


Abstract
Pulmonary diseases offer many targets for oligonucleotide therapeutics. However, effective delivery of oligonucleotides to the lung is challenging. For example, splicing mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) affect a significant cohort of Cystic Fibrosis (CF) patients. These individuals could potentially benefit from treatment with splice switching oligonucleotides (SSOs) that can modulate splicing of CFTR and restore its activity. However, previous studies in cell culture used oligonucleotide transfection methods that cannot be safely translated in vivo. In this report, we demonstrate effective correction of a splicing mutation in the lung of a mouse model using SSOs. Moreover, we also demonstrate effective correction of a CFTR splicing mutation in a pre-clinical CF patient-derived cell model. We utilized a highly effective delivery strategy for oligonucleotides by combining peptide-morpholino (PPMO) SSOs with small molecules termed OECs. PPMOs distribute broadly into the lung and other tissues while OECs potentiate the effects of oligonucleotides by releasing them from endosomal entrapment. The combined PPMO plus OEC approach proved to be effective both in CF patient cells and in vivo in the mouse lung and thus may offer a path to the development of novel therapeutics for splicing mutations in CF and other lung diseases.
INTRODUCTION
Lung diseases are one of the largest health burdens and are a main cause of mortality and morbidity worldwide (1). Yet, non-communicable and chronic pulmonary problems are often treated symptomatically without addressing pathogenesis. In this report we present a novel platform for delivering therapeutic oligonucleotides to directly correct splicing defects in the lung and have selected cystic fibrosis (CF) as a model to test this delivery approach. Additionally, our work may have potential therapeutic implications in various pulmonary and non-pulmonary diseases that involve splicing defects.
The cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP-regulated chloride channel that serves a critical role in the regulation of epithelial salt and water transport (2,3). Loss-of-function mutations in CFTR result in CF a multi-organ disease that involves chronic pulmonary pathologies, pancreatic insufficiency, GI obstruction, and male infertility (4). In the airways, the loss of CFTR-mediated chloride transport can result in morbidity and death as a result of dehydrated, adherent mucus leading to chronic infection and inflammation (5,6). Until recently only lung transplant and symptomatic therapies were available for CF but there are now small molecule modulators that, at the protein level, affect the trafficking and function of certain CFTR mutants (7,8). Modulator combinations such as the recently approved double combination Orkambi® and triple drug Trikafta® can provide substantial benefit to a large percentage of CF patients (9).
However, a significant subpopulation of CF patients cannot be effectively treated with modulator drugs. For example, approximately 11% of CF patients have CFTR splicing defects, the most common being the 3849+10kb C→T mutation (legacy name, a.k.a. CFTR splicing mutation c.3718-2477C→T) (10). Splicing mutations may be homozygous or be paired with other CFTR defects such as the common deletion-F508 trafficking mutation (3), channel gating mutations, or stop mutations. Although modulator drugs have been approved for certain CFTR splicing mutations (11,12) they do not address the underlying molecular defect. For patients where homozygous splicing mutations or a combination of splice and stop codon mutations result in very little CFTR protein production (13,14), the modulator drugs are unlikely to be effective and other therapies will be needed.
Splicing defects in CF patients should be correctable through use of splice switching oligonucleotides (SSOs) (15–19). These molecules modulate interactions between pre-mRNA and the spliceosome and can be used to attain correction of splicing defects, as well as to increase or decrease gene expression (16,20). The most widely used SSO chemistry involves a negatively charged phosphorothioate (PS) backbone and pendant nucleosides with 2′O-Me, methoxyethoxy (MOE) or locked nucleic acid (LNA) modifications (21). An alternative is to use SSOs with an uncharged phosphorodiamidate morpholino (PMO) backbone (22,23). Recently SSOs have entered the clinic with FDA approval of a PS-MOE SSO for treatment of spinal muscular atrophy and of two PMO SSOs for treatment of Duchenne muscular dystrophy (24). Thus, CFTR splicing mutations would seem to be appropriate targets for SSOs.
The first report of SSO-mediated correction of splicing in cells transfected with mutant CFTR constructs dates back >20 years (18). More recently Igreja et al. (25) used 2′O-Me PS SSOs to correct splicing in a non-physiological HEK293 cell model expressing a mutant CFTR and measured ion fluxes as well as correction at the RNA level. Michaels et al .(26) performed a very thorough study of CFTR splicing correction in patient-derived cells harboring the 3849+10kb C->T mutation. This study demonstrated correction of the endogenous mutant CFTR RNA and provided detailed measurements of restoration of CFTR ion fluxes. In this case, PMO SSOs were utilized; however, the PMOs were delivered to cells using a cationic transfection agent.
Correcting CF splicing mutations in patients implies efficient delivery of oligonucleotides to lung and other epithelial tissues that are difficult to transfect (27). To date none of the reports on correction of CFTR splicing can be directly projected to CF therapeutics since they all relied on delivery methods that are not suitable for in vivo use. Here, we have addressed the delivery problem inherent in treatment of CF splicing mutations through a novel combination of two powerful oligonucleotide delivery technologies. First, we utilize SSOs that are peptide-morpholino oligomer conjugates (PPMOs) (28). These molecules have a broad tissue distribution and, when given systemically, they can produce oligonucleotide effects in extra-hepatic tissues (29,30). Second, we make use of oligonucleotide enhancing compounds (OECs). These are small molecules, discovered through high throughput screening, which selectively release oligonucleotides from non-productive entrapment in endosomal compartments (19,31,32). Thus, OECs allow oligonucleotides to access the cytosol and nucleus providing substantial enhancement of pharmacological effects. In this report, we describe use of the novel PPMO plus OEC strategy to efficiently correct splicing in the lung in an animal model, as well as in patient-derived primary airway epithelial cells encoding an important CFTR splicing mutation.
MATERIALS AND METHODS
Biological resources
Cell models
HeLa EGFP654 cells
This previously described cell line (33) contains a stably transfected enhanced green fluorescent protein (EGFP) splicing reporter that can be activated by SSOs. The EGFP construct encodes the same mutated intron as the mouse model described below. (Repository: ATCC).
HBEC
Patient-derived, well-differentiated primary Human Bronchial Epithelial Cells (HBEC) harboring the 3849 + 10 kb C→T splicing mutation were cultured as described (3,34). Passage 2 cells were used in these studies. Patient cells were procured and used in accordance with the University of North Carolina Office of Research Ethics Biomedical Institutional Review Board approved protocol No. 03–1396. (Repository: UNC Marsico Lung Institute, not a cell line).
UNCCF8T cells
These cells were created by lentiviral addition of hTERT and Bmi-1 to bronchial epithelial cells derived from a patient donor homozygous for the CFTR splicing mutation 3849 + 10 kb C→T using methods previously described for non-CF and F508del homozygous donors (35), with some modifications. For this novel cell line, passage 1 cryopreserved cells were expanded using the conditionally reprogrammed cell culture method (34) and cells were transduced with a novel vector expressing both hTERT and Bmi-1 from a single virus, and also expressing a puromycin resistance module and copepod GFP fusion protein. The cells have expanded growth capacity and polarize and differentiate in air-liquid interface cultures as previously reported for the other genotypes and are suitable for Ussing chamber studies. Passage 5–7 were used in these studies, showing no significant physiologic changes on epithelial differentiation and CFTR expression (Repository: UNC MLI, not a cell line).
EGFP654 MTEC
Well-differentiated, primary Mouse Tracheal Epithelial Cells (MTEC) were derived from the EGFP654 mouse (see below). These cells were developed in the Kreda lab by adapting a published protocol (36). Cells were differentiated on collagen-coated permeable inserts to air-liquid interface into cilia and mucus producing epithelial cell layers; passages 0–2 were used in these studies with no significant physiologic differences in morphology and EGFP expression levels. (Repository: UNC MLI, not a cell line).
Animal model
The EGFP654 transgenic mouse expresses the EGFP protein coding sequence interrupted by the intron of human beta globin encoding the 654 splicing mutation; the mutant EGFP RNA is expressed in all cell types and is non functional (37). Upon splicing correction with a specific oligonucleotide, functional EGFP is expressed (19,32) (repository: UNC).
Oligonucleotides and OECs
Morpholino oligonucleotides (PMOs) were purchased from Genetools Inc. and were then conjugated with peptide to form PPMOs. Conjugation of peptide and purification of the PPMO conjugates were carried out as previously described (29). Identity of the PPMO conjugates was confirmed by mass spectrometry. PCR primers and probes, were purchased from Integrated DNA Technologies. The oligonucleotide and peptide sequences used in this study are shown in Supplementary Table S1. The Oligonucleotide Enhancing Compounds (OECs) UNC7938 and UNC2383 were synthesized by ChemoGenics BioPharma as previously described (31) and were purified by liquid chromatography with chemical identify confirmed by mass spectrometry. The compounds were used in their acid salt form (UNC7938B, UNC2383C).
In vivo studies in the EGFP654 animal model
For the in vivo studies we utilized the EGFP654 transgenic mouse that expresses a mutant EGFP RNA in all cell types (37). All animal procedures were conducted in compliance with guidelines of the UNC Laboratory Animal Medicine Department and with federal guidelines (UNC IACUC # 18-265). EGFP654 mice were maintained by the UNC Animal Studies Core; this group also performed all injections of materials. Both male and female mice were used in the studies. For experiments, various doses of ‘naked’ PPMO654 or of OEC or both were administered via tail vein injection. After incubation periods ranging from 4h to 22 days mice were euthanized and tissues collected. Tissue samples were either quickly frozen on dry ice for analysis by RT-PCR or were fixed in 10% buffered formalin for immunohistochemistry. Blood samples were analyzed for indicators of renal and hepatic toxicity by the UNC Animal Clinical Chemistry Core facility. These procedures were similar to those we have previously described (19,32). Further experimental details are provided in the figure legends. For airway cultures, mice were euthanized and tracheas were excised and processed in sterile conditions for epithelial cell isolation and culture (see above).
EGFP654 MTEC and HeLa EGFP654 cell culture experiments
HeLa EGFP654 cells were cultured in 96-well plates. Cells were exposed to PPMO during an overnight incubation, rinsed, and subsequently exposed to OEC, usually for 2 h. After rinsing cells were incubated for either 16 or 24 h after which EGFP fluorescence was quantitated using a Varioskan imaging unit. Viability was assessed using the CellTiter Blue assay (Promega) according to manufacturer's instructions.
Well-differentiated primary EGFP654 MTEC cultured at air-liquid interfaces on 6.5 mm diameter permeable supports were incubated with PPMOs and OECs in concentrations and for periods of time indicated in the figures. PPMOs and OECs were administered ‘naked’ in complete medium using the following volumes: 30 μl for apical and 200 μl for basolateral administrations in MTEC. Two treatment modalities were utilized: sequential and co-administration of PPMO and OEC. Sequential treatment was performed by incubating cells overnight with PPMO followed by 2hs incubation with OEC. PPMO and OEC co-administration was performed for 6 h, which was the optimal incubation time. For the mucus experiments, a preparation of concentrated mucus isolated from human airway cells (a kind gift of Dr B Button, UNC) was diluted to different concentrations in complete medium. PPMO and OECs were pre-incubated 2 h at 37ºC with the mucus preparation before adding to the surface of MTEC cultures that were pre-washed with PBS to eliminate the endogenous mucus layer. Lumenal additions were performed in 30 ul, while other incubations parameters were as described above. For all experiments, after PPMO/OEC incubations, cells were further cultured in complete growth medium for 24–48 h. For the assessment of EGFP expression, cells were analyzed for EGFP fluorescence levels in a Tecan plate scanner or an Azure Scanning System. After fluorescence quantification, cells were fixed and stained with propidium iodide to label nuclei and re-scanned for cell number normalization. In some experiments, cells were also analyzed for EGFP mRNA expression using RT-PCR techniques described below.
RT-PCR analysis
RT-PCR studies in tissues were performed following our previous work (19,32). Messenger RNA was isolated by the UNC CF Center Molecular Biology Core or in our labs. The cDNA synthesis and PCR reactions were performed using specific primers framing the EGFP654 mutation (EGFP reporter cells and tissues) or CFTR 3849+10kb mutation (human CF cells), as well as GAPDH for normalization. The WT band in human CFTR studies was identified by size comparing with the band produced by normal human cells and was subjected to sequencing to confirm identification. PCR gels stained with ethidium bromide or Gel Green were imaged in a high resolution scanner for densitometry analysis of the mutant and corrected PCR bands. Percent correction was estimated as 100 x WT/(WT + mut).
Immunostaining and confocal microscopy analyses
Tissues were processed and immunostained using an antibody against EGFP as we previously described (19). Co-staining was performed with an antibody against tubulin to reveal cilia and identify ciliated cells as we previously described (38). Confocal Microscopy was performed as described (19). For tissue cultures, live cells on permeable supports were briefly stained with supravital dye Cell Mask® (Thermo Fisher Sci.), which stains apical membrane and cilia when added to polarized epithelial layers. Cell cultures were subjected to live imaging in a Leica SP5 with the stage enclosed in an environmentally-controlled chamber; 63× Leica lenses and two independent laser sources were used; scanning parameters were constant across experimental specimens. Picture mounting was performed in Adobe Photoshop. Live imaging of freshly excised organs was performed in a Leica DMRB epifluorescence microscope with a 10× lenses and a low-resolution camera.
Ussing chamber experiments
For studies of topical administration of ‘naked’ oligonucleotides and OECs, HBEC and UNCCF8T cells were cultured on 12 mm Millicell permeable supports (Millipore) and in air-liquid interface for 2–3 weeks before Ussing chamber analyses. Drugs were administered as indicated in the figures. PPMO and OECs were administered, either apically or basolaterally, 72 h before analysis, while a combination of CFTR modulators similar to Trikafta® (VX-445 2 μM, VX-661 3 μM, VX-770 1μM) were administered both apically and basolaterally 48 h before Ussing chamber studies. All reagents were added in complete growth medium; cells were washed the evening before analysis. Ussing chamber analyses followed protocols optimized in the Gentzsch lab (34,39,40). Briefly, changes in short-circuit current (ΔIsc in μA/cm2) were measured in Ussing chambers in a bilateral Krebs bicarbonate-Ringers solution at 37ºC. Amiloride (100 μM, Sigma-Aldrich) was added to the apical bath to inhibit the epithelial sodium channel ENaC. Bilateral addition of forskolin (10 μM, Sigma-Aldrich) followed to stimulate CFTR channel activity. CFTR inhibitor-172 (10 μM, Sigma-Aldrich) was then apically introduced to inhibit CFTR (as a CFTR control). UTP (100 μM, GE Healthcare) response for Ca2+ channel activity was assessed as an internal control. Transepithelial resistance (in Ω·cm2) was monitored to assess monolayer integrity, and it is also a good indicator of epithelial health/cytotoxicity. After completing Ussing chamber studies, cells were processed for CFTR mRNA expression using techniques described above and PCR primers described in Supplementary Table S1.
Formulation of PPMO and OECs
Stock solutions of PPMO were prepared in deionized water. OECs were dissolved in DMSO at 40°C for preparation of stock solutions. Dilutions of OECs for experiments were made in DMSO and then added to complete medium immediately before use. Solutions of PPMO and OEC for in vivo administration were prepared in 6% PEG400 plus 20% DMSO, which was also used as vehicle in control animals.
Statistics
Based on our previous experience, mouse groupings of 3–4 animals for the in vivo studies and four cultures per experimental condition for the in vitro studies were deemed sufficient for statistical power. Differences between means were assessed with ANOVA and post-hoc analysis or two sample Student's t test and two-tailed alternative hypothesis.
RESULTS
OECs increase PPMO delivery in cell culture
We had previously shown that Oligonucleotide Enhancing Compounds (OECs) could improve the pharmacological effects of a variety of anionic oligonucleotides and conjugates (19,31,32). However, we had not tested OECs with peptide conjugates of morpholino oligomers (PPMOs) that have a net positive charge and that also have some innate endosome escape capability. To address this issue we initially utilized HeLa EGFP654 cells that are stably transfected with an EGFP reporter whose coding sequence is interrupted by an aberrantly spliced intron, resulting in failure to produce mature EGFP mRNA and protein (33). We synthesized PPMO654 (Supplementary Table S1), a SSO designed to correct the splicing defect thus allowing EGFP production. HeLa EGFP654 cells were treated with PPMO654 in combination or in the absence of OEC UNC7938 or OEC UNC2383. As seen in Figure 1A increasing concentrations of PPMO alone led to a progressive increase in EGFP expression. The figure also shows that a 5 base mismatched PPMO was completely inactive. The effect of a fixed concentration of PPMO was substantially enhanced by increasing concentrations of OEC UNC7938 (Figure 1B, white circle symbols) or OEC UNC2383 (Figure 1C, D, white circle symbols). However, the presence of the OEC did not augment the effect of a mismatched PPMO (Figure 1B, white square symbols) and provided only a minor enhancement of the effect of PMO654, which lacks the peptide moiety (Figure 1B, white triangle symbols). As seen in Figure 1C. the concentration-effect profile of the PPMO was substantially enhanced in the presence of a fixed concentration of OEC, with the greatest enhancement seen at low PPMO concentrations. Viability/cytotoxicity analyzed concurrently with EGFP expression (see Methods) was not significantly affected within the range of concentration of PPMO and OEC utilized in these studies (Figure 1B, D, black square symbols).
Figure 1.

Effect and Toxicity of PPMOs and OECs in Cell Culture. (A–D) PMO, PPMO and OEC dose responses. (A) HeLaEGFP654 cells were incubated overnight with various concentrations of splice correcting PPMO654 or with a control PPMO having a 5 base mismatch (PPMO-MM). The cells were rinsed and further incubated and then processed for EGFP fluorescence and viability as described in Methods. (B) Cells were incubated with 0.5μM PPMO654, PPMO-MM or PMO654 and further incubated with various concentrations of OEC UNC7938. After rinsing and further incubation, EGFP fluorescence and viability were determined. (C) The concentration of PPMO654 was varied plus or minus subsequent treatment with 10 μM OEC UNC2383. (D) Subsequent to treatment with 0.5 μM PPMO654, cells were treated with various concentrations of OEC UNC2383. Data in A-D are expressed as mean ± SD, n = 4 (error bars fall within symbols). (E) PPMO intracellular distribution. HeLaEGFP654 cells were incubated with lissamine-PPMO654 overnight followed by vehicle or 10 μM OEC UNC7938 for 1 h and then cells were observed by microscopy. The upper images are phase contrast while the lower images are superposition of fluorescence from lissamine-PPMO (red) and Hoechst nuclear stain (blue). Lissamine-PPMO654 accumulated in endosomal compartments but after OEC treatment the PPMO re-localized into the nuclei, bar = 10 μm.
We also utilized a 3′-lissamine-labeled PPMO654 to image the intracellular distribution of PPMO in the presence or absence of an OEC (Figure 1E). As seen, OEC treatment caused a substantial re-distribution of the PPMO from cytosolic vesicles to the nucleus. Thus, despite major differences between the two types of molecules, the OECs act on cationic PPMOs in a manner similar to their previously observed effects on anionic oligonucleotides (32) and can promote release of PPMOs from endosomes, increase delivery to the nucleus, and substantially enhance splice correction in cultured cells.
The combination of PPMO and OEC substantially increases oligonucleotide delivery and activity in airway epithelial cells
We developed a new cell model of well-differentiated primary mouse tracheal epithelial cells (MTEC) derived from the splicing reporter mouse model (EGFP654 mouse, see below). The EGFP654 MTEC recapitulate airway epithelial features including mucus and cilia production and other physiological barriers to oligonucleotide delivery. Splicing in the EGFP654 mouse-derived cells can be corrected using PPMO654, the same PPMO as used in the HeLa EGFP654 cells. Thus, prior to in vivo testing, we utilized EGFP654 MTEC to evaluate certain key issues of oligonucleotide delivery in a physiologically-relevant model of airway epithelia.
The cells were differentiated on permeable supports at air-liquid interface to mimic the in vivo situation. We then examined treatment via the basolateral side, thus mimicking systemic administration, or via the apical side mimicking intra-pulmonary administration. We also examined the effect of apical mucus on administration by the apical route. As seen in Figure 2A through D, the OEC substantially enhanced the splice-correcting effect of the PPMO when both were administered either by the apical or basolateral routes. As in the case of the HeLa EFGP654 model of Figure 1C, the effect of the OEC was greater at lower concentrations of PPMO. Significant enhancement of EGFP expression was observed with as little as 0.3 μM PPMO in the presence of the OEC. The basolateral route usually produced a stronger response (>70% MTEC expressed EGFP 48hs post-treatment). The difference in the effects of apical and basolateral administration is to be expected based on the fact that these distinct membrane domains have different plasma membrane surface area and patterns of endocytosis (41–43). Moreover, since the exact basolateral surface area is difficult to calculate it is thus difficult to administer the same dose of drug/cell membrane area to the apical and basolateral sides.
Figure 2.
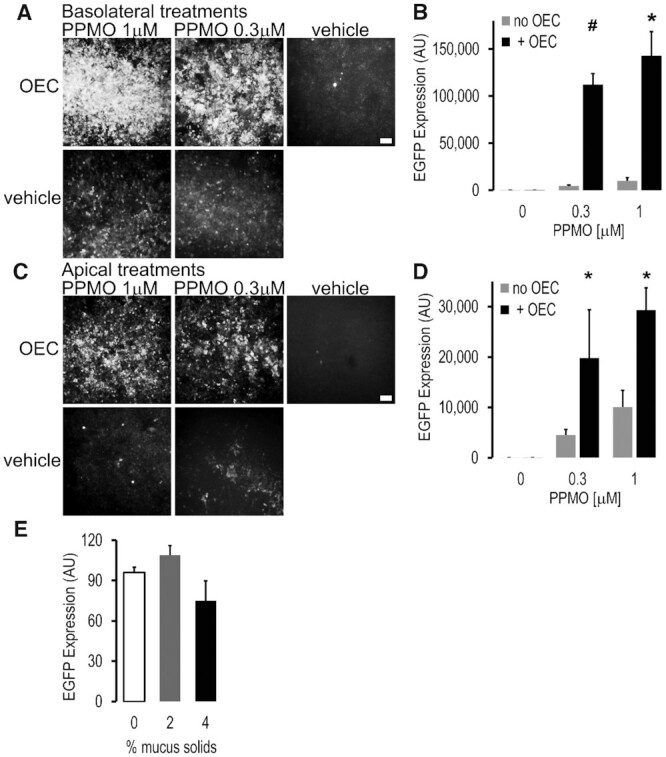
Correction of a splicing mutation in mouse-derived primary airway cells using PPMO plus OEC. Primary mouse tracheal epithelial cells derived from the EGFP654 mouse (EGFP654 MTEC) were differentiated on permeable supports under air-liquid interface for 2–3 weeks as described in Methods. (A–D) PPMO plus OEC effects. The SSO PPMO654 and OEC7938 were administered once in a bolus either into the basolateral (A, B) or apical (C, D) compartments; PPMO (0.3 and 1 μM, 16 h) followed by OEC7938 (10 μM, 2 h). MTEC were subjected to microscopy and fluorometric analysis 48 h post-treatment. Live MTEC were imaged with constant settings in a Leica DMRB fluorescent microscope using a 10x magnification, bar = 100 μm (A, C), and quantitated in a Azure fluorimeter scanner (B, D). All wells have similar total cell numbers; mean ± SD; n = 3; *P < 0.01 and #P < 0.05 versus no OEC. (E) Effect of apical mucus on PPMO plus OEC delivery in EGFP654 MTEC. PPMO (0.5 μM) and OEC (10 μM) were administered apically mixed in a small (30ul) bolus of human mucus at 2 and 4% solids’ concentration; 0 = cell medium bolus; other experimental conditions were as before. Fluorescence was quantified in a Tecan system with a control set at 100%; mean ± SD, n = 4. Lumenal mucus did not greatly affect delivery of PPMO and OEC to the airway epithelial cells and thus splicing correction and EGFP expression was observed.
In similar experiments, the corrective effect of PPMO plus OEC administration was examined at longer times after a single treatment. Thus EGFP fluorescence persisted after 14 and 22 days post treatment, and was higher for the PPMO plus OEC combination versus the PPMO alone at all points examined (Supplementary Figure S1).
Since over-production of airway mucus is an issue in many lung diseases such as CF (5,6) we also examined the effect of human airway mucus on apical delivery of PPMO654. We used exogenous mucus to control the concentration in a pathophysiologically relevant manner. As seen in Figure 2E, mucus concentrations up to 4% solids [typical of CF (5,6)] had little deleterious effect on EGFP induction when PPMO and OEC were applied via the apical route. Thus onetime treatment with the PPMO plus OEC combination is highly effective in splice correction even in airway disease-mimicking conditions.
The combination of PPMO and OEC increases oligonucleotide delivery and activity in murine lung
Currently there is no animal model for CFTR splicing mutations to test our oligonucleotide strategy. However, we have available a valuable animal model for studying the delivery of SSOs to CF-relevant tissues. The EGFP654 transgenic mouse incorporates an EGFP reporter whose coding sequence is interrupted by an intron that is aberrantly spliced resulting in failure to produce mature EGFP mRNA and protein. However, successful in vivo delivery of an appropriate SSO will correct splicing leading to restoration of message and protein expression in the lung and other tissues (19,32,37). This mouse model allows both quantitation through measurement of tissue RNA levels and cell-level topography through fluorescence microscopy of tissue sections.
Based on the abovementioned studies with mouse tracheal epithelial cells (EGFP654 MTEC), we tested the PPMO654 plus OEC combination in EGFP654 mice. We decided to focus on systemic administration based on the results with the MTEC suggesting that the basolateral route, which mimics systemic delivery, was very effective. Based on our earlier experience (19,32), our initial studies in mice involved pre-administration of PPMO654 followed several hours later by administration of the OEC. At intervals thereafter mice were euthanized and lung tissues evaluated for splice correction of EGFP mRNA by RT-PCR and/or for expression of EGFP by fluorescence microscopy.
As seen in Figure 3A and E, in samples obtained 48h after in vivo treatment, PPMO654 alone caused low level correction of mRNA splicing and EGFP expression in the lung, while the addition of OEC UNC7938 substantially increased these effects. Thus the in vivo effect generally paralleled that seen in cell culture (Figures 1 and 2, above). Immunostaining of EGFP and confocal microscopy analyses showed that significant EGFP expression was observed in several epithelial cell types including ciliated cells, secretory cells and basal cells (e.g. stem cells) (Figure 3B, C). Cells in the epithelial layer express EGFP at different levels in an apparent random and mosaic-like pattern (Figure 3B). Other cell types showed EGFP expression such as alveolar cells (Figure 3A) and cells in the muscle layer in the bronchi (Figure 3D).
Figure 3.
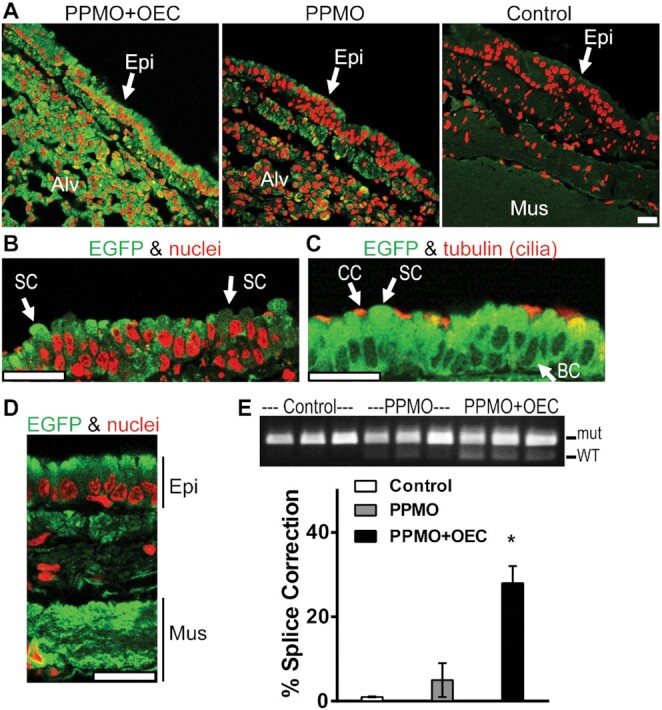
PPMO plus OEC effects on EGFP expression in murine lung. EGFP654 mice received PPMO654 at 12 mg/kg IV on three successive days and thereafter one cohort received OEC (UNC7938) IV at 15 mg/kg once (n = 3–4 per group). After 48 h, mice were euthanized, tissues recovered and analyzed by immunostaining with antibodies to EGFP and confocal microscopy; RT-PCR analysis was performed from rapidly frozen tissues. (A–D) Lung distribution of EGFP expression. (A) Images of EGFP immunostaining (green) and nuclear staining (red, propidium iodide) of fixed lung sections from mice treated with PPMO plus OEC, PPMO only, or vehicle; images were acquired with constant settings in a Leica SP5 confocal microscope with a 63× immersion lens. In PPMO plus OEC-treated mice, EGFP expression levels were elevated in the cells of the surface epithelium (Epi) and in the alveolar cells (Alv) compared to PPMO controls. (B) High magnification, low intensity image from PPMO plus OEC cohort to reveal EGFP expression patterns in cells displaying the morphology of secretory cells (SC). (C) Lung sections were co-stained for EGFP and for cilia with tubulin antibodies (red) to identify ciliated cells (CC) as well as cells displaying the morphology of secretory cells (SC) and basal cells (BC). (D) Image from PPMO plus OEC cohort to reveal EGFP expression in the airway muscle layer (Mus) underlying the surface epithelial cell layer (Epi). All bars = 20 μm. (E) PCR analysis. RT-PCR gel at 48 h post-treatment (top); WT designates the splice corrected form of EGFP RNA while mut is the splice mutant. The band intensity quantification performed as described in Materials and Methods is shown at the bottom; *P < 0.01 versus no OEC.
It was also possible to directly visualize EGFP fluorescence in freshly excised, unfixed organs (Supplementary Figure S2). Clinical chemistry data from blood samples from this experiment showed no significant changes in liver enzymes or kidney function (Supplementary Table S2) which were within normal published values (44). Mouse behavior appeared normal during the experiment as did post-euthanasia lung morphology.
The results of a similar in vivo experiment studying the duration of the splice correction effect after a single treatment with PPMO and OEC are shown in Figure 4. Substantial splice correction was seen not only at 2 days but also at 7 days and 22 days post-treatment (4A) indicating that the oligonucleotide effect was of long duration not only in vitro but also in vivo. Quantitation of the RT-PCR results is provided in 4B showing >70% splice correction at 48h with PPMO plus OEC and ∼40% after 7 days. We currently do not have an explanation for the apparent differences in RNA correction efficiency at 48 h post-treatment between experiments in Figures 3 and 4. Further in vivo pharmacodynamic studies are required to address the source of these differences.
Figure 4.
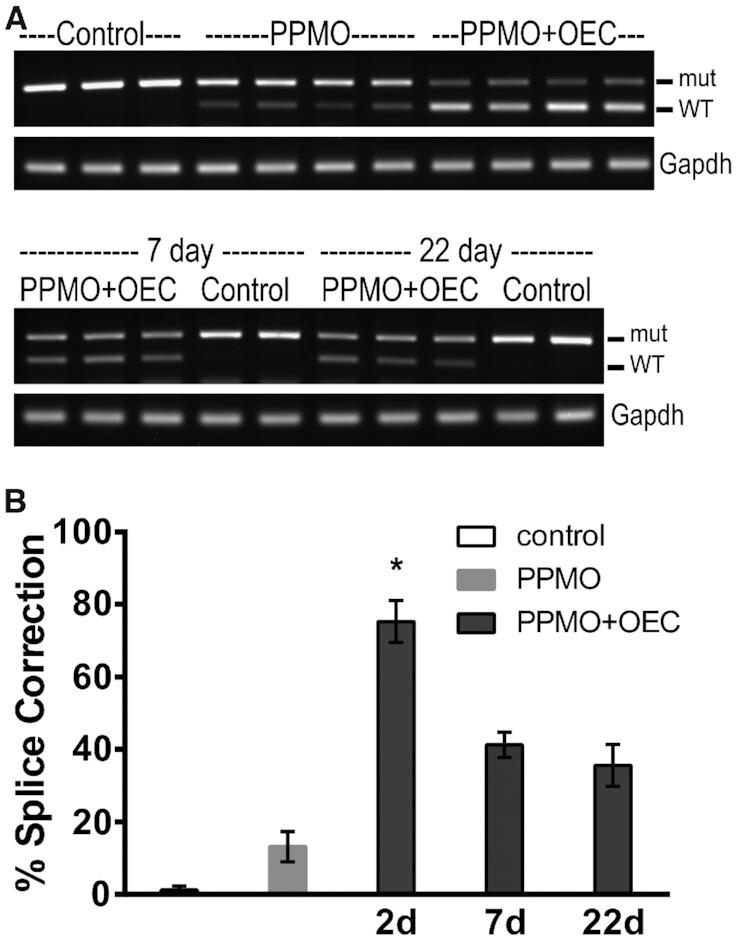
Extended splice correction effect of PPMO plus OEC in murine lung. EGFP654 mice were retained as controls or received PPMO654 at 5 mg/kg IV on two successive days and thereafter one cohort received OEC (UNC7938) IV at 15 mg/kg once (n = 3–4 per group). After 2, 7 or 22 days mice were euthanized, lung tissues recovered and analyzed by RT-PCR as described in Methods. (A) PCR Gel analysis at 48 h (top) and 7 and 22 days. (B) Quantitation of splice correction as described in Methods; shown vehicle and PPMO at 48 h; *P < 0.01 versus no OEC.
Experiments illustrated in Figures 3 and 4 indicate that in vivo administration of the PPMO SSO plus OEC combination provides a robust and durable correction of splicing in relevant lung epithelial cell types without detectable systemic toxicity.
Co-administration of PPMO and OECs is effective in the lung
In our work to date we typically pre-loaded the endosomal compartment with SSO and then triggered endosomal release with the OEC (19,32). However, this approach might be inconvenient for future therapeutic development. Thus, we explored the feasibility of co-administration of the SSO and OEC, both in cell culture and in vivo. As seen in Figure 5A, B, both sequential and co-administration of PPMO plus OEC to HeLa and MTEC cell models produced comparable increases in EGFP expression indicating that the co-administration approach is effective in these cells. In most experiments, co-administration attained maximal effects by 4 to 6 h after treatment. Both co-administration and sequential treatments affected airway epithelial cells in a similar pattern. Thus the confocal microscopy xy and xz scan sections in Figure 5B illustrate that both ciliated and non-ciliated airway epithelial cells expressed EGFP after either treatment. As expected, different levels of EGFP expression were observed across the epithelial cell layer in a seemingly random and mosaic pattern.
Figure 5.
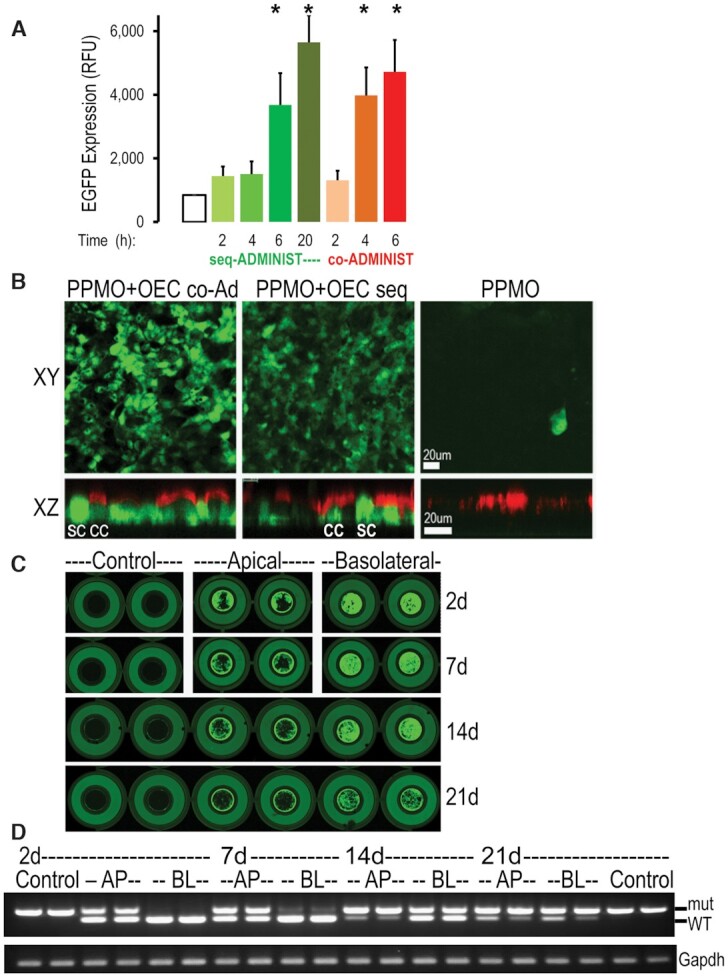
Co-administration of PPMO and OEC in epithelial cells. (A, B) Efficacy of co-administration. Cells were treated with PPMO654 (1 μM) plus OEC (10 μM) by either sequential or co-administration. (A) EGFP expression quantification in HeLa EGFP654 cells incubated for 2–20 h; n = 3, *P < 0.01 versus control; 4 or 6 h co-administration and 6 or 20 h sequential are not significantly different. (B) Well-differentiated primary EGFP654 MTEC (see Methods) were imaged live by confocal microscopy (top, xy and bottom, xz confocal planes) showing that 20 h sequential and 6 h co-administration treatments were effective in eliciting correction and expression of EGFP in both ciliated epithelial cells (CC, red cilia stained with Cell Mask®) and non-ciliated secretory cells (SC). Note: expression of EGFP at different levels in a random pattern; bar = 20 μm. (C, D) Extended splice correction effect of PPMO plus OEC in EGFP654 MTEC. Well-differentiated cells were treated once with PPMO plus OEC by co-administration for 6 h as above. (C) Fluorescence in live cultures was measured 2d, 7, 14 and 21 days post-treatment in an Azure Scanner System using identical parameters in all the scanning sessions (intensity, resolution, and height scan position) (n = 4). Note: the outer circle is the holding well (no cells) while the inner circle (6.5 mm diameter) is the permeable insert containing the live MTEC; cells were 100% confluent and well differentiated at all time points and exhibited a mosaic pattern of EGFP expression levels. (D) Parallel cultures were subjected to RNA isolation and RT-PCR analysis as before (n = 2). Co-administration of PPMO and OEC via the basolateral (BL) and apical (AP) sides was efficient and had extended effects in correcting the EGFP splicing defect in airway epithelial cell cultures.
In EGFP654 MTEC, the co-administration of PPMO plus OEC (Figure 5C, D) had prolonged effects as was the case with the sequential administration of both components (see Supplementary Figure S1). Thus, significant EGFP expression (5C) and splice correction (5D) were observed at 2, 7, 14 and 21 days after a single co-administration of PPMO654 plus OEC.
Next, we tested the co-administration strategy in vivo in EGFP654 mice. Figure 6 demonstrates that systemic co-administration of PPMO654 plus OEC resulted in strong splice correction in the lung at 48h, comparable in magnitude to sequential administration. Co-administration of PPMO654 was performed using two different OECs (UNC7938, UNC2383) with similar results. Clinical chemistry data from this experiment showed no indication of significant impairment of liver or kidney function (Supplementary Table S3). In a separate experiment we also measured clinical chemistry parameters 6 days after treatment by co-administration (Supplementary Table S4). Although there was considerable variation in the parameters tested, the values for the three toxicity evaluations (Supplementary Tables S2–S4) were all within normal ranges for several murine strains (44).
Figure 6.
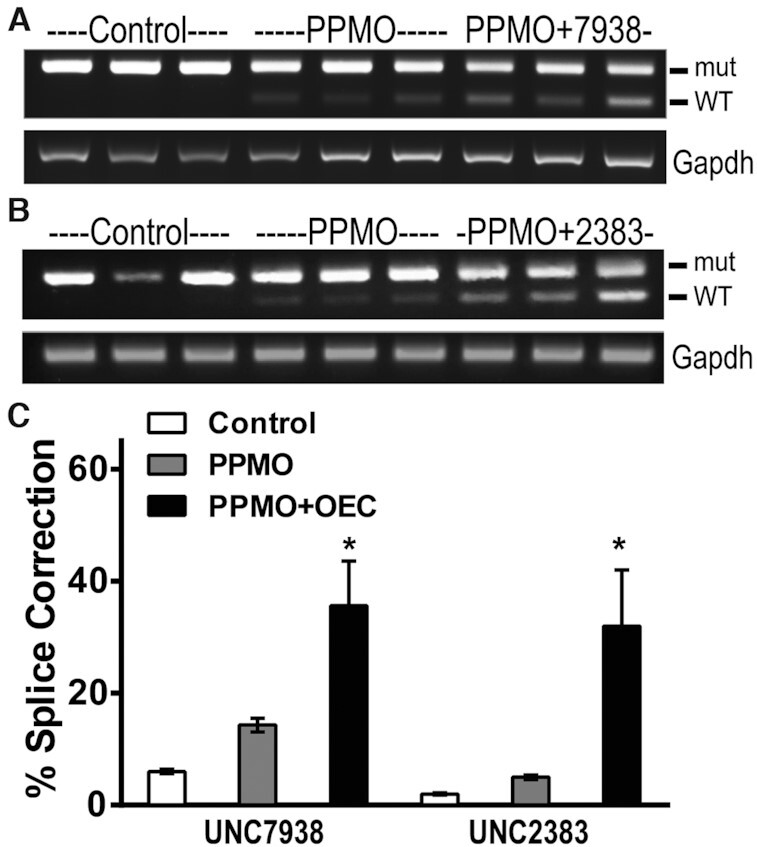
In vivo co-administration of PPMO and OEC. EGFP654 mice received a single IV co-injection of PPMO654 at 7.5 mg/kg plus OEC UNC7938 or UNC2383 at 15 and 5 mg/kg respectively (n = 3-4 per group). After 48 h, mice were euthanized, and lung tissues recovered and analyzed by RT-PCR as before. (A, B) RT-PCR gel of lung tissues at 48 h post-treatment. (C) Lung RT-PCR band quantification, *P < 0.02 versus no OEC. Co-administration of PPMO and OEC is efficient in correcting the splicing defect in EGFP in vivo in lung epithelia.
In this report, we have focused on the correction of splicing in the airway epithelia and lung. However, extra-pulmonary organs were also corrected in vivo. Both sequential (Supplementary Figure S2) and co-administration (Supplementary Figure S3) of PPMO654 and OEC (UNC7938 or UNC2383) produced significant splice correction in kidney at 48 h post-treatment. Therefore either sequential or co-administration strategies are possible with PPMOs and OECs, allowing a flexible approach to possible therapeutic use.
Combination of PPMO and OEC efficiently corrects CFTR activity in cystic fibrosis patient-derived airway epithelial cells
Studies in the EGFP654 mice indicated that systemic treatment with a PPMO plus OEC combination can attain substantial splice correction in CF–relevant tissues such as the lung. However, it is essential to demonstrate that this approach is effective in cells from cystic fibrosis (CF) patients. Well-differentiated, primary human bronchial epithelial cells (HBEC) provide the most physiologically relevant (pre-clinical) model currently available. Thus we synthesized PPMO3849 (Supplementary Table S1) to correct the 3849+10 kb C→T splicing defect in CF transmembrane conductance regulator (CFTR) RNA and tested it in patient-derived HBEC homozygous for that mutation. HBEC differentiated on permeable supports at air-liquid interfaces (see Methods) were treated with naked PPMO plus OEC and/or with a Trikafta®-like CFTR modulator combination. PPMO3849 and OEC were added through the apical or basolateral sides. CFTR ion channel activity was measured via Ussing chamber analysis, while correction of CFTR RNA splicing was examined by RT-PCR (see Materials and Methods). The Ussing chamber analysis of Figure 7A demonstrates that PPMO plus OEC treatment resulted in significant restoration of CFTR ion transport activity in the CF cells to levels comparable to wild type CFTR (Supplementary Figure S4). Thus, the post-amiloride forskolin peak was >10 and >6 times higher for the basolateral and apical PPMO plus OEC treatments compared to vehicle controls. Importantly, the triple combination of CFTR modulators (in the clinic, Trikafta®) failed to correct these cells at a dose known to be effective in correcting cells with the frequent delF508 mutation (34), presumably because of the lack of protein production in the 3849+10 kb C→T cells. PPMO plus OEC treatments did not affect ciliary beating or cell morphology (ad-hoc observations), or transepithelial resistances (TER, Supplementary Figure S4) an index of epithelial integrity and absence of cytotoxic effects. In these studies, CFTR modulators did not have any additional effect over PPMO3849 plus OEC treatment.
Figure 7.
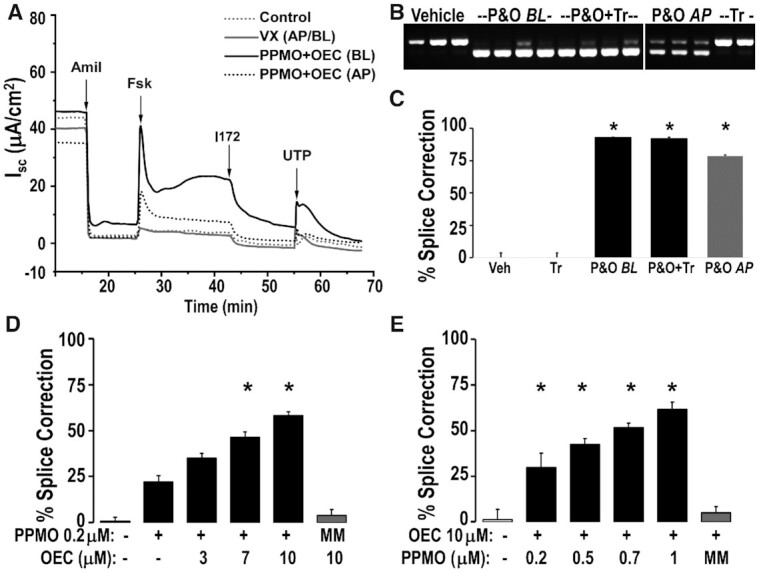
CFTR activity restored using PPMO plus OEC but not a CFTR modulator combination (Trikafta®) in HBEC from a 3849+10 kb C→T homozygous CF patient. Patient-derived HBEC passage 2 differentiated on permeable supports and under air-liquid interface for 21 days were treated with PPMO3849 (20hs, 1 μM) and OEC UNC7938 (2 h, 10 μM). PPMO & OEC (P&O) were added either basolateral (BL, 500 μl) or apical (AP, 30μl); vehicle or a combination of VX-445 2μM, VX-661 3 μM, VX-770 1 μM (similar to Trikafta®, Tr, 48 h) was added basolateral + apically; see Methods. (A) CFTR functional correction by Ussing chamber analysis. Representative trace diagram of short circuit currents (Isc) versus time is shown, see Methods for details; n = 4; Amil = Amiloride, Fsk = forskolin, I172 = CFTR inhibitor 172, UTP = uridine triphosphate. (B, C) RT-PCR analysis. HBEC were subjected to RNA analysis after Ussing chamber studies; RT-PCR gel (B) and gel band quantification (C); n = 3–4, * = P < 0.01 versus vehicle; the lower band is the correctly spliced CFTR (WT). Significant CFTR correction was observed in response to PPMO plus OEC treatment. (D, E) OEC and PPMO dose–responses in patient-derived cells. Patient-derived UNCCF8T cells differentiated on permeable supports under air-liquid interfaces for 2 weeks were treated once with PPMO3849 in combination with OEC co-administered basolaterally for 6h at the concentrations indicated in the figures. PPMO654 (MM) was used as a non-targeted control at 0.2 μM (D) and 1 μM (E). RT-PCR analysis and quantification was performed 48 h post-treatment as described above; n = 4; *P < 0.05 versus no OEC (D) and *P < 0.02 versus vehicle (E).
As seen in Figure 7B and C restored CFTR function was paralleled by substantial correction of the splicing defect at the mRNA level. Thus, basolateral and apical PPMO plus OEC treatments elicited ∼93% and ∼78% mRNA correction, respectively (vehicle and CFTR modulator treated cells displayed 0.1–4.1% mRNA correction). Basolateral treatment appeared more efficacious than apical addition, consistent with the EGFP654 MTEC experiments (Figure 2, above). Thus, a single treatment of the PPMO3849 plus OEC combination can provide correction of a splicing defect in human cells that are largely refractory to CFTR modulator drugs.
In further studies we used the growth-enhanced patient-derived cell line UNCCF8T homozygous for the 3849+10 kb C→T mutation to explore dose-response relationships for the PPMO plus OEC combination (details for the creation of the UNCCF8T cells are depicted in Supplementary Figure S5). As seen in Figure 7D there was a progressive increase in the degree of splice correction as the OEC concentration was increased from zero to 10 μM at fixed PPMO concentration (PP, 0.2 μM). The process was highly selective in that the correction effect was PPMO3849 dose dependent (Figure 7E) while a PPMO that is irrelevant to the 3849+10 kb C→T mutation (MM) had no effect even in the presence of 10 μM OEC (Figure 7D, E). Thus the PPMO plus OEC approach can effectively correct RNA splicing and restore CFTR ion channel activity in primary cultures of human airway cells that currently are the best pre-clinical model available for CF splicing mutations.
DISCUSSION
The development of small molecules that modulate the function of the cystic fibrosis transmembrane conductance regulator (CFTR) protein has dramatically changed the therapeutic landscape for cystic fibrosis (CF) (8,12,45–48). However, there remain cohorts of patients who fail to express significant levels of CFTR protein and who are thus refractory to the modulator drugs. These cohorts include patients with mutations that produce premature termination codons or severe splicing defects. In the case of splicing errors, it has been clear for some time that these could potentially be corrected with splice switching oligonucleotides (SSOs). Recent work with SSOs in cellular models of CF splicing mutations have reinforced this possibility (14,25,26). However progress in converting this concept to a therapeutic approach has been slow. A major challenge to the use of SSOs to treat CF is in vivo delivery to extra-hepatic tissues. The SSO must effectively reach CF-relevant tissues and once there it must overcome the endosomal escape barrier that constrains all oligonucleotide therapeutics (49). In this report we describe a novel approach to this problem using a combination of peptide-morpholino oligomer conjugates (PPMOs) and small molecules (OECs) that promote endosomal release. Therapeutic use of PPMOs has been extensively explored in the context of animal models including those for Duchene muscular dystrophy, myotonic dystrophy and spinal muscular atrophy where they have been found to be effective and well tolerated (23,28,30,50–52). PPMOs have a broad biodistribution including to CF-relevant tissues such as lung as our data and that of others (28,29) suggest. Although PPMOs have some innate endosome escape capability, our work indicates that this can be substantially enhanced via use of OECs.
We have demonstrated that systemic administration of a PPMO SSOs plus an OEC can attain substantial in vivo correction of splicing in the lung of the EGFP654 mouse model. This was achieved without indication of significant toxicity and was sustained for at least three weeks after a single treatment. Importantly, by co-administration of the PPMO and OEC we attained effects comparable to our established sequential administration approach. Immunochemical studies showed that all of the major cell types of the lung responded to the treatment with induction of EGFP. Additionally, this approach also allowed splice correction in other CF disease-relevant issues such as kidney.
We tested our oligonucleotide approach in patient-derived bronchial epithelial cells (HBEC) that have the 3849+10 kb C→T mutation since this is the most frequent splicing mutation in CF (10). We tested cells derived directly from a patient (HBEC) as well as our novel growth-enhanced UNCCF8T cells generated from patient cells. We cultured the cells in the most stringent conditions to reduce epithelial permeability to oligonucleotides and we did not enhance CFTR expression with medium additives. Both cell models receiving a single treatment with the PPMO SSO plus OEC combination displayed substantial correction of aberrant RNA splicing (∼93% and ∼78% for basolateral and apical treatments, respectively). Furthermore, PPMO plus OEC treatment elicited the production of fully functional CFTR with ion channel activity in the treated CF HBEC cells comparable to that found in normal cells. Transepithelial resistances were not significantly affected by PPMO/OEC treatments suggesting a lack of cytotoxicity issues.
Recently, Michaels et al (26) showed correction of the 3849+10 kb C→T CFTR splicing mutation with a morpholino oligomer. A 3-fold increase in CFTR channel activity was reported using prolonged PMO doses that were >40 times higher than the dose of PPMO used here; only after 16 days of high dose, continuous PMO treatment did the CFTR activity reach values found in normal cells. In contrast, our present studies showed that one treatment with a relatively low dose of peptide-conjugated PMO plus an OEC produced CFTR activity in the wild type range.
Our data show, as also reported by Michaels et al. (26), that current CF modulator drugs that act at the protein level have minimal effects in patient-derived cells with the 3849+10 kb C→T splicing mutation, while SSOs can restore CFTR function. Thus SSOs may offer a major advantage for some patients with this mutation. However, PMOs cannot be translated into lung therapeutics without addressing their poor delivery in tissues and cells. In this work, we harness the advantages of morpholino oligomers (stability and lack of toxicity), by conjugating the PMO with a peptide and administering it with a small molecule to overcome tissue and cell distribution and endosomal escape. To summarize our results, the PPMO plus OEC delivery strategy proved effective to correct splicing defects in lung epithelia in vivo using an informative animal model, as well as in the most physiologically relevant cell model for CF splicing studies.
It should be noted that the PPMO plus OEC combination described here is certainly not the only oligonucleotide-based approach for pulmonary diseases. Thus, there have been numerous studies directed at various molecular targets in the lung, many of which utilized delivery of oligonucleotides via the airways (53). A particularly interesting approach from investigators at Ionis Pharmaceuticals addressed CF using antisense oligonucleotides that target ENaC, an epithelial sodium channel that is involved in CF pathogenesis (54,55). Aerosol delivery was used so as to attain a localized pulmonary effect and to minimize deleterious effects of the ENaC ASOs in other organs such as kidney. However, CF is a multi-organ disease and thus the aerosol delivery approach may not address all aspects of CF pathogenesis, whereas the PPMO plus OEC approach can attain systemic effects. Thus, both the ENaC ASOs and the PPMO plus OEC approach may be of value depending on the therapeutic context.
The current studies potentially provide a foundation for future clinical development of the PPMO plus OEC approach for correction of splicing mutations in CF and other lung diseases. However, some issues remain to be addressed. Since these are prototype molecules, we are also pursuing medicinal chemistry studies for the OECs and the peptide component of the PPMO. Because the OEC and PPMO are chemically distinct they are likely to have distinct pharmacokinetic, pharmacodynamic and toxicity profiles. This suggests that it may be possible to optimize the therapeutic window by altering the ratio of the two components, similar to other combination therapies with two or three different CFTR modulators (e.g. Orkambi®, Trikafta®). Thus, further development of the PPMO plus OEC strategy will require a fuller understanding of dose-response relationships as well as detailed investigation of both the therapeutic and safety consequences of multiple administrations and long-term use. Additionally, we are exploring the obvious possibility of intra-pulmonary administration of the PPMO plus OEC combination. Our in vitro studies in patient-derived cells and mouse-derived tracheal cells are very encouraging showing that the apical delivery of both PPMO and OEC is effective in correcting splicing defects. Moreover, conditions of concentrated mucus mimicking the CF milieu in the lumen of airway epithelial cultures did not greatly affect the delivery of PPMOs and OECs. Similar data was described for other oligonucleotide chemistries in lung epithelial (56). Because CF is a systemic disease that affects multiple organs, systemic administration may be most beneficial. Daily dosing of current CFTR modulators have beneficial systemic effects and importantly in the maintanance/improvement of BMI (8,45–48,57–60). However, airway administration might blunt possible systemic toxicities of the PPMO plus OEC, as well as focusing their therapeutic effects in the lung of individuals with CF or other severe pulmonary disease, and thus this approach should be explored. Despite these complexities, both the cell-based and the in vivo results presented here suggest that the PPMO SSO plus OEC combination may have substantial potential as a therapy for severe splicing mutations in CF as well as in other diseases that may be addressed by control of splicing (61,62).
DATA AVAILABILITY
There are no on-line data files associated with this manuscript. Access to the original data is available by request.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Lisa Morton and Dr Wanda O’Neal for their help in optimizing mRNA isolations, Dr Barbara Grubb and Troy Rogers for their help in the initial measuring of CFTR activity, Leslie Fulcher for her advice in optimizing HBEC culture conditions, and Deanna Clemmer, Katherine Patton and Elisabeth Burnor for their help in mouse work and culturing MTEC. We are very grateful to Dr Brian Button for his kind gift of human mucus samples and the access to his microscopy and scanning equipment. We would like to thank Drs Jon Moulton and James Summerton from GENE TOOLS for their help in the design and synthesis of the PMOs. We would like to acknowledge Dr Jianwen Que, who provided the backbone for the lentivirus to create the UNCCF8T cells. We are grateful to Dr. Richard Boucher for his support. We are humbled and grateful to the patients who donated their airway epithelial cells and the patient from whom HeLa cells were isolated. Animals were cared for by the UNC Lineberger Center Animal Studies Core. Histology specimens have been processed in the UNC Lineberger Center Animal Histopathology Core. Blood analyses were performed in the UNC Animal Histopathology and Laboratory Medicine Core.
Contributor Information
Yan Dang, Marsico Lung Institute/Cystic Fibrosis Center, The University of North Carolina at Chapel Hill, 6009 Thurston Bowles Bldg, Chapel Hill NC 27599-7248, USA.
Catharina van Heusden, Marsico Lung Institute/Cystic Fibrosis Center, The University of North Carolina at Chapel Hill, 6009 Thurston Bowles Bldg, Chapel Hill NC 27599-7248, USA.
Veronica Nickerson, Marsico Lung Institute/Cystic Fibrosis Center, The University of North Carolina at Chapel Hill, 6009 Thurston Bowles Bldg, Chapel Hill NC 27599-7248, USA.
Felicity Chung, Marsico Lung Institute/Cystic Fibrosis Center, The University of North Carolina at Chapel Hill, 6009 Thurston Bowles Bldg, Chapel Hill NC 27599-7248, USA.
Yang Wang, Marsico Lung Institute/Cystic Fibrosis Center, The University of North Carolina at Chapel Hill, 6009 Thurston Bowles Bldg, Chapel Hill NC 27599-7248, USA.
Nancy L Quinney, Marsico Lung Institute/Cystic Fibrosis Center, The University of North Carolina at Chapel Hill, 6009 Thurston Bowles Bldg, Chapel Hill NC 27599-7248, USA.
Martina Gentzsch, Marsico Lung Institute/Cystic Fibrosis Center, The University of North Carolina at Chapel Hill, 6009 Thurston Bowles Bldg, Chapel Hill NC 27599-7248, USA.
Scott H Randell, Marsico Lung Institute/Cystic Fibrosis Center, The University of North Carolina at Chapel Hill, 6009 Thurston Bowles Bldg, Chapel Hill NC 27599-7248, USA.
Hong M Moulton, Department of Biomedical Sciences, Carlson College of Veterinary Medicine, Oregon State University, Corvallis, OR 97331, USA.
Ryszard Kole, Department of Pharmacology, The University of North Carolina at Chapel Hill, 4010 Genetic Medicine Bldg, Chapel Hill, NC 27599, USA.
Aiguo Ni, Initos Pharmaceuticals, LLC, Chapel Hill, NC 27514, USA.
Rudolph L Juliano, Initos Pharmaceuticals, LLC, Chapel Hill, NC 27514, USA.
Silvia M Kreda, Marsico Lung Institute/Cystic Fibrosis Center, The University of North Carolina at Chapel Hill, 6009 Thurston Bowles Bldg, Chapel Hill NC 27599-7248, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Cystic Fibrosis Foundation [KREDA16G0, KREDA19I0, KREDA20G0 to S.M.K., Y.D., C.vH., V.N., F.C.]; National Institutes of Health (Small Business Technology Transfer NIH [1 R41 TR002692-01 to R.L.J., S.M.K., Y.D., V.N.]; Small Business Innovation Research [R44 TR002692-02 to R.L.J., S.M.K., Y.D., C.V.H., H.M.]; UNC-CF Center Cores (to S.H.R., Y.W., M.G., N.Q.) were partially supported by grants from the Cystic Fibrosis Foundation [BOUCHE19R0]; National Institutes of Health [P30DK065988]; UNC Lineberger Center Animal Studies Core was supported in part by a National Cancer Institute Center Core Support Grant [NIH CA16086]. Funding for open access charge: NIH [R44 TR002692-02].
Conflict of interest statement. S.M.K., R.K. and R.L.J. have ownership interests in Initos Pharmaceuticals LLC a company that has licensed the OEC compounds from the University of North Carolina.
REFERENCES
- 1. Viegi G., Maio S., Fasola S., Baldacci S.. Global burden of chronic respiratory diseases. J. Aerosol. Med. Pulm. Drug Deliv. 2020; 33:171–177. [DOI] [PubMed] [Google Scholar]
- 2. Cutting G.R. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat. Rev. Genet. 2015; 16:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kreda S.M., Mall M., Mengos A., Rochelle L., Yankaskas J., Riordan J.R., Boucher R.C.. Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol. Biol. Cell. 2005; 16:2154–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boucher R.C. Muco-Obstructive lung diseases. N. Engl. J. Med. 2019; 380:1941–1953. [DOI] [PubMed] [Google Scholar]
- 5. Anderson W.H., Coakley R.D., Button B., Henderson A.G., Zeman K.L., Alexis N.E., Peden D.B., Lazarowski E.R., Davis C.W., Bailey S.et al.. The relationship of mucus concentration (Hydration) to mucus osmotic pressure and transport in chronic bronchitis. Am. J. Respir. Crit. Care Med. 2015; 192:182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Button B., Anderson W.H., Boucher R.C.. Mucus hyperconcentration as a unifying aspect of the chronic bronchitic phenotype. Ann. Am. Thorac. Soc. 2016; 13(Suppl. 2):S156–S162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gentzsch M., Mall M.A.. Ion channel modulators in cystic fibrosis. Chest. 2018; 154:383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guimbellot J., Sharma J., Rowe S.M.. Toward inclusive therapy with CFTR modulators: progress and challenges. Pediatr. Pulmonol. 2017; 52:S4–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bear C. A therapy for most with cystic fibrosis. Cell. 2020; 180:211. [DOI] [PubMed] [Google Scholar]
- 10. Cystic Fibrosis Centre at the Hospital for Sick Children in Toronto 2011; https://www.sickkids.ca/en/care-services/centres/cystic-fibrosis-centre/.
- 11. Cystic Fibrosis Foundation News: CFTR modulators 2020; https://www.cff.org/Research/Developing-New-Treatments/CFTR-Modulator-Types/.
- 12. Kerem E., Cohen-Cymberknoh M., Tsabari R., Wilschanski M., Reiter J., Shoseyov D., Gileles-Hillel A., Pugatsch T., Davies J.C., Short C.et al.. Ivacaftor in people with cystic fibrosis and a 3849+10kb C →T or D1152H residual function mutation. Ann. Am. Thorac. Soc. 2020; 18:433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Castellani C., Cuppens H., Macek M. Jr, Cassiman J.J., Kerem E., Durie P., Tullis E., Assael B.M., Bombieri C., Brown A.et al.. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 2008; 7:179–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oren Y.S., Pranke I.M., Kerem B., Sermet-Gaudelus I.. The suppression of premature termination codons and the repair of splicing mutations in CFTR. Curr. Opin. Pharmacol. 2017; 34:125–131. [DOI] [PubMed] [Google Scholar]
- 15. Bennett C.F., Baker B.F., Pham N., Swayze E., Geary R.S.. Pharmacology of antisense drugs. Annu. Rev. Pharmacol. Toxicol. 2017; 57:81–105. [DOI] [PubMed] [Google Scholar]
- 16. Kole R., Krainer A.R., Altman S.. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012; 11:125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martinovich K.M., Shaw N.C., Kicic A., Schultz A., Fletcher S., Wilton S.D., Stick S.M.J.M., Pediatrics C. The potential of antisense oligonucleotide therapies for inherited childhood lung diseases. Mol. Cell Pediatr. 2018; 5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Friedman K.J., Kole J., Cohn J.A., Knowles M.R., Silverman L.M., Kole R.. Correction of aberrant splicing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by antisense oligonucleotides. J. Biol. Chem. 1999; 274:36193–36199. [DOI] [PubMed] [Google Scholar]
- 19. Wang L., Ariyarathna Y., Ming X., Yang B., James L.I., Kreda S.M., Porter M., Janzen W., Juliano R.L.. A novel family of small molecules that enhance the intracellular delivery and pharmacological effectiveness of antisense and splice switching oligonucleotides. ACS Chem. Biol. 2017; 12:1999–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Havens M.A., Hastings M.L.. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016; 44:6549–6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shen X., Corey D.R.. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018; 46:1584–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Summerton J.E. Invention and early history of morpholinos: from pipe dream to practical products. Methods Mol. Biol. 2017; 1565:1–15. [DOI] [PubMed] [Google Scholar]
- 23. Erdos M.R., Cabral W.A., Tavarez U.L., Cao K., Gvozdenovic-Jeremic J., Narisu N., Zerfas P.M., Crumley S., Boku Y., Hanson G.et al.. A targeted antisense therapeutic approach for Hutchinson-Gilford progeria syndrome. Nat. Med. 2021; 27:536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ruger J., Ioannou S., Castanotto D., Stein C.A.. Oligonucleotides to the (gene) rescue: FDA approvals 2017–2019. Trends Pharmacol. Sci. 2020; 41:27–41. [DOI] [PubMed] [Google Scholar]
- 25. Igreja S., Clarke L.A., Botelho H.M., Marques L., Amaral M.D.. Correction of a cystic fibrosis splicing mutation by antisense oligonucleotides. Hum. Mutat. 2016; 37:209–215. [DOI] [PubMed] [Google Scholar]
- 26. Michaels W.E., Bridges R.J., Hastings M.L.. Antisense oligonucleotide-mediated correction of CFTR splicing improves chloride secretion in cystic fibrosis patient-derived bronchial epithelial cells. Nucleic Acids Res. 2020; 48:7454–7467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kreda S.M., Pickles R.J., Lazarowski E.R., Boucher R.C.. G-protein-coupled receptors as targets for gene transfer vectors using natural small-molecule ligands. Nat. Biotechnol. 2000; 18:635–640. [DOI] [PubMed] [Google Scholar]
- 28. Jearawiriyapaisarn N., Moulton H.M., Buckley B., Roberts J., Sazani P., Fucharoen S., Iversen P.L., Kole R.. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther. 2008; 16:1624–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amantana A., Moulton H.M., Cate M.L., Reddy M.T., Whitehead T., Hassinger J.N., Youngblood D.S., Iversen P.L.. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug. Chem. 2007; 18:1325–1331. [DOI] [PubMed] [Google Scholar]
- 30. Hammond S.M., Hazell G., Shabanpoor F., Saleh A.F., Bowerman M., Sleigh J.N., Meijboom K.E., Zhou H., Muntoni F., Talbot K.et al.. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. PNAS. 2016; 113:10962–10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Juliano R.L., Wang L., Tavares F., Brown E.G., James L., Ariyarathna Y., Ming X., Mao C., Suto M.. Structure-activity relationships and cellular mechanism of action of small molecules that enhance the delivery of oligonucleotides. Nucleic Acids Res. 2018; 46:1601–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang B., Ming X., Cao C., Laing B., Yuan A., Porter M.A., Hull-Ryde E.A., Maddry J., Suto M., Janzen W.P.et al.. High-throughput screening identifies small molecules that enhance the pharmacological effects of oligonucleotides. Nucleic Acids Res. 2015; 43:1987–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sazani P., Astriab-Fischer A., Kole R.. Effects of base modifications on antisense properties of 2′-O-methoxyethyl and PNA oligonucleotides. Antisense Nucleic Acid Drug Dev. 2003; 13:119–128. [DOI] [PubMed] [Google Scholar]
- 34. Gentzsch M., Boyles S.E., Cheluvaraju C., Chaudhry I.G., Quinney N.L., Cho C., Dang H., Liu X., Schlegel R., Randell S.H.. Pharmacological rescue of conditionally reprogrammed cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2017; 56:568–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fulcher M.L., Gabriel S.E., Olsen J.C., Tatreau J.R., Gentzsch M., Livanos E., Saavedra M.T., Salmon P., Randell S.H.. Novel human bronchial epithelial cell lines for cystic fibrosis research. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009; 296:L82–L91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. You Y., Brody S.L.. Culture and differentiation of mouse tracheal epithelial cells. Methods Mol. Biol. 2013; 945:123–143. [DOI] [PubMed] [Google Scholar]
- 37. Sazani P., Gemignani F., Kang S.H., Maier M.A., Manoharan M., Persmark M., Bortner D., Kole R.. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat. Biotechnol. 2002; 20:1228–1233. [DOI] [PubMed] [Google Scholar]
- 38. Kreda S.M., Gentzsch M.. Imaging CFTR protein localization in cultured cells and tissues. Methods Mol. Biol. 2011; 742:15–33. [DOI] [PubMed] [Google Scholar]
- 39. Cholon D.M., Quinney N.L., Fulcher M.L., Esther C.R. Jr, Das J., Dokholyan N.V., Randell S.H., Boucher R.C., Gentzsch M. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014; 6:246ra296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gentzsch M., Ren H.Y., Houck S.A., Quinney N.L., Cholon D.M., Sopha P., Chaudhry I.G., Das J., Dokholyan N.V., Randell S.H.et al.. Restoration of R117H CFTR folding and function in human airway cells through combination treatment with VX-809 and VX-770. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016; 311:L550–L559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sheff D.R., Daro E.A., Hull M., Mellman I.. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J. Cell Biol. 1999; 145:123–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Apodaca G. Endocytic traffic in polarized epithelial cells: role of the actin and microtubule cytoskeleton. Traffic. 2001; 2:149–159. [DOI] [PubMed] [Google Scholar]
- 43. Kreda S.M., Okada S.F., van Heusden C.A., O’Neal W., Gabriel S., Abdullah L., Davis C.W., Boucher R.C., Lazarowski E.R.. Coordinated release of nucleotides and mucin from human airway epithelial Calu-3 cells. J. Physiol. 2007; 584:245–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Serfilippi L.M., Pallman D.R., Russell B.. Serum clinical chemistry and hematology reference values in outbred stocks of albino mice from three commonly used vendors and two inbred strains of albino mice. Contemp. Top. Lab. Anim. Sci. 2003; 42:46–52. [PubMed] [Google Scholar]
- 45. Clancy J.P., Cotton C.U., Donaldson S.H., Solomon G.M., VanDevanter D.R., Boyle M.P., Gentzsch M., Nick J.A., Illek B., Wallenburg J.C.et al.. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019; 18:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Keating D., Marigowda G., Burr L., Daines C., Mall M.A., McKone E.F., Ramsey B.W., Rowe S.M., Sass L.A., Tullis E.et al.. VX-445-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 2018; 379:1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Davies J.C., Moskowitz S.M., Brown C., Horsley A., Mall M.A., McKone E.F., Plant B.J., Prais D., Ramsey B.W., Taylor-Cousar J.L.et al.. VX-659-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 2018; 379:1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rowe S.M., Daines C., Ringshausen F.C., Kerem E., Wilson J., Tullis E., Nair N., Simard C., Han L., Ingenito E.P.et al.. Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N. Engl. J. Med. 2017; 377:2024–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H.. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:230–237. [DOI] [PubMed] [Google Scholar]
- 50. Gait M.J., Arzumanov A.A., McClorey G., Godfrey C., Betts C., Hammond S., Wood M.J.A. Cell-penetrating peptide conjugates of steric blocking oligonucleotides as therapeutics for neuromuscular diseases from a historical perspective to current prospects of treatment. Nucleic Acid Ther. 2019; 29:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klein A.F., Varela M.A., Arandel L., Holland A., Naouar N., Arzumanov A., Seoane D., Revillod L., Bassez G., Ferry A.et al.. Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice. J. Clin. Invest. 2019; 129:4739–4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nikan M., Tanowitz M., Dwyer C.A., Jackson M., Gaus H.J., Swayze E.E., Rigo F., Seth P.P., Prakash T.P.. Targeted delivery of antisense oligonucleotides using neurotensin peptides. J. Med. Chem. 2020; 63:8471–8484. [DOI] [PubMed] [Google Scholar]
- 53. Liao W., Dong J., Peh H.Y., Tan L.H., Lim K.S., Li L., Wong W.F.. Oligonucleotide therapy for obstructive and restrictive respiratory diseases. Molecules. 2017; 22:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao C., Crosby J., Lv T., Bai D., Monia B.P., Guo S.. Antisense oligonucleotide targeting of mRNAs encoding ENaC subunits alpha, beta, and gamma improves cystic fibrosis-like disease in mice. J. Cyst. Fibros. 2019; 18:334–341. [DOI] [PubMed] [Google Scholar]
- 55. Crosby J.R., Zhao C., Jiang C., Bai D., Katz M., Greenlee S., Kawabe H., McCaleb M., Rotin D., Guo S.et al.. Inhaled ENaC antisense oligonucleotide ameliorates cystic fibrosis-like lung disease in mice. J. Cyst. Fibros. 2017; 16:671–680. [DOI] [PubMed] [Google Scholar]
- 56. Brinks V., Lipinska K., de Jager M., Beumer W., Button B., Livraghi-Butrico A., Henig N., Matthee B.. The cystic fibrosis-like airway surface layer is not a significant barrier for delivery of eluforsen to airway epithelial cells. J. Aerosol Med. Pulm. Drug Deliv. 2019; 32:303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Heijerman H.G.M., McKone E.F., Downey D.G., Van Braeckel E., Rowe S.M., Tullis E., Mall M.A., Welter J.J., Ramsey B.W., McKee C.M.et al.. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet North Am. Ed. 2019; 394:1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McNamara J.J., McColley S.A., Marigowda G., Liu F., Tian S., Owen C.A., Stiles D., Li C., Waltz D., Wang L.T.et al.. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2–5 years with cystic fibrosis homozygous for F508del-CFTR: an open-label phase 3 study. Lancet Respir. Med. 2019; 7:325–335. [DOI] [PubMed] [Google Scholar]
- 59. Middleton P.G., Mall M.A., Dřevínek P., Lands L.C., McKone E.F., Polineni D., Ramsey B.W., Taylor-Cousar J.L., Tullis E., Vermeulen F.et al.. Elexacaftor–Tezacaftor–Ivacaftor for cystic fibrosis with a single Phe508del allele. N. Engl. J. Med. 2019; 381:1809–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rosenfeld M., Cunningham S., Harris W.T., Lapey A., Regelmann W.E., Sawicki G.S., Southern K.W., Chilvers M., Higgins M., Tian S.et al.. An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2–5 years (KLIMB). J. Cyst. Fibros. 2019; 18:838–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Montes M., Sanford B.L., Comiskey D.F., Chandler D.S.. RNA splicing and disease: animal models to therapies. Trends Genet. 2019; 35:68–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Scotti M.M., Swanson M.S.. RNA mis-splicing in disease. Nat. Rev. Genet. 2016; 17:19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There are no on-line data files associated with this manuscript. Access to the original data is available by request.