


Abstract
The TREX-TAP pathway is vital for mRNA export. For spliced mRNA, the TREX complex is recruited during splicing; however, for intronless mRNA, recruitment is sequence dependent. However, the export of cytoplasmic long noncoding RNA (lncRNA) is poorly characterized. We report the identification of a cytoplasmic accumulation region (CAR-N) in the intronless lncRNA, NKILA. CAR-N removal led to strong nuclear retention of NKILA, and CAR-N insertion promoted the export of cDNA transcripts. In vitro RNP purification via CAR-N, mass spectrometry, and siRNA screening revealed that SRSF1 and SRSF7 were vital to NKILA export, and identified a cluster of SRSF1/7 binding sites within a 55 nucleotide sequence in CAR-N. Significant nuclear enrichment of NKILA was observed for NKILA lacking CAR-N or the cluster of binding sites in knock-in models. Depletion of TREX-TAP pathway components resulted in strong nuclear retention of NKILA. RNA and protein immunoprecipitation verified that SRSF1/7 were bound to NKILA and interacted with UAP56 and ALYREF. Moreover, NKILA lacking CAR-N was unable to inhibit breast cancer cell migration. We concluded that the binding of SRSF1/7 to clustered motifs in CAR-N facilitated TREX recruitment, promoting the export of NKILA, and confirmed the importance of NKILA localization to its function.
INTRODUCTION
The correct localization of RNA is vital to its function. In the human genome, 95% of protein-coding genes contain multiple exons; the remaining 5% are genes with a single exon (1). All mRNAs encoded by multi-exon or single exon genes must be exported from the nucleus to the cytoplasm for translation. It is now widely accepted that the TREX-TAP pathway is fundamental for the export of transcripts from multi-exon genes, with the TREX complex recruited during splicing (2). Specifically, the cap-binding complex (CBC) binds the cap structure after transcription and the CBC component CBP80 interacts with the TREX component ALYREF during splicing to recruit TREX complex to the 5′-end of spliced mRNA; then, ALYREF interacts with the TAP/p15 (NXF1:NXT1) dimer at the nuclear pore, leading to the export of spliced mRNA from 5′ to 3′ (3,4). More recently, PABPN1-dependent ALYREF binding at the 3′-end of mRNA has been reported (5) and such interactions link 3′-end processing and mRNA export to enhance the export of non-polyadenylated histone mRNA (6). ALYREF and other TREX components have also been shown to interact with spliced mRNA in an exon junction complex (EJC)- and CBC-dependent manner (7). In contrast, in S. cerevisiae, the Prp19 complex is essential for stable TREX recruitment at actively transcribed intron-containing as well as intronless genes (8). Apart from ALYREF, serine/arginine-rich (SR) proteins, such as 9G8/SRSF7 and SRp20/SRSF3, have been reported to serve as adaptors in mRNA export that facilitate the recruitment of general export factors (9–11).
In contrast to the extensive knowledge of the export mechanisms of spliced mRNAs, much less is known about the export of naturally intronless mRNAs in humans. Indeed, the question of how these mRNAs are exported efficiently in the absence of splicing has remained unsolved for many decades. Our studies using four naturally intronless reporters from humans support a model of sequence-dependent recruitment of export machinery, in which cytoplasmic accumulation regions (CARs) critical for export were identified in the coding region of the transcripts (12,13). The CAR or the consensus sequence, CAR-E, facilitates the export of naturally intronless mRNAs via the TREX-TAP pathway (12,13). Other studies exploring the mechanisms for unspliced RNA export have focused on viral RNAs and identified the importance of RNA sequences/motifs. At present, the motifs mapped include the post-transcriptional regulatory element (PRE), constitutive transport element (CTE), Rev-response element (RRE), direct repeat (DR), and pre-mRNA processing enhancer (PPE). These motifs have been reported to interact mostly with the mRNA export receptor TAP/NXF1 or, in selected cases, with CRM1 (14–18), often in the presence of additional adaptors. For example, PRE utilizes the adaptor ICP27 to bind TAP/NXF1 (19) and the binding of hnRNP-L to PPE enhances export (20). Likewise, CTE-RNA recognition is facilitated by NXF1:NXT1 dimerization (21) and recruitment of ZC3H18 to the sub-element of PRE is essential for the association of TREX and RNA (22) to stimulate nuclear export.
The recent emergence of a large number of lncRNAs has provided further challenges in studying the mechanisms of RNA localization. Unlike mRNAs, lncRNAs do not encode proteins and are reported to have fewer exons (23). Initially, lncRNAs were believed to be localized in the nucleus (24), but their location was eventually clarified to be predominantly cytoplasmic. The analysis of human and Drosophila subcellular compartments revealed that approximately ∼75% of lncRNAs were enriched in the cytoplasmic fractions (25). However, several extensively studied lncRNAs, including XIST and MEG3, predominantly localize in the nucleus even though they are spliced; such a dilemma can be explained by the presence of sequences/motifs in the lncRNAs that facilitate their retention in the nucleus. The localization and stability of XIST is dependent on sequences scattered throughout the RNA, with a 5′ element that is vital to its correct localization and transcriptional silencing (26). For MEG3, the nuclear retention element is mapped and the element can recruit U1 small nuclear ribonucleoprotein (snRNP) components to retain MEG3 in the nucleus (27). The interaction of U1 snRNP with an RNA motif has also been linked with chromatin retention of other noncoding RNAs (28). In addition, a short pentamer, AGCCC, in BORG (29) and a longer repeating region in FIRRE have been reported to be vital for their localization, respectively (30). C-rich motifs derived from Alu repeats have been shown to govern lncRNA nuclear localization by recruiting HNRNPK (31). A C-rich nuclear enrichment pattern was also reported to be responsible for the nuclear localization of several human lncRNAs (32).
Like mRNAs, there are also mono-exonic lncRNAs, including NEAT1, MALAT1, NORAD and NKILA (33–36). NKILA is a cytoplasmic intronless lncRNA comprising 2615 nucleotides. It is reported to be a key factor in breast cancer metastasis and inflammation, and acts as a negative feedback regulator of NF-kappaB (34). However, the export mechanism of NKILA is still undetermined. Here, we have reported that similar to naturally intronless protein-coding genes, the nuclear export of NKILA is sequence dependent. A CAR vital to NKILA export, which we named CAR-N, was mapped, and functioned in both natural and heterologous contexts. Further, we identified SRSF1 and SRSF7 as trans factors recruited to CAR-N that facilitated TREX recruitment by interacting with UAP56 and ALYREF to promote NKILA export. In addition, we uncovered evidence to show that NKILA was also exported by the TREX-TAP pathway and that the removal of CAR-N impaired its ability to suppress cell migration. Overall, these results, in addition to those from previous studies, favor the sequence-dependent export of intronless mRNAs and lncRNAs via the TREX-TAP pathway.
MATERIALS AND METHODS
Constructs and antibodies
The full-length NKILA plasmid was a gift, kindly provided by Prof. Erwei Song of Sun Yat-sen University. NKILA truncation and deletion constructs were PCR-amplified from the full-length NKILA plasmid and inserted into the pcDNA3 vector (Invitrogen) at the KpnI and XhoI sites. To construct a chimeric reporter containing CAR-N with β-globin or GAS5 cDNA, overlap PCR was performed and the products were inserted into the pcDNA3 vector at the KpnI and XhoI sites. As a negative control for CAR-N, a sequence of similar length was amplified from the full-length NKILA plasmid and cloned using the same strategy. All constructs were sequenced before transfection. The primers used for constructs are listed in Supplementary Table S1.
Antibodies targeted to SRSF1 (ProteinTech 12929–2-AP), SRSF7 (ProteinTech 11044-1-AP), DHX9 (ProteinTech 17721-1-AP), HNRNPC (ProteinTech 11760-I-AP), PRPF8 (ProteinTech 11171-I-AP), SRSF9 (ProteinTech 17926-I-AP), U2AF2 (Santa Cruz-48804), E-cadherin (CST-14472), vimentin (ProteinTech 10366–1-AP), and β-actin (Origene-TA811000), all used at a 1:1000 dilution, and to tubulin (Sigma T9026; 1:10 000 dilution), were used for western blotting. Antibodies targeted to ALYREF, UAP56, TAP/NXF1, THOC2 and PRPF19 were gifts from the Reed Lab of Harvard Medical School. For immunoprecipitation (IP), SRSF7 (ab137247) and IgG (ab172730) antibodies were used. For western blotting after IP, a conformation-specific mouse anti-rabbit IgG (Cell Signaling 3678) secondary antibody was used.
Cell culture and transfection
MCF-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS; Life Technologies). For transient transfection, cells were grown to 70–80% confluency and treated with 1 μg plasmid DNA in Lipofectamine 2000 (Invitrogen). For siRNA treatment, cells at 30%–35% confluency were transfected with siRNA duplexes in Lipo3000 (Invitrogen). Non-targeting siRNA was used as the negative control. All siRNAs were manufactured by RiboBio, China, and the siRNA sequences and IDs are listed in Supplementary Table S2.
RNA-fluorescence in situ hybridization (FISH)
RNA-FISH was performed as previously described (13). Briefly, at 24 h after plasmid transfection, the cells were fixed with 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100. After washing in 1× SSC/50% formamide, the cells were incubated with the probe overnight at 37°C. DAPI was used for nuclear staining. The FISH probe sequence was 5′-AAGGCACGGGGGAGGGGCAAACAACAGATGGCTGGCAACTAGAAGGCAC AGTCGAGGCTGATCAGCGGGT. The FISH probe was prelabeled at the 5′-end with Alexa Fluor 546 NHS ester and purified by HPLC. Fluorescence was detected using a DMI8 microscope (Leica).
For endogenous RNA-FISH of NKILA, probe sets against NKILA and GAPDH were purchased from Biosearch Technologies, Inc., Petaluma, CA. MCF-7 cells were hybridized with the NKILA FISH probe set labeled with Quasar 570 dye in accordance with the manufacturer's instructions. Briefly, the cells were fixed with 4% PFA, permeabilized with 70% ethanol, and then incubated with the probe and hybridization buffer for ∼16 h at 37°C. GAPDH was used as the positive control.
Preparation of nuclear extracts
The nuclear and cytoplasm of cells were separated as described by Folco et al. (37). Briefly, MCF-7 cells at 90% confluency were detached using a cell scraper and then treated with hypotonic buffer. The swelled cells were lysed using a Dounce homogenizer. Low- and high-salt buffers were used to extract the nuclear fraction and dialyzed in buffer (20 mM HEPES, 100 mM KCL, 0.2 mM EDTA, and 20% glycerol) using MINI dialysis units (Thermo).
RNP purification in vitro
For in vitro RNP purification, the plasmid construct containing CAR-N or the control sequence driven by the T7 promoter was linearized using XhoI and the products were used as a template for RNA transcription. Transcription was performed with biotin-UTP at 37°C for 2 h using a mixture of UTP (2 mM) and ATP/CTP/GTP (all at 4 mM) with a UTP:biotin-16-UTP ratio of ∼12:1. Transcribed RNA was purified using a Mini Quick Spin Column (Roche) after DNase I treatment. RNA (1 μg) was incubated with 200 μl MCF-7 nuclear extract in RNA binding buffer (0.5% NP40, 50 mM KCl, 10 mM HEPES and 1.5 mM MgCl2) for 30 min at 30°C, and then 50 μl of streptavidin agarose resin (Thermo) was added and rotated at room temperature for ∼50 min. Finally, the proteins were eluted in protein gel loading buffer.
Protein immunoprecipitation
For protein IP, ALYREF and IgG antibodies or a mixture of SRSF1 and SRF7 antibodies were crosslinked to protein A beads (GE Healthcare) with DMP. The beads were incubated overnight at 4°C with nuclear extract from MCF-7 cells. The mixture was then washed in buffer (0.1% Triton X-100 in PBS) and the proteins were eluted in protein gel loading buffer.
RNA immunoprecipitation
For RIP, full-length NKILA or NKILA lacking the cluster of SRSF1 and SRSF7 binding sites (55 nt) was transfected into MCF-7 cells. Nuclear extracts were prepared 12 h after transfection. The mixture of SRSF1 and SRSF7 antibodies was crosslinked to protein A beads (GE Healthcare), incubated with nuclear extract, and washed with buffer (20 mM HEPES, 0.1% Triton X-100, 250 mM KCl, 2.5 mM EDTA and 5 mM DTT). RNA was then purified for use in RT-PCR by phenol/chloroform extraction and ethanol precipitation.
Inducible stable cell lines
The Flp-In T-Rex technique was used to establish doxycycline (Dox)-inducible stable cell lines. Full-length NKILA, NKILA-del-CAR-N and NKILA-del-55nt SR1/7 cluster were subcloned into the pcDNA5/FRT/TO vector (Invitrogen). The vectors were co-transfected with pOG44 plasmid encoding the recombinase and selected for by hygromycin. A final concentration of 1 μg/mL Dox was added to induce expression for 24 h and single-cell clone with good expression was selected and then expanded.
qRT-PCR
Total RNA was purified using TRIzol and treated with DNase I (Promega). 1 μg of DNA-free total RNA was reverse transcribed using M-MLV reverse transcriptase (Promega). For separation of nuclear and cytoplasmic fractions, the extraction kit (Beyotime China, P0027) was used. For RT-qPCR, PCR was performed with SYBR qPCR Q711 kit (Vazyme China, Q711-03) and Mx3005P Real-Time PCR System (Agilent). The thermocycling protocol was 95°C for 30 s followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. NKILA expression level was normalized to the level of 5srRNA in each fraction. The primer sequences are listed in Supplementary Table S1. Cyto/Nuc ratio was calculated using ΔCt values. All experiments were performed in triplicates.
Mass spectrometry
Mass spectrometric analysis was performed at the core facility of Tsinghua University. In-gel digestion was performed after catalase activity and proteins were identified as previously described (38). Briefly, 5 mM dithiothreitol (DTT) was added to reduce protein disulfide bonds and 11 mM iodoacetamide was added for alkylation. Sequencing grade-modified trypsin with 50 mM ammonium bicarbonate was used overnight at 37°C for in-gel digestion. An aqueous solution of 1% trifluoroacetic acid in 50% acetonitrile was used for peptide extraction and the volume was reduced using a SpeedVac. An Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Scientific) was used for the final peptide analysis. In Supplementary Table S3, contaminants such as keratin, tubulin, translation, and ribosome-associated proteins have been excluded and proteins enriched in CAR-N RNP relative to the control are listed.
Migration assay
For the migration assay, the cells were cultured in flat-bottomed plates and treated with TGF-β1 (Peprotech 100-21) at a final concentration of 10 ng/ml. After a monolayer was formed, a wound was created manually in ∼90–95% confluent cells using a 200 μl pipette tip. The medium was discarded to remove the detached cells and fresh medium was added. Images at 0 h were captured immediately after wound creation; then, the cells were incubated at 37ºC in a 5% CO2 incubator and images were captured at the indicated time points by a DMI4000B (Leica). The wound area was quantified by ImageJ and represented as a percentage migration relative to the gap area at 0 h.
Quantification and statistical analysis
The intensity of the RT-PCR bands was quantified by ImageJ and presented after normalization to the loading control. For statistical analysis, data from three independent experiments were compared with the control group by ANOVA and presented graphically using Prism 8 with specific P-values shown in each figure. The SRSF1 and SRSF7-binding motifs in CAR-N were predicted using the online tool RBPmap (http://rbpmap.technion.ac.il) by the algorithm based on the weighted-rank (WR) approach, as previously described (39). The WR reflects the propensity of suboptimal motifs to cluster around the significant motif. Briefly, the WR score was compared with the mean WR score of a pre-defined genomic region, the Z-score was the deviation of the WR score from the mean of background data sets, and the P-value represents the probability of obtaining a specific Z-score considering a normal distribution.
RESULTS
NKILA is predominantly localized in the cytoplasm
To determine the cellular localization of endogenous NKILA, the expression of NKILA transcript was analyzed by RT-PCR in MCF-7 cellular fractions. The expression of NKILA was detected in both the cytoplasmic and nuclear compartments: relatively, 65% was present in the cytoplasm and 35% was present in the nucleus (Figure 1A). Efficient nuclear and cytoplasmic fractionation was confirmed by western blotting with UAP56 and tubulin as the nuclear and cytoplasmic markers, respectively (Figure 1B).
Figure 1.

Intronless lncRNA NKILA is predominantly localized in the cytoplasm. (A) RT-PCR analysis of endogenous NKILA expression in the nucleus and cytoplasm of MCF-7 cells. Cyt: cytoplasm, Nuc: nucleus, +rt: with reverse transcriptase, -rt: without reverse transcriptase. (B) Western blotting analysis to check the efficiency of nuclear and cytoplasmic separation. Tubulin and UAP56 were used as the cytoplasmic and nuclear markers, respectively. (C) Schematic of NKILA and MEG3 reporter constructs. Digits indicate the nucleotide numbers. The position of the CMV promoter and the BGH polyA signal is marked. The sequence of the probe binding site is indicated by a red line. (D) RNA-FISH to show localization of RNA transcribed from the respective DNA reporter constructs after transfection. DAPI staining was used to identify the nucleus. Scale bar = 10 μm.
To explore the mechanism of intronless NKILA export, full-length NKILA cDNA cloned in the pcDNA3 vector was used as a reporter (Figure 1C). The NKILA reporter construct was transfected in MCF-7 cells and RNA-FISH was performed to determine the subcellular localization of the reporter transcripts. As shown in Figure 1D, NKILA reporter transcripts were predominantly observed in cytoplasm. As a control, the lncRNA MEG3 reporter was also transfected and the transcripts were predominantly located in the nucleus (Figure 1C and D). MEG3 was used as a control as it has been shown to be an lncRNA located predominantly in the nucleus (27). To confirm the localization of endogenous NKILA, endogenous RNA-FISH was performed with a probe set targeting NKILA, and GAPDH was used a positive control. We observed no signal from the sample with no addition of the probe, whereas both NKILA and GAPDH were predominantly cytoplasmic (Supplementary Figure S1A). We confirmed the FISH signal observed for NKILA by depleting or overexpressing NKILA. As shown in Supplementary Figure S1B, knockdown of NKILA led to the loss of FISH signal and NKILA overexpression resulted in a striking enhancement of fluorescence. The knockdown efficiency of siRNA targeting NKILA was confirmed by RT-PCR (Supplementary Figure S1C).
To exclude the possibility of cryptic splicing of NKILA transcripts, NKILA was transfected into MCF-7 cells and RT-PCR was performed with two pairs of primers spanning the full NKILA sequence. Products of the expected sizes were observed without noticeable cryptic splicing (Supplementary Figure S2A and B). Similarly, no obvious cryptic splicing in endogenous NKILA was observed using the two primer sets spanning the NKILA sequence (Supplementary Figure S2C). Together, these results suggested that NKILA was mostly unspliced cytoplasmic transcript at reporter and endogenous levels, and that the NKILA reporter could be used to examine the export mechanism of NKILA.
NKILA contains a cytoplasmic accumulation region (CAR) facilitating nuclear export
To test the hypothesis that intronless NKILA also contained a CAR vital to its export, as described for naturally intronless mRNAs (12,13), a series of truncated NKILA constructs were generated (Figure 2A, a–n) and transfected into MCF-7 cells. Next, RNA-FISH was performed to determine the localization of transcripts from each construct (Figure 2B, a–n).
Figure 2.

Identification of the cytoplasmic accumulation region in intronless lncRNA NKILA. (A) Schematic of the NKILA constructs and deletions. The numbers indicate the nucleotide position. Dotted lines represent the deleted sequence. (B) RNA-FISH images that show the subcellular distribution of NKILA fragments transcribed from the constructs shown in A. Scale bar = 10 μm. (C) The percentage of cells with a cytoplasmic signal (n = 3, 200–300 cells were counted for each transfection). ***P < 0.001. Transcripts with predominantly nuclear signal are labeled in red.
While truncation of different lengths at the 3′-end of NKILA had no significant impact on the cytoplasmic localization of NKILA transcripts (Figure 2A–C, a–d), removal of the 5′-end of NKILA led to striking nuclear retention: only 10% of cells had a cytoplasmic FISH signal, as compared with 90% of cells with the wild-type construct (Figure 2A–C, e–f). Further fine mapping revealed a region spanning ∼200 nucleotides between 251 and 450 nt of NKILA was vital for its cytoplasmic accumulation (Figure 2A–C, g–m); removal of this region resulted in significant export defect, with only 5% of cells showing a cytoplasmic FISH signal (Figure 2A–C, m). In addition, transcripts from this region alone were mostly localized in the cytoplasm, with 80% of cells showing a cytoplasmic FISH signal as compared with 90% in the control (Figure 2A–C, n). These data supported that intronless NKILA transcripts contained a CAR vital for cytoplasmic accumulation; subsequently, we named this CAR as CAR-N (Cytoplasmic Accumulation Region in NKILA).
CAR-N promotes nuclear export of cDNA transcripts
To examine whether CAR-N promoted the export of nuclear transcripts, CAR-N was inserted at the 5′-end of β-globin cDNA or lncRNA GAS5 cDNA, and a region of the same length from the 3′-end of NKILA (control sequence CS; NKILA nt 1773–1973) was inserted at the same position as a control (Figure 3A). Transfection followed by RNA-FISH revealed that CAR-N promoted the export of both cDNA transcripts. Specifically, the insertion of CAR-N into β-globin cDNA resulted in 60% of cells displaying cytoplasmic accumulation of transcripts, compared with ∼18% in the control (P < 0.001), and the insertion of CAR-N to GAS5 cDNA resulted in 90% of cells displaying cytoplasmic accumulation of the transcripts, compared with 5% in the control (P < 0.001) (Figure 3B and C). In contrast, insertion of the control sequence had no or little effect on the cellular localization of cDNA transcripts (Figure 3B and C). To exclude the possibility of cryptic splicing after CAR-N insertion, RT-PCR was performed and all the transcripts were of the expected size (Figure 3D), which indicated that the enhanced export was due to the insertion of CAR-N itself. These results suggested that CAR-N could facilitate the nuclear export of cDNA derived from mRNA, as well as lncRNA.
Figure 3.

CAR-N from NKILA promotes the export of nuclear cDNA transcripts derived from mRNA and lncRNA. (A) Schematic of β-globin cDNA and GAS5 cDNA constructs with or without insertion. CAR-N: cytoplasmic accumulation region in NKILA with nt 251–450, CS: Control Sequence (NKILA nt 1773–1973). (B) RNA-FISH images showing subcellular distribution of RNA transcribed from the constructs shown in A. Scale bar = 10 μm. (C) Percentage of cells with predominant cytoplasmic signal after the transfection of respective constructs (n = 3, 200–300 cells were counted for each transfection), ***P < 0.001, ns: non-significant P-value. (D) RT-PCR analysis was used to determine cryptic splicing. PC: positive control, PCR using plasmid DNA as a marker for the full-length product. +RT: with reverse transcriptase, –RT: negative control without reverse transcriptase.
SRSF1 and SRSF7 recruitment to CAR-N is required for efficient export of NKILA via the TREX-TAP pathway
To determine whether export of NKILA lncRNA also occurred via the TREX-TAP pathway, the TREX components UAP56, THOC2, ALYREF and TAP were depleted by treatment of MCF-7 cells with gene-specific siRNA. The knockdown efficiency of each siRNA was confirmed by western blotting analysis (Figure 4A). As shown in Figure 4B and C, depletion of UAP56 or TAP resulted in severe export defects with almost complete nuclear retention of NKILA reporter transcripts, depletion of THOC2 blocked nuclear export in approximately 88% of cells, whereas the depletion of ALYREF led to moderate nuclear retention in ∼50% of cells (Figure 4B and C). The lower nuclear retention of NKILA after ALYREF depletion may arise from a low knockdown efficiency or the presence of proteins with functional redundancy of ALYREF, such as SR proteins or UIF (40,41).
Figure 4.

Depletion of TREX/TAP/SRSF1/SRSF7 blocks export of the NKILA reporter. (A) Western blot showing the relative level of proteins in MCF-7 cells after transfection of siRNA targeting UAP56 (and its homolog URH49), TAP, THOC2, and ALYREF. Tubulin was used as the loading control. (B) RNA-FISH showing the localization of NKILA in knockdown cells. Scale bar = 5 μm. C) Percentage of cells with predominant cytoplasmic signal after transfection of appropriate siRNA (n = 3, 200–300 cells were counted for each transfection). ***P < 0.001. (D) Silver staining of RNP from CAR-N and control sequence after separation on SDS-PAGE. (E) Western blot showing the relative level of SRSF1 and SRSF7 in MCF-7 cells after treatment with appropriate siRNA. (F) Quantification of SRSF1 and SRSF7 protein level on the western blot in E (n = 3), ***P < 0.001. (G) RNA-FISH for NKILA localization in cells treated with siRNA. Scale bar = 10 μm. (H) Percentage of cells with apparent cytoplasmic signal in RNA-FISH (n = 3, 200–300 cells were counted in each transfection). ***P < 0.001.
To identify the trans factors recruited by CAR-N that function in NKILA export, RNPs from CAR-N or the control sequence in vitro was purified using biotin-streptavidin affinity purification. Proteins enriched in each RNP were separated using SDS-PAGE (Figure 4D). Mass spectrometric analysis detected a total of 118 proteins enriched in CAR-N RNP (Supplementary Table S3) and several of the proteins identified were confirmed by western blotting analysis (Supplementary Figure S3A). Gene ontology (GO) enrichment analysis revealed 13 proteins in RNA transport/localization/export categories (Supplementary Table S4). Screening of these proteins with siRNA revealed that SRSF1 and SRSF7 were required for the export of NKILA reporter transcripts; specifically, depletion of SRSF1 led to export blockage of the NKILA reporter in 60% of cells and depletion of SRSF7 led to blockage of nuclear export in 80% of cells (Figure 4E–H). Co-knockdown of SRSF1 and SRSF7 resulted in striking nuclear retention of NKILA in approximately 90% of cells (Figure 4E–H). To exclude off-target effects of siRNA, a second set of siRNA against SRSF1 or SRSF7 was used and a consistent phenotype was observed (Supplementary Figure S3B and C). Transfection of siRNA targeting other proteins into MCF-7 cells caused no or minor effect on the localization of NKILA reporter transcripts (Supplementary Figure S3D and E). The smaller effect of the depletion of different proteins (specifically, ALYREF and SRSF1) compared with the deletion of CAR-N may result from relatively low knockdown efficiency or multiple proteins interacting simultaneously with CAR-N to mediate the effect. Together, these results indicated that the recruitment of SRSF1 and SRSF7 to CAR-N was required to export NKILA via the TREX-TAP pathway.
Removal of cluster of SRSF 1/7-binding motifs resulted in the nuclear retention of NKILA
To minimize the cis-element sufficient to NKILA export, SRSF1 and SRSF7 binding sites within CAR-N were predicted using the online tool RBPmap (39). A cluster for overlapping sites of SRSF1 and SRSF7 was predicted within a small region (363–418 nt) of NKILA, with a total of three SRSF1 motifs and six SRSF7 motifs (Figure 5A). Intriguingly, removal of this cluster of SRSF1/7 motifs led to striking nuclear retention of NKILA, comparable with the deletion of 251–450 nt of NKILA (Figure 5B–D). This result further supported that SRSF1 and SRSF7 were recruited to CAR-N in a sequence-dependent manner and were critical for NKILA export.
Figure 5.

Removal of SRSF1 and SRSF7 sites led to severe nuclear retention of NKILA. (A) Predicted binding sites and motifs for SRSF1 and SRSF7 in CAR-N. (B) Schematic of NKILA reporter with cluster of SRSF1/7 sites deleted. (C) RNA-FISH images for NKILA localization of RNA transcribed from the indicated reporter. Scale bar = 5 μm. (D) Percentage of cells with apparent cytoplasmic location in RNA-FISH (n = 3, 200–300 cells were counted in each transfection). ***: P < 0.001. (E) RT-PCR of NKILA expression in the cytoplasmic and nuclear fractions of cells from knock-in cell lines expressing full-length NKILA, NKILA lacking CAR-N, and NKILA lacking SR1/7 binding sites. (F) Quantification of RT-PCR from two separate clones of each cell line (n = 2). *P < 0.05.
To further evaluate the impact of removal of CAR-N or the cluster of SRSF1/7 binding sites on NKILA localization, inducible FRT-HeLa stable cell lines were established using the Flp-In technique with plasmids of full-length NKILA, NKILA lacking CAR-N, and NKILA lacking the cluster of SRSF1/7 binding sites. RT-PCR analysis of RNA from the nuclear and cytoplasmic compartments indicated that the removal of these sequences led to significant enrichment of NKILA transcripts in the nucleus (Figure 5E). Specifically, the removal of CAR-N led to an increase from 38% to 63% of nuclear NKILA, and the removal of SRSF1/7 binding sites led to a similar increase to 60% (P < 0.05 in both cases) (Figure 5E and F), which further illustrated the importance of these sequences in the export of NKILA transcripts.
Depletion of TREX components or SRSF1/7 resulted in nuclear accumulation of endogenous NKILA
To clarify whether TREX components also functioned in the export of endogenous NKILA, MCF-7 cells were transfected with siRNA specific to UAP56, TAP, THOC2, or ALYREF followed by nucleo-cytoplasmic separation. RT-PCR analysis revealed that depletion of these components led to a moderate enrichment of endogenous NKILA in the nucleus. Specifically, knockdown of UAP56, TAP, THOC2, and ALYREF resulted in an increase in endogenous NKILA in the nucleus from 33% to 58%, 57%, 55% and 43%, respectively (Figure 6A–C).
Figure 6.

Depletion of TREX and SRSF 1/7 resulted in nuclear accumulation of endogenous NKILA. (A) Western blot showing relative level of TREX/TAP proteins in MCF-7 cells after transfection of appropriate siRNA. Tubulin was used as a loading control. (B) RT-PCR analysis of the relative distribution of endogenous NKILA. 5S rRNA was used as a loading control. Cyt: cytoplasm, Nuc: nucleus. (C) Quantification of NKILA in the nucleus as shown in B (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001. (D) Western blot showing the relative level of SRSF1/7 proteins after transfection of appropriate siRNA. (E) Quantification of SRSF1 and SRSF7 protein level on the western blot in D (n = 3), ***P < 0.001. (F) RT-PCR analysis of the distribution of endogenous NKILA in the nuclear and cytoplasmic fractions. (G) Quantification of NKILA in the nucleus as shown in F (n = 3). ***P < 0.001. (H) qRT-PCR showing the cytoplasmic/nuclear ratio of NKILA in each sample (n = 3). ***P < 0.001.
Similar analysis was performed for SRSF1 and SRSF7 depletion; knockdown of SRSF1 or SFSR7 individually led to a moderate increase in endogenous NKILA in the nucleus, from 37% to 50% and 52%, respectively, whereas simultaneous depletion of SRSF1 and SRSF7 resulted in further enrichment, to 60%, of endogenous NKILA in the nucleus (Figure 6D–G). To quantify the effect of protein depletion on NKILA export further, qRT-PCR analysis was performed of RNAs from both nuclear and cytoplasmic compartments after siRNA treatment. Consistent with the RT-PCR results, knockdown of each of the proteins led to a significant decrease of NKILA in the cytoplasm (Figure 6H). Together, these results supported that SRSF1 and SRSF7 were crucial for export of endogenous NKILA via the TREX-TAP pathway.
SRSF1 and SRSF7 binding to clustered motifs in CAR-N facilitated TREX recruitment
To build further links between the predicted cluster of SRSF1/7 binding sites in CAR-N, SRSF1/7 and TREX recruitment, RIP was performed using mixture of SRSF1 and SRSF7 antibodies after transfection of full-length NKILA or NKILA lacking cluster of SRSF1/7 binding sites to MCF7 cells. As shown in Figure 7A and B, a strong signal for NKILA was detected by RT-PCR analysis after RIP with SRSF1/7 antibodies compared with the control. Moreover, removal of the cluster of SRSF1/7 binding sites led to a ∼40% reduction in NKILA in RIP (P<0.01). Next, protein IP was performed and, as shown in Figure 7C, UAP56 and ALYREF were enriched in SRSF1/7 IP, and SRSF1/7 were enriched in ALYREF IP. The results were further verified by mass spectrometric analysis of the IP samples, with enrichment in total peptides of UAP56, ALYREF, and SRSF1/7 detected in SRSF1/7 and ALYREF IP compared with the control (Figure 7D). The full list of proteins detected by mass spectrometry is shown in Supplementary Tables S5 and S6. Unexpectedly, co-knockdown SRSF1 and SRSF7 resulted in a reduction of NKILA in RIP with ALYREF and TAP antibody (Figure 7E and F). Together, these results suggested that SRSF1 and SRSF7 bound on clustered motifs in CAR-N facilitated the recruitment of TREX.
Figure 7.

SRSF1/7 interacts with NKILA and components of the TREX complex. (A) RT-PCR analysis of NKILA in RIP with SRSF1/7 antibodies after full-length NKILA and NKILA lacking SRSF1/7 binding sites were transfected into MCF-7 cells. IgG was used as the control. (B) Quantification of RT-PCR bands in A from two independent experiments. ***P < 0.001, **P < 0.01. (C) Western blot showing the relative enrichment after protein IP. IgG was used as the control antibody. Nuclear extract from MCF-7 cells was used for IP with ALYREF and SRSF1/7 antibodies and UAP56, ALYREF, SRSF1 and SRSF7 were checked by western blotting analysis. (D) Total peptide numbers identified by mass spectrometry in IP samples. (E) RT-PCR analysis of NKILA in RIP performed with the ALYREF antibody after treatment of MCF-7 cells with siNC and siSRSF1/7. Right panel showing the mean of NKILA detected from two independent experiments. *P < 0.05. (F) As for E, except RIP was performed with the TAP antibody. *P < 0.05.
NKILA lacking CAR-N failed to suppress cell migration
NKILA is reported to suppress breast cancer cell migration by masking the phosphorylation motifs of IκB to inhibit NF-κB activation (34,42). To test whether NKILA localization affected cell migration, MCF-7 cells were treated with TGF-β and, after overexpression of full-length NKILA and NKILA lacking CAR-N, cell migration assays were performed. As shown in Figure 8A and B, the overexpression of full-length NKILA significantly suppressed migration of MCF-7 cells, with less than 12% and 20% wound area healed at 24 and 48 h, respectively, compared with 40% and 60% healing following overexpression of NKILA lacking CAR-N. Consistent with these results, significant increase in E-cadherin expression and decrease in vimentin expression were observed in cells transfected with full-length NKILA, whereas the overexpression of NKILA lacking CAR-N showed a small effect on the expression of E-cadherin and vimentin (Figure 8C). These results support that correct NKILA localization is vital for its function to suppress cancer cell migration.
Figure 8.

Functional impairment of cell migration suppression for NKILA lacking CAR-N. (A) Wound healing in cell migration assay in MCF-7 cells treated with TGF-β. (B) Quantification of cell migration at the indicated time points relative to 0 h (n = 3), ***P < 0.001, **P < 0.01. (C) Western blot showing the relative expression of E-cadherin and vimentin; β-actin is used as the loading control.
DISCUSSION
Different classes of RNA have been reported to have distinct mechanisms of nuclear export. The export of microRNAs is mediated by exportin-5 (43) and rRNAs are exported via the CRM1-Nmd3 pathway (44,45), whereas exportin-t is vital for the nuclear export of tRNAs (46) and snRNAs are exported via the CRM1-PHAX pathway (47,48). Spliced mRNAs are exported via the TREX-TAP pathway and splicing plays a fundamental role in the export (11), with the TREX complex recruited to the 5′-end of the transcripts during splicing via the interaction between the cap-binding complex component CBP80 and TREX component ALYREF (3,4). The TREX-TAP pathway is also used in the export of naturally intronless mRNAs in humans (12); the recruitment of export machinery is sequence-dependent in the absence of splicing, with the CAR vital to the export identified in the coding region (12,13).
In this study, we aimed to determine how intronless lncRNA NKILA was exported. Intriguingly, consistent with our previous report of CARs in naturally intronless mRNAs, a cytoplasmic accumulation region (CAR-N) of ∼200 nt near the 5′-end of NKILA was mapped that was vital to NKILA export. Removal of this CAR-N led to a striking decrease in NKILA export, and insertion of CAR-N upstream of β-globin cDNA or lncRNA GAS5 cDNA promoted export of the transcripts. Previously, it was reported that β-globin mRNA contained a nuclear retention element that could be overcome by increasing the length of the mRNA (49). However, in our study, the insertion sequence of similar length to CAR-N from the 3′-end of NKILA did not promote nuclear export of the reporter transcripts when compared with CAR-N, suggesting that the export of these reporters was not due to the increase in sequence length.
NKILA has been reported as an intronless lncRNA localized in the cytoplasm (34). Although the potential 5′- and 3′-splice sites were not recognized by constitutive splice site prediction, the UCSC genome browser showed a spliced NKILA variant from EST-generated data with the intron spans from nucleotides 433 to 1079. To clarify if there was cryptic NKILA splicing in our system, two sets of primers were used in the RT-PCR analysis for the reporter and endogenous transcripts, with one primer specifically designed to detect potential splicing variants. Although we detected no significant cryptic splicing in NKILA, caution should be taken as cryptic splice sites could be activated under certain conditions.
It is well known that RNA localization is the basis for its function; however, this is also true for lncRNAs. For example, the lncRNA MALAT1 is localized to nuclear speckles and NEAT1 is associated with paraspeckles and both are functionally active at these sites (50). The amusing accumulation of lncRNA lacking translation potential in the cytoplasm is generally considered an indication of cytoplasmic roles. For example, cytoplasmic NORAD sequesters PUMILO protein for genomic stability (33), a subset of cytoplasmic lncRNA regulates mRNA translation (51) or base pairing with Alu element in mRNA leading to mRNA decay (52). For the intronless lncRNA NKILA, it was reported to block IKB phosphorylation in the cytoplasm (34). More recently, NKILA was shown to suppress the TGF-β–induced epithelial-mesenchymal transition by blocking migration in breast cancer (42). In our study, we have shown that the action of NKILA as a suppressor of migration was largely dependent on its cytoplasmic accumulation as the deletion of CAR-N retained NKILA in the nucleus and impaired its ability to suppress migration in MCF-7 breast cancer cells. We also tried to delete CAR-N or the cluster of SRSF1/7 binding sites in NKILA from the genome of MCF-7 cells using the CRISPR technique, but no single cell colony expanded, further implying the vital role of NKILA in the proliferation of MCF-7 cells. Surprisingly, Liu et al. (2015) (34) identified a region between 300–500 nt of NKILA that was responsible for interaction with p65; this region largely overlapped with the CAR-N (251–450 nt) mapped in this study. Moreover, they also showed that NKILA co-localized with p65 in the cytoplasm of MCF-7 cells. In our study, mass spectrometric analysis of the purification of RNP from CAR-N using the nuclear extract from MCF-7 cells did not identify p65, which further supported that the binding of NKILA with p65 is indeed cytoplasmic in nature.
Moreover, we showed the export defects in NKILA by knockdown of TREX components UAP56, THOC2, and to a lesser extent, by ALYREF. It should be noted that in our study, siRNAs targeting UAP56 and its homolog URH49 were used for UAP56 knockdown as they were previously reported to achieve efficient blockage of the export of wild type β-globin transcripts only with co-depletion of these two proteins (53). Although the moderate effect of ALYREF depletion may come from a low knockdown efficiency, it is most likely due to proteins with functional redundancy of ALYREF. For example, previous studies suggested that ALYREF was dispensable for mRNA export in both drosophila and C. elegans (54,55). There is no doubt that ALYREF is a major adaptor between mRNA and TAP; however, several SR proteins, including 9G8/SRSF7 and SRp20/SRSF3, can also act as a bridge between mRNA and the TAP receptor (40). UIF is another mRNA export adaptor that works together with ALYREF (41). SR proteins (9G8/SRSF7, SRp20/SRSF3, and ASF/SF2/SRSF1) have been reported to interact with the same domain of NXF1/TAP as ALYREF, which may lead to competitive interactions (40). Moreover, a peptide was identified adjacent to the RRMs of SRp20 and 9G8 critical for interaction with TAP (9). Most recently, it has been reported that speckle association of intronless transcripts is mediated by SR proteins that enhance TREX recruitment for enhanced export of unspliced transcripts (56).
We previously proposed a model of sequence-dependent export of intronless mRNA in the absence of splicing, in which CAR and the consensus CAR-E could eventually recruit mRNA export machinery (12,13). However, the proteins that bind to CAR-E to initiate TREX recruitment have not been identified. A later study reported that a sub-element in PRE can enhance intronless mRNA export by recruiting TREX via ZC3H18 (22). Most recently, sequences featuring G/C-rich, 5′-biased, and m6A-modified regions have been reported to promote NXF1/TAP-dependent export of intronless RNAs (57). In this study, we identified SRSF1 and SRSF7 as trans factors vital to NKILA export that were recruited by the cluster of binding sites of these two proteins in CAR-N. Together, SRSF1 and SRSF7 facilitate TREX recruitment by interacting with UAP56 and ALYREF to promote NKILA export via the TREX-TAP pathway (proposed model in Figure 9). Thus, the data from this study provide direct and solid evidence to support the model of sequence-dependent RNA export.
Figure 9.
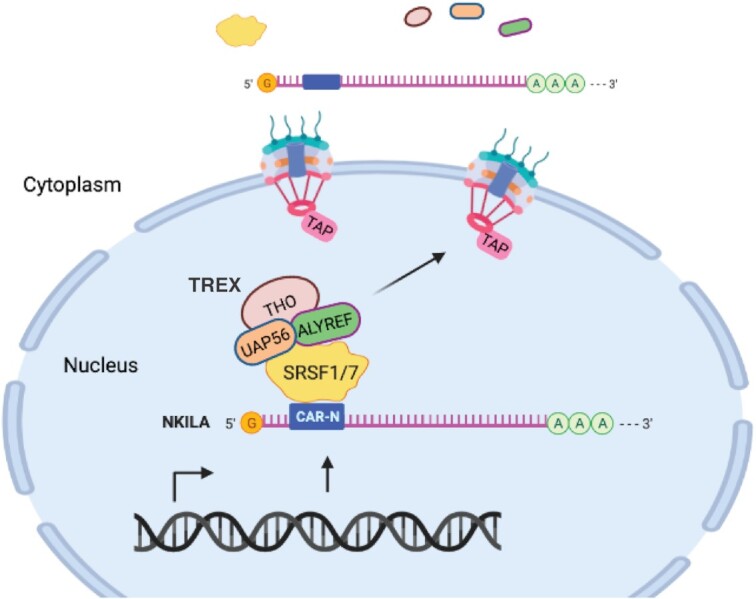
Model for the export of the intronless lncRNA NKILA. CAR-N with cluster of SRSF1/7 binding sites in NKILA recruits SRSF1 and SRSF7, which facilitate TREX recruitment by interaction with UAP56 and ALYREF and mediate the export of NKILA via the TREX-TAP pathway. (Created with Biorender.com)
With the evidence accumulated so far, this model can be further extended to sequence-dependent RNA localization in several ways. (i) Sequence-dependent RNA export: CARs and the consensus element CAR-E are vital for the export of intronless HSPB3, IFNβ1, IFNα1 and c-JUN transcripts in humans (12,13). Recently, 5′-biased regions have been reported to play a significant role in export of single exon transcripts, including the intronless lncRNA, NORAD (57). More interestingly, independent verification of the correlation of CAR-E and export supported its function in nuclear export and even implied the involvement of such motifs in the export of spliced RNAs (57). (ii) Sequence-dependent RNA retention in the nucleus: our previous study of the nuclear retention of spliced lncRNA MEG3 revealed an NRE of ∼350 nt interacting with the U1 snRNP components SNRPA, SNRNP70, and SNRPD2 to retain MEG3 in the nucleus, and the removal of the NRE or depletion of these proteins both led to cytoplasmic localization of MEG3 (27). A specific motif or region was mapped for the nuclear retention of spliced lncRNA BORG and FIRRE (29,30). The localization of intronless lncRNA MALAT1 to nuclear speckles depends on two separate regions of 1 kb and 600 nt in MALAT1, which can interact with RNPS1 to facilitate its localization (58). In future studies, it will be of interest to investigate whether such sequences share consensus motifs and the trans factors recruited.
DATA AVAILABILITY
Lists of proteins identified by mass spectrometry are included in supplementary data.
Supplementary Material
Contributor Information
Misbah Khan, Institute of Cancer Stem Cell, Cancer Center, Dalian Medical University, 9 West Section, Lvshun South Rd, Dalian 116044, P.R. China.
Shuai Hou, Institute of Cancer Stem Cell, Cancer Center, Dalian Medical University, 9 West Section, Lvshun South Rd, Dalian 116044, P.R. China; School of Food Science and Technology, Dalian Polytechnic University, Dalian 1160343, P.R. China.
Sikandar Azam, Institute of Cancer Stem Cell, Cancer Center, Dalian Medical University, 9 West Section, Lvshun South Rd, Dalian 116044, P.R. China; Department of Microbial Pathogenesis and Immunology, Texas A&M Health Science Center, Bryan, USA.
Haixin Lei, Institute of Cancer Stem Cell, Cancer Center, Dalian Medical University, 9 West Section, Lvshun South Rd, Dalian 116044, P.R. China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31670823 to H.L.]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Shabalina S.A., Ogurtsov A.Y., Spiridonov A.N., Novichkov P.S., Spiridonov N.A., Koonin E.V.. Distinct patterns of expression and evolution of intronless and intron-containing mammalian genes. Mol. Biol. Evol. 2010; 27:1745–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kohler A., Hurt E.. Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. Mol. Cell Biol. 2007; 8:761–773. [DOI] [PubMed] [Google Scholar]
- 3. Cheng H., Dufu K., Lee C.S., Hsu J.L., Dias A., Reed R.. Human mRNA export machinery recruited to the 5′ end of mRNA. Cell. 2006; 127:1389–1400. [DOI] [PubMed] [Google Scholar]
- 4. Masuda S., Das R., Cheng H., Hurt E., Dorman N., Reed R.. Recruitment of the human TREX complex to mRNA during splicing. Genes Dev. 2005; 19:1512–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shi M., Zhang H., Wu X.D., He Z.S., Wang L.T., Yin S.Y., Tian B., Li G.H., Cheng H.. ALYREF mainly binds to the 5′ and the 3′ regions of the mRNA in vivo. Nucleic Acids Res. 2017; 45:9640–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fan J., Wang K., Du X., Wang J.S., Chen S.L., Wang Y.M., Shi M., Zhang L., Wu X.D., Zheng D.H.et al.. ALYREF links 3-end processing to nuclear export of non-polyadenylated mRNAs. EMBO J. 2019; 38:e99910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gromadzka A.M., Steckelberg A.L., Singh K.K., Hofmann K., Gehring N.H.. A short conserved motif in ALYREF directs cap- and EJC-dependent assembly of export complexes on spliced mRNAs. Nucleic Acids Res. 2016; 44:2348–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chanarat S., Seizl M., Strasser K.. The Prp19 complex is a novel transcription elongation factor required for TREX occupancy at transcribed genes. Gene Dev. 2011; 25:1147–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hargous Y., Hautbergue G.M., Tintaru A.M., Skrisovska L., Golovanov A.P., Stevenin J., Lian L.Y., Wilson S.A., Allain F.H.. Molecular basis of RNA recognition and TAP binding by the SR proteins SRp20 and 9G8. EMBO J. 2006; 25:5126–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang Y., Steitz J.A.. Splicing factors SRp20 and 9G8 promote the nucleocytoplasmic export of mRNA. Mol. Cell. 2001; 7:899–905. [DOI] [PubMed] [Google Scholar]
- 11. Reed R., Cheng H.. TREX, SR proteins and export of mRNA. Curr. Opin. Cell Biol. 2005; 17:269–273. [DOI] [PubMed] [Google Scholar]
- 12. Lei H., Dias A.P., Reed R.. Export and stability of naturally intronless mRNAs require specific coding region sequences and the TREX mRNA export complex. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:17985–17990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lei H., Zhai B., Yin S., Gygi S., Reed R.. Evidence that a consensus element found in naturally intronless mRNAs promotes mRNA export. Nucleic Acids Res. 2013; 41:2517–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hammarskjold M.L. Constitutive transport element-mediated nuclear export. Curr. Top. Microbiol. Immunol. 2001; 259:77–93. [DOI] [PubMed] [Google Scholar]
- 15. Liu X., Mertz J.E.. HnRNP L binds a cis-acting RNA sequence element that enables intron-dependent gene expression. Genes Dev. 1995; 9:1766–1780. [DOI] [PubMed] [Google Scholar]
- 16. Paca R.E., Ogert R.A., Hibbert C.S., Izaurralde E., Beemon K.L.. Rous sarcoma virus DR posttranscriptional elements use a novel RNA export pathway. J. Virol. 2000; 74:9507–9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pollard V.W., Malim M.H.. The HIV-1 Rev protein. Annu. Rev. Microbiol. 1998; 52:491–532. [DOI] [PubMed] [Google Scholar]
- 18. Zang W.Q., Li B., Huang P.Y., Lai M.M., Yen T.S.. Role of polypyrimidine tract binding protein in the function of the hepatitis B virus posttranscriptional regulatory element. J. Virol. 2001; 75:10779–10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen I.H., Sciabica K.S., Sandri-Goldin R.M.. ICP27 interacts with the RNA export factor Aly/REF to direct herpes simplex virus type 1 intronless mRNAs to the TAP export pathway. J. Virol. 2002; 76:12877–12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guang S., Felthauser A.M., Mertz J.E.. Binding of hnRNP L to the pre-mRNA processing enhancer of the herpes simplex virus thymidine kinase gene enhances both polyadenylation and nucleocytoplasmic export of intronless mRNAs. Mol. Cell. Biol. 2005; 25:6303–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aibara S., Katahira J., Valkov E., Stewart M.. The principal mRNA nuclear export factor NXF1:NXT1 forms a symmetric binding platform that facilitates export of retroviral CTE-RNA. Nucleic Acids Res. 2015; 43:1883–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chi B., Wang K., Du Y., Gui B., Chang X., Wang L., Fan J., Chen S., Wu X., Li G.et al.. A sub-element in PRE enhances nuclear export of intronless mRNAs by recruiting the TREX complex via ZC3H18. Nucleic Acids Res. 2014; 42:7305–7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G.et al.. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012; 22:1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khalil A.M., Guttman M., Huarte M., Garber M., Raj A., Rivea Morales D., Thomas K., Presser A., Bernstein B.E., van Oudenaarden A.et al.. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:11667–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benoit Bouvrette L.P., Cody N.A.L., Bergalet J., Lefebvre F.A., Diot C., Wang X., Blanchette M., Lecuyer E.. CeFra-seq reveals broad asymmetric mRNA and noncoding RNA distribution profiles in Drosophila and human cells. RNA. 2018; 24:98–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wutz A., Rasmussen T.P., Jaenisch R.. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 2002; 30:167–174. [DOI] [PubMed] [Google Scholar]
- 27. Azam S., Hou S., Zhu B., Wang W., Hao T., Bu X., Khan M., Lei H.. Nuclear retention element recruits U1 snRNP components to restrain spliced lncRNAs in the nucleus. RNA Biol. 2019; 16:1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yin Y., Lu J.Y., Zhang X., Shao W., Xu Y., Li P., Hong Y., Cui L., Shan G., Tian B.et al.. U1 snRNP regulates chromatin retention of noncoding RNAs. Nature. 2020; 580:147–150. [DOI] [PubMed] [Google Scholar]
- 29. Zhang B., Gunawardane L., Niazi F., Jahanbani F., Chen X., Valadkhan S.. A novel RNA motif mediates the strict nuclear localization of a long noncoding RNA. Mol. Cell. Biol. 2014; 34:2318–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hacisuleyman E., Goff L.A., Trapnell C., Williams A., Henao-Mejia J., Sun L., McClanahan P., Hendrickson D.G., Sauvageau M., Kelley D.R.et al.. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014; 21:198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lubelsky Y., Ulitsky I.. Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Nature. 2018; 555:107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shukla C.J., McCorkindale A.L., Gerhardinger C., Korthauer K.D., Cabili M.N., Shechner D.M., Irizarry R.A., Maass P.G., Rinn J.L.. High-throughput identification of RNA nuclear enrichment sequences. EMBO J. 2018; 37:e98452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee S., Kopp F., Chang T.C., Sataluri A., Chen B., Sivakumar S., Yu H., Xie Y., Mendell J.T.. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell. 2016; 164:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu B., Sun L., Liu Q., Gong C., Yao Y., Lv X., Lin L., Yao H., Su F., Li D.et al.. A cytoplasmic NF-kappaB interacting long noncoding RNA blocks IkappaB phosphorylation and suppresses breast cancer metastasis. Cancer Cell. 2015; 27:370–381. [DOI] [PubMed] [Google Scholar]
- 35. Naganuma T., Hirose T.. Paraspeckle formation during the biogenesis of long non-coding RNAs. RNA Biol. 2013; 10:456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sun Y., Ma L.. New insights into long non-coding RNA MALAT1 in cancer and metastasis. Cancers (Basel). 2019; 11:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Folco E.G., Lei H., Hsu J.L., Reed R.. Small-scale nuclear extracts for functional assays of gene-expression machineries. J. Vis. Exp. 2012; 64:4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li X., Tao J., Han J., Hu X., Chen Y., Deng H., Zhang G., Hu X., Mi K.. The gain of hydrogen peroxide resistance benefits growth fitness in mycobacteria under stress. Protein Cell. 2014; 5:182–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Paz I., Kosti I., Ares M. Jr, Cline M., Mandel-Gutfreund Y.. RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014; 42:W361–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang Y., Gattoni R., Stevenin J., Steitz J.A.. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol. Cell. 2003; 11:837–843. [DOI] [PubMed] [Google Scholar]
- 41. Hautbergue G.M., Hung M.L., Walsh M.J., Snijders A.P.L., Chang C.T., Jones R., Ponting C.P., Dickman M.J., Wilson S.A.. UIF, a new mRNA export adaptor that works together with REF/ALY, requires FACT for recruitment to mRNA. Curr. Biol. 2009; 19:1918–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu W., Chen F., Cui X., Yang L., Chen J., Zhao J., Huang D., Liu J., Yang L., Zeng J.et al.. LncRNA NKILA suppresses TGF-beta-induced epithelial-mesenchymal transition by blocking NF-kappaB signaling in breast cancer. Int. J. Cancer. 2018; 143:2213–2224. [DOI] [PubMed] [Google Scholar]
- 43. Lund E., Guttinger S., Calado A., Dahlberg J.E., Kutay U.. Nuclear export of microRNA precursors. Science. 2004; 303:95–98. [DOI] [PubMed] [Google Scholar]
- 44. Ho J.H., Kallstrom G., Johnson A.W.. Nmd3p is a Crm1p-dependent adapter protein for nuclear export of the large ribosomal subunit. J. Cell Biol. 2000; 151:1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thomas F., Kutay U.. Biogenesis and nuclear export of ribosomal subunits in higher eukaryotes depend on the CRM1 export pathway. J. Cell Sci. 2003; 116:2409–2419. [DOI] [PubMed] [Google Scholar]
- 46. Kutay U., Lipowsky G., Izaurralde E., Bischoff F.R., Schwarzmaier P., Hartmann E., Gorlich D.. Identification of a tRNA-specific nuclear export receptor. Mol. Cell. 1998; 1:359–369. [DOI] [PubMed] [Google Scholar]
- 47. Ohno M., Segref A., Bachi A., Wilm M., Mattaj I.W.. PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell. 2000; 101:187–198. [DOI] [PubMed] [Google Scholar]
- 48. Fornerod M., Ohno M., Yoshida M., Mattaj I.W.. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997; 90:1051–1060. [DOI] [PubMed] [Google Scholar]
- 49. Akef A., Lee E.S., Palazzo A.F.. Splicing promotes the nuclear export of beta-globin mRNA by overcoming nuclear retention elements. RNA. 2015; 21:1908–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen L.L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016; 41:761–772. [DOI] [PubMed] [Google Scholar]
- 51. Yoon J.H., Abdelmohsen K., Srikantan S., Yang X., Martindale J.L., De S., Huarte M., Zhan M., Becker K.G., Gorospe M.. LincRNA-p21 suppresses target mRNA translation. Mol. Cell. 2012; 47:648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gong C., Maquat L.E.. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature. 2011; 470:284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dias A.P., Dufu K., Lei H.X., Reed R.. A role for TREX components in the release of spliced mRNA from nuclear speckle domains. Nat. Commun. 2010; 1:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Longman D., Johnstone I.L., Caceres J.F.. The Ref/Aly proteins are dispensable for mRNA export and development in Caenorhabditis elegans. RNA. 2003; 9:881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gatfield D., Izaurralde E.. REF1/Aly and the additional exon junction complex proteins are dispensable for nuclear mRNA export. J. Cell Biol. 2002; 159:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang K., Wang L., Wang J., Chen S., Shi M., Cheng H.. Intronless mRNAs transit through nuclear speckles to gain export competence. J. Cell Biol. 2018; 217:3912–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zuckerman B., Ron M., Mikl M., Segal E., Ulitsky I.. Gene architecture and sequence composition underpin selective dependency of nuclear export of long RNAs on NXF1 and the TREX complex. Mol. Cell. 2020; 79:251–267. [DOI] [PubMed] [Google Scholar]
- 58. Miyagawa R., Tano K., Mizuno R., Nakamura Y., Ijiri K., Rakwal R., Shibato J., Masuo Y., Mayeda A., Hirose T.et al.. Identification of cis- and trans-acting factors involved in the localization of MALAT-1 noncoding RNA to nuclear speckles. RNA. 2012; 18:738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Lists of proteins identified by mass spectrometry are included in supplementary data.