


Abstract
Metal-induced genes are usually transcribed at relatively low levels under normal conditions and are rapidly activated by heavy metal stress. Many of these genes respond preferentially to specific metal-stressed conditions. However, the mechanism by which the general transcription machinery discriminates metal stress from normal conditions and the regulation of MTF-1-meditated metal discrimination are poorly characterized. Using a focused RNAi screening in Drosophila Schneider 2 (S2) cells, we identified a novel activator, the Drosophila gawky, of metal-responsive genes. Depletion of gawky has almost no effect on the basal transcription of the metallothionein (MT) genes, but impairs the metal-induced transcription by inducing the dissociation of MTF-1 from the MT promoters and the deficient nuclear import of MTF-1 under metal-stressed conditions. This suggests that gawky serves as a ‘checkpoint’ for metal stress and metal-induced transcription. In fact, regular mRNAs are converted into gawky-controlled transcripts if expressed under the control of a metal-responsive promoter, suggesting that whether transcription undergoes gawky-mediated regulation is encrypted therein. Additionally, lack of gawky eliminates the DNA binding bias of MTF-1 and the transcription preference of metal-specific genes. This suggests a combinatorial control of metal discrimination by gawky, MTF-1, and MTF-1 binding sites.
INTRODUCTION
Cells grow well only under a narrow range of conditions. For cells to survive, a variety of genes are conditionally expressed to adapt to different cellular and stress conditions (for review, see (1)). For example, metal homeostasis (i.e. the proper balance of metals inside cells) is controlled by many metal-induced genes that encode proteins to maintain the appropriate concentrations of heavy metals by importing, storing, expelling, or sequestering metals. The responses of metal-induced genes are often rapid and robust to meet the challenges of heavy metals in the environment. As an important aspect of metal-responsive gene regulation, the transcriptional outputs of these genes are often regulated and coordinated by various regulatory factors (for review, see (2)).
Metal-responsive genes usually contain multiple copies of metal-responsive elements (MREs) that are short DNA sequence motifs and typically present in the upstream regulatory sequences. MREs are recognized by the metal regulatory transcription factor-1 (MTF-1), and play important roles in transcription activation of nearby genes (for review see (2–4)). The core consensus sequence of MREs is TGCRCNC (R = A or G, N = any nucleotide). The sixth nucleotide of the core MRE can dictate MTF-1 binding bias and metal-specific transcription during different metal stresses, demonstrating a mechanism of metal discrimination (5). However, it is unclear whether there are additional factors that contribute to the regulation of these events.
As a member of the nucleocytoplasmic shuttling proteins, MTF-1 has been reported to act as the major transcription factor involved in defense against different heavy metals (for review see (2–4)). Based on the degree of influence that heavy metals have on biological systems, they can be divided into two groups: essential and non-essential metals (for review see (6)). Essential metals, such as copper, represent integral components of various proteins in appropriate amounts but are often toxic at high concentrations. Therefore, the concentration range by which essential metals activate MTF-1 is relatively high. In contrast, non-essential or toxic metals, such as cadmium, are simply toxic even in trace amounts (for review, see (2,6)). One class of evolutionarily conserved genes that are controlled by MTF-1 is the metallothionein (MT) family. MT can be stimulated by heavy metals and encodes a type of small, Cys-rich and metal-binding proteins (6–10). Zinc-bound MT proteins quickly respond to heavy metals in the environment by the release of zinc that can be sensed and bound to MTF-1. Metal-activated MTF-1 then moves into the nucleus where it recognizes MREs of the MT genes and recruits a series of transcriptional co-activators to coordinate MT expression (11–13).
Even though this model can explain some aspects of metal-responsive gene activation and the functions of MTF-1 in metal homeostasis, it is particularly important to note that MTF1 is not only responsive to metal overload, but also contributes to the regulation of many other categories of genes. First, MTF-1 mediates the expression of membrane metal transporters across species (14–20). For example, the zinc transporters, such as Slc30a1 (14), Slc30a2 (15) and Slc39a10 (14,16), and the iron transporter Slc40a1 (17) are target genes of MTF-1 in mouse. Also, the zinc transporter gene ZnT35C (18), the ferritin genes (18), and the copper transporter gene Ctr1B (19) have been reported to be activated in an MTF-1-dependent manner in Drosophila. Second, in defense against redox stress, MTF-1 activates the expression of selenoprotein 1 (Sepw1) which encodes an antioxidant protein (14) but represses the transcription activities of the redox sensing selenoprotein H (SELH) gene and the thioredoxin reductase 2 (Txnrd2) gene (21). Third, in response to hypoxic stress, placenta growth factor (PlGF) is highly induced via MTF-1 and its cofactors in human and mouse (22,23). Fourth, MTF-1 is involved in the transcriptional regulation of certain genes, such as tear lipocalin (LCN1) (24) and complement factor B (CFB) (25), without an obvious connection to a stress response (14,24,25). Firth, MTF-1 also plays essential biological functions in embryonic development and cell differentiation (26–28). Mouse embryos lacking MTF-1 show embryonic lethality probably due to the dysregulation of γ-glutamylcysteine synthetase (γ-GCS), an essential MTF-1 target gene in glutathione biosynthesis (27). A recent study demonstrated that MTF-1 regulates myoblast differentiation via direct interaction with the myogenic gene promoter (28). These studies suggest that MTF-1 is an important factor involved in the regulation of a diverse set of genes, thereby protecting cells from various stressors. Therefore, to exert its functions precisely, MTF-1 may rely on other proteins to dictate its distinct functions.
Nevertheless, depletion or overexpression of MTF-1 significantly affects the expression of metal-responsive genes under both normal and metal-stressed conditions, indicating that both the basal and metal-induced transcription of metal-responsive genes are controlled by MTF-1 (19,29). However, the DNA binding patterns of MTF-1 in unstressed and copper treated cells are similar to each other (5,30,31). These findings suggest that the process of genome-reading and transcription activation is more complicated than currently appreciated, and indicate the requirement of additional regulatory factors that assist MTF-1 and the general transcriptional machinery to distinguish between normal and metal-stressed conditions.
Using RNAi screening and rescue strategies, we identified that the Drosophila gawky is required for the transcription activation of the MT genes in response to metal stress, but is bypassed under normal conditions. Gawky is a Gly/Trp-rich protein that is usually thought to act as a molecular scaffold bringing together various RNA decay enzymes and trigging RNA degradation in P-bodies (32–36). Here, we provided evidence that gawky translocates into the nucleus and binds to the MT promoters during metal stress. With further experiments, we found that metal-activated MTF-1 is imported into the nucleus and recruited to the promoter regions of the MT genes in a gawky-dependent manner. Lastly, we demonstrated that gawky is important for metal discrimination by influencing the DNA binding bias of MTF-1 and the transcription preference of metal-specific genes, suggesting a combinatorial control of metal discrimination by MTF-1 binding sequences and gawky protein.
MATERIALS AND METHODS
Drosophila cell culture and initial assays of metal treatment
We used Drosophila Schneider 2 (S2) cells, one of the most commonly used cell lines derived from Drosophila melanogaster embryos, as a representative model for the eukaryotic system based on their ease of use, less functional redundancy, and availability of comprehensive genome-wide information. In this study, S2 cells were cultured at 25°C with Schneider's Drosophila medium (Sigma, S9895) plus 10% fetal bovine serum (HyClone, SH30910.03) and 1% penicillin streptomycin (Thermo Fisher Scientific, 15140122).
Heavy metals, such as copper, serve as essential micronutrients for cells at an appropriate concentration, while other metals, such as cadmium, are toxic even at a low concentration (for review, see (2,6)). Therefore, we chose copper sulfate (CuSO4) and cadmium chloride (CdCl2) as inducers of heavy metal stresses in this study. To determine the appropriate working concentration and time point for metal treatment in our initial experiments, a total of 1.5 × 106 S2 cells in each well of the 12-well plates were maintained for 2 days and treated with different concentrations of CuSO4 or CdCl2 for various time points between 0 and 24 h before collection. RNA was purified using TRIzol (Thermo Fisher Scientific, 15596018) according to the manufacturer's instructions. The expression of endogenous MtnA mRNA was used as a readout to examine the induction level of each condition. To test the effect of each condition on cell growth, the number of cells was subsequently counted using Bright-Line™ Hemacytometer (Sigma, Z359629).
Double-strand RNAs and RNAi
The information of double-strand RNAs (dsRNAs) used in this study can be found in Drosophila RNAi Screening Center (DRSC; https://www.flyrnai.org/cgi-bin/DRSC_gene_lookup.pl) except gawky dsRNA #4 and #5 that were designed in our lab (Supplementary Table S1; Supplementary Screening Data). The DRSC ID, if any, of each dsRNA is provided in Supplementary Table S1 and Supplementary Screening Data. For dsRNA preparation, dsRNAs were generated by in vitro transcription (MEGAscript kit, Thermo Fisher Scientific, AM1334) of PCR templates containing the T7 promoter sequence on both ends.
For RNAi experiments in S2 cells, a total of 1.5 × 106 S2 cells in each well of the 12-well plates were bathed with 8 μg of the indicated dsRNA for 3 days. To induce metal stress, a final concentration of 500 μM CuSO4 or 50 μM CdCl2 was added for the final 12 h before collection.
Plasmids and stable cell line construction
Plasmid constructs were generated by inserting the indicated sequences into a pMK33/pMtHy-based vector (https://www.addgene.org/69911/) as previously described (35,37–40). Cloning details for all plasmids are provided in Supplementary Plasmid Information. To generate a stable cell line, 1 μg of the indicated expression plasmid was introduced into a total of 0.5 × 106 S2 cells in each well of the 12-well plates using Effectene transfection reagent (QIAGEN, 301425) according to the manufacturer's instructions. The transfected S2 cells were then maintained by selection with 150 μg/ml hygromycin B for 3 weeks.
Rescue assay
For gawky rescue experiments, a total of 1.5 × 106 S2 cells in each well of the 12-well plates were first bathed with 8 μg of gawky dsRNA (4 μg of #4 + 4 μg of #5) targeting 5′ and 3′ UTR of the endogenous gawky transcript on day 1. The gawky rescue plasmid (0.1, 0.5 or 1 μg) (Supplementary Plasmid Information) that is insensitive to dsRNA treatment was then introduced on day 2. To increase the proportion of the cells which got transfected, we took advantage of the hygromycin resistance cassette and maintained gawky-rescued cells with 150 μg/ml hygromycin B in the media for another 2 days. To induce copper stress, a final concentration of 500 μM CuSO4 was added for the final 12 h before harvest. RNA was purified using TRIzol according to the manufacturer's instructions. The expression of MT mRNA in unstressed or copper treated cells was measured by real-time RT-PCR.
Nuclear run-on assay
Nuclear run-on was performed as previously described, with minor modifications (41). A total of 2 × 107 S2 cells in T75 flasks were treated with 40 μg of the indicated dsRNA for 3 days. To induce copper stress, a final concentration of 500 μM CuSO4 was added for the final 12 h. After harvest, cells were washed twice with 5 ml of 1 × PBS (phosphate buffer saline pH 7.4: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 1.8 mM KH2PO4), resuspended in 10 ml of ice-cold Solution I (150 mM KCl, 4 mM MgOAc, 10 mM Tris–HCl pH 7.4, and 80 units/ml RNase inhibitor [Beyotime, R0102]), and incubated on ice for 10 min. The pellets were collected by centrifugation at 1000g for 3 min at 4°C, washed with 3 ml of ice-cold Solution I, resuspended with slow pipetting in 1 ml of ice-cold Solution II (Solution I + 0.5% NP40 (v/v)), and incubated on ice for 10 min. Samples were added to the top of 4 ml of 0.6 M sucrose in the presence of 80 units/ml RNase Inhibitor, separated by centrifugation at 2000g for 10 min at 4°C, and washed with 2 ml of ice-cold Solution I. The final pellets (nuclei) were resuspended in 100 μl of fresh prepared nuclear run-on buffer (50 mM Tris–HCl pH 7.5, 5 mM MgCl2, 150 mM KCl, 0.1% sarkosyl (w/v), 10 mM DTT and 80 units/ml RNase Inhibitor), and 4 μl of NTP (2.5 mM ATP, GTP, CTP and BrUTP) was added. The nuclear run-on mixture was incubated at 28°C for 5 min. Nuclear RNA was isolated by TRIzol and resuspended into 500 μl of fresh prepared RNA immunoprecipitation buffer (20 mM Tris–HCl pH 7.5, 200 mM NaCl, 2.5 mM MgCl2, 0.5% NP-40 (v/v), 10% glycerol (v/v) and 80 units/ml RNase Inhibitor). BrUTP-labeled and unlabeled RNA was separated using anti-BrdU (Abcam, ab1893) and Dynabeads® Protein G (Thermo Fisher Scientific, 10003D) according to the manufacturer's instructions. The purified nascent RNA was converted to complementary DNA for real-time PCR.
Nuclear and cytoplasmic fractionation
Cellular fractionation of S2 cells was performed as described in our previous works (35,37,38). In brief, after RNAi and/or metal treatment, cells were washed twice with 1 ml of 1× PBS and resuspended with slow pipetting in 1 ml of ice-cold lysis buffer A (10 mM Tris–HCl pH 8, 140 mM NaCl, 1.5 mM MgCl2, 0.5% IGEPAL CA-630 (v/v) and 1 mM dithiothreitol). Samples were separated by centrifugation at 1000g for 3 min at 4°C. The supernatant was saved as the cytoplasmic fraction, and the pellets (nuclei) were resuspended in 1.1 ml of ice-cold lysis buffer B (1 ml of lysis buffer A + 100 μl of detergent [3.3% (w/v) sodium deoxycholate, 6.6% (v/v) Tween 40]) with slow vortexing for 10 s. After 10 min incubation on ice, the pellets were collected by centrifugation at 1000g for 3 min and washed with 1 ml of lysis buffer A. The final pellets were saved as the nuclear fraction. Efficient fractionation of nuclear and cytoplasmic proteins was verified by testing the protein levels of HDAC1 and α-Tubulin by western blotting experiments.
Fluorescence in situ hybridization (FISH)
For FISH probe preparation, RNA probe was generated by in vitro transcription (MEGAscript kit, Thermo Fisher Scientific, AM1334) of PCR templates containing the T7 promoter sequence, labeled with Alexa Fluor 488 dye using ULYSIS Nucleic Acid Labeling Kit (Thermo Fisher Scientific, U21650), and denatured at 65°C for 5 min. The sequences of RNA probes are provided in Supplementary Table S1.
After RNAi and/or copper treatment, cells were resuspended, and 0.5 × 105 cells were seeded onto the coverslip in a well of the 12-well plates for 1 h before FISH assays. Considering that S2 cells are roughly spherical, and adheres weakly to cell culture substrate, coverslips for microscopy were coated with concanavalin A (Con A; Solarbio, C8110) to promote attachment and spreading before use (42).
FISH assay was performed as described in our previous work (41). In brief, cells on ConA-coated coverslips were treated with 4% paraformaldehyde for 10 min at room temperature, denatured for 10 min at 80°C, and incubated with the denatured RNA probes in the presence of 30 ng/μl human Cot-1 DNA (Thermo Fisher Scientific, 15279011) at 42°C for 16 h. Coverslips (cell side up) were washed with 2× SSC (saline sodium citrate pH 7.0: 300 mM NaCl and 30 mM sodium citrate) twice at 45°C and then subjected to fluorescence signal detection using a confocal laser scanning microscopy (Leica TCS, SP8). The intensity of the fluorescence signal was analyzed using Image J software.
Immunofluorescence staining
To visualize the subcellular localization of gawky protein under normal or metal-stressed conditions, regular S2 cells or FLAG-tagged gawky stable cells were treated with 500 μM CuSO4 or 50 μM CdCl2 for 12 h. To visualize the subcellular localization of MTF-1 protein upon gawky depletion, regular S2 cells were treated with gawky dsRNA for 3 days, and 500 μM CuSO4 or 50 μM CdCl2 was added for the final 12 h. These cells were then resuspended, and 0.5 × 105 of them were seeded onto the ConA-coated coverslip in a well of the 12-well plates for 1 h before immunofluorescence staining assays. Immunofluorescence staining was performed as previously described (35). Coverslips (cell side up) were incubated with an antibody against gawky (anti1)*, MTF-1**, or FLAG-tagged protein (Beyotime, AF519) for 12 h at 4°C followed by incubating with a fluorescence secondary antibody (Abcam, ab150116 or ab150077) at room temperature for 2 h. Fluorescence signals were captured with a confocal laser scanning microscopy (Leica TCS, SP8). Line profiles were analyzed using Image-Pro Plus 6.0 software, and the intensity of the nuclear fluorescence signal was analyzed using Image J software according to refs (43,44). * Anti-gawky (anti1) was purified from rabbit antisera, raised against amino acids 969–982 of gawky conjugated to KLH. ** Anti-MTF-1 was purified from rabbit antisera, raised against amino acids 136–152 of MTF-1 conjugated to KLH.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using ChIP Assay Kit (Beyotime, P2078) according to the manufacturer's instructions. After RNAi and/or metal treatment, cells were washed with ice-cold 1 × PBS containing 1 mM PMSF (phenylmethanesulfonyl fluoride), fixed with 1% formaldehyde, and sonicated in SDS lysis buffer (1% SDS (w/v), 10 mM EDTA, 50 mM Tris-HCl pH 8.1, and 1 mM PMSF) to obtain chromatin solution containing 200–500 bp DNA fragments. The chromatin solution was precleared and immunoprecipitated with an antibody against gawky (anti1), MTF1, or FLAG-tagged protein (Beyotime, AF519) for 8 h at 4°C. To rule out the potential non-specific effects, a non-specific IgG (Beyotime, A7028) was also included as a negative control. Eluted DNA was then subjected to real-time PCR to examine the enriched genomic DNA regions. The binding of gawky or MTF-1 was defined as levels enriched over the input DNA (% input).
Reverse transcription and real-time PCR
For reverse transcription of RNA extracts, complementary DNA was synthesized using PrimeScript RT Master Mix kit (Takara, RR036A). Real-time PCR was then performed using FastSYBR Mixture (CWBIO, CW0955M) according to the manufacturer's instructions.
Ct values from all total RNA samples were normalized to rp49 mRNA expression. Ct values from all nascent RNA samples were normalized to Act42A mRNA expression. Ct values from all ChIP samples were normalized to input as described in the above section (% input). All real-time PCR primer sequences are provided in Supplementary Tables S2 and S3. The MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) (45) checklist is provided in Supplementary Table S4.
Co-immunoprecipitation (co-IP)
In this study, whole-cell extracts, nuclear and cytoplasmic fractions were used in co-IP assays. Whole-cell extracts were prepared using RIPA buffer (50mM Tris–HCl pH 7.4, 150mM NaCl, 0.1% SDS (w/v), 1% sodium deoxycholate (w/v) and 1% Triton X-100 (v/v)). To remove DNA, whole-cell extracts were digested with 20 units/ml DNase I (QIAGEN, 79254) for 30 min at room temperate before co-IP assays (46). Nuclear and cytoplasmic fraction were prepared as described in the nuclear and cytoplasmic fractionation section. Samples were then precleared and immunoprecipitated with Protein A+G Agarose beads (Beyotime, P2055-50) bound to an antibody against gawky (anti1), MTF1, FLAG (Beyotime, AF519) or HA (Beyotime, AF0039) in the presence of 1× Protease Inhibitor Cocktail (Beyotime, P1045) for 8 h at 4°C. IP samples were washed six times with RIPA buffer and subjected to western blotting analyses to examine the associated proteins.
Western blotting
Protein extracts from nuclear fractions, cytoplasmic fractions, whole cells or IP samples were subjected to western blotting analyses. The same amount of protein from each sample was separated on NuPAGE 4–12% Bis–Tris gel (Thermo Fisher Scientific, NP0329BOX). The gel was then transferred to polyvinylidene fluoride membranes (Bio-Rad, 1620177). Membranes were processed following the ECL western blotting protocol (Thermo Fisher Scientific, EI9051), as described previously (37,47). These antibodies were used: anti-HDAC1 (Abcam, ab1767, 1:1000 dilution), anti-α-Tubulin (Sigma, T6074, 1:10 000 dilution), anti-Lam (Beyotime, AF5222, 1:300 dilution), anti-FLAG (Beyotime, AF519, 1:1000 dilution), anti-HA (Beyotime, AF0039, 1:1000 dilution), anti-gawky (anti1, 1:250 dilution; anti2*, 1:250 dilution), and anti-MTF-1 (1: 500 dilution). Blots were viewed with a Bio-Rad ChemiDoc Imaging System and quantified using ImageJ software. * Anti-gawky2 (anti2) was purified from rabbit antisera, raised against amino acids 928–941 of gawky conjugated to KLH. Considering that the specificity of anti1 is better than anti2, anti2 was only used to confirm the knockdown efficiency of gawky dsRNAs.
Statistical analyses
The values reported in each graph represent averages of at least three independent experiments. ‘n’ describes the number of biological replicates in each figure legend. Statistical significance for comparisons of means was assessed by Student's t-test. Statistical details and error bars (± SEM) are defined in each figure legend: P < 0.01 (**) and P <0.05 (*).
RESULTS
Initial assay conditions and RNAi screening
In order to determine the appropriate working concentration and time point for copper treatment in our experiments, Drosophila S2 cells were treated with different concentrations of copper sulfate over time (0–24 h), and the metal-induced expression range of endogenous MtnA mRNA was then measured by real-time RT-PCR. Consistent with previous results (5,48,49), MtnA mRNA expression drastically increased upon copper treatment, and reached a peak level at 500 μM copper sulfate (Figure 1A). Furthermore, the copper-induced MtnA mRNA accumulated with time and reached a plateau at 12 h of induction (Figure 1A). Finally, the growth rate of S2 cells grown in the presence of 500 μM copper sulfate was similar to the growth rate under normal conditions (Figure 1B). Based on the maximal induction level of MtnA in our system, 500 μM copper sulfate and 12 h time point were chosen for subsequent experiments, except where indicated.
Figure 1.

Initial assay conditions and RNAi screening. (A) The induction levels of endogenous MtnA under different copper-stressed conditions. S2 cells were treated with different concentrations of CuSO4 (10 μM, 50 μM, 500 μM or 1 mM) for the indicated amounts of time (0, 2, 4, 8, 12, 18 or 24 h), and the expression range of MtnA mRNA was then measured by real-time RT-PCR following RNA isolation. Data were normalized to the unstressed sample and are shown as mean ± SEM. n = 3. (B) The effect of different concentrations of copper on cell growth. S2 cells were treated with different concentrations of CuSO4 (10 μM, 50 μM, 500 μM or 1 mM) for the indicated amounts of time (0, 2, 4, 8, 12, 18 or 24 h), the number of copper treated cells was counted at different time points. The growth rate of unstressed cells was also measured as a control. Data are shown as mean ± SEM. n = 3. (C, D) RNAi screening results of 127 genes. After RNAi treatment, the basal (no treatment, NT) or copper-induced (Cu2+) expression of MtnA (C) and MtnD (D) was measured by real-time RT-PCR following RNA isolation. To induce copper stress, S2 cells were treated with 500 μM CuSO4 for the final 12 h before collection. Loss of candidates such as MTF-1 (marked in green) resulted in a large reduction in MtnA and MtnD expression regardless of whether cells were stressed with copper or not. Instead, gawky (marked in red) was found to function solely under copper-stressed conditions (for details, see Supplementary Screening Data). Data were normalized to the β-gal dsRNA sample and are shown as Log2 fold change. n = 3. (E, F) The effect of gawky on MtnA and MtnD activation at different concentrations of copper. After RNAi treatment, S2 cells were treated with increasing concentrations of CuSO4 (1 μM, 10 μM, 50 μM, 500 μM, or 1 mM) for the final 12 h before collection. The copper-induced expression of MtnA (E) and MtnD (F) was measured by real-time RT-PCR following RNA isolation. Data were normalized to the β-gal dsRNA sample and are shown as mean ± SEM. n = 3. ∗∗P < 0.01; ∗P < 0.05.
To identify factor(s) that control heavy metal-induced transcription program, we took advantage of a focused RNAi screening strategy to individually knock down 127 genes with known effects on transcription in S2 cells for 3 days. S2 cells were unstressed or treated with 500 μM copper sulfate for the final 12 h, and the levels of MtnA and MtnD mRNAs were then assessed by real-time RT-PCR (Figure 1C and D). We found that many regulators of metal-responsive genes, such as the TATA-binding protein (TBP), the TBP-associated factors (TAFs), MTF-1, the Integrator complex, and the MED complex, are required for MtnA and MtnD expression under both normal and metal-stressed conditions (Figure 1C and D; Supplementary Screening Data). Especially, MTF-1 was among the most potent positive regulators identified regardless of whether cells were stressed with copper or not, which is in line with previous studies (19,29). The results indicate that these factors are involved in not only the transcription activation of metal-responsive genes in defense against metal stress, but also their basal transcription under unstressed conditions.
Instead, gawky was found to be the only candidate of the 127 genes screened in this study that solely controls the transcription activation of MtnA and MtnD upon copper treatment (Figure 1C and D; Supplementary Screening Data). Depletion of gawky resulted in a large decrease in the copper-induced expression of MtnA and MtnD, but had almost no effect on their basal transcription. Gawky is an essential component of P-body (also known as cytoplasmic processing body) and miRNA repressor complex (mi-RISC) as it facilitates P-body formation and acts as a scaffold bringing together mi-RISC and various RNA decay factors, as well reviewed in refs (36,50). Interestingly, knockdown of other P-body subunits or miRNA pathway components had a very limited effect on MtnA and MtnD mRNA levels, suggesting that the phenotype probably was not a result of an indirect effect due to loss of post-transcriptional control of another regulator, and thus gawky may function in MT activation in a P-body or miRNA pathway independent manner (Supplementary Figure S1A and B; Supplementary Screening Data).
To further determine whether gawky exerts similar roles in MtnA and MtnD activation at different concentrations of copper, we applied real-time RT-PCR to assess the levels of MtnA and MtnD mRNA in gawky-depleted cells treated with increasing concentrations of copper sulfate. Unexpectedly, compared to the control (β-gal) dsRNA treated cells, knockdown of gawky did not result in a significant reduction in MtnA and MtnD expression at a low concentration of copper (e.g. 1 μM) (Figure 1E and F). Unlike what was observed with MtnA (Figure 1E), depletion of gawky only reduced MtnD expression at higher concentrations of copper above 50 μM (Figure 1F). This suggests that MtnA expression is more sensitive to gawky depletion in the presence of copper. As another metal stress inducer, cadmium is more toxic than copper (Supplementary Figure S1C and D). Upon examining the effect of gawky depletion on MtnA and MtnD expression in the presence of increasing concentrations of cadmium chloride, we similarly found that loss of gawky had little effect on MtnA and MtnD expression at a low concentration of cadmium (e.g. 10 nM; Supplementary Figure S1E and F). In contrast to copper treatment, MtnD expression was found to be more sensitive to gawky depletion in response to cadmium stress (Supplementary Figure S1F). Taken together, we identified gawky as a potent activator of MtnA and MtnD during heavy metal stress and found that the effect of gawky on MtnA and MtnD activation depends on the concentrations of heavy metals in the environment.
Loss of gawky impairs the transcription activities of the MT genes during metal stress
Considering that there are five endogenous MT genes (MtnA, MtnB, MtnC, MtnD and MtnE) that can be robustly activated by heavy metals in the Drosophila system (Supplementary Figure S2A and B) (7,8), we next examined whether gawky exerts a general or limited role in regulating MT transcription by RNAi, nuclear run-on, and rescue experiments (Figure 2A). To rule out the potential off-target effects of RNAi technique such as increasing cellular ATP levels (51), we employed five independent gawky dsRNAs to silence gawky in S2 cells (Figure 2B–D; Supplementary Figure S2C). To induce copper stress, a final concentration of 500 μM copper sulfate was added to the media for the final 12 h. The levels of five endogenous MT mRNAs were then examined by real-time RT-PCR and FISH assays. Indeed, we found that depletion of gawky significantly down-regulated the metal-induced transcription of all the MT genes, but had almost no effect on the basal transcription under unstressed conditions (Figure 2E-G).
Figure 2.

Gawky is required for the metal-induced transcription but not the basal transcription of the MT genes. (A) Flow chart of the experimental setup for the data presented in (E, H and I). (B) Targeted positions of five independent dsRNAs that were used for RNAi depletion of gawky (for details, see Supplementary Table S1). (C, D) The knockdown efficiency of gawky dsRNAs. (C) S2 cells were treated with the indicated dsRNAs for 3 days, and western blotting was then applied to measure the expression of gawky protein with two different antibodies. Considering that the specificity of anti1 is better than that of anti2, anti1 was selected for the following experiment. (D) The level of gawky protein was quantified using ImageJ from three independent western blotting experiments. Data were normalized to the β-gal dsRNA sample and are shown as mean ± SEM. n = 3. ∗∗P < 0.01; ∗P < 0.05. (E–H) S2 cells were treated with the indicated gawky dsRNAs for 3 days, and 500 μM copper was added for the final 12 h to induce metal stress. (E–G) MT expression was measured by real-time RT-PCR using RNA extracts from whole cells (E) or was visualized by FISH assays (F, G). Representative images are shown. Scale bars, 12 μm. (H) The transcription activities were also measured by real-time RT-PCR using nascent RNA extracts from nuclear run-on assays. Data were normalized to the β-gal dsRNA sample and are shown as mean ± SEM. n = 3. ∗∗P < 0.01; ∗P < 0.05. (I) S2 cells were first treated with the gawky UTR dsRNAs (#4+5), and the rescue plasmid which is insensitive to dsRNA treatment was introduced to restore gawky expression. Real-time RT-PCR was then applied to quantify MT expression in unstressed or copper treated cells using RNA extracts from whole cells. Data were normalized to the β-gal dsRNA sample and are shown as mean ± SEM. n = 3. ∗∗P < 0.01; ∗P < 0.05.
To further verify whether the phenotype resulted from a change in the transcription activities of the MT genes, we extracted nuclei from unstressed or copper treated gawky-depleted cells and performed nuclear run-on experiments with BrUTP so that newly-synthesized transcripts could be purified by anti-BrUTP-bound magnetic beads. As expected, we confirmed that the transcription levels of five MT genes were significantly decreased in the absence of gawky upon copper treatment, but did not change under unstressed conditions (Figure 2H).
Also, we generated an Act5C promoter-driven gawky expression plasmid that contains the coding sequence and is insensitive to the dsRNAs targeting 5′ or 3′ UTR of the endogenous gawky transcript (Supplementary Plasmid Information). Given the fact that RNAi works on the entire culture, while the efficiency of plasmid transfection is relatively low in Drosophila cell culture, we took advantage of the hygromycin resistance cassette and maintained gawky-rescued cells for 2 days by selection with 150 μg/ml hygromycin B to increase the proportion of the cells which got transfected in rescue experiments (Figure 2I; Supplementary Figure S2D–F). As observed, the copper-induced expression of MtnA was rescued in a dose-dependent manner (Supplementary Figure S2D–F), suggesting a concentration effect of gawky. Intriguingly, when gawky-depleted cells were transfected with 1 μg of rescue plasmid, the levels of the other four copper-induced MT mRNAs were even restored to levels higher than the control, indicating perhaps a larger effect in the fraction which got transfected (Figure 2I).
Collectively, these results strongly demonstrate that gawky is necessary for full levels of MT activation during heavy metal stress, but not under normal conditions. Therefore, we concluded that gawky serves as a ‘checkpoint’ for metal stress and metal-induced transcription.
Gawky translocates to the nucleus in response to metal stress
Considering that the transcription cycle of eukaryotic genes is a highly regulated process that occurs in the nucleus, we therefore hypothesized that gawky may regulate the transcription activation of the MT genes in the nucleus during metal stress. To compare the subcellular localization of gawky protein under unstressed and metal-stressed conditions, nuclear and cytoplasmic proteins were isolated from unstressed or metal treated S2 cells and measured by western blotting. Under all conditions, the majority of gawky protein was found in the cytoplasm. However, during metal stress, there was a 6-fold increase in gawky protein level in the nucleus. This phenotype was not due to an overall change in total gawky expression (Figure 3A and B), and was further confirmed by immunofluorescence and confocal microscopy analyses at the single-cell level (Figure 3C–E). Next, we repeated these experiments using a S2 cell line stably expressing FLAG-tagged gawky (Supplementary Figure S3A). Similarly, FLAG-tagged gawky was found to significantly accumulate in the nucleus during metal stress (Supplementary Figure S3B–I). These data thus indicate that heavy metal stress leads to changes in the nucleocytoplasmic distribution of gawky protein.
Figure 3.

Gawky moves into the nucleus and interacts with the MT promoters in response to metal stress. (A, B) S2 cells were treated with 500 μM copper or 50 μM cadmium for 12 h. (A) Proteins from nuclei (Nuc), cytoplasm (Cyto), or whole-cell extracts (Total) were subjected to western blotting analyses. HDAC1 served as a marker for the nuclear fraction. α-Tubulin served as a marker for the cytoplasmic fraction. Representative blots are shown. n = 3. (B) The level of gawky protein was quantified using ImageJ from three independent western blotting experiments. The Nuc/Cyto ratio (the relative nuclear gawky level divided by the relative cytoplasmic gawky level) of gawky was also calculated. Data were normalized to the unstressed sample and are shown as mean ± SEM. n = 3. (C–E) S2 cells were treated with 500 μM copper or 50 μM cadmium for 12 h and seeded on ConA-covered coverslips for the final 1 h. (C) Confocal microscopy analyses of gawky were performed with anti-gawky (anti1). Representative images are shown. Scale bars, 5 μm. n = 3. (D) Line profiles of fluorescence intensities in (C) showing changes in gawky subcellular localization. (E) Statistics of nuclear gawky signal in each condition. Data were normalized to the unstressed sample and are shown as mean ± SEM. n = 50 cells for each condition. ∗∗P < 0.01; ∗P < 0.05. (F) ChIP-real-time PCR analysis of the interaction between gawky and the MT promoters in unstressed cells or after metal treatment for 12 h. The ChIP data generated by a non-specific antibody were also included. Gawky binding sites were defined as regions enriched over the input DNA. The MT and Act5C loci with the locations of ChIP amplicons are shown below. Data are shown as mean ± SEM. n = 3.
In fact, upon the prediction of the metal ion-binding residues of gawky protein using Metal Ion-Binding Site Prediction and Docking Server (Supplementary Figure S4A), a comprehensive and user friendly web prediction server for evaluation of metal ion-binding sites (52), we characterized four potential docking positions for copper ion and two potential docking positions for cadmium ion. The predicted copper- or cadmium-bound 3D structures of gawky are also provided accordingly (Supplementary Figure S4B and C). The prediction suggests that metal ion might be the direct trigger for nuclear translocation of gawky protein.
Gawky is recruited to the MT promoters in a metal-dependent manner
Upon examining the transcription levels of the MT genes, we demonstrated that gawky solely controls MT activation under metal-stressed conditions, but is bypassed under normal conditions. To further explore the underlying basis for this distinct regulation of MT activation, we examined whether gawky interacts with the MT loci under normal or metal-stressed conditions by ChIP assays (Figure 3F). The ChIP assays were performed on biological replicates using anti-gawky (anti1). To rule out the possibility that highly active promoter DNA might be artificially precipitated in ChIP assays (53–56), a non-specific antibody was also included as a negative control. Copper or cadmium was added for the final 12 h, and sites within the promoter and transcription regions of the MT genes or the house-keeping Act5C gene were examined by real-time PCR subsequently with a series of primer sets. Gawky binding was defined as levels enriched over the input DNA. As observed, gawky was generally present at the promoter regions (0–400 bp) upstream of the transcriptional start site of all the MT genes, but not at the Act5C locus, in both normal and metal treated cells (Figure 3F). Intriguingly, the interaction between gawky and the MT promoters was drastically elevated in defense against metal stress (Figure 3F). A similar phenotype was observed in the FLAG-tagged gawky stable cell line (Supplementary Figure S5). Taken together, these findings provide evidence that the transcription activation of the MT genes in response to heavy metal stress couples with increased nuclear pool of gawky and stronger interaction between gawky and the MT promoters.
Gawky requirement for transcription activation depends on the promoter
Based on these proof-of-concept experiments in hand, we hypothesized that the regulatory effect of gawky on transcription activation may depend on the promoter context. To test this model, a series of FLAG-tagged firefly luciferase expression plasmids under control of the MT promoters or the Act5C promoter were generated and used for stable cell line construction (Figure 4A). As expected, the levels of firefly luciferase mRNA (Supplementary Figure S6A) and protein (Supplementary Figure S6B) were robustly increased during copper stress only under control of the MT promoters. We noticed that the level of activation in these stable cells was relatively reduced compared with the endogenous MT loci. This is likely due to the overexpression of MT promoter-driven mRNAs through basal transcription in these stable cells. Next, we treated the stable cell lines with two independent dsRNAs to reduce gawky expression for 3 days, and copper was added to induce metal stress for the final 12 h. Real-time RT-PCR was used to measure the levels of firefly luciferase mRNAs. In comparison to β-gal dsRNA treated cells, the mRNA levels of MT promoter-driven firefly luciferase were significantly down-regulated in gawky dsRNA treated cells during copper stress, but not under unstressed conditions (Figure 4B), which is consistent with the data generated from the endogenous loci (Figure 2). Western blotting experiments further confirmed that the copper-induced expression of MT promoter-driven firefly luciferase dropped to ∼10–50% at the protein level (Figure 4C and D), when gawky was depleted from these stable cells. In contrast, no detectable changes were found under unstressed conditions (Figure 4C; Supplementary Figure S6C and D). Furthermore, the expression of Act5C promoter-driven firefly luciferase was largely unaffected by gawky depletion in either unstressed or copper-stressed cells (Figure 4B–D; Supplementary Figure S6C and D). Collectively, these findings demonstrate that the fate of undergoing gawky-mediated transcriptional regulation is encrypted in the promoter context.
Figure 4.

The regulatory effect of gawky on transcription depends on the promoter. (A) Schematic representation of FLAG-tagged firefly luciferase expression plasmids. The MT promoters or the Act5C promoter was inserted upstream of the transcriptional start site. Stable cell lines were generated using these plasmids. (B–D) S2 cell lines stably expressing FLAG-tagged firefly luciferase were treated with two independent gawky dsRNAs for 3 days followed by the addition of 500 μM copper for the final 12 h to induce metal stress. (B) The expression of firefly luciferase mRNA was subjected to real-time RT-PCR analyses following RNA isolation. Data were normalized to the β-gal dsRNA sample and are shown as mean ± SEM. n = 3. ∗∗P < 0.01; ∗P < 0.05. (C) Firefly luciferase protein was subjected to western blotting analyses using equal amounts of protein extracts from whole cells. α-Tubulin served as a loading control. Representative blots are shown. n = 3. (D) The protein level of the copper-induced firefly luciferase in (C) was quantified using ImageJ from three independent western blotting experiments. To provide better representation of the levels of non-induced proteins, we increased the amount of protein used for western blotting analyses (see Supplementary Figure S6C and D). Data were normalized to the β-gal dsRNA sample and shown as mean ± SEM. ∗∗P < 0.01; ∗P < 0.05. n = 3. (E) Schematic representation of dati expression plasmids that can generate linear RNAs (exon 1, exon 2, and exon 3 are joined in a linear order) and circular RNAs (the end of exon 2 is joined to the beginning of exon 2). The MT promoters or the dati promoter was inserted upstream of the transcriptional start site. (F, G) After gawky RNAi, S2 cells were transfected with different dati expression plasmids for 2 days followed by the addition of 500 μM copper for the final 12 h to induce metal stress. The level of nascent linear (F) or circular (G) dati RNA was measured by real-time RT-PCR using RNA extracts from nuclear run-on experiments. Data were normalized to the β-gal dsRNA sample and are shown as mean ± SEM. n = 3. ∗∗P < 0.01; ∗P < 0.05.
Gawky-mediated transcription activation is in a canonical splicing or backsplicing independent manner
It is well-known that thousands of mRNA precursors (pre-mRNAs) can be alternatively spliced to generate mature linear RNAs via canonical splicing (splicing sites are joined in a linear order) or circular RNAs via backsplicing (a splicing donor is joined to an upstream splicing acceptor) (57–61). To evaluate whether different splicing patterns affect gawky-mediated control of transcription activation, we applied our previously described Drosophila dati expression plasmid system that can generate a linear RNA as well as a circular RNA (35,37). The MT promoters or the dati promoter was inserted upstream to drive the expression of plasmid-derived dati. (Figure 4E). These expression plasmids were transiently introduced into gawky-depleted cells, and the transcription activities were examined by measuring the levels of nascent linear and circular dati RNAs using nuclear run-on technique. Consistent with figure 4A–D, for MT promoter-driven dati expression plasmids, both linear and circular RNAs were significantly reduced upon gawky depletion during copper stress, but not under unstressed conditions. No obvious changes in the levels of dati promoter-driven RNAs were detected, which served as negative controls (Figure 4F and G). These results thus indicate that gawky-mediated transcription activation is in a canonical splicing or backsplicing independent manner.
Gawky recruits MTF-1 to the MT promoters to activate transcription in response to metal stress
Since MTF-1 acts as the major transcription factor involved in metal homeostasis (Supplementary Figure S7) (for review, see (2–4)), we reasoned that the regulatory effect of gawky on MT activation is related to MTF-1. To examine whether gawky interacts with MTF-1 under unstressed or metal-stressed conditions, co-IP was first performed using whole-cell extracts with anti-gawky (anti1), anti-MTF-1, or a negative IgG followed by western blotting analyses. Little interaction between gawky and MTF-1 was detected in unstressed cells, while the interaction was strongly enhanced during copper stress (Figure 5A). We next wanted to determine if the interaction is DNA-dependent. To address this, we repeated co-IP experiments using whole-cell extracts treated with DNase I. The metal-induced interaction was still observed in the presence of DNase I, indicating that gawky interacts with MTF-1 in a DNA-independent manner (Figure 5B). In addition, we applied biochemical fractionation to prepare nuclear and cytoplasmic extracts from copper treated cells. Upon co-IP analyses, gawky was found to interact with MTF-1 not only in the nucleus but also in the cytoplasm (Figure 5C and D), complementing the result in figure 5B. These phenotypes were further confirmed in FLAG-tagged gawky stable cells transfected with a HA-tagged MTF1 expression plasmid (Supplementary Figure S8). Taken together, the results presented here suggest that the interaction between gawky and MTF-1 depends on the presence of heavy metals in the environment, and gawky may only have a regulatory effect on MTF-1 in response to metal stress.
Figure 5.

Gawky is a partner of metal-activated MTF1 and is required for MTF1 recruitment to the MT promoters during metal stress. (A–D) Gawky binds to MTF-1 during metal stress. (A) The interaction between gawky and MTF-1 under normal or copper-stressed conditions was assayed by co-IP with an antibody against gawky (anti1), MTF-1, or a negative IgG using whole-cell extracts. (B) To examine whether the copper-induced interaction is in a DNA dependent manner, co-IP was performed using whole-cell extracts in the presence of DNase I. (C, D) To examine whether gawky interacts with MTF-1 in the nucleus or cytoplasm, co-IP was performed using nuclear or cytoplasmic extracts. All samples were subjected to western blotting analyses. α-Tubulin or Lam served as a negative control. Representative blots are shown. n = 3. (E) To examine the effect of gawky on MTF1 recruitment to the MT promoters, S2 cells were treated with two independent gawky dsRNAs for 3 days, and 500 μM copper was added for the final 12 h to induce metal stress. MTF1 recruitment to the MT genes was then measured using ChIP-real-time PCR. The ChIP data generated by a non-specific antibody were also included. MTF-1 binding sites were defined as regions enriched over the input DNA. The loci of the MT genes with the locations of ChIP amplicons are shown below. Data are shown as mean ± SEM. n = 3.
To further determine whether gawky is involved in MTF-1 recruitment to the MT genes, S2 cells were treated with two independent gawky dsRNAs or β-gal dsRNA followed by copper treatment for the final 12 h. MTF-1 ChIP experiments were performed with anti-MTF-1 or a negative IgG. The interaction between MTF-1 and the MT genes was subsequently assayed by real-time PCR with a series of primer sets positioned at the promoter and transcription regions of the MT genes. MTF-1 binding sites were defined as regions enriched over the input DNA (Figure 5E). Notably, we found that depletion of gawky resulted in the dissociation of MTF-1 from the MT promoters during copper stress (Figure 5E), suggesting that gawky is required for MTF-1 recruitment to the MT promoters. Next, we performed gawky ChIP experiments to examine whether depletion of MTF-1 affects gawky recruitment to the MT genes during copper stress. In contrast, loss of MTF-1 had little effect on the interaction between gawky and the MT promoters (Supplementary Figure S9). Furthermore, we repeated these assays using FLAG-tagged MTF-1 stable cells (Supplementary Figure S10A and B) and FLAG-tagged gawky stable cells (Supplementary Figure S10C), and similar results were observed. Taken together, these data indicate that gawky is required for MTF-1 recruitment to the MT promoters in response to metal stress; however, gawky recruitment to the MT promoters is in an MTF-1 independent manner.
Gawky controls nuclear import of MTF-1 in response to metal stress
It has been well established that MTF-1 is a metal-sensing and nucleocytoplasmic shuttling protein with significant nuclear import in metal-stressed cells, as well reviewed in refs (2–4). The result of MTF-1 localization analysis generated from S2 cells (Figure 3A) or FLAG-tagged MTF-1 stable cells (Supplementary Figure S11A and B) supported the previous study in Drosophila system (5). On the other hand, upon examining the interaction between gawky and MTF-1 as shown in the above section, we surprisingly found that gawky interacts with MTF-1 in both the nuclear and cytoplasmic fraction. Therefore, we hypothesized that gawky may also exert its regulatory effect on the subcellular localization of MTF-1. To test the hypothesis, the level of MTF-1 protein in the nucleus and cytoplasm was then examined by western blotting (Figure 6A and B; Supplementary Figure S11C and D) and immunofluorescence staining assays (Figure 6C and D). As shown, loss of gawky resulted in a large decrease in the nuclear pool of MTF-1 during metal stress, but had almost no effect under unstressed conditions. In contrast, knockdown of MTF-1 did not affect the nucleocytoplasmic distribution of gawky (Supplementary Figure S12). The result suggests that nuclear translocation of gawky is independent of MTF-1, thereby excluding the possibility that gawky is carried into the nucleus by a metal-activated version of MTF-1.
Figure 6.

Gawky controls nuclear import of metal-activated MTF-1. (A–D) S2 cells were treated with 500 μM copper or 50 μM cadmium for 12 h. (A, B) Proteins from nuclei (Nuc), cytoplasm (Cyto), or whole-cell extracts (Total) were subjected to western blotting analyses. HDAC1 served as a marker for the nuclear fraction. α-Tubulin served as a marker for the cytoplasmic fraction. Representative blots are shown. The level of MTF-1 protein was quantified using ImageJ from three independent western blotting experiments. The Nuc/Cyto ratio of MTF-1 was also calculated. Data were normalized to the β-gal dsRNA sample and are shown as mean ± SEM. n = 3. (C, D) Confocal microscopy analyses of MTF-1 were performed with anti-MTF-1. Representative images are shown. Scale bars, 8 μm. n = 3. (D) Statistics of nuclear MTF-1 signal in each condition. Data were normalized to the β-gal dsRNA sample and are shown as mean ± SEM. n = 40 cells for each condition. ∗∗P < 0.01; ∗P < 0.05.
Gawky modulates MTF-1-mediated metal discrimination
The alteration in the sequence-specific binding preference of MTF-1 is determined by the specific metal encountered and can lead to a unique response. For example, copper stress results in Cu MRE recognition by MTF-1 and copper-preferred transcription activation, while cadmium stress leads to Cd MRE recognition and cadmium-preferred transcription activation (5). To determine whether gawky plays a role in MTF-1-mediated metal discrimination, we generated an MRE plasmid system in which four direct repeats of the Cu or Cd MREs, arranged in tandem arrays, were inserted upstream of the dati promoter (Figure 7A; Supplementary Plasmid Information). The Cu or Cd MRE plasmid was introduced into S2 cells for 2 days, and copper or cadmium was added for the final 12 h. The plasmid containing only the dati promoter was also tested as a negative control. ChIP-real-time PCR was assayed to measure the association between MTF-1 and the MRE or dati promoter. Real-time RT-PCR was then performed to measure the transcription response of the MRE plasmid using whole-cell RNA extracts. The data in figure 7 were processed to better demonstrate the metal-specific response by plotting the data to a non-preferred condition that is set to 1 as previously described by Sims et al. (5). In agreement with Sims et al.'s work, the metal-specific transcription of the Cu or Cd MRE plasmid corresponded to the MTF-1 binding preference (Figure 7B–D; Supplementary Figure S13A–C) (5). However, when gawky was depleted from metal-stressed cells, MTF-1 no longer showed the strong binding preference for the Cu or Cd MRE promoter, and the strong transcription preference of the Cu or Cd MRE plasmid was eliminated (Figure 7B–D; Supplementary Figure S13A–C).
Figure 7.

The regulatory effect of gawky on MTF-1-mediated metal discrimination. (A) Schematic representation of MRE plasmids. Four direct repeats of Cu or Cd MREs were inserted upstream of the dati promoter. (B–D) MRE plasmids were introduced into gawky dsRNA treated S2 cells for 2 days followed by the addition of 500 μM copper or 50 μM cadmium for the final 12 h to induce metal stress. (B) ChIP assay was performed with anti-MTF1 to examine its recruitment to the MRE containing promoter. (C, D) Real-time RT-PCR was performed to measure the levels of plasmid-derived dati mRNA (C) and circular dati RNA (D). The non-preferred condition is set to 1. Data are shown as mean ± SEM. n = 3. ∗∗P < 0.01; ∗P < 0.05. The same data were also processed in the way to separate the effect on the activation level from the metal specificity (see Supplementary Figure S13A–C). (E, F) S2 cells were treated with two independent gawky dsRNAs for 3 days followed by the addition of 500 μM copper or 50 μM cadmium for the final 12 h to induce metal stress. (E) ChIP assay was performed with anti-MTF1 to examine the MTF1 binding preference of six endogenous metal-specific genes. (F) The transcription preference was measured by real-time RT-PCR following RNA isolation. The non-preferred condition is set to 1. Data are shown as mean ± SEM. n = 3. ∗∗P < 0.01; ∗P < 0.05. The same data were also processed in the way to separate the effect on the activation level from the metal specificity (see Supplementary Figure S13D and E).
Upon the observation of the MRE plasmid system, we reasoned that gawky may also influence the activation and discrimination of endogenous metal-specific genes. To test this, six endogenous candidates with metal-specific MTF-1 binding preferences were selected from the previously described annotation (5). ChIP analyses revealed that the endogenous cadmium-specific genes (CG5964, 18w, and CG6234) were preferentially bound to MTF-1 under cadmium treatment, whereas the endogenous copper-specific genes (CG9220, beat-VB and sha) were preferentially bound to MTF-1 under copper treatment (Figure 7E; Supplementary Figure S13D), which is consistent with the previous study (5). Furthermore, the MTF-1binding preference was substantially impaired by RNAi depletion of gawky (Figure 7E; Supplementary Figure S13D). Consequently, these genes no longer generated the strong transcription preference under specific metal treatment (Figure 7F; Supplementary Figure S13E).
Taken together, these results indicate that loss of gawky eliminates the metal-specific transcription preference by impairing MTF-1 binding preference. We therefore concluded that gawky is required for MTF-1-mediated metal discrimination.
DISCUSSION
Cells survive under metal-stressed conditions by coordinating the outputs of metal-responsive genes (1,5,30,31,39,62–64). Therefore, the regulation of the basal and metal-induced transcription of these genes must be different, suggesting a pathway through which the general transcriptional machinery discriminates metal stress from normal conditions. In the present study, the data lend credence to a novel mechanism whereby gawky quickly responses to the heavy metal environment by moving into the nucleus where it directs MTF-1 to MREs to stimulate transcription of metal-induced genes. In contrast, under normal conditions, gawky is bypassed from the basal transcription. These results suggest that gawky is a ‘checkpoint’ for metal stress and a ‘hallmark’ of metal-induced transcription (Figure 8A). Moreover, cells have specific responses in adaption to different heavy metals. Our work shows that gawky and metal-specific MREs combinatorially regulate metal discrimination by influencing the MTF-1 binding bias of metal-specific genes (Figure 8B).
Figure 8.
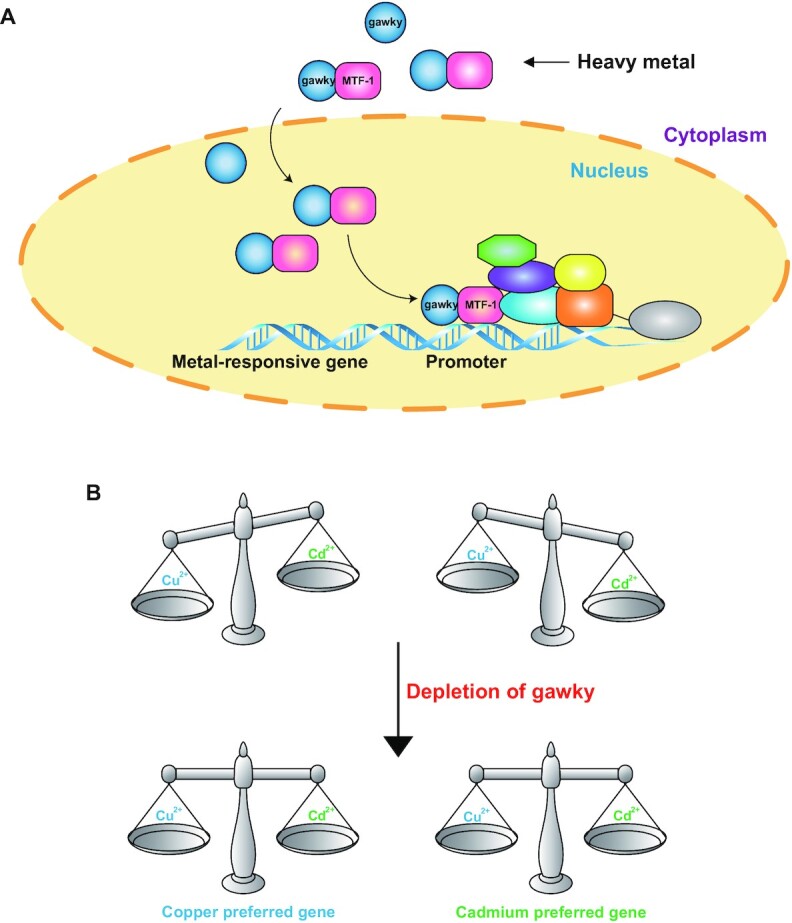
A working model for the regulatory effects of gawky on metal-induced transcription. (A) In defense of heavy metals, gawky activates metal-responsive genes in concert with metal-activated MTF-1. (B) Loss of gawky eliminates the metal-specific transcription preference by impairing the DNA binding bias of MTF-1.
Interaction partners of MTF-1 (Regulatory co-factors of MTF-1)
MTF-1 is an important transcription factor of many stress-induced genes, thereby protecting cells from diverse stressors. Co-factors of MTF-1 may determine its distinct functions. Concerning metal stress, considerable studies have attempted to elucidate the mechanism of metal-responsive transcriptional regulation by characterizing the interaction partners of MTF-1 (30,65–70). The activities of these partners must be balanced to keep the appropriate expression of metal-responsive genes. For example, two basal transcription regulators - Mediator (MED) and TFIID have been reported to combinatorially control the transcription activation of the MT genes via MTF-1 in Drosophila. TFIID inhibits the transcription initiation of the MtnA gene, while MED stimulates its transcription (30). The coactivator/histone acetyltransferase p300 and the transcription factor Sp1 have been found to bind to the acidic activation domain of MTF-1 in human and mouse. Both p300 and Sp1 contribute to the transcriptional regulation of the MT genes by chromatin remodeling (67,68). Upstream stimulatory factor 1 (USF1) activates the basal expression of mouse metallothionein 1 in concert with MTF-1 via direct interaction with the metallothionein 1 promoter (71). In our study, we identified a new MTF-1 partner, gawky, that binds to MTF-1 in a metal-dependent manner, possibly through the induction of structural changes in MTF-1 (31). Considering that MTF-1 is required for both metal inducible and constitutive transcription in various cellular contexts (Figure 1C and D; Supplementary Figure S7) (5,19,30,31), identification of MTF-1′s interaction partners will provide novel perspectives into MTF-1-mediated transcriptional regulation.
MREs and metal-specific transcription
Recognition of specific DNA motifs by DNA binding factors is an important mechanism to control transcription activation (72–75). A subset of DNA binding factors choose different motifs depending on specific cellular contexts (5,76). For example, the recruitment of MTF-1 shows a preferential response to copper or cadmium through a single nucleotide in the core MRE (5,30). The core MRE containing a purine in the sixth nucleotide position is preferred by MTF-1 during copper stress, whereas a pyrimidine is preferred during cadmium stress (5). Our work presented here is unique in that it provides additional insights into the important role of gawky in regulating the MTF-1 binding bias of metal-specific MREs (Figure 7; Supplementary Figure S13), hence revealing a novel mechanism of DNA recognition in Drosophila.
The novel roles of gawky
Gawky is evolutionarily conserved from C. elegan to human (32,77–79). It has long been assumed that gawky, as a RNA decay factor, is only involved in post-transcriptional regulation, such as deadenylation, decapping, or degradation of RNAs, in the cytoplasmic fraction (32–35,80,81). However, it is now becoming increasingly clear that gawky may function in chromatin silencing, splicing, and transcriptional regulation in the nucleus (79,82–84), indicating that gawky is a nucleocytoplasmic shuttling protein (79,85). Indeed, human homologs of gawky with nuclear export signal (NES) mutations are predominantly localized in the nucleus (79). In line with human data, our results show that metal stress induces nuclear import of gawky (Figure 3A–E; Supplementary Figure S3B–I). We also demonstrated that gawky is important for MTF-1 nuclear import (Figure 6; Supplementary Figure S11C and D) and MTF-1 recruitment to promoters of metal-responsive genes (Figure 5E; Supplementary Figure S10B) in response to metal stress, but MTF-1 does not affect gawky subcellular localization (Supplementary Figure S12) or its recruitment (Supplementary Figure S9B and S10C). Therefore, it is not likely that gawky is carried into the nucleus by a metal-activated version of MTF-1. Furthermore, using a metal ion-binding prediction server, gawky protein was predicted to have a strong metal ion-binding potential (Supplementary Figure S4), suggesting that gawky may act as an intracellular metal sensor and signal transducer in Drosophila. It is thus more likely that the direct binding of metal ions to gawky triggers its nuclear import. However, the question whether gawky could directly sense intracellular metal will be validated in the future through specialized techniques for protein structure analyses, such as metal-affinity column chromatography. Besides, factors that affect the subcellular localization of gawky under specific metal-stressed conditions may provide additional levels of metal-specific control. Further studies are still needed to clarify these questions.
In conclusion, our findings indicate that metal stress induces gawky-mediated transcriptional control that is precluded under normal conditions, and provide key insights into how gawky, MTF-1, and MREs combinatorially control metal-responsive transcription as well as metal discrimination, pointing towards a novel mechanism of gene expression regulation in metal homeostasis (Figure 8).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Michael G. Kearse, Emmanuel Enoch Dzakah, Hassaan Awan, Fei Xia, Liang Chen, Shuai Wei, Jianwen Chen, the members of the Shan Lab (University of Science and Technology of China), the Wilusz Lab (University of Pennsylvania), the Kearse Lab (Ohio State University) and the Xu Lab (Chongqing University) for discussions or technical supports.
Author contributions: C.H. conceived this project and supervised its execution. R.J., Z.S., J.L., Z.L. and G.S. performed experiments, analyzed data, or provided experimental material. C.H. wrote the manuscript with input from the other authors.
Contributor Information
Ruirui Jia, School of Life Sciences, Chongqing University, Chongqing 401331, China; Center of Plant Functional Genomics, Institute of Advanced Interdisciplinary Studies, Chongqing University, Chongqing 401331, China.
Zhenxing Song, School of Life Sciences, Chongqing University, Chongqing 401331, China; Center of Plant Functional Genomics, Institute of Advanced Interdisciplinary Studies, Chongqing University, Chongqing 401331, China.
Jiamei Lin, School of Life Sciences, Chongqing University, Chongqing 401331, China; Center of Plant Functional Genomics, Institute of Advanced Interdisciplinary Studies, Chongqing University, Chongqing 401331, China.
Zhengguo Li, School of Life Sciences, Chongqing University, Chongqing 401331, China; Center of Plant Functional Genomics, Institute of Advanced Interdisciplinary Studies, Chongqing University, Chongqing 401331, China.
Ge Shan, School of Basic Medical Sciences, Division of Life Science and Medicine, University of Science and Technology of China, Hefei 230027, China.
Chuan Huang, School of Life Sciences, Chongqing University, Chongqing 401331, China; Center of Plant Functional Genomics, Institute of Advanced Interdisciplinary Studies, Chongqing University, Chongqing 401331, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [32070633]; Natural Science Foundation of Chongqing, China [cstc2019jcyj-msxmX0085]; Innovation Support Program for Overseas Returned Scholars of Chongqing, China [cx2019142]; Fundamental Research Funds for the Central Universities of China [2020CDJQY-A076]; 100 Talent Program of Chongqing University [0304001104433]. Funding for open access charge: National Natural Science Foundation of China [32070633].
Conflict of interest statement. None declared.
REFERENCE
- 1. Vihervaara A., Duarte F.M., Lis J.T.. Molecular mechanisms driving transcriptional stress responses. Nat. Rev. Genet. 2018; 19:385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gunther V., Lindert U., Schaffner W.. The taste of heavy metals: gene regulation by MTF-1. Biochim. Biophys. Acta. 2012; 1823:1416–1425. [DOI] [PubMed] [Google Scholar]
- 3. Takahashi S. Positive and negative regulators of the metallothionein gene (review). Mol Med Rep. 2015; 12:795–799. [DOI] [PubMed] [Google Scholar]
- 4. Giedroc D.P., Chen X., Apuy J.L.. Metal response element (MRE)-binding transcription factor-1 (MTF-1): structure, function, and regulation. Antioxid. Redox. Signal. 2001; 3:577–596. [DOI] [PubMed] [Google Scholar]
- 5. Sims H.I., Chirn G.W., Marr M.T. 2nd. Single nucleotide in the MTF-1 binding site can determine metal-specific transcription activation. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16516–16521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lynes M.A., Kang Y.J., Sensi S.L., Perdrizet G.A., Hightower L.E.. Heavy metal ions in normal physiology, toxic stress, and cytoprotection. Ann. N. Y. Acad. Sci. 2007; 1113:159–172. [DOI] [PubMed] [Google Scholar]
- 7. Egli D., Domenech J., Selvaraj A., Balamurugan K., Hua H., Capdevila M., Georgiev O., Schaffner W., Atrian S.. The four members of the Drosophila metallothionein family exhibit distinct yet overlapping roles in heavy metal homeostasis and detoxification. Genes Cells. 2006; 11:647–658. [DOI] [PubMed] [Google Scholar]
- 8. Atanesyan L., Gunther V., Celniker S.E., Georgiev O., Schaffner W.. Characterization of MtnE, the fifth metallothionein member in Drosophila. J. Biol. Inorg. Chem. 2011; 16:1047–1056. [DOI] [PubMed] [Google Scholar]
- 9. Miles A.T., Hawksworth G.M., Beattie J.H., Rodilla V.. Induction, regulation, degradation, and biological significance of mammalian metallothioneins. Crit. Rev. Biochem. Mol. Biol. 2000; 35:35–70. [DOI] [PubMed] [Google Scholar]
- 10. Klaassen C.D., Liu J., Choudhuri S.. Metallothionein: an intracellular protein to protect against cadmium toxicity. Annu. Rev. Pharmacol. Toxicol. 1999; 39:267–294. [DOI] [PubMed] [Google Scholar]
- 11. Waldron K.J., Rutherford J.C., Ford D., Robinson N.J.. Metalloproteins and metal sensing. Nature. 2009; 460:823–830. [DOI] [PubMed] [Google Scholar]
- 12. Bittel D., Dalton T., Samson S.L., Gedamu L., Andrews G.K.. The DNA binding activity of metal response element-binding transcription factor-1 is activated in vivo and in vitro by zinc, but not by other transition metals. J. Biol. Chem. 1998; 273:7127–7133. [DOI] [PubMed] [Google Scholar]
- 13. Koizumi S., Suzuki K., Ogra Y., Gong P., Otuska F.. Roles of zinc fingers and other regions of the transcription factor human MTF-1 in zinc-regulated DNA binding. J. Cell. Physiol. 2000; 185:464–472. [DOI] [PubMed] [Google Scholar]
- 14. Wimmer U., Wang Y., Georgiev O., Schaffner W.. Two major branches of anti-cadmium defense in the mouse: MTF-1/metallothioneins and glutathione. Nucleic Acids Res. 2005; 33:5715–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo L., Lichten L.A., Ryu M.S., Liuzzi J.P., Wang F., Cousins R.J.. STAT5-glucocorticoid receptor interaction and MTF-1 regulate the expression of ZnT2 (Slc30a2) in pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:2818–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lichten L.A., Ryu M.S., Guo L., Embury J., Cousins R.J.. MTF-1-mediated repression of the zinc transporter Zip10 is alleviated by zinc restriction. PLoS One. 2011; 6:e21526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Troadec M.B., Ward D.M., Lo E., Kaplan J., De Domenico I.. Induction of FPN1 transcription by MTF-1 reveals a role for ferroportin in transition metal efflux. Blood. 2010; 116:4657–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yepiskoposyan H., Egli D., Fergestad T., Selvaraj A., Treiber C., Multhaup G., Georgiev O., Schaffner W.. Transcriptome response to heavy metal stress in Drosophila reveals a new zinc transporter that confers resistance to zinc. Nucleic Acids Res. 2006; 34:4866–4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Selvaraj A., Balamurugan K., Yepiskoposyan H., Zhou H., Egli D., Georgiev O., Thiele D.J., Schaffner W.. Metal-responsive transcription factor (MTF-1) handles both extremes, copper load and copper starvation, by activating different genes. Gene Dev. 2005; 19:891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burke R., Commons E., Camakaris J.. Expression and localisation of the essential copper transporter DmATP7 in Drosophila neuronal and intestinal tissues. Int. J. Biochem. Cell Biol. 2008; 40:1850–1860. [DOI] [PubMed] [Google Scholar]
- 21. Stoytcheva Z.R., Vladimirov V., Douet V., Stoychev I., Berry M.J.. Metal transcription factor-1 regulation via MREs in the transcribed regions of selenoprotein H and other metal-responsive genes. Biochim. Biophys. Acta. 2010; 1800:416–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Green C.J., Lichtlen P., Huynh N.T., Yanovsky M., Laderoute K.R., Schaffner W., Murphy B.J.. Placenta growth factor gene expression is induced by hypoxia in fibroblasts: A central role for metal transcription factor-1. Cancer Res. 2001; 61:2696–2703. [PubMed] [Google Scholar]
- 23. Cramer M., Nagy I., Murphy B.J., Gassmann M., Hottiger M.O., Georgiev O., Schaffner W.. NF-kappaB contributes to transcription of placenta growth factor and interacts with metal responsive transcription factor-1 in hypoxic human cells. Biol. Chem. 2005; 386:865–872. [DOI] [PubMed] [Google Scholar]
- 24. Lichtlen P., Wang Y., Belser T., Georgiev O., Certa U., Sack R., Schaffner W.. Target gene search for the metal-responsive transcription factor MTF-1. Nucleic Acids Res. 2001; 29:1514–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kindermann B., Doring F., Budczies J., Daniel H.. Zinc-sensitive genes as potential new target genes of the metal transcription factor-1 (MTF-1). Biochem. Cell. Biol. 2005; 83:221–229. [DOI] [PubMed] [Google Scholar]
- 26. Wang Y., Wimmer U., Lichtlen P., Inderbitzin D., Stieger B., Meier P.J., Hunziker L., Stallmach T., Forrer R., Rulicke T.et al.. Metal-responsive transcription factor-1 (MTF-1) is essential for embryonic liver development and heavy metal detoxification in the adult liver. FASEB J. 2004; 18:1071–1079. [DOI] [PubMed] [Google Scholar]
- 27. Gunes C., Heuchel R., Georgiev O., Muller K.H., Lichtlen P., Bluthmann H., Marino S., Aguzzi A., Schaffner W.. Embryonic lethality and liver degeneration in mice lacking the metal-responsive transcriptional activator MTF-1. EMBO J. 1998; 17:2846–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tavera-Montanez C., Hainer S.J., Cangussu D., Gordon S.J.V., Xiao Y., Reyes-Gutierrez P., Imbalzano A.N., Navea J.G., Fazzio T.G., Padilla-Benavides T.. The classic metal-sensing transcription factor MTF1 promotes myogenesis in response to copper. FASEB J. 2019; 33:14556–14574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heuchel R., Radtke F., Georgiev O., Stark G., Aguet M., Schaffner W.. The Transcription Factor Mtf-1 Is Essential for Basal and Heavy Metal-Induced Metallothionein Gene-Expression. EMBO J. 1994; 13:2870–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marr M.T. 2nd, Isogai Y., Wright K.J., Tjian R.. Coactivator cross-talk specifies transcriptional output. Genes Dev. 2006; 20:1458–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marr S.K., Pennington K.L., Marr M.T.. Efficient metal-specific transcription activation by Drosophila MTF-1 requires conserved cysteine residues in the carboxy-terminal domain. Biochim. Biophys. Acta. 2012; 1819:902–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Behm-Ansmant I., Rehwinkel J., Doerks T., Stark A., Bork P., Izaurralde E.. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006; 20:1885–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chekulaeva M., Mathys H., Zipprich J.T., Attig J., Colic M., Parker R., Filipowicz W.. miRNA repression involves GW182-mediated recruitment of CCR4-NOT through conserved W-containing motifs. Nat. Struct. Mol. Biol. 2011; 18:1218–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fabian M.R., Cieplak M.K., Frank F., Morita M., Green J., Srikumar T., Nagar B., Yamamoto T., Raught B., Duchaine T.F.et al.. miRNA-mediated deadenylation is orchestrated by GW182 through two conserved motifs that interact with CCR4-NOT. Nat. Struct. Mol. Biol. 2011; 18:1211–1217. [DOI] [PubMed] [Google Scholar]
- 35. Jia R., Xiao M.S., Li Z., Shan G., Huang C.. Defining an evolutionarily conserved role of GW182 in circular RNA degradation. Cell Discov. 2019; 5:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Niaz S., Hussain M.U.. Role of GW182 protein in the cell. Int. J. Biochem. Cell Biol. 2018; 101:29–38. [DOI] [PubMed] [Google Scholar]
- 37. Huang C., Liang D., Tatomer D.C., Wilusz J.E.. A length-dependent evolutionarily conserved pathway controls nuclear export of circular RNAs. Genes Dev. 2018; 32:639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Song Z., Jia R., Tang M., Xia F., Xu H., Li Z., Huang C.. Antisense oligonucleotide technology can be used to investigate a circular but not linear RNA-mediated function for its encoded gene locus. Sci China Life Sci. 2021; 64:784–794. [DOI] [PubMed] [Google Scholar]
- 39. Tatomer D.C., Elrod N.D., Liang D., Xiao M.S., Jiang J.Z., Jonathan M., Huang K.L., Wagner E.J., Cherry S., Wilusz J.E.. The Integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev. 2019; 33:1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiao M.S., Wilusz J.E.. An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3' ends. Nucleic Acids Res. 2019; 47:8755–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Z., Huang C., Bao C., Chen L., Lin M., Wang X., Zhong G., Yu B., Hu W., Dai L.et al.. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015; 22:256–264. [DOI] [PubMed] [Google Scholar]
- 42. Rogers S.L., Rogers G.C.. Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nat. Protoc. 2008; 3:606–611. [DOI] [PubMed] [Google Scholar]
- 43. Wang Y., Hu S.B., Wang M.R., Yao R.W., Wu D., Yang L., Chen L.L.. Genome-wide screening of NEAT1 regulators reveals cross-regulation between paraspeckles and mitochondria. Nat. Cell Biol. 2018; 20:1145–1158. [DOI] [PubMed] [Google Scholar]
- 44. Liu X., Wang X., Li J., Hu S., Deng Y., Yin H., Bao X., Zhang Q.C., Wang G., Wang B.et al.. Identification of mecciRNAs and their roles in the mitochondrial entry of proteins. Sci. China Life Sci. 2020; 63:1429–1449. [DOI] [PubMed] [Google Scholar]
- 45. Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L.et al.. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009; 55:611–622. [DOI] [PubMed] [Google Scholar]
- 46. Pascual-Garcia P., Jeong J., Capelson M.. Nucleoporin Nup98 associates with Trx/MLL and NSL histone-modifying complexes and regulates Hox gene expression. Cell Rep. 2014; 9:433–442. [DOI] [PubMed] [Google Scholar]
- 47. Huang C., Wang X., Liu X., Cao S., Shan G.. RNAi pathway participates in chromosome segregation in mammalian cells. Cell Discov. 2015; 1:15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bunch T.A., Grinblat Y., Goldstein L.S.. Characterization and use of the Drosophila metallothionein promoter in cultured Drosophila melanogaster cells. Nucleic Acids Res. 1988; 16:1043–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Silar P., Theodore L., Mokdad R., Erraiss N.E., Cadic A., Wegnez M.. Metallothionein Mto gene of Drosophila melanogaster: structure and regulation. J. Mol. Biol. 1990; 215:217–224. [DOI] [PubMed] [Google Scholar]
- 50. Braun J.E., Huntzinger E., Izaurralde E.. The role of GW182 proteins in miRNA-mediated gene silencing. Adv. Exp. Med. Biol. 2013; 768:147–163. [DOI] [PubMed] [Google Scholar]
- 51. Mohr S.E., Hu Y., Rudd K., Buckner M., Gilly Q., Foster B., Sierzputowska K., Comjean A., Ye B., Perrimon N.. Reagent and data resources for investigation of RNA binding protein functions in Drosophila melanogaster cultured cells. G3 (Bethesda). 2015; 5:1919–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lin Y.F., Cheng C.W., Shih C.S., Hwang J.K., Yu C.S., Lu C.H.. MIB: metal ion-binding site prediction and docking server. J. Chem. Inf. Model. 2016; 56:2287–2291. [DOI] [PubMed] [Google Scholar]
- 53. Teytelman L., Thurtle D.M., Rine J., van Oudenaarden A.. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:18602–18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krebs W., Schmidt S.V., Goren A., De Nardo D., Labzin L., Bovier A., Ulas T., Theis H., Kraut M., Latz E.et al.. Optimization of transcription factor binding map accuracy utilizing knockout-mouse models. Nucleic Acids Res. 2014; 42:13051–13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Waldminghaus T., Skarstad K.. ChIP on Chip: surprising results are often artifacts. BMC Genomics. 2010; 11:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jain D., Baldi S., Zabel A., Straub T., Becker P.B.. Active promoters give rise to false positive ‘Phantom Peaks’ in ChIP-seq experiments. Nucleic Acids Res. 2015; 43:6959–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li Z., Kearse M.G., Huang C.. The nuclear export of circular RNAs is primarily defined by their length. RNA Biol. 2019; 16:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wilusz J.E. Circular RNAs: unexpected outputs of many protein-coding genes. RNA Biol. 2017; 14:1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wilusz J.E. A 360 degrees view of circular RNAs: From biogenesis to functions. Wiley Interdiscip Rev RNA. 2018; 9:e1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen L., Huang C., Wang X.L., Shan G.. Circular RNAs in Eukaryotic Cells. Curr. Genomics. 2015; 16:312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu K., Chen D., Wang Z., Ma J., Zhou J., Chen N., Lv L., Zheng Y., Hu X., Zhang Y.et al.. Annotation and functional clustering of circRNA expression in rhesus macaque brain during aging. Cell Discov. 2018; 4:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wright K.J., Marr M.T. 2nd, Tjian R.. TAF4 nucleates a core subcomplex of TFIID and mediates activated transcription from a TATA-less promoter. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:12347–12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pennington K.L., Marr S.K., Chirn G.W., Marr M.T. 2nd. Holo-TFIID controls the magnitude of a transcription burst and fine-tuning of transcription. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:7678–7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee W., Haslinger A., Karin M., Tjian R.. Activation of transcription by two factors that bind promoter and enhancer sequences of the human metallothionein gene and SV40. Nature. 1987; 325:368–372. [DOI] [PubMed] [Google Scholar]
- 65. Giot L., Bader J.S., Brouwer C., Chaudhuri A., Kuang B., Li Y., Hao Y.L., Ooi C.E., Godwin B., Vitols E.et al.. A protein interaction map of Drosophila melanogaster. Science. 2003; 302:1727–1736. [DOI] [PubMed] [Google Scholar]
- 66. Vardanyan A., Atanesyan L., Egli D., Raja S.J., Steinmann-Zwicky M., Renkawitz-Pohl R., Georgiev O., Schaffner W.. Dumpy-30 family members as determinants of male fertility and interaction partners of metal-responsive transcription factor 1 (MTF-1) in Drosophila. BMC Dev. Biol. 2008; 8:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li Y., Kimura T., Huyck R.W., Laity J.H., Andrews G.K.. Zinc-induced formation of a coactivator complex containing the zinc-sensing transcription factor MTF-1, p300/CBP, and Sp1. Mol. Cell. Biol. 2008; 28:4275–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ogra Y., Suzuki K., Gong P., Otsuka F., Koizumi S.. Negative regulatory role of Sp1 in metal responsive element-mediated transcriptional activation. J. Biol. Chem. 2001; 276:16534–16539. [DOI] [PubMed] [Google Scholar]
- 69. Lin M.C., Liu Y.C., Tam M.F., Lu Y.J., Hsieh Y.T., Lin L.Y.. PTEN interacts with metal-responsive transcription factor 1 and stimulates its transcriptional activity. Biochem. J. 2012; 441:367–377. [DOI] [PubMed] [Google Scholar]
- 70. Saydam N., Steiner F., Georgiev O., Schaffner W.. Heat and heavy metal stress synergize to mediate transcriptional hyperactivation by metal-responsive transcription factor MTF-1. J. Biol. Chem. 2003; 278:31879–31883. [DOI] [PubMed] [Google Scholar]
- 71. Andrews G.K., Lee D.K., Ravindra R., Lichtlen P., Sirito M., Sawadogo M., Schaffner W.. The transcription factors MTF-1 and USF1 cooperate to regulate mouse metallothionein-I expression in response to the essential metal zinc in visceral endoderm cells during early development. EMBO J. 2001; 20:1114–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dynan W.S., Tjian R.. Control of eukaryotic messenger RNA synthesis by sequence-specific DNA-binding proteins. Nature. 1985; 316:774–778. [DOI] [PubMed] [Google Scholar]
- 73. Mitchell P.J., Tjian R.. Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science. 1989; 245:371–378. [DOI] [PubMed] [Google Scholar]
- 74. Holmes M.C., Tjian R.. Promoter-selective properties of the TBP-related factor TRF1. Science. 2000; 288:867–870. [DOI] [PubMed] [Google Scholar]
- 75. D’Alessio J.A., Ng R., Willenbring H., Tjian R.. Core promoter recognition complex changes accompany liver development. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:3906–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Meijsing S.H., Pufall M.A., So A.Y., Bates D.L., Chen L., Yamamoto K.R.. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009; 324:407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ding L., Spencer A., Morita K., Han M.. The developmental timing regulator AIN-1 interacts with miRISCs and may target the argonaute protein ALG-1 to cytoplasmic P bodies in C. elegans. Mol. Cell. 2005; 19:437–447. [DOI] [PubMed] [Google Scholar]
- 78. Eystathioy T., Chan E.K., Tenenbaum S.A., Keene J.D., Griffith K., Fritzler M.J.. A phosphorylated cytoplasmic autoantigen, GW182, associates with a unique population of human mRNAs within novel cytoplasmic speckles. Mol. Biol. Cell. 2002; 13:1338–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nishi K., Nishi A., Nagasawa T., Ui-Tei K.. Human TNRC6A is an Argonaute-navigator protein for microRNA-mediated gene silencing in the nucleus. RNA. 2013; 19:17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Piao X., Zhang X., Wu L., Belasco J.G.. CCR4-NOT deadenylates mRNA associated with RNA-induced silencing complexes in human cells. Mol. Cell. Biol. 2010; 30:1486–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rehwinkel J., Behm-Ansmant I., Gatfield D., Izaurralde E.. A crucial role for GW182 and the DCP1:DCP2 decapping complex in miRNA-mediated gene silencing. RNA. 2005; 11:1640–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Matsui M., Chu Y., Zhang H., Gagnon K.T., Shaikh S., Kuchimanchi S., Manoharan M., Corey D.R., Janowski B.A.. Promoter RNA links transcriptional regulation of inflammatory pathway genes. Nucleic Acids Res. 2013; 41:10086–10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hicks J.A., Li L., Matsui M., Chu Y., Volkov O., Johnson K.C., Corey D.R.. Human GW182 paralogs are the central organizers for RNA-mediated control of transcription. Cell Rep. 2017; 20:1543–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu J., Liu Z., Corey D.R.. The requirement for GW182 scaffolding protein depends on whether Argonaute is mediating translation, transcription, or splicing. Biochemistry. 2018; 57:5247–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kalantari R., Hicks J.A., Li L., Gagnon K.T., Sridhara V., Lemoff A., Mirzaei H., Corey D.R.. Stable association of RNAi machinery is conserved between the cytoplasm and nucleus of human cells. RNA. 2016; 22:1085–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.