

Supplemental Digital Content is available in the text.
Keywords: atherosclerosis, cardiovascular diseases, endothelial cells, gene expression, macrophages, microRNA
Abstract
Objective:
Atherosclerosis is the underlying cause of most cardiovascular diseases. The main cell types associated with disease progression in the vascular wall are endothelial cells, smooth muscle cells, and macrophages. Although their role in atherogenesis has been extensively described, molecular mechanisms underlying gene expression changes remain unknown. The objective of this study was to characterize microRNA (miRNA)-related regulatory mechanisms taking place in the aorta during atherosclerosis.
Approach and Results:
We analyzed the miRNA expression changes in primary human aortic endothelial cells and human umbilical vein endothelial cells, human aortic smooth muscle cells, and macrophages (CD14+) under various proatherogenic stimuli by integrating GRO-seq, miRNA-seq, and RNA-seq data. Despite the highly cell-type-specific expression of multi-variant primary miRNAs, the majority of mature miRNAs were found to be common to all cell types and dominated by 2 to 5 abundant miRNA species. We demonstrate that transcription contributes significantly to the mature miRNA levels although this is dependent on miRNA stability. An analysis of miRNA effects in relation to target mRNA pools highlighted pathways and targets through which miRNAs could affect atherogenesis in a cell-type-dependent manner. Finally, we validate miR-100-5p as a cell-type specific regulator of inflammatory and HIPPO-YAP/TAZ-pathways.
Conclusions:
This integrative approach allowed us to characterize miRNA dynamics in response to a proatherogenic stimulus and identify potential mechanisms by which miRNAs affect atherogenesis in a cell-type-specific manner.
Highlights.
Pri-miRNAs (primary-microRNAs) transcription and transcriptional start site usage is highly cell-type specific and minimally affected by the proatherogenic stimulus.
Mature miRNA expression patterns are similar among cells types and poorly correlated to primary miRNA expression. Furthermore, 2 to 5 miRNAs are shown to account for over 50% of the total miRNA expression in the cells.
Pri-miRNA expression under single and combined proatherogenic stimulus provide evidence of similar transcription factor enrichment at regulated transcriptional start sites.
miR-100-5p silences the HIPPO pathway, leading to the activation of the YAP/TAZ cascade in endothelial cells.
TNFα-, IFNβ-, and hypoxia-pathway mediators are positively enriched among the miR-100-5p regulated genes in human aortic smooth muscle cells but negatively enriched in human umbilical vein endothelial cells. Similarly, miR-100-5p regulated genes in human umbilical vein endothelial cells were enriched for genes involved in proliferation whereas in human aortic smooth muscle cells the genes were enriched for apoptosis.
Early events in atherogenesis take place in the endothelium, which forms the inner surface of the vascular wall (Figure IA in the Data Supplement). Endothelial dysfunction increases the vascular permeability promoting lipid accumulation into the vessel wall, and increases local oxidative stress leading to the formation of oxidized phospholipids, which have been shown to accumulate in the atherosclerotic plaques.1 The formation of oxidized lipids stimulates chemokine production, which attracts monocytes to the site, and induces their differentiation into macrophages. After the ingestion of oxidized lipids, macrophages turn into foam cells that perpetuate vascular remodeling. Eventually, the local release of inflammatory mediators induces proliferation and migration of vascular smooth muscle cells, resulting in vessel wall thickening and hypoxia, which further stimulates intraplaque angiogenesis and facilitates the progression of the disease.1,2 Although the general proatherogenic changes in endothelial cell, macrophage, and smooth muscle cell function have been described, the mechanisms leading to these changes have not been characterized. This is mainly due to a lack of knowledge on how transcriptional programs differ between cell types and a lack of comprehensive gene expression data which integrates different levels of gene regulation.
MicroRNAs (miRNAs) are small noncoding RNAs that repress gene expression post-transcriptionally in the cytoplasm3 but may also mediate noncanonical roles to regulate gene expression in the nucleus,4 as well as protein function.5 miRNA biogenesis begins with primary miRNA (pri-miRNA) transcription from the genome, followed by cleavage with the DGCR8 (DiGeorge syndrome critical region 8)/Drosha complex into a hairpin-shaped loop structure called precursor-miRNA (pre-miRNA; Figure IC in the Data Supplement). Pre-miRNA is exported from the nucleus to the cytoplasm and further processed by Dicer into 2 single-stranded mature miRNAs.6 The primary role of miRNAs is to fine-tune cellular functions and maintain tissue homeostasis, but in pathological states their effects become more pronounced and play more decisive roles.6 miRNA therapeutics that manipulate cellular miRNA levels have already entered clinical trials,6 although many aspects of miRNA function remain elusive.
Knowledge on the roles that miRNAs play in gene regulatory networks has increased significantly in the past 2 decades following the initial miRNA discovery.7,8 However, most profiling studies have been performed at the tissue/organ level, which makes it difficult to discern the actual causes of the altered signals between different tissue types and patient and control samples. Tissues are heterogeneous collections of cell types and the observed changes in the miRNA profiles can, for instance, arise from the differences in the cell composition of the samples rather than from changes in miRNA expression in specific cell types.9 Similarly, the miRNA expression arising from cell types that are found essentially in all organs, such as endothelial cells, red blood cells, and fibroblasts, can be misinterpreted as ubiquitous without knowledge on cellular miRNA expression profiles.10 Having cell-type-specific knowledge on miRNA expression patterns assures that functional miRNA studies are performed in appropriate cell types and will thus be of biological relevance.
Recent efforts11–13 to clarify the global miRNA expression patterns in human primary cells are filling the gaps in present knowledge, but the cellular miRNA expression patterns under disease-causing stimuli remain unknown. Although both miRNA and mRNA profiling studies have actively been conducted in various disease-relevant contexts, integration of different levels of genomics data is rarely done to elucidate miRNA expression and function. Importantly, mature miRNAs are processed from primary transcripts (pri-miRNAs), and their expression is controlled at the transcriptional and posttranscriptional levels. However, how regulation at multiple levels achieves precise control remains elusive.
In this study, we aimed to address these questions by investigating the miRNA expression profiles in atherosclerosis-modeling primary cell types, namely human aortic endothelial cell (HAEC) and human umbilical vein endothelial cell (HUVECs), aortic smooth muscle cells (HASMCs), and macrophages (CD14+), subjected to proatherogenic stimuli (Figure IB in the Data Supplement) to shed light on the miRNA-related regulatory mechanisms that could contribute to atherosclerosis. To achieve this, we generated transcriptomics data sets measuring nascent primary miRNA transcription (global run-on sequencing [GRO-seq]), mature microRNAs (miRNA-seq), and target mRNAs (ribosome depleted RNA-seq; Figure IC in the Data Supplement). Using this integrative approach, we were able to establish regulatory networks among the proatherogenic-stimuli-responsive miRNAs and their target genes and to identify potential players driving proatherogenic changes in a cell-type-specific manner.
Materials and Methods
Cell Cultures and Treatments
HUVECs were isolated from 6 different donors. HUVECs were extracted from umbilical cords obtained from the maternity ward of Kuopio University Hospital (used at passage 4–7) or purchased from Lonza (passage 9). This study has been performed according to the recommendations of the Research Ethics Committee of the Hospital District of Northern Savo, Kuopio, Finland. Informed written consent was received from all the participants, and the experiments were performed according to the relevant guidelines and regulations. HUVECs were cultivated in Endothelial Cell Basal Medium (EBM; Lonza) with recommended supplements (EGM SingleQuot Kit Supplements and Growth Factors, Lonza) after a fibronectin-gelatin coating (10 µg/mL fibronectin [Sigma, St Louis, MO] and 0.05% gelatin). Each HUVEC donor was analyzed as a separate replicate.
HAECs from different donors were obtained from Lonza and cultivated in Endothelial Cell Basal Medium (Lonza) with recommended supplements (EGM SingleQuot Kit Supplements & Growth Factors, Lonza) after fibronectin-gelatin coating (10 µg/mL fibronectin [Sigma, St Louis, MO] and 0.05% gelatin). HASMCs from different donors were obtained from Lonza and cultivated in medium 231 (Thermofisher, Carlsbad, CA) supplemented with smooth muscle growth supplement. Human monocytes from different donors were obtained from Lonza. The monocytes were cultivated in Roswell Park Memorial Institute (RPMI) 1640 Medium, supplemented with 10% FBS, 100 μg/mL streptomycin, 100 U/mL penicillin, 2 mmol/L glutamine, 1% Na-pyruvate, 1% NEAA (Non-Essential Amino Acid cell culture supplement), and supplemented with rHu M-CSF (50 ng/mL; ThermoFisher Scientific) to differentiate them into macrophages. The monocyte-derived macrophages will be referred to as CD14+ macrophages throughout the study.
To mimic an intermediate atherosclerotic state, we treated cells with oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC) and hypoxia for 7 hours. Oxidized phospholipids have been reported to accumulate after disruption of the endothelial barrier, contributing to vascular inflammation.14–17 In addition, hypoxia is present in atherosclerotic lesions stimulating proatherosclerotic processes.18 A 7-hour timepoint was chosen to capture both early and late transcriptional responses to stimulus.19 Moreover, short exposure to hypoxia tends to promote cell survival and growth, while prolonged exposure to hypoxia leads to cell death.20 Hypoxia was achieved using a Ruskinn InvivO2 400 hypoxia workstation (Baker Ruskinn, Bridgend, Wales) in the presence of 1% O2 and 5% CO2. OxPAPC was generated from 1-palmitoyl-2-archidonoyl-sn-glycero-3-phophocholine (PAPC, 10 mg/mL [Avanti Polar Lipids Inc, Alabaster, AL]). PAPC was exposed to air for 40 hours, then dissolved in chloroform and stored at −70 °C. At the start of the oxPAPC stimulation, chloroform was evaporated with nitrogen gas, and lipids were resuspended in growth medium to achieve a concentration of 30 μg/mL.
GRO-Seq, RNA-Seq, and miRNA-Seq
For the GRO-seq, HUVECs, HAECs, HASMCs, and CD14+ macrophages were treated with a medium containing 1% FBS with or without oxPAPC (30 μg/mL) for 6 hours under normoxia or hypoxia. Cells were washed twice with cold PBS and incubated on ice with 10 mL of swelling buffer (10 mmol/L Tris-HCl, 2 mmol/L MgCl2, 3 mmol/L CaCl2, and 2 U/mL SUPERase inhibitor [Thermofisher, Carlsbad, CA]) for 5 minutes. The cells were scraped and centrifuged at 400g for 10 minutes and resuspended in 50 μL of swelling buffer supplemented with 10% glycerol. Next, 500 μL of swelling buffer containing 10% glycerol and 1% Igepal was added drop by drop while gently vortexing. The nuclei were washed twice with a lysis buffer (10 mL of swelling buffer containing 0.5% igepal and 10% glycerol), and once with 1 mL of a freezing buffer (50 mmol/L Tris-HCl pH 8.3, 40% glycerol, 5 mmol/L MgCl2, and 0.1 mmol/L EDTA). Finally, the nuclei were counted, centrifuged at 900g for 6 minutes to be further resuspended into a concentration of 5 million nuclei per 100 μL of freezing buffer, snap-frozen in liquid nitrogen, and stored at −80 °C until run-on reaction. The run-on reaction and library preparation were performed as described in Bouvy-Liivrand et al.21 Libraries were amplified using 13 to 15 cycles, size-selected (180–350 bp) from 10% TBE gels (Life Technologies) and sequenced (single end 50 bases) using an Illumina HiSeq 2000 at EMBL GeneCore (Heidelberg, Germany).
RNA-seq samples were obtained 1 hour after the collection of the GRO-seq samples from the matching samples (reflecting the lag in nascent transcription and mature RNAs). The cells were treated for 10 minutes with cycloheximide (0.1 mg/mL) to stop mRNA translation,22–24 subsequently washed with PBS and scraped into a lysis buffer (1x Mammalian Polysome Buffer [Epicentre, Madison, Wisconsin], 1% Triton X-100, 1 mmol/L DTT, 250 U/mL SUPERase Inhibitor, 7.1 U/ml Turbo DNase [ThermoFisher Scientific, Waltham, MA] and 0.1 mg/mL Cycloheximide) on ice. To confirm the complete lysis of the cells, they were drawn up and expelled 4× through a sterile 22 to 25 gauge needle. The cleared whole cell lysate was further treated with 10% SDS, snap-frozen in liquid nitrogen, and stored at −80 °C. Larger mRNAs (>200 nt), from the total RNA, were purified with a Zymo RNA Clean and Conc kit (Zymo Research, Irvine, CA), and rRNAs were eliminated using the Ribo-Zero Gold rRNA Removal Kit (Illumina, San Diego, CA). The RNA was then fragmentated (RNA fragmentation reagent, Thermofisher) and dephosphorylated. The libraries were prepared as described in Bouvy-Liivrand et al21 for GRO-seq but omitting the anti-BrUTP pulldown. The libraries were amplified by 11 to 16 cycles, size selected (190–350 bp), and quantified (Qubit dsDNA HS Assay Kit on a Qubit fluorometer, Thermofisher, Carlsbad, CA). Sequencing (single-end 50 bases) was performed with an Illumina Hi-Seq2000 at EMBL GeneCore (Heidelberg, Germany).
For the RNA-seq gene expression quantification of the HASMC and HUVECs samples transfected with miRNA mimic control and miR-100-5p treated with hypoxia+oxPAPC, the total RNA was isolated using an RNeasy Mini Kit (QIAGEN). Libraries were prepared using the QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina (Lexogen) according to the manufacturer’s instructions. For each sample, 250 ng of total RNA was used for the library preparation, and the libraries were sequenced using a read length of 78 bases (single-end) on an Illumina NextSeq 500 sequencer. The average sequencing depth was 6.3×106 reads per library (range, 5.5–7.3×106).
A small RNA-fraction (17–200 bases) was obtained by adding ethanol to the flow through from the total RNA extracted from the RNA Clean and Concentrator (Zymo Research, Irvin, CA) kit and passing the RNA through a new column. The libraries were prepared at the Finnish Microarray and Sequencing Centre Turku Centre for Biotechnology (Turku, Finland) using the TruSeq small RNA-seq protocol or at Exiqon (Vedbæk, Denmark) with the small RNA-Seq protocol.
miRNA Overexpression and Silencing (Transfection)
HUVECs were seeded on 6-well plates and transfected at 70% confluence using Oligofectamine (Invitrogen, Carlsbad, CA). The following oligonucleotides were employed: MISSION miRNA Mimics Negative Control No. 1 (HMC0002, Sigma-Aldrich, Saint-Louis, MO), hsa-miR-100-5p mimic (HMI0023, Sigma-Aldrich, Saint-Louis, MO), MISSION Synthetic microRNA (miRNA) Inhibitors Negative control I (NCSTUD001, Sigma-Aldrich, Saint-Louis, MO), and hsa-miR-100-5p inhibitor (HSTUD0023, Sigma-Aldrich, Saint-Louis, MO). Mimic miRNAs were used at a final concentration of 25 nmol/L, while inhibitor miRNAs were used at 1 nmol/L. Medium supplements were added 4 hours posttransfection, and on the next day the cells were washed with PBS and a fresh EBM medium with full supplements was added. RNA isolation, Western blot, and miRNA expression analyses were performed 48 hours after transfections.
miRNA Purification
HUVEC miRNAs and mRNAs were separated using an RNeasy Mini Kit (QIAGEN, Hilden, Germany). The cells were disrupted using 350 μL of buffer RLT, followed by 350 μL of 70% ethanol. The samples were then transferred to an RNeasy Mini spin column and centrifuged for 15 s at 8000g. The flow through was kept to extract the miRNAs while the column was loaded with the larger RNAs (>200 nt). The miRNA fraction was diluted using a 0.65 volume of 100% ethanol and loaded into a new RNEasy Mini Kit (QIAGEN, Hilden, Germany) column. The column was then washed using 2 steps of RPE buffer (RNEasy Mini Kit [QIAGEN, Hilden, Germany]) of 700 μL and 500 μL, respectively, followed by 500 μL of 80% ethanol. Each washing step was separated by a centrifugation step at 8000g, and the flow through was discarded. The miRNAs were then eluted in RNAse-free water.
miRNA cDNA Synthesis
The miRNA fraction was reverse transcribed using a miRCURY LNA Universal microRNA polymerase chain reaction (PCR; Exiqon, Vedbaek, Denmark) kit according to manufacturer’s protocol for individual assays.
Quantitative PCR Analysis
A reverse-transcribed miRNA fraction was diluted 80-fold in RNAse-free water and measured in 10 μL PCR reactions according to the protocol for the miRCURY LNA Universal microRNA PCR (Exiqon, Vedbaek, Denmark) and the ExiLENT SYBR Green master mix (Exiqon, Vedbaek, Denmark) using a LightCycler 480 Real-Time PCR System (Roche, Basel, Switzerland). Levels of the miRNAs were determined using a miRCURY LNA miRNA PCR assay primer mix (Exiqon, Vedbaek, Denmark) of miR-100-5p and SNORD48. Data analysis was done using the Roche LC software for Cp determination (using the second derivative method) and for a melting curve analysis. SNORD48 was used for the normalization of the miRNA levels.
For mRNA detection upon miR-100-5p silencing and overexpression, quantitative PCR was performed on the Applied Biosystems StepOne Plus TM system using SYBR Green ER master mix (Invitrogen) and the following conditions: 10 minutes at 95 °C, then 40 cycles of 15 seconds at 95 °C, 15 seconds at 60 °C, and 30 seconds at 72 °C. The RPLP0 and ATP5F1 housekeeping genes were used for the normalization of the mRNA levels. Primer sequences used are listed in Table I in the Data Supplement. Data was checked for normal distribution before performing statistical tests. Paired Student t test (2-tailed) was used for data that followed normal distribution and equal variance. Otherwise, nonparametric Mann-Whitney test was used. P<0.05 was used to define a significant difference between the groups.
Western Blot Sample Preparation
HUVEC samples were collected using an NE-PER Nuclear and Cytoplasmic Extraction kit (ThermoFisher Scientific, Waltham, MA) following the manufacturer’s protocol for an adherent cell culture. A PhosSTOP Phosphatase Inhibitor Cocktail (Roche, Basel, Switzerland) and cOmplete, EDTA-free Protease Inhibitor Cocktail (Roche, Basel, Switzerland) were used to replace the BupH Phosphate (ThermoFisher Scientific, Waltham, MA), and the Thermo Scientific Halt Protease Inhibitor Cocktail (ThermoFisher Scientific, Waltham, MA), respectively. Protein concentrations were assessed using a Pierce BCA Protein Assay Kit (ThermoFisher Scientific, Waltham, MA) according to manufacturer’s protocol. Equal amounts of proteins were loaded into stain-free gels (Bio-Rad, Hercules, CA) from each sample (10 µg of nuclear and 15 µg of cytoplasmic proteins in HUVEC and 5 µg of proteins in all HASMC samples). For total protein measurement, the stain-free gel was activated with 2.5 minutes exposure to UV-light before protein transfer to membrane. For detecting and normalizing the amount of nuclear TAZ (transcriptional co-activator with PDZ-binding motif) protein, primary antibodies YAP (Yes-associated protein)/TAZ and histone H3 (Cell Signaling Technology, Danvers, MA) were used. Similarly, antibodies for phospho-TAZ and β-actin (Cell Signaling Technology, Danvers, MA) were used to determine the cytoplasmic retention of TAZ. HRP (Horseradish peroxidase) conjugates were used as secondary antibodies. Detection of antigen-antibody complexes was performed with a PIERCE ECL Western Blotting Substrate (Thermo Fisher Scientific, Waltham, MA) and ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA). A quantitative analysis of the immunoblot target protein bands were performed with ImageLab 6.0 software (Bio-Rad, Hercules, CA). For HUVEC samples, the adjusted volume intensity values of target proteins were normalized with the same values of the loading controls (Histone H3, β-actin). For HASMC samples, the target protein intensity values were obtained with an automatic total protein normalization tool. The loading control balanced values were normalized to the respective treatment control.
Data Analysis
Reads obtained after library preparation were poly(A) trimmed and low-quality reads were discarded. After the quality control, the miRNA reads were trimmed to a final size of 21 bp. For GRO-seq, reads passing quality control were mapped to the hg19 genome using Bowtie.25 The Bowtie parameters allowed up to 2 mismatches and reporting only one alignment for each read. RNA-seq and miRNA-seq reads were aligned to the GRCh37/hg19 reference genome using STAR v2.5.4b.26 STAR parameters followed ENCODE standard options for a long RNA-Seq pipeline and small RNA-Seq pipeline.27 Tag directories were generated with the fragment length set to 75 and for the RNA-seq, the maximum number of tags per base pair was set to 3. Raw counts were quantified using Homer V4.928 and the analyze Repeats routine using the standard option, or a customized bed file containing the annotations from mirbase v2229–31 and the custom pri-miRNA coordinates (Table III in the Data Supplement).21 The fold change was calculated after filtering low expressed transcripts (RPKM >0.5 in at least 3 samples) for each cell type to improve the sensitivity and the precision of the differential expression analysis.32 Differential expression was calculated using EdgeR33,34 or DESeq235 for each cell type individually.
For RNA-seq gene expression quantification of HASMC and HUVEC samples transfected with miRNA mimic control and miR-100-5p, reads were first trimmed for poly(A), Illumina adapter, and low-quality bases using cutadapt (version 2.836). Subsequently, the nf-core RNA-Seq pipeline (version 1.4.237) was used to align the reads to the GRCh37/hg19 human genome with the STAR aligner and count the reads in transcripts according to the Ensembl GRCh37 release 87 gene annotations. The following gene biotypes were retained in the gene expression matrix: protein coding, lincRNA, and antisense. To filter out lowly expressed genes, the filterByExpr function of the EdgeR package (version 3.24.333,34) was used (minimum count: 5; minimum total count: 15). For each cell type, all genes were ranked by the response to miR-100-5p (relative to mimic control) based on the Wald statistic calculated by the DESeq2 package (version 1.22.235) using the default parameters. Gene set enrichment analysis of the ranked lists was performed using the fgsea package (version 1.8.038) with the Hallmark gene sets obtained from the Molecular Signatures Database (release 7.139).
Pri-miRNA Transcript Variant Annotation and Quantification
Primary miRNA transcript coordinates were identified from GRO-seq data using a custom de novo detection pipeline.21 To achieve this, GRO-seq, and matching ChIP-seq and CAGE-seq data were assembled for 27 human cell types. Briefly, nonmappable coordinates, exons of coding genes, and ribosomal RNA regions were removed from pri-miRNA transcripts before quantification using BEDTools40 (subtractBed) to exclude regions known to cause problems in quantification of GRO-seq data.
Next, primary transcripts were identified de novo from GRO-seq nascent transcripts using the HOMER software suite program findPeaks.pl, with the -groseq -uniqmap options, as well as 3 parameter settings for varying detection sensitivity and specificity. Transcription start site (TSS) coordinates were then assigned based on CAGE-seq peaks within ±500 bp of the detected transcripts and collapsed for unique across cell types. The TSS status was further supported by promoter histone methylation levels: all putative TSSs had to exhibit >10 CPM H3K4me3 and 5-fold H3K4me3> H3K4me1. Transcript end coordinates were assigned based on de novo transcripts clustered by adjacency and a change point analysis. The final de novo coordinates represent nonoverlapping regions between TSSs across individual loci thus allowing the investigation of transcription originating from different TSSs per locus. Pre-miRNA locations were assigned based on GENCODE (v19) and miRBase (v20) existing annotations and subsequently the overlap of de novo coordinates was used to define candidate pri-miRNA transcripts. To obtain transcript variant (TV)-specific expression values, the nonoverlapping pieces of each pri-miRNA transcript were quantified using HOMER (analyzeRepeats.pl with parameters -strand + -noadj -noCondensing -pc 3). The lengths of the quantified region and total read counts per sample were used to report normalized signal levels (RPKM). The contribution of each TSS (named TSSi) to the overall transcriptional activity in a given locus was determined by subtracting the signal level at the upstream element (named TSSi+1), based on the RPKM (reads per kilobase of transcript per million mapped reads) values: RPKMi=RPKMi–RPKM(i+1).
The differential expression of the primary miRNA transcripts between the different cell types was calculated using DESeq.
Super-enhancer locations were obtained from previous reports.41,42 The distance between the super-enhancers’ location and the mature miRNAs were calculated using the HOMER annotatePeaks algorithm using the -pdist option.
To detect the transcription factor enriched under the studied stimuli, we pooled the tag directories from each cell type separately under all stimuli. All transcribed regions were identified using HOMER v4.9 (http://homer.ucsd.edu/homer/43) with the command findPeaks.pl with the option -groseq. The expression of each transcript was further quantified using the HOMER command analyzeRepeats.pl for all the tag directories. The transcript start sites (±200 bp) of each transcript displaying a fold change above 1 between different conditions were further analyzed to detect the enriched transcription factor using CiiiDER.44 To achieve this, the hg19 coordinates were converted to hg38 using the convertcoordinates.pl tool from HOMER and the fasta-sequences were extracted using BEDTools40 (getfasta).
Analysis of miRNA Targets and Function
To study the biological effects of the expressed miRNAs, the MicroRNA Target Filter of Ingenuity Pathway Analysis (IPA, Qiagen Redwood City, www.qiagen.com/ingenuity) and miRWalk (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/) were used. MicroRNA Target Filter uses experimentally determined microRNA targets using miRecords45 and TarBase46 alongside with manual curation from published literature. IPA’s target filter was used to analyze the full lists of the expressed miRNAs with their expressional changes under proatherogenic stimuli for one cell type at a time. On the contrary, miRWalk has been shown to present better accuracy and precision than other available programs.47 The data sets were coupled with their respective RNA-seq data sets and filtered to include those miRNA target genes that showed opposite expression trends from the miRNAs and thus could be under canonical posttranscriptional regulation. The addition of this correlation between miRNAs and their potential targets allows us to be more accurate in the miRNA target definition. We further filtered the targets to contain those most relevant to cardiovascular disease. Then, we compiled the target lists with their expression changes and entered them into IPA’s Core Analysis, and finally continued to perform an IPA Comparison Analysis to bring all the data sets together into one analysis.
To study the function and interaction of a selected miRNA set, we used miRWalk47,48 and compiled putative target lists for the miRNAs. We included targets, which were present in at least 3 of the 6 target prediction databases for 5′ UTR targets, or 4 of 12 target prediction databases for 3′ UTR and coding sequence targets. The list was further filtered to contain only those targets that were differentially expressed (RNA-seq/GRO-seq exhibiting a false discovery rate [FDR] <0.05) under proatherogenic stimuli compared with control conditions. These targets were uploaded to IPA for core analysis, and a subsequent comparison analysis to determine the canonical pathways and functions that were affected by the proatherogenic stimuli and the selected miRNAs.
Results
Promoter Usage and Transcript Variant Expression of pri-miRNAs Is Cell-Type-Dependent
We first generated GRO-Seq data from HAECs, HUVECs, HASMCs, and CD14+ macrophages to compare the nascent pri-miRNA transcription profiles between the 3 cell types. Here, the cell-type specificity could arise from either pri-miRNA (1) being expressed only in one of the studied cell types or (2) exhibiting alternative usage of TSS and thus resulting in pri-miRNAs of different lengths. To evaluate these options, we used our previously described pri-miRNA transcript variant annotation tool.21 Altogether, we identified 781 pri-miRNAs expressed in at least one condition in all replicates of a single cell type (RPKM >0.5). Despite most pri-miRNAs being expressed in all cell types, the majority of them (681/781, 87%) were differentially expressed (FDR <0.05) between the cell types under all conditions and for 453 (67%) pri-miRNAs the difference was over 10-fold in at least one pair-wise comparison of cell types (Figure 1A and 1B). Of these, 24% (110/453) exhibited alternative TSS usage as exemplified by a miR-23b/27b/24-1 cluster (Figure 1C and 1D). We did not observe clear stimuli-specific patterns in TSS usage and transcript variant expressions, which suggests that the expression of pri-miRNAs is mainly regulated in a cell-type-specific manner (Figure 1C, Figure II in the Data Supplement). Altogether, our data provides evidence that pri-miRNA transcription and TSS usage is highly cell-type specific and minimally affected by the proatherogenic stimulus.
Figure 1.

Promoter usage and transcript variant expression of pri-miRNAs is cell-type-dependent. A, The numbers of pri-miRNAs expressed (false discovery rate [FDR] <0.05) in all cell types and the fraction of differentially expressed pri-miRNAs (FDR <0.05 and fold change >10 comparing 2 cell types). B, Fraction of DE (FDR <0.05) pri-miRNAs exhibiting multiple transcript variants (TVs). C, Heatmap showing transcript variant (TV) expression in log2 (RPKM [reads per kilobase of transcript per million mapped reads]) in different cell types and conditions. The contribution of each transcriptional start site (TSS) to the overall transcriptional activity in a given locus was determined by subtracting the signal level at the upstream element. D, An MIR23B cluster is shown as an example of cell-type-specific promoter usage and transcript variant expression, as 3 of the cell types (human umbilical vein endothelial cell [HUVEC], human aortic endothelial cell [HAEC], and human aortic smooth muscle cell [HASMC]) express 2 TVs, namely TV2 and TV3, and macrophages (CD14) only one of the TVs (TV3). University of California Santa Cruz (UCSC) genome browser shot demonstrating global run-on sequencing (GRO-seq) signal around the miRNA locus.49 E, Row normalized expression (Row z-score) of cell-type specific pri-miRNAs separated according to the cell-type of origin. The rows are hierarchically clustered using the Euclidian distance and complete linkage clustering. **pri-miR-3179-2/3180-2/3670-2/6511a-2/6511a-3/3180-3/6511a-4_TV3. AOPEP indicates aminopeptidase O.
Although the majority of pri-miRNAs were differentially expressed over 10-fold, only 84 pri-miRNAs were exclusively expressed (RPKM <0.5 in the 3 other cell types) in a given cell type (Figure 1E). These are exemplified by previously described MIR143, MIR145 (which are essential for HASMC functions),50–53 MIR126, endothelial cell-specific miRNA regulating vascular inflammation54 and MIR342, that promotes macrophage-driven inflammation.55,56 However, high similarity in the pri-miRNA expression was seen between HUVECs and HAECs, as only 6 pri-miRNAs were differentially expressed including MIR339, MIR3194, MIR3938, MIR10B, and MIR196B for HUVEC and MIR320E for HAEC. Altogether, this analysis provides a resource for investigation of pri-miRNA transcription in atherosclerosis-associated cell types.
Majority of Expressed miRNAs Are Shared Between Cell Types and Dominated by the Expression of Few miRNAs
Next, we set out to compare the cell-type-specificity of nascent pri-miRNAs to the mature miRNA. To this end, we analyzed the miRNA profiles of HUVECs, HAECs, HASMCs, and CD14+ macrophages in response to 7 hours proatherogenic stimuli (oxPAPC, hypoxia, and oxPAPC under hypoxia) in matching samples. Similarly to pri-miRNAs, a large fraction (45%) of the ≈500 miRNAs were expressed in all the cell types, irrespective of conditions (Figure 2A, Figure III and Table II in the Data Supplement). However, a small subgroup of nonuniformly expressed miRNAs (from Figure IIIA and IIIB in the Data Supplement) were able to distinguish the cell types from each other (Figure 2B). Among these miRNAs, only 30% to 50% were identified as cell-type specific at the level of pri-miRNA transcription (Figure IVA in the Data Supplement) and the expression levels exhibited poor correlation (Figure 2C). This relation was further supported by the observation that only 14% (12/84) of the 84 cell-type exclusive pri-miRNAs (from Figure 1E) were expressed in a cell-type specific manner on a mature miRNA level (Figure IVB in the Data Supplement). Altogether, our data provides evidence that miRNA expression is strongly controlled at the level of posttranscriptional regulation. This was clearly demonstrated at the level of miRNA clusters such as miRNAs arising from MIR3613 host gene, namely miR-15a-3p, miR-15a-5p, miR-16-1-3p, miR-16-5p, miR-3613-5p, and miR-3613-3p, which are transcribed as one pri-miRNA, but results in several mature miRNAs with very different expression values (Figure 2D).
Figure 2.

Majority of expressed miRNAs are shared between cell types and dominated by the expression of few miRNAs. A, Basally expressed miRNAs. The number of expressed miRNAs (counts per million reads mapped [CPM] >1 and present in all samples of a condition) in each cell type in basal conditions and under proatherogenic stimuli (hypoxia, oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine [oxPAPC], and hypoxia+oxPAPC) is plotted (mean, SD, n=2 for each condition). B, Heatmap for selected cell-type-specific miRNAs. Values are plotted as logCPM. ND indicates not detected. C, Spearman correlation between the expression of cell-type specific pri-miRNAs and the corresponding expression of mature miRNA in normoxia. Paired Student t test (2-tailed) was used to calculate the correlation’s P value. D, Endothelial cell specific MIR3613 host gene cluster expression detected using global run-on sequencing (GRO-seq) and miR-seq. Cluster miRNAs are transcribed as one pri-miRNA but result in several mature miRNAs with different expression values. Average expression of all treatments in log2 (RPKM [reads per kilobase of transcript per million mapped reads]; left) and log2 (fold change [FC]) comparing different stimuli to the control (right) are shown. E, The number of miRNAs that contribute to at least 50% of the total miRNA expression in each cell type/condition (mean, SD, n=2). F, Percentage of top 10 miRNAs expressed in each cell type. The values represent averages of all treatments and basal conditions for each cell type. G, Violin plot showing the distance in log10 (base pair) between the mature microRNA location and the closest super-enhancer’s peak. Cell-type-specific mature miRNAs are the top 10 miRNAs in Figure 3F, while Others are all the microRNAs considered expressed (>1 CPM in at least 2 samples. The violin plots were created using the vioplot package [version 0.3.2] in R [version 3.5.2]). H, University of California Santa Cruz (UCSC) genome browser shot images of MIR21. Normalized tag counts are shown for H3K27ac ChIP-Seq tracks. Black bars represent the super-enhancer position for each cell type. HAEC indicates human aortic endothelial cell; HASMC, human aortic smooth muscle cell; HUVEC, human umbilical vein endothelial cell; PTRH2, peptidyl-TRNA hydrolase 2; RPS6KB1, ribosomal protein S6 kinase B1; TUBD1, tubulin delta 1; and VMP1, vacuole membrane protein 1.
The Functional Annotation of the Mammalian Genome project recently published11 that miRNA expression levels differ highly and are extremely skewed, with about 5 miRNAs being responsible of half of the total miRNA expression in a given sample. In line with this, 2 to 5 miRNAs were confirmed to be the source of roughly 50% of the total miRNA expression in the cells (Figure 2E). The number was the lowest in HUVECs where only 2 miRNAs, namely miR-126-3p and miR-21-5p, contributed to 56% of the total miRNA expression in basal conditions and under stimuli. Overall, the top 10 miRNAs expressed contributed to 73% to 88% of the total miRNA expression and were largely invariant to stimulus (Figure 2F, Figure VA and VI in the Data Supplement). Thus, a surprisingly small set of miRNAs, many of which are ubiquitously expressed, account for significant differences in the miRNA profiles between different cell types and states. A comparison of the top 10 miRNAs arising from different cell types revealed the most similar profiles in the endothelial subtypes, HUVECs and HAECs, and the least similarity between the macrophages (CD14) and the rest of the cell types (HUVECs, HAECs, and HASMC; Figure 2F, Figures VB and VI in the Data Supplement). Interestingly, we also observed that the top 10 miRNAs were associated with closer proximity to super-enhancers, suggesting a potential mechanism accounting for their high expression level (Figure 2G). To this end, miR-21 had a super-enhancer detected in all the cell types analyzed here (Figure 2H).
High Similarity of Pri-miRNA Expression Changes in Response to Single or Combined Treatment
Next, we investigated the effect of different stimuli on the expression of pri-miRNAs. Altogether, 569, 430, and 515 pri-miRNAs were differentially expressed (GRO-seq, FDR <0.05) upon hypoxia, oxPAPC, or combined treatment, respectively. Altogether a larger fraction of DE pri-miRNA was shared between the cell types (shared pri-miRNAs—hypoxia: 27.5%, OxPAPC: 3.5%, HypoOxP: 20.7% [Figure 3A and Figure VIIA in the Data Supplement]), compared with nascent mRNA expressions where the larger fraction was cell-type specific (shared nascent mRNAs—hypoxia: 16.02%, OxPAPC: 1.37%, HypoOxP: 12.4% [Figure VIIC in the Data Supplement]). However, for both pri-miRNAs and mRNAs, the single stimulus responsive transcripts were also differentially expressed by the other single stimulus and by the hypoxia+oxPAPC combination (Figure 3A, Figures VIIB, VIID, VIIIA, and VIIIB in the Data Supplement). This suggests that transcriptional changes in the hypoxia+oxPAPC group are provoked by either oxPAPC or hypoxia, or that all the treatments have a similar direction of effect. To test these options, we extracted stimuli-specific signatures (Figure 3A and 3B) by assigning the differentially expressed pri-miRNAs (GRO-seq, FDR <0.05) in each data set into 7 groups: (1–3) oxPAPC, hypoxia, and hypoxia+oxPAPC only, which contained pri-miRNAs that were upregulated in one group and downregulated in the others (or vice versa); (4–6) combinations of the 3 (oxPAPC and hypoxia/oxPAPC and hypoxia+oxPAPC/hypoxia and hypoxia+oxPAPC), which contained pri-miRNAs that were upregulated in 2 of the groups and downregulated in the third (or vice versa); and (7) pri-miRNAs similarly expressed under all 3 stimuli (upregulated/downregulated in all). In a given data set, one pri-miRNA could only be assigned to one group, for example, in oxPAPC-treated cells, pri-miRNAs belonging to the oxPAPC only group were extracted first, then pri-miRNAs belonging to combinations and all categories. Additionally, pri-miRNA was required to be significantly differentially expressed in the treatment group that was under investigation and only the direction of expression (up/no change/down) was noted in the other treatment groups. The results showed that the majority of the pri-miRNAs were expressed similarly under all stimuli, and only ≈11% of the pri-miRNAs were found to respond stimuli specifically (Figure 3B and 3C, Figure VIIIC in the Data Supplement). Taken together, our results suggest that hypoxia, oxPAPC, and combined treatment exhibit surprisingly similar directions of effect on pri-miRNA expressions and thus likely involve similar regulatory mechanisms.
Figure 3.

High similarity of primary microRNA (pri-miRNA) expression changes in response to single or combined treatment. A, Overlap of differentially expressed pri-miRNAs under all stimuli (top) and for all cell types (bottom) studied. B, Prevalence of the assigned miRNA groups is shown. Differentially expressed pri-miRNAs (global run-on sequencing [GRO-seq], FDR <0.05) in each data set have been assigned to one of the 7 groups. Each miRNA has been assigned to a group and each miRNA can only be present in one of the groups. C, Log2FC of the pri-miRNA expression under the studied stimuli compared with the control. Heatmaps were generated using Graphpad Prism 8. D, Enrichment analysis showing a selection of over-represented and under-represented motifs at the transcriptional start site (TSS) of transcripts regulated at least 2-fold by each stimulus. The plots were generated using CiiiDER.44 ARNT indicates aryl hydrocarbon receptor nuclear translocator; EGR1, early growth response 1; HAEC, human aortic endothelial cell; HASMC, human aortic smooth muscle cell; HIF1A, hypoxia-inducible factor 1 subunit alpha; HUVEC, human umbilical vein endothelial cell; KLF, Krüppel-like factor; oxPAPC, oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine; and TV, transcript variant.
To further analyze the commonalities in the transcriptional mechanisms due to hypoxia and oxPAPC, we analyzed the de novo motif enrichment at the TSS of regulated transcripts (fold change >2 in at least one comparison in at least one cell type) for each stimulus. We identified 679, 140, and 172 enriched motifs for hypoxia, oxPAPC and hypoxia+oxPAPC, respectively. On the contrary, 1329 and 361 motifs for hypoxia, oxPAPC, and hypoxia+oxPAPC, respectively, were depleted compared with the background. In line with the pri-miNA expression profiles, most of the enriched motifs contributing to the combined response were shared between hypoxia and oxPAPC stimuli separately (Figure 3D, Table III in the Data Supplement). Indeed, only 2 motifs were specific in hypoxia+oxPAPC, suggesting that most motifs were also enriched in the single stimulus responsive TSSs. Among the top enriched motifs in hypoxia+oxPAPC, we identified previously validated signal responsive TFs (transcription factors) such as ARNT (aryl hydrocarbon receptor nuclear translocator) mediating the hypoxia response57 or Zfp148 (zinc-finger protein 148) and ERG-1 (early growth response 1) mediating the response to oxidative stress,17,58 as well as many members of the KLF (Krüppel-like factor) family of TFs that have been shown to contribute to proatherogenic gene expression changes.59 A comparison of the single stimuli demonstrated that only 1 motif was specific to oxPAPC, whereas hypoxia exhibited a larger set of stimulus-specific motifs. To provide further evidence of the involvement of the predicted TFs in the regulation of miRNAs, we used TransmiR v2.0.60,61 that makes use of literature-curated TF–miRNA regulation data. This allowed confirmation of the KLFs, HIF1a (hypoxia-inducible factor 1 subunit alpha)/ARNT, and EGR1 in the regulation of miRNAs (Figure IX in the Data Supplement). Altogether, we present evidence that there is high similarity in the pri-miRNA responses to the proatherogenic stimuli between the cell types, which could be partly due to the sharing of many TFs that mediate the responses to hypoxia and oxidative stress.
MicroRNA Target Genes Are Associated With Atherosclerosis-Related Functions
Most studies comparing atherosclerosis-associated changes in miRNA expression profiles have been performed at the level of bulk tissue, leaving the cell of origin of the differential signals unclear. To address this knowledge gap and to analyze the relevance of our miRNAs in the disease context, we collected information of the differentially expressed miRNAs from all published studies conducted in human atherosclerotic plaques to date62–66 and analyzed their miRNA expression level and stimulus response in each of the 3 cell types (Figure 4A). Importantly, a highly cell type-specific expression pattern of the mature miRNAs was observed as exemplified by macrophage-specific expression of miR-223-3/5p and endothelial-specific expression of miR-99a-5p, miR-126-3p, miR-146a-3p, and miR-196a/b-5p. Furthermore, >85% (45/52) of the pri-miRNAs were found differentially expressed (FDR <0.05) in at least one condition supporting the disease relevance of our findings. These findings suggest that several of the disease-relevant miRNAs respond to proatherogenic stimulus at transcriptional level and their expression can be assigned to one cell type predicting the potential cell type of action.
Figure 4.

MicroRNA (miRNA) target genes are associated with atherosclerosis-related functions. A, Heatmap of row-normalized expression of miRNAs found deregulated in human atherosclerotic plaques62–66 (left) and the log2 fold change [FC] of those miRNAs compared with normoxia in global run-on sequencing (GRO-seq; right). B, Diseases and functions from IPA’s Comparison Analysis for miRNA targets in oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine [oxPAPC]-treated cells under hypoxia. The heatmaps were generated using Graphpad Prism 8. C, Mechanistic summary of the proatherogenic functions of the miRNA target genes in the various cell types and their possible interaction with each other in the plaque environment. D, Selected canonical pathways from IPA’s Comparison Analysis for targets of miR-21-5p, miR-22-3p, miR-100-5p, miR-34a-5p, and miR-92a-3p in oxPAPC-treated cells under hypoxia. Heatmaps were generated using Graphpad Prism 8. FDR indicates false discovery rate; HAEC, human aortic endothelial cell; HASMC, human aortic smooth muscle cell; HUVEC, human umbilical vein endothelial cell; IL, interleukin; INF, interferon; PDGF, platelet-derived growth factor; TF, transcription factor; and TGF, transforming growth factor.
Although the mature miRNA profiles in different cell types or under proatherogenic stimuli did not show drastic differences (Figure III in the Data Supplement), the miRNA functions could differ as the target gene pools are not the same between the cell types, with about 5% of the mRNAs being exclusively expressed in one cell type or exhibiting over 8-fold difference in expression level (Figure XA and XB in the Data Supplement). To study the biological effects of the expressed miRNAs further in all cell types under proatherogenic stimuli, we used the IPA MicroRNA Target Filter that relies on experimentally validated and predicted mRNA targets. Inspection of the results of the Diseases and Functions section revealed several changes in the cellular functions that indicate the entering and maintenance of a proatherogenic state, such as increased endothelial activation, vascular permeability and cellular movement, and migration of the cells (Figure 4B). In addition to the prodisease changes, the overall results showed clear indications of changes that would, in the plaque environment, increase the vulnerability of the plaque and the susceptibility to adverse cardiac events, such as increased smooth muscle cell death and decreased proliferation, increased neovascularization, and increased platelet activation and thrombus formation.67 Overall, there was a high similarity in the directionality of the effect (z-score) for the majority of the functions, suggesting that the majority of the effects mediated by the target mRNAs are concordant between the cell types. However, activation of the cell migration, vascularization, and inhibition of apoptosis was more predominant in endothelial cells compared with HASMCs and CD14+ macrophages. Cell-type-specificity was also evident from the target mRNA changes, as shown for the atherosclerosis signaling molecules in Figure XC in the Data Supplement. A schematic of the possible interactions of the different cell types and signaling molecules within the plaque environment is provided in Figure 4C. The full table of miRNAs and their predicted targets can be found in the Table IV in the Data Supplement.
miR-100-5p Promotes Atherogenesis-Associated Cellular Events by Regulating the HIPPO Pathway and Inflammation
To find miRNAs with significant effects on target gene expression and the genes most affected by the miRNA function, we filtered the data based on target numbers and by targeting miRNAs, respectively. The miRNAs with the highest target numbers in the data and genes with the highest number of miRNAs targeting them are summarized in Figure XI in the Data Supplement. For HUVECs, HAECs, and HASMCs, the miRNA lists mostly consist of miRNAs which are downregulated, and thus their targets are upregulated. For CD14+ the trend is the opposite. Overall, the miRNAs on the lists are very lowly expressed in the cells, and therefore, may not have strong biological effects, as argonaute (AGO)-loading has been suggested to be dominated by the most abundant miRNAs. Indeed, by selecting the 5 of the most highly expressed miRNAs, namely miR-21-5p, miR-100-5p, miR-22-5p, miR-34a-5p, and miR-92a-3p, it was possible to largely recapitulate the predicted functional effects on the cell types (Figure 4D and Figure XD in the Data Supplement). Here, HIPPO signaling was found to be repressed by miRNA target prediction with the strongest effect detected in endothelial cells. A recent study suggests that YAP/TAZ activation in endothelial cells plays a causal role in the initiation and progression of atherosclerosis,68 and the overexpression of miR-100-5p has been associated with vulnerable plaque phenotypes.65,69,70 However, since the target mRNA predictions are solely based on bioinformatic predictions with limited accuracy, we sought to experimentally modulate miR-100-5p expression to validate the predicted miRNA-mediated effects on the YAP/TAZ pathway (Figure 5A, Figures XIIA and XIII in the Data Supplement). The results confirmed that in HUVECs the overexpression of miR-100-5p introduces a trend towards increased nuclear TAZ levels in proatherogenic conditions, whereas miR-100-5p silencing reduced the nuclear TAZ levels. The opposite was true for miR-100-5p silencing. However, in HASMCs, the trend was opposite, with the overexpression of miR-100-5p leading to a decrease in nuclear TAZ level and an increase in phosphorylated TAZ.
Figure 5.
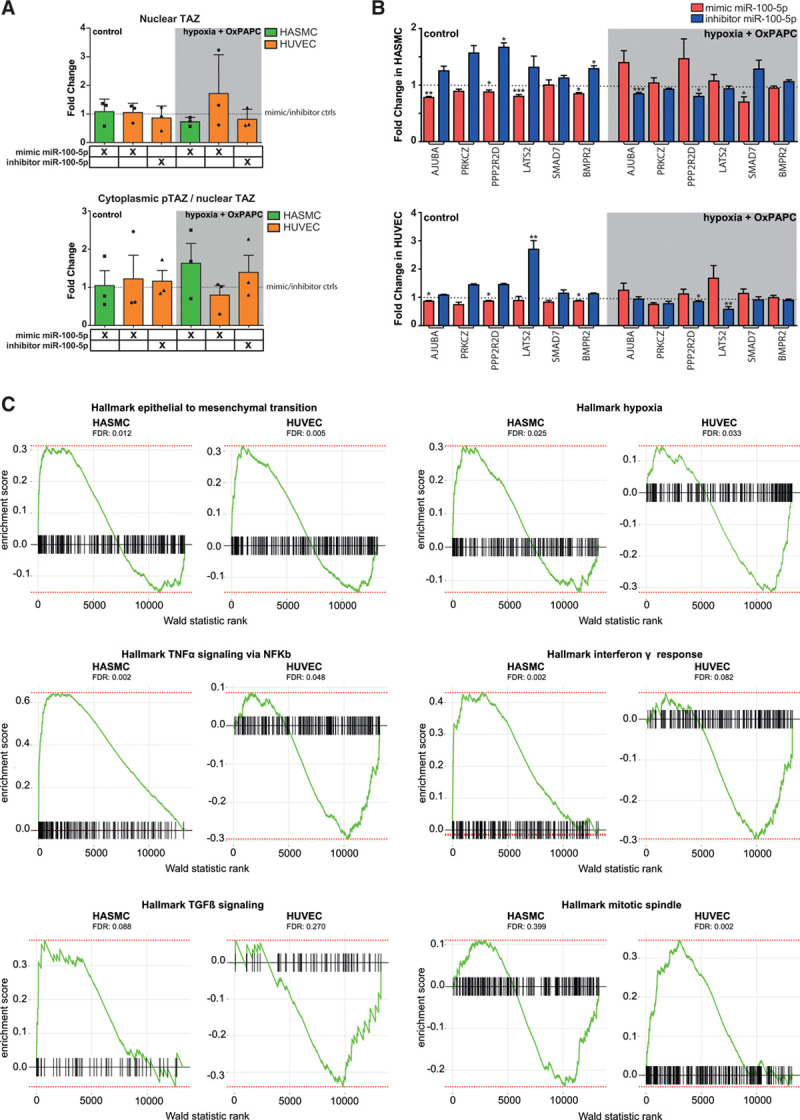
miR-100-5p promotes atherogenesis-associated cellular events by regulating the HIPPO pathway and inflammation. A, Western blot quantitation for nuclear TAZ normalized to Histone H3 (human umbilical vein endothelial cell [HUVEC]) or total protein (human aortic smooth muscle cell [HASMC]), and for cytoplasmic phosphorylated TAZ (pTAZ) and nuclear TAZ ratio from miR-100-5p inhibitor and mimic transfected cells. Cytoplasmic pTAZ was normalized to β-actin (HUVEC) or total protein (HASMC). All results are presented relative to the respective control values with the SD. n=3. B, Gene expression in HASMC (top) and HUVEC (bottom) samples transfected with miR-100-5p mimic (red) or inhibitor (blue) under normoxia or hypoxia+oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (OxPAPC) relative to respective controls. Data was checked for normal distribution before performing statistical tests. Paired Student t test (2-tailed) was used for data that followed normal distribution and equal variance. Otherwise, nonparametric Mann-Whitney test was used (mean±SD, n=6, *P≤0.05; **P≤0.01; and ***P≤0.001). C, GSEA analysis of selected hallmark gene sets after ranking the sequencing results of miR-100-5p overexpression using Wald statistics in HASMCs and HUVECs. FDR indicates false discovery rate; TAZ, transcriptional co-activator with PDZ-binding motif; TGFβ, transforming growth factor beta; and TNFα, tumor necrosis factor-alpha.
The evolutionary conserved HIPPO pathway is a key regulator of cell fate in response to biochemical and biophysical cues.68,71,72 In addition to controlling organ development and growth as well as tissue homeostasis, the HIPPO pathway regulates metabolic processes at the cellular and organismal levels in both physiological processes and metabolic disease states, also in atherosclerosis. Silencing of the HIPPO signaling pathway leads to dephosphorylation and subsequent activation of the Yes-associated protein and Transcriptional Co-Activator With PDZ-Binding Motif (YAP and TAZ) further towards proproliferative and proinflammatory responses in cells, which promote atherogenesis. Thus, we next sought to investigate if the effect of miR-100-5p on the HIPPO pathway was dependent on stimulus or the cell type. To achieve this, we analyzed effects of miR-100-5p activation or inhibition on target mRNA expression in HUVECs and HASMCs under a control conditions and hypoxia+oxPAPC combination stimulus. Based on miRWalk-database,47,48 the miR-100-5p was predicted to regulate the expression of several regulators of the HIPPO-YAP/TAZ cascade, including the pathway regulators AJUBA, BMPR2, LATS2, PPP2R2D, PRKCZ, and SMAD7. The results demonstrated highly similar regulation of the target mRNAs upon control conditions, whereas combination treatment revealed cell-type specific regulation of LATS2 (HUVEC only), AJUBA (HASMC only), and SMAD7 (HASMC only; Figure 5B). In addition, the effect of miR-100-5p on the SMAD7 expression was only evident upon combined treatment in HASMCs. Interestingly, the effect of miR-100-5p on LATS2 and AJUBA expression was opposite to what would be expected from the canonical miRNA regulation under the combination stimulus, suggesting that the effect was indirect. Altogether, this suggests that miR-100-5p displays both cell-type and treatment-specific control of the HIPPO pathway regulators.
The HIPPO pathway and miR-100-5p have both been shown to exhibit extensive crosstalk with pathways involved in proliferation, angiogenesis, and inflammation.73–75 To provide an unbiased view of the cell-type specific effects of miR-100-5p, we performed RNA-Seq profiling in HUVECs and HASMCs transfected with miR-100-5p mimic under hypoxia+oxPAPC stimulus (Figure XIIB and Table V in the Data Supplement). To determine the biological pathways of the genes affected by the overexpression of miR-100-5p, we performed a Gene Set Enrichment Analysis of the genes ranked by FDR and the effect direction (Walk statistic rank; Table VI in the Data Supplement). The top biological pathway, epithelial to mesenchymal transition, was highly enriched among the upregulated genes in both cell types. However, the hallmark gene sets related to hypoxia, TNFα (tumor necrosis factor-alpha) signaling via NFκb (nuclear factor kappa B subunit 1), and IFNγ (interferon gamma) response exhibited positive enrichment in HASMCs, while a negative enrichment was seen in HUVECs (Figure 5C). This signifies that miR-100-5p could have an opposite effect on these 2 signaling pathways, with the upregulated genes in HASMCs and downregulated genes in HUVECs showing gene set enrichment. In contrast, only HUVECs demonstrated significant (FDR <0.1) positive enrichment for genes associated with proliferation (Mitotic spindle) and negative enrichment for mTORC (mechanistic target of rapamycin kinase) signaling and ROS (reactive oxygen species) pathway whereas in HASMCs genes related to TGF-β (transforming growth factor beta 1) signaling and apoptosis were positively enriched (Figure 5C and Figure XIIC in the Data Supplement). Our results suggest that complex networks of miRNA-mRNA interactions could exert opposing roles in different cell types of the same tissue. Importantly, we provide the first evidence of the participation miR-100-5p in the regulation of the HIPPO pathway in endothelial cells and its antagonistic role in inflammatory signaling in vascular cell types.
Discussion
In this study, the mechanisms and signaling pathways associated with proatherogenic stimulus were studied in disease-associated cell types by obtaining and integrating different levels of next-generation sequencing data, with a special focus on the miRNA expression and function under proatherogenic stimuli. First, we analyzed the pri-miRNA expression levels and TSS usage in HAECs, HUVECs, HASMCs, and CD14+ macrophages. In line with several previous studies,21,76,77 our results provide strong evidence that the transcription of pri-miRNA and the TSS usage is largely cell-type specific. In addition, we demonstrate that the TSS usage is poorly affected by stimuli, further suggesting that the TSS activation is performed through cell-type specific regulatory elements that largely preexist before the stimulus.78
In our study, we also demonstrate that the correlation between pri-miRNA and mature miRNA expression was low for the cell types and stimulus, suggesting that miRNA expression is further regulated in a posttranscriptional manner. This is in line with a recent study based on metabolic labeling of nascent RNAs demonstrating that many pri-miRNAs indeed fail to generate mature miRNAs.79 Likely reason for this is the extensive regulation of miRNA biogenesis that influence miRNA processing and turnover. Interestingly, previous studies have shown miRNA processing to play a stronger role over expression in determining the level of mature miRNAs. The microprocessor complex transforms pri-miRNA into pre-miRNA and the efficiency seems to be determined by motifs within the pri-miRNA (such as the GC dinucleotide motif within the miR-100 pri-miRNA), in addition to secondary structural features, as well as microprocessor cofactors.80 Moreover, accessory proteins can bind and regulate the pri-miRNA or the microprocessor complex, and other regulators such as noncoding RNAs are also involved in the regulation.81 Alternatively, mature miRNA levels can also be regulated during the nuclear export and at the level of cytoplasmatic processing,80 nuclear import, and subcellular localization5,82,83 and by the availability of the proteins participating in their biogenesis and stability such as DICER and AGO.84 However, we chose 1-hour difference between GRO-Seq and miRNA-Seq based on previous studies showing that mRNA level temporally lag behind the corresponding changes in transcription rates by ≈15 to 30 minutes85 thus allowing enough time for processing to occur. Still, we cannot exclude the possibility that the short time difference chosen partly contributes to the poor correlation observed between pri-miRNA and miRNA levels. Our analysis also demonstrates that the highest expressed miRNAs are found close to super-enhancers, which could contribute to the higher expression levels. Supporting this, a recent report has suggested that super-enhancers enable nascent pri-miRNA transcription, and facilitate Drosha/DGCR8 recruitment and pri-miRNA processing to boost cell-specific miRNA production.86 Moreover, miRNA decay constitutes another mechanism by which the levels or miRNA are regulated. A recent study based on a pulse-chase approach on metabolic RNA labeling enabled the study of the heterogeneity of miRNAs half-lives. miRNA decay is achieved by modifying the miRNA ends, either by adding or trimming the nucleotides, as well as through interaction with the target mRNAs. Interestingly, miR-100 was classified as a fast decaying miRNA, which would make it possible to act in specific cellular responses that require a rapid change in the level of miRNA.87 Since microprocessor regulation and miRNA decay are affected by physiological processes, future studies focused on the pri-miRNA regulation in atherogenesis are needed.
Our analysis identified several cell-type-selective miRNAs such as miR-142 and mir-223 for macrophages, miR-126-3p and miR-126-5p for vascular endothelial cells, and miR-143 and miR-145 for vascular smooth muscle cells, which have previously been shown to regulate essential functions and differentiation of these cell types.51,88–91 Still, the majority of the miRNA expression was dominated by a few miRNAs that were largely shared between the cell types. This is in line with previous studies demonstrating that on average 5 miRNAs contribute to half of the total miRNA expression in a given sample.11 Given that the most abundant miRNAs have been suggested to dominate posttranscriptional target gene regulation by AGO proteins, these miRNAs may also play critical roles in disease pathogenesis.92,93 Indeed, miR-21 and miR-22 have been shown to be increased in several pathologies, such as cancer and cardiac hypertrophy, respectively.94,95
To our knowledge, this is the first study to characterize the combined response to hypoxia and oxidized lipids in the main cell types representative of atherosclerosis. To our surprise, most of the pri-miRNAs and the direction of regulation were the same for the single and combined stimulus. These results suggest that hypoxia, oxPAPC, and combined treatment have a similar direction of effect on pri-miRNA regulation in a given cell type and are, thus, likely to involve similar transcriptional regulatory mechanisms. By analyzing the motifs around the TSSs of those pri-miRNAs under different stimuli, we were able to identify stimulus-specific motifs and show that the combination of stimuli is solely the result of the interaction between the 2 stimuli-specific responses. Among the top enriched motifs, ARNT, also known as HIF1-β (hypoxia-inducible factor 1, beta subunit), participates in the hypoxia response by binding to HIF1α. Although it is extensively known that HIF1-β is constitutively expressed, some studies have revealed that it can be upregulated by hypoxia in a cell-type specific manner.57 In addition, ARNT has been shown to be involved in the oxidative stress response through the activation of NRF2 (nuclear factor erythroid 2-related factor 2)96 and recent studies have shown significant crosstalk between the NRF2 and HIF1α signaling pathways.97–99 Another enriched motif, the Zfp148 transcription factor, has been demonstrated to promote cell proliferation under oxidative stress conditions and its deficiency confers protection against atherosclerosis in vivo.58 Moreover, oxPAPC has also been shown to induce an atherogenic EGR-1 expression in vivo, contributing to an atherosclerosis progression.17 Under combined hypoxia and oxPAPC, several members of the KLF family were also among the top enriched motifs. Particularly, KLF2, 4, 6, and 11 have been shown to participate in different biological functions involved in cardiovascular diseases, such as regulation of inflammation, angiogenesis, and thrombosis in endothelial and smooth muscle cells.59,100 Still, direct experimental validation of the transcription factors binding near the miRNA TSS are still needed to validate our findings.
By analyzing the target genes of the miRNAs, we identified the HIPPO pathway as one of the main targets of the 5 most highly expressed miRNAs. To confirm this prediction, we silenced and overexpressed miR-100-5p in HUVECs and HASMCs. Our data demonstrates that miR-100-5p exerts a highly cell and stimulus-specific regulation of the HIPPO pathway. This was exemplified by the nuclear localization of TAZ upon miR-100-5p overexpression in HUVECs but not in HASMCs and the HASMC-specific regulation of SMAD7 (mothers against decapentaplegic homolog 7) under combination treatment. Recent evidence positions SMAD7 as a central coordinator of crosstalk between HIPPO- and TGF-β signaling101,102 where SMAD7 has been shown to suppress the TGF-β signaling. This provides one potential mechanism that could explain the cell type-specific upregulation of TGF-β pathway hallmark genes in HASMCs that warrants further research.
Importantly, a recent study demonstrated extensive crosstalk between mTORC and HIPPO pathways by showing that when the HIPPO pathway is active, mTORC1 signaling is turned off and vice versa.102 In line with the cell-type-specific regulation of the HIPPO pathway, the genes related to mTORC pathway were only enriched among the miR-100 downregulated genes in HUVECs. Repression of mTORC signaling has been further mechanistically linked to an anti-inflammatory reaction to atherosclerotic stimuli in HUVECs under miR-100 overexpression.70 To this end, we also demonstrate a significant representation of TNFα-, IFNβ-, and hypoxia-pathway mediators among the downregulated genes in HUVECs whereas the opposite was seen in HASMCs. Our findings could also provide a partial explanation to the different functional effects previously reported for miR-100, where inhibition of miR-100 had a significant stimulatory effect on HASMC migration whereas no effect was seen in HUVECs.75 In summary, our study identifies miR-100-5p as a prominent player mediating the cell-type specific regulation of inflammation and hypoxia associated genes. Still, we acknowledge that our work is limited to in vitro experiments, and we cannot directly infer mechanisms of atherogenesis from our results. Future analysis and perturbation of cell-type specific miRNA expression in the tissue context is hoped to address these limitations.
Altogether, our work reveals a greater complexity in miRNA regulation than previously known and provides a resource for investigations on cell-type specific differences in miRNA transcription in the vascular wall. This information could be used to further characterize the atherosclerosis relevant regulatory networks and serve as the basis for future development of cell-targeted therapeutics.104,105
Acknowledgments
We would like to thank the EMBL Sequencing Service GeneCore Sequencing Facility (EMBL, http://www.genecore.embl.de) for the Next-Generation Sequencing (NGS) library sequencing and the University of Eastern Finland (UEF) Bioinformatics Center for server infrastructure. Sequencing data from this study has been submitted to the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus under accession number GSE136813 and GSE154427. The public data sets for CHiP-seq can be found under accession number GSE4428841 and GSE5264242 and the miRNA profiling of human umbilical vein endothelial cells under normoxia and hypoxia (24 hours) can be found under accession number GSE26497.106 The super-enhancers were obtained from GSE51522107 and GSE52642.42
Sources of Funding
This work was supported by the Academy of Finland (Grants Nos. 287478, 294073, and 319324 to M.U. Kaikkonen, 321535 and 328835 to J.P Laakonen), the Emil Aaltonen Foundation (to S. Linna-Kuosmanen), the Finnish Foundation for Cardiovascular Research (to S. Linna-Kuosmanen, P.R. Moreau and M.U. Kaikkonen), the Maud Kuistila Memorial Foundation (to S. Linna-Kuosmanen and P.R. Moreau), the Orion Research Foundation (to S. Linna-Kuosmanen), the Jane and Aatos Erkko Foundation (to M.U. Kaikkonen), the Sigrid Jusélius Foundation (to S. Linna-Kuosmanen and M.U. Kaikkonen) and Ella and Georg Ehrnrooth Foundation (to J.P. Laakkonen). This research was supported by the Doctoral Programme in Molecular Medicine (DPMM). This project was partly funded by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant No. 802825 to M.U. Kaikkonen, 931683 to S. Ylä-Herttuala). Funding for open access is provided by Academy of Finland.
Disclosures
None.
Supplemental Materials
Online Figures I–XII
Online Tables I–VI
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- CD14+
- macrophages
- DGCR8
- DiGeorge syndrome critical region 8
- GRO-seq
- global run-on sequencing
- HAECs
- human aortic endothelial cell
- HASMCs
- human aortic smooth muscle cell
- HRP
- horseradish peroxidase
- HUVECs
- human umbilical vein endothelial cell
- miRNA
- microRNA
- oxPAPC
- oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine
- PCR
- polymerase chain reaction
- pre-miRNA
- precursor miRNA
- pri-miRNA
- primary microRNA
- TF
- transcription factor
- TSS
- transcriptional start site
- TV
- transcript variant
S. Linna-Kuosmanen and M.U. Kaikkonen contributed equally.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.121.315579.
For Sources of Funding and Disclosures, see page 2164.
References
- 1.Gimbrone MA, Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sedding DG, Boyle EC, Demandt JAF, Sluimer JC, Dutzmann J, Haverich A, Bauersachs J. Vasa vasorum angiogenesis: key player in the initiation and progression of atherosclerosis and potential target for the treatment of cardiovascular disease. Front Immunol. 2018;9:706. doi: 10.3389/fimmu.2018.00706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu H, Lei C, He Q, Pan Z, Xiao D, Tao Y. Nuclear functions of mammalian MicroRNAs in gene regulation, immunity and cancer. Mol Cancer. 2018;17:64. doi: 10.1186/s12943-018-0765-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao M, Li J, Li W, Wang Y, Wu F, Xi Y, Zhang L, Ding C, Luo H, Li Y, et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol. 2017;14:1326–1334. doi: 10.1080/15476286.2015.1112487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santovito D, Egea V, Bidzhekov K, Natarelli L, Mourão A, Blanchet X, Wichapong K, Aslani M, Brunßen C, Horckmans M, et al. Noncanonical inhibition of caspase-3 by a nuclear microRNA confers endothelial protection by autophagy in atherosclerosis. Sci Transl Med. 2020;12:eaaz2294. doi: 10.1126/scitranslmed.aaz2294 [DOI] [PubMed] [Google Scholar]
- 6.Kuosmanen S. MicroRNAs in endothelial aging and atherosclerosis [PhD Thesis]. Kuopio, Finland: University of Eastern Finland; 2017.
- 7.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y [DOI] [PubMed] [Google Scholar]
- 8.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4 [DOI] [PubMed] [Google Scholar]
- 9.Kent OA, McCall MN, Cornish TC, Halushka MK. Lessons from miR-143/145: the importance of cell-type localization of miRNAs. Nucleic Acids Res. 2014;42:7528–7538. doi: 10.1093/nar/gku461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halushka MK, Fromm B, Peterson KJ, McCall MN. Big strides in cellular MicroRNA expression. Trends Genet. 2018;34:165–167. doi: 10.1016/j.tig.2017.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Rie D, Abugessaisa I, Alam T, Arner E, Arner P, Ashoor H, Åström G, Babina M, Bertin N, Burroughs AM, et al. ; FANTOM Consortium. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat Biotechnol. 2017;35:872–878. doi: 10.1038/nbt.3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Juzenas S, Venkatesh G, Hübenthal M, Hoeppner MP, Du ZG, Paulsen M, Rosenstiel P, Senger P, Hofmann-Apitius M, Keller A, et al. A comprehensive, cell specific microRNA catalogue of human peripheral blood. Nucleic Acids Res. 2017;45:9290–9301. doi: 10.1093/nar/gkx706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCall MN, Kim MS, Adil M, Patil AH, Lu Y, Mitchell CJ, Leal-Rojas P, Xu J, Kumar M, Dawson VL, et al. Toward the human cellular microRNAome. Genome Res. 2017;27:1769–1781. doi: 10.1101/gr.222067.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee S, Birukov KG, Romanoski CE, Springstead JR, Lusis AJ, Berliner JA. Role of phospholipid oxidation products in atherosclerosis. Circ Res. 2012;111:778–799. doi: 10.1161/CIRCRESAHA.111.256859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsimikas S, Mallat Z, Talmud PJ, Kastelein JJ, Wareham NJ, Sandhu MS, Miller ER, Benessiano J, Tedgui A, Witztum JL, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56:946–955. doi: 10.1016/j.jacc.2010.04.048 [DOI] [PubMed] [Google Scholar]
- 16.Tsimikas S, Brilakis ES, Miller ER, McConnell JP, Lennon RJ, Kornman KS, Witztum JL, Berger PB. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005;353:46–57. doi: 10.1056/NEJMoa043175 [DOI] [PubMed] [Google Scholar]
- 17.Furnkranz A, Schober A, Bochkov VN, Bashtrykov P, Kronke G, Kadl A, Binder BR, Weber C, Leitinger N. Oxidized phospholipids trigger atherogenic inflammation in murine arteries. Arterioscler Thromb Vasc Biol. 2005;25:633–638. doi: 10.1161/01.ATV.0000153106.03644.a0 [DOI] [PubMed] [Google Scholar]
- 18.Sluimer JC, Gasc JM, van Wanroij JL, Kisters N, Groeneweg M, Sollewijn Gelpke MD, Cleutjens JP, van den Akker LH, Corvol P, Wouters BG, et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol. 2008;51:1258–1265. doi: 10.1016/j.jacc.2007.12.025 [DOI] [PubMed] [Google Scholar]
- 19.Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen G, Zhang W, Li YP, Ren JG, Xu N, Liu H, Wang FQ, Sun ZJ, Jia J, Zhao YF. Hypoxia-induced autophagy in endothelial cells: a double-edged sword in the progression of infantile haemangioma? Cardiovasc Res. 2013;98:437–448. doi: 10.1093/cvr/cvt035 [DOI] [PubMed] [Google Scholar]
- 21.Bouvy-Liivrand M, Hernández de Sande A, Pölönen P, Mehtonen J, Vuorenmaa T, Niskanen H, Sinkkonen L, Kaikkonen MU, Heinäniemi M. Analysis of primary microRNA loci from nascent transcriptomes reveals regulatory domains governed by chromatin architecture. Nucleic Acids Res. 2017;45:9837–9849. doi: 10.1093/nar/gkx680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klinge S, Voigts-Hoffmann F, Leibundgut M, Arpagaus S, Ban N. Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6. Science. 2011;334:941–948. doi: 10.1126/science.1211204 [DOI] [PubMed] [Google Scholar]
- 23.Pestova TV, Hellen CU. Translation elongation after assembly of ribosomes on the Cricket paralysis virus internal ribosomal entry site without initiation factors or initiator tRNA. Genes Dev. 2003;17:181–186. doi: 10.1101/gad.1040803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6:209–217. doi: 10.1038/nchembio.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, Khatun J, Lajoie BR, Landt SG, Lee BK, Pauli F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34Database issueD140–D144. doi: 10.1093/nar/gkj112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36Database issueD154–D158. doi: 10.1093/nar/gkm952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39Database issueD152–D157. doi: 10.1093/nar/gkq1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sha Y, Phan JH, Wang MD. Effect of low-expression gene filtering on detection of differentially expressed genes in RNA-seq data. Annu Int Conf IEEE Eng Med Biol Soc. 2015;2015:6461–6464. doi: 10.1109/EMBC.2015.7319872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal. 2011;17:10. [Google Scholar]
- 37.Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020;38:276–278. doi: 10.1038/s41587-020-0439-x [DOI] [PubMed] [Google Scholar]
- 38.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov M, Sergushichev A. Fast gene set enrichment analysis. biorXiv. 2016. Preprint posted online October 22, 2019. doi: 10.1101/060012 [Google Scholar]
- 39.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaikkonen MU, Niskanen H, Romanoski CE, Kansanen E, Kivelä AM, Laitalainen J, Heinz S, Benner C, Glass CK, Ylä-Herttuala S. Control of VEGF-A transcriptional programs by pausing and genomic compartmentalization. Nucleic Acids Res. 2014;42:12570–12584. doi: 10.1093/nar/gku1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gearing LJ, Cumming HE, Chapman R, Finkel AM, Woodhouse IB, Luu K, Gould JA, Forster SC, Hertzog PJ. CiiiDER: a tool for predicting and analysing transcription factor binding sites. PLoS One. 2019;14:e0215495. doi: 10.1371/journal.pone.0215495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37Database issueD105–D110. doi: 10.1093/nar/gkn851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, Vlachos IS, Tastsoglou S, Kanellos I, Papadimitriou D, Kavakiotis I, Maniou S, Skoufos G, et al. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018;46D1D239–D245. doi: 10.1093/nar/gkx1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dweep H, Sticht C, Pandey P, Gretz N. miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform. 2011;44:839–847. doi: 10.1016/j.jbi.2011.05.002 [DOI] [PubMed] [Google Scholar]
- 48.Dweep H, Gretz N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat Methods. 2015;12:697. doi: 10.1038/nmeth.3485 [DOI] [PubMed] [Google Scholar]
- 49.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hergenreider E, Heydt S, Tréguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14:249–256. doi: 10.1038/ncb2441 [DOI] [PubMed] [Google Scholar]
- 51.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009;16:1590–1598. doi: 10.1038/cdd.2009.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boettger T, Beetz N, Kostin S, Schneider J, Krüger M, Hein L, Braun T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest. 2009;119:2634–2647. doi: 10.1172/JCI38864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci USA. 2008;105:1516–1521. doi: 10.1073/pnas.0707493105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wei Y, Nazari-Jahantigh M, Chan L, Zhu M, Heyll K, Corbalán-Campos J, Hartmann P, Thiemann A, Weber C, Schober A. The microRNA-342-5p fosters inflammatory macrophage activation through an Akt1- and microRNA-155-dependent pathway during atherosclerosis. Circulation. 2013;127:1609–1619. doi: 10.1161/CIRCULATIONAHA.112.000736 [DOI] [PubMed] [Google Scholar]
- 56.Santovito D, Egea V, Weber C. Small but smart: MicroRNAs orchestrate atherosclerosis development and progression. Biochim Biophys Acta. 2016;186112pt B2075–2086. doi: 10.1016/j.bbalip.2015.12.013 [DOI] [PubMed] [Google Scholar]
- 57.Mandl M, Depping R. Hypoxia-inducible aryl hydrocarbon receptor nuclear translocator (ARNT) (HIF-1β): is it a rare exception? Mol Med. 2014;20:215–220. doi: 10.2119/molmed.2014.00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sayin VI, Khan OM, Pehlivanoglu LE, Staffas A, Ibrahim MX, Asplund A, Agren P, Nilton A, Bergström G, Bergo MO, et al. Loss of one copy of Zfp148 reduces lesional macrophage proliferation and atherosclerosis in mice by activating p53. Circ Res. 2014;115:781–789. doi: 10.1161/CIRCRESAHA.115.304992 [DOI] [PubMed] [Google Scholar]
- 59.Fan Y, Lu H, Liang W, Hu W, Zhang J, Chen YE. Krüppel-like factors and vascular wall homeostasis. J Mol Cell Biol. 2017;9:352–363. doi: 10.1093/jmcb/mjx037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J, Lu M, Qiu C, Cui Q. TransmiR: a transcription factor-microRNA regulation database. Nucleic Acids Res. 2010;38Database issueD119–D122. doi: 10.1093/nar/gkp803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tong Z, Cui Q, Wang J, Zhou Y. TransmiR v2.0: an updated transcription factor-microRNA regulation database. Nucleic Acids Res. 2019;47D1D253–D258. doi: 10.1093/nar/gky1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parahuleva MS, Lipps C, Parviz B, Hölschermann H, Schieffer B, Schulz R, Euler G. MicroRNA expression profile of human advanced coronary atherosclerotic plaques. Sci Rep. 2018;8:7823. doi: 10.1038/s41598-018-25690-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raitoharju E, Lyytikäinen LP, Levula M, Oksala N, Mennander A, Tarkka M, Klopp N, Illig T, Kähönen M, Karhunen PJ, et al. miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis. 2011;219:211–217. doi: 10.1016/j.atherosclerosis.2011.07.020 [DOI] [PubMed] [Google Scholar]
- 64.Cipollone F, Felicioni L, Sarzani R, Ucchino S, Spigonardo F, Mandolini C, Malatesta S, Bucci M, Mammarella C, Santovito D, et al. A unique microRNA signature associated with plaque instability in humans. Stroke. 2011;42:2556–2563. doi: 10.1161/STROKEAHA.110.597575 [DOI] [PubMed] [Google Scholar]
- 65.Maitrias P, Metzinger-Le Meuth V, Massy ZA, M’Baya-Moutoula E, Reix T, Caus T, Metzinger L. MicroRNA deregulation in symptomatic carotid plaque. J Vasc Surg. 2015;62:1245–50.e1. doi: 10.1016/j.jvs.2015.06.136 [DOI] [PubMed] [Google Scholar]
- 66.Markus B, Grote K, Worsch M, Parviz B, Boening A, Schieffer B, Parahuleva MS. Differential expression of MicroRNAs in endarterectomy specimens taken from patients with asymptomatic and symptomatic carotid plaques. PLoS One. 2016;11:e0161632. doi: 10.1371/journal.pone.0161632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Allahverdian S, Chaabane C, Boukais K, Francis GA, Bochaton-Piallat ML. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. 2018;114:540–550. doi: 10.1093/cvr/cvy022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan KL, Li YJ, Chien S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA. 2016;113:11525–11530. doi: 10.1073/pnas.1613121113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang R, Dong LD, Meng XB, Shi Q, Sun WY. Unique MicroRNA signatures associated with early coronary atherosclerotic plaques. Biochem Biophys Res Commun. 2015;464:574–579. doi: 10.1016/j.bbrc.2015.07.010 [DOI] [PubMed] [Google Scholar]
- 70.Pankratz F, Hohnloser C, Bemtgen X, Jaenich C, Kreuzaler S, Hoefer I, Pasterkamp G, Mastroianni J, Zeiser R, Smolka C, et al. MicroRNA-100 suppresses chronic vascular inflammation by stimulation of endothelial autophagy. Circ Res. 2018;122:417–432. doi: 10.1161/CIRCRESAHA.117.311428 [DOI] [PubMed] [Google Scholar]
- 71.Ardestani A, Lupse B, Maedler K. Hippo signaling: key emerging pathway in cellular and whole-body metabolism. Trends Endocrinol Metab. 2018;29:492–509. doi: 10.1016/j.tem.2018.04.006 [DOI] [PubMed] [Google Scholar]
- 72.Zhou Q, Li L, Zhao B, Guan KL. The hippo pathway in heart development, regeneration, and diseases. Circ Res. 2015;116:1431–1447. doi: 10.1161/CIRCRESAHA.116.303311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ben Mimoun S, Mauviel A. Molecular mechanisms underlying TGF-ß/Hippo signaling crosstalks – Role of baso-apical epithelial cell polarity. Int J Biochem Cell Biol. 2018;98:75–81. [DOI] [PubMed] [Google Scholar]
- 74.Oceandy D, Amanda B, Ashari FY, Faizah Z, Azis MA, Stafford N. The cross-talk between the TNF-α and RASSF-hippo signalling pathways. Int J Mol Sci. 2019;20:E2346. doi: 10.3390/ijms20092346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grundmann S, Hans FP, Kinniry S, Heinke J, Helbing T, Bluhm F, Sluijter JP, Hoefer I, Pasterkamp G, Bode C, et al. MicroRNA-100 regulates neovascularization by suppression of mammalian target of rapamycin in endothelial and vascular smooth muscle cells. Circulation. 2011;123:999–1009. doi: 10.1161/CIRCULATIONAHA.110.000323 [DOI] [PubMed] [Google Scholar]
- 76.Liu Q, Wang J, Zhao Y, Li CI, Stengel KR, Acharya P, Johnston G, Hiebert SW, Shyr Y. Identification of active miRNA promoters from nuclear run-on RNA sequencing. Nucleic Acids Res. 2017;45:e121. doi: 10.1093/nar/gkx318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang TC, Pertea M, Lee S, Salzberg SL, Mendell JT. Genome-wide annotation of microRNA primary transcript structures reveals novel regulatory mechanisms. Genome Res. 2015;25:1401–1409. doi: 10.1101/gr.193607.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–154. doi: 10.1038/nrm3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bedi K, Paulsen MT, Wilson TE, Ljungman M. Characterization of novel primary miRNA transcription units in human cells using Bru-seq nascent RNA sequencing. NAR Genom Bioinform. 2020;2:lqz014. doi: 10.1093/nargab/lqz014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Conrad T, Marsico A, Gehre M, Orom UA. Microprocessor activity controls differential miRNA biogenesis In Vivo. Cell Rep. 2014;9:542–554. doi: 10.1016/j.celrep.2014.09.007 [DOI] [PubMed] [Google Scholar]
- 81.Creugny A, Fender A, Pfeffer S. Regulation of primary microRNA processing. FEBS Lett. 2018;592:1980–1996. doi: 10.1002/1873-3468.13067 [DOI] [PubMed] [Google Scholar]
- 82.Pitchiaya S, Heinicke LA, Park JI, Cameron EL, Walter NG. Resolving subcellular miRNA trafficking and turnover at single-molecule resolution. Cell Rep. 2017;19:630–642. doi: 10.1016/j.celrep.2017.03.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weinmann L, Höck J, Ivacevic T, Ohrt T, Mütze J, Schwille P, Kremmer E, Benes V, Urlaub H, Meister G. Importin 8 is a gene silencing factor that targets argonaute proteins to distinct mRNAs. Cell. 2009;136:496–507. doi: 10.1016/j.cell.2008.12.023 [DOI] [PubMed] [Google Scholar]
- 84.Gibbings D, Mostowy S, Jay F, Schwab Y, Cossart P, Voinnet O. Selective autophagy degrades DICER and AGO2 and regulates miRNA activity. Nat Cell Biol. 2012;14:1314–1321. doi: 10.1038/ncb2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 2011;29:436–442. doi: 10.1038/nbt.1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Suzuki HI, Young RA, Sharp PA. Super-enhancer-mediated RNA processing revealed by integrative MicroRNA network analysis. Cell. 2017;168:1000.e15–1014.e15. doi: 10.1016/j.cell.2017.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marzi MJ, Ghini F, Cerruti B, de Pretis S, Bonetti P, Giacomelli C, Gorski MM, Kress T, Pelizzola M, Muller H, et al. Degradation dynamics of microRNAs revealed by a novel pulse-chase approach. Genome Res. 2016;26:554–565. doi: 10.1101/gr.198788.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shrestha A, Mukhametshina RT, Taghizadeh S, Vásquez-Pacheco E, Cabrera-Fuentes H, Rizvanov A, Mari B, Carraro G, Bellusci S. MicroRNA-142 is a multifaceted regulator in organogenesis, homeostasis, and disease. Dev Dyn. 2017;246:285–290. doi: 10.1002/dvdy.24477 [DOI] [PubMed] [Google Scholar]
- 89.Yuan X, Berg N, Lee JW, Le TT, Neudecker V, Jing N, Eltzschig H. MicroRNA miR-223 as regulator of innate immunity. J Leukoc Biol. 2018;104:515–524. doi: 10.1002/JLB.3MR0218-079R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schober A, Nazari-Jahantigh M, Wei Y, Bidzhekov K, Gremse F, Grommes J, Megens RT, Heyll K, Noels H, Hristov M, et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat Med. 2014;20:368–376. doi: 10.1038/nm.3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008;15:272–284. doi: 10.1016/j.devcel.2008.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, Evans MJ, Sachidanandam R, Brown BD. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods. 2012;9:840–846. doi: 10.1038/nmeth.2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bosson AD, Zamudio JR, Sharp PA. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell. 2014;56:347–359. doi: 10.1016/j.molcel.2014.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li S, Liang Z, Xu L, Zou F. MicroRNA-21: a ubiquitously expressed pro-survival factor in cancer and other diseases. Mol Cell Biochem. 2012;360:147–158. doi: 10.1007/s11010-011-1052-6 [DOI] [PubMed] [Google Scholar]
- 95.Huang ZP, Wang DZ. miR-22 in cardiac remodeling and disease. Trends Cardiovasc Med. 2014;24:267–272. doi: 10.1016/j.tcm.2014.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ma Q, Kinneer K, Bi Y, Chan JY, Kan YW. Induction of murine NAD(P)H:quinone oxidoreductase by 2,3,7,8-tetrachlorodibenzo-p-dioxin requires the CNC (cap “n” collar) basic leucine zipper transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2): cross-interaction between AhR (aryl hydrocarbon receptor) and Nrf2 signal transduction. Biochem J. 2004;377:205–213. doi: 10.1042/BJ20031123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lacher SE, Levings DC, Freeman S, Slattery M. Identification of a functional antioxidant response element at the HIF1A locus. Redox Biol. 2018;19:401–411. doi: 10.1016/j.redox.2018.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ji W, Wang L, He S, Yan L, Li T, Wang J, Kong AT, Yu S, Zhang Y. Effects of acute hypoxia exposure with different durations on activation of Nrf2-ARE pathway in mouse skeletal muscle. PLoS One. 2018;13:e0208474. doi: 10.1371/journal.pone.0208474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johansson K, Cebula M, Rengby O, Dreij K, Carlström KE, Sigmundsson K, Piehl F, Arnér ES. Cross talk in HEK293 cells between Nrf2, HIF, and NF-κB activities upon challenges with redox therapeutics characterized with single-cell resolution. Antioxid Redox Signal. 2017;26:229–246. doi: 10.1089/ars.2015.6419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Raitoharju E, Oksala N, Lehtimäki T. MicroRNAs in the atherosclerotic plaque. Clin Chem. 2013;59:1708–1721. doi: 10.1373/clinchem.2013.204917 [DOI] [PubMed] [Google Scholar]
- 101.Qin Z, Xia W, Fisher GJ, Voorhees JJ, Quan T. YAP/TAZ regulates TGF-β/Smad3 signaling by induction of Smad7 via AP-1 in human skin dermal fibroblasts. Cell Commun Signal. 2018;16:18. doi: 10.1186/s12964-018-0232-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nakao A, Okumura K, Ogawa H. Smad7: a new key player in TGF-beta-associated disease. Trends Mol Med. 2002;8:361–363. doi: 10.1016/s1471-4914(02)02376-6 [DOI] [PubMed] [Google Scholar]
- 103.Gan W, Dai X, Dai X, Xie J, Yin S, Zhu J, Wang C, Liu Y, Guo J, Wang M, et al. LATS suppresses mTORC1 activity to directly coordinate Hippo and mTORC1 pathways in growth control. Nat Cell Biol. 2020;22:246–256. doi: 10.1038/s41556-020-0463-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026 [DOI] [PubMed] [Google Scholar]
- 105.Bader AG. miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet. 2012;3:120. doi: 10.3389/fgene.2012.00120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Voellenkle C, Rooij Jv, Guffanti A, Brini E, Fasanaro P, Isaia E, Croft L, David M, Capogrossi MC, Moles A, et al. Deep-sequencing of endothelial cells exposed to hypoxia reveals the complexity of known and novel microRNAs. RNA. 2012;18:472–484. doi: 10.1261/rna.027615.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.