


Abstract
The current understanding of how overall principles of translational control govern the embryo-to-adult transition in mammals is still far from comprehensive. Herein we profiled the translatomes and transcriptomes of six tissues from the mice at embryonic and adult stages and presented the first report of tissue- and stage-specific translational landscape in mice. We quantified the extent of gene expression divergence among different expression layers, tissues and stages, detected significant changes in gene composition and function underlying these divergences and revealed the changing architecture of translational regulation. We further showed that dynamic translational regulation can be largely achieved via modulation of translational efficiency. Translational efficiency could be altered by alternative splicing (AS), upstream and downstream open reading frames (uORFs and dORFs). We revealed AS-mediated translational repression that was exerted in an event type-dependent manner. uORFs and dORFs exhibited mutually exclusive usage and the opposing effects of translational regulation. Furthermore, we discovered many novel microproteins encoded by long noncoding RNAs and demonstrated their regulatory potential and functional relevance. Our data and analyses will facilitate a better understanding of the complexity of translation and translational regulation across tissue and stage spectra and provide an important resource to the translatome research community.
INTRODUCTION
Mammalian tissues in a species show extreme functional diversity despite having nearly identical genome sequences. Their unique physiological functions are achieved through the precise orchestration of spatiotemporal changes in gene expression. The quantification of gene expression across diverse tissues and developmental stages is vital for understanding the molecular and mechanistic principles underlying morphogenesis. Transcriptomic studies have characterized the gene expression profile in a variety of mammalian tissues during development and have thus revealed the complexity and dynamics of the transcriptome (1,2). Proteomic studies have resolved the molecular details of proteome variations in different mammalian tissues and thus extended our understanding of the spatiotemporal programs of protein expression (3,4).
Transcription and translation are the two major steps in gene expression. During translation, ribosomes perform protein synthesis to ensure that the genetic information contained in mRNA is successfully translated into the proteins (5). Although transcriptomic and proteomic analyses have provided great biological insights into tissue specificity and physiological relevance of tissues in development, gene translation profiles during the embryo-to-adult transition have not been systematically investigated. Exhilaratingly, ribosome profiling (Ribo-seq) enables genome-wide quantitative measurements of gene translation at nucleotide resolution (6), thereby facilitating decoding principles of gene translation. By pinpointing ribosomes during translation, this technique allows a detailed analysis of the ribosome density on individual RNAs, the characterization of canonical translation events and the identification of cryptic noncanonical open reading frames (ORFs), such as upstream ORFs (uORFs) in 5′UTRs, downstream ORFs (dORFs) in 3′UTRs and small ORFs (smORFs) in lncRNAs (7–12) Thus, a multi-tissue and multi-stage survey of the translational landscape will provide insight into key translational contents and regulatory responses underlying the generation and maintenance of tissue and stage specificity.
In this study, we performed a genome-wide translatome survey of six mouse tissues at embryonic and adult stages, by combining ribosome profiling with RNA sequencing, to comprehensively investigate tissue- and stage-specific gene translation and translational regulation. We first quantified the extent of gene expression divergence among different expression layers, tissues and stages. We then characterized tissue- and stage-specific patterns of gene expression to understand how changes in the gene composition and function across tissues and stages relate to the regulatory architecture underlying expression divergence. Furthermore, we dissected the contributions of transcriptional and translational controls to tissue and stage differences and illuminated dynamic changes in the translational efficiency that was optimized for the translation of tissue‐ and stage-specific genes by AS-, uORF- and dORF-mediated translational regulatory mechanisms. Additionally, we detected pervasive translation of lncRNA to demonstrate their multi-specificity character, regulatory potentials and functional relevance. Our analyses presented a broad overview of tissue- and stage-specific translational landscape and provided novel insights into the general principles of dynamic gene regulatory programs in mice.
MATERIALS AND METHODS
Tissue collection
Wild-type C57BL/6 mice were purchased from the Guangdong Medical Experimental Animal Center (Guangdong, China; License No: SCXK (YUE) 2018–0002). Brain, heart, kidney, liver, lung and retinal tissues were harvested separately from embryonic (E15.5) and adult (P42) C57BL/6 mice and immediately snap-frozen in liquid nitrogen. All experimental procedures were approved by the Animal Ethics Committee of the Zhongshan Ophthalmic Center, Sun Yat-sen University (Guangzhou, China; License No: SYXK (YUE) 2018–0189), in accordance with institutional animal welfare guidelines and the Animal Protection Law of China.
Library preparation and sequencing
Frozen tissue samples were lysed using 1 ml of mammalian lysis buffer (200 μl of 5× Mammalian Polysome Buffer, 100 μl of 10% Triton X-100, 10 μl of DTT (100 mM), 10 μl of DNase I (1 U/μl), 2 μl of cycloheximide (50 mg/ml), 10 μl of 10% NP-40 and 668 μl of nuclease-free water). After incubation for 20 min on ice, the lysates were cleared by centrifugation at 10 000 × g and 4°C for 3 min. For each tissue and replicate sample, the lysate was divided into 300- and 100-μl aliquots. For the 300-μl aliquots of clarified lysates, 5 units of ARTseq Nuclease were added to each A260 lysate, and the mixtures were incubated for 45 min at room temperature. Nuclease digestion was stopped by the addition of 15 μl of SUPERase·In RNase Inhibitor (Ambion). Subsequently, the lysates were applied to Sephacryl S-400 HR spin columns (GE Healthcare Life Sciences), and ribosome-protected fragments were purified using the Zymo RNA Clean & Concentrator-25 kit (Zymo Research). Ribosomal RNA was depleted using the Ribo-Zero magnetic kit (Epicentre). Sequencing libraries of ribosome-protected fragments (RPFs) were generated using the ARTseq™ Ribosome Profiling Kit (Epicentre, RPHMR12126), according to the manufacturer’s instructions. From the 100-μl aliquots of clarified lysates, poly(A)+ RNAs were extracted and purified, and sequencing libraries of poly(A)+ RNAs were then generated using the VAHTSTM mRNA-seq v2 Library Prep Kit from Illumina (Vazyme Biotech, NR601-01) according to the manufacturer’s instructions. The resulting 48 barcoded libraries were pooled and sequenced using an Illumina HiSeq 2500 instrument in single-end mode.
Sequencing data preprocessing
The raw sequence reads were demultiplexed using CASAVA (v1.8.2), and the 3′-end adapter was clipped using Cutadapt (v1.8.1) (with the parameters ‘-a AGATCGGAAGAGCACACGTCTGAACTCCAGTCA -match-read-wildcards -m 6′). Low-quality sequences were trimmed using Sickle (v1.33) (with the parameters ‘-q 20′). The trimmed reads were filtered by length based on the ranges [25, 34] for ribosome-associated footprints and [20, 50] for mRNA. The retained reads that mapped to reference mouse rRNAs or tRNAs were then removed, and the remaining reads were aligned to the mouse reference genome (downloaded from GENCODE, Release M18: GRCm38.p6) using Tophat2 (v2.0.14) (13) with the following command: ‘tophat2 -g 20 -N 2 –transcriptome-index [index_file] -G [gtf_file] [fastq_file] -o [output_directory]’. Only those uniquely mapped reads were extracted for gene expression determination. The number of reads per gene was counted using the Subread R package-featureCounts (v1.6.2) (14). To avoid differences in library composition across samples, the raw counts for all Ribo-seq and RNA-seq samples were combined together and normalized against the reference to yield a pool-based size factor using the DESeq2 R package (15), and the normalized counts were further converted to transcripts per kilobase million (TPM) values.
Triplet periodicity analysis
Three-nucleotide (3-nt) periodicity is a well-known intrinsic property of genuine translation. Triplet periodicity and metagene analysis were performed to evaluate the quality of Ribo-seq experiments. Briefly, footprint profiles within coding sequences (CDSs) of canonical protein-coding genes were produced by assigning ribosomal P-sites to each nucleotide position per codon, that is, reading frames 1, 2 and 3. The average footprint density of metagene profiles along the CDS was calculated by dividing the number of P-sites in each of the three reading frames by the total number of P-sites within the CDS. In contrast to the RNA-seq reads that mapped evenly to the three sub-codon positions, the ribosome-associated footprints mapped primarily to the first nucleotide of the codon, that is, reading frame 1.
Detection of actively translated ORFs
Canonical and noncanonical ORF detection was performed using Ribo-TISH (v0.2.1) (16) with the longest strategy under the default threshold setting, which uses a frame test based on the nonparametric Wilcoxon rank-sum test to determine the significance of 3-nt periodicity in the P-site signals along an ORF. Notably, to increase the statistical power of the ORF identification, the aligned BAM files for two replicates of each tissue were merged together with ‘samtools merge’ (v1.6), and only those uniquely mapped reads were used in the Ribo-TISH analysis. The final set of actively translated ORFs with an AUG-start codon followed by an in-frame stop codon in annotated transcripts was stringently filtered based on the requirement of a minimum length of 18 nucleotides and the expression of the ORF-containing gene at an above-background level. uORFs were defined as ORFs originating from the 5′UTRs of annotated protein-coding genes (that is, with TisType: 5′UTR); dORFs were defined as ORFs originating from the 3′UTRs of annotated protein-coding genes (i.e. with TisType: 3′UTR), and smORFs were defined as ORFs originating from annotated long noncoding genes.
Defining expressed genes
To determine putative genes expressed at levels that are significantly higher than the background levels, a half-Gaussian distribution of expression values (log2(TPM)) for each sample was fitted through kernel density estimation using the ks R package (http://cran.r-project.org/web/packages/ks/ks.pdf). The half-Gaussian was then mirrored to full Gaussian distribution. A 2-fold standard deviation below the mean of the distribution was chosen as the minimum threshold for gene expression. Different threshold values, which were defined in a sample-specific manner, were used to filter low-abundance genes. Genes below the threshold in any one replicate of each tissue were filtered out in the subsequent analysis. Additionally, the translated genes were further required to contain actively translated ORFs.
Calculation of expression divergence
The divergence of expression profiles between a pair of tissues was measured based on the Euclidean distance (root mean squared deviation) (17), which was defined as follows:
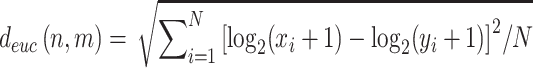 |
where xi and yi represent the normalized TPM values of gene i in two samples from tissues n and m, and N represents the number of protein-coding genes used in the comparison of global gene expression patterns (here, N is 17 488). Notably, for a pair of tissues, there were four-way combinations of samples due to two replicates of each tissue. The larger value of the Euclidean distance, the greater the divergence is, indicating higher dissimilarity.
Gene classification
The different omics-based analyses have allowed the classification of the mouse protein-coding genes with regard to tissue-restricted expression. In line with this, we took advantage of the algorithm provided in the Human Protein Atlas (18) to define tissue-specific genes that can be grouped as follows: (i) ‘tissue-enriched’ genes, defined as genes showing at least 5-fold higher expression in one tissue compared with all other tissues; (ii) ‘group-enriched’ genes, defined as genes showing at least 5-fold higher expression in a group of tissues (2–5) compared with all other tissues; (iii) ‘expressed-in-all’ genes, defined as genes expressed in all analyzed tissues; (iv) ‘not detected’ genes, defined as genes not present in any of the analyzed tissues; and (v) ‘mixed’ genes defined as genes not belonging to any part of the other categories. The RNA-seq- and Ribo-seq-based classifications of all mouse protein-coding genes were conducted using the TissueEnrich R package (19) with a modification: ‘maxNumberOfTissues = 5′, which took a tabulated matrix of TPM values (averaged over replicates of each tissue) with genes as rows and tissues as columns as the input.
Tissue specificity analysis
The tissue specificity of each gene was estimated using the τ index (20) as follows:
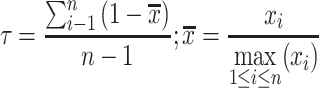 |
where n represents the number of tissue types and xi represents the average TPM value of the gene between two replicates in tissue i. This index varies on a scale from 0 to 1, where 0 indicates ubiquity and 1 indicates specificity.
Gene ontology (GO)-based enrichment analysis
All GO annotations for Mouse Genome Informatics (MGI) were extracted from the ‘mgi.gaf.gz’ file (v2.1 and release date 9 October 2019) that was downloaded from the Gene Ontology homepage (http://current.geneontology.org/products/pages/downloads.html). After assigning all genes to GO terms, only those GO terms for biological processes containing at least five genes were retained for functional enrichment analysis. In total, 17 596 genes assigned to 4896 GO terms were included in this analysis. The hypergeometric distribution was further used to determine whether a GO term was overrepresented in a given gene set. After multiple testing corrections using the Benjamini–Hochberg (BH) approach, those GO terms with a false discovery rate (FDR) below 5% were determined to be statistically significant.
Differential gene expression analysis
To allow proper comparisons among the RNA-seq and Ribo-seq data, raw read counts obtained at the exon level using featureCounts were combined together and normalized against the reference to yield a pool-based size factor, and the resulting data were used for differential expression analysis with the DESeq2 R package (15). A gene was considered to be significantly differentially transcribed or translated if it met the following criteria: (i) the FDR was controlled at the 5% level, and (ii) the absolute fold-change (FC) threshold was set to the most typical cutoff value of 2 (FC > 2 and FC < 1/2). After characterizing concordant and discordant changes in transcription and translation, we defined three distinct patterns of differential genes, as done previously (21), which represent different modes of regulation: ‘mRNA+RPF_both’, which indicated concordant differential expression in both transcription and translation, representing transcriptional forwarding; ‘mRNA_only’, which indicated differential expression in transcription but not in translation, representing translational buffering, and ‘RPF_only’, which indicated differential expression in translation but not in transcription, representing translational reinforcing.
Principal component analysis
To dissect the main contributing layer of gene expression regulation (transcriptional forwarding, translational buffering and translational reinforcement) for each of the coregulatory functional arrangements, principal component analysis (PCA) was performed, as described in a previous report (22). For each arrangement, we calculated the relative fractions of previously defined differential genes with three different modes of regulation that were used as the input for the PCA. The prcomp and fviz_pca_biplot functions from the factoextra R package (https://cran.r-project.org/web/packages/factoextra/index.html) were used for the PCA and visualizing the output of the PCA, respectively. The placement of each cluster in the PCA plot was based on the directionality of three layers of gene expression regulation.
Estimation of translational efficiency
Translational efficiency (TE), defined as the rate of protein production per mRNA (6), was calculated for a given gene as the ratio of TPM values of Ribo-seq to RNA-seq reads within the annotated CDS region. Notably, it was not a direct measure of protein output but ribosome density, and ribosome density per mRNA was used as a proxy for relative translational efficiency. Given a high degree of TE correlations between two replicate samples of each tissue (mean Pearson’s correlation coefficient, r = 0.911), the TE values were averaged between replicates for each gene in the subsequent analysis. TE range for each tissue was calculated as the ratio of 97.5% to the 2.5% quantile of the TE values.
Analysis of differential translational efficiency
The changes in the TE of a gene between different tissues within the same stage and within the same tissue between different stages were assessed using the DESeq2 R package (15) with a threshold of 0.05 to control the FDR and an absolute FC > 2. A table of raw read counts within the whole CDS regions obtained using featureCounts was used as the input for this analysis.
Detection of alternative splicing
Alternative splicing events were identified in RNA-seq data by the VAST-TOOLS pipeline (v2.4.0; https://github.com/vastgroup/vast-tools) (23). Briefly, the clean reads were first mapped to genome assemblies using Bowtie to obtain unmapped reads, and these were then aligned to a predefined splice junction library (the mm10 VastDB library). Unique exon-exon junctions (EEJ) were generated to derive measurements of exon inclusion levels using the metric ‘Percent Spliced In’ (PSI), which utilized all hypothetically possible EEJ combinations from annotated and de novo splice sites, including cassette, mutually exclusive and microexon events (24).
Sample preparation for liquid chromatography-tandem mass spectrometry (LC-MS/MS)
Mouse brain or liver tissue separately at E15.5 and P42 was homogenized in lysis buffer (8 M urea, 100 mM Tris-HCl, 0.5% sodium deoxycholate, pH 8.0) with 1× protease inhibitor (EDTA-free, Roche) by using an automated homogenizer (Bertin Technologies). The temperature of the cooling unit chamber was controlled at 4°C during homogenization. After centrifugation at 16 000 g, 4°C for 20 min, the supernatant fraction was collected and adjusted to 2.0 mg/ml with BCA assay. About 100 μg of each sample was aliquoted for subsequent protein digestion. Samples were reduced with 5 mM dithiothreitol at room temperature for 30 min, followed by 15 mM iodoacetamide alkylation in the dark for another 30 min. Then samples were diluted in 50 mM ammonium bicarbonate to reach 1 M urea concentration followed by Lys-C digestion (Mass Spectrometry Grade, Wako) with a final enzyme-to-protein ratio 1:100 (w/w) at 25°C for 6 h and trypsin digestion (Sequencing Grade, Promega) with a final enzyme-to-protein ratio of 1:50 for 12 h at 25°C. Digestion was stopped by adding 1% formic acid (FA). Next, the sample was desalted with a C18 Sep-Pak cartridge (Waters), dried by a vacuum centrifuge and then resuspended in 0.1% FA. Notably, two biological replicates for each tissue were prepared for LC-MS/MS analysis.
LC-MS/MS analysis and differential protein expression analysis
LC-MS/MS analyses were performed on an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific) coupled with an EASY-nLC™ 1200 System (Thermo Fisher Scientific) with C18 analytical column. Mobile phases A and B consist of 0.1% FA in water and 0.1% FA in 80% ACN, respectively. A 150 min gradient at a flow rate of 300 nL/min was used. Mobile phases B was increased to 11% at 10 min, 30% at 100 min, 45% at 125 min, 100% at 140 min and held for 10 min. Data were collected in data-dependent acquisition (DDA) mode with HCD fragmentation at TopN mode. The resolution was set at 120 000 for MS1 and 30 000 for MS2 with 54 ms maximum injection time. Of note, each biological replicate was run in two technical replicates.
All resulting spectra were searched against UniProt/Swiss-Prot mouse protein database (August 2020, 17 020 entries) using MaxQuant software (v1.6.15.0) (25). The following parameters were used for the search: a mass tolerance of 10 ppm for precursor ions, ±0.02 Da for fragment ions, carbamidomethylation of cysteine as a fixed modification, oxidation of methionine and protein N-terminal acetylation as variable modifications. Two miscleavages were allowed for the trypsin digest, and a maximum of three variable modifications was allowed per peptide. An FDR of 0.01 was set as a threshold for peptide- and protein-level identifications. The differential protein expression analysis was then performed using the DEP R package (26), where the MaxQuant ‘proteinGroups.txt’ file was used as an input.
Western blot analysis
Mouse brain tissues were grounded with liquid nitrogen and lysed with Cell Disruption Buffer (Invitrogen PARIS Kit). Proteins were separated by sodium dodecylsulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to 0.2 μm polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% nonfat milk for 1 h and incubated with indicated primary antibodies overnight at 4°C. After washing to remove the unbound primary antibody, the membranes were incubated with secondary antibody for 1 h. The anti-ATF2 (OriGene, #TA316504), anti-CEBPD (OriGene, #TA322658) and anti-ATF5 (OriGene, #TA312342) primary antibodies were used at 1:500 dilution, the anti-β-Tublin (CST, #2128) and anti-GAPDH (Proteintech, #60004–1-Ig) primary antibody were used at 1:2000 dilution, and the anti-rabbit IgG (CST, #7074) and anti-mouse IgG (CST, #7076) secondary antibody were used at 1:5000 dilution. Then, the membranes were washed with TBST and western blotting signals were developed using Immobilon Western Chemiluminescent HRP Substrate (Millipore) and imaged with Alliance Q9 system (UVITEC).
Luciferase reporter assay of uORF-mediated regulation
To validate the regulatory effects of uORFs on downstream CDS translation under different conditions, we chose an uORF (chr15:80255680–80256381) from the Atf4 gene that was detected in all the tissues. The 5′UTR of Atf4 was fused to the firefly luciferase (Fluc) ORF and further cloned into pcDNA3.1 (pcDNA3.1–5′UTR-Fluc). Briefly, the 5′UTR sequence was amplified by polymerase chain reaction (PCR) from mouse Brain cDNA, and the Fluc ORF sequence was also amplified by PCR from pmirGlo (Promega). The fusion 5′UTR-Fluc sequence was then inserted between the 5′BamHI and 3′XhoI restriction sites by seamless cloning strategy using ClonExpress® Ultra One Step Cloning Kit (Vazyme, # C115). The uORF-mutant 5′UTR sequence was amplified from the wild-type 5′UTR sequence by overlap extension PCR using mutation primers (see Supplementary Table S10), and the mutant plasmid was generated by the same strategy as wild-type plasmid. The sequences of the wild-type and mutant plasmids were verified by Sanger sequencing. For dual luciferase assay, one million Neuro-2A cells were co-transfected with 2250 ng of pcDNA3.1–5′UTR-Fluc (wild-type or mutant) and 250 ng of transfection control Renilla luciferase (Rluc) plasmid pRL-TK (Promega). At 24 h post-transfection, cells were passaged to 24-well plates, cultured in the media containing 10% and 1% fetal bovine serum (FBS) for 48 h, respectively. The Fluc and Rluc luminescence were measured using the Dual-Luciferase Reporter Assay System (Promega, #E1910) and further, the FLuc/RLuc luminescence ratio was calculated for comparative analysis.
Proteomic validation of translated noncanonical ORFs
Two public proteomics data (accession number: PXD009909 and download link: https://phosphomouse.hms.harvard.edu/data/) and an in-house proteomics data (accession number: PXD025201) were used to detect protein products encoded by noncanonical ORFs. Notably, the first public dataset included samples from five of our analyzed tissue types, and the second public dataset included samples of another tissue type. The raw data files were analyzed using MaxQuant software (v1.6.15.0) (25) against a custom-made database, which combined all mouse sequences from UniProt/Swiss-Prot (August 2020) with sequences derived from u/dORFs and smORFs, based on the target-decoy strategy (Reverse) with the standard search parameters with the following exceptions: (i) the peptide-level FDR was set to 5% and the protein-level FDR was excluded; (ii) the minimal peptide length was set to six amino acids; and (iii) a maximum of two missed cleavages was allowed. In total, 451 uORF-, 113 dORF- and 263 smORF-encoded peptides were supported by at least one unique peptide, respectively.
In vitro translation experiments
Plasmid constructs
To generate 3xFlag fusion protein constructs, smORF sequences with endogenous pseudo 5′UTRs (defined as the upstream of the smORF start codon) were amplified by RT-PCR and then cloned into the pcDNA3.1–3xFlag vector, which is a homemade plasmid from pcDNA3.1(+) (Invitrogen). A mutation construct (5′UTR-ORFmut-3xFlag) in which the smORF start codon was mutated to ATT was generated using a Mut Express II Fast Mutagenesis Kit V2 (Vazyme). The wild-type and mutant plasmids were verified by Sanger sequencing. The designed sequences used in this study were listed in Supplementary Table S10.
In vitro translation (IVT)
Both wild-type and mutant plasmids were transfected into Neuro-2A cells using Lipofectamine 3000 reagent (Invitrogen), and 48 h later, the cells were harvested and resuspended in RIPA buffer (Beyotime) with protease inhibitor cocktail (Roche). The cellular lysates were denatured at 85°C for 5 min and then separated on 16.5% Tricine gels for 1 h at 30 V and then for 4 h at 100 V. The proteins were then electroblotted onto a polyvinylidene fluoride (PVDF) membrane (Millipore), and the PVDF membranes were then blocked in 5% nonfat dry milk in TBST for 1 h. Western blotting was performed using anti-Flag (1:1000) (Sigma) or anti-GAPDH (1:5000) (Proteintech) primary antibodies, and the membranes were incubated with secondary antibodies conjugated to horseradish peroxidase (anti-mouse from CST, 1:10 000) for 1 h. The Western blotting signals were developed using Immobilon Western Chemiluminescent HRP Substrate (Millipore) and imaged with ChemiDoc™ Imaging Systems (Bio-Rad).
Functional annotation of lncRNA-derived peptides
Conserved domain and protein homology detection
Each of the putative smORF-encoded peptides (SEPs) was queried against the Conserved Domain Database (CDD) using a web Batch CD-Search tool (27) with the default parameters. In total, 192 SEPs could be assigned at least one known CDD domain. All mouse protein-coding transcript translation sequences were downloaded from GENCODE (Release M18: GRCm38.p6), and the sequences of proteins composed of <100 amino acids (aa) were further retrieved as the set of known small proteins. Each SEP was then queried against these known small proteins using BLASTp software (v2.7.1+) (28) with a hit e-value threshold of 0.0001. In total, 91 SEPs were identified to have recognizable homologs of these known small proteins.
Subcellular localization prediction
The localization of each SEP was predicted using DeepLoc (v1.0) (29) with default parameters. These SEPs were classified into 10 different localizations, including the nucleus (n = 287), cytoplasm (n = 113), extracellular (n = 697), mitochondrial (n = 782), cell membrane (n = 32), endoplasmic reticulum (n = 29), plastid (n = 55), Golgi apparatus (n = 17), lysosome/vacuole (n = 3) and peroxisome (n = 3). Meanwhile, the subcellular localization also included an additional label, where S indicates soluble (n = 1775) and M indicates membrane (n = 268).
Transmembrane helix and signal peptide prediction
Transmembrane and secreted SEPs were predicted using the web applications TMHMM 2 (http://www.cbs.dtu.dk/services/TMHMM/) and SignalP-5.0 (http://www.cbs.dtu.dk/services/SignalP/) with the default parameters. The predictions provided the most likely location and orientation of the transmembrane helices in the sequence as well as the presence of signal peptides and the location of their cleavage sites in the proteins. A total of 150 SEPs were predicted to be either transmembrane and/or secreted, of which 91 were solely transmembrane, 47 were solely secreted and 12 were both transmembrane and secreted.
Coexpressed genomic neighboring protein-coding genes
Each lncRNA with smORF was assigned to its nearest protein-coding gene using bedtools (v2.25.0), and then each protein-coding gene assigned to a translated lncRNA was matched to its immediately neighboring protein-coding gene, which was used as a control, as described in a previous study (1). Pearson’s expression correlation between lncRNA–mRNA and mRNA–mRNA pairs was computed in all samples in our dataset. Candidate coexpressed lncRNA–mRNA pairs were identified as those with correlation coefficients >0.75 and in which each lncRNA–mRNA correlation was significantly higher than the corresponding mRNA–mRNA control, tested using the function paired.r from the psych R package (v1.9.11) with a threshold of 0.05 to control the FDR. The protein-coding genes of these pairs were used for GO enrichment analysis.
RESULTS
Transcriptional and translational profiles of mouse embryonic and adult tissues
To obtain a global view of gene translation and translational regulation in mammalian embryonic and adult tissues, we performed Ribo-seq and RNA sequencing (RNA-seq) to profile six tissues from wild-type C57BL/6 mice at embryonic day (E) 15.5 and postnatal day (P) 42 (Figure 1A). These tissues included ectoderm-derived brain and retinal tissues, mesoderm-derived heart and kidney tissues, and endoderm-derived liver and lung tissues. In total, the Ribo-seq experiments yielded >2.58 billion raw reads, with an average of ∼107 million reads per library, and the RNA-seq experiments yielded >1.19 billion raw reads, with an average of ∼50 million reads per library (Supplementary Table S1). The ribosome-protected fragments (RPFs) obtained from the Ribo-seq analyses showed a predominant length of 29–30 nucleotides (Supplementary Figure S1a), which is the known fragment size protected by 80S ribosomes. On average, 76.4% of the RPFs were mapped to annotated coding sequences (CDSs), whereas 10.2% were mapped to 3′UTRs, 7.3% were mapped to intronic sequences (introns), and 6.1% were mapped to 5′UTRs. Compared with RNA-seq reads, Ribo-seq reads had a strong preference for CDS and 5′ untranslated regions (UTRs) (Supplementary Figure S1b), reflecting a hallmark of good translation-specific data. Metagene analysis of the RPFs mapped to annotated CDS regions revealed a characteristic three-nucleotide (3-nt) periodic subcodon pattern and a striking bias toward the translated frame, with 79% of the RPFs accumulated in the first frame (Supplementary Figure S1c). As expected, the RNA-seq data did not show 3-nt periodicity or frame preference (Supplementary Figure S1d). Our experiments were highly reproducible, as indicated by nearly perfect Pearson’s correlation coefficients (r) between the two replicate samples of each tissue (mean r = 0.965 and 0.986 for Ribo-seq and RNA-seq, respectively) (Supplementary Figure S1e). Principal component analysis (PCA) showed a distinct separation of both the Ribo-seq and RNA-seq data among different stages and tissues (Supplementary Figure S1f). Overall, these results demonstrated that our sequencing data were of high quality.
Figure 1.

Global expression patterns between different tissues and stages. (A) A brief overview of the experimental design. Detailed step-by-step protocols for the Ribo-seq and RNA-seq experiments can be found in the Materials and Methods section. (B) Boxplots of the Euclidean expression distance (root mean squared deviation, see Materials and Methods section for more details) between different tissues at the same stage and between different stages for the same tissue. Each point represents the Euclidean expression distance between a pair of tissues. The between-group differences were compared using a Wilcoxon rank-sum test, and the P-values are shown. The boxplots show the medians, first quartiles and third quartiles; the lines extend to the furthest value within 1.5 of the interquartile range, and the gray points represent the mean values.
Global shifts in gene expression across tissues and stages
Gene expression divergence has been proposed as a phenotypic trait reflecting the evolution of gene regulation and characterizing dissimilarity between tissues within the same species (17). Therefore, using the generated Ribo-seq and RNA-seq datasets, we investigated divergence in gene expression between and within embryonic tissues and their corresponding adult tissues. To do this, the Euclidean distance metric was applied to quantify gene expression divergence between a pair of tissues (see Materials and Methods section).
We first examined gene expression divergence between different tissues (inter-tissue) within the same stage (Figure 1B, left). The inter-tissue expression divergence at the RPF level was observed to be always significantly less than that at the mRNA level. The median expression distance between tissues at the RPF level relative to that at the mRNA level was 83% and 85% for the embryonic and adult stages, respectively. This indicated the existence of substantial posttranscriptional buffering of gene expression, suggesting that posttranscriptional regulation of gene expression, particularly critical translational control, might diminish the inter-tissue expression divergence in transcription. Moreover, we also observed that the inter-tissue expression divergence at the adult stage was significantly greater than that at the embryonic stage, with a 1.29-fold increase at the mRNA level and a 1.32-fold increase at the RPF level in terms of median expression distance, which was consistent with the fact that tissues are more similar early in development and then become increasingly distinct.
Then, we examined gene expression divergence within the same tissue (intra-tissue) between different stages (Figure 1B, right). A similar pattern was seen again, where the median expression distance between tissues was around 16% smaller at the RPF level than at the mRNA level. Collectively, the translatomes were more similar not only between different tissues at the same stage but also between different stages for the same tissue than the transcriptomes. In addition, regardless of expression layers, the intra-tissue expression divergence was generally found to be slightly less than the inter-tissue expression divergence at the embryonic stage (median expression distance: 1.58 versus 1.62 and 1.33 versus 1.35 at the mRNA and RPF levels, respectively) and much less than that at the adult stage (median expression distance: 1.58 versus 2.09 and 1.33 versus 1.78 at the mRNA and RPF levels, respectively). This consequently demonstrated that gene expression patterns were more similar between different stages for the same tissue than between different tissues at the same stage.
Changing patterns of gene and pathway contents underlying expression divergence
We then sought to characterize the genes and pathways underlying the expression divergence and analyze their changing patterns. To this end, we classified all the protein-coding genes into five major categories separately based on their transcriptional and translational levels in six tissues, namely, ‘tissue-enriched’, ‘group-enriched’, ‘expressed-in-all’, ‘mixed’ and ‘not-expressed’ (Figure 2A and Supplementary Table S2; see Materials and Methods section). After characterizing the composition and fraction of genes in each category, we found a dramatic difference in gene composition of each category between the mRNA and RPF levels (Figure 2B), although the majority (>90%) of all expressed genes in these two levels were shared (Supplementary Figure S2a). The most dramatic difference was observed for the mixed category, followed by group-enriched, tissue-enriched, and expressed-in-all categories. On the other hand, we found that during the embryo-to-adult transition, some categories exhibited obvious changes in gene fraction (Figure 2A). There was a general trend for the tissue-enriched category to have an increased gene fraction and the expressed-in-all category to have a decreased gene fraction. As a result of this shift, enhanced tissue specificity of gene expression was observed in the adult (Supplementary Figure S2b).
Figure 2.

Genes and pathways underlying gene expression divergences. (A) Classification of mouse protein-coding genes based on their expression levels in all six tissues, done with the TissueEnrich R package (see Materials and Methods section for more details). The pie charts show the numbers and percentages of genes in each category. (B) Overlap of protein-coding genes for each category between the mRNA and RPF levels. (C) Venn diagrams showing GO term overlap across different expression layers and stages separately for each category. (D) Relative fractions of specific and shared GO terms for each category of genes between the mRNA and RPF levels. (E) An example for translation/translational regulation-related GO terms, uniquely enriched in the expressed-in-all category of genes at the RPF level. (F) Relative fractions of specific and shared GO terms for each category of genes between the embryonic and adult stages. (G) UpSet plot showing the number of specific and shared GO terms enriched in retina-specific genes. The 20 adult-specific GO terms are highlighted in the red rectangle, where the majority are associated with light-induced transformations or responses. For panels C, D, F and G, GO enrichment analysis was performed using a hypergeometric test, with an FDR of 5% used to determine significant terms.
Along with these changes, gene ontology (GO) enrichment analysis revealed extensive differences in the function of the resulting genes (Figure 2C and Supplementary Table S3; see Materials and Methods section). The comparison of GO terms enriched for each category of genes at the RPF level with those at the mRNA level showed that the mixed category had the largest functional differences between the two levels, followed by group-enriched, tissue-enriched and expressed-in-all categories (Figure 2D). The RPF level made an important contribution to the uncoupling functional profiles, contributing an average of 27% (range 6–36%) specific terms. For instance, some GO terms enriched in the expressed-in-all category, including ‘formation of translation preinitiation complex’, ‘IRES-dependent viral translational initiation’, ‘ribosome disassembly’ and ‘miRNA mediated inhibition of translation’, were uniquely found at the RPF level (Figure 2E). On the other hand, the comparison of GO terms enriched for each category at the embryonic stage with those at the adult stage further revealed stage-dependent enrichment, with an average of 73% (range 43–100%) terms uniquely found in a single stage (Figure 2F). For instance, the retina-specific genes of adult mice were greatly involved in light stimulus-related functions, such as ‘detection of light stimulus involved in ‘visual perception’, ‘sensory perception of light stimulus’ and ‘cellular response to light stimulus’ (Figure 2G). These functions were related to enhancing visual functions, which might be a result of increases in visual stimuli and interactions with light that promote neural plasticity in the retina after eye-opening. The lung-specific genes of adult mice were more significantly involved in immunity-related functions compared with their embryonic counterparts, which was likely a manifestation of immune maturation during the postnatal period. Altogether, these results demonstrated the importance and significance of the translatome in ensuring proper tissue architecture and functionality.
Regulatory changes contributing to tissue- and stage-specific gene expression
We next attempted to access the contributions of regulatory changes to divergent gene expression. To infer regulatory change from gene expression, we performed differential gene expression analysis across different tissues and stages (see Materials and Methods section). On average, thousands of tissue- or stage-specific differentially expressed genes (DEGs) were detected, but showing profound uncoupling differential changes between the mRNA and RPF levels (Figure 3A; Supplementary Figures S3–4 and Supplementary Table S4). Based on their differential patterns, we classified these DEGs into three different types representing three modes of regulation, namely, forwarding (mRNA+PRF_both), buffering (mRNA_only) and reinforcing (RPF_only) (see Materials and Methods section). The results showed that differences in transcription were not always forwarded to the RPF level and on average, and >24% of differentially transcribed genes were translationally buffered (Figure 3B). In addition, translational reinforcement could also influence gene expression independently, with at least 20% of DEGs found in translation that was not observed at the mRNA level, which emphasized the complexity of translational regulation in modulating gene expression. Combining with proteomics data from brain and liver, we found that the majority (>65%) of differential expression across tissues and stages in protein could be traced back to transcription and translation, of which about 7–39% were as a result of translational buffering and reinforcement, further highlighting the importance of translational regulation in controlling protein synthesis (Supplementary Figure S5a; see Materials and Methods section). Taking differentially translated genes-Atf2 (reinforcing) and Atf5 (buffering) between stages in the brain as an example, we used western blot analysis to confirm that the specific changes in translation could be reflected at the protein level (Figure 3C and Supplementary Figure S5b; see Materials and Methods section).
Figure 3.

Relative contributions of transcriptional and translational regulatory changes. (A) Differential gene expression analysis across different stages for the same tissue, performed with the DESeq2 package from Bioconductor in R (adjusted P-value < 0.05, absolute log2-fold change >1). Each point represents a gene. Differentially expressed genes (DEGs) are classified as mRNA+PRF_both (orange), mRNA_only (blue) and RPF_only (purple), representing three different modes of regulation: transcriptional forwarding, translational buffering and translational reinforcing. (B) Numbers of differentially expressed genes with distinct regulatory modes (forwarding, buffering and reinforcing) between pairwise-tissue comparisons. The between-group differences were compared using a Wilcoxon rank-sum test, and the P-values are shown. (C) Western blot analysis of Atf2 and Atf5 proteins in the embryonic and adult brain. Quantitative results are presented in bar plots, shown on the right side of the panel, where the differences were compared using a Student’s t-test, and the P-values are shown. (D) Scatter plots of the principal component analysis results showing the manifestations of individual arrangements within the global regulatory programs (see Materials and Methods section for more details). Each numbered point represents a coregulatory functional term, and its position along each axis indicates the relative contribution of transcriptional and translational regulation to the overall differential patterns. The detailed information on each GO term can be found in Supplementary Table S5. (E) Barplots showing the manifestations of individual functional arrangements within the global regulatory programs, where two of the brain-specific GO terms (#28 and #83) are highlighted in the panel (F). (F) Examples of brain-specific coregulatory functional arrangements. The pie chart shows the relative fractions of differentially expressed genes with distinct regulatory modes. The assigned GO term names and corresponding P-values are given.
Given that coregulated genes have been found to often share functional properties, for the detected DEGs, we next applied GO enrichment analysis to delineate potential coregulatory functional arrangements. This analysis yielded an average of 78 and 65 significantly enriched GO terms in each inter- and intra-tissue comparison, respectively (Supplementary Table S5). The DEGs across tissues at the same stage were found to be mainly involved in the functional maintenance of organs, and the DEGs between stages for the same tissue were mainly related to different physiological stages. To dissect the relative contribution of different modes of regulation to the overall differential pattern of each function, we performed a principal component analysis (see Materials and Methods section), which revealed the manifestations of individual functions within the global regulatory programs: (i) some were subjected to major transcriptional or translational regulation, whereas others were subjected to their combinatorial regulation (Figure 3D; Supplementary Figures S6 and 7) and (ii) many functions were orchestrated in tissue- and stage-specific regulatory contexts (Figure 3E). Looking at the brain as an example, its specific arrangements necessary for proper brain function ‘neurotransmitter receptor localization to postsynaptic specialization membrane’ (#28) was primarily under the regulation of transcriptional forwarding, and the ‘gamma-aminobutyric acid signaling pathway’ (#83) was primarily under the regulation of translational reinforcement (Figure 3F), which might allow rapid changes of cellular signaling possibly through modulating their translational efficiencies, enabling an immediate response. Overall, the importance of translational regulation was highlighted in terms of its contribution to the high diversity of translational states, thereby allowing for precise control of gene expression patterns.
Translational efficiency achieving dynamic range control of gene expression
Controlling translational efficiency (TE) is frequently used as a means of translational regulation, and thus we quantified relative TE per gene for each tissue and decoded the patterns of TE changes. Considering that the overall TE changes could be due to different mRNAs present in different tissues and stages, we only focused on the protein-coding genes shared by all embryonic and adult tissues. Comparative analysis of global TE distribution across tissues for the same stage showed clear differences, which were, however, of markedly smaller scale between embryonic tissues than between adult tissues (Figure 4A). This was further indicated by the observation of a sharp difference in the number of differential TE genes (average number: 465 versus 1162) (see Materials and Methods section). Comparative analysis of global TE distribution across stages for the same tissue further showed significantly enhanced TEs in the adult, with an average of 3226 genes exhibiting intra-tissue TE differences (Figure 4A). Notably, the enhanced TEs in the adult might be associated with changes in poly(A) tail length because, in the embryo, short poly(A) tails are necessary to repress translation until the appropriate stage of development is reached (30). This was further supported by the observations that (i) multi-subunits of the major deadenylase responsible for the efficient processive shortening of poly(A) tails, CCR4-NOT complex, were significantly more highly expressed in embryonic tissues than in adult tissues (Figure 4B), and (ii) the 3′UTRs of genes with differential TE upregulation in adult tissues were longer (Figure 4C and Supplementary Figure S8a). Moreover, compared to embryonic tissues, adult tissues exhibited relatively wider ranges of TE (Figure 4D). For instance, the TE range spanned up to 217-fold in the adult liver, whereas the range in the corresponding embryonic liver spanned only 23-fold. A broad TE range might allow for greater flexibility in the control of gene translation. In addition, we observed a considerably narrow spread of TEs versus transcriptional abundances for all the tissues (Figure 4E and Supplementary Figure S8b), which indicated that the effects exerted by transcriptional regulation on gene expression outputs were diluted by modulating the TE. To some extent, this could also partially explain the effect size of the inter-tissue expression divergence at the RPF level becoming significantly smaller than that at the mRNA level, as described in our previous section. Collectively, these results illuminated dynamic changes in the translational efficiency that was optimized for the translation of tissue‐ and stage-specific genes.
Figure 4.

Analysis of translational efficiency (TE) and alternative splicing (AS)-mediated translational regulation. (A) Cumulative distribution of TEs of protein-coding genes shared by all embryonic and adult tissues (N = 9659). The comprison of cross-tissue TE distributions was performed using a Kolmogorov–Smirnov test, and all P-values were statistically significant, except for one with P = 0.295 between the embryonic heart and kidney tissues. The TE range, defined as the ratio of the 97.5% to the 2.5% quantile of the TEs, was given for each tissue. The inserted Box-Wisher plots show the numbers of differential TE genes upon differential TE analysis across different tissues at the same stage and different stages for the same tissue (adjusted P-value < 0.05, absolute log2-fold change >1). The difference significance of gene numbers was computed using a Wilcoxon rank-sum test. (B) Comparison of expression levels of CCR4-NOT complex subunits between embryonic and adult tissues. The expression level differences were compared using a Wilcoxon rank-sum test, and ***: P< 0.001; **: P< 0.01; *: P< 0.05; and ns: no significant. (C) 3′UTR length distribution of differential TE genes in each tissue between the adult and embryonic stages. The differentially upregulated and downregulated TE genes in adult tissue are represented by orange and blue colors, respectively. The distributions were compared using a Kolmogorov–Smirnov test. (D) Comparison of TE ranges between the embryonic and adult tissues. The differences in the TE range were compared using a Wilcoxon rank-sum test. (E) Scatter plot of the adult-to-embryo ratio of transcriptional abundance versus TEs for all expressed protein-coding genes. The corresponding density curves are plotted on the margins. The dotted lines of the same colors represent the 2.5 and 97.5 percentiles of each variable, and the corresponding fold-change range is indicated. The coefficients and P-values for the variables in a linear regression model are presented in the left upper corner. (F) Summary of AS events detected in all six tissues. (G) Comparison of the translatable transcript number for AS and non-AS genes. The P-value obtained by a Wilcoxon rank-sum test is given. (H) Cumulative distribution of CDS TEs in AS genes versus non-AS genes, showing that AS significantly reduced the efficiency of gene translation. (I) Magnitude of AS-mediated translational repression for different types of AS events. The TE differences were compared using a Wilcoxon rank-sum test. (J) Venn diagrams showing AS event overlapping between different tissues at the same stage. (K) Relative fractions of specific and shared AS events for each tissue type between different stages. The brain is shown in panels C, E, G, H and I, and the other tissues are shown in Supplementary Figures S8 and S9.
Alternative splicing-dependent modulation of translational output
Given the prevalence of alternative splicing (AS) in multicellular eukaryotes and its importance in post-transcriptional gene expression control (31), we comprehensively detected all major types of AS events from our RNA-seq datasets, including individual and complex combinations of cassette exons and microexons (AltEx), Alternative 5′ and 3′ splice site choices (Alt5 and Alt3, respectively), and intron retention (IR), and examined the effect of AS events on gene translation (see Materials and Methods section). In total, we identified 12 716 AS events in 5160 protein-coding genes, with IR being the most common AS event accounting for 32% of the total AS events, followed by AltEx (31%), Alt3 (22%) and Alt5 (15%) (Figure 4F and Supplementary Table S6). On average, 2591 and 2315 AS events were identified in each tissue of the embryo and adult, respectively. Although AS could significantly increase coding diversity within genes (Figure 4G and Supplementary Figure S9a), by comparing TEs of protein-coding genes with and without AS events, we observed a consistent trend where AS dramatically reduced the efficiency of the resulting gene translation (Figure 4H and Supplementary Figure S9b). The magnitude of AS-mediated translational repression was further shown to be generally associated with the types of AS events, with IR AS events causing the most prominent repressive effect and AltEx AS events causing the least prominent repressive effect (Figure 4I and Supplementary Figure S9c). Notably, of the identified AS events, the majority were occurred exclusively in one tissue and a single stage (Figure 4J,K), suggesting the existence of tissue- and stage-specific AS-dependent translational regulation. Overall, these results demonstrated that AS represents a widespread and universal translation regulatory mechanism, making tissue- and stage-specific contributions to the translational output.
uORFs and dORFs fine-tuning translational output
To further determine the underlying regulatory elements modulating TE changes, we specifically searched for actively translated ORFs within the 5′- and 3′-UTRs of protein-coding genes (see Materials and Methods section). In total, we identified 6702 unique uORFs in 4297 protein-coding genes across all the tissues, with a median length of 25 codons. In addition to thousands of uORFs, we only identified a total of 830 unique dORFs in 637 protein-coding genes, with a median length of 41 codons (Figure 5A). Using mass spectrometry (MS)-based proteomics data, we further provided directly in vivo evidence for translation of 451 uORFs and 113 dORFs, confirming the stable expression of hundreds of u/dORF-encoded peptides (Supplementary Table S7).
Figure 5.

uORF- and dORF-mediated translational regulation. (A) Number of u/dORFs and u/dORF-containing genes detected in each tissue. (B) Venn diagrams showing uORF overlapping between different tissues at the same stage. (C) Prevalence of uORFs in each category of genes, with higher prevalence in the expressed-in-all category of genes and lower prevalence in the tissue-enriched category of genes. (D) Relative fractions of specific and shared uORFs for each tissue type between different stages. (E) Luciferase reporter assay of uORF-mediated regulation. Here an uORF of the Atf4 gene that underwent active translation in all tissue types was selected to construct wild-type and mutant reporter vectors (see Materials and Methods section for more details). The difference significance was obtained using a Wilcoxon rank-sum test, and the P-values are shown. (F) Cumulative distribution of CDS TEs in uORF-containing genes versus those lacking uORFs. Here, uORF-containing genes were grouped by their number of uORFs, showing that the number of uORFs was associated with the extent of decrease in CDS TEs. The TE differences were compared using a Wilcoxon rank-sum test. (G) Heat map for the enriched GO terms with a frequency of >2 across tissues. Bright yellow color represents significant enriched GO term. (H) Overlap between genes with uORFs and dORFs, showing a trend of mutually exclusive usage. The brain is shown in panel F, and the other tissues are shown in Supplementary Figure S10.
We found large variations in the number of uORFs among tissues and stages, ranging from 682 in the adult heart to 2534 in the embryonic kidney. Notably, this varying number of uORFs was not induced by sequencing depths (Supplementary Figure S10a), which should be an actual reflection of uORF usage patterns. Of the identified uORFs, many (47% in the embryo and 61% in the adult) were detected exclusively in one tissue, showing some degree of tissue specificity, but some were commonly detected in multiple tissues (Figure 5B), which might be associated with a high prevalence of uORFs in the expressed-in-all category of genes (Figure 5C). For each tissue type, 80–85% of uORFs were detected exclusively in a single stage (Figure 5D), showing a strong stage specificity. These findings would thus imply that uORF-mediated translational regulation might occur in a tissue- and stage-specific manner. Furthermore, we provided experimental evidence for condition-dependence of uORF-mediated translational regulation, demonstrating that uORFs, acting as repressors of downstream CDS translation, exerted distinct repressive effects under different conditions (Figure 5E). In addition, uORF-mediated regulation of downstream CDS translation did not always confer the repressive effect (Supplementary Figure S10b). For instance, in the embryonic brain uORFs significantly repressed translation of their downstream CDSs, whereas in the adult brain uORFs significantly enhanced translation of their downstream CDSs (Figure 5F), which could be achieved by either leaky scanning or translational re-initiation (32,33). GO enrichment analysis revealed that uORF-containing genes were not only enriched for many tissue-specific biological functions, but also frequently enriched for basic cellular processes (Supplementary Table S8), particularly those involved in posttranslational modifications such as ‘protein polyubiquitination’, ‘protein glycosylation’ and ‘protein phosphorylation’ (Figure 5G), which suggested that uORFs are regularly used in protein modification processes to tune proteomic diversity.
Similar to uORFs, dORFs also had a high prevalence in the expressed-in-all category of genes (Supplementary Figure S10c) and showed tissue- and stage-specific usage (Supplementary Figure S10d,e). Interestingly, dORFs and uORFs did not tend to simultaneously present in the same gene, exhibiting a mutually exclusive pattern of usage (Figure 5H). Contrary to uORFs, dORFs significantly enhanced the translation of their corresponding CDSs. This trend was consistently observed in all embryonic tissues but not in all adult tissues (Supplementary Figure S10f), suggesting that the enhancive effect of dORFs on their CDS translation might also be condition dependent. The functional importance of dORFs in each tissue was further examined, but regrettably, they did not exhibit enrichment for any particular functions possibly due to their much smaller size.
Pervasive translation of long noncoding RNAs
Apart from the uORFs and dORFs, translation can also occur in small ORFs (smORFs) within putative long noncoding RNAs (lncRNAs). Herein, we identified a total of 2023 actively translated smORFs in 1034 lncRNAs, with >92% of smORFs originating from lincRNAs, antisense transcripts and processed transcripts (Figure 6A and Supplementary Table S7). The capacity for these smORFs to produce a stably translated protein was further accessed by two independent methods, including that (i) MS-based proteomics data provided directly in vivo evidence for peptide products translated from 263 of the 2023 smORFs, and (2) in vitro translation experiments showed peptide generation for 3 out of 10 randomly chosen smORFs, namely, Lsmem2, RP23-83I13.10 and RP23-52N2.1 (Figure 6B). Pervasive translation of lncRNAs would open the possibility that lncRNAs are a source of cryptic translation events with functional roles.
Figure 6.

lncRNA translation. (A) Number of actively translated smORFs detected in each tissue by using Ribo-TISH. The smORF distribution by locus biotypes of the GENCODE lncRNA annotation (Release M18) is shown below, where the lncRNAs have been reclassified into nine distinct locus biotypes based on their location with respect to protein-coding genes, including lincRNA, macro_lncRNA, antisense, bidirectional_promoter_lncRNA, 3prime_overlapping_ncRNA, processed_transcript, sense_intronic, sense_overlapping and TEC (to be experimentally confirmed) (see definitions of detail on these biotypes at https://www.gencodegenes.org/pages/biotypes.html). (B) In vitro experiments for validating smORF translation. Molecular weights of micropeptides are indicated in kilodaltons (kDa). (C) Fraction of translated lncRNAs in each lncRNA class, where classification of lncRNAs was based on their expression levels in the six tissues, done with the TissueEnrich R package. (D) Venn diagrams showing translated lncRNA overlapping between different tissues at the same stage. (E) Relative fractions of specific and shared translated lncRNAs for each tissue between different stages. (F) A brief overview of functional characterization of smORF-encoded peptides (SEPs). (G) Enriched GO terms for protein-coding genes significantly correlated with their neighboring translated lncRNAs, determined by GO enrichment analysis (n = 166; P-value < 0.01, hypergeometric test).
Given that determining a gene’s pattern of expression is a key step toward understanding its function, we next analyzed the patterns of lncRNA translation across tissues and stages. On average, 403 and 407 smORFs were detected in each tissue of the embryo and adult, respectively. Despite no statistically significant difference in the average number of smORFs between two stages (P = 0.937, Wilcoxon rank-sum test), during the embryo-to-adult transition, we found a significant change in translational pattern, where the translated fraction in the tissue-enriched category of lncRNAs increased from 23% to 40% whereas this fraction in the expressed-in-all category of lncRNAs decreased from 38% to 21% (P = 1.89e-08, Fisher’s exact test; Figure 6C). Coinciding with these changes, translation of lncRNAs showed an increased tissue-specificity in the adult (Figure 6D). The fraction of tissue-specific translated lncRNAs was nearly 1.27-fold higher in the adult than in the embryo (66% versus 52%). Moreover, translation of lncRNAs also had stage-specificity, with an average of 67% (range 63–72%) detected exclusively in a single stage (Figure 6E).
To gain insight into the potential translation functions, we subsequently performed functional annotation on these putative smORF-encoded peptides (SEPs) (Figure 6F; see Materials and Methods section). Their peptide sequences were first queried separately against the CDD domains and the annotated known small proteins in the mouse genome, which revealed that only a small subset (220) were relevant to the well-characterized protein domains or small proteins, meaning that the majority (>89%) of the 2023 SEPs were novel. Subcellular localization prediction revealed that these SEPs were primarily localized to the mitochondria (782, 38.7%), followed by the extracellular (697, 34. 5%), nucleus (287, 14.2%) and cytoplasm (113, 5.6%). Notably, of these SEPs, 1755 (86.8%) were predicted to be soluble. This also partially explained the lower validation rate in the proteome and IVT assay. Nevertheless, 150 SEPs were further predicted to be either transmembrane and/or secreted, suggesting that they could act as potential mediators of cell–cell communication given that cellular communication is typically mediated by the secretion of small diffusible signaling molecules or through direct cell–cell contact (34). GO enrichment analysis on the coexpressed adjacent protein-coding genes with these translated lncRNAs (see Materials and Methods section) showed that they were frequently enriched for functions associated with tissue development, differentiation and morphogenesis (Figure 6G and Supplementary Table S9), indicating their potential functional importance. Overall, these results revealed that the translation of at least some, if not all, lncRNAs could produce stable peptides with potential regulatory roles in vivo.
DISCUSSION
A comprehensive translatome profile of the tissue types and stage types can reveal the enormous diversity in gene translation and its regulation associated with tissue and stage. In this study, our comparative translatome analysis provided many novel insights into translational regulation that modulates the dynamics of gene expression and physiological functions in a tissue- and stage-specific manner. The functional characterization of genes and pathways underlying the divergences in gene expression within and between embryonic tissues and their corresponding adult tissues further enhanced our understanding of the molecular basis of tissue physiology. We observed the incomplete coupling between biological functions of transcribed and translated genes, highlighting the significance of translation and its regulation in ensuring the proper functioning of tissue at different life stages. Notably, translational profiling will enable a better definition of ‘housekeeping genes’, as supported by the observation that approximately 9.3% of ubiquitously transcribed genes are not included in the set of ubiquitously translated genes. This change is likely subject to a disallowance of regulation during the process transition from transcription to translation.
Tight regulatory controls of transcription and translation play an important role in the development of the embryo and the maintenance of adult tissues. We note that although translational up- and downregulation may have very different mechanistic underpinnings and functional outcomes, distinguishing between their regulatory effects would require enhanced knowledge from the regulators, which can be precisely obtained through perturbation experiments. Controlling TEs is frequently used as a means of translational regulation, which delivers dynamic and context-specific TE changes, thereby influencing quantitative differences in gene outputs in different tissues and life stages. uORF- and dORF-mediated translational regulation represents another important layer for the manipulation of gene expression. We revealed that many uORFs provide functionally important repression of the downstream CDS translation in a dose-dependent manner, in line with previous reports (35,36). However, some uORFs exert different regulatory effects on translation of the downstream CDSs in adult tissues, demonstrating the complexities of uORF-mediated translational regulation (33). Behind the complexities, many properties may contribute to an uORF’s role in translational regulation, including the length of the 5′UTR, the secondary structure and GC content, as well as the strength of the surrounding Kozak context, the uORF length, and conservation, which have been substantially discussed in detail in previous reports (32,37). Apart from uORFs encoding regulatory peptides, some uORFs have been reported to encode for proteins with functions independent of the control of the downstream CDS (38). In addition to uORFs, translation of dORFs in the 3′-UTRs represents a new translation regulatory mechanism, enhancing translation of their corresponding CDSs (11), but notably, dORFs did not always confer translational enhancement, which might also be condition dependent.
The identification of actively translated smORFs in lncRNAs broadens our understanding of coding ability in the mouse genome. We revealed a notable number of translated lncRNAs that exhibit unanticipated tissue- and stage-specificity. However, as demonstrated in our previous study (39), translatable lncRNAs are also obviously different from annotated protein-coding genes, with markedly distinguishing properties, including expression, structural, sequence, evolutionary and functional features, which underpin the mysteries surrounding the biology of lncRNAs. Although our analysis revealed pervasive translated lncRNAs in different tissues and stages, translatable lncRNAs are not necessarily detectable peptides. Indeed, only a portion of the translated products was validated by different strategies, including in vivo peptide detection by mass spectrometry and in vitro translation experiments. One possibility is that some peptides likely escape detection due to extremely low expression abundance (40), and another possibility is that some peptides are likely degraded during proofreading through nonsense-mediated decay (41). In addition, detectable peptides are not necessarily functional peptides. To understand the functional potential of these putative peptides, we used integrative annotation approaches and pointed to their potential functional relevance. Nevertheless, understanding the full repertoire of translated lncRNAs and their biological functions remains challenging. The translated details of individual lncRNAs and their biological functions also need to be further experimentally validated.
In summary, our analyses will facilitate a better understanding of how tissue-specific and stage-specific phenotypes are achieved through the precise control of gene translation and its regulation and our data may serve as a valuable resource for future research in the field of translatomics.
DATA AVAILABILITY
All raw Ribo-seq and RNA-seq data generated in this study have been submitted to the NCBI Gene Expression Omnibus under accession number GSE94982. The generated LC-MS/MS data have been deposited in the PRIDE database under accession number PXD025201.
Supplementary Material
ACKNOWLEDGEMENTS
Author Contributions: H.W.W. and Z.X. designed the study and wrote the manuscript. J.Q.Y. and N.T. performed the RNA-seq and Ribo-seq experiments. Q.Z. and Y.Y. performed the MS-based proteome experiment. C.Y.C performed western blot and luciferase reporter assay. N.T. performed in vitro translation experiments. H.W.W., Y.W., H.H.L., C.C.C., and M.Z.X. performed the bioinformatics analyses and result explanations. All named authors read and approved the final manuscript.
Contributor Information
Hongwei Wang, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, China.
Yan Wang, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, China.
Jiaqi Yang, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, China.
Qian Zhao, State Key Laboratory of Chemical Biology and Drug Discovery, Department of Applied Biology and Chemical Technology, Hong Kong Polytechnic University, Hong Kong 999077, China.
Nan Tang, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, China.
Congying Chen, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, China.
Huihui Li, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, China.
Chichi Cheng, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, China.
Mingzhe Xie, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, China.
Yang Yang, State Key Laboratory of Chemical Biology and Drug Discovery, Department of Applied Biology and Chemical Technology, Hong Kong Polytechnic University, Hong Kong 999077, China.
Zhi Xie, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31871302 to Z.X. (in part)]; Joint Research Fund for Overseas Natural Science of China [31829002 to Z.X.]. Funding for open access charge: National Natural Science Foundation of China [31871302 to Z.X. (in part)]; Joint Research Fund for Overseas Natural Science of China [31829002 to Z.X.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Sarropoulos I., Marin R., Cardoso-Moreira M., Kaessmann H.. Developmental dynamics of lncRNAs across mammalian organs and species. Nature. 2019; 571:510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cardoso-Moreira M., Halbert J., Valloton D., Velten B., Chen C., Shao Y., Liechti A., Ascencao K., Rummel C., Ovchinnikova S.et al.. Gene expression across mammalian organ development. Nature. 2019; 571:505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou Q., Liu M., Xia X., Gong T., Feng J., Liu W., Liu Y., Zhen B., Wang Y., Ding C.et al.. A mouse tissue transcription factor atlas. Nat. Commun. 2017; 8:15089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A.et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 5. Sonenberg N., Hinnebusch A.G.. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009; 136:731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ingolia N.T. Ribosome profiling: new views of translation, from single codons to genome scale. Nat. Rev. Genetics. 2014; 15:205–213. [DOI] [PubMed] [Google Scholar]
- 8. Brar G.A., Weissman J.S.. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol. 2015; 16:651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ingolia N.T. Ribosome Footprint profiling of translation throughout the genome. Cell. 2016; 165:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ingolia N.T., Hussmann J.A., Weissman J.S.. Ribosome profiling: global views of translation. Cold Spring Harb. Perspect. Biol. 2019; 11:a032698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu Q., Wright M., Gogol M.M., Bradford W.D., Zhang N., Bazzini A.A.. Translation of small downstream ORFs enhances translation of canonical main open reading frames. EMBO J. 2020; 39:e104763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li H., Xie M., Wang Y., Yang L., Xie Z., Wang H.. 2021) riboCIRC: a comprehensive database of translatable circRNAs. Genome Biol. 22:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liao Y., Smyth G.K., Shi W.. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019; 47:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang P., He D., Xu Y., Hou J., Pan B.F., Wang Y., Liu T., Davis C.M., Ehli E.A., Tan L.et al.. Genome-wide identification and differential analysis of translational initiation. Nat. Commun. 2017; 8:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Glazko G., Mushegian A.. Measuring gene expression divergence: the distance to keep. Biol. Direct. 2010; 5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim M.S., Pinto S.M., Getnet D., Nirujogi R.S., Manda S.S., Chaerkady R., Madugundu A.K., Kelkar D.S., Isserlin R., Jain S.et al.. A draft map of the human proteome. Nature. 2014; 509:575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jain A., Tuteja G.. TissueEnrich: tissue-specific gene enrichment analysis. Bioinformatics. 2019; 35:1966–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yanai I., Benjamin H., Shmoish M., Chalifa-Caspi V., Shklar M., Ophir R., Bar-Even A., Horn-Saban S., Safran M., Domany E.et al.. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005; 21:650–659. [DOI] [PubMed] [Google Scholar]
- 21. Schafer S., Adami E., Heinig M., Rodrigues K.E.C., Kreuchwig F., Silhavy J., van Heesch S., Simaite D., Rajewsky N., Cuppen E.et al.. Translational regulation shapes the molecular landscape of complex disease phenotypes. Nat. Commun. 2015; 6:7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Heesch S., Witte F., Schneider-Lunitz V., Schulz J.F., Adami E., Faber A.B., Kirchner M., Maatz H., Blachut S., Sandmann C.L.et al.. The translational landscape of the human heart. Cell. 2019; 178:242–260. [DOI] [PubMed] [Google Scholar]
- 23. Tapial J., Ha K.C.H., Sterne-Weiler T., Gohr A., Braunschweig U., Hermoso-Pulido A., Quesnel-Vallieres M., Permanyer J., Sodaei R., Marquez Y.et al.. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res. 2017; 27:1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Irimia M., Weatheritt R.J., Ellis J.D., Parikshak N.N., Gonatopoulos-Pournatzis T., Babor M., Quesnel-Vallieres M., Tapial J., Raj B., O’Hanlon D.et al.. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014; 159:1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 26. Zhang X., Smits A.H., van Tilburg G.B., Ovaa H., Huber W., Vermeulen M.. Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat. Protoc. 2018; 13:530–550. [DOI] [PubMed] [Google Scholar]
- 27. Lu S., Wang J., Chitsaz F., Derbyshire M.K., Geer R.C., Gonzales N.R., Gwadz M., Hurwitz D.I., Marchler G.H., Song J.S.et al.. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020; 48:D265–D268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 29. Almagro Armenteros J.J., Sonderby C.K., Sonderby S.K., Nielsen H., Winther O.. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics. 2017; 33:3387–3395. [DOI] [PubMed] [Google Scholar]
- 30. Subtelny A.O., Eichhorn S.W., Chen G.R., Sive H., Bartel D.P.. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature. 2014; 508:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee Y., Rio D.C.. Mechanisms and regulation of alternative Pre-mRNA splicing. Annu. Rev. Biochem. 2015; 84:291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Young S.K., Wek R.C.. Upstream open reading frames differentially regulate gene-specific translation in the integrated stress response. J. Biol. Chem. 2016; 291:16927–16935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang H., Wang Y., Lu J.. Function and evolution of upstream ORFs in eukaryotes. Trends Biochem. Sci. 2019; 44:782–794. [DOI] [PubMed] [Google Scholar]
- 34. Dang Y., Grundel D.A.J., Youk H.. Cellular dialogues: cell-cell communication through diffusible molecules yields dynamic spatial patterns. Cell Syst. 2020; 10:82–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnstone T.G., Bazzini A.A., Giraldez A.J.. Upstream ORFs are prevalent translational repressors in vertebrates. EMBO J. 2016; 35:706–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bazzini A.A., Johnstone T.G., Christiano R., Mackowiak S.D., Obermayer B., Fleming E.S., Vejnar C.E., Lee M.T., Rajewsky N., Walther T.C.et al.. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014; 33:981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Somers J., Poyry T., Willis A.E.. A perspective on mammalian upstream open reading frame function. Int. J. Biochem. Cell Biol. 2013; 45:1690–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Samandi S., Roy A.V., Delcourt V., Lucier J.F., Gagnon J., Beaudoin M.C., Vanderperre B., Breton M.A., Motard J., Jacques J.F.et al.. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. eLife. 2017; 6:e27860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang H., Wang Y., Xie S., Liu Y., Xie Z.. Global and cell-type specific properties of lincRNAs with ribosome occupancy. Nucleic Acids Res. 2017; 45:2786–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang S., Mao C., Liu S.. Peptides encoded by noncoding genes: challenges and perspectives. Signal Transduct. Target Ther. 2019; 4:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Makarewich C.A., Olson E.N.. Mining for Micropeptides. Trends Cell Biol. 2017; 27:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw Ribo-seq and RNA-seq data generated in this study have been submitted to the NCBI Gene Expression Omnibus under accession number GSE94982. The generated LC-MS/MS data have been deposited in the PRIDE database under accession number PXD025201.