


Abstract
DNA methylation, chromatin-binding proteins, and DNA looping are common components regulating genomic imprinting which leads to parent-specific monoallelic gene expression. Loss of methylation (LOM) at the human imprinting center 2 (IC2) on chromosome 11p15 is the most common cause of the imprinting overgrowth disorder Beckwith-Wiedemann Syndrome (BWS). Here, we report a familial transmission of a 7.6 kB deletion that ablates the core promoter of KCNQ1. This structural alteration leads to IC2 LOM and causes recurrent BWS. We find that occupancy of the chromatin organizer CTCF is disrupted proximal to the deletion, which causes chromatin architecture changes both in cis and in trans. We also profile the chromatin architecture of IC2 in patients with sporadic BWS caused by isolated LOM to identify conserved features of IC2 regulatory disruption. A strong interaction between CTCF sites around KCNQ1 and CDKN1C likely drive their expression on the maternal allele, while a weaker interaction involving the imprinting control region element may impede this connection and mediate gene silencing on the paternal allele. We present an imprinting model in which KCNQ1 transcription is necessary for appropriate CTCF binding and a novel chromatin conformation to drive allele-specific gene expression.
INTRODUCTION
Genomic imprinting is the parent-specific monoallelic expression of a subset of mammalian genes. Many imprinted genes play a role in fetal or neonatal development (1,2). In general, paternally-expressed genes promote growth leading to larger offspring, while maternally-expressed genes promote growth restriction and smaller birth weight (2). Marking of the paternal and maternal alleles to differentiate between the two is achieved through epigenetic mechanisms, including DNA methylation and histone post-translational modifications, which are established in germ cells while the genomes are in separate compartments (3). Misregulation of the epigenetic marks and/or expression of imprinted loci often leads to growth or developmental disorders (4).
Beckwith–Wiedemann Syndrome (BWS, OMIM 130650) is one such human imprinting disorder. BWS has recently been revised as the Beckwith–Wiedemann Spectrum (BWSp), which comprises a range of fetal and neonatal overgrowth phenotypes, including macroglossia, omphalocele, organomegaly and embryonal tumors (5–7). The incidence of BWS has been estimated at 1/10 000 live births (8). A small percentage (∼3%) of patients are diagnosed with BWS due to a structural abnormality within the KCNQ1 gene (5,9,10). Deletions, translocations, and duplications encompassing the imprinting control region (ICR) at the centromeric KCNQ1OT1:TSS-DMR, also known as imprinting center 2 (IC2), located on chromosome 11p15 have been reported (9,11–16). More rarely, structural abnormalities outside of the ICR are observed (17–19).
Approximately half of all patients with BWS are molecularly diagnosed with isolated loss of methylation (LOM) at IC2 (5). Within this imprinting cluster, cyclin-dependent kinase inhibitor 1C (CDKN1C) and potassium voltage-gated channel subfamily Q member 1 (KCNQ1) are maternally-expressed, while the non-coding KCNQ1 antisense transcript KCNQ1OT1 is paternally-expressed in the embryo proper (20–24). A maternally-methylated CpG island located at the 5′ end of KCNQ1OT1 and within intron 10 of the KCNQ1 gene acts as the differentially methylated region (DMR) for this ICR (22,25–27). Maintenance of this methylation and, subsequently, imprinted gene expression is also dependent on the parent-specific gene expression of both KCNQ1 and KCNQ1OT1 (20,28–33).
While aspects of the mechanism that regulates this locus have been investigated, for example the Kcnq1ot1 transcript recruitment of Dnmt1, Ezh2, Prc2 and G9a (31,34–36), the sequential steps responsible for organizing and establishing the IC2 imprint have yet to be elucidated. As such, interest in investigating the importance of the three-dimensional (3-D) organization at this locus has been growing. Premature termination of Kcnq1ot1 was found to alter chromatin conformation capture (3C) interactions in the mouse heart (31,37,38) and, more directly, Kcnq1ot1 silencing abrogates a long-range interaction between the DMR and the Kcnq1 promoter (36).
One major player in the long-range organization and transcription of imprinted clusters is CCCTC-Binding Factor (CTCF) (39). Within the telomeric 11p15 domain of H19-ICR, also known as imprinting center 1 (IC1), CTCF plays a role in methylation maintenance (40), imprinted gene expression (41), and the 3D conformation underlying this domain (42). Although its exact role in IC2 imprinting is as yet unknown, CTCF binding across the region has been previously reported. CTCF binds to CDKN1C to modulate cell-specific expression (43–45) and potentially to the DMR itself in a methylation-dependent manner (44–46). Within KCNQ1 intron 2, there are two additional sites of CTCF occupancy; single nucleotide polymorphisms (SNPs) at these sites reduce CTCF binding affinity and confer risk of IC2 LOM (47) and the region has been suggested as a potential CDKN1C enhancer (48). Lopez Abad and colleagues demonstrated this region interacts with CDKN1C in human placenta (49). Recently, a study by Rovina et al. demonstrated that interaction between the DMR and the KCNQ1 intron 2 CTCF sites is significantly reduced in BWS patient cell lines (50). Together, these results suggest that CTCF performs an important function in organizing the imprinted locus and may precede initial deposition of DNA methylation or play a role in maintaining DNA methylation.
Here, we report a familial case of BWS transmitted through a deletion at the 5′ end of the KCNQ1 gene, outside of the ICR. Similar to the cases reported by Beygo et al. (19) and Demars et al. (17), the structural abnormality a distance away from the DMR leads to LOM at the DMR. We investigate the CTCF occupancy outside of the IC2 DMR and its role in chromatin organization of this imprinted domain.
MATERIALS AND METHODS
Patient samples
Samples and clinical information were collected under a previously established Institutional Review Board protocol (IRB 13-010658) at the Children's Hospital of Philadelphia. Consent was obtained from all patients and/or their guardians to collect clinical information and samples. Skin samples were collected from patients and fibroblasts were cultured as previously described (51,52). Briefly, skin samples were split, with one section chemically disrupted using collagenase and the other mechanically disrupted using a scalpel blade to mince. Both explants were seeded into a T25 flask and fed with RPMI skin media (RPMI with fetal bovine serum, penicillin–streptomycin antibiotic, and a final concentration of 2 mM l-glutamine). Flasks were incubated at 37°C for up to one month, with periodic media changes. Successful explant cultures were trypsinized and passaged for sustained growth, then frozen down and stored in liquid nitrogen. Clinical testing for BWS was performed at the University of Pennsylvania Genetic Diagnostic Laboratory as previously described (53).
Fibroblast and placenta/amniocyte samples collected from patients clinically diagnosed with BWS due to isolated LOM at IC2 and not caused by a structural alteration are identified as BWS LOM. Control fibroblast samples were collected from patients who were not diagnosed with BWS and are identified as controls 1–3. Control placenta samples were collected from the birth of BWS LOM patients 3–5 siblings that themselves did not present with BWS features. Placenta tissue was flash frozen for storage at –80°C. Methylation results are reported for amniocytes in place of placenta where such a test was clinically performed and results were available for research use to conserve limited tissue samples.
Whole genome sequencing (WGS)
Genomic DNA (gDNA) was isolated from control 3 and III-3 fibroblasts using the AllPrep DNA/RNA Micro Kit (QIAGEN) per manufacturer's instructions, then quantified using the Qubit dsDNA HS Assay kit (Invitrogen). Libraries were prepared at the Children's Hospital of Philadelphia Center for Applied Genomics using Enzymatic Fragmentation and Twist Universal Adapter System (Twist Bioscience) with 200 ng of gDNA input as per the manufacturer instructions with the following modifications: 2 PCR cycles were used instead of the recommended 8; 14 min of fragmentation time was applied instead of the recommended 22 min. The TapeStation 4200 (Agilent) was used to quality check the libraries, which were then sequenced on the NovaSeq SP platform (Illumina) across two lanes to achieve 30× coverage. Reads were deduplexed and quality assessed using FastQC (Andrews, S. (2010) FastQC: a quality control tool for high throughput sequence data). Mapping was performed using standard BWA MEM variables (54) to the human hg19 assembly. Reads from both lanes were pooled and visualized in .bam and .bedgraph formats using IGV (55).
IC2 expression analysis
RNA was extracted from fibroblasts and placenta using the AllPrep DNA/RNA Micro Kit (QIAGEN) per manufacturer's instructions. cDNA was synthesized using the iScript™ cDNA Synthesis kit (Bio-Rad) with a consistent amount of RNA (500 ng for placenta and 70 ng for fibroblasts), assessed by the Qubit RNA HS Assay kit (Invitrogen) for each sample. Primers were manufactured by Integrated DNA Technologies and the primer sequences are listed in Supplementary Table S1. qRT-PCR gene expression quantification was performed using iTAQ SYBR (Bio-Rad) on the Bio-Rad CFX96 Touch Real-Time PCR Detection System with the following condition changes: 58°C annealing/extension. Results of the qPCR were analyzed using the Δ ΔCT method (56).
Publicly available data usage
CTCF chromatin immunoprecipitation sequencing (ChIP-seq) data available through the ENCODE project (57,58) was viewed using the UCSC genome browser (59) from the following UCSC accession codes for human hg19 build: hESC1 wgEncodeEH000085 for CTCF, wgEncodeEH000106 for H3K4me1, wgEncodeEH000086 for H3K4me3, wgEncodeEH000997 for H3K27ac, wgEncodeEH000074 for H3K27me3; and for mouse mm9 build: wgEncodeEM001954 for CH12 CTCF. Mouse allele-specific CTCF binding ChIP-seq files (GSM862560 and GSM862561) (60) were accessed through NCBI GEO, downloaded through NCBI SRA run selector, and BAM files were viewed directly with IGV (55). BAM files were also converted to bedgraph for viewing on UCSC Genome Browser through the use of Bedtools2 (61). Hi-C data available through the ENCODE project was accessed through the 3D Genome Browser (62) from Lieberman-raw for the GM12878 human hg19 build and CH12 mouse mm9 build (63).
Chromatin immunoprecipitation (ChIP)
ChIP was performed on chromatin isolated from patient-derived fibroblasts using the MAGnify Chromatin Immunoprecipitation System (Invitrogen) as per manufacturer recommendation. Chromatin was sonicated using the Covaris ME220 with the following parameters: PIP 75, DF 5%, CPB 200, 6°C setpoint for 16 min total, then precipitated using anti-CTCF (ab70303; Abcam) and rabbit IgG. To quantify CTCF occupancy, qPCR was performed with primers listed in Supplementary Table S1 using iTAQ SYBR (Bio-Rad) on the Bio-Rad CFX96 Touch Real-Time PCR Detection System with the following conditions: 58°C annealing/extension. Results of the qPCR were analyzed using the ΔΔCT method (56).
Sanger sequencing
Sanger sequencing was used to perform allele-specificity experiments. PCR products were amplified from one of three templates as specified: (i) gDNA isolated from fibroblasts using the AllPrep DNA/RNA Micro Kit (QIAGEN) per manufacturer's instructions, (ii) cDNA created as described in the expression analysis and (iii) ChIP DNA generated in the CTCF immunoprecipitation. Amplicons were run on an agarose gel and subsequently gel extracted using the Gel Extraction kit (QIAGEN) per manufacturer's instructions. Sequencing was carried out on the Applied Biosystems 3730xl DNA Analyzer platform (Thermo Fisher). Primers were manufactured by Integrated DNA Technologies and the primer sequences are listed in Supplementary Table S1.
Capture C
Capture-C was performed according to the protocol from Davies et al. (64) using the restriction enzyme DpnII (NEB) with the following modifications. After each step, the concentration and size distribution of the samples was determined using the Bioanalyzer 2100 (Agilent) unless otherwise noted. After cross-linking, samples were sonicated using the Covaris S220 with the following parameters: PIP 5, DF 10%, CPB 200, 7°C setpoint for two 60 s cycles. NEBNext Multiplex Oligos for Illumina (NEB) were used in adaptor ligation with the following modifications: 0.1× TE buffer was used for all elution steps, LoBind PCR plates (Eppendorf) were used for dA-Tailing of End Repaired DNA and subsequent steps, adaptor ligated DNA was size selected for 200 bp fragments using AMPure XP beads (Beckman Coulter), and a total of 6 cycles were employed for PCR enrichment of the adaptor ligated DNA. Adaptor ligated DNA samples were mixed together in equal amounts by mass to obtain two separate pools of 1.5–2ug: one pool containing three samples and the other containing four.
Probes, whose sequences are listed in Supplementary Table S1, were obtained as 4 nmol biotinylated oligonucleotide IDT ultramers and were resuspended to a final concentration of 2.9 uM. Equimolar amounts of these probes were then pooled to use for the oligonucleotide capture. The capture was performed with the SeqCap EZ Library Kit (Roche) as per manufacturer's instructions, with the following modifications: Capture Beads provided in the SeqCap Pure Capture Bead Kit and KAPA HiFi HotStart ReadyMix from the SeqCap EZ Accessory Kit V2 were used. Two rounds of capture were performed with only 75% (up to 2 ug) of captured material used in the second capture and the hybridization was performed for 24 h instead of 64–72 h as in the first capture. Before sequencing, the concentration, quality, and size of the final captured material was determined by Qubit dsDNA HS Assay (Invitrogen) and the Bioanalyzer 2100 (Agilent). Samples were sequenced on the HiSeq platform (Illumina) with a 2 × 150 bp run format.
Paired-end reads were trimmed with Trim Galore (version 0.4.4_dev) (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) using default parameters. FLASH (v1.2.11) (65) was then used to merge paired-end reads from the same fragment, allowing the reads to overlap to a maximum of 150 bp (‘-M 150′). Fragments were digested with DpnII using the Python script implemented by the CCanalyser3 pipeline (64). The digested fragments were mapped to hg19 reference genome using Bowtie (version 1.2.2) (66), during which reads with more than two alignments were suppressed and only the best alignment was reported (‘-m 2 –best’). The paired-end relationship between the reads were re-established, PCR duplicates were removed, and read enrichment was quantified by the CCanalyser3 pipeline. Contact sites in each sample were defined by fourSig (67). Signal for each captured region was quantified on each bigWig file generated from the pipeline using bwtool (v1.0) (68). cis- and trans-interactions were filtered using Bedtools2 (61) to capture signals common between biological replicates within each group. Figures depicting these cis- and trans-interactions were generated using the R packages Sushi (69) and Circlize (70), respectively.
RESULTS
Characterization of a case of familial BWS
The index patient or proband (III-3) was a female born at 26 6/7 weeks to a 29-year old (II-7) by natural conception (Figure 1). There was a maternal history of a first trimester spontaneous miscarriage (Figure 1). An omphalocele and enlarged adrenal glands were detected in the second trimester leading to BWS testing on amniocytes that showed IC2 LOM (Table 1). Birth weight was 1.16 kg (75th percentile) and postnatal exam demonstrated macroglossia, ear crease, omphalocele, but no lateralized overgrowth, features all consistent with the diagnosis of BWS. Placental pathology showed mesenchymal dysplasia and initial abdominal ultrasound showed cortical renal cysts. The patient passed away at 2 days due to complications of prematurity. Additionally, the maternal half-aunt of the proband, through a common grandfather II-10, was found to carry a fetus (III-14) with a prenatal diagnosis of placentomegaly, nephromegaly, and micrognathia, without polyhydramnios on a 22-week anatomy ultrasound (Figure 1). Given the likelihood of severe BWS based on the index case, the family opted for termination.
Figure 1.

Familial inheritance of a KCNQ1 5′ deletion. Three generation pedigree of the familial transmission of BWS. Arrow indicates proband (III-3). Affected family members in black shapes (individuals III-3, III-4 and III-14); dots in the shape indicate unaffected deletion carriers (individuals II-7 and II-10). These carriers and affected family members are related through a common maternal father and grandfather, respectively (individual I-4).
Table 1.
Methylation levels at IC1 and IC2 in controls, BWS LOM patients and IC2 deletion family members. Normal range for IC1 is 45–55% and for IC2 is 46–54% as per reported error rate
| Sample type | IC1% | IC2% | Deletion carrier | |
|---|---|---|---|---|
| Control1 | BLOOD | 49.49 | 51.43 | N/A |
| Control2 | SKIN | 49.85 | 50.27 | N/A |
| Control3 | SKIN | 50.55 | 50.72 | N/A |
| BWS LOM1 | BLOOD | 50.91 | 2.43 | N/A |
| BWS LOM2 | BLOOD | 48.16 | 0.05 | N/A |
| BWS LOM3 | BLOOD | 48.36 | 0.05 | N/A |
| BWS LOM3 | AMNIOCYTES | 53.13 | 2.78 | N/A |
| BWS LOM4 | PLACENTA | 50.00 | 0.02 | N/A |
| BWS LOM5 | AMNIOCYTES | 49.03 | 22.54 | N/A |
| Familial deletion I-4 | BLOOD | 51.04 | 50.87 | Likely germline mosaic |
| Familial deletion II-6 | BLOOD | 52.49 | 51.01 | No |
| Familial deletion II-7 | BLOOD | 49.55 | 50.87 | Yes |
| Familial deletion II-10 | BLOOD | 51.04 | 50.54 | Yes |
| Familial deletion II-11 | BLOOD | 49.39 | 50.44 | No |
| Familial deletion III-2 | BLOOD | 43.73 | 50.36 | No |
| Familial deletion III-3 | BLOOD | 51.01 | 0.01 | Yes |
| Familial deletion III-3 | AMNIOCYTES | 49.12 | 0.04 | Yes |
| Familial deletion III-14 | SKIN | 51.52 | 0.01 | Yes |
| Familial deletion III-14 | PLACENTA | 51.86 | 0.01 | Yes |
Methylation testing on chromosome 11p15 was performed on family members (Table 1). All samples were found to have normal methylation, ∼50%, at the telomeric H19-ICR (IC1) (Table 1). While most family members also presented with normal methylation at IC2, again ∼50%, samples from both the proband III-3 and individual III-14 presented with complete depletion of methylation at this centromeric domain (Table 1). However, as LOM is usually a mosaic somatic event and not likely to present with familiar transmission, we used Agilent custom microarrays as previously described (53) to verify the previous MS-MLPA copy number results. Using this technique, a small 6.8 kb deletion was identified (chr11: 2 466 678–2 473 512 [grch37/hg19]) (Supplementary Figure S1A). To more finely map the breakpoints of this deletion, we performed Whole Genome Sequencing (WGS) on fibroblasts derived from patient III-3 as well as fibroblasts derived from a non-BWS patient, control3 (Table 1). Read counts were decreased by approximately 50% along the chr11:2 466 050–2 473 630 interval in the III-3 sample (Supplementary Figure S1B) that are unlikely to be a sequencing artifact based on the mapped read count in the control3 profile (data not shown). This result indicates that the full deletion stretches nearly 7.6 kb from approximately 200 bp upstream of the KCNQ1 transcription start site (TSS) and into its first intron, thereby spanning the KCNQ1 core promoter (Figure 2).
Figure 2.

Diagram of KCNQ1, KCNQ1OT1, and CDKN1C within the IC2 domain. (A) UCSC genome browser view of hg19 chr11:2 443 460–2 921 770 including ENCODE tracks of histone modifications and DNase hypersensitivity (HS) sites observed in H1-hESC samples. (B) A stylized diagram of the IC2 domain demonstrating the parent-of-origin gene expression in scale with the UCSC view. In both panels. The documented BWS-causing KCNQ1 5′ deletion is indicated by the gold box. Protein-coding genes (KCNQ1 and CDKN1C) antisense transcript (KCNQ1OT1) are indicated in purple. CpG islands within the region are displayed in green, while CTCF binding sites are highlighted in aqua. Capture C anchors and ChIP-qPCR primer sites are indicated by Region1-5 labels. Scale bar indicates 100 kb.
Transcription of genes within the centromeric 11p15 imprinted domain
As the observed familial deletion appears to ablate the core promoter of KCNQ1 (Figure 2), we wanted to determine the impact of this structural abnormality on its transcription level. Transcription levels of the maternally-expressed KCNQ1 are low in fibroblast samples, which are used in subsequent experiments; to effectively assess its expression by quantitative reverse transcription PCR (qRT-PCR), we used placenta, a tissue with moderate transcript abundance (71) (Figure 3A). For this assay, BWS LOM patient 3, 4 and 5 placentae are represented in the BWS LOM group (Table 1, Figure 3A). Controls used in this assay were collected from siblings of BWS LOM 3, 4 and 5 who did not present with BWS (Figure 3A). Approximately 50% decreased expression of KCNQ1 was observed in BWS LOM samples relative to the non-syndromic sibling samples. In the III-3 KCNQ1 5′ deletion sample, ∼85% reduced KCNQ1 expression was observed (Figure 3A). These results indicate that maternal KCNQ1 transcription is disrupted further in individual III-3 carrying the maternal deletion as compared to the isolated LOM patients. Some of the moderate KCNQ1 transcription detected in III-3 placenta may be due to maternal contamination in tissue processing, but a study in mouse demonstrated a small amount of transcription is detected from the paternal allele in late-term placenta (34). In either event, KCNQ1 expression is effectively maternally-silenced by the deletion at the 5′ end of this gene.
Figure 3.

Expression of KCNQ1, CDKN1C, and KCNQ1OT1 imprinted genes within IC2. Relative expression of IC2 imprinted genes was assessed by qRT-PCR and normalized to ACTB and RPLP0 levels. For LOM, N = 3; for control, N = 3. Error bars represent CI. (A) Expression of maternally-expressed KCNQ1 in placenta is reduced in BWS LOM cells and is further disrupted in III-3 cells carrying the maternal allele deletion relative to control samples from BWS LOM siblings. (B) Expression of maternally-expressed CDKN1C in fibroblasts is similarly aberrantly repressed in both BWS LOM and III-3 cells relative to unrelated non-BWS controls. (C) Expression of paternally-expressed KCNQ1OT1 in fibroblasts is increased in III-3 cells carrying the KCNQ1 5′ deletion relative to unrelated non-BWS controls. (D) Sanger sequencing trace of a KCNQ1OT1 SNP, rs231362 designated by the black box, in genomic (top) and complementary (bottom) DNA isolated from BWS LOM1 fibroblasts demonstrates biallelic expression. (E) Sanger sequencing trace of the boxed rs231362 SNP in genomic (top) and complementary (bottom) DNA isolated from III-3 KCNQ1 5′ deletion fibroblasts demonstrates biallelic expression.
Additionally, maternal methylation at the IC2 DMR, located at the 5′ end of KCNQ1OT1, was affected by the KCNQ1 5′ deletion (Table 1). As this methylation is important in the imprinted expression of KCNQ1, KCNQ1OT1, and CDKN1C, we wanted to determine the expression status of KCNQ1OT1 and CDKN1C as well (Figure 2B). We determined imprinting maintenance of CDKN1C and KCNQ1OT1 in fibroblasts for BWS LOM patients 1, 2, 3 and III-3 (Table 1), as this cell type was used in subsequent experiments in this study (Figure 3B-C). For these qRT-PCR experiments, controls were derived from unrelated non-BWS patients (Table 1, Figure 3B and C). Transcription of the maternally-expressed CDKN1C was ablated, with 7% and 4% expression in BWS LOM and III-3 fibroblasts respectively, compared to that of the control group (Figure 3B). Conversely, transcription of the paternally-expressed KCNQ1-antisense KCNQ1OT1, which initiates from the ICR, is more than doubled in fibroblasts from patient III-3 although expressed at similar levels between BWS LOM and control groups (Figure 3C). Due to loss of maternal methylation in both BWS LOM and III-3 KCNQ1 5′ deletion samples, we wanted to determine whether expression of KCNQ1OT1 was biallelic, irrespective of transcript abundance (Figure 2B). We performed Sanger sequencing to assay for allele-specific polymorphisms in KCNQ1OT1 transcripts (Figure 3D and E). In the sample isolated from BWS LOM1 (Table 1), a single nucleotide polymorphism (SNP), designated rs231362, was captured at the genomic DNA (gDNA) level (Figure 3D). The SNP was also observed in KCNQ1OT1 complementary DNA (cDNA) (Figure 3D), indicating biallelic expression of this antisense RNA. The rs231362 polymorphism was also detected in III-3 fibroblasts at the gDNA and cDNA levels (Figure 3E), suggesting loss of maternal KCNQ1 expression causes reactivation of the maternal KCNQ1OT1 antisense transcript as a result of the deletion.
CTCF occupancy within the centromeric 11p15 imprinted domain
We wanted to understand the mechanism of this allele-specific expression disruption and hypothesized that CTCF binding across the region may play a role. CTCF has been shown to bind at CDKN1C (43,47), but observations as to whether it binds at the ICR have been conflicting (30,36,46,60,72). While the importance and consistency of occupancy at the DMR is questionable, other CTCF binding sites have been observed across the imprinted domain through chromatin immunoprecipitation sequencing (ChIP-seq) experiments (Figure 2) and in targeted ChIP studies (47,49).
As the orthologs of KCNQ1, KCNQ1OT1 and CDKN1C are also subject to genomic imprinting in the mouse (21,25,73), we wanted to determine the landscape of CTCF binding across the orthologous domain. Again, murine occupancy of CTCF on the ICR has previously been investigated and results include the finding that CTCF binds biallelically (72), to only the paternal allele (46), or not at all (60). Outside of the ICR, murine ChIP-seq profiles indicate CTCF binds upstream of Kcnq1, within the Kcnq1 intron2, and upstream of Cdkn1c (Supplementary Figure S2A). To ascertain whether this binding is allele-specific in the mouse, we used a publicly available CTCF ChIP-seq dataset (60). In the previously published work, Prickett and colleagues determined that CTCF does not bind appreciably to the ICR of IC2, but our visualization of their data indicates CTCF binds to sites upstream of Kcnq1, strongly within the Kcnq1 gene body, and upstream of Cdkn1c (Supplementary Figure S2A). To determine whether this binding was monoallelic or biallelic, we visualized SNPs present within these binding sites (Supplementary Figure S2B). Variants were observed in approximately equal proportion by sequencing, indicating equal interaction of CTCF with both alleles (Supplementary Figure S2B).
To determine whether CTCF binds to similar sites across the human IC2 domain and whether this binding was disrupted by the KCNQ1 5′ deletion, termed Region2 (Figure 2), we performed chromatin immunoprecipitation followed by quantitative PCR (ChIP-qPCR). Comparable to the sites observed in mouse, we targeted CTCF sites near the deletion upstream of KCNQ1 termed Region1 as well as within KCNQ1 intron 2 termed Region3 (Figure 2). In addition, we assessed CTCF binding at the ICR termed Region4, and a site ∼500 kb from the deletion at the 5′ end of CDKN1C termed Region5 (Figure 2). While CTCF was detected at the Region4 ICR, the level of occupancy at this site was relatively low across the control1-3, BWS LOM1-3 and III-3 fibroblast samples (Figure 4A). Additionally, there were no appreciable differences in CTCF binding level between groups at this site (Figure 4A). CTCF binding sites at Regions 1, 3 and 5 all demonstrated greater relative enrichment, with the most CTCF detected at the Region3 binding motif in controls (Figure 4A). Similar levels of occupancy were detected at Regions 1 and 3 between the control and BWS LOM groups (Figure 4A). While there was some increased CTCF binding to Region5 in BWS LOM samples relative to control, this did not reach a 2-fold increase (Figure 4A). Decreased CTCF binding was observed in the III-3 deletion fibroblasts with 50–60% of control-level enrichment at both Regions 1 and 3 (Figure 4A).
Figure 4.

CTCF binding across the IC2 deletion. ChIP-qPCR results of CTCF occupancy relative to the non-imprinted ANXA9 locus. For BWS LOM, N = 3; for unrelated non-BWS controls, N = 3. Error bars represent CI. (A) CTCF binding at Regions 1, 3, 4 and 5 relative to the control level of binding at the Region4 ICR. Regions 1, 3 and 5 have higher CTCF occupancy than that at Region4. Relative to controls, III-3 fibroblasts carrying the KCNQ1 5′ deletion demonstrate reduced CTCF binding Regions 1 and 3. (B) Sanger sequencing trace of the rs67439072 SNP designated by the black box in genomic DNA isolated from III-3 KCNQ1 5′ deletion fibroblasts located less than 100 bp from the Region1 CTCF site. (C) Sanger sequencing trace of the same boxed SNP in ChIP DNA isolated from III-3 KCNQ1 5′ deletion fibroblasts.
As the III-3 KCNQ1 5′ deletion is only present on the maternal allele, we wanted to determine whether CTCF occupancy was also monoallelic in this fibroblast sample. We performed Sanger sequencing on amplicons generated from gDNA and from the CTCF ChIP DNA to detect allelic SNPs (Figure 4B and C). In this sample, a SNP was detected within the Region1 CTCF site, designated rs67439072, at the genomic level (Figure 4B). Visualization of the same sequence in the ChIP DNA indicated the SNP was still present, suggesting the capture of both alleles by the CTCF immunoprecipitation (Figure 4C). There was a bias towards capture of the C allele, which may indicate preferential loss of CTCF on one allele, but certainly not complete loss of CTCF in an allele-specific manner (Figure 4C). Overall, these results indicate that high biallelic CTCF binding occurs at sites across the domain and the structural abnormality observed in the III-3 samples may somehow influence this binding.
Cis and trans interactions with the centromeric 11p15 imprinted domain
It has been well-established that CTCF can direct DNA looping and chromatin organization (39). More specifically, CTCF has been observed or proposed to play a role in small-scale interactions across IC2 (36,37,43,49). We performed Capture-C to better understand interactions stemming from the CTCF binding sites, the KCNQ1 5′ deletion itself, or the ICR (Figure 5A). First, interactions in the control 1–3 fibroblasts were observed between Region1, Region3 and Region5, with limited connectivity to Region4 (Figure 5B). Interactions between all of these regions are almost completely abrogated in the BWS LOM 1–3 fibroblast samples (Figure 5C). Lastly, in the III-3 deletion fibroblasts, while a few connections between Region4 and Regions1/3/5 remain, the long-range interactions from Region1/3 to Region5 are no longer captured (Figure 5D). Interestingly, these results suggest that the connectivity between the 5′ end of KCNQ1 and the 5′ end of CDKN1C, and to a lesser extent to the ICR, is important to the regulation of imprinting within this domain.
Figure 5.
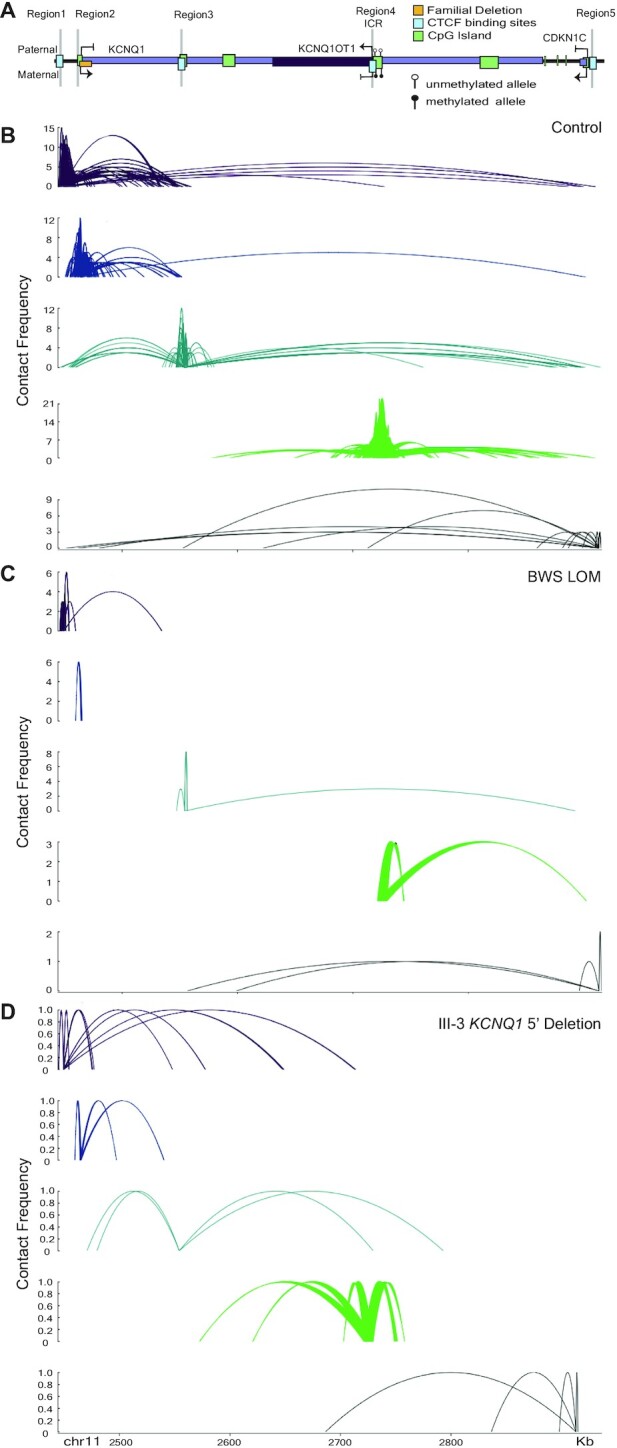
Interaction profiles between 5′ KCNQ1, IC2, and 5′ CDKN1C change after BWS LOM and KCNQ1 5′ deletion. For LOM, N = 3; for control, N = 3. Splines represent interactions commonly observed between biological replicates. Contact frequency indicates the number of times an interaction was observed. (A) Simplified locus map indicating the probes for interaction capture. (B) cis interactions across the IC2 domain in control fibroblasts (N = 3) demonstrate interactions between Region1, Region3 and Region5. (C) cis interactions across the IC2 domain in BWS LOM fibroblasts are reduced, notably connectivity between Region1 and Region5 is absent. (D) cis interactions across the IC2 domain in III-3 KCNQ1 5′ deletion fibroblasts are further reduced in strength compared to BWS LOM, again lacking connectivity between Region1 and Region5.
Recently, Rovina et al. (50) reported an interaction between IC1 and IC2 in a limited number of patient lymphoblastoid cell lines. To determine whether we observed such an interaction in our patient fibroblast lines, we expanded the window of interactions. In the control fibroblast lines, Region1 was the only anchor to form long-range interactions that extended to the 5′ region of IGF2; there were no interactions observed between the Region4 IC2 anchor and IC1 (Supplementary Figure S3B). Trends in the BWS LOM and III-3 deletion samples were similar to those observed within the IC2 domain (Supplementary Figure S3C and D). The BWS LOM fibroblast group lost many connections with all anchors and specifically between Region1 and IC1 (Supplementary Figure S3C), while the III-3 IC2 deletion fibroblasts maintained some connection between Region1/3 and the 5′ end of IGF2 (Supplementary Figure S3D). Neither of these samples displayed any interactions between Region4 IC2 and IC1 (Supplementary Figure S3C and D). Although we did not confirm the reciprocal interaction using a probe at IC1 to interrogate IC2, these results were supported by Hi-C data that demonstrate a contact depletion indicated by white coloration in a stripe initiating from the middle of the IC1 domain (Supplementary Figure S2A). Further, we wanted to determine whether these aspects of domain organization were conserved in the mouse, as with the CTCF binding. The contact depletion separating the imprinted domains was even more pronounced in the mm9 data (Supplementary Figure S4).
While we did not observe interactions between IC1 and IC2, we thought there may be trans interactions, or associations between other chromosomes and this imprinted domain important to the imprinted expression regulation of IC2 and the etiology of BWS. As such, we first considered interactions between the ICR at Region4 with other chromosomes (Figure 6). Control interactions common among the biological replicates were limited and, as expected, the number of these interactions was severely reduced in the BWS LOM group (Figure 6, Supplementary Table S2). Surprisingly, the number of interactions with this anchor in III-3 KCNQ1 5′ deletion fibroblasts was increased both overall as well as the number of chromosomes with which interactions were observed (Figure 6, Supplementary Table S2). This pattern of interaction decreases among the BWS LOM group and increases in the III-3 deletion sample consistently across Region2, Region3, and Region5 probes (Supplementary Figure S5B–D, Supplementary Table S2). Of all the anchors, Region1 had the greatest number of overall interactions and the control and BWS LOM groups presented with a similar quantity of interactions, indicating that a number of these interactions are unlikely to be caused by or to influence imprinting (Supplementary Figure S5A). Together, these results across all anchors point to the importance of previously unexplored trans interactions and whole genome organization in regulating genomic imprinting at IC2.
Figure 6.

Trans interactions of IC2 ICR across the genome. Trans interactome of the ICR Region4 probe are displayed. Interactions across chromosome 11 are not shown. Common features of control fibroblasts (N = 3) profile are shown in green to establish an average interactome of the ICR when imprinting is maintained. Common features of BWS LOM fibroblasts (N = 3) are shown in yellow and indicate that, when imprinting is disrupted, fewer trans interactions are made. The profile of III-3 fibroblasts carrying the KCNQ1 5′ deletion on the maternal allele is shown in blue and demonstrates a greater number as well as novel interactions across the genome when imprinting is disrupted and the structure of the imprinted domain is altered.
DISCUSSION
DNA binding proteins, histone post-translational modifications, and chromatin conformation have been shown to play a role in cell-specific and developmental genome regulation (74). Loci subject to parent-of-origin gene expression are also regulated by these epigenetic factors, wherein the maternal and paternal alleles demonstrate distinct intra- and interdomain profiles (75). Here, we investigate aspects of the human centromeric 11p15 imprinted domain epigenetic landscape. Alterations to the structure or ICR-DMR methylation of this domain have been associated with BWSp, a human overgrowth spectrum most commonly caused by the resultant loss of maternal expression of CDKN1C.
Part of this profiling includes our presentation of the first trans-interactome of IC2 in control and BWS patient fibroblast samples (Figure 6, Supplementary Figure S5). As the control profile is comprised of distinct maternal and paternal allele contacts, we were unsurprised to observe fewer interactions in the BWS LOM group which lost allele-specific regulation (Figure 6, Supplementary Figure S5). Additionally, as the III-3 interactome was not filtered against another biological sample, some of the increased number of interactions in this profile are likely background contacts (Figure 6, Supplementary Figure S5). Although many 11p15 contacts with other chromosomes are likely due to nuclear organization and association of transcriptionally active or inactive domains (76), some may represent functional elements that influence the parent-specific gene expression within IC2. None of the control trans-Region5 CDKN1C interactions lost in BWS samples displayed the combinatorial histone and DNA binding factor landscape to suggest a potential CDKN1C enhancer function (Supplementary Table S2) (57–59). However, one trans interaction we noted was that between Region1/Region4 and human chromosome 2p22.3 within intron4 of LINC00486 (Figure 6, Supplementary Figure S4A, Supplementary Table S2). This contact was observed in the control samples, but not in the BWS samples (Figure 6, Supplementary Figure S4A, Supplementary Table S2). Furthermore, while this long, intergenic, non-coding RNA has not been well-characterized, the interaction contact point overlapped with the signal for histone modifications including H3K4me1 and H3K27ac, as well as binding of CTCF, bHLH transcription factor cMYC, and RNA polymerase II (57–59). In combination, these factors may indicate an enhancer element (77), although whether it acts as such in trans on IC2 was not within the scope of this study and requires further exploration. Additionally, while an imprinting gene network has been previously proposed to describe the co-regulation of loci subject to parent-specific gene expression (78,79), we did not observe any connections between any of the IC2 domain anchors and other imprinted loci (Supplementary Table S2).
We report a case of familial BWS caused by the maternal transmission of a 7.6 kB deletion at the 5′ end of KCNQ1 that causes KCNQ1 silencing (Figure 3A) and LOM downstream at the ICR (Table 1). Along with the structural abnormalities reported by Beygo et al. (19) and Demars et al. (17), this deletion is the third observed alteration outside of the ICR-DMR that impacts its methylation status. To determine if long-range intradomain interactions mediated by the chromatin organizer CTCF act within IC2, we profiled CTCF binding and chromatin conformation in control, BWS LOM, and KCNQ1 5′ deletion samples (Figure 4–5). To identify common features of IC2 imprint disruption, we compared the III-3 KCNQ1 5′ deletion profile to that of the BWS LOM features. While previous studies investigating the CTCF binding profile across the domain have focused on the occupancy at the Region4 ICR (30,36,46,60,72), we find that of the four annotated CTCF binding sites we considered, the control occupancy at the ICR was lowest (Figure 4A). The highest control CTCF binding was observed within Region3 (Figure 4A).
Although our KCNQ1 5′ deletion did not encompass any CTCF sites, the two most proximal binding sites, Region1 and Region3, demonstrated reduced CTCF occupancy in this fibroblast sample (Figure 4A). It is possible that KCNQ1 transcription is required to open Region3 within its intron 2 to allow for CTCF binding to maintain maternal CDKN1C expression. The CTCF motif and occupancy at Region3 have been previously investigated in BWS and human imprinting mechanism studies. Risk for IC2 LOM has been correlated to SNP presence within the Region3 CTCF binding sequence (47). Based on epigenetic histone marks, this site was also identified as a potential CDKN1C enhancer (Figure 2) (48). The familial duplication reported by Demars et al. (17) encompassed this CTCF site and caused IC2 LOM, which resulted in decreased CDKN1C expression. Furthermore, an interaction between this Region3 and CDKN1C was observed in placenta by Lopez-Abad et al. (49). We confirm this physical connection in control fibroblast samples and further conclude that the interaction includes the CTCF site at Region1 nearly 450 kB from CDKN1C (Figure 5A-B). It is possible that the presence of a SNP within the binding site impacted CTCF affinity for Region1 in the III-3 sample, although this does not completely explain the capture of both alleles in the ChIP DNA Sanger sequencing (Figure 4C). The interaction between Region1 and Region 3 may also reinforce CTCF binding at both sites; the decrease in CTCF binding at Region3 may influence occupancy at Region1 thereby causing its decrease in III-3 KCNQ1 5′ deletion cells (Figure 4A). These long-range connections are depleted in both isolated LOM and KCNQ1 5′ deletion samples (Figure 5C-D), implying their importance in the parent-of-origin transcription across the domain.
We also observed an interaction between Regions 3/4 and 4/5 that occurred at approximately half the frequency of the control Region1/3/5 connection (Figure 5). This conformation became apparent in the KCNQ1 5′ deletion and BWS LOM profiles, respectively, which suggests these interactions were not abrogated by the BWS-associated maternal allele dysregulation. As such, we propose an IC2 looping model wherein CTCF cooperatively mediates the maternal high-strength Regions1/3/5 interaction to bring together the KCNQ1 and CDKN1C TSSs and drive their allele-specific expression (Figure 7). On the paternal allele, the CTCF site at Region4 loops with Region3 or Region5 at a low-frequency, which we propose prevents the formation of the Region1/3/5 loop (Figure 7). Importantly, the results of this study and our model demonstrate that allele-specific maintenance of the centromeric 11p15 domain is not limited to the ICR itself, but involves epigenetic elements across the region.
Figure 7.

Proposed model of intra-domain looping in IC2 maintenance of imprinting. (A) On the maternal allele, interactions connect the 5′ end of KCNQ1, KCNQ1 intron2 and the 5′ end of CDKN1C, which promotes expression of both KCNQ1 and CDKN1C. This looping excludes the ICR. (B) On the paternal allele, low frequency contacts between the ICR and the 5′ end of KCNQ1 and KCNQ1 intron2 or the 5′ end of CDKN1C, but not all three. The result is expression of KCNQ1OT1. (C) On the maternal allele carrying the KCNQ1 5′ deletion, this interaction does not organize correctly. This may be caused by transcription disruption of KCNQ1, but leads to an interaction profile resembling the paternal allele and loss of CDKN1C expression.
However, one question presented by these interactome findings is how the familial KCNQ1 5′ deletion, which lies between but does not encompass Regions1/3 can abrogate CTCF binding and alter the ICR methylation status. Imprinting methylation marks are established during gametogenesis when chromatin is compacted for cell division and is not generally organized by topological domain (3,74). Therefore, it is unlikely that CTCF-mediated loops are responsible for the constitutive failure to establish IC2-ICR methylation in the III-3 KCNQ1 5′ deletion patient. Previous studies in mouse and human have demonstrated the importance of the sense and antisense transcription of KCNQ1 and KCNQ1OT1, respectively (20,28–33). Several studies report the silencing ability of KCNQ1OT1 in a bidirectional manner across the domain (28,31,33,80–83). Yet, one role of antisense transcription at other imprinted loci is promoter competition (84), and the Kcnq1ot1 transcript does not proceed through, and therefore cause interference with, the Kcnq1 promoter (35,85). Interestingly, it has been noted that knockdown of the Kcnq1ot1 transcript post-transcriptionally does not disrupt the imprint (86). A study by Golding et al. suggests that paternal transcription initiation or chromatin opening is more important than the antisense transcript itself (86). The results we present in our study support this conclusion in that both BWS LOM and III-3 KCNQ1 5′ deletion samples demonstrate biallelic expression of KCNQ1OT1 although this is not accompanied by an increased transcript abundance in the case of the BWS LOM group (Figure 3C–E).
From the sense perspective, truncation of Kcnq1 transcription prior to reaching the DMR resulted in near complete LOM and biallelic Kcnq1ot1 transcription (33). The human deletion reported by Beygo et al. (19) also maps upstream of the ICR-DMR, encompasses the KCNQ1 TSS, and leads to ICR LOM similar to the smaller deletion reported in this study (Figure 2A). They propose abrogated KCNQ1 transcription during oocyte development leads to the imprint failure (19). Our study supports this conclusion and we further report resultant increased KCNQ1OT1 expression as a result of the structural alteration (Figure 3A,C), suggesting a KCNQ1/KCNQ1OT1 antagonistic effect even without promoter competition. Further studies in human and hybrid mouse models are required to better understand the interplay and role of sense versus antisense transcription as a regulator of allele-specific methylation deposition during gametogenesis, as well as in methylation maintenance in somatic tissues.
DATA AVAILABILITY
Data are accessible in dbGAP (phs002408.v1.p1).
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the patients and their family for their participation in the BWS registry and this study. We thank Matthew Deardorff for discussion. We thank Peter Papenhause for discussion and additional testing of some of the clinical samples. We also thank Christina Gonzalez-Gandolfi for help with the figures.
Authors contributions: N.S.S.N. designed and performed experiments, analyzed data and drafted the manuscript; D.F.D., J.H. and E.T. executed experiments; Y.L. analyzed data; R.W. discussed the case, performed experiments, and analyzed data; A.G. discussed the case, designed and performed experiments and analyzed data. N.E. conceptualized the study, designed experiments, analyzed data and revised the manuscript. J.M.K. conceptualized the study, evaluated the patients, designed experiments, analyzed data, and drafted and revised the manuscript.
Contributor Information
Natali S Sobel Naveh, Division of Human Genetics, Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA.
Daniel F Deegan, Fels Institute for Cancer Research and Molecular Biology, Temple University School of Medicine, Philadelphia, PA 19140, USA.
Jacklyn Huhn, Fels Institute for Cancer Research and Molecular Biology, Temple University School of Medicine, Philadelphia, PA 19140, USA.
Emily Traxler, Division of Human Genetics, Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA.
Yemin Lan, Epigenetics Institute, Department of Cell and Developmental Biology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Rosanna Weksberg, Division of Clinical and Metabolic Genetics, Genetics and Genome Biology, Hospital for Sick Children, and Institute of Medical Science, University of Toronto, Toronto, Canada.
Arupa Ganguly, Department of Genetics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA.
Nora Engel, Fels Institute for Cancer Research and Molecular Biology, Temple University School of Medicine, Philadelphia, PA 19140, USA.
Jennifer M Kalish, Division of Human Genetics, Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA; Department of Genetics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA; Department of Pediatrics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Alex's Lemonade Stand Foundation (to J.M.K.); Margaret Q. Landenberger Foundation; St. Baldrick's Foundation Scholar Award; Damon Runyon Cancer Research Foundation; NIH [K08CA193915]. Funding for open access charge: Damon Runyon Cancer Research Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Plasschaert R.N., Bartolomei M.S.. Genomic imprinting in development, growth, behavior and stem cells. Development. 2014; 141:1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crespi B.J. Why and how imprinted genes drive fetal programming. Front Endocrinol (Lausanne). 2019; 10:940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abramowitz L.K., Bartolomei M.S.. Genomic imprinting: recognition and marking of imprinted loci. Curr. Opin. Genet. Dev. 2012; 22:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kalish J.M., Jiang C., Bartolomei M.S.. Epigenetics and imprinting in human disease. Int. J. Dev. Biol. 2014; 58:291–298. [DOI] [PubMed] [Google Scholar]
- 5. Brioude F., Kalish J.M., Mussa A., Foster A.C., Bliek J., Ferrero G.B., Boonen S.E., Cole T., Baker R., Bertoletti M.et al.. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat. Rev. Endocrinol. 2018; 14:229–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duffy K.A., Cielo C.M., Cohen J.L., Gonzalez-Gandolfi C.X., Griff J.R., Hathaway E.R., Kupa J., Taylor J.A., Wang K.H., Ganguly A.et al.. Characterization of the Beckwith-Wiedemann spectrum: Diagnosis and management. Am. J. Med. Genet. C Semin. Med. Genet. 2019; 181:693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang K.H., Kupa J., Duffy K.A., Kalish J.M.. Diagnosis and management of Beckwith-Wiedemann syndrome. Front Pediatr. 2019; 7:562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mussa A., Russo S., De Crescenzo A., Chiesa N., Molinatto C., Selicorni A., Richiardi L., Larizza L., Silengo M.C., Riccio A.et al.. Prevalence of Beckwith-Wiedemann syndrome in North West of Italy. Am. J. Med. Genet. A. 2013; 161A:2481–2486. [DOI] [PubMed] [Google Scholar]
- 9. Algar E., Dagar V., Sebaj M., Pachter N.. An 11p15 imprinting centre region 2 deletion in a family with Beckwith Wiedemann syndrome provides insights into imprinting control at CDKN1C. PLoS One. 2011; 6:e29034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heide S., Chantot-Bastaraud S., Keren B., Harbison M.D., Azzi S., Rossignol S., Michot C., Lackmy-Port Lys M., Demeer B., Heinrichs C.et al.. Chromosomal rearrangements in the 11p15 imprinted region: 17 new 11p15.5 duplications with associated phenotypes and putative functional consequences. J. Med. Genet. 2018; 55:205–213. [DOI] [PubMed] [Google Scholar]
- 11. Gurrieri F., Zollino M., Oliva A., Pascali V., Orteschi D., Pietrobono R., Camporeale A., Coll Vidal M., Partemi S., Brugada R.et al.. Mild Beckwith-Wiedemann and severe long-QT syndrome due to deletion of the imprinting center 2 on chromosome 11p. Eur. J. Hum. Genet. 2013; 21:965–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zollino M., Orteschi D., Marangi G., De Crescenzo A., Pecile V., Riccio A., Neri G.. A case of Beckwith-Wiedemann syndrome caused by a cryptic 11p15 deletion encompassing the centromeric imprinted domain of the BWS locus. J. Med. Genet. 2010; 47:429–432. [DOI] [PubMed] [Google Scholar]
- 13. Baskin B., Choufani S., Chen Y.A., Shuman C., Parkinson N., Lemyre E., Micheil Innes A., Stavropoulos D.J., Ray P.N., Weksberg R.. High frequency of copy number variations (CNVs) in the chromosome 11p15 region in patients with Beckwith-Wiedemann syndrome. Hum. Genet. 2014; 133:321–330. [DOI] [PubMed] [Google Scholar]
- 14. Chiesa N., De Crescenzo A., Mishra K., Perone L., Carella M., Palumbo O., Mussa A., Sparago A., Cerrato F., Russo S.et al.. The KCNQ1OT1 imprinting control region and non-coding RNA: new properties derived from the study of Beckwith-Wiedemann syndrome and Silver-Russell syndrome cases. Hum. Mol. Genet. 2012; 21:10–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beygo J., Burger J., Strom T.M., Kaya S., Buiting K.. Disruption of KCNQ1 prevents methylation of the ICR2 and supports the hypothesis that its transcription is necessary for imprint establishment. Eur. J. Hum. Genet. 2019; 27:903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niemitz E.L., DeBaun M.R., Fallon J., Murakami K., Kugoh H., Oshimura M., Feinberg A.P.. Microdeletion of LIT1 in familial Beckwith-Wiedemann syndrome. Am. J. Hum. Genet. 2004; 75:844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Demars J., Rossignol S., Netchine I., Lee K.S., Shmela M., Faivre L., Weill J., Odent S., Azzi S., Callier P.et al.. New insights into the pathogenesis of Beckwith-Wiedemann and Silver-Russell syndromes: contribution of small copy number variations to 11p15 imprinting defects. Hum. Mutat. 2011; 32:1171–1182. [DOI] [PubMed] [Google Scholar]
- 18. Kaltenbach S., Capri Y., Rossignol S., Denjoy I., Soudee S., Aboura A., Baumann C., Verloes A.. Beckwith-Wiedemann syndrome and long QT syndrome due to familial-balanced translocation t(11;17)(p15.5;q21.3) involving the KCNQ1 gene. Clin. Genet. 2013; 84:78–81. [DOI] [PubMed] [Google Scholar]
- 19. Beygo J., Joksic I., Strom T.M., Ludecke H.J., Kolarova J., Siebert R., Mikovic Z., Horsthemke B., Buiting K.. A maternal deletion upstream of the imprint control region 2 in 11p15 causes loss of methylation and familial Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2016; 24:1280–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee M.P., DeBaun M.R., Mitsuya K., Galonek H.L., Brandenburg S., Oshimura M., Feinberg A.P.. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:5203–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee M.P., Hu R.J., Johnson L.A., Feinberg A.P.. Human KVLQT1 gene shows tissue-specific imprinting and encompasses Beckwith-Wiedemann syndrome chromosomal rearrangements. Nat. Genet. 1997; 15:181–185. [DOI] [PubMed] [Google Scholar]
- 22. Smilinich N.J., Day C.D., Fitzpatrick G.V., Caldwell G.M., Lossie A.C., Cooper P.R., Smallwood A.C., Joyce J.A., Schofield P.N., Reik W.et al.. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:8064–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hatada I., Inazawa J., Abe T., Nakayama M., Kaneko Y., Jinno Y., Niikawa N., Ohashi H., Fukushima Y., Iida K.et al.. Genomic imprinting of human p57KIP2 and its reduced expression in Wilms' tumors. Hum. Mol. Genet. 1996; 5:783–788. [DOI] [PubMed] [Google Scholar]
- 24. Matsuoka S., Thompson J.S., Edwards M.C., Bartletta J.M., Grundy P., Kalikin L.M., Harper J.W., Elledge S.J., Feinberg A.P.. Imprinting of the gene encoding a human cyclin-dependent kinase inhibitor, p57KIP2, on chromosome 11p15. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:3026–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mitsuya K., Meguro M., Lee M.P., Katoh M., Schulz T.C., Kugoh H., Yoshida M.A., Niikawa N., Feinberg A.P., Oshimura M.. LIT1, an imprinted antisense RNA in the human KvLQT1 locus identified by screening for differentially expressed transcripts using monochromosomal hybrids. Hum. Mol. Genet. 1999; 8:1209–1217. [DOI] [PubMed] [Google Scholar]
- 26. Du M., Zhou W., Beatty L.G., Weksberg R., Sadowski P.D.. The KCNQ1OT1 promoter, a key regulator of genomic imprinting in human chromosome 11p15.5. Genomics. 2004; 84:288–300. [DOI] [PubMed] [Google Scholar]
- 27. Beatty L., Weksberg R., Sadowski P.D.. Detailed analysis of the methylation patterns of the KvDMR1 imprinting control region of human chromosome 11. Genomics. 2006; 87:46–56. [DOI] [PubMed] [Google Scholar]
- 28. Mancini-Dinardo D., Steele S.J., Levorse J.M., Ingram R.S., Tilghman S.M.. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006; 20:1268–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Valente F.M., Sparago A., Freschi A., Hill-Harfe K., Maas S.M., Frints S.G.M., Alders M., Pignata L., Franzese M., Angelini C.et al.. Transcription alterations of KCNQ1 associated with imprinted methylation defects in the Beckwith-Wiedemann locus. Genet. Med. 2019; 21:1808–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schultz B.M., Gallicio G.A., Cesaroni M., Lupey L.N., Engel N.. Enhancers compete with a long non-coding RNA for regulation of the Kcnq1 domain. Nucleic Acids Res. 2015; 43:745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mohammad F., Mondal T., Guseva N., Pandey G.K., Kanduri C.. Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development. 2010; 137:2493–2499. [DOI] [PubMed] [Google Scholar]
- 32. Essinger C., Karch S., Moog U., Fekete G., Lengyel A., Pinti E., Eggermann T., Begemann M.. Frequency of KCNQ1 variants causing loss of methylation of Imprinting Centre 2 in Beckwith-Wiedemann syndrome. Clin Epigenetics. 2020; 12:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singh V.B., Sribenja S., Wilson K.E., Attwood K.M., Hillman J.C., Pathak S., Higgins M.J.. Blocked transcription through KvDMR1 results in absence of methylation and gene silencing resembling Beckwith-Wiedemann syndrome. Development. 2017; 144:1820–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Umlauf D., Goto Y., Cao R., Cerqueira F., Wagschal A., Zhang Y., Feil R.. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat. Genet. 2004; 36:1296–1300. [DOI] [PubMed] [Google Scholar]
- 35. Pandey R.R., Mondal T., Mohammad F., Enroth S., Redrup L., Komorowski J., Nagano T., Mancini-Dinardo D., Kanduri C.. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 2008; 32:232–246. [DOI] [PubMed] [Google Scholar]
- 36. Zhang H., Zeitz M.J., Wang H., Niu B., Ge S., Li W., Cui J., Wang G., Qian G., Higgins M.J.et al.. Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus. J. Cell Biol. 2014; 204:61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Korostowski L., Raval A., Breuer G., Engel N.. Enhancer-driven chromatin interactions during development promote escape from silencing by a long non-coding RNA. Epigenet. Chromatin. 2011; 4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Korostowski L., Sedlak N., Engel N.. The Kcnq1ot1 long non-coding RNA affects chromatin conformation and expression of Kcnq1, but does not regulate its imprinting in the developing heart. PLoS Genet. 2012; 8:e1002956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Franco M.M., Prickett A.R., Oakey R.J.. The role of CCCTC-binding factor (CTCF) in genomic imprinting, development, and reproduction. Biol. Reprod. 2014; 91:125. [DOI] [PubMed] [Google Scholar]
- 40. Schoenherr C.J., Levorse J.M., Tilghman S.M.. CTCF maintains differential methylation at the Igf2/H19 locus. Nat. Genet. 2003; 33:66–69. [DOI] [PubMed] [Google Scholar]
- 41. Engel N., Thorvaldsen J.L., Bartolomei M.S.. CTCF binding sites promote transcription initiation and prevent DNA methylation on the maternal allele at the imprinted H19/Igf2 locus. Hum. Mol. Genet. 2006; 15:2945–2954. [DOI] [PubMed] [Google Scholar]
- 42. Kurukuti S., Tiwari V.K., Tavoosidana G., Pugacheva E., Murrell A., Zhao Z., Lobanenkov V., Reik W., Ohlsson R.. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:10684–10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Battistelli C., Busanello A., Maione R.. Functional interplay between MyoD and CTCF in regulating long-range chromatin interactions during differentiation. J. Cell Sci. 2014; 127:3757–3767. [DOI] [PubMed] [Google Scholar]
- 44. Du M., Beatty L.G., Zhou W., Lew J., Schoenherr C., Weksberg R., Sadowski P.D.. Insulator and silencer sequences in the imprinted region of human chromosome 11p15.5. Hum. Mol. Genet. 2003; 12:1927–1939. [DOI] [PubMed] [Google Scholar]
- 45. Rodriguez B.A., Weng Y.I., Liu T.M., Zuo T., Hsu P.Y., Lin C.H., Cheng A.L., Cui H., Yan P.S., Huang T.H.. Estrogen-mediated epigenetic repression of the imprinted gene cyclin-dependent kinase inhibitor 1C in breast cancer cells. Carcinogenesis. 2011; 32:812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fitzpatrick G.V., Pugacheva E.M., Shin J.Y., Abdullaev Z., Yang Y., Khatod K., Lobanenkov V.V., Higgins M.J.. Allele-specific binding of CTCF to the multipartite imprinting control region KvDMR1. Mol. Cell. Biol. 2007; 27:2636–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Demars J., Shmela M.E., Khan A.W., Lee K.S., Azzi S., Dehais P., Netchine I., Rossignol S., Le Bouc Y., El-Osta A.et al.. Genetic variants within the second intron of the KCNQ1 gene affect CTCF binding and confer a risk of Beckwith-Wiedemann syndrome upon maternal transmission. J. Med. Genet. 2014; 51:502–511. [DOI] [PubMed] [Google Scholar]
- 48. Cerrato F., De Crescenzo A., Riccio A.. Looking for CDKN1C enhancers. Eur. J. Hum. Genet. 2014; 22:442–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lopez-Abad M., Iglesias-Platas I., Monk D.. Epigenetic characterization of CDKN1C in placenta samples from non-syndromic intrauterine growth restriction. Front Genet. 2016; 7:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rovina D., La Vecchia M., Cortesi A., Fontana L., Pesant M., Maitz S., Tabano S., Bodega B., Miozzo M., Sirchia S.M.. Profound alterations of the chromatin architecture at chromosome 11p15.5 in cells from Beckwith-Wiedemann and Silver-Russell syndromes patients. Sci. Rep. 2020; 10:8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barch M. The AGT Cytogenetics Laboratory Manual. 1991; 1st ednNY: Raven Press. [Google Scholar]
- 52. Rooney D.E., Czepulkowski B.H.. Human Cytogenetics: A Practical Approach. 1986; 1st ednWashington DC: Oxford University Press. [Google Scholar]
- 53. Baker S.W., Duffy K.A., Richards-Yutz J., Deardorff M.A., Kalish J.M., Ganguly A.. Improved molecular detection of mosaicism in Beckwith-Wiedemann Syndrome. J. Med. Genet. 2020; 58:178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 May 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 55. Robinson J.T., Thorvaldsdottir H., Wenger A.M., Zehir A., Mesirov J.P.. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017; 77:e31–e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 57. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Consortium E.P., Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A.et al.. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020; 583:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prickett A.R., Barkas N., McCole R.B., Hughes S., Amante S.M., Schulz R., Oakey R.J.. Genome-wide and parental allele-specific analysis of CTCF and cohesin DNA binding in mouse brain reveals a tissue-specific binding pattern and an association with imprinted differentially methylated regions. Genome Res. 2013; 23:1624–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang Y., Song F., Zhang B., Zhang L., Xu J., Kuang D., Li D., Choudhary M.N.K., Li Y., Hu M.et al.. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 2018; 19:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O.et al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Davies J.O., Telenius J.M., McGowan S.J., Roberts N.A., Taylor S., Higgs D.R., Hughes J.R.. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat. Methods. 2016; 13:74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Magoc T., Salzberg S.L.. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011; 27:2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Langmead B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinformatics. 2010; 10.1002/0471250953.bi1107s32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Williams R.L. Jr, Starmer J., Mugford J.W., Calabrese J.M., Mieczkowski P., Yee D., Magnuson T.. fourSig: a method for determining chromosomal interactions in 4C-Seq data. Nucleic Acids Res. 2014; 42:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pohl A., Beato M.. bwtool: a tool for bigWig files. Bioinformatics. 2014; 30:1618–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Phanstiel D.H., Boyle A.P., Araya C.L., Snyder M.P.. Sushi.R: flexible, quantitative and integrative genomic visualizations for publication-quality multi-panel figures. Bioinformatics. 2014; 30:2808–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gu Z., Gu L., Eils R., Schlesner M., Brors B.. circlize Implements and enhances circular visualization in R. Bioinformatics. 2014; 30:2811–2812. [DOI] [PubMed] [Google Scholar]
- 71. Fagerberg L., Hallstrom B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K.et al.. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics. 2014; 13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lin S., Ferguson-Smith A.C., Schultz R.M., Bartolomei M.S.. Nonallelic transcriptional roles of CTCF and cohesins at imprinted loci. Mol. Cell. Biol. 2011; 31:3094–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hatada I., Mukai T.. Genomic imprinting of p57KIP2, a cyclin-dependent kinase inhibitor, in mouse. Nat. Genet. 1995; 11:204–206. [DOI] [PubMed] [Google Scholar]
- 74. Zheng H., Xie W.. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019; 20:535–550. [DOI] [PubMed] [Google Scholar]
- 75. Noordermeer D., Feil R.. Differential 3D chromatin organization and gene activity in genomic imprinting. Curr. Opin. Genet. Dev. 2020; 61:17–24. [DOI] [PubMed] [Google Scholar]
- 76. Fritz A.J., Sehgal N., Pliss A., Xu J., Berezney R.. Chromosome territories and the global regulation of the genome. Genes Chromosomes Cancer. 2019; 58:407–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Spicuglia S., Vanhille L.. Chromatin signatures of active enhancers. Nucleus. 2012; 3:126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Arima T., Kamikihara T., Hayashida T., Kato K., Inoue T., Shirayoshi Y., Oshimura M., Soejima H., Mukai T., Wake N.. ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res. 2005; 33:2650–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Varrault A., Gueydan C., Delalbre A., Bellmann A., Houssami S., Aknin C., Severac D., Chotard L., Kahli M., Le Digarcher A.et al.. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell. 2006; 11:711–722. [DOI] [PubMed] [Google Scholar]
- 80. Kanduri C., Thakur N., Pandey R.R.. The length of the transcript encoded from the Kcnq1ot1 antisense promoter determines the degree of silencing. EMBO J. 2006; 25:2096–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Thakur N., Tiwari V.K., Thomassin H., Pandey R.R., Kanduri M., Gondor A., Grange T., Ohlsson R., Kanduri C.. An antisense RNA regulates the bidirectional silencing property of the Kcnq1 imprinting control region. Mol. Cell. Biol. 2004; 24:7855–7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shin J.Y., Fitzpatrick G.V., Higgins M.J.. Two distinct mechanisms of silencing by the KvDMR1 imprinting control region. EMBO J. 2008; 27:168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mohammad F., Pandey R.R., Nagano T., Chakalova L., Mondal T., Fraser P., Kanduri C.. Kcnq1ot1/Lit1 noncoding RNA mediates transcriptional silencing by targeting to the perinucleolar region. Mol. Cell. Biol. 2008; 28:3713–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. O’Neill M.J. The influence of non-coding RNAs on allele-specific gene expression in mammals. Hum. Mol. Genet. 2005; R113–R120.14. [DOI] [PubMed] [Google Scholar]
- 85. Redrup L., Branco M.R., Perdeaux E.R., Krueger C., Lewis A., Santos F., Nagano T., Cobb B.S., Fraser P., Reik W.. The long noncoding RNA Kcnq1ot1 organises a lineage-specific nuclear domain for epigenetic gene silencing. Development. 2009; 136:525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Golding M.C., Magri L.S., Zhang L., Lalone S.A., Higgins M.J., Mann M.R.. Depletion of Kcnq1ot1 non-coding RNA does not affect imprinting maintenance in stem cells. Development. 2011; 138:3667–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are accessible in dbGAP (phs002408.v1.p1).