane [14]. Trafficking is mediated by masking an endoplasmic reticulum (ER) retention signal on the α1 subunit by the β subunit‟s BID [15]. The four isoforms of the β subunit interact uniquely with α1, enabling VSCCs to function differently in various tissues [16].
Of the three auxiliary subunits, the function of the γ subunit is least clear. Originally thought to only be present in skeletal muscle, γ has wide tissue distribution. All eight isoforms (γ1–8) of the γ subunit are glycoproteins with four transmembrane domains (~32 kDa) and an intracellular amino terminus [17–21]. The exact functions of the γ subunit remain unclear but appear to vary based upon the isoform and tissue type. γ1 modulates the biophysical properties, but not membrane trafficking, in skeletal muscle [22], while γ1–4 and γ7 alter the activation and/or inactivation kinetics of Ca2+ currents in neural cells [23, 20, 21]. Continued research will elucidate additional function(s) of the γ subunits.
The α2δ subunit has four isoforms (α2δ1–4) with varying tissue distribution and functions (Table 1) [24–27]. The α2 (~150 kDa) and δ (17–25 kDa) subunits are encoded by a single gene (Cacna2d), which is post-translationally cleaved and then relinked together by a disulfide-linkage [28]. The α2 portion is entirely extracellular while δ secures the subunit within the cell membrane through a glycosyl-phosphatidylinositol (GPI) anchor [28, 29]. In addition to heavily glycosylated cysteine residues, α2δ has three functional domains including a Von Willebrand Factor A (VWA) domain [28, 30] and two Cache domains [30–32]. The VWA domain initiates binding of α2 to extracellular proteins through its metal ion-dependent adhesion site (MIDAS) [33], while the Cache domains further support extracellular interactions [30]. Most importantly, the VWA and Cache domains of α2δ stabilize and interact with the α1 subunit, enabling modulation of α1 by α2δ.
The α2δ subunit regulates α1 pore functions including gating kinetics, membrane density, and interactions with extracellular ligands. The mechanism(s) regulating α2δ trafficking of α1 to the membrane is unclear, but interactions with the MIDAS motif appears essential [34, 35]. Binding of extracellular ligands to α2δ subunits can also alter channel activity. In particular, α2δ1 and α2δ2 bind gabapentinoids including gabapentin and pregabalin [36], which bind with greatest affinity to α2δ1 [37, 38]. These drugs decrease channel currents [39, 40] and membrane localization [40], decreasing pain receptor activation and overall neural activity.
VSCC Nomenclature
Though structurally similar, there are ten mammalian VSCC variants, each with distinct properties (Table 2) [41–43]. VSCCs are categorized in several ways including date of discovery, activation threshold, channel conductance/latency, cell-specificity, and subunit isoforms. These nomenclature paradigms have been condensed into three families: L-type VSCCs (CaV1.1–4; α1S, α1C, α1D, α1F), N/P/Q/R-Type VSCCs (CaV2.1–3; α1A, α1B, α1E), and T-Type Channels (CaV3.1–3; α1G, α1H, α1I) (Fig. 2) [44].
Table 2.
Structure, Function, and Pharamacological Properlties of VSCC
| Calcium Channel Family | α1 Subunit | Channel Blocker | Primary Tissues | Primary Function |
|---|---|---|---|---|
| L-Type | Cav1.1 | DHPs, PHAs, BNZs | Skeletal muscle | Excitation-contraction coupling in smooth muscle, regulation of transcription |
| Cav1.2 | DHPs, PHAs, BNZs | Smooth and cardia muscle, Brain, pituitary, adrenal | Excitation-contraction coupling in cardiac and smooth muscle, endocrine secretion, neuronal Ca2+ transients in cell bodies and dendrites, regulation of enzyme activity, regulation of transcription | |
| Cav1.3 | DHPs, PHAs, BNZs | Brain, pancreas, kidney, ovary, adrenal, cochlea | Endorine secretion, cardiac packing, neuronal Ca2+ transients in cell bodies and dendrites, auditory transduction | |
| Cav1.4 | DHPs, PHAs, BNZs | Retina | Visual transduction | |
| N-Type | Cav2.1 | ω-CTx-GVIZ | Brain, nervous system | Neurotransmitter release, Dendritic Ca2+ transients |
| P/Q-Type | Cav2.2 | ω-Agatoxin | Brain, nervous system | Neurotransmitter release, Dendritic Ca2+ transients |
| R-Type | Cav2.3 | SNX-482 | Brain, cochlea, pituitary, retina, cardiac muscle | Neurotransmitter release, Dendritic Ca2+ transients |
| T-Type | Cav3.1 | Ni2+, Cd2+, KTx, mibefradil, NNC55–0395 | Brain, nervous system | Pacemaking and rhymic firing |
| Cav3.2 | Ni2+, Cd2+, KTx, mibefradil, NNC55–0396 | Brain, cardiac muscle, kidney, liver | Pacemaking and rhymic firing | |
| Cav3.3 | Ni2+, Cd2+, KTx. mibefradil, NNC55–0397 | Brain |
Abbreviations: BNZ, benzothiazepines; DHP, Dihydropyridines; KTx, Scorpin toxin Kurtoxin; MibefradilPHA, Phenylalkylamines; SNX-482, synthetic toxin from Tarantula Hysterocrates gigas; ω-CTx-GVIZ, ω-conotoxin GVIA from cone snail Conus geographus
Figure 2. Phylogeny of Voltage-Sensitive Calcium Channel α1 Subunit.
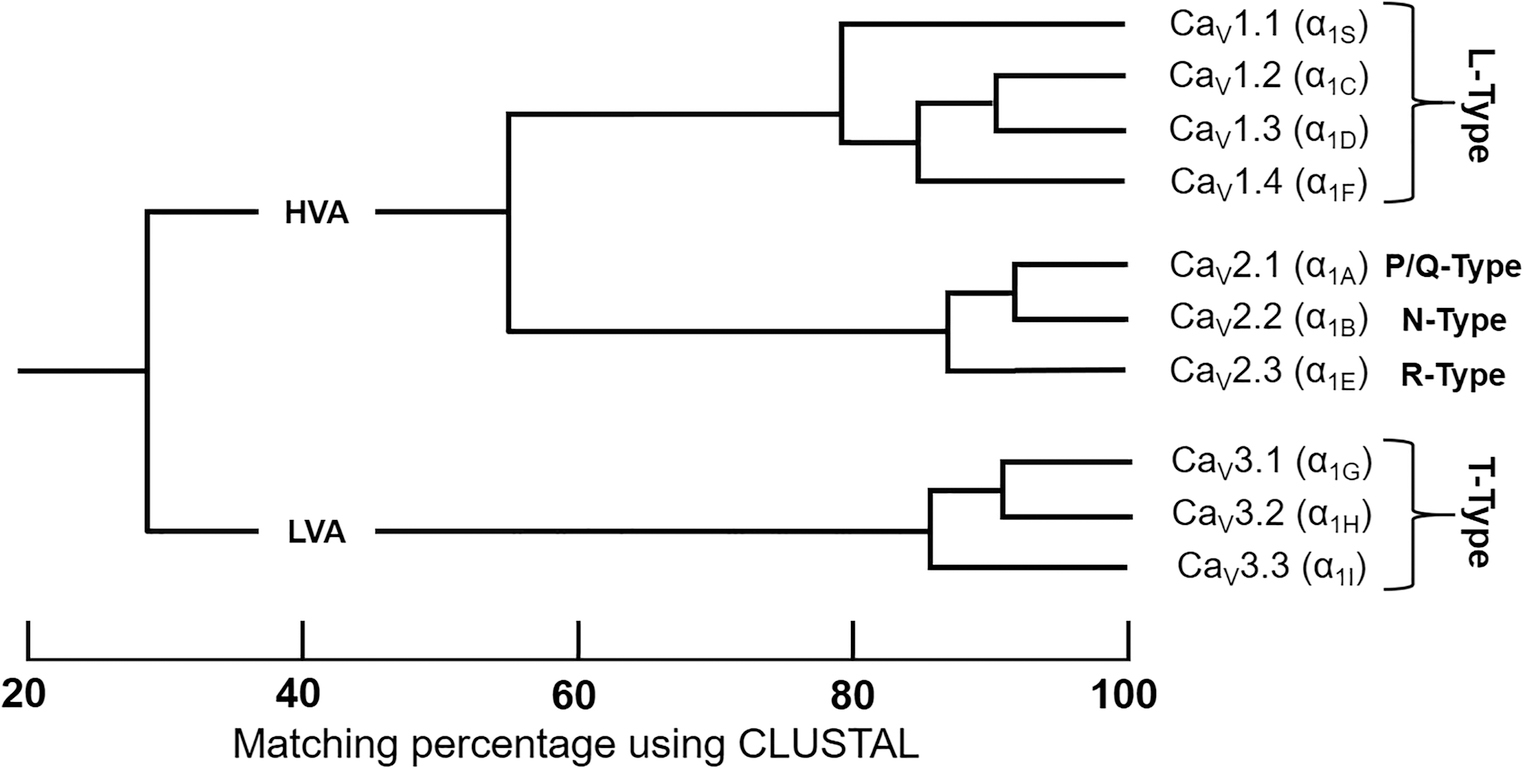
Only the membrane-spanning segments and the pore loops (~350 amino acids) were compared. By only comparing sequencing pairs, three distinct VSCCs families can be identified with intrafamily sequence identities above 80% (CaV1.x, CaV2.x, CaV3.x). High voltage-activated (HVA); low voltage-activated (LVA). Adapted from Ertel EA et al [44] & Dolphin AC [43].
Though L- & T-type channels are found in a variety of tissues, N, P/Q, and R-type are largely expressed in neurons. L-Type (CaV1.x) VSCCs exhibit high voltage activation, large single channel conductance, slow voltage-dependent inactivation, and are the most widely distributed. These properties lend L-Type VSCCs to remain open longer, thus being designated as “L” type due to their „long lasting‟ characteristics [45]. L-Type VSCCs are susceptible to Ca2+ binding reagents including dihydropyridines, phenylalkylamines, and benzothiazepines, often referred to as Ca2+ channel blockers [46].
In contrast, T-Type (Cav3.x) VSCCs are activated at more negative membrane potentials and have a very fast voltage-dependent inactivation causing small single channel conductance [45, 47–49]. Compared to L-type channels, voltage-dependence for activation of T-type VSCCs is shifted 20–30 mV in the hyperpolarized direction, resulting in faster (τ ~ 15–30 ms vs. 2,000 ms) and more complete inactivation with a more negative membrane potential (~ −50 mV vs. 0 mV) and slower deactivation than L-type VSCCs [50, 51]. As such, T-Type VSCCs are well-suited for rhythmic firing action potentials and are distinguished by their frequent generation of transient (T-type) Ca2+ peaks. Unlike other VSCC families, T-type VSCCs are insensitive to Ca2+ binding reagents, spider or snake venoms, but are antagonized by inorganic cations, such as Ni2+ and Cd2+, the scorpion toxin kurtoxin, the organic inhibitor mibefradil [50], and its analog NNC 55–0396 [52].
N, P/Q, and R-type VSCCs (Cav2.x) have an intermediate voltage dependence and rate of inactivation, more negative and faster than L-type but more positive and slower than T-type. Though electrophysiologically similar and located predominately in neurons, the N, P/Q, and R-type channels differ based upon voltage dependence and sensitivity to specialized toxins including spider toxin ω-agatoxin IVA, cone snail peptide ω-conotoxin GVIA, and related peptides [53, 54, 45, 55, 56].
Due to their ability to sense membrane voltage changes, VSCCs were once thought to function only in excitable tissues such as muscle and neurons. However, it has become clear that these channels play vital roles in the skeleton where they influence calcium signaling, bone homeostasis, and mechanical responses.
Skeletal Functions of VSCCs
VSCCs regulate skeletal development, bone turnover, and mechanotransduction. While most studies focused on the function of L-Type VSCCs in osteoblasts, recent work established an important role of T-type VSCCs in osteocytes [57–59] even though the exact mechanism(s) by which T-type channels regulate bone remain unclear. Additionally, little is known regarding the function of VSCCs in osteoclasts, though recent studies suggest both direct and indirect roles of VSCCs in osteoclast activity.
L-type VSCCs regulate osteoblast proliferation and differentiation
The presence of VSCCs in bone was first discovered in primary murine osteoblasts using patch clamp techniques. These studies showed that a rapid, transient, increase in intracellular Ca2+ was required for osteoblast function [60]. These channels were identified as the CaV1.2 (α1c) isoform [61, 62] where both in vivo and in vitro studies confirmed CaV1.2 as the primary channel in osteoblasts [63–71]. Additional work showed that the VSCC complex in osteoblasts lacked a γ subunit [72]. By employing L-type Ca2+ channel blockers, such as nifedipine, verapamil, and diltiazem, numerous studies revealed that L-type VSCCs regulate osteoblast differentiation, proliferation, and mechanosensitivity [73–75, 62, 57, 71].
Calcium channel blockers decreased Ca2+ influx and subsequent downstream signaling pathways in osteoblasts, which helped delineate the function of VSCCs in osteoblasts. Ca2+ influx increased osteocalcin release in osteosarcoma cells, which was further enhanced by the L-type agonist BAY K8644 and suppressed by nifedipine [62, 61]. Nifedipine also impaired osteogenesis in vivo, resulting in decreased bone formation rates, cancellous bone volume, epiphyseal growth plate thickness, and humeral length [76]. While use of Ca2+ channel blockers established the essential function of VSCCs in bone, the cellular mechanisms remained elusive.
When extracellular Ca2+ concentrations increase locally, as during bone resorption, osteoblast-like cells (MC3T3-E1) increase Ca2+ influx and intracellular Ca2+ signaling, triggering a number of responses including increased osteopontin (OPN) expression [77]. OPN is a matrix protein synthesized by osteoblasts during osteoid formation, and is essential for mineralization and remodeling [78]. Inhibition of L-type VSCCs decreased OPN expression and activity [79], suggesting that VSCCs regulate bone formation and remodeling by altering expression of matrix proteins.
In addition to influencing osteoblast matrix production, L-type VSCCs support osteoblast proliferation. Several soluble factors, including epidermal growth factor (EGF) and purinergic signals (i.e. ATP), which activate intracellular signaling by binding P2Y receptors, enhance osteoblast proliferation. Inhibition of L-type VSCCs blocked both EGF [65] and ATP-induced increases in osteoblast proliferation [80]. In the latter case, L-type inhibition impaired c-jun N-terminal kinase-1 (JNK1) phosphorylation [80], demonstrating that VSCC-mediated Ca2+ influx facilitates ATP-stimulated activation of JNK1 to mediate osteoblast proliferation.
While increased osteoblast proliferation provides more matrix-secreting cells, entry into the osteoblast lineage from mesenchymal precursors (MSCs) is essential for osteogenesis. Deletion or inhibition of L-type VSCCs impairs osteogenic allocation of MSCs. The osteoblast markers osteoprotegerin (OPG) [81] and Runx2 [82] are decreased following L-type VSCC inhibition or siRNA knockdown of Cav1.2 (α1C), leading to decreased alkaline phosphatase and alizarin red staining of MSCs [82]. Additional work demonstrated that Cav1.2-mediated changes in MSC osteogenesis was induced through the Wnt/β-catenin pathway, as Cav1.2 knockdown decreased GSK-3β phosphorylation and inactivated β-catenin [83]. Further work showed that expression of Cav1.2 decreased in transgenic mice exhibiting an advanced-age phenotype. These impairments in osteogenic differentiation of MSCs were rescued by overexpression or activation of Cav1.2 [83], suggesting loss of VSCCs expression/activity may explain some of the skeletal changes seen in elderly individuals.
More recent in vivo work further delineated the function of CaV1.2 in bone formation. While global deletion of CaV1.2 impairs cardiac muscle and is embryonic lethal [84], a novel CaV1.2 haploinsufficient model with ~80% reduction in Cacna1C expression (the gene encoding CaV1.2) showed impairments in bone development and formation, affecting both the shape and size of the mandible in zebrafish and mice [85]. A comparable but modest impairment in bone formation also was found in CaV1.3 KO mice, decreasing body weight, femur mass, and midshaft cross-sectional area. Deletion of CaV1.3 simultaneously increased CaV1.2 expression, possibly explaining the attenuated effect of CaV1.3 deletion on bone formation [86]. This compensation further suggests an essential role of CaV1.2 in bone formation.
Additional work using a Timothy syndrome, gain-of-function mutation in Cacna1C and the Cre/Lox system allowed conditional overexpression of CaV1.2 in cells expressing early mesenchymal and osteogenic lineages under the control of Prx1, Col2a1, and Col1a1 promoters. Overexpression of CaV1.2 in osteogenic cells increased cortical and trabecular bone at multiple sites, which by 12 months of age had nearly closed the marrow cavity. This high bone mass resulted from increased osteoblast differentiation as shown by widespread nodule formation, increased expression of osteogenic markers (Alpl, Runx2, Sp7), excess bone formation, and decreased osteoclast differentiation [71, 87]. Taken together, these studies demonstrate that Ca2+-mediated regulation of osteogenic gene expression as well as osteoblast proliferation and differentiation is controlled by L-type VSCCs.
L-type VSCCs Regulate Mechanotransduction
Bone is sensitive to mechanical signals, relying on physical cues to direct bone modeling and remodeling. As one of the first measurable responses of bone cells to mechanical force is a rapid and transient increase in intracellular Ca2+ [88, 89]; tight regulation of Ca2+ influx is a key component of anabolic signaling in bone. Early work used pharmacological inhibitors of L-type VSCCs to decrease Ca2+ influx and the anabolic response to loading. A single dose of verapamil or nifedipine suppressed load-induced bone formation in rodents by 50–75% [68]. Importantly, global deletion of CaV1.3 did not alter osteogenic responses following ulnar loading [86], suggesting CaV1.2 (α1C) may be the primary driver of osteoblast mechanotransduction. However, L-type inhibitors did not completely eliminate anabolic responses to loading [68, 90], suggesting that other VSCC types influence mechanical bone adaptation.
While in vivo studies demonstrated the tissue-level consequences of VSCC inhibition, in vitro work uncovered the cell-specific, mechanical functions of VSCCs. Using osteoblasts isolated from murine long bones, confocal microscopy enabled visualization of stretch-induced Ca2+ influx, which correlated with changes in OPG and OPN expression [91]. These stretch-induced responses were eliminated in Ca2+-free media or following nifedipine treatment, but further enhanced with VSCC agonists. Using optical tweezers to mechanically stimulate human osteoblasts, it was found that nifedipine blocked mechanically-induced L-type Ca2+ influx [92]. This response was not seen in chondrocytes or osteocytic cells, again further suggesting that osteoblasts utilize channels that are distinct from other mesenchymal cells.
As VSCCs are inherently sensitive to changes in membrane electrical potential, the mechanism(s) by which these channels regulate bone responses is debatable. One possibility is that mechanical stimuli directly activate “mechanosensitive channels” such as transient receptor cation channels (i.e., TRPV4) causing a small, local membrane depolarization (Fig. 3A). This local membrane change then is sufficient to open VSCCs generating Ca2+ influx and downstream responses that further increase intracellular Ca2+ concentrations [89]. It also is possible that the VSCCs are directly responsive to mechanical stimuli, not requiring other channels, a notion supported by the ability of auxiliary VSCC subunits to interact with the ECM [93] and the internal cytoskeleton [94].
Figure 3. Role of L-type VSCCs in Osteoblast Mechanotransduction.
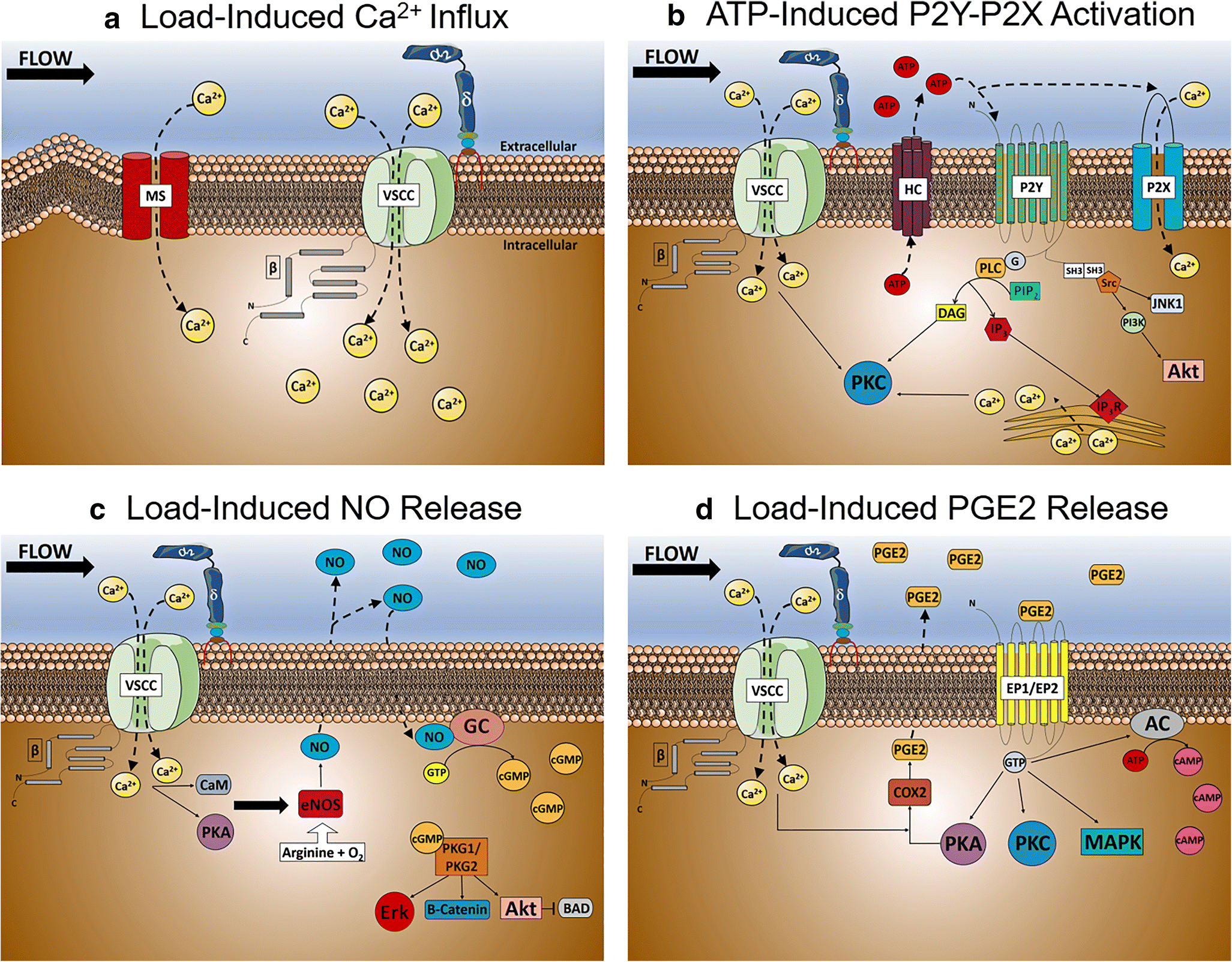
A) Calcium influx following load-induced shear stress in osteoblasts. Following a relatively small, but rapid influx of calcium via mechanosensitive channels, the plasma membrane is depolarized triggering VSCCs to further increasing intracellular calcium influx. B) Load-induced ATP release following calcium influx by osteoblast VSCCs. Increases in intracellular calcium concentrations by VSCCs increases ATP efflux through hemichannels, activating the ATP-gated purinergic P2X and P2Y receptors to increase P2X-dependent calcium influx and release of endoplasmic reticulum calcium stores. C) Load-induced Nitric Oxide (NO) release by osteoblast VSCCs. Endothelial nitric oxide synthase (eNOS) increases NO expression following VSCC calcium influx and activation by calmodulin (CaM) and protein kinase A (PKA), causing NO-induced guanylate cyclase activation and increases in cyclic guanosine monophosphate (cGMP) intracellular concentrations. D) Load-induced Prostaglandin E2 (PGE2) release by VSCCs in osteoblasts. Following VSCC calcium influx and PKA activation, PGE2 is synthesized by cyclooxygenase-2 (COX-2) and prostanoid synthases, causing an increase in PGE2 release and the activation of Prostaglandin E1/E2 Receptors (EP1/EP2). ATP, Adenosine triphosphate; AC; Adenylate cyclase; BAD, Bcl-2-associated death promoter; cAMP, Cyclic adenosine monophosphate; JNK1, c-Jun N-terminal kinase; DAG, diacylglycerol; eNOS, endothelial nitric oxide synthase; Erk, Extracellular-signal-regulated kinase; IP3, Inositol triphosphate; IP3R, Inositol triphosphate receptor; MAPK, Mitogen-activated protein kinase; PI3K, Phosphoinositide 3-kinases; PLC, Phospholipase C; PIP2, Phosphoinositol-4,5-bisphosphate; PKA, Protein kinase A; Akt, Protein kinase B; PKC, Protein kinase C; PKG1/PKG2, Protein kinase G; Src, Src kinase; SH3, Src homology 3 domain
Once Ca2+ enters the cell, several signaling pathways are activated, including release of adenosine triphosphate (ATP), prostaglandin E2 (PGE2), and nitric oxide (NO) (Fig. 3B–D). ATP acts in an autocrine manner, further increasing Ca2+ influx by binding the purinergic P2X receptor [95], and releasing ER Ca2+ stores through cleavage of phosphoinositol-4,5-bisphosphate into diacylglycerol and inositol triphosphate (IP3) via phospholipase C (PLC) [96, 97]. ATP-induced increases in intracellular Ca2+ require L-Type VSCCs, as inhibition of L-type channels in osteoblasts suppress ATP release and Ca2+ influx [68, 96]. VSCC-mediated ATP release activates several pathways including MAPK, protein kinase C (PKC), and Src signaling in osteoblasts to stimulate anabolic responses including ERK1/2 phosphorylation [98, 99]. Mechanically-induced ERK1/2 phosphorylation not only is crucial for osteogenesis, but also osteoblast proliferation, differentiation [100–104], and anabolic responses to loading [105, 106]. In fact, mechanically-induced activation of ERK1/2 is necessary for TGFβ-mediated osteogenic differentiation of MSCs [107] as well as downregulation of RANKL and upregulation of NO synthase (NOS) [108].
Mechanically-induced release of PGE2 [109–112] is VSCC-dependent [96], and serves an important role in osteoblast mechanotransduction [113, 114]. PGE2 is synthesized by cyclooxygenase-2 (COX-2) [115] and osteoblast-specific prostanoid synthases [116] following load-induce activation of protein kinase A (PKA) [117]. PGE2 elicits anabolic responses in osteoblasts in response to mechanical stimuli including activation of cAMP [118], PKA, PKC, and MAPK signaling [119–121], ultimately increasing osteoblast proliferation and differentiation [122, 116, 109, 123].
An important regulator of homeostatic processes, NO is synthesized by NOS following increases in intracellular Ca2+. A labile, water-soluble molecule, NO production is increased following mechanical loading in bone explants [124] and osteoblasts [125]. Importantly, VSCC inhibition restricts mechanically-stimulated NO release [124], dramatically reducing anabolic responses to loading [126]. NO is generated by one of three isoforms of NOS, neuronal NOS (nNOS or NOS1), endothelial NOS (eNOS or NOS3), and inducible NOS (iNOS or NOS2) [127–129]. Of these isoforms, eNOS plays the most significant role in load-induced bone formation [130–132, 125, 133–135]. Once synthesized, NO interacts with guanylate cyclase to generate cGMP and increase the activity of cGMP-dependent protein kinases (PKG1 and PKG2) [136, 131]. This increase in PKG1 and PKG2 activity elicits an anti-apoptotic and pro-proliferative effect in osteoblasts, activating MEK/Erk and Akt signaling pathways [137, 138], inactivating the pro-apoptotic protein BAD [139], stimulating the Wnt/ β-catenin signaling pathway [138], and increasing expression of Runx2 and Fos genes which collectively promotes osteoblast proliferation, differentiation, and survival [138, 140, 141].
Mechanical loading increases Ca2+ influx in osteoblasts predominantly through L-type VSCCs, leading to release of ATP, PGE2, and NO, which activate downstream signals to stimulate osteoblast proliferation, differentiation, and bone formation. While L-type channels predominate responses in osteoblasts, this is not true for all bone cells.
Transition from L-type to T-type VSCCs: Increased evidence for VSCC role in osteocytes
Osteoblasts are characterized by a “plump”, cuboidal morphology attributed largely to an extensive amount of ER and golgi (Fig. 4A) [142]. These secretory organelles enable the high output of ECM necessary for bone formation and remodeling. Influx of Ca2+ through the plasma membrane accounts for only a small amount of the Ca2+ required to initiate intracellular signaling events. The greatest contributor to the change in cytosolic [Ca2+] is Ca2+-induced Ca2+ release from the ER. Thus, it is likely that osteoblasts require the long-lasting activation times of L-type channels to enable full release of Ca2+ from ER stores and high output of ECM.
Fig. 4: Predominate VSCC composition coincides with the morphology and function of osteoblasts and osteocytes.
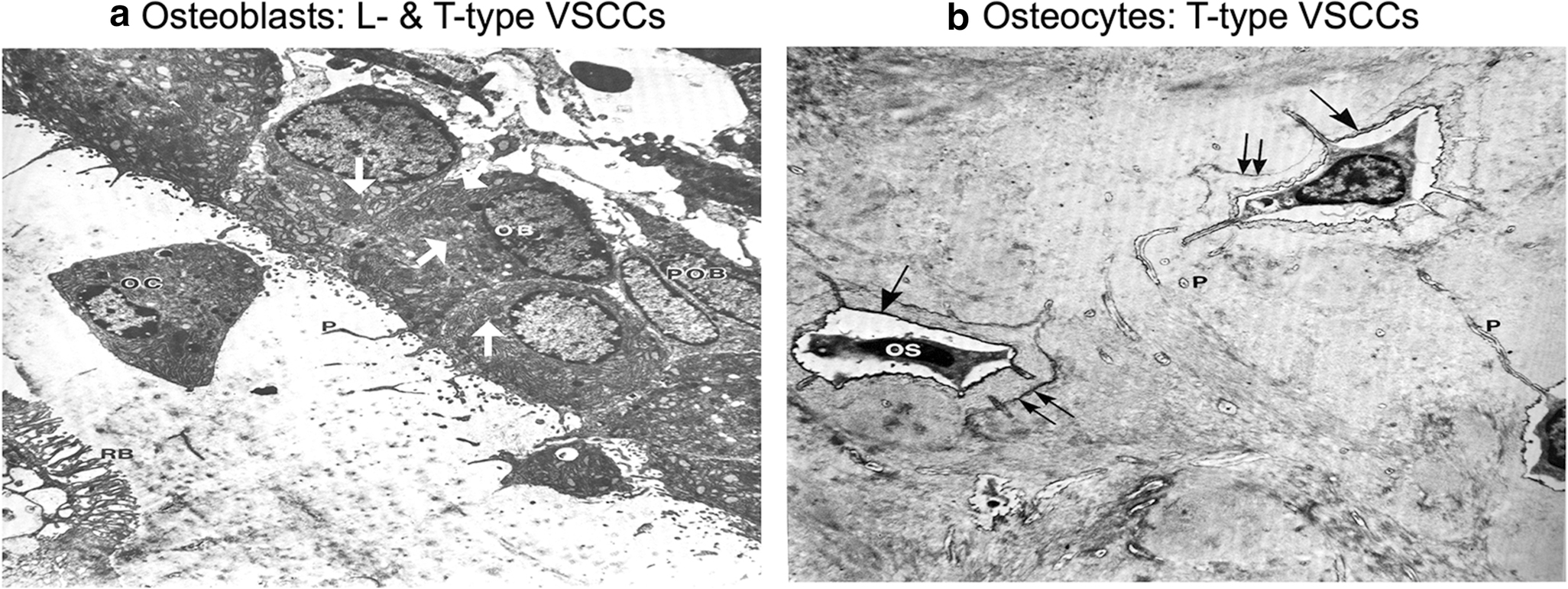
A). The enlarged ER and golgi secretory organelles (white arrows) of osteoblasts (OB) gives its characteristic “plump”, cuboidal morphology and high output of extracellular matrix. The predominate CaV1.2 L-type VSCCs in osteoblasts have a large single channel conductance and slow voltage-dependent inactivation, allowing for „long-lasting‟ calcium influx, the full release of ER calcium stores, and the stimulus necessary for bone formation and remodeling. B) With its flat, stellate shape, mature osteocytes (OS) have much smaller secretory organelles in comparison to osteoblasts. By shifting from expression of L-type to T-type VSCCs during differentiation, the presence of CaV3.2 VSCCs in osteocytes enables faster and more complete channel inactivation than L-type VSCCs, allowing osteocytes to retain its sensitivity to calcium influx without generating cytotoxic Ca2+ levels. Figures adapted from Butler TW [142].
As osteoblasts are buried in matrix and differentiate into osteocytes (Fig. 4B), expression of L-type channels is downregulated leaving T-type VSCCs to perform necessary functions. This transition is observed in immunostaining images where L-type (CaV1.2) and T-type (CaV3.2 and CaV3.1) channels are predominantly expressed at sites of active bone formation in osteoblasts throughout murine skeletal development [143, 144]. However, CaV1.2 expression decreases as T-type CaV3.2 VSCC expression increases upon differentiation into osteocytes [143–145, 59]. Subsequently, regulation of VSCC-dependent Ca2+ influx and subsequent intracellular responses in osteocytes is mediated by T-type and not L-type VSCCs.
In contrast to the extensive ER/golgi of osteoblasts, osteocyte cell bodies are much smaller with very little secretory machinery (Fig. 4B). As osteocytes are terminally differentiated, preventing the death of cells that control both osteoblast and osteoclast activity is crucial for maintaining tight regulation of bone remodeling. Excessive concentrations of cytosolic Ca2+ induces apoptosis [146]. Thus, one possible explanation for the transition from long-lasting (L-type) to transient (T-type) VSCCs in osteocytes is to limit Ca2+ influx to prevent cell death. Recent work applied an overload of mechanical stimuli to osteoblasts resulting in highly elevated intracellular Ca2+ concentrations and subsequent cell death. Importantly, treatment with the L-type inhibitor verapamil restricted apoptosis due to excessive cytosolic Ca2+ [147]. Similar effects were seen in renal and neural cells where blocking L-type VSCCs attenuated apoptosis [148, 149]. These studies support the hypothesis that osteocytes down regulate L-type VSCC expression to prevent excessive cytosolic Ca2+ concentrations as a means of self-preservation and ultimately to maintain bone remodeling capacity.
T-type VSCCs and osteocyte mechanotransduction
Shifting the predominant VSCC from L- to T-type in osteocytes facilitates lower Ca2+ permeability and current potential, enabling osteocytes to retain sensitivity to external stimuli without generating cytotoxic Ca2+ levels. Just as L-type channels enable mechanical responses in osteoblasts, T-type VSCCs serve a similar and distinct function in osteocytes. T-type VSCCs are uniquely sensitive to fluid shear vs. membrane strain. In one study, fluid shear induced repetitive spike-like peaks of Ca2+ in nearly all osteocytes (~97%), while a lesser number of osteoblasts responded to the same mechanical stimulation, only showing a single large Ca2+ peak followed by a few smaller Ca2+ peaks [150]. Furthermore, osteocytes showed a greater number of Ca2+ peaks with lower latency periods, which maintained their magnitude throughout the loading cycle compared to osteoblasts [144, 145].
While these in vitro studies highlight differences in Ca2+ responses of osteoblasts and osteocytes, recent work used intravital imaging to delineate osteocyte responses to loading. Use of a transgenic mouse expressing GCaMP under the direction of Dmp1-Cre enabled real-time visualization of osteocyte Ca2+ influx in the 3rd metatarsal of mice while subjected to mechanical loading [151]. The study examined strains ranging from 250–3,000 micro-strain and frequencies of 0.5–2 Hz. The number of responding osteocytes incrementally increased with increasing magnitudes and frequencies, with a more exponential increase at 2 Hz. However, the Ca2+ signal intensity within responding osteocytes remained consistent and unaltered with increasing magnitudes and frequencies, suggesting a threshold to mechanically-induced Ca2+ influx. This study suggests that the thresholding capacity of osteocytes limits Ca2+ influx, possibly to prevent Ca2+ cytotoxicity and further supporting the function of T-type VSCCs, but also shows that the response to mechanical loading is regulated by the recruitment of osteocytes and not total Ca2+ influx.
In addition to the work demonstrating differences in Ca2+ responses of osteoblasts and osteocytes, several studies confirm that inhibition of T-type VSCCs impairs osteocyte responses to mechanical stimuli. In one study, treatment with NNC55–0396, a T-type VSCC inhibitor, decreased fluid shear-induced Ca2+ influx in osteocytic cells; decreasing the amplitude and number of Ca2+ peaks as well as total number of responding osteocytes. In contrast, treatment with the L-type inhibitor nifedipine decreased anabolic responses to mechanical loading in osteoblasts, but elicited no change in Ca2+ influx [144, 145] nor the release of PGE2 and NO to loading in osteocytes [124]. T-type VSCC inhibition of in situ osteocytes prior to mechanical stimulation also showed similar decreases in total number of Ca2+ responses [152]. Unpublished work from our group demonstrated that deletion of CaV3.2 in mice impairs bone formation and decreases responses to mechanical stimuli, further highlighting the skeletal function of T-type VSCCs.
As mentioned earlier, VSCCs are multi-subunit complexes composed of auxiliary subunits which influence properties of the channel pore. In particular, the large extracellular region of the α2δ1 subunit provides a means for VSCCs to interact with the ECM or ligands. While there are four genes that encode unique α2δ proteins, we demonstrated that α2δ1 is the predominantly expressed isoform in osteocytes, and even though α2δ1 does not bind T-type channels in all tissues, it associates with CaV3.2 in osteocytes [93]. Furthermore, we showed that knockdown of α2δ1 in osteocytic cells dramatically impaired mechanically induced ATP release. The α2δ1 subunit also was necessary for activation of ERK1/2 signaling in response to mechanical signals in osteocytes [93].
Purinergic signaling mediates the response of bone to mechanical stimuli and is initiated by Ca2+ signaling. VSCC-mediated Ca2+ influx results in ATP release, which subsequently activates purinergic receptors releasing IP3 by activation of PLC. This cascade enables release of Ca2+ stores from the ER. Mechanical loading in osteocytes releases ER Ca2+ stores; however, following the initial Ca2+ spike, subsequent intracellular Ca2+ peaks only occur after recovery of ER Ca2+ stores, as nearly 85% of all intracellular Ca2+ peaks were associated with a reciprocal decrease in ER Ca2+ stores in MLO-Y4 cells [145, 152]. As such, direct inhibition of P2R/PLC, or the prevention of ER Ca2+ store repletion decreases mechanically-induced Ca2+ oscillations in MLO-Y4 cells [145] and osteocytes in situ [152, 153]. In contrast, osteoblasts do not require full recovery of ER Ca2+ stores for repetitive mechanical responses, possibly due to their larger ER structure [145]. Therefore, mechanical responses of osteocytes depend on purinergic signaling mediated by T-type VSCCs [145, 152, 144].
Calcium channel functions in osteoclasts
In contrast to mesenchymal osteoblasts and osteocytes, the function of VSCCs in hematopoietic osteoclasts is less clear. While several studies suggest that osteoclasts lack VSCCs [71, 154], others show that these channels are produced in osteoclasts [155]. Additionally, osteoclast differentiation [156] and OPG/RANKL/ANK axis [157] require MAPK/ERK signaling, both of which are influenced by Ca2+ influx. In fact, RANKL-mediate osteoclastogenesis is accompanied by increases in Ca2+ influx [158]. Furthermore, deletion of Ca2+-sensing receptors impairs osteoclast differentiation and activity as well as induces apoptosis [159, 160]. These data support an essential function of Ca2+ signaling in osteoclasts.
Recent in vivo and in vitro data show L-type VSCCs are highly expressed in osteoclasts and influence osteoclastogenesis. Immunostaining and qPCR analyses revealed CaV1.3 expression in primary murine osteoclasts [155], which increased 4-fold upon ovariectomy, suggesting that upregulation of CaV1.3 is associated with increased osteoclast activity. Additionally, over-expression of CaV1.3 in RAW264.7 cells increased osteoclast number and expression of numerous osteoclast-specific genes which were decreased upon Cav1.3 knockdown [155]. While data delineating the function of VSCCs in osteoclasts is limited, the dependence of osteoclasts on oscillating Ca2+ concentrations for proper function [159–161] warrants further investigation to study VSCCs influence on osteoclast-mediate bone resorption.
While evidence for direct functions of VSCCs in osteoclasts remains unclear, several studies suggest that channel activity in other bone cells has downstream effects on osteoclasts. One such example is the increased release of OPG by osteoblasts following activation of VSCCs, which leads to decreased osteoclast activity and survival [91, 96, 81, 71, 87, 162]. Likewise, inhibition or deletion of CaV1.2 increased RANKL-induced osteoclast activity predominantly due to reduced OPG release [71, 87, 81].
Another example of indirect regulation of VSCCs on osteoclasts is the increased release of NO following VSCC activation. The anti-resorptive effects of skeletal loading are modulated at least in part by NO production from osteoblasts and osteocytes [112, 163], which decreases RANKL [164, 106]. Additional work showed that inhibition of NOS or L-type VSCCs prior to mechanical loading abrogated the anti-osteoclastogenic effects of loading [165, 105, 106, 68]. These data indicate that mechanical loading regulates release of NO from osteoblasts/cytes, which then influences osteoclast activity and subsequent bone remodeling.
Taken together, these studies suggest that the primary function of VSCCs within the skeleton are mediated through osteoblasts and osteocytes, which then influence osteoclast activity. However, VSCCs may regulate osteoclast function directly in ways that have yet to be determined.
Conclusions
All cells are designed with various receptors and channels to enable interaction with the external environment. As skeletal formation and remodeling rely on spatial, hormonal, and mechanical signals, tight regulation of the membrane electrochemical gradient provides a mechanism by which cells use Ca2+ influx to trigger various signaling events. VSCCs allow bone cells to rapidly respond to voltage changes at the plasma membrane. Furthermore, bone cells take advantage of the specific properties afforded by different types of VSCCs, allowing matrix producing osteoblasts to respond to stimuli differently than osteocytes. While the coordinated responses of bone cells are regulated in part by VSCCs, many questions remain regarding other classes of molecules or pharmacological agents which influence these channels in musculoskeletal tissues. Understanding these questions provides a rich field for future investigation.
Acknowledgments
Funding: 1R01AR074473–01 to WRT, MCFC, and AGR; R15AR069943–01 to WRT, and 1F32AR074893–01 to CSW
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflicts of Interest: None
References
- 1.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57(4):411–25. doi:57/4/411 [pii] 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 2.Yu FH, Catterall WA. The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE. 2004;2004(253):re15. doi:stke.2532004re15 [pii] 10.1126/stke.2532004re15. [DOI] [PubMed] [Google Scholar]
- 3.Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol. 2003;13(3):298–307. doi:S0959438803000667 [pii]. [DOI] [PubMed] [Google Scholar]
- 4.Garcia K, Nabhani T, Garcia J. The calcium channel alpha2/delta1 subunit is involved in extracellular signalling. J Physiol. 2008;586(3):727–38. doi:jphysiol.2007.147959 [pii] 10.1113/jphysiol.2007.147959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi M, Seagar MJ, Jones JF, Reber BF, Catterall WA. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc Natl Acad Sci U S A. 1987;84(15):5478–82. doi: 10.1073/pnas.84.15.5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castellano A, Wei X, Birnbaumer L, Perez-Reyes E. Cloning and expression of a neuronal calcium channel beta subunit. J Biol Chem. 1993;268(17):12359–66. [PubMed] [Google Scholar]
- 7.Hullin R, Singer-Lahat D, Freichel M, Biel M, Dascal N, Hofmann F et al. Calcium channel beta subunit heterogeneity: functional expression of cloned cDNA from heart, aorta and brain. EMBO J. 1992;11(3):885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perezreyes E, Schneider T. Calcium Channels - Structure, Function, and Classification. Drug Develop Res. 1994;33(3):295–318. doi:DOI 10.1002/ddr.430330311. [DOI] [Google Scholar]
- 9.Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel beta-subunit binds to a conserved motif in the I-II cytoplasmic linker of the alpha 1-subunit. Nature. 1994;368(6466):67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- 10.Ellinor PT, Zhang JF, Randall AD, Zhou M, Schwarz TL, Tsien RW et al. Functional expression of a rapidly inactivating neuronal calcium channel. Nature. 1993;363(6428):455–8. doi: 10.1038/363455a0. [DOI] [PubMed] [Google Scholar]
- 11.Williams ME, Feldman DH, McCue AF, Brenner R, Velicelebi G, Ellis SB et al. Structure and functional expression of alpha 1, alpha 2, and beta subunits of a novel human neuronal calcium channel subtype. Neuron. 1992;8(1):71–84. doi: 10.1016/0896-6273(92)90109-q. [DOI] [PubMed] [Google Scholar]
- 12.Varadi G, Lory P, Schultz D, Varadi M, Schwartz A. Acceleration of activation and inactivation by the beta subunit of the skeletal muscle calcium channel. Nature. 1991;352(6331):159–62. doi: 10.1038/352159a0. [DOI] [PubMed] [Google Scholar]
- 13.De Waard M, Pragnell M, Campbell KP. Ca2+ channel regulation by a conserved beta subunit domain. Neuron. 1994;13(2):495–503. doi:0896-6273(94)90363-8 [pii]. [DOI] [PubMed] [Google Scholar]
- 14.Brice NL, Berrow NS, Campbell V, Page KM, Brickley K, Tedder I et al. Importance of the different beta subunits in the membrane expression of the alpha1A and alpha2 calcium channel subunits: studies using a depolarization-sensitive alpha1A antibody. Eur J Neurosci. 1997;9(4):749–59. doi: 10.1111/j.1460-9568.1997.tb01423.x. [DOI] [PubMed] [Google Scholar]
- 15.Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T et al. The I-II loop of the Ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron. 2000;25(1):177–90. doi:S0896-6273(00)80881-8 [pii]. [DOI] [PubMed] [Google Scholar]
- 16.Dolphin AC. Beta subunits of voltage-gated calcium channels. J Bioenerg Biomembr. 2003;35(6):599–620. doi: 10.1023/b:jobb.0000008026.37790.5a. [DOI] [PubMed] [Google Scholar]
- 17.Chu PJ, Robertson HM, Best PM. Calcium channel gamma subunits provide insights into the evolution of this gene family. Gene. 2001;280(1–2):37–48. doi:S0378-1119(01)00738-7 [pii]. [DOI] [PubMed] [Google Scholar]
- 18.Burgess DL, Gefrides LA, Foreman PJ, Noebels JL. A cluster of three novel Ca2+ channel gamma subunit genes on chromosome 19q13.4: evolution and expression profile of the gamma subunit gene family. Genomics. 2001;71(3):339–50. doi: 10.1006/geno.2000.6440 S0888-7543(00)96440-1 [pii]. [DOI] [PubMed] [Google Scholar]
- 19.Klugbauer N, Dai S, Specht V, Lacinova L, Marais E, Bohn G et al. A family of gamma-like calcium channel subunits. FEBS Lett. 2000;470(2):189–97. doi:S0014-5793(00)01306-5 [pii]. [DOI] [PubMed] [Google Scholar]
- 20.Rousset M, Cens T, Restituito S, Barrere C, Black JL 3rd, McEnery MW et al. Functional roles of gamma2, gamma3 and gamma4, three new Ca2+ channel subunits, in P/Q-type Ca2+ channel expressed in Xenopus oocytes. J Physiol. 2001;532(Pt 3):583–93. doi: 10.1111/j.1469-7793.2001.0583e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moss FJ, Viard P, Davies A, Bertaso F, Page KM, Graham A et al. The novel product of a five-exon stargazin-related gene abolishes Ca(V)2.2 calcium channel expression. EMBO J. 2002;21(7):1514–23. doi: 10.1093/emboj/21.7.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arikkath J, Chen CC, Ahern C, Allamand V, Flanagan JD, Coronado R et al. Gamma 1 subunit interactions within the skeletal muscle L-type voltage-gated calcium channels. J Biol Chem. 2003;278(2):1212–9. doi: 10.1074/jbc.M208689200 M208689200 [pii]. [DOI] [PubMed] [Google Scholar]
- 23.Kang MG, Chen CC, Felix R, Letts VA, Frankel WN, Mori Y et al. Biochemical and biophysical evidence for gamma 2 subunit association with neuronal voltage-activated Ca2+ channels. J Biol Chem. 2001;276(35):32917–24. doi: 10.1074/jbc.M100787200 M100787200 [pii]. [DOI] [PubMed] [Google Scholar]
- 24.Klugbauer N, Lacinova L, Marais E, Hobom M, Hofmann F. Molecular diversity of the calcium channel alpha2delta subunit. J Neurosci. 1999;19(2):684–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barclay J, Balaguero N, Mione M, Ackerman SL, Letts VA, Brodbeck J et al. Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. Journal of Neuroscience. 2001;21(16):6095–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin N, Yagel S, Momplaisir ML, Codd EE, D’Andrea MR. Molecular cloning and characterization of the human voltage-gated calcium channel alpha(2)delta-4 subunit. Molecular Pharmacology. 2002;62(3):485–96. doi:UNSP 1542/1003845 DOI 10.1124/mol.62.3.485. [DOI] [PubMed] [Google Scholar]
- 27.Wycisk KA, Zeitz C, Feil S, Wittmer M, Forster U, Neidhardt J et al. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am J Hum Genet. 2006;79(5):973–7. doi:Doi 10.1086/508944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jay SD, Sharp AH, Kahl SD, Vedvick TS, Harpold MM, Campbell KP. Structural characterization of the dihydropyridine-sensitive calcium channel alpha 2-subunit and the associated delta peptides. J Biol Chem. 1991;266(5):3287–93. [PubMed] [Google Scholar]
- 29.Davies A, Kadurin I, Alvarez-Laviada A, Douglas L, Nieto-Rostro M, Bauer CS et al. The alpha2delta subunits of voltage-gated calcium channels form GPI-anchored proteins, a posttranslational modification essential for function. Proc Natl Acad Sci U S A. 2010;107(4):1654–9. doi: 10.1073/pnas.0908735107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu J, Yan Z, Li Z, Yan C, Lu S, Dong M et al. Structure of the voltage-gated calcium channel Cav1.1 complex. Science. 2015;350(6267):aad2395. doi: 10.1126/science.aad2395. [DOI] [PubMed] [Google Scholar]
- 31.Bannister RA, Beam KG. Ca(V)1.1: The atypical prototypical voltage-gated Ca(2)(+) channel. Biochimica et biophysica acta. 2013;1828(7):1587–97. doi: 10.1016/j.bbamem.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dolphin AC. The alpha2delta subunits of voltage-gated calcium channels. Biochimica et biophysica acta. 2013;1828(7):1541–9. doi: 10.1016/j.bbamem.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 33.Whittaker CA, Hynes RO. Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell. 2002;13(10):3369–87. doi: 10.1091/mbc.e02-05-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canti C, Nieto-Rostro M, Foucault I, Heblich F, Wratten J, Richards MW et al. The metal-ion-dependent adhesion site in the Von Willebrand factor-A domain of alpha2delta subunits is key to trafficking voltage-gated Ca2+ channels. Proc Natl Acad Sci U S A. 2005;102(32):11230–5. doi:0504183102 [pii] 10.1073/pnas.0504183102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tran-Van-Minh A, Dolphin AC. The alpha2delta ligand gabapentin inhibits the Rab11-dependent recycling of the calcium channel subunit alpha2delta-2. J Neurosci. 2010;30(38):12856–67. doi: 10.1523/JNEUROSCI.2700-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem. 1996;271(10):5768–76. doi: 10.1074/jbc.271.10.5768. [DOI] [PubMed] [Google Scholar]
- 37.Davies A, Douglas L, Hendrich J, Wratten J, Tran Van Minh A, Foucault I et al. The calcium channel alpha2delta-2 subunit partitions with CaV2.1 into lipid rafts in cerebellum: implications for localization and function. J Neurosci. 2006;26(34):8748–57. doi: 10.1523/JNEUROSCI.2764-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su TZ et al. Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc Natl Acad Sci U S A. 2006;103(46):17537–42. doi:0409066103 [pii] 10.1073/pnas.0409066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin DJ, McClelland D, Herd MB, Sutton KG, Hall MD, Lee K et al. Gabapentin-mediated inhibition of voltage-activated Ca2+ channel currents in cultured sensory neurones is dependent on culture conditions and channel subunit expression. Neuropharmacology. 2002;42(3):353–66. doi: 10.1016/s0028-3908(01)00181-2. [DOI] [PubMed] [Google Scholar]
- 40.Hendrich J, Van Minh AT, Heblich F, Nieto-Rostro M, Watschinger K, Striessnig J et al. Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc Natl Acad Sci U S A. 2008;105(9):3628–33. doi:0708930105 [pii] 10.1073/pnas.0708930105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hess P Calcium channels in vertebrate cells. Annu Rev Neurosci. 1990;13:337–56. doi: 10.1146/annurev.ne.13.030190.002005. [DOI] [PubMed] [Google Scholar]
- 42.Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3(8):a003947. doi: 10.1101/cshperspect.a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol. 2016;594(19):5369–90. doi: 10.1113/JP272262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E et al. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25(3):533–5. doi:S0896-6273(00)81057-0 [pii]. [DOI] [PubMed] [Google Scholar]
- 45.Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature. 1985;316(6027):440–3. doi: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- 46.Reuter H Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 1983;301(5901):569–74. doi: 10.1038/301569a0. [DOI] [PubMed] [Google Scholar]
- 47.Carbone E, Lux HD. A low voltage-activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature. 1984;310(5977):501–2. doi: 10.1038/310501a0. [DOI] [PubMed] [Google Scholar]
- 48.Swandulla D, Armstrong CM. Fast-deactivating calcium channels in chick sensory neurons. J Gen Physiol. 1988;92(2):197–218. doi: 10.1085/jgp.92.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fedulova SA, Kostyuk PG, Veselovsky NS. Two types of calcium channels in the somatic membrane of new-born rat dorsal root ganglion neurones. J Physiol. 1985;359:431–46. doi: 10.1113/jphysiol.1985.sp015594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lacinova L, Klugbauer N, Hofmann F. Low voltage activated calcium channels: from genes to function. Gen Physiol Biophys. 2000;19(2):121–36. [PubMed] [Google Scholar]
- 51.Perez-Reyes E Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83(1):117–61. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- 52.Huang L, Keyser BM, Tagmose TM, Hansen JB, Taylor JT, Zhuang H et al. NNC 55–0396 [(1S,2S)-2-(2-(N-[(3-benzimidazol-2-yl)propyl]-N-methylamino)ethyl)-6-fluo ro-1,2,3,4-tetrahydro-1-isopropyl-2-naphtyl cyclopropanecarboxylate dihydrochloride]: a new selective inhibitor of T-type calcium channels. J Pharmacol Exp Ther. 2004;309(1):193–9. doi: 10.1124/jpet.103.060814 jpet.103.060814 [pii]. [DOI] [PubMed] [Google Scholar]
- 53.McCleskey EW, Fox AP, Feldman DH, Cruz LJ, Olivera BM, Tsien RW et al. Omega-conotoxin: direct and persistent blockade of specific types of calcium channels in neurons but not muscle. Proc Natl Acad Sci U S A. 1987;84(12):4327–31. doi: 10.1073/pnas.84.12.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple Types of Neuronal Calcium Channels and Their Selective Modulation. Trends in Neurosciences. 1988;11(10):431–8. doi:Doi 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 55.Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15(4):2995–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9(1):85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- 57.Bergh JJ, Shao Y, Puente E, Duncan RL, Farach-Carson MC. Osteoblast Ca(2+) permeability and voltage-sensitive Ca(2+) channel expression is temporally regulated by 1,25-dihydroxyvitamin D(3). Am J Physiol Cell Physiol. 2006;290(3):C822–31. [DOI] [PubMed] [Google Scholar]
- 58.Paic F, Igwe JC, Nori R, Kronenberg MS, Franceschetti T, Harrington P et al. Identification of differentially expressed genes between osteoblasts and osteocytes. Bone. 2009;45(4):682–92. doi: 10.1016/j.bone.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thompson WR, Majid AS, Czymmek KJ, Ruff AL, Garcia J, Duncan RL et al. Association of the alpha(2)delta(1) subunit with Ca(v)3.2 enhances membrane expression and regulates mechanically induced ATP release in MLO-Y4 osteocytes. J Bone Miner Res. 2011;26(9):2125–39. doi: 10.1002/jbmr.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chesnoy-Marchais D, Fritsch J. Voltage-gated sodium and calcium currents in rat osteoblasts. J Physiol. 1988;398:291–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guggino SE, Lajeunesse D, Wagner JA, Snyder SH. Bone remodeling signaled by a dihydropyridine- and phenylalkylamine-sensitive calcium channel. Proc Natl Acad Sci U S A. 1989;86(8):2957–60. doi: 10.1073/pnas.86.8.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grygorczyk C, Grygorczyk R, Ferrier J. Osteoblastic cells have L-type calcium channels. Bone and mineral. 1989;7(2):137–48. [DOI] [PubMed] [Google Scholar]
- 63.Gu Y, Preston MR, el Haj AJ, Hamid J, Zamponi GW, Howl J et al. Osteoblasts derived from load-bearing bones of the rat express both L- and T-like voltage-operated calcium channels and mRNA for alpha 1C, alpha 1D and alpha 1G subunits. Pflugers Arch. 1999;438(4):553–60. [DOI] [PubMed] [Google Scholar]
- 64.el Haj AJ, Walker LM, Preston MR, Publicover SJ. Mechanotransduction pathways in bone: calcium fluxes and the role of voltage-operated calcium channels. Medical & biological engineering & computing. 1999;37(3):403–9. [DOI] [PubMed] [Google Scholar]
- 65.Loza JC, Carpio LC, Bradford PG, Dziak R. Molecular characterization of the alpha1 subunit of the L type voltage calcium channel expressed in rat calvarial osteoblasts. J Bone Miner Res. 1999;14(3):386–95. [DOI] [PubMed] [Google Scholar]
- 66.Barry EL. Expression of mRNAs for the alpha 1 subunit of voltage-gated calcium channels in human osteoblast-like cell lines and in normal human osteoblasts. Calcif Tissue Int. 2000;66(2):145–50. [DOI] [PubMed] [Google Scholar]
- 67.Li B, Chik CL, Taniguchi N, Ho AK, Karpinski E. 24,25(OH)2 vitamin D3 modulates the L-type Ca2+ channel current in UMR 106 cells: involvement of protein kinase A and protein kinase C. Cell calcium. 1996;19(3):193–200. [DOI] [PubMed] [Google Scholar]
- 68.Li J, Duncan RL, Burr DB, Turner CH. L-type calcium channels mediate mechanically induced bone formation in vivo. J Bone Miner Res. 2002;17(10):1795–800. [DOI] [PubMed] [Google Scholar]
- 69.Meszaros JG, Karin NJ, Farach-Carson MC. Voltage-sensitive calcium channels in osteoblasts: mediators of plasma membrane signalling events. Connective tissue research. 1996;35(1–4):107–11. [DOI] [PubMed] [Google Scholar]
- 70.Wang XT, Nagaba S, Nagaba Y, Leung SW, Wang J, Qiu W et al. Cardiac L-type calcium channel alpha 1-subunit is increased by cyclic adenosine monophosphate: messenger RNA and protein expression in intact bone. J Bone Miner Res. 2000;15(7):1275–85. doi: 10.1359/jbmr.2000.15.7.1275. [DOI] [PubMed] [Google Scholar]
- 71.Cao C, Ren Y, Barnett AS, Mirando AJ, Rouse D, Mun SH et al. Increased Ca2+ signaling through CaV1.2 promotes bone formation and prevents estrogen deficiency-induced bone loss. JCI Insight. 2017;2(22). doi: 10.1172/jci.insight.95512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bergh JJ, Shao Y, Akanbi K, Farach-Carson MC. Rodent osteoblastic cells express voltage-sensitive calcium channels lacking a gamma subunit. Calcif Tissue Int. 2003;73(5):502–10. doi: 10.1007/s00223-002-0016-y. [DOI] [PubMed] [Google Scholar]
- 73.Meszaros JG, Karin NJ, Akanbi K, Farach-Carson MC. Down-regulation of L-type Ca2+ channel transcript levels by 1,25-dihyroxyvitamin D3. Osteoblastic cells express L-type alpha1C Ca2+ channel isoforms. J Biol Chem. 1996;271(51):32981–5. [DOI] [PubMed] [Google Scholar]
- 74.Duncan RL, Akanbi KA, Farach-Carson MC. Calcium signals and calcium channels in osteoblastic cells. Semin Nephrol. 1998;18(2):178–90. [PubMed] [Google Scholar]
- 75.Zhang J, Ryder KD, Bethel JA, Ramirez R, Duncan RL. PTH-induced actin depolymerization increases mechanosensitive channel activity to enhance mechanically stimulated Ca2+ signaling in osteoblasts. J Bone Miner Res. 2006;21(11):1729–37. doi: 10.1359/jbmr.060722. [DOI] [PubMed] [Google Scholar]
- 76.Duriez J, Flautre B, Blary MC, Hardouin P. Effects of the calcium channel blocker nifedipine on epiphyseal growth plate and bone turnover: a study in rabbit. Calcif Tissue Int. 1993;52(2):120–4. [DOI] [PubMed] [Google Scholar]
- 77.Jung H, Best M, Akkus O. Microdamage induced calcium efflux from bone matrix activates intracellular calcium signaling in osteoblasts via L-type and T-type voltage-gated calcium channels. Bone. 2015;76:88–96. doi: 10.1016/j.bone.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 78.Neve A, Corrado A, Cantatore FP. Osteocalcin: skeletal and extra-skeletal effects. J Cell Physiol. 2013;228(6):1149–53. doi: 10.1002/jcp.24278. [DOI] [PubMed] [Google Scholar]
- 79.Wu X, Itoh N, Taniguchi T, Nakanishi T, Tanaka K. Requirement of calcium and phosphate ions in expression of sodium-dependent vitamin C transporter 2 and osteopontin in MC3T3-E1 osteoblastic cells. Biochimica et biophysica acta. 2003;1641(1):65–70. [DOI] [PubMed] [Google Scholar]
- 80.Katz S, Boland R, Santillan G. Purinergic (ATP) signaling stimulates JNK1 but not JNK2 MAPK in osteoblast-like cells: contribution of intracellular Ca2+ release, stress activated and L-voltage-dependent calcium influx, PKC and Src kinases. Arch Biochem Biophys. 2008;477(2):244–52. [DOI] [PubMed] [Google Scholar]
- 81.Bergh JJ, Xu Y, Farach-Carson MC. Osteoprotegerin expression and secretion are regulated by calcium influx through the L-type voltage-sensitive calcium channel. Endocrinology. 2004;145(1):426–36. [DOI] [PubMed] [Google Scholar]
- 82.Wen L, Wang Y, Wang H, Kong L, Zhang L, Chen X et al. L-type calcium channels play a crucial role in the proliferation and osteogenic differentiation of bone marrow mesenchymal stem cells. Biochemical and biophysical research communications. 2012;424(3):439–45. doi: 10.1016/j.bbrc.2012.06.128. [DOI] [PubMed] [Google Scholar]
- 83.Fei D, Zhang Y, Wu J, Zhang H, Liu A, He X et al. Cav 1.2 regulates osteogenesis of bone marrow-derived mesenchymal stem cells via canonical Wnt pathway in age-related osteoporosis. Aging Cell. 2019;18(4):e12967. doi: 10.1111/acel.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Seisenberger C, Specht V, Welling A, Platzer J, Pfeifer A, Kuhbandner S et al. Functional embryonic cardiomyocytes after disruption of the L-type alpha1C (Cav1.2) calcium channel gene in the mouse. J Biol Chem. 2000;275(50):39193–9. doi: 10.1074/jbc.M006467200. [DOI] [PubMed] [Google Scholar]
- 85.Ramachandran KV, Hennessey JA, Barnett AS, Yin X, Stadt HA, Foster E et al. Calcium influx through L-type CaV1.2 Ca2+ channels regulates mandibular development. J Clin Invest. 2013;123(4):1638–46. doi: 10.1172/JCI66903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li J, Zhao L, Ferries IK, Jiang L, Desta MZ, Yu X et al. Skeletal phenotype of mice with a null mutation in Cav 1.3 L-type calcium channel. J Musculoskelet Neuronal Interact. 2010;10(2):180–7. [PubMed] [Google Scholar]
- 87.Cao C, Oswald AB, Fabella BA, Ren Y, Rodriguiz R, Trainor G et al. The CaV1.2 L-type calcium channel regulates bone homeostasis in the middle and inner ear. Bone. 2019;125:160–8. doi: 10.1016/j.bone.2019.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ajubi NE, Klein-Nulend J, Alblas MJ, Burger EH, Nijweide PJ. Signal transduction pathways involved in fluid flow-induced PGE(2) production by cultured osteocytes. Am J Physiol-Endocrinol Metab. 1999;276(1):E171–E8. [DOI] [PubMed] [Google Scholar]
- 89.Hung CT, Allen FD, Pollack SR, Brighton CT. Intracellular Ca2+ stores and extracellular Ca2+ are required in the real-time Ca2+ response of bone cells experiencing fluid flow. J Biomech. 1996;29(11):1411–7. doi: 10.1016/0021-9290(96)84536-2. [DOI] [PubMed] [Google Scholar]
- 90.Samnegard E, Cullen DM, Akhter MP, Kimmel DB. No effect of verapamil on the local bone response to in vivo mechanical loading. J Orthop Res. 2001;19(2):328–36. doi: 10.1016/S0736-0266(00)90005-6. [DOI] [PubMed] [Google Scholar]
- 91.Walker LM, Publicover SJ, Preston MR, Said Ahmed MA, El Haj AJ. Calcium-channel activation and matrix protein upregulation in bone cells in response to mechanical strain. J Cell Biochem. 2000;79(4):648–61. [DOI] [PubMed] [Google Scholar]
- 92.Walker LM, Holm A, Cooling L, Maxwell L, Oberg A, Sundqvist T et al. Mechanical manipulation of bone and cartilage cells with ‘optical tweezers’. FEBS Lett. 1999;459(1):39–42. [DOI] [PubMed] [Google Scholar]
- 93.Thompson WR, Majid AS, Czymmek KJ, Ruff AL, García J, Duncan RL et al. Association of the α2δ1 subunit with Cav3.2 enhances membrane expression and regulates mechanically induced ATP release in MLO-Y4 osteocytes. Journal of Bone and Mineral Research. 2011;26(9):2125–39. doi: 10.1002/jbmr.437.• This was the first work demonstrating a function of auxilliary subunits of VSCCs in bone. This manuscript showed that the α2δ1 subunit associates with T-type (CaV3.2) regulating both trafficking of the pore-subunit to the plasma membrane and mechanically-induced ATP release in osteocytes.
- 94.Shao Y, Czymmek KJ, Jones PA, Fomin VP, Akanbi K, Duncan RL et al. Dynamic interactions between L-type voltage-sensitive calcium channel Cav1.2 subunits and ahnak in osteoblastic cells. American Journal of Physiology - Cell Physiology. 2009;296(5):C1067–C78. doi: 10.1152/ajpcell.00427.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li J, Liu D, Ke HZ, Duncan RL, Turner CH. The P2X7 nucleotide receptor mediates skeletal mechanotransduction. J Biol Chem. 2005;280(52):42952–9. doi: 10.1074/jbc.M506415200. [DOI] [PubMed] [Google Scholar]
- 96.Genetos DC, Geist DJ, Liu D, Donahue HJ, Duncan RL. Fluid shear-induced ATP secretion mediates prostaglandin release in MC3T3-E1 osteoblasts. J Bone Miner Res. 2005;20(1):41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Duncan RL, Turner CH. Mechanotransduction and the functional response of bone to mechanical strain. Calcif Tissue Int. 1995;57(5):344–58. doi: 10.1007/bf00302070. [DOI] [PubMed] [Google Scholar]
- 98.Liu D, Genetos DC, Shao Y, Geist DJ, Li J, Ke HZ et al. Activation of extracellular-signal regulated kinase (ERK1/2) by fluid shear is Ca(2+)- and ATP-dependent in MC3T3-E1 osteoblasts. Bone. 2008;42(4):644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Katz S, Boland R, Santillan G. Modulation of ERK 1/2 and p38 MAPK signaling pathways by ATP in osteoblasts: involvement of mechanical stress-activated calcium influx, PKC and Src activation. Int J Biochem Cell Biol. 2006;38(12):2082–91. doi: 10.1016/j.biocel.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 100.Jaiswal RK, Jaiswal N, Bruder SP, Mbalaviele G, Marshak DR, Pittenger MF. Adult human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by mitogen-activated protein kinase. J Biol Chem. 2000;275(13):9645–52. doi: 10.1074/jbc.275.13.9645. [DOI] [PubMed] [Google Scholar]
- 101.Gallea S, Lallemand F, Atfi A, Rawadi G, Ramez V, Spinella-Jaegle S et al. Activation of mitogen-activated protein kinase cascades is involved in regulation of bone morphogenetic protein-2-induced osteoblast differentiation in pluripotent C2C12 cells. Bone. 2001;28(5):491–8. doi: 10.1016/s8756-3282(01)00415-x. [DOI] [PubMed] [Google Scholar]
- 102.Lai CF, Chaudhary L, Fausto A, Halstead LR, Ory DS, Avioli LV et al. Erk is essential for growth, differentiation, integrin expression, and cell function in human osteoblastic cells. J Biol Chem. 2001;276(17):14443–50. doi: 10.1074/jbc.M010021200. [DOI] [PubMed] [Google Scholar]
- 103.Lou J, Tu Y, Li S, Manske PR. Involvement of ERK in BMP-2 induced osteoblastic differentiation of mesenchymal progenitor cell line C3H10T1/2. Biochemical and biophysical research communications. 2000;268(3):757–62. doi: 10.1006/bbrc.2000.2210. [DOI] [PubMed] [Google Scholar]
- 104.Mathov I, Plotkin LI, Sgarlata CL, Leoni J, Bellido T. Extracellular signal-regulated kinases and calcium channels are involved in the proliferative effect of bisphosphonates on osteoblastic cells in vitro. J Bone Miner Res. 2001;16(11):2050–6. doi: 10.1359/jbmr.2001.16.11.2050. [DOI] [PubMed] [Google Scholar]
- 105.Fan X, Rahnert JA, Murphy TC, Nanes MS, Greenfield EM, Rubin J. Response to mechanical strain in an immortalized pre-osteoblast cell is dependent on ERK1/2. J Cell Physiol. 2006;207(2):454–60. doi: 10.1002/jcp.20581. [DOI] [PubMed] [Google Scholar]
- 106.Rubin J, Murphy TC, Fan X, Goldschmidt M, Taylor WR. Activation of extracellular signal-regulated kinase is involved in mechanical strain inhibition of RANKL expression in bone stromal cells. J Bone Miner Res. 2002;17(8):1452–60. doi: 10.1359/jbmr.2002.17.8.1452. [DOI] [PubMed] [Google Scholar]
- 107.Arita NA, Pelaez D, Cheung HS. Activation of the extracellular signal-regulated kinases 1 and 2 (ERK1/2) is needed for the TGFbeta-induced chondrogenic and osteogenic differentiation of mesenchymal stem cells. Biochem Biophys Res Commun. 2011;405(4):564–9. doi: 10.1016/j.bbrc.2011.01.068. [DOI] [PubMed] [Google Scholar]
- 108.Rubin J, Murphy TC, Zhu L, Roy E, Nanes MS, Fan X. Mechanical strain differentially regulates endothelial nitric-oxide synthase and receptor activator of nuclear kappa B ligand expression via ERK1/2 MAPK. J Biol Chem. 2003;278(36):34018–25. doi: 10.1074/jbc.M302822200. [DOI] [PubMed] [Google Scholar]
- 109.Thorsen K, Kristoffersson AO, Lerner UH, Lorentzon RP. In situ microdialysis in bone tissue. Stimulation of prostaglandin E2 release by weight-bearing mechanical loading. J Clin Invest. 1996;98(11):2446–9. doi: 10.1172/JCI119061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Batra NN, Li YJ, Yellowley CE, You L, Malone AM, Kim CH et al. Effects of short-term recovery periods on fluid-induced signaling in osteoblastic cells. J Biomech. 2005;38(9):1909–17. doi: 10.1016/j.jbiomech.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 111.Donahue TL, Haut TR, Yellowley CE, Donahue HJ, Jacobs CR. Mechanosensitivity of bone cells to oscillating fluid flow induced shear stress may be modulated by chemotransport. J Biomech. 2003;36(9):1363–71. doi: 10.1016/s0021-9290(03)00118-0. [DOI] [PubMed] [Google Scholar]
- 112.Bakker AD, Soejima K, Klein-Nulend J, Burger EH. The production of nitric oxide and prostaglandin E(2) by primary bone cells is shear stress dependent. J Biomech. 2001;34(5):671–7. doi: 10.1016/s0021-9290(00)00231-1. [DOI] [PubMed] [Google Scholar]
- 113.Jee WS, Ma YF. The in vivo anabolic actions of prostaglandins in bone. Bone. 1997;21(4):297–304. doi: 10.1016/s8756-3282(97)00147-6. [DOI] [PubMed] [Google Scholar]
- 114.Yao W, Jee WS, Zhou H, Lu J, Cui L, Setterberg R et al. Anabolic effect of prostaglandin E2 on cortical bone of aged male rats comes mainly from modeling-dependent bone gain. Bone. 1999;25(6):697–702. doi: 10.1016/s8756-3282(99)00220-3. [DOI] [PubMed] [Google Scholar]
- 115.Myers LK, Bhattacharya SD, Herring PA, Xing Z, Goorha S, Smith RA et al. The isozyme-specific effects of cyclooxygenase-deficiency on bone in mice. Bone. 2006;39(5):1048–52. doi: 10.1016/j.bone.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 116.Raisz LG. Prostaglandins and bone: physiology and pathophysiology. Osteoarthritis Cartilage. 1999;7(4):419–21. doi: 10.1053/joca.1998.0230. [DOI] [PubMed] [Google Scholar]
- 117.Wadhwa S, Choudhary S, Voznesensky M, Epstein M, Raisz L, Pilbeam C. Fluid flow induces COX-2 expression in MC3T3-E1 osteoblasts via a PKA signaling pathway. Biochemical and biophysical research communications. 2002;297(1):46–51. doi: 10.1016/s0006-291x(02)02124-1. [DOI] [PubMed] [Google Scholar]
- 118.Raisz LG. Physiologic and pathologic roles of prostaglandins and other eicosanoids in bone metabolism. J Nutr. 1995;125(7 Suppl):2024S–7S. doi: 10.1093/jn/125.suppl_7.2024S. [DOI] [PubMed] [Google Scholar]
- 119.Markovic T, Jakopin Z, Dolenc MS, Mlinaric-Rascan I. Structural features of subtype-selective EP receptor modulators. Drug Discov Today. 2017;22(1):57–71. doi: 10.1016/j.drudis.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 120.Minamizaki T, Yoshiko Y, Kozai K, Aubin JE, Maeda N. EP2 and EP4 receptors differentially mediate MAPK pathways underlying anabolic actions of prostaglandin E2 on bone formation in rat calvaria cell cultures. Bone. 2009;44(6):1177–85. doi: 10.1016/j.bone.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 121.Blackwell KA, Raisz LG, Pilbeam CC. Prostaglandins in bone: bad cop, good cop? Trends Endocrinol Metab. 2010;21(5):294–301. doi: 10.1016/j.tem.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kawaguchi H, Pilbeam CC, Harrison JR, Raisz LG. The role of prostaglandins in the regulation of bone metabolism. Clin Orthop Relat Res. 1995(313):36–46. [PubMed] [Google Scholar]
- 123.Weinreb M, Rutledge SJ, Rodan GA. Systemic administration of an anabolic dose of prostaglandin E2 induces early-response genes in rat bones. Bone. 1997;20(4):347–53. doi: 10.1016/s8756-3282(97)00011-2. [DOI] [PubMed] [Google Scholar]
- 124.Rawlinson SC, Pitsillides AA, Lanyon LE. Involvement of different ion channels in osteoblasts’ and osteocytes’ early responses to mechanical strain. Bone. 1996;19(6):609–14. [DOI] [PubMed] [Google Scholar]
- 125.Johnson DL, McAllister TN, Frangos JA. Fluid flow stimulates rapid and continuous release of nitric oxide in osteoblasts. The American journal of physiology. 1996;271(1 Pt 1):E205–8. doi: 10.1152/ajpendo.1996.271.1.E205. [DOI] [PubMed] [Google Scholar]
- 126.Turner CH, Takano Y, Owan I, Murrell GAC. Nitric oxide inhibitor L-NAME suppresses mechanically induced bone formation in rats. Am J Physiol-Endoc M. 1996;270(4):E634–E9. [DOI] [PubMed] [Google Scholar]
- 127.Helfrich MH, Evans DE, Grabowski PS, Pollock JS, Ohshima H, Ralston SH. Expression of nitric oxide synthase isoforms in bone and bone cell cultures. J Bone Miner Res. 1997;12(7):1108–15. doi: 10.1359/jbmr.1997.12.7.1108. [DOI] [PubMed] [Google Scholar]
- 128.Wimalawansa SJ. Nitric oxide and bone. Ann N Y Acad Sci. 2010;1192:391–403. doi: 10.1111/j.1749-6632.2009.05230.x. [DOI] [PubMed] [Google Scholar]
- 129.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329(27):2002–12. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 130.Klein-Nulend J, van Oers RF, Bakker AD, Bacabac RG. Nitric oxide signaling in mechanical adaptation of bone. Osteoporos Int. 2014;25(5):1427–37. doi: 10.1007/s00198-013-2590-4. [DOI] [PubMed] [Google Scholar]
- 131.Kalyanaraman H, Schall N, Pilz RB. Nitric oxide and cyclic GMP functions in bone. Nitric Oxide. 2018;76:62–70. doi: 10.1016/j.niox.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fox SW, Chambers TJ, Chow JW. Nitric oxide is an early mediator of the increase in bone formation by mechanical stimulation. The American journal of physiology. 1996;270(6 Pt 1):E955–60. doi: 10.1152/ajpendo.1996.270.6.E955. [DOI] [PubMed] [Google Scholar]
- 133.Aguirre J, Buttery L, O’Shaughnessy M, Afzal F, Fernandez de Marticorena I, Hukkanen M et al. Endothelial nitric oxide synthase gene-deficient mice demonstrate marked retardation in postnatal bone formation, reduced bone volume, and defects in osteoblast maturation and activity. Am J Pathol. 2001;158(1):247–57. doi: 10.1016/S0002-9440(10)63963-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bergula AP, Haidekker MA, Huang W, Stevens HY, Frangos JA. Venous ligation-mediated bone adaptation is NOS 3 dependent. Bone. 2004;34(3):562–9. doi: 10.1016/j.bone.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 135.Armour KE, Armour KJ, Gallagher ME, Godecke A, Helfrich MH, Reid DM et al. Defective bone formation and anabolic response to exogenous estrogen in mice with targeted disruption of endothelial nitric oxide synthase. Endocrinology. 2001;142(2):760–6. doi: 10.1210/endo.142.2.7977. [DOI] [PubMed] [Google Scholar]
- 136.Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62(3):525–63. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rangaswami H, Schwappacher R, Marathe N, Zhuang S, Casteel DE, Haas B et al. Cyclic GMP and protein kinase G control a Src-containing mechanosome in osteoblasts. Sci Signal. 2010;3(153):ra91. doi: 10.1126/scisignal.2001423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rangaswami H, Schwappacher R, Tran T, Chan GC, Zhuang S, Boss GR et al. Protein kinase G and focal adhesion kinase converge on Src/Akt/beta-catenin signaling module in osteoblast mechanotransduction. J Biol Chem. 2012;287(25):21509–19. doi: 10.1074/jbc.M112.347245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Marathe N, Rangaswami H, Zhuang S, Boss GR, Pilz RB. Pro-survival effects of 17beta-estradiol on osteocytes are mediated by nitric oxide/cGMP via differential actions of cGMP-dependent protein kinases I and II. J Biol Chem. 2012;287(2):978–88. doi: 10.1074/jbc.M111.294959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lin IC, Smartt JM Jr., Nah HD, Ischiropoulos H, Kirschner RE. Nitric oxide stimulates proliferation and differentiation of fetal calvarial osteoblasts and dural cells. Plast Reconstr Surg. 2008;121(5):1554–66; discussion 67–9. doi: 10.1097/PRS.0b013e31816c3bd7. [DOI] [PubMed] [Google Scholar]
- 141.Afzal F, Polak J, Buttery L. Endothelial nitric oxide synthase in the control of osteoblastic mineralizing activity and bone integrity. J Pathol. 2004;202(4):503–10. doi: 10.1002/path.1536. [DOI] [PubMed] [Google Scholar]
- 142.Butler TW. The Chemistry and Biology of Mineralized Tissues. Hormone Responsiveness of Bone Cell Populations. Birmingham, Alabama: Ebsco Media, Inc. ; 1984. [Google Scholar]
- 143.Shao Y, Alicknavitch M, Farach-Carson MC. Expression of voltage sensitive calcium channel (VSCC) L-type Cav1.2 (alpha1C) and T-type Cav3.2 (alpha1H) subunits during mouse bone development. Dev Dyn. 2005;234(1):54–62. doi: 10.1002/dvdy.20517. [DOI] [PubMed] [Google Scholar]
- 144.Lu XL, Huo B, Chiang V, Guo XE. Osteocytic network is more responsive in calcium signaling than osteoblastic network under fluid flow. Journal of Bone and Mineral Research. 2012;27(3):563–74. doi: 10.1002/jbmr.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Brown GN, Leong PL, Guo XE. T-Type voltage-sensitive calcium channels mediate mechanically-induced intracellular calcium oscillations in osteocytes by regulating endoplasmic reticulum calcium dynamics. Bone. 2016;88:56–63. doi: 10.1016/j.bone.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Verkhratsky A. Calcium and cell death. Subcell Biochem. 2007;45:465–80. [DOI] [PubMed] [Google Scholar]
- 147.Liu L, Li H, Cui Y, Li R, Meng F, Ye Z et al. Calcium Channel Opening Rather than the Release of ATP Causes the Apoptosis of Osteoblasts Induced by Overloaded Mechanical Stimulation. Cell Physiol Biochem. 2017;42(2):441–54. doi: 10.1159/000477592. [DOI] [PubMed] [Google Scholar]
- 148.Brewer LD, Thibault V, Chen KC, Langub MC, Landfield PW, Porter NM. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci. 2001;21(1):98–108. doi:21/1/98 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tanaka T, Nangaku M, Miyata T, Inagi R, Ohse T, Ingelfinger JR et al. Blockade of calcium influx through L-type calcium channels attenuates mitochondrial injury and apoptosis in hypoxic renal tubular cells. J Am Soc Nephrol. 2004;15(9):2320–33. doi: 10.1097/01.ASN.0000138287.46849.82. [DOI] [PubMed] [Google Scholar]
- 150.Lu XL, Huo B, Chiang V, Guo XE. Osteocytic network is more responsive in calcium signaling than osteoblastic network under fluid flow. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2012;27(3):563–74. doi: 10.1002/jbmr.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lewis KJ, Frikha-Benayed D, Louie J, Stephen S, Spray DC, Thi MM et al. Osteocyte calcium signals encode strain magnitude and loading frequency in vivo. Proc Natl Acad Sci U S A. 2017;114(44):11775–80. doi: 10.1073/pnas.1707863114.• Lewis and colleagues used intravital imaging to delineate the responses of Ca2+ influx in osteocytes in response to in vivo mechanical loading. Modulation of the frequency and magnitude of loading in real-time provided critical insight into the function of Ca2+ signaling in bone.
- 152.Jing D, Baik AD, Lu XL, Zhou B, Lai X, Wang L et al. In situ intracellular calcium oscillations in osteocytes in intact mouse long bones under dynamic mechanical loading. FASEB J. 2014;28(4):1582–92. doi: 10.1096/fj.13-237578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ishihara Y, Sugawara Y, Kamioka H, Kawanabe N, Kurosaka H, Naruse K et al. In situ imaging of the autonomous intracellular Ca(2+) oscillations of osteoblasts and osteocytes in bone. Bone. 2012;50(4):842–52. doi: 10.1016/j.bone.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 154.Kajiya H, Okamoto F, Fukushima H, Takada K, Okabe K. Mechanism and role of high-potassium-induced reduction of intracellular Ca2+ concentration in rat osteoclasts. Am J Physiol Cell Physiol. 2003;285(2):C457–66. [DOI] [PubMed] [Google Scholar]
- 155.Fan P, Hu N, Feng X, Sun Y, Pu D, Lv X et al. Cav1.3 is upregulated in osteoporosis rat model and promotes osteoclast differentiation from preosteoclast cell line RAW264.7. J Cell Physiol. 2019;234(8):12821–7. doi: 10.1002/jcp.27937. [DOI] [PubMed] [Google Scholar]
- 156.Suda T, Tanaka S, Takahashi N. Macrophage colon-stimulating factor (M-CSF) is essential for differentiation rather than proliferation of osteoclast progenitors. Osteoporos Int. 1993;3 Suppl 1:111–3. doi: 10.1007/bf01621881. [DOI] [PubMed] [Google Scholar]
- 157.Takayanagi H, Kim S, Taniguchi T. Signaling crosstalk between RANKL and interferons in osteoclast differentiation. Arthritis Res. 2002;4 Suppl 3:S227–32. doi: 10.1186/ar581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3(6):889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 159.Mentaverri R, Yano S, Chattopadhyay N, Petit L, Kifor O, Kamel S et al. The calcium sensing receptor is directly involved in both osteoclast differentiation and apoptosis. FASEB J. 2006;20(14):2562–4. doi: 10.1096/fj.06-6304fje. [DOI] [PubMed] [Google Scholar]
- 160.Boudot C, Saidak Z, Boulanouar AK, Petit L, Gouilleux F, Massy Z et al. Implication of the calcium sensing receptor and the Phosphoinositide 3-kinase/Akt pathway in the extracellular calcium-mediated migration of RAW 264.7 osteoclast precursor cells. Bone. 2010;46(5):1416–23. doi: 10.1016/j.bone.2010.01.383. [DOI] [PubMed] [Google Scholar]
- 161.Kaji H, Sugimoto T, Kanatani M, Chihara K. High extracellular calcium stimulates osteoclast-like cell formation and bone-resorbing activity in the presence of osteoblastic cells. J Bone Miner Res. 1996;11(7):912–20. doi: 10.1002/jbmr.5650110707. [DOI] [PubMed] [Google Scholar]
- 162.Ng PY, Brigitte Patricia Ribet A, Pavlos NJ. Membrane trafficking in osteoclasts and implications for osteoporosis. Biochem Soc Trans. 2019;47(2):639–50. doi: 10.1042/BST20180445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bacabac RG, Smit TH, Mullender MG, Dijcks SJ, Van Loon JJ, Klein-Nulend J. Nitric oxide production by bone cells is fluid shear stress rate dependent. Biochemical and biophysical research communications. 2004;315(4):823–9. doi: 10.1016/j.bbrc.2004.01.138. [DOI] [PubMed] [Google Scholar]
- 164.Fan X, Roy E, Zhu L, Murphy TC, Ackert-Bicknell C, Hart CM et al. Nitric oxide regulates receptor activator of nuclear factor-kappaB ligand and osteoprotegerin expression in bone marrow stromal cells. Endocrinology. 2004;145(2):751–9. doi: 10.1210/en.2003-0726. [DOI] [PubMed] [Google Scholar]
- 165.Turner CH, Owan I, Jacob DS, McClintock R, Peacock M. Effects of nitric oxide synthase inhibitors on bone formation in rats. Bone. 1997;21(6):487–90. doi: 10.1016/s8756-3282(97)00202-0.• This work demonstrated that osteoporosis accompanies increased expression of L-type VSCCs. Knockdown of CaV1.3 inhibits osteoclast differentiation and activity.