

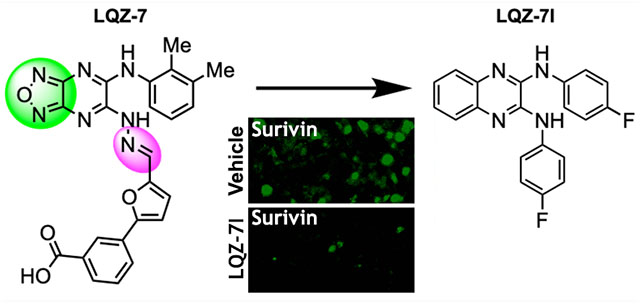
Abstract
Survivin, a homodimeric member of the Inhibitor of Apoptosis Protein (IAP) family, is required for cancer cell survival and overexpressed in almost all solid tumors. However, targeting survivin has been challenging due to its “undruggable” nature. Recently, we used a novel approach to target the dimerization interface and identified inhibitors of two scaffolds that can directly bind to and inhibit survivin dimerization. One of the scaffolds, represented by the compound LQZ-7, contains an undesirable labile hydrazone linker and a potentially nonfunctional furazanopyrazine ring that we attempted to eliminate in this study. We found one compound, 7I, that is more active than the parent compound, LQZ-7, and when given orally effectively inhibits xenograft tumor growth and induces survivin loss in tumors. These findings indicate that 7I with a stable linker and a quinoxaline ring can be used as a lead for further optimization of this novel class of survivin inhibitors.
Graphical Abstract
INTRODUCTION
Survivin (BIRC5), a 16.5 kDa homodimeric protein,1 is a member of the Inhibitor of Apoptosis Protein (IAP) family with a single BIR domain, a zinc-finger fold, and an extended C-terminal helical coiled coil.2–4 Ectopic overexpression of survivin inhibits both intrinsic and extrinsic apoptosis pathways in cell lines and animal models and has been suggested to contribute to treatment resistance.5–8 Interestingly, survivin is expressed at undetectable or low levels in normal adult tissues, while it has been shown to be overexpressed in almost all solid tumors.9 Molecular probes such as antisense oligonucleotides, ribozymes, siRNAs, and dominant negative mutants all resulted in caspase-dependent cell death and increased drug-induced apoptosis.10–16 Thus, survivin has become a significant and attractive drug target.17
However, survivin has been considered “undruggable” due to lack of known enzymatic activities, and the majority of drug discovery studies targeting survivin have avoided targeting the protein directly.17,18 Recently, using a combination of computational analysis and in silico screening, we have identified the first direct small molecule inhibitor of survivin targeting the residues Leu98 and Phe101 in the dimerization interface of survivin to inhibit survivin dimerization.19 The initial hit inhibitor, LQZ-7, upon binding to the survivin dimeric interface, causes exposure of the hydrophobic dimerization core and leads to protein misfolding and subsequent degradation in the proteasome.
In the current study, we further investigated the scaffold of LQZ-7 and synthesized five novel analogues with a goal to improve its properties and assess whether removal of the undesirable labile hydrazone linker and a potentially nonfunctional furazanopyrazine is possible. Of these 5 analogues, one compound LQZ-7I (7I), showed significantly improved activity and is the focus of this work. The data obtained utilizing 7I in both in vitro and in vivo studies highlights its potential as a lead for further development, which may yield a potential cancer therapeutic by targeting the survivin protein directly.
RESULTS
Design and Synthesis of Novel LQZ-7 Analogues.
To investigate the structure–activity relationship of LQZ-7 for lead identification and creation of better and novel survivin inhibitors, we first performed molecular dynamic simulation analysis of LQZ-7 bound to the dimeric interface of survivin. As shown in Figure 1A,B, LQZ-7 has three important interactions with survivin: (a) a H-bond between an aniline NH group of LQZ-7 and Glu94 of survivin; (b) an interaction between the substituted aniline in LQZ-7 and Phe93, Phe101, and Leu98 in the hydrophobic pocket of survivin via π−π stacking and hydrophobic interactions; and (c) a H-bond between the carboxylic acid of LQZ-7 and Trp10 of survivin. This analysis shows clearly that the furazanopyrazine ring did not contribute to the binding to survivin. It is also noteworthy that LQZ-7 has a labile hydrazone linker, which is undesirable. Thus, for the new synthesis, we attempted to remove the hydrazone linker and to replace the furazanopyrazine with a quinoxaline ring. To this end, five analogues were synthesized by two nucleophilic aromatic substitutions of dichloroquinoxaline with the corresponding amine or amide nucleophiles (Figure 1C). All five analogues, LQZ-7G to LQZ-7K (7G–7K), retain the two predicted critical interactions with survivin and have the labile hydrazone linker replaced.
Figure 1.

Analysis of LQZ-7 binding to survivin and synthesis of LQZ-7 analogues. (A) Molecular dynamic simulation analysis of LQZ-7 bound to the dimeric interface of survivin. (B) Critical interactions between LQZ-7 and survivin. (C) Scheme of LQZ-7 analogue (quinoxaline derivatives) synthesis.
Compound 7I Has Enhanced Cytotoxicity Compared to the Parent Compound LQZ-7.
These five newly synthesized LQZ-7 analogs were first tested for their cytotoxicity against prostate cancer cell lines C4-2 and PC-3 in comparison with their parent compound LQZ-7 using methylene blue assay. Dose–response survival curves were generated following 72 h treatments (Figure S1), which were used to derive their IC50 values. As shown in Figure 2A,B, the analogue 7I has improved cytotoxicity with an IC50 of 3.1 μM against C4-2 cells and 4.8 μM against PC-3 cells compared with the parent compound LQZ-7, which has IC50 values of 9.1 and 8.1 μM, respectively. The other compounds performed worse with higher IC50 values compared with LQZ-7.
Figure 2.

Characterization of LQZ-7 analogues. (A, B) IC50 of LQZ-7 and its analogues in C4-2 (A) and PC-3 (B) cells derived from dose–response survival curves (Figure S1) as determined using methylene blue assay. (C, D) SEAP reporter activity determined using mammalian two hybrid assay in the presence of LQZ-7 or its analogues. Vector and p53/large T antigen were used as negative and positive controls for the assay, respectively (*p < 0.05; **p < 0.01; ***p < 0.001).
Compound 7I Inhibits Survivin Dimerization.
As this series of survivin inhibitors were designed to exert their action through interaction with the dimeric interface and disruption of survivin dimerization, we next assessed their activity in inhibiting survivin dimerization using a mammalian two hybrid assay by fusing one survivin molecule to the GAL4-DNA-binding domain and another to the VP16 activation domain. Dimerization of survivin activates the expression of the secreted embryonic alkaline phosphatase (SEAP) reporter gene, which can be monitored. As shown in Figure 2C, 7I significantly decreased the SEAP reporter expression, while 7H slightly inhibited and the other compounds did not reduce reporter expression, consistent with their limited cytotoxicity against C4-2 and PC-3 cells. Thus, 7I was chosen for further studies.
To ensure that the 7I inhibition of survivin dimerization in the above two hybrid assay is specific to survivin and to eliminate any potential nonspecific effect, we tested the effect of newly synthesized 7I on the dimerization of p53 with the large T antigen using the same mammalian two-hybrid assay and including the vector control. As shown in Figure 2D, both 7I and the parent compound LQZ-7 significantly inhibited survivin dimerization. However, they had no effect on p53–large T antigen dimerization or the basal reporter expression from vector alone, suggesting that the 7I inhibition of survivin dimerization may be specific. Furthermore, 7I is significantly more effective than LQZ-7 in inhibiting survivin dimerization, consistent with the difference in their IC50 values against different prostate cancer cell lines. These findings also suggest that 7I may inhibit cell survival via inhibiting survivin dimerization.
Survivin Expression Level Correlates with 7I IC50 in Different Prostate Cancer Cell Lines.
To determine if 7I inhibits cell survival via inhibiting survivin, we performed an association analysis of survivin expression with 7I IC50 in five prostate cancer cell lines. Figure 3A shows the relative survivin level in five prostate cancer cell lines assessed using Western blot analyses. Figure 3B shows the IC50 of 7I in these cell lines derived from dose-dependent survival curves (Figure S2). Association analysis showed that the IC50 of 7I strongly correlates with the survivin expression level with a correlation coefficient R2 = 0.79 (Figure 3C). This finding suggests that the cancer cells with higher level of survivin expression are more resistant to 7I and that 7I likely exerts its cytotoxicity via survivin.
Figure 3.
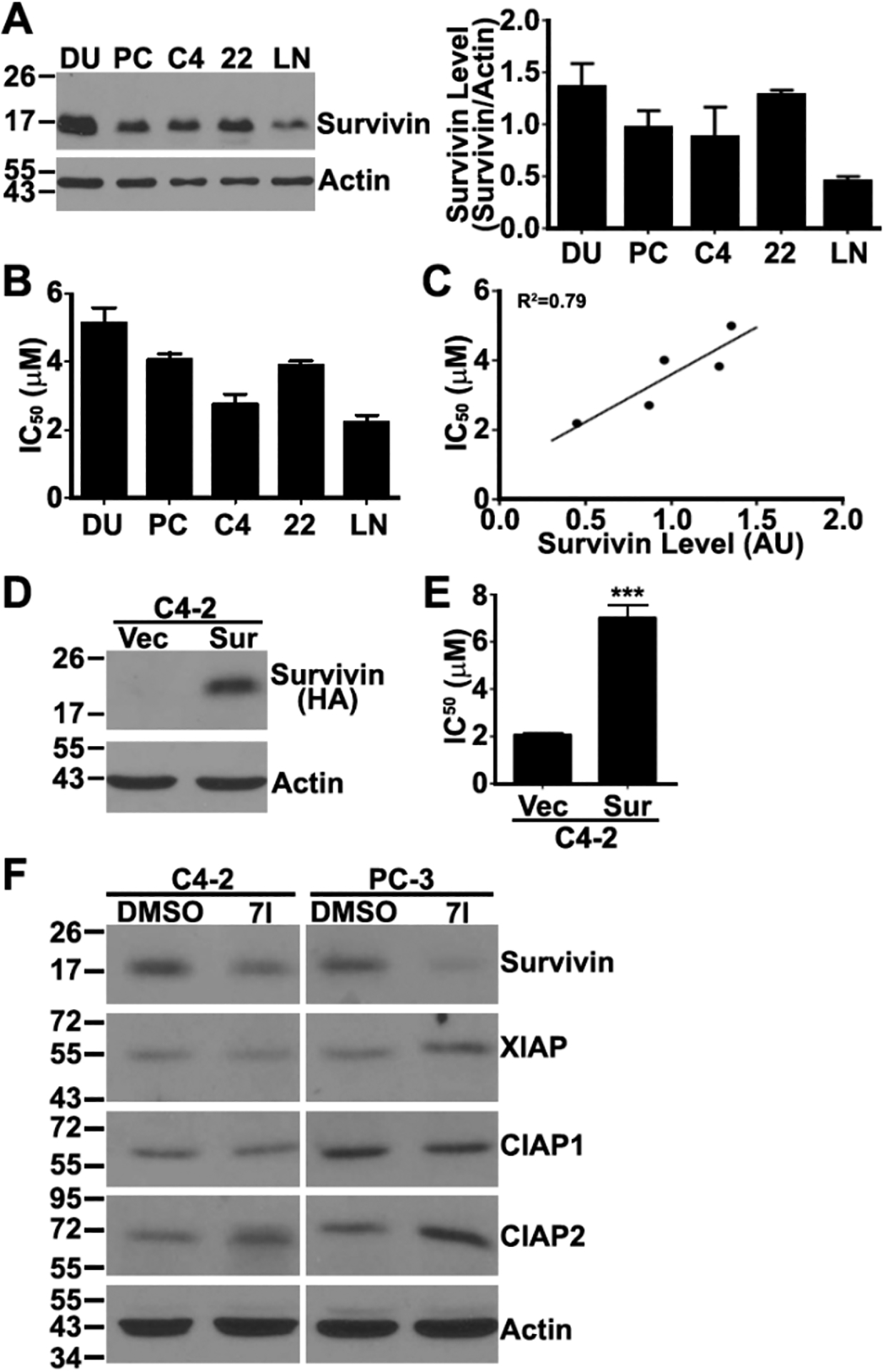
(A) Relative survivin protein level in different human prostate cancer cell lines determined using Western blot analyses. (B) IC50 of 7I in different human prostate cancer cell lines derived from dose-dependent survival curves (Figure S2) as determined using methylene blue assay. (C) Association of 7I IC50 with survivin expression level in different human prostate cancer cell lines. (D, E) Expression of HA-tagged survivin determined using Western blot analysis (D) and IC50 of 7I derived from survival curves (Figure S3) as determined using methylene blue assay (E) in stable C4-2 clone with overexpression of HA-tagged survivin (Sur) and vector-transfected (Vec) control cells. (***p < 0.001). (F) Effect of 7I on the expression of survivin, XIAP, CIAP1, and CIAP2 in C4-2 and PC-3 cells as determined using Western blot analysis.
Survivin Overexpression Decreases Cellular Sensitivity to 7I.
To corroborate the above findings and to further investigate if 7I inhibition of cancer cell survival is via survivin, we generated a stable C4-2 clone with overexpression of HA-tagged survivin (Figure 3D) and tested the effect of survivin overexpression on 7I cytotoxicity. As shown in Figure 3E and Figure S3, survivin overexpression significantly increased cellular resistance to 7I with an IC50 of ~7 μM compared with ~2 μM in vector-transfected control clone, further supporting the conclusion that 7I likely works via inhibiting survivin.
Selectivity of 7I.
As discussed above, 7I may selectively inhibit survivin dimerization. To ensure that 7I is selective to survivin, we tested 7I’s effect on other members of the IAP family by taking advantage of its induction of target loss (see below). For this purpose, we performed Western blot analysis to determine the expression level of other IAPs including XIAP, CIAP1, and CIAP2 in addition to survivin following 7I treatment in C4-2 and PC-3 cells. As shown in Figure 3F, 7I treatment reduced the expression of survivin. However, it did not reduce the expression of XIAP, CIAP1, and CIAP2. Thus, 7I may be selective to its intended target, survivin.
Compound 7I Induces Survivin Degradation in a Proteasome-Dependent Manner.
Previously, it has been shown that the parent compound LQZ-7 induces survivin degradation via a proteasome-dependent mechanism by inhibiting survivin dimerization. Because 7I inhibited survivin dimerization as shown above, we hypothesized that 7I will also induce survivin degradation. To test this hypothesis, we examined survivin expression level in C4-2 and PC-3 cells following 7I treatments. As shown in Figure 4A, 7I treatments induced time-dependent survivin loss in both cell lines. We next performed a cycloheximide-chase experiment following 7I treatment to determine the half-life of survivin. As shown in Figure 4B,C, 7I treatments reduced the half-life of survivin from 2.2 and 2.3 h to 50 and 25 min in C4-2 and PC-3 cells, respectively. Thus, 7I induces survivin degradation in both cell lines.
Figure 4.
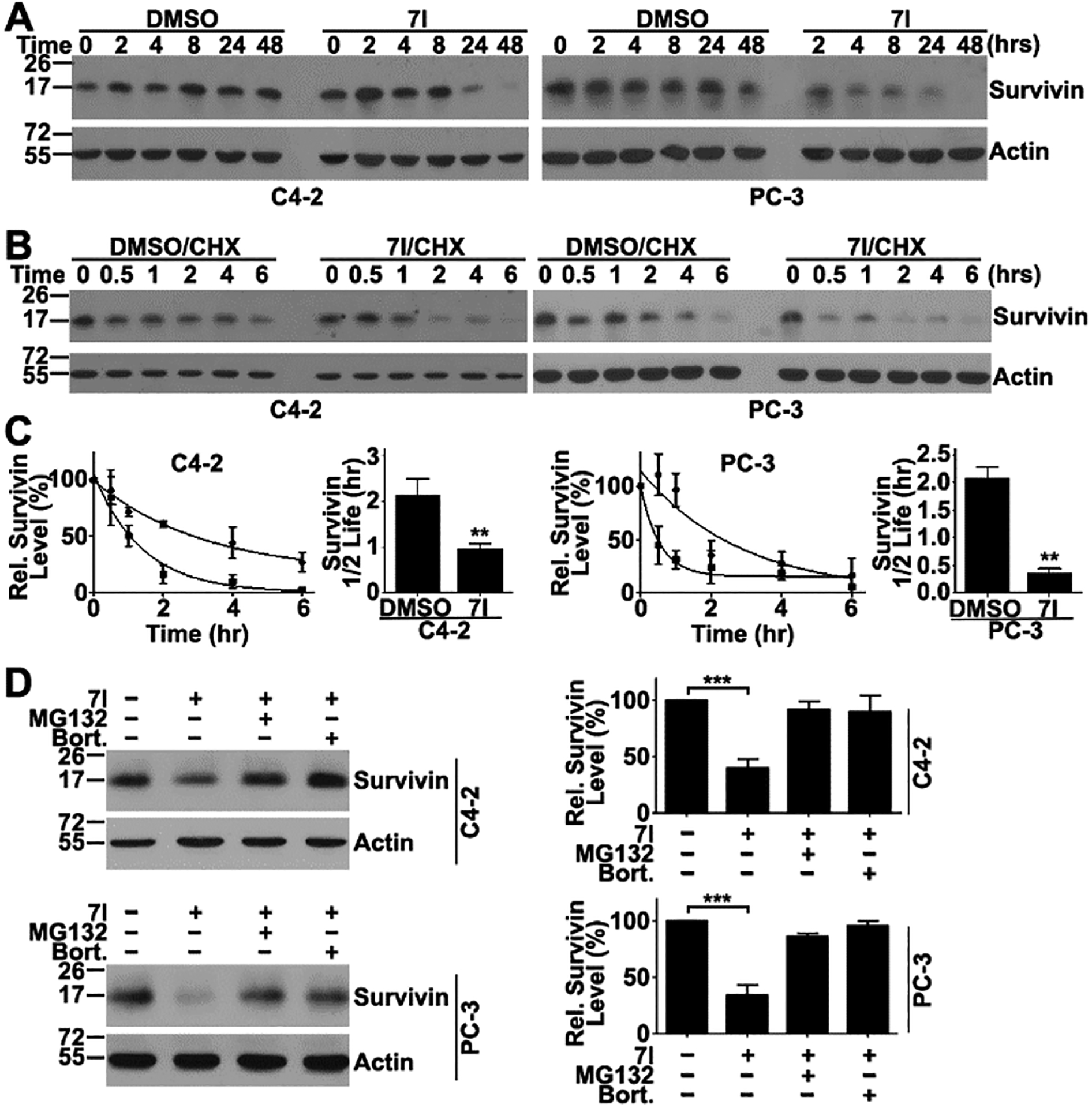
Compound 7I induces survivin degradation in the proteasome. (A) Time course of 7I-induced survivin loss in C4-2 and PC-3 cell lines determined using Western blot analysis. (B, C) Effect of 7I on survivin half-life. C4-2 and PC-3 cells were pretreated with cycloheximide (CHX) followed by chasing for different times in the presence of 7I or DMSO control. Cells were lysed for Western blot analysis of remaining survivin (B) and quantification of gel densities from three independent experiments (C). (D) Effect of proteasome inhibitors MG132 and bortezomib on 7I-induced survivin degradation as determined using Western blot analysis. Actin was used as a loading control. Quantification was from three independent experiments (**p < 0.01; ***p < 0.001).
To determine if 7I-induced survivin degradation occurs via the proteasome, we performed a rescue experiment by pretreating cells using proteasome inhibitors MG132 and bortezomib for 1 h prior to 7I treatment. As shown in Figure 4D, both MG132 and bortezomib significantly reversed 7I-induced survivin loss. Thus, 7I likely induces survivin degradation via the proteasome.
Compound 7I Induces Apoptosis.
As discussed above, depleting survivin or inhibiting survivin using dominant negative forms of survivin cause spontaneous apoptosis in cancer cells, and the parent compound LQZ-7 also induces apoptosis.20 We, thus, tested if 7I would induce apoptosis by inhibiting survivin. For this purpose, we first performed Western blot analysis of cleaved caspase 3 in C4-2 and PC-3 cells following 7I treatments. As shown in Figure 5A,B, 7I induced cleavage of caspase 3 in a dose dependent manner in both cell lines. We also performed annexin V staining of C4-2 and PC-3 cells as another indicator of apoptosis following 7I treatments. As shown in Figure 5C, 3 μM 7I induced ~48% and ~39% apoptosis in C4-2 and PC-3 cells, respectively.
Figure 5.
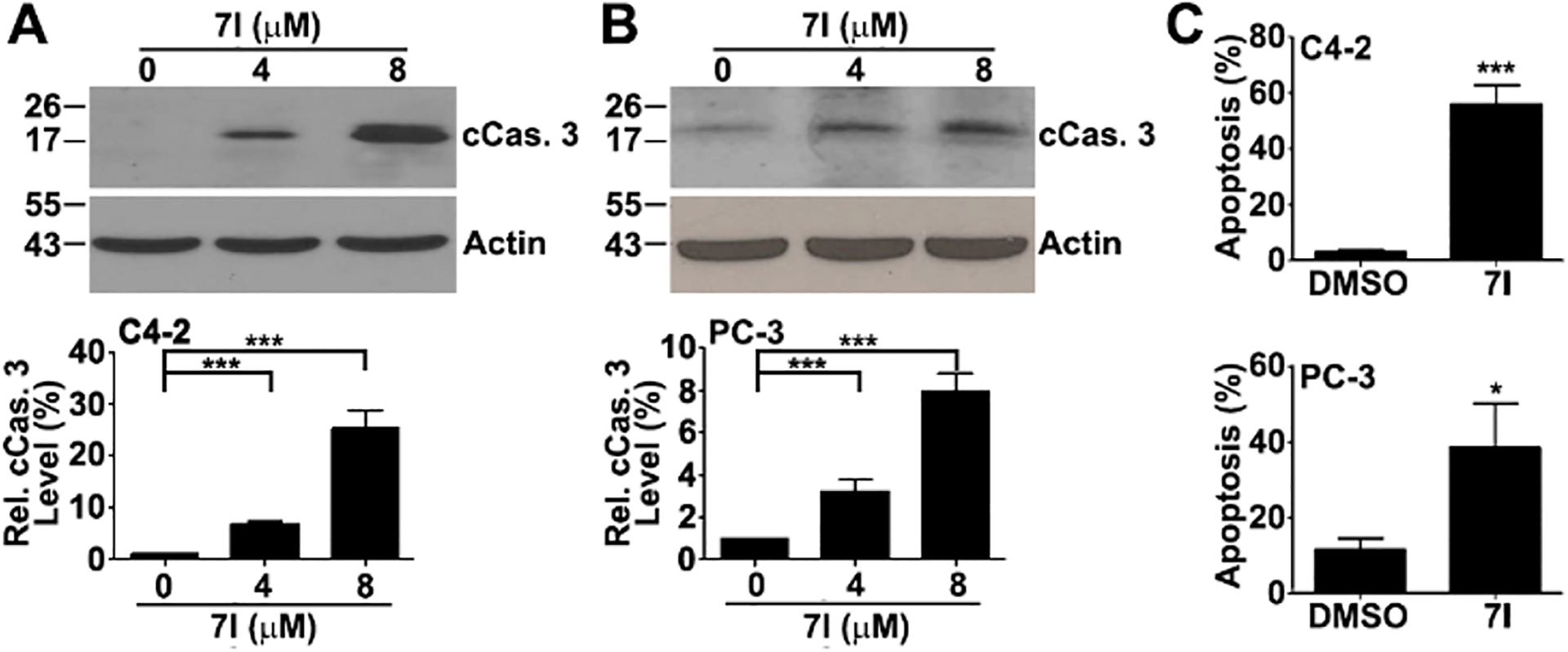
Compound 7I induction of apoptosis. (A, B) Western blot analysis of cleaved caspase 3 in C4-2 (A) and PC-3 (B) cells following 7I treatment. Quantifications were from three independent experiments. (C) Compound 7I-induced apoptosis as determined using Annexin V staining in C4-2 and PC-3 cells (*p < 0.05; ***p < 0.001).
Compound 7I Inhibits Growth of PC-3 Xenograft Tumors by Inhibiting Survivin.
We finally determined 7I’s in vivo target inhibition and if it is active in suppressing tumor growth using a PC-3 xenograft model. For in vivo analysis, 7I was dissolved in corn oil for oral administration. As shown in Figure 6A, 7I treatment at 100 mg/kg significantly suppressed tumor growth without any notable adverse effect on the mice as indicated by lacking changes in body weight (Figure 6B) or in wet weight of major organs at the end of the study (Figure S4). The xenograft tumors in the 7I-teatment group tended to be smaller and paler in color than those of the control group (Figure 6C,D). Thus, 7I may be effective in inhibiting tumor growth with little toxicity.
Figure 6.
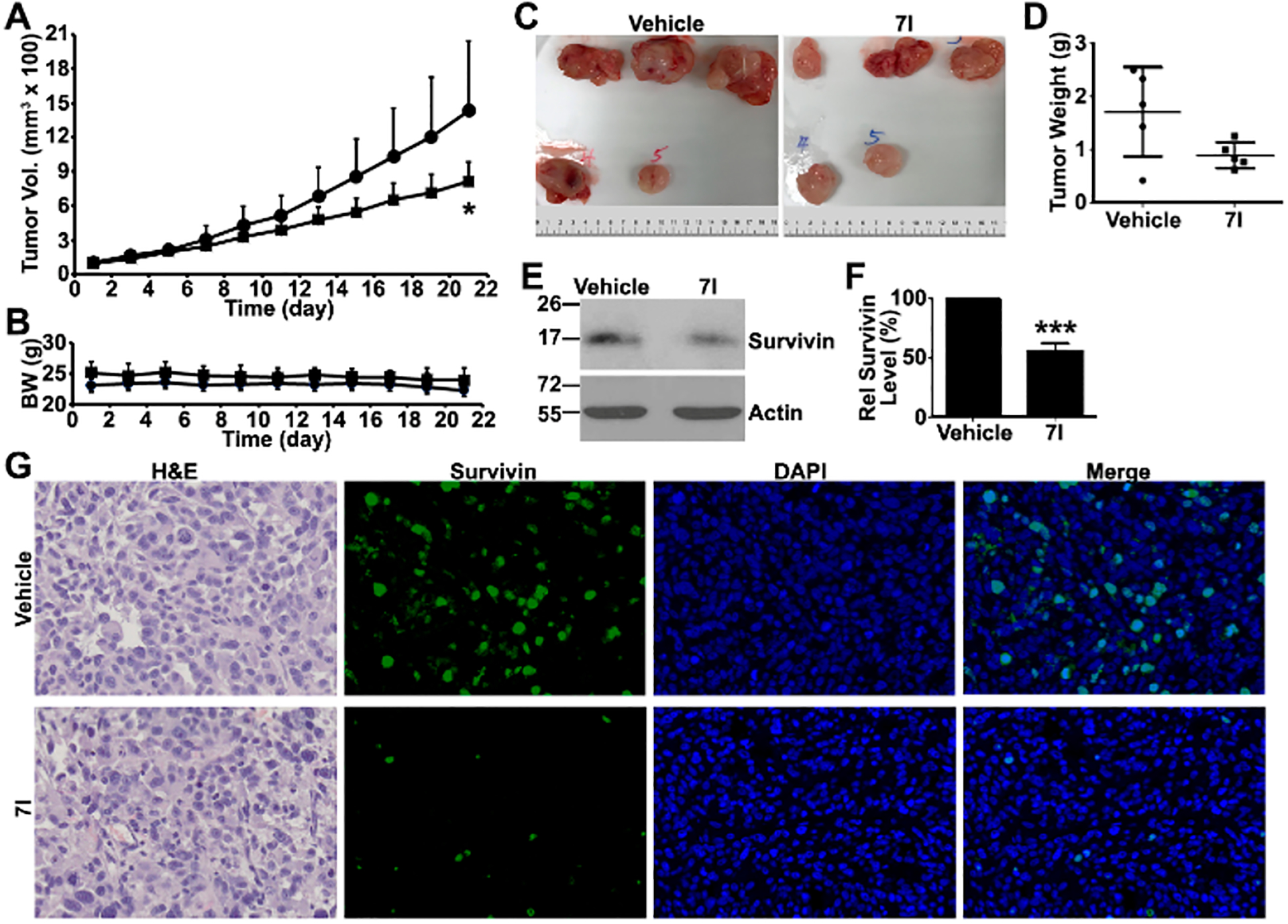
Compound 7I in vivo efficacy and target inhibition. (A, B) Effect of 7I on ectopic xenograft tumor growth and body weight in male NSG mice. (■) 7I; (●) vehicle. (C) Gross anatomy of xenograft tumors. (D) Final wet weight of xenograft tumors. (E, F) Western blot analysis of in vivo survivin target inhibition by 7I treatment. (G) Immunofluorescence staining of survivin in xenograft tumors (*p < 0.05; ***p < 0.001).
We finally examined if 7I inhibits the survivin target in vivo in the treatment group by performing Western blot and immunofluorescence staining analyses of xenograft tumors. As shown in Figure 6E–G, survivin expression was drastically reduced in the xenograft tumors of the treatment group compared with that of the control group. These findings suggest that 7I inhibits xenograft tumor growth possibly by inhibiting survivin dimerization and inducing survivin degradation.
DISCUSSION AND CONCLUSION
In this study, we synthesized five analogues of LQZ-7 to investigate the possibility of eliminating the labile hydrazone and replacing the furazanopyrazine with a quinoxaline, resulting in an analogue, 7I, that is more active than the parent compound LQZ-7 and soluble in corn oil. Compound 7I is also selective to survivin and induces apoptosis and survivin degradation in a proteasome-dependent manner. As a result, 7I inhibits cancer cell survival and suppresses xenograft tumor growth.
Of the 5 newly synthesized analogues, only 7I shows better activity in inducing survivin loss and inhibiting cancer cell survival compared with the parent compound LQZ-7. We presented several lines of evidence that strongly support the conclusion that 7I selectively inhibits survivin by inducing survivin loss, leading to inhibition of cancer cell survival, and induction of apoptosis. Thus, 7I is unlikely to exert its actions as a potential PAINS molecule. These findings also confirmed our previous observation on LQZ-7 in inducing survivin degradation by potentially binding to the dimeric interface of survivin.
Although the other 4 analogues have similar structure to 7I, they do not have activity in inhibiting survivin dimerization. Their IC50 values against prostate cancer cell lines are also high, consistent with their lack of survivin inhibition. In our previous study, several commercially available analogues of LQZ-7 (LQZ-7A–E) have also been studied with variable activities in inducing survivin degradation and in inhibiting cancer cell survival19 (Table 1). It is noteworthy that LQZ-7E (7E) is the only compound that does not have the benzene ring, which is essential for the π−π interaction with survivin, and it displayed a complete loss of the ability to induce survivin degradation and inhibited cancer cell survival with an IC50 of 60 μM. The other analogues had variable activities in inducing survivin degradation and in inhibiting cancer cell survival.
Table 1.
Characteristics of LQZ-7 Series of Survivin-Targeting Inhibitorsc
| Name | Structure | IC50 (μM) | Dimerization Inhibition | Survivin Degradation | |
|---|---|---|---|---|---|
| PC-3 | C4-2 | ||||
| LQZ-7b | 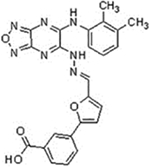 |
8.1±0.5 | 9.1±1.6 | + | + |
| LQZ-7Aa |  |
7.2±2.7 | ND | − | |
| LQZ-7Ba |  |
3.3±0.2 | ND | + | |
| LQZ-7Ca |  |
3.5±0.0 | ND | + | |
| LQZ-7Da |  |
2.6±0.3 | ND | − | |
| LQZ-7Ea | 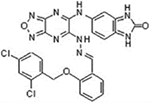 |
60.3±1.0 | ND | − | |
| LQZ-7Gb |  |
20.7± 2.0 | 14.3±1.4 | − | ND |
| LQZ-7Hb | 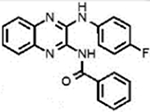 |
19.7±0.4 | 12.7±1.7 | − | ND |
| LQZ-7Ib | 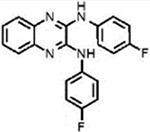 |
4.8±0.1 | 3.1±0.2 | + | + |
| LQZ-7Jb | 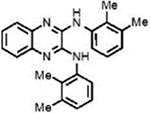 |
24.8±1.8 | 17.6±3.4 | − | ND |
| LQZ-7Kb | 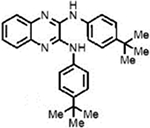 |
18.2±0.2 | 18.6±0.9 | − | ND |
The data for LQZ-7A–E were from the previous study.19 The IC50 was determined using MTT assay.
The data for LQZ-7G–K were from this study. The IC50 was determined using methylene blue assay.
ND = not determined.
While we do not have enough information to dissect a detailed structure–activity relationship of these compounds, we feel that with the data obtained in this set of experiments we have accomplished the goal of this study, which was to eliminate the undesirable labile hydrazone linker and potentially nonfunctional furazanopyrazine ring. Indeed, the activity of 7I suggests that the labile hydrazone linker and the furazanopyrazine ring of LQZ-7 are replaceable. This study will serve as a proof-of-concept study for future studies continuing to modify 7I without the labile hydrazone linker and furazanopyrazine ring, which may lead to identification of more potent compounds. One of the potential modification sites includes the nitrogen linker predicted to interact with Glu94 of survivin. Converting the nitrogen to oxygen may help reveal if the hydrogen bonding is important for 7I–survivin interaction. The benzene ring predicted to interact with survivin via π−π stacking is another potential site for further study. We are currently working toward this direction.
Because survivin has been shown to contribute to drug resistance by inhibiting drug-induced apoptosis.5–8, it will be of interest to assess the effectiveness of 7I in combination with chemotherapy for synergism in further suppression of xenograft tumor growth greater than 7I treatment alone. One possible combination for the castration resistant prostate cancer is docetaxel, which is a frontline treatment for this disease. Although very challenging, it will also be of interest to investigate the binding affinity of 7I versus survivin dimerization affinity and to investigate if 7I inhibits survivin dimerization on or off rate.
Future studies are also warranted to explore the complex upstream regulatory pathways that tightly control survivin expression to assess the effectiveness of our inhibitors for cancer cells with different genetic backgrounds. For example, we note a trend that 7I cytotoxicity correlates with p53 status in the five different prostate cancer cells lines. The cell lines with mutant p53 or p53 null (PC-3 and DU-145) tend to not only have higher survivin expression as one would expect but also higher 7I IC50. C4-2 and LNCaP with functional p53 had lower survivin protein levels and lower 7I IC50. Interestingly, 22RV-1 cells, which are known to have both full and truncated versions of p53, behave similarly to the cells with mutant p53 in this study. Although DNA-damaging agents are not the standard of care for castration resistant prostate cancers, information like this may help guide us in what context and disease state these survivin inhibitors may be most effective. Other upstream regulators of survivin, such as STAT3 and NF-κB, and their potential relationship with the cytotoxicity of our survivin dimerization inhibitors may also need to be investigated.
It is also noteworthy that although castration resistant prostate cancer was the disease focus of this study, survivin inhibitors may be also developed against other difficult-to-treat cancers such as glioblastoma since survivin expression is up-regulated in essentially all solid tumors. It is our long-term goal to expand and apply our knowledge in developing this class of unique survivin inhibitors to test their effectiveness in other difficult-to-treat cancers.
In a previous publication,19 we detailed the discovery of another LQZ-7 analogue, LQZ-7F (7F). The interesting feature of 7F compared to LQZ-7 and 7I is that it was the only compound identified in any of our screenings that had a different backbone scaffold. This compound displayed a locked backbone conformation consisting of 2 five-membered rings and 2 six-membered rings that maintained key interactions with survivin including (a) and H-bond between the primary amine group of 7F and Glu94 of survivin and (b) π−π stacking and hydrophobic interactions between the tetracyclic furazanopyrazine ring of 7F and the hydrophobic residues Trp10 and Phe93 in addition to the dimerization core residues Leu98 and Phe101. Compound 7F displayed more potent inhibition of cancer cells and had a strong positive correlation between survivin expression and cytotoxicity in cancer cell lines. More importantly, in an in vivo xenograft model, 7F treatment via ip injection significantly inhibited the growth of xenograft tumors.19
In conclusion, there are two structural scaffolds of inhibitors targeting the dimerization interface of survivin, represented by 7F and 7I, which effectively inhibit survivin dimerization, induce survivin degradation in the proteasome, and induce apoptosis of prostate cancer cells. They both also effectively inhibit xenograft tumor growth and reduce survivin level in vivo. Thus, both 7F and 7I can be used as leads of different scaffolds for further optimization.
EXPERIMENTAL SECTION
Materials.
Antibodies against HA tag (H6908) and actin (A5316) and the Annexin V-FITC apoptosis detection kit were obtained from MilliporeSigma. The antibody against survivin (2808) and the Metafectene Pro transfection reagent were purchased from Cell Signaling Technology and Bion-tex, respectively. The enhanced chemiluminescence reagent was from GE Healthcare. The matchmaker mammalian two hybrid assay kit and the Great Escape SEAP Chemiluminescence assay kit were purchased from Clontech. Cell culture media and fetal bovine serum were from Corning and Applied Biosystems-Life Technologies, respectively. All other chemicals were purchased from Sigma or Fisher Scientific.
Cell Lines.
The human prostate cancer cell lines C4-2, PC-3, Du145, LNCAP, and 22Rv1 were purchased from and authenticated by ATCC. All cell lines were maintained at 37 °C in 5% CO2 and grown in RPMI with 10% fetal bovine serum.
To generate C4-2 cells with stable expression of HA-tagged survivin, 2 × 106 cells were plated in a 10 cm dish and transfected with 10 μg of pcDNA3.1 vector control or HA-survivin cDNA cloned in pcDNA3.1 vector using Metafectene pro as we previously described.19 At 48 h after transfection, cells were harvested and replated in a fresh 10 cm dish at 2 × 105 cells/dish followed by selection using 0.8 mg/mL G418 with media and G418 changed every 72 h until colonies were visible for cloning. The stable clones were maintained with 0.2 mg/mL G418.
Construct Engineering.
For the mammalian two hybrid assay, we engineered two constructs encoding GAL4–survivin and VP16–survivin fusion proteins using pBIND and pACT vectors, respectively, in the ClonTech Matchmaker Mammalian Two Hybrid Assay kit21 as instructed by the manufacturer. Briefly, the cDNA encoding the open reading frame of survivin was amplified by PCR using primers 5′-AAGAATTCGGCGGCGGCGGCGGCGATGGGTGCCCCG-ACGTTGCCC-3′ (forward) and 5′-AACTCGAGTCAATCCATG-GCAGCCAGCTGCTC-3′ (reverse) and cloned into pGEM-T easy vector (Promega). The resulting plasmid was then digested with EcoRI, blunt ended, and digested again using XhoI to release the PCR insert, which was then cloned into pBIND and pACT digested with BamH1, blunt-ended, and then digested with Sal1. The resulting plasmids, pBIND-survivin and pACT-survivin, were validated by sequencing.
Survivin Mammalian Two Hybrid Assay.
The mammalian two hybrid assay was performed using the ClonTech Matchmaker Mammalian Two Hybrid Assay kit with the two newly engineered fusion plasmids as described above. Briefly, DU145 cells were seeded in 12-well plates at 1 × 105 cells/well and cultured for 24 h followed by transfection using pBIND-survivin and pACT-survivin (0.45 μg/well) along with the pGSEAP reporter plasmid (0.09 μg/well) and firefly luciferase reporter plasmid (30 ng/well). Two days after transfection, media was changed and cells were treated with DMSO, LQZ-7, or LQZ-7 analogues for 24 h followed by determination of SEAP activity using the Takara ClonTech SEAP Great Escape chemiluminescence assay kit 2.0. Luciferase activity was also measured as a control for transfection efficiency.
Methylene Blue Survival Assay.
The methylene blue survival assay was performed as previously described.22 Briefly, prostate cancer cells were seeded at 2000–3500 cells per well and treated with various concentrations of LQZ-7 or its analogues for 72 h. Media Technology and Biontex, respectively. The enhancedwas then removed, and cells were fixed with methanol for 30 min and subsequently stained with 100 μL of 1% methylene blue (diluted in 10 mM borate buffer) for 1 h. The cells were then washed 3 times with 10 mM borate buffer and then allowed to air-dry for 30 min. One hundred microliters of 100% ethanol/0.1 M HCl (1:1) was added to each well to dissolve the methylene blue stain, and OD650nm was measured. The data were analyzed using GraphPad Prism software to generate fitted curves and IC50.
Survivin Half-Life Determination.
The effect of 7I on survivin half-life was performed as previously described.19,23 Briefly, PC-3 or C4-2 cells were pretreated with 10 μM cycloheximide for 1 h followed by incubation with or without 10 μM 7I for different times (0–6 h). The cells were then harvested for Western Blot analysis of survivin.
Annexin V Apoptosis Assay.
The apoptosis assay was performed as detailed in the Annexin V-FITC kit from BioVision. Briefly, cells were treated with 3 μM 7I or DMSO control for 48 h followed by harvest, washing, and staining with annexin V-FITC antibody and propidium iodide. The stained cells were then analyzed using flow cytometry with excitation at 488 and 530 nm.
In Vivo Efficacy.
In vivo studies using animals were approved by IACUC at Indiana University School of Medicine. For the in vivo efficacy study, 3 × 106 PC-3 cells in media were implanted in the hind flanks of 6-week old NSG male mice. After the tumor volume reached approximately 100 mm3 in volume, the mice were randomized into two groups (5 mice/group). The mice were given 200 μL of either vehicle (90% corn oil/10% DMSO) or 7I (100 mg/kg) via oral gavage every other day for a total of ten treatments. The body weight and tumor size were measured every 2 days. At the end of the study mice were sacrificed, and tumors were excised for Western blot and IHC analysis of survivin expression.
Statistical Analysis.
All statistical analysis was performed using the program GraphPad Prism. Three independent experiments were performed for each study. Data presented is the mean ± standard deviation unless otherwise noted. All p-values were calculated using the Prism program via Student t tests or one-way ANOVA followed by post hoc test depending on the number of groups being compared.
Chemical Synthesis and Methods.
To access the quinoxaline diamine derivatives (7I, 7J, and 7K), commercially available 1,4-dihydro-2,3-quinoxalinedione (1, Figure 1C) was used as the starting point. Treating 1,4-dihydro-2,3-quinoxalinedione (1) with 4-fluoroaniline (2) or 2,3-dimethyl aniline (3) in refluxing ethanol afforded 7I and 7J, respectively. When 4-tert-butyl aniline (4) was used, the reaction needed to be heated in DMSO to give the desired product 7K. When a 1:1 mixture of 2,3-quinoxalinedione (1) and 4-fluoroaniline (2) was treated with AlCl3 in DCE at an elevated temperature, monosubstituted product 5 was obtained, which further underwent another nucleophilic substitution with either 6a or 6b to afford 7G and 7H, respectively.
All chemical reactions were performed using standard syringe techniques under argon. Starting materials and reagents were used as received from commercial suppliers. Acetonitrile (CH3CN), methanol (MeOH), and toluene were purified by passing the previously degassed solvents through activated alumina columns. Dichloromethane (CH2Cl2) and tetrahydrofuran (THF) were distilled prior to use.
All compounds were purified using flash chromatography with silica gel (230–400 mesh). Thin layer chromatography (TLC) was performed using glass-backed silica plates (Silicycle). NMR spectra were recorded on a Bruker ARX-400 spectrometer or AV-500 spectrometer at room temperature. Chemical shifts δ (in ppm) are given in reference to the solvent signal. 1H NMR data are reported as follows: chemical shifts (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quin = quintuplet, m = multiplet, br = broad), coupling constant (Hz), and integration. 13C NMR data are reported in terms of chemical shift and multiplicity. IR data were recorded on a Thermo Nicolet Nexus 470 FTIR. Electronspray ionization mass spectrometry data for compound characterization were determined using an Advion Expression CMS instrument. The purity was confirmed to be >95% using Waters ACQUITY UPC2 system.
General Procedure for the Synthesis of Quinoxaline Amine (5).
To a flame-dried round-bottom flask charged with a stir bar, commercially available 2,3-dichloroquinoxaline (1) (170 mg, 0.85 mmol, 1.0 equiv), 4-fluoroaniline (2) (0.081 mL, 0.85 mmol, 1.0 equiv), AlCl3 (125.3 mg, 0.94 mmol, 1.1 equiv), and DCE (4.25 mL, 0.2M) were added sequentially. The reaction mixture was then stirred at 80 °C for 16 h. After this period, the reaction was cooled to room temperature and quenched with ice-cold water (10 mL) while stirring. Stirring was continued for an additional 10 min. The reaction was then extracted with EtOAc (three times), and the combined organic layers were washed with ice-cold water two times. The organics were then dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography using hexanes–EtOAc (49:1 to 20:1) to give the pure product as a yellow solid. Yield: 92.2 mg, 40%. All spectroscopic data matched that previously reported in the literature.1
General Procedure for the Synthesis of Quinoxaline Amine Carboxamides 7G and 7H from Intermediate (5).
To a suspension of NaH (60% in mineral oil, 14.4 mg, 0.36 mmol, 2.0 equiv) in 0.8 mL of DMF was added the desired benzamide (0.36 mmol, 2.0 equiv) The reaction mixture was stirred at room temperature for 1 h. After the suspension turned light yellow, 3-chloro-N-(4-fluorophenyl)-quinoxalin-2-amine (5, 50 mg, 0.18 mmol, 1.0 equiv) in DMF (0.2 mL) was added slowly, and the solution turned dark red instantly. The reaction mixture was then stirred at 150 °C for 4 h and then cooled to room temperature before it was quenched with ice-cold water (3 mL). The reaction mixture was then extracted with EtOAc (three times), and the combined organic layers were dried over anhydrous MgSO4 and concentrated under reduced pressure. The crude product was then purified by column chromatography to give a bright yellow solid.
N-(3-((4-Fluorophenyl)amino)quinoxalin-2-yl)benzo[d][1,3]-dioxole-5-carboxamide (7G).
Analog 7G was synthesized from corresponding intermediate 5 and 6a. Column chromatography was performed using a DCM/hexanes (3:1) mixture to give the pure product (yellow solid, 5% yield). 1H NMR (500 MHz, CDCl3): δ = 9.04 (s, 1H), 8.01–7.94 (m, 3H), 7.77 (d, J = 1.6 Hz, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.42 (ddd, J = 8.3, 6.6, 2.1 Hz, 1H), 7.38–7.31 (m, 2H), 7.14 (t, J = 8.7 Hz, 2H), 6.93 (d, J = 8.2 Hz, 1H), 6.09 (s, 2H). 13C NMR (125 MHz, CDCl3): δ = 179.12, 159.85, 157.92, 151.43, 147.84, 145.96, 145.19, 135.15, 134.81, 131.33, 126.66, 126.59, 125.90, 125.22, 124.75, 121.23, 116.84, 115.81, 115.63, 109.30, 107.95, 101.78. IR (neat, cm−1): ν = 3319, 2921, 1593, 1546, 1506, 1483, 1437, 1382, 1356, 1292, 1255, 1209, 1192, 1156, 1104, 1038, 926, 832, 808. MS (ESI): m/z 403.1 found for C22H16FN4O3[M + H]+.
N-(3-((4-Fluorophenyl)amino)quinoxalin-2-yl)benzamide (7H).
Analog 7H was synthesized from corresponding intermediate 5 and 6b. Column chromatography was performed using a hexanes/EtOAc (5.6:1) mixture to give the pure product (yellow solid, 58% yield). 1H NMR (500 MHz, CDCl3): δ = 9.06 (s, 1H), 8.30 (d, J = 7.5 Hz, 2H), 7.98–7.89 (m, 2H), 7.98–7.89 (m, 1H), 7.61–7.55 (m, 1H), 7.51 (t, J = 7.5 Hz, 2H), 7.43–7.37 (m, 1H), 7.35–7.27 (m, 1H), 7.11 (t, J = 8.5 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ = 180.15, 159.91, 157.98, 146.00, 145.41, 136.90, 135.37, 134.95, 132.64, 129.44, 128.48 (2C), 126.80, 126.76, 126.00, 124.72, 121.20, 121.14, 117.01, 115.89, 115.71. IR (neat, cm−1): ν = 3316, 3066, 1592, 1551, 1506, 1483, 1448, 1413, 1385, 1356, 1314, 1291, 1210, 1156, 1124, 1100, 1068, 1024, 921, 864, 831. MS (ESI): m/z 359.2 found for C21H16FN4O[M + H]+.
General Procedure for the Synthesis of Quinoxaline-2,3-Diamines (7I and 7J).
To a flame-dried round bottomed flask, commercially available 2,3-dichloroquinoxaline (1, 200 mg, 1.0 mmol, 1.0 equiv), substituted anilines (2 or 3, 2.2 mmol, 2.2 equiv), and ethanol (5.0 mL, 0.2 M) were added sequentially. The reaction mixture was then refluxed for 16 h. After reaching room temperature, the solvent was slowly removed under reduced pressure. The resulting solid was dissolved in EtOAc (10 mL), washed with water and brine, dried over anhydrous MgSO4 and concentrated under reduced pressure to give the crude product. The crude product was then purified by column chromatography.
N2,N3-Bis(4-fluorophenyl)quinoxaline-2,3-diamine (7I).
Analog 7I was synthesized from commercially available 1 and 4-fluoroaniline (2). Column chromatography was performed using a hexanes/EtOAc (20:1 to 3:1) mixture to give the pure product (yellow solid, 52% yield). 1H NMR (500 MHz, DMSO-d6): δ = 9.06 (s, 2H), 7.95–7.75 (m, 4H), 7.53 (dd, J = 6.1, 3.5 Hz, 2H), 7.33 (dd, J = 6.1, 3.4 Hz, 2H), 7.24 (t, J = 8.9 Hz, 4H). 13C NMR (125 MHz, DMSO-d6): δ = 158.74 (2C), 156.84 (2C), 141.19, 141.16, 136.49, 136.13, 125.34, 125.15, 125.11, 122.37, 122.31, 115.29 (2C), 115.26 (2C), 115.11, 115.09 (2C). IR (neat, cm−1): ν = 3386, 2925, 1644, 1616, 1574, 1537, 1501, 1449, 1410, 1328, 1301, 1212, 1155, 1118, 1098, 1012, 923, 830. MS (ESI): m/z 349.2 found for C20H15F2N4[M + H]+.
N2,N3-Bis(2,3-dimethylphenyl)quinoxaline-2,3-diamine (7J).
Analog 7J was synthesized from commercially available 1 and 2,3-dimethylaniline (3). Column chromatography was performed using a hexanes/EtOAc (49:1 to 20:1) mixture to give the pure product (light brown solid, 16% yield). 1H NMR (500 MHz, DMSO-d6): δ = 8.52 (s, 2H), 7.38–7.23 (m, 4H), 7.22–7.12 (m, 4H), 7.09 (d, J = 7.6 Hz, 2H), 2.32 (s, 6H), 2.11 (s, 6H). 13C NMR (125 MHz, DMSO-d6): δ 142.59 (2C), 137.72 (2C), 137.15 (2C), 136.62 (2C), 132.27 (2C), 126.88 (2C), 125.47 (2C), 125.05 (2C), 124.30 (2C), 124.16 (2C), 20.29 (2C), 14.56 (2C). IR (neat, cm−1): ν = 3312, 2916, 1640, 1614, 1567, 1541, 1463, 1381, 1351, 1324, 1258, 1215, 1181, 1119, 1089, 1060, 990, 927, 896, 779. MS (ESI): m/z 369.2 found for C24H25N4[M + H]+.
General Procedure for the Synthesis of Quinoxaline-2,3-diamine (7K).
A mixture of 2,3-dichloroquinoxaline (1, 50 mg, 0.25 mmol, 1.0 equiv), 4-tert-butylaniline (4, 1.25 mmol, 5.0 equiv) and DMSO (2.5 mL, 0.1 M) in a flame-dried round-bottomed flask was heated at 130 °C for 2.5 h. After this period, the reaction mixture was cooled to room temperature, diluted with EtOAc (2.5 mL), and poured into ice–water (10 mL). The organic phase was then washed with ice-cold water two times, dried over MgSO4, and concentrated under reduced pressure. The crude product was then purified by column chromatography using hexanes/EtOAc (20:1) to give the pure product.
N2,N3-Bis(4-(tert-butyl)phenyl)quinoxaline-2,3-diamine (7K).
Light brown oil, 23% yield. 1H NMR (500 MHz, DMSO-d6): δ = 8.97 (s, 2H), 7.82 (d, J = 8.7 Hz, 4H), 7.52 (dd, J = 6.1, 3.4 Hz, 2H), 7.42 (d, J = 8.7 Hz, 4H), 7.32 (dd, J = 6.1, 3.4 Hz, 2H), 1.32 (s, 18H). 13C NMR (125 MHz, DMSO-d6): δ 144.96 (2C), 141.12 (2), 137.55 (2C), 136.19 (2C), 125.24 (4C), 124.93 (4C), 120.35 (4C), 34.06 (2C), 31.31 (6C). IR (neat, cm−1): ν = 3385, 2958, 2902, 2865, 1640, 1613, 1586, 1570, 1513, 1450, 1408, 1361, 1330, 1315, 1294, 1266, 1245, 1191, 1144, 1111, 1016, 940, 858, 831. MS (ESI): m/z 425.3 found for C28H33N4[M + H]+.
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank the University of Toledo Integrated Core Facilities for histology analysis of xenograft tumors.
Funding
This work was supported in part by the DOD-CDMRP Grant PC131242 (J.T.Z.) and NIH Grants R01 GM127656 (J.Y.L.) and R35 GM128570 (M.D.). M.D. thanks the NIH, Grant P30 CA023168, for supporting shared NMR resources to Purdue Center for Cancer Research.
ABBREVIATIONS
- IAP
inhibitor of apoptosis protein
- XIAP
X-linked IAP
- CIAP
cellular IAP
- STAT3
signal transducer and activator of transcription 3
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- BIRC5
baculoviral IAP repeat containing 5
- HA
hemagglutinin
- SEAP
secreted alkaline phosphatase
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00475.
Dose–response curves for all compounds for survival of human prostate cancer cell lines C4-2 and PC-3 and for 7I for cell lines DU145, PC-3, C4-2, 22RV-1, and LNCaP and C4-2 stably overexpressing survivin, mean wet weight of major organs in 7I treated mice, and purity analysis of 7I (PDF)
Molecular formula strings (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00475
The authors declare no competing financial interest.
Contributor Information
Robert Peery, Department of Pharmacology and Toxicology, Indiana University School of Medicine, Indianapolis, Indiana 46202, United States.
Kwaku Kyei-Baffour, Department of Chemistry and Center for Cancer Research, Purdue University, West Lafayette, Indiana 47907, United States.
Zizheng Dong, Department of Pharmacology and Toxicology, Indiana University School of Medicine, Indianapolis, Indiana 46202, United States; Department of Cancer Biology, University of Toledo College of Medicine and Life Sciences, Toledo, Ohio 43614, United States.
Jianguo Liu, Department of Cancer Biology, University of Toledo College of Medicine and Life Sciences, Toledo, Ohio 43614, United States.
Jing-Yuan Liu, Department of Pharmacology and Toxicology, Indiana University School of Medicine, Indianapolis, Indiana 46202, United States; Department of Medicine, University of Toledo College of Medicine and Life Sciences, Toledo, Ohio 43614, United States.
Jian-Ting Zhang, Department of Pharmacology and Toxicology, Indiana University School of Medicine, Indianapolis, Indiana 46202, United States; Department of Cancer Biology, University of Toledo College of Medicine and Life Sciences, Toledo, Ohio 43614, United States.
REFERENCES
- (1).Verdecia MA; Huang H; Dutil E; Kaiser DA; Hunter T; Noel JP Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat. Struct. Biol 2000, 7, 602–608. [DOI] [PubMed] [Google Scholar]
- (2).Grossman D; Kim PJ; Blanc-Brude OP; Brash DE; Tognin S; Marchisio PC; Altieri DC Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J. Clin. Invest 2001, 108, 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Salvesen GS; Duckett CS IAP proteins: blocking the road to death’s door. Nat. Rev. Mol. Cell Biol 2002, 3, 401–410. [DOI] [PubMed] [Google Scholar]
- (4).Chantalat L; Skoufias DA; Kleman JP; Jung B; Dideberg O; Margolis RL Crystal structure of human survivin reveals a bow tie-shaped dimer with two unusual alpha-helical extensions. Mol. Cell 2000, 6, 183–189. [PubMed] [Google Scholar]
- (5).Tamm I; Wang Y; Sausville E; Scudiero DA; Vigna N; Oltersdorf T; Reed JC IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res 1998, 58, 5315–520. [PubMed] [Google Scholar]
- (6).Grossman SA; Ye X; Peereboom D; Rosenfeld MR; Mikkelsen T; Supko JG; Desideri S; Adult Brain Tumor Consortium. Phase I study of terameprocol in patients with recurrent high-grade glioma. Neuro Oncol 2012, 14, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Suzuki A; Ito T; Kawano H; Hayashida M; Hayasaki Y; Tsutomi Y; Akahane K; Nakano T; Miura M; Shiraki K Survivin initiates procaspase 3/p21 complex formation as a result of interaction with Cdk4 to resist Fas-mediated cell death. Oncogene 2000, 19, 1346–1353. [DOI] [PubMed] [Google Scholar]
- (8).Rodel F; Hoffmann J; Grabenbauer GG; Papadopoulos T; Weiss C; Gunther K; Schick C; Sauer R; Rodel C High survivin expression is associated with reduced apoptosis in rectal cancer and may predict disease-free survival after preoperative radiochemotherapy and surgical resection. Strahlenther. Onkol 2002, 178, 426–435. [DOI] [PubMed] [Google Scholar]
- (9).Schmidt SM; Schag K; Muller MR; Weck MM; Appel S; Kanz L; Grunebach F; Brossart P Survivin is a shared tumor-associated antigen expressed in a broad variety of malignancies and recognized by specific cytotoxic T cells. Blood 2003, 102, 571–576. [DOI] [PubMed] [Google Scholar]
- (10).Li F; Ackermann EJ; Bennett CF; Rothermel AL; Plescia J; Tognin S; Villa A; Marchisio PC; Altieri DC Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat. Cell Biol 1999, 1, 461–466. [DOI] [PubMed] [Google Scholar]
- (11).Chen J; Wu W; Tahir SK; Kroeger PE; Rosenberg SH; Cowsert LM; Bennett F; Krajewski S; Krajewska M; Welsh K; Reed JC; Ng SC Down-regulation of survivin by antisense oligonucleotides increases apoptosis, inhibits cytokinesis and anchorage-independent growth. Neoplasia 2000, 2, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kasof GM; Gomes BC Livin, a novel inhibitor of apoptosis protein family member. J. Biol. Chem 2001, 276, 3238–3246. [DOI] [PubMed] [Google Scholar]
- (13).Olie RA; Simoes-Wust AP; Baumann B; Leech SH; Fabbro D; Stahel RA; Zangemeister-Wittke U A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res 2000, 60, 2805–2809. [PubMed] [Google Scholar]
- (14).Pennati M; Colella G; Folini M; Citti L; Daidone MG; Zaffaroni N Ribozyme-mediated attenuation of survivin expression sensitizes human melanoma cells to cisplatin-induced apoptosis. J. Clin. Invest 2002, 109, 285–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Williams NS; Gaynor RB; Scoggin S; Verma U; Gokaslan T; Simmang C; Fleming J; Tavana D; Frenkel E; Becerra C Identification and validation of genes involved in the pathogenesis of colorectal cancer using cDNA microarrays and RNA interference. Clin. Cancer Res 2003, 9, 931–46. [PubMed] [Google Scholar]
- (16).Choi KS; Lee TH; Jung MH Ribozyme-mediated cleavage of the human survivin mRNA and inhibition of antiapoptotic function of survivin in MCF-7 cells. Cancer Gene Ther 2003, 10, 87–95. [DOI] [PubMed] [Google Scholar]
- (17).Peery RC; Liu JY; Zhang JT Targeting survivin for therapeutic discovery: past, present, and future promises. Drug Discovery Today 2017, 22, 1466–1477. [DOI] [PubMed] [Google Scholar]
- (18).Li F; Aljahdali I; Ling X Cancer therapeutics using survivin BIRC5 as a target: what can we do after over two decades of study? J. Exp. Clin. Cancer Res 2019, 38, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Qi J; Dong Z; Liu J; Peery RC; Zhang S; Liu JY; Zhang JT Effective targeting of the survivin dimerization interface with small-molecule inhibitors. Cancer Res 2016, 76, 453–62. [DOI] [PubMed] [Google Scholar]
- (20).Zhang M; Mukherjee N; Bermudez RS; Latham DE; Delaney MA; Zietman AL; Shipley WU; Chakravarti A Adenovirus-mediated inhibition of survivin expression sensitizes human prostate cancer cells to paclitaxel in vitro and in vivo. Prostate 2005, 64, 293–302. [DOI] [PubMed] [Google Scholar]
- (21).Fagan R; Flint KJ; Jones N Phosphorylation of E2F-1 modulates its interaction with the retinoblastoma gene product and the adenoviral E4 19 kDa protein. Cell 1994, 78, 799–811. [DOI] [PubMed] [Google Scholar]
- (22).Beebe JD; Lee MS; McClelland C Chiasmal and Retrochiasmal Disorders. Int. Ophthalmol. Clin 2019, 59, 59–81. [DOI] [PubMed] [Google Scholar]
- (23).Peng H; Dong Z; Qi J; Yang Y; Liu Y; Li Z; Xu J; Zhang JT A Novel Two Mode-Acting Inhibitor of ABCG2-Mediated Multidrug Transport and Resistance in Cancer Chemotherapy. PLoS One 2009, 4, No. e5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.