

Abstract
Clinical data from ADA-SCID patients registered in the U.S. Immunodeficiency Network (USIDNet) Repository were analyzed. Sixty-four ADA-SCID patients born between 1981 and 2017 had clinical data entered by their local (or home) enrolling institution. Median age at diagnosis was 1 month for those with a positive family history and 3 months for those without a prior family history, with some diagnosed at birth and one as late as 9 years of age. Overall survival was 79.7%, which increased to 94.1% since 2010. These patients had multiple infections and pulmonary, gastrointestinal, and neurological complications. The majority received enzyme replacement therapy (ERT) at some time, including 88% of those born since 2010. Twenty-six patients underwent allogeneic hematopoietic stem cell transplant (HSCT). HSCT successfully supported survival (17/26, 65%) using a variety of cell sources (bone marrow, mobilized peripheral blood, and cord blood) from sibling, family and unrelated donors. Nineteen patients underwent autologous HSCT with gene therapy (GT) using retroviral and lentiviral vectors and all are surviving. The prognosis for patients with ADA-SCID has continued to improve but these patients do have multiple early and potentially long-term conditions that require medical monitoring and management.
Keywords: Adenosine deaminase–deficient severe combined immune deficiency, ADA-SCID, immunodeficiency, hematopoietic stem cell transplant, gene therapy, US Immunodeficiency Network (USIDNet)
Introduction
Adenosine deaminase (ADA) deficiency (OMIM #102700) is an autosomal recessive genetic defect responsible for a form of severe combined immunodeficiency (SCID) characterized by severe deficiencies in T, B, and NK cells. Clinical onset typically occurs within the first several months of life, although there is heterogeneity in presentation, with some individuals having relatively late-onset presentation depending on their particular genotype [1–3]. ADA, which is ubiquitously expressed by mammalian tissues and found in the serum, is required for the processing and excretion of byproducts from DNA metabolism by converting adenosine (Ado) and deoxyadenosine (dAdo) to inosine (Ino) and 2′-deoxyinosine (dIno). The accumulation of dAdo nucleotides (dAXP), especially dATP, negatively affects the viability, differentiation, and function of cells, particularly lymphoid cells in the thymus which are rapidly dividing and undergoing selection. Given the ubiquitous nature of the enzyme, it is not surprising ADA deficiency is a systemic purine metabolic disorder with multiple systemic manifestations affecting the pulmonary, skeletal, dermatologic, and gastrointestinal systems. [4]
Treatment options for ADA-SCID include enzyme replacement therapy (ERT) with ADA conjugated to polyethylene glycol (PEG-ADA), allogeneic hematopoietic stem cell transplant (HSCT), and now gene therapy (GT) with autologous HSCT [5]. While PEG-ADA is life-saving and readily available, immune function may decline long term. Transplantation from human leukocyte antigen (HLA)–matched sibling donors without cytoreductive conditioning remains the standard of care for ADA-SCID, although there have been excellent outcomes with 100% survival in the more than 70 ADA-SCID patients treated with gene therapy. [6, 7]
Although ADA deficiency accounts for approximately 10–15% of all SCID cases in the USA [8, 9], it remains a rare disease with an overall prevalence of approximately 1 in 500,000 live births. Therefore, it is likely that only centers with specialized immunologists and bone marrow transplant physicians see enough patients to gain significant experience in the evaluation and management of these patients. For this reason, reports from multicenter databases remain an important resource for physicians who care for patients with ADA-SCID. This is particularly important in an era when treatments such as gene therapy with autologous transplant are available, with most affected individuals transplanted and hospitalized only briefly at a specialized center, receiving the remainder of their care with their local immunologist. Here, we report on the treatment and long-term outcomes of 64 patients with ADA-SCID reported to the US Immunodeficiency Network (USIDNet) Registry.
Methods
The USIDNet is an NIH-funded (U24 AI086037) research consortium of the Immune Deficiency Foundation (IDF) that collects information on patients with primary immune deficiencies in North America. After patient consent and medical records, which have been confirmed by the treating physician, are uploaded online by enrolling institutions, the Registry was queried by investigators in April 2019, who were provided de-identified patient data for further analysis. No minimum follow-up period from diagnosis was required for entry into the database. Comparison of multiple survival curves was performed by log-rank test for trend on GraphPad Prism with statistical significance set at p < 0.05. Graphs were created using GraphPad Prism v7.0 (La Jolla, CA).
Results
Demographics
A total of 64 patients were registered with ADA-deficient SCID in the USIDNet Registry at the time of query with relatively equal gender distribution (35 male, 29 female). (Table 1) The five centers to enter the largest number of patients were the National Institutes of Health (n = 21), University of California San Francisco Benioff Children’s Hospital (n = 12), Duke University Medical Center (n = 4), Mt. Sinai Hospital (n = 3), and Cincinnati Children’s Hospital Medical Center (n = 3). Patients were born between 1981 and 2017 with ages ranging from 2 to 38 years at the time of last follow-up. The diagnosis of ADA-SCID was based on molecular analyses (genetic [n = 10] or erythrocyte deoxyadenosine nucleotide levels [n = 19]) or family history in the majority of patients. The longest follow-up period reported is 26 years. The median age at diagnosis was 0.3 years for the entire cohort, decreasing to 0.1 years at time of diagnosis for patients born after 2010. Of patients for whom family history was recorded, 24% (14/58) had a family history of ADA-SCID. A known family history of ADA-SCID did not appear to affect the age at diagnosis; the average age at time of diagnosis was 0.77 ± 1.5 years versus 0.81 ± 1.4 years (p = 0.07) in those without and with family history, respectively (Fig. 1). Ethnicity/race was not reported for 7 patients; of the remainder, 60% (34/57) were Caucasian, 24% (13/57) were African American, 7% (4/57) were Hispanic, 2% (1/57) were Asian, and 9% (5/57) were mixed race.
Table 1.
Patient demographics (N = 64)
| Characteristic | Number of patients |
|---|---|
| Gender | 35 male; 29 female |
| Year of birth | 1981–2017 |
| Median age of symptom onset (years) | 0.2 (range 0–8) |
| Median age at diagnosis (years) | 0.3 (range 0–9) |
| + Family history, n (% known) | 14 (24%) |
| Race/ethnicity, n (% known) | |
| Caucasian | 34 (60%) |
| African American | 13 (24%) |
| Hispanic | 4 (7%) |
| Asian | 1 (2%) |
| Mixed race | 5 (9%) |
| Not reported | 7 |
Fig. 1.
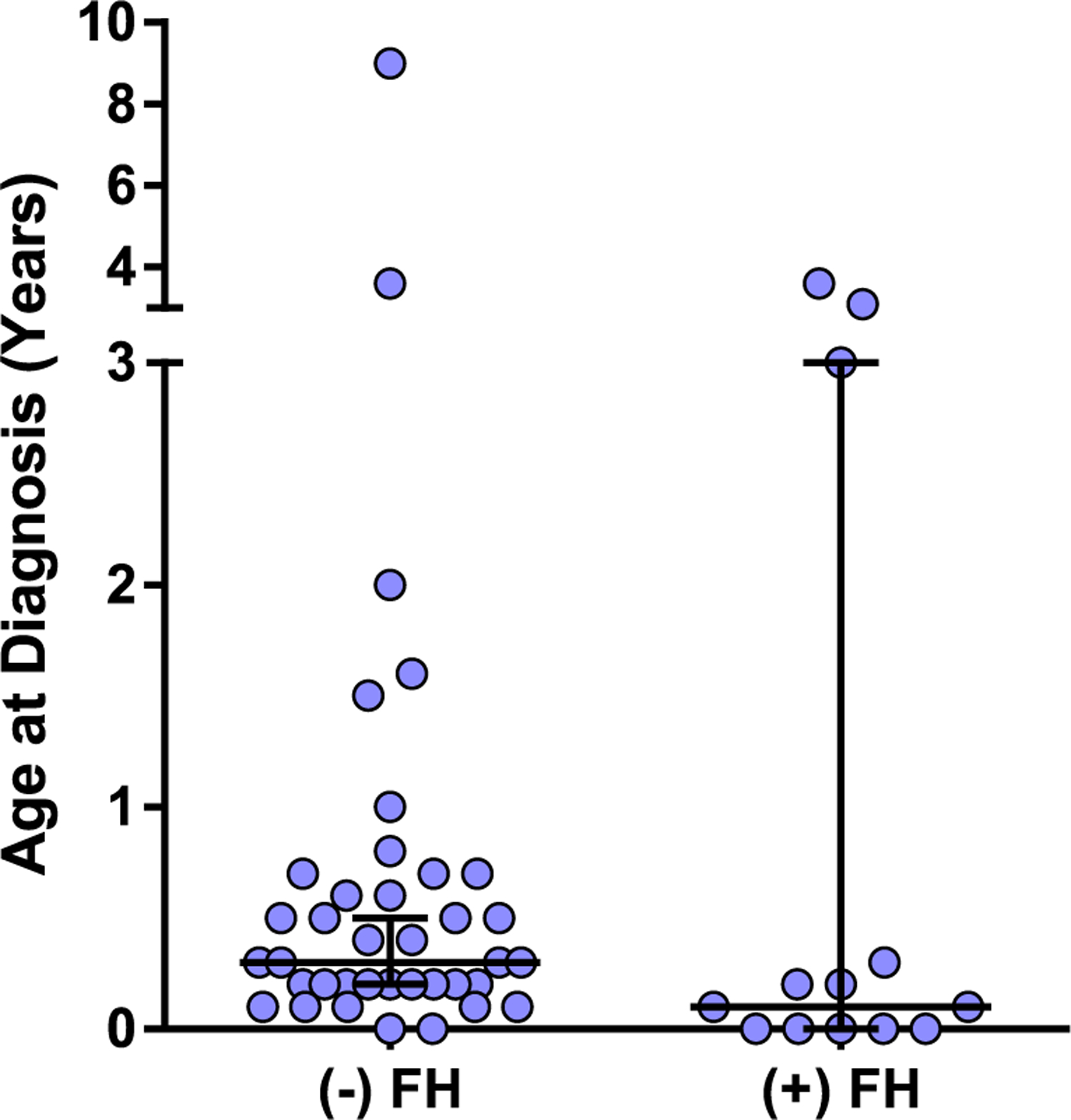
Age at diagnosis by family history. The ages at which patients were diagnosed are shown, divided into those with no prior family history (FH) and those with known family history of SCID. Median ± 95% confidence intervals are shown. Average age of diagnosis was 0.77 ± 1.5 years versus 0.81 ± 1.4 years in those without versus with family history, respectively. (p = 0.07, Mann-Whitney)
Overall Survival
Overall survival was 80% (51/64), with survival by decade shown in Fig. 2. Thirteen of the registered patients are deceased (Table 2) with age at death ranging from 0.2 to 23 years (median 1.9 years). The cause of death was known in all but one patient who was lost to follow-up. Listed causes of death were most commonly infectious (n = 7) or due to transplant-related organ toxicities (n = 4). The two older patients who died were an 8.3-year-old who was status post haplo-identical HSCT at 1.3 years and died from parainfluenza infection and a 23-year-old treated only with ERT who died of pneumonia. Nine of 13 patients who died had received HSCT, with the cause of death in 6 due to transplant-related problems such as disseminated infection, multiple organ failure, and veno-occlusive disease. Known causes of death for the remaining patients included disseminated infection, pneumonia, parainfluenza, CMV pneumonitis, and uncontrolled hemolytic anemia, with 3 of these patients treated with ERT.
Fig. 2.
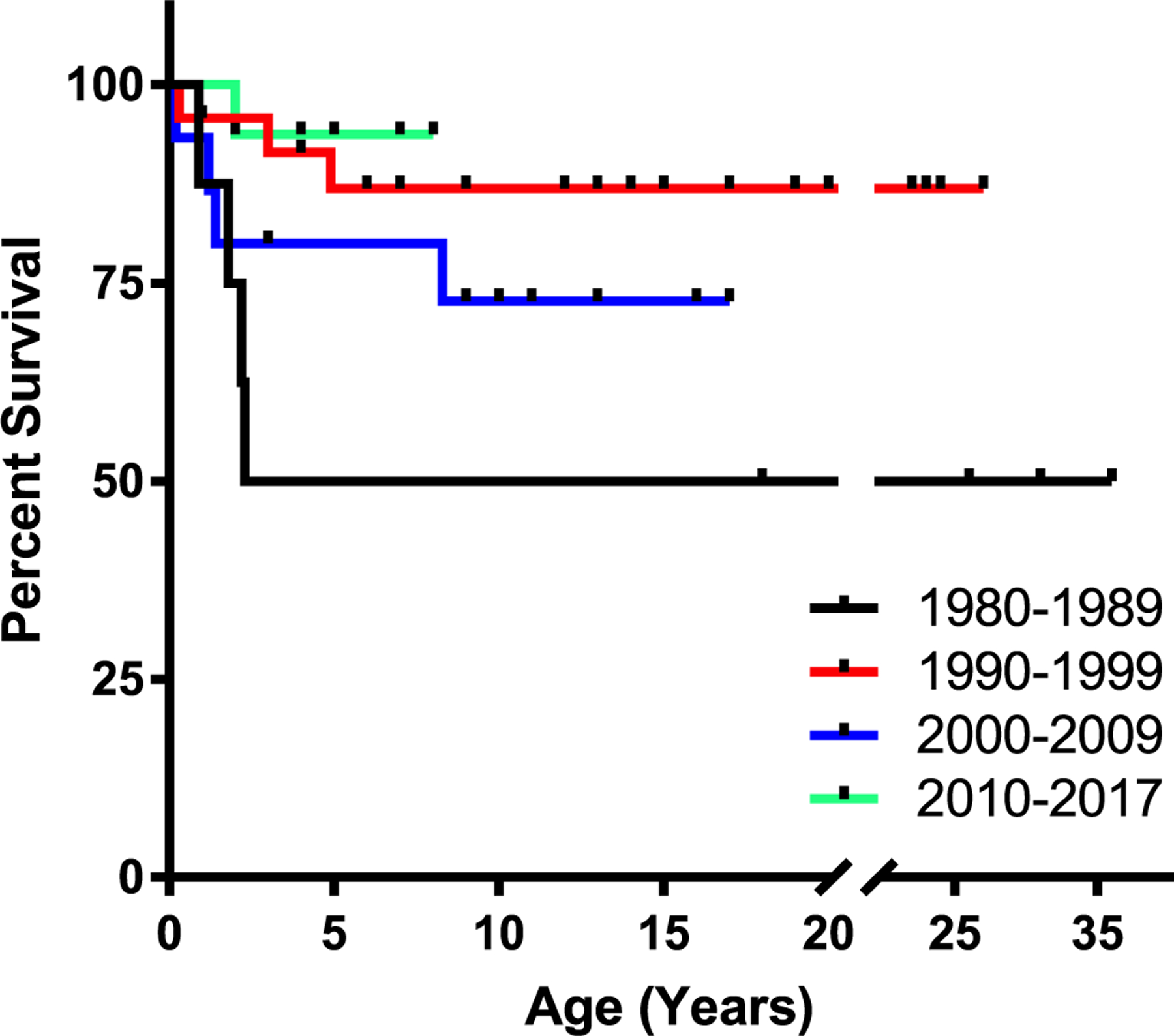
Survival by decade. Kaplan-Meier survival curves for ADA-SCID patients separated by decade of birth (p = 0.065; log-rank test). p < 0.05 (Bonferroni) when comparing overall survival rates for 1980–1989 versus 1990–1999 and 1980–1989 versus 2010–2017. For the time period 1980–1989: n = 8 (St = 0.5), 1990–1999: n = 24 (St = 0.88), 2000–2009: n = 15 (St = 0.73), 2010–2017: n = 17 (St = 0.94)
Table 2.
Listed causes of death
| Causes of death | ERT | HSCT | Age at death (years) |
|---|---|---|---|
| Infection-disseminated | Yes | 2.2 | |
| Infection-disseminated, transplant-related problem(s) | Yes | 0.9 | |
| Infection-disseminated, transplant-related problem(s) | Yes | 1.8 | |
| Parainfluenza | Yes | Yes | 8.3 |
| Pneumonia | Yes | No | 23 |
| Post-BMT, multiple organ failure, CHF, hepatic failure | Yes | Yes | 4.9 |
| Respiratory failure due to CMV pneumonitis | Unknown | Yes | 1.4 |
| Septic shock and respiratory failure, cardiorespiratory failure | No | 0.2 | |
| Transplant-related problem(s) | Yes | 0.3 | |
| Transplant-related problem(s) | Yes | 3 | |
| Uncontrolled hemolytic anemia | Yes | No | 1.2 |
| Unknown; lost to Follow-up | Yes | No | |
| Veno-occlusive liver disease/GVHD | Yes | Yes | 2 |
| Mean | 4.1 |
ERT, enzyme replacement therapy; HSCT, hematopoietic stem cell transplant; CHF, congestive heart failure; CMV, cytomegalovirus; GVHD, graft versus host disease
Infectious Complications
There were a total of 187 infections reported in 50 patients (Fig. 3). The most commonly reported infection was lower respiratory tract infections (LRTI) including pneumonia, with 44 episodes occurring in 32 patients and 3 episodes of bronchiolitis in 3 patients. Sixty percent (28/47) of these episodes occurred before or at diagnosis of immunodeficiency. Specific organisms were reported in approximately half of the cases; Pneumocystis jirovecii pneumonia was the most commonly reported organism (n = 12), with only one case occurring in the last decade. Various bacterial (Haemophilus influenzae, Moraxella catarrhalis, pseudomonas, methicillin-resistant Staphylococcus aureus, serratia, Mycobacterium tuberculosis) and viral (parainfluenza type 3, rhinovirus, RSV, and cytomegalovirus) etiologies were also recorded. Following LRTIs, skin and soft tissues infections (SSTI) and upper respiratory tract infections (URI) were the next most frequently reported. SSTIs were caused by bacterial, fungal, and viral organisms ranging from superficial dermatitis to cellulitis and abscesses. There were 21 episodes of URIs, 11 episodes of otitis media, 10 episodes of sinusitis, and 1 episode of pharyngitis in 20 patients. Infectious diarrhea was also frequently reported, with 19 reported episodes in 15 patients. Commonly isolated organisms included Clostridium difficile in 5 patients and rotavirus in 7 (Table 3).
Fig. 3.
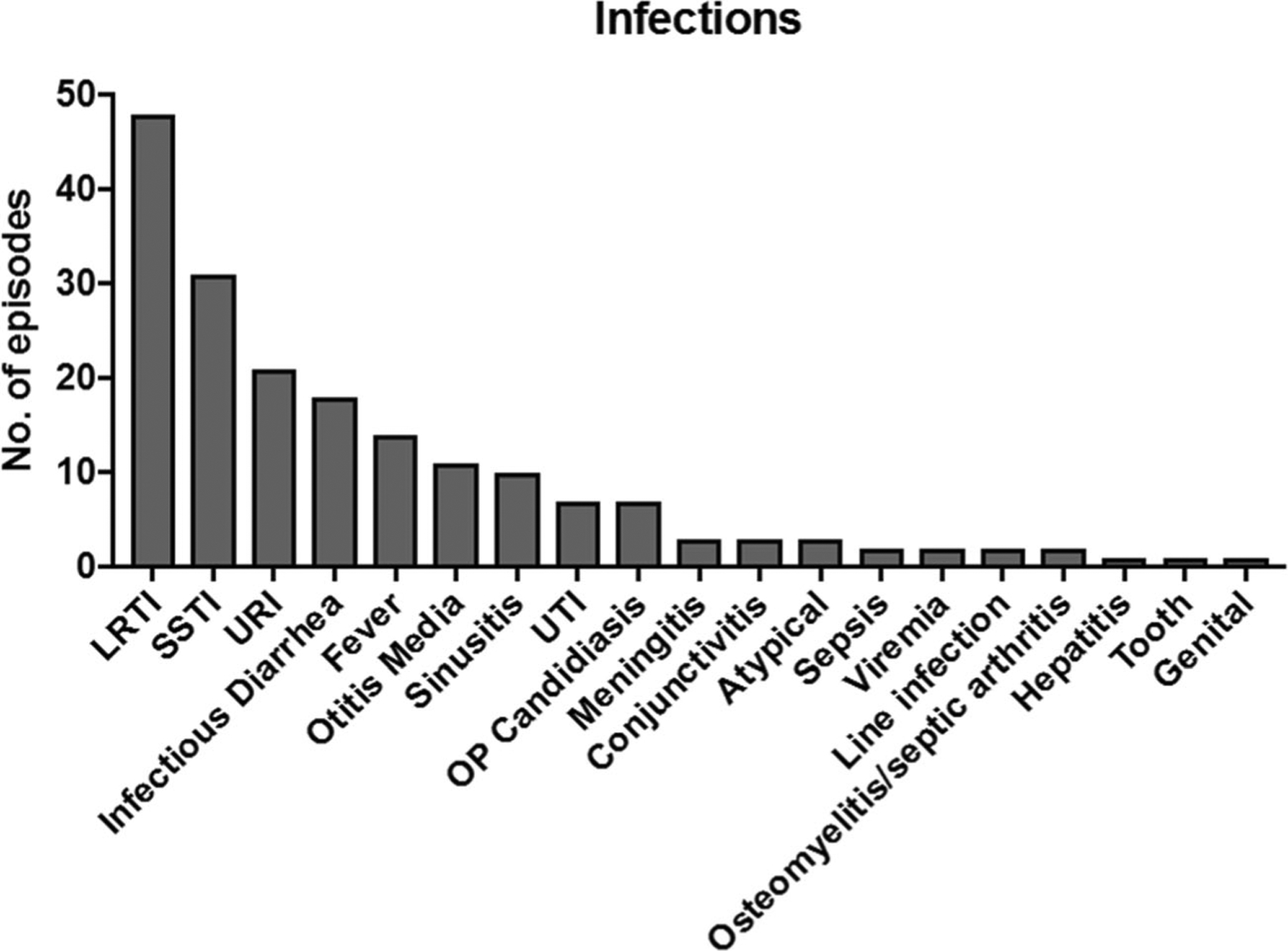
The types of infectious episodes reported are shown. LRTI, lower respiratory tract infections; SSTI, skin and soft tissue infections; URI, upper respiratory infections; UTI, urinary tract infections; OP, oropharyngeal
Table 3.
Infectious complications and reported organisms
| Infection type | Organisms (number of patients) |
|---|---|
| Lower respiratory tract infection (pneumonia, bronchiolitis) | Pneumocystis jirovecii (14), Haemophilus influenzae (1), Moraxella catarrhalis (1), pseudomonas (1), serratia (1), Mycobacterium tuberculosis (1), Streptococcus pneumoniae (1), Klebsiella pneumoniae (1), MRSA (1) rhinovirus (1), parainfluenza 3 (1), RSV (1), influenza (1), CMV (1) |
| Skin and soft tissue infection | Staphylococcus aureus (1), MRSA (1), Candida albicans (3), molluscum contagiosum (2), Tinea corporis (4), varicella (3), HPV (1) |
| Upper respiratory infections (URI, otitis media, sinusitis, pharyngitis) | Stenotrophomonas multiphilia (1), Staphylococcus aureus (1), Haemophilus influenzae (1), MRSA (1) |
| Infectious diarrhea | Rotavirus (7), Clostridium difficile (5), giardia (2), norovirus (1) |
| Otitis Media | Staphylococcus aureus (1) |
| Sinusitis | Haemophilus influenzae (1), MRSA (1) |
| Urinary tract infection | Enterovirus (1), enterococcus (1), candida (1) |
| Meningitis | Enterovirus (2), pseudomonas (1) |
| Sepsis | Shigella (1) |
| Viremia | EBV (1), CMV (1) |
| Line Infection | Enterobacter cloacae (1), candida (1) |
| Osteomyelitis | MSSA |
Non-infectious Complications
Multiple chronic medical conditions were reported in this cohort (Table 4). Forty-five percent (n = 29) of patients had one or more neurological conditions including hearing loss, developmental delay, learning and/or coordination disability, and attention-deficit/hyperactivity disorder (ADHD). Sixty-seven percent (n = 43) had gastrointestinal conditions, with diarrhea, poor growth, and gastroesophageal reflux being the most common. Thirty percent (n = 19) had one or more pulmonary conditions manifesting as asthma, bronchiectasis, bronchiolitis obliterans, and interstitial lung disease. A total of 12 patients had dermatofibrosarcoma protuberans, 12 had neutropenia, and 10 had autoimmune phenomena such as anemia, thrombocytopenia, and diabetes mellitus.
Table 4.
Non-infectious complications (% of all registrants)
| n = 29—one or more neurological conditions (45%) |
| 18 hearing deficit/loss (6 hearing only) (28%) |
| 15 developmental delay/learning disability (23%) |
| 5 attention-deficit/hyperactivity disorder (ADHD) (8%) |
| 3 coordination disability (5%) |
| n = 19—one or more pulmonary conditions (30%) |
| 11 asthma (17.2%) |
| 6 bronchiectasis |
| 2 Bronchiolitis obliterans |
| 7 interstitial lung disease |
| n = 22—failure to thrive (FTT) or unintended weight loss |
| n = 13—gastro-esophageal reflux |
| n = 12—dermatofibrosarcoma protuberans |
| n = 12—neutropenia |
| n = 6—short stature |
| n = 5—autoimmune hemolytic anemia |
Enzyme Replacement Therapy
Enzyme replacement therapy with PEGylated bovine ADA enzyme was used in 70% of patients (45/64). Treatment with PEG-ADA increased by decade, with 88% of ADA-SCID patients born between 2010 and 2017 treated with ERT at some point in their lives (Fig. 4). At the time of last follow-up, 28 patients remained on ERT, 7 were off PEG-ADA, and ERT status was unspecified in the remainder.
Fig. 4.
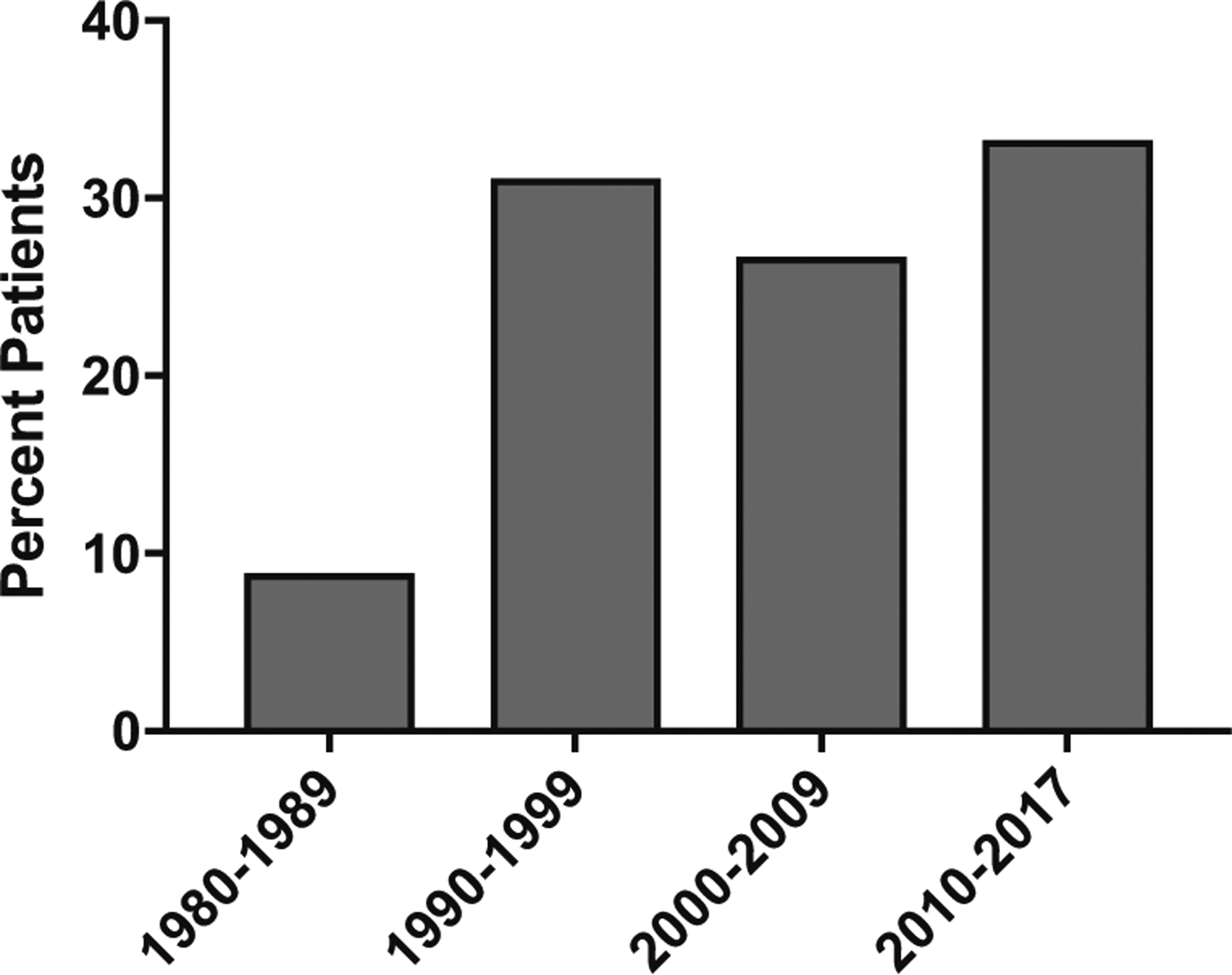
The percentages of patients who received ADA enzyme replacement therapy (ERT) by decade of birth
Allogeneic Hematopoietic Stem Cell Transplant
A total of 26 patients underwent allogeneic hematopoietic stem cell transplant (HSCT) with bone marrow grafts used for 20 (77%) and cord blood for 1 (4%), while cell source was unknown for 8 (31%). Survival after allogeneic HSCT was summarized by decade of birth (Fig. 5a), with overall survival highest in the most recent time period. Overall survival was 65% for those who underwent allogeneic HSCT, with survival by cell source shown in Fig. 5b. Conditioning regimens were unknown or not reported for the majority of transplanted patients; of the 9 in whom it was reported, 6 received no conditioning, 1 received reduced-intensity conditioning (RIC), and 3 received myeloablative conditioning. Acute graft-versus-host disease occurred in 3 individuals and was limited to the skin in two of these patients.
Fig. 5.
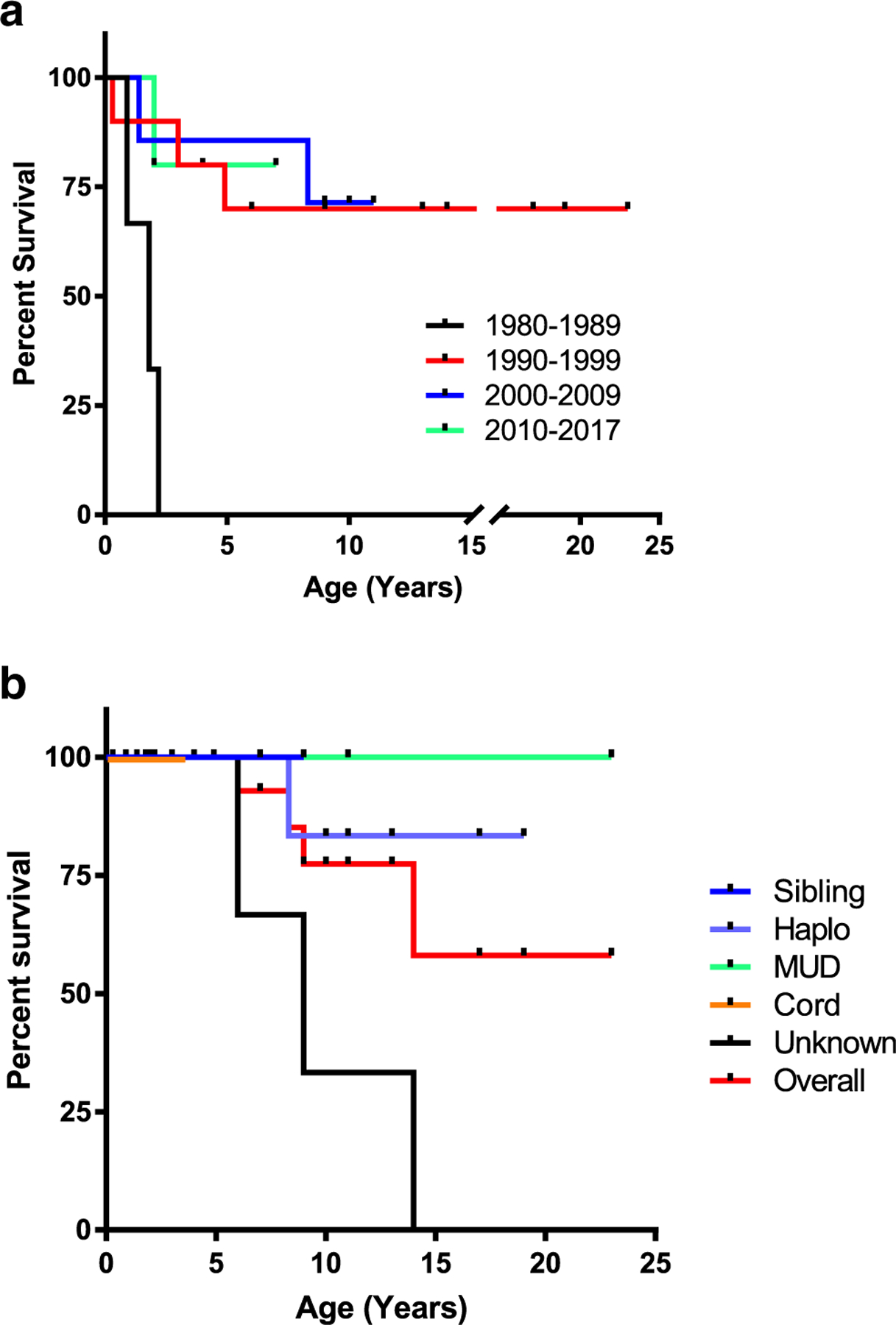
Survival after allogeneic hematopoietic stem cell transplant (HSCT). Kaplan-Meier curves for ADA-SCID patient undergoing HSCT by a decade of birth (p = 0.0048; log-rank test). For time period 1980–1989: n = 3 (St = 0), 1990–1999: n = 10 (St = 0.70), 2000–2009: n = 7 (St = 0.71), and 2010–2017: n = 6 (St = 0.83). b Survival after allogeneic HSCT shown by donor type (p = 0.1447; log-rank test). For sibling donor: n = 3 (St = 1), haplo donor: n = 6 (St = 0.83), MUD donor: n = 5 (St = 1), cord donor: n = 1 (St = 1), unknown donor source: n = 11 (St = 0.73), and overall n = 26 (St = 0.85)
Gene Therapy
A total of 19 patients were reported to be treated with gene therapy and autologous transplant between 1990 and 2016 (Fig. 6). Besides one patient who received gene-modified autologous T lymphocytes without conditioning and achieved only partial, short-term correction in humoral and cellular immunity, the remainder received autologous hematopoietic stem cells that were gene-modified. Thirteen patients were treated with bone marrow CD34+ stem cells, 3 with mobilized peripheral blood CD34+ cells, 1 using umbilical cord blood, and an unspecified stem cell source in 2. Between 1990 and 2011, 10 patients were reported who received gene therapy with gamma-retroviral vectors; between 2014 and 2016, 2 were treated with gamma-retroviral vectors and 7 with lentiviral vectors.
Fig. 6.
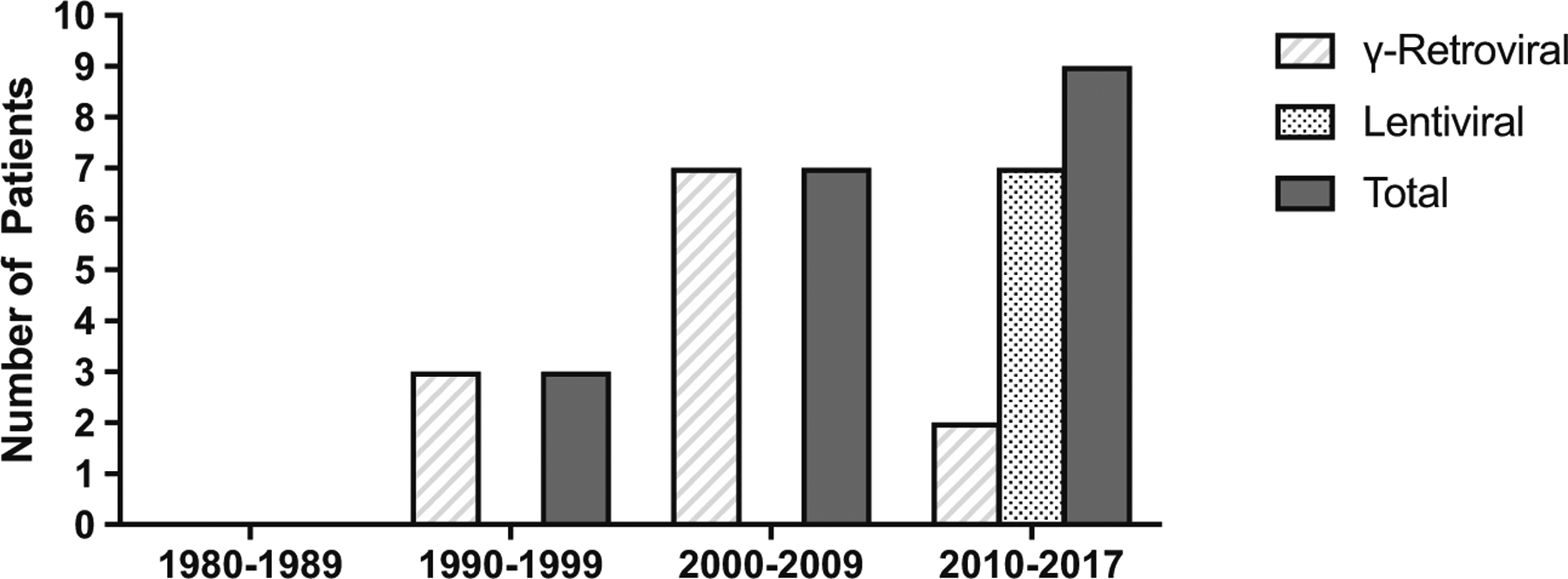
Use of gene therapy (GT) for ADA-SCID by decade. The types of gene transfer vector used are shown for gamma-retroviral vectors and lentiviral vectors
The overall survival of patients undergoing gene therapy was 100%. Between 1990 and 2002, four patients were treated with gene therapy and did not receive cytoreductive conditioning, and as expected, did not have correction of their disease process. One patient who received minimal-intensity conditioning (MIC) in the gamma-retroviral era did not achieve disease correction. Four patients who received MIC or RIC achieved partial disease correction, two who received RIC attained full correction, and conditioning and disease outcomes were not reported for 8 patients. The degree of disease correction was defined by criteria from individual treating institutions.
Discussion
Adenosine deaminase deficiency is an autosomal recessive form of SCID that accounts for 10–15% of all cases of human SCID. ADA is a ubiquitously expressed enzyme required for the degradation of purine nucleoside metabolites; lack of the enzyme profoundly affects T, B, and NK lymphocytes and may have associated organ toxicities. Affected children generally present within the first few months of life from severe infections, if not detected earlier by newborn screening, although there are some who have milder disease and present later in life depending on their particular genetic mutation. In this cohort, there was approximately equal gender distribution with a majority that were Caucasian followed by African American, reflecting the racial composition of the USA.
As newborn screening (NBS) for SCID through quantification of T cell receptor excision circles has been performed in all states in the USA since December 2018, it is now even more critical to assess patient outcomes in light of earlier diagnosis. In this USIDNet data set, infectious episodes represented a common cause of morbidity and mortality. Thirty-one of 64 patients had one or more infections diagnosed either before or at the time of SCID diagnosis; 10 of these patients had a diagnosis of ADA-SCID at 2.5 months of age or younger. Within the 13 patients who died in our cohort, the most commonly listed cause was infectious complications. Knowing that transplant outcomes for SCID are excellent in the absence of active infection [11], it is important to continue to follow clinical outcomes in an era of very early diagnosis through NBS.
As ADA is expressed in all mammalian tissues, deficiency of the enzyme is known to affect organs outside of the immune system. Specifically, some ADA-SCID patients have been reported to have central nervous system abnormalities ranging from sensorineural hearing loss and developmental delay to cortical blindness, seizure disorder, and motor dysfunction, some of which were associated with abnormal findings on imaging and EEG [10, 12]. These neurologic manifestations of ADA deficiency were not necessarily related to infections, use of CNS-toxic drugs, transplant status, or perinatal/neonatal events. The exact mechanism of neurologic abnormalities in ADA-SCID is not known, but ADA expression levels in the brain are amongst the highest, and it has been observed in some cohorts to be correlated with the severity of dATP accumulation at the time of diagnosis [13]. Based on work in the ADA−/− mouse model, administration of PEG-ADA does not necessarily ameliorate neurologic disease, since PEGylated proteins cannot leave the circulation to enter the brain [14]. Therefore, the elimination of metabolites in the CNS is relatively slower and relies on peripheral detoxification from enzyme replacement. In this USIDNet cohort, 44% of patients (n = 28) had one or more neurologic and/or behavioral conditions including hearing deficit/loss, developmental delay/learning disability, ADHD, seizures, and coordination disability. These conditions were observed both before and after the immune deficiency was diagnosed, and several of these patients had neurologic manifestations even though the diagnosis of ADA-SCID was made shortly after birth.
Standard therapy for ADA-SCID includes enzyme replacement therapy and HSCT. ERT can be life-saving and restore immune function in a relatively short period of time but has been associated with suboptimal immune reconstitution long-term [7]. In the upcoming years, it may become even more clear which definitive therapy approaches (allogeneic HSCT versus gene therapy with autologous transplant) are most appropriate for patients with ADA-SCID. In May 2016, the first ex vivo retroviral-based stem cell gene therapy for ADA-SCID was approved by the European Commission, and it is expected that a lentiviral-based therapy, which possesses a better vector integration profile and the ability to enter non-dividing stem cells, will soon be available in the USA. Both retro- and lentiviral-based gene therapy approaches for ADA-SCID require reduced-intensity cytoreductive conditioning, such as with busulfan alone (e.g., 4–6 mg/kg), to create room in the bone marrow niche. At the time of this manuscript preparation, there were no open lentiviral clinical trials for ADA-SCID as the therapy undergoes regulatory approval from the FDA for market application. A retroviral vector therapy developed at TIGET in Milan has been licensed in the European Union as Strimvelis, now marketed by Orchard Therapeutics. In an age when gene therapy is already an approved treatment modality, it may be the perfect time to address the two most common causes of death in our transplanted cohort, infection and transplant-related complications.
There are several limitations to the current study. As with many retrospective, multi-center studies, a major limitation is the reliance on data entered by physicians from various institutions which may be subject to recall biases and inaccuracies. Due to the nature of data collection, missing or contradictory information is often difficult to query and is eliminated from final data analysis or categorized as “unknown.” For instance, the infection data was simply reported for before and after diagnosis of ADA-SCID without additional granularity in regard to timing, correlation with interventions, etc. In addition, molecular testing for ADA was not reported for a small number of patients, leading to potential inclusion of a few patients with other forms of SCID. Furthermore, while the current study represents the largest cohort of ADA-deficient SCID patient to date, the sample size may still be insufficient to highlight rare outcomes in these patients.
Although outcomes for ADA-SCID have significantly improved in recent years, there remains a critical need for continued follow-up to assess outcomes with earlier diagnosis through newborn screening, improved HSCT approaches, and the increasing use of gene therapy. Additionally, routine implementation of more standardized neurodevelopmental assessments across a large group of ADA-SCID patients can better define disease-related complications and potentially discriminate relative responses to different treatments.
Acknowledgments
We acknowledge Hannah Wright (USIDNet Research Data Analyst) and Julieann Magnusson (USIDNet Project Manager) for their expertise in data management and aid in obtaining and analyzing the data. We also thank Dr. Mary Ellen Conley and Dr. Vivian Hernandez-Trujillo for providing additional patient information through chart review.
Footnotes
Conflict of Interest The authors declare that they have no conflict of interest.
References
- 1.Santisteban I, Arredondo-Vega FX, Kelly S, Mary A, Fischer A, Hummell DS, et al. Novel splicing, missense, and deletion mutations in seven adenosine deaminase-deficient patients with late/delayed onset of combined immunodeficiency disease: contribution of genotype to phenotype. J Clin Invest. 1993;92(5):2291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shovlin CL, et al. Adult presentation of adenosine deaminase deficiency. Lancet. 1993;38(6):1253–65. [DOI] [PubMed] [Google Scholar]
- 3.Umetsu DT, Schlossman CM, Ochs HD, Hershfield MS. Heterogeneity of phenotype in two siblings with adenosine deaminase deficiency. J Allergy Clin Immunol. 1994;93(2):543–50. [DOI] [PubMed] [Google Scholar]
- 4.Bradford KL, Moretti FA, Carbonaro-Sarracino DA, Gaspar HB, Kohn DB. Adenosine deaminase (ADA)-deficient severe combined immune deficiency (SCID): molecular pathogenesis and clinical manifestations. J Clin Immunol. 2017;37(7):626–37. [DOI] [PubMed] [Google Scholar]
- 5.Kohn DB, Hershfield MS, Puck JM, Aiuti A, Blincoe A, Gaspar HB, et al. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J Allergy Clin Immunol. 2019;143(3):852–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cicalese MP, Ferrua F, Castagnaro L, Rolfe K, de Boever E, Reinhardt RR, et al. Gene therapy for adenosine deaminase deficiency: a comprehensive evaluation of short- and medium-term safety. Mol Ther. 2018;26(3):917–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kohn DB, Gaspar HB. How we manage adenosine deaminase-deficient severe combined immune deficiency (ADA SCID). J Clin Immunol. 2017;37(4):351–6. [DOI] [PubMed] [Google Scholar]
- 8.Buckley RH, Schiff RI, Schiff SE, Markert ML, Williams LW, Harville TO, et al. Human severe combined immunodeficiency: genetic, phenotypic, and functional diversity in one hundred eight infants. J Pediatr. 1997;130(3):378–87. [DOI] [PubMed] [Google Scholar]
- 9.Kwan A, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2015;312(7):729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nofech-Mozes Y, Blaser SI, Kobayashi J, Grunebaum E, Roifman CM. Neurologic abnormalities in patients with adenosine deaminase deficiency. Pediatr Neurol. 2007;37(3):218–21. [DOI] [PubMed] [Google Scholar]
- 11.Pai S-Y, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med. 2014;371(5):434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hönig M, et al. Patients with adenosine deaminase deficiency surviving after hematopoietic stem cell transplantation are at high risk of CNS complications. Blood. 2007;109(8):3595–602. [DOI] [PubMed] [Google Scholar]
- 13.Rogers MH, Lwin R, Fairbanks L, Gerritsen B, Gaspar HB. Cognitive and behavioral abnormalities in adenosine deaminase deficient severe combined immunodeficiency. J Pediatr. 2001;139(1):44–50. [DOI] [PubMed] [Google Scholar]
- 14.Sauer AV, et al. Alterations in the brain adenosine metabolism cause behavioral and neurological impairment in ADA-deficient mice and patients. Sci Rep. 2016;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]