

Abstract
There is increasing understanding of the genetic basis to dilated cardiomyopathy and in this review, we offer a practical primer for the practising clinician. We aim to help all clinicians involved in the care of patients with dilated cardiomyopathy to understand the clinical relevance of the genetic basis of dilated cardiomyopathy, introduce key genetic concepts, explain which patients and families may benefit from genetic testing, which genetic tests are commonly performed, how to interpret genetic results, and the clinical applications of results. We conclude by reviewing areas for future research in this dynamic field.
Keywords: Genetics, Dilated cardiomyopathy, Heart
Graphical Abstract

DCM genetics- what the practising clinician needs to know.
Introduction
Dilated cardiomyopathy (DCM) affects up to 1 in 250 people and is the leading global indication for heart transplantation. It is characterized by left ventricular (LV) dilatation and systolic impairment. There is increasing understanding of the genetic basis to this disease and in this review, we offer a practical primer for the practising clinician. We aim to help all clinicians involved in the care of patients with DCM understand the clinical relevance of the genetic basis of DCM, which patients and families may benefit from genetic testing, which genetic tests are commonly performed, how to interpret genetic results, and the clinical applications of results (Graphical abstract).
Dilated cardiomyopathy definition
According to the revised 2016 European Society of Cardiology position statement, DCM is defined as LV or biventricular systolic dysfunction and dilatation that is not explained by abnormal loading conditions or coronary artery disease.1 Systolic dysfunction is defined by abnormal LV ejection fraction (LVEF) measured using any modality, and LV dilatation is defined as LV end-diastolic volumes or diameters greater than two standard deviations from age, gender, and body surface area-adjusted nomograms.1
Dilated cardiomyopathy prevalence
Prevalence data for DCM are variable. Historic estimates from the Olmsted County cohort prior to the widespread availability of echocardiography suggested the disease affected 1 in 2700 individuals.2 In the absence of large contemporary population studies, estimated DCM prevalence has more recently been revised based on triangulation of data from hypertrophic cardiomyopathy, heart failure, and asymptomatic LV dysfunction, yielding estimates of up to 1 in 250 individuals,3 though this estimate likely represents an upper bound.
Genetic basis of dilated cardiomyopathy: key concepts
Dilated cardiomyopathy is recognized as familial in 20–30% of cases.4 Approximately 40% of these families have an identifiable monogenic cause,3 or at least a rare variant of large effect size as the primary determinant of risk. Higher estimates of sensitivity for genetic testing have been reported (from 46% to 73% in one study5) but these estimates are likely inflated by enrichment for familial cases, and likely confounded by false positives due to insufficient control for background population variation in the genes studied. In reality, the genetic contribution to DCM risk is not solely attributable to individual DNA variants of large effects, as in a Mendelian model; rather many variants with individually small effects on disease risk likely contribute to the observed heritability. There is also increasing recognition of wider genetic or environmental modifiers,6 as discussed later in this article. Similarly, a seemingly identifiable aetiology for DCM (such as alcohol exposure) does not preclude a relevant genetic predisposition to disease that may influence management.7
In this review, we focus on DCM presenting to the adult cardiologist. The clinical and genetic evaluation of DCM presenting in early childhood, which is much rarer, is overlapping but distinct—representing a mixture of early presentations of those same entities that present in adults, alongside molecularly distinct entities. We direct the reader to other resources for comprehensive reviews of paediatric cardiomyopathies.8–10
We next review some key genetic concepts as relevant to DCM.
The continuum from monogenic to polygenic disease
Since variants with large effects on disease risk are under strong negative selection, they are depleted from the population. Genetic contributions to disease can therefore come from individually rare variants of potentially large effect size, from common variants each of small effect size, and from variants lying on the spectrum spanning those extremes. Variants may act alone or in combination to predispose to disease. Diseases with a genetic component may have various architectures, including monogenic, polygenic, oligogenic, and multifactorial (Figure 1). Dilated cardiomyopathy is genetically heterogeneous, with an important proportion behaving as monogenic diseases, but many cases are best understood by a more complex genetic model.
Figure 1.

Genetic architectures. Diseases with a genetic component may have monogenic, oligogenic, or polygenic architectures, or a multifactorial process. Our current understanding of the genetic architecture of dilated cardiomyopathy (DCM) supports a likely monogenic and oligogenic basis, though this may be revised as our understanding of common genetic variants develops.
Modes of inheritance
Most monogenic forms of DCM follow an autosomal dominant inheritance pattern, although X-linked, autosomal recessive, and mitochondrial inheritance are observed, particularly in paediatric populations.11
Genetic architecture of monogenic dilated cardiomyopathy
The genetic architecture of presumed ‘monogenic’ DCM has emerged to be particularly complex, with more than 60 genes12 purported to be associated with DCM and/or included in diagnostic test panels. However, not all implicated genetic loci are statistically robust. Some genes were reported in studies of candidate genes without adequate control populations.13 Subsequently, many genes suggested on the basis of candidate gene studies do not show association with disease in larger case-control studies.14 As a more critical evaluation of the genes linked to DCM continues,15 we expect that many purported DCM genes will be refuted as has happened with hypertrophic cardiomyopathy.16 , 17
The genes reported to be associated with DCM and/or included on diagnostic test panels span diverse biological pathways, including components of the sarcomere, cytoskeletal and desmosomal proteins, and mitochondrial proteins amongst others (key examples given in Figure 2). This is more heterogeneous than the genetic aetiology of other cardiomyopathies such as hypertrophic cardiomyopathy and suggests that DCM may manifest as a final common pathway for a number of processes.
Figure 2.

Genes most robustly associated with monogenic dilated cardiomyopathy, grouped by their location within the cardiomyocyte. For clarity, genes associated with syndromic forms of dilated cardiomyopathy have not been included. Graphics created by Elfy Chiang (elfylandstudios.com).
In addition to this locus heterogeneity, there is marked allelic heterogeneity, whereby different variants in the same gene can cause a similar phenotype. Of note, different variants in the same gene can also produce contrasting phenotypes (e.g. different variants in MYH7, with distinct molecular effects, cause hypertrophic cardiomyopathy and DCM18)
Penetrance and expressivity
Many genetic variants in DCM exhibit incomplete and age-related penetrance and variable expressivity. Penetrance is defined as the proportion of individuals carrying a variant who develop the disease phenotype, and incomplete penetrance means that not all individuals who carry a particular genetic variant will develop disease. Penetrance is typically age related in DCM—an individual carries the genetic variant from conception, but usually does not develop the DCM phenotype until middle age (>40 years old), or may not manifest at all. Penetrance is essentially unknown for many DCM variants and is an area of active research. Variable expressivity refers to heterogeneity in the severity and diversity of the resulting phenotype. For example, considering two individuals within a family carrying exactly the same genetic variant, one may have severe DCM with ventricular tachyarrhythmias and advanced heart failure requiring heart transplantation, while the other may be minimally symptomatic with only mildly impaired cardiac function. This raises the possibility that additional modifiers, either genetic, epigenetic, or environmental, contribute to the phenotype.
Unknowns in dilated cardiomyopathy genetics
There are a number of unanswered questions in our understanding of the genetics of DCM. One puzzle is the relatively low genetic diagnostic yield even in familial DCM. This most likely represents cases with genetic architectures that are harder to study—strongly genetic, but not monogenic. These include oligogenic and polygenic combinatorial models where genetic susceptibility may be determined by the cumulative effect of multiple common genetic variants,19 , 20 as opposed to a near Mendelian inheritance driven by a strong monogenic component. These additional variants may produce additive effects (where a cumulative burden of these more common variants contributes to disease), or non-additive interactions (where more common variants modify the effect of a rare variant). Gene–environment interactions21 likely also modify the effect of both rare and common variants.
Genetic studies in DCM have largely focused on more interpretable variants that directly alter protein-coding sequence, but there is increasing evidence that variation in areas of the genome that are not assessed using current standard methods, such as non-coding regulatory variants and larger structural variants (e.g. copy number changes) may also contribute to genetic risk in DCM.22–24 Studies of many complex traits have shown that common variants with small effect sizes are often non-coding variants, likely serving regulatory functions.
It is also likely that there are as yet undiscovered genes contributing to monogenic DCM, but these likely account for a relatively small proportion of genetically unexplained cases.
Updates on genetic testing
Genetic testing methodologies
A number of methodologies have been used to understand the genetics of human disease and their application as relevant to DCM is reviewed in detail in Supplementary material online (Table 1).
Table 1.
Advantages and limitations of sequencing technologies used in clinical practice
| Sequencing of single gene or individual variant | Panel sequencing | Whole-exome sequencing | Whole-genome sequencing | |
|---|---|---|---|---|
| Role | Confirmatory testing in a family member of variant detected in proband through other technique | Majority of diagnostic cardiomyopathy testing | Limited role in diagnostic testing in adult-onset DCM | Limited role in diagnostic testing in adult-onset DCM |
| Advantages | Focused testing of variant of interest—reduced cost and time | Good coverage of genes robustly linked to disease (i.e. genes most likely to yield clinically actionable data) | Is not limited to genes previously linked to disease—potential to identify novel variants in new genes of interest when applied in families or cohorts with sufficient power to establish a new gene–disease relationship (rare) |
|
| Limitations | Not appropriate for diagnostic testing in the proband |
|
|
|
DCM, dilated cardiomyopathy.
Confirmatory vs. predictive genetic testing
Currently, genetic testing is performed in DCM patients who have evidence of the disease. This is known as confirmatory or diagnostic testing. The results of this testing may direct management for the patient or be used to guide family (cascade) screening.
Predictive genetic testing is the use of a genetic test in an asymptomatic person to predict future risk of disease.25 However, the presence of a single monogenic variant is not sufficiently indicative of developing disease so in practice this refers to cascade genetic testing. In the context of DCM, this is most useful in families where someone has genetically explained DCM (the identification of a pathogenic/likely pathogenic variant), where genetic testing can identify family members who do not carry the genetic predisposition and can therefore be safely discharged without ongoing surveillance. For unaffected relatives who do carry a familial variant the predictive value is less clear. These individuals are at risk and remain under surveillance, but it is not possible to precisely quantify the risk of developing overt disease given our current limited understanding of disease penetrance.
Why is the genetic test result useful and who benefits?
A genetic test may have implications for the management of the individual tested, or their family members. Historically the main beneficiaries have been family members who can be stratified for ongoing surveillance, or reassured and confidently discharged, on the basis of predictive testing and this remains the major benefit of testing today. While only a proportion of test results have direct implications for management of the proband, this is likely to become increasingly informative.
Benefits for the patient
For the DCM patient, the diagnosis is largely made based on clinical and imaging data. However, in a limited number of patients, genetic testing may assist in confirming the diagnosis when there is diagnostic uncertainty. For example, if there is a dual pathology under consideration such as sarcoid disease, the presence of an alternate genetic diagnosis can inform a decision to desist with more extensive and lower yield diagnostic testing (e.g. endomyocardial biopsy for isolated cardiac sarcoid).26 We would recommend this application of genetic testing in diagnostic uncertainty be limited to expert centres with multidisciplinary input, given the challenges in interpreting genetic variants in the absence of a confirmed phenotypic diagnosis.
There is great promise for the results of genetic testing to inform precision medicine through risk stratification and targeted therapies. At present, however, in DCM, there are only a few genes in which identified pathogenic variants may change management, reviewed in later sections. The most notable is LMNA (lamin), where variants are associated with high rates of conduction disease, atrial and ventricular arrhythmias, and sudden cardiac death. This may lower the threshold for primary prevention implantable cardioverter-defibrillator (ICD) implantation, and encourage enhanced rhythm surveillance.
With improved genotype-phenotype correlations studied in larger multicentre cohorts with long-term follow-up data, we expect the list of genes that will lead to a change in patient management to evolve, and these are discussed in more detail below.
Benefits for the family and cascade screening
The main benefit of cascade genetic screening is identification of family members not at risk who can be safely discharged from ongoing surveillance as previously described. Cascade genetic screening can also identify asymptomatic affected family members and pre-symptomatic unaffected carriers of pathogenic variants that were identified in the proband. It is important that family members are appropriately counselled prior to undertaking genetic testing particularly with regard to reduced penetrance and variable expressivity, meaning that even if they carry a pathogenic variant, they may never develop the phenotype and if they do, the severity of that phenotype may vary. Unaffected carriers should be counselled regarding symptoms and signs of incipient DCM and they will undergo more regular clinical surveillance—every 1–3 years according to the European Society of Cardiology (ESC) guidelines27 or every 3–5 years according to the American Heart Association guidelines.28 Future remote monitoring devices able to exclude or detect a DCM phenotype early based upon machine learning analysis of combined clinical and electrocardiographic characteristics, used by the generalist or the family members themselves, may reduce this burden of cardiological surveillance.
Using genetic information to guide reproduction options
Providing information regarding recurrence or transmission risk of DCM is an important benefit of genetic testing for both the proband and their family members. The identification of a disease-causing variant in a prospective parent may also be used to inform prenatal genetics, such as pre-implantation genetic diagnosis, which involves in vitro fertilization with embryo selection or editing.29 , 30 This is an invasive process with high attrition at each stage, requiring oocyte retrieval, in vitro fertilization, genetic testing of embryos, and implantation of pathogenic variant negative embryos. All patients with a robust diagnosis with a clear pathogenic/likely pathogenic variant should be aware of this option. Chorionic villus sampling and amniocentesis are other examples of prenatal genetic diagnosis methodologies. With all of these options, it is vital patients have adequate pre-test genetic counselling, to ensure they have sufficient information and support to make an informed decision. This includes being able to understand the potential implications of test results, exploring the decision-making in response to the results, and supporting communication to other at-risk relatives.
Which dilated cardiomyopathy patient should I refer for genetic testing?
What do the guidelines say?
At present, there is no consensus recommendation for genetic testing of all patients with DCM. The general principles are for genetic testing to be used when it may change management (of the patient or family members), and when it is cost-effective. International guidelines differ and it is worth noting that some have not been updated since pivotal discoveries were made (e.g. recognition of the important contribution of titin-truncating variants in 2012), and sequencing costs have fallen markedly, both leading to evolution of the cost-benefit ratio of genetic testing since some of these guidelines were written.
The American College of Medical Genetics and Genomics (ACMG) recommends genetic testing for patients with cardiomyopathy in two scenarios: (i) for a confirmed affected individual; if in a family then the most clearly affected family member and (ii) cascade testing of at-risk family members for pathogenic and likely pathogenic variants.31 The ACMG recommend genetic testing at the time of a new cardiomyopathy diagnosis, but it can also be conducted any time following diagnosis. Genetic testing can also be thought of as a continual process, with testing being repeated years after an initial negative test, as new genetic discovery comes to light.
In slight contrast, the 2010 ESC position statement on genetic testing in cardiomyopathies states that ‘In most patients with a definite clinical diagnosis, there is no confirmatory role for routine genetic testing. The main role of genetic testing in this context is to provide predictive diagnosis in first-degree relatives’.27 In practice, we interpret this to mean that genetic testing can be performed whenever there are relatives who might benefit from a family diagnosis—e.g. relatives who could be discharged from ongoing follow-up who would otherwise be under clinical surveillance.
The European guidelines do recommend genetic testing for the diagnosis of a particular cardiomyopathy in the presence of features suggesting a specific genetic aetiology that would influence management (e.g. conduction defects in suspicion of LMNA cardiomyopathy).
The US Heart Rhythm Society and the European Heart Rhythm Association issued a consensus statement in 2011 recommending DCM genetic testing only for patients with DCM and significant cardiac conduction disease and/or a family history of premature unexpected sudden death.32 They did not explicitly recommend genetic testing for patients with familial DCM but suggested testing could be useful for these patients to confirm the diagnosis, recognize those at highest risk of arrhythmia, and to facilitate cascade screening.32
Genetic testing is not recommended for the diagnosis of a borderline cardiomyopathy. However, the prevalence and prognostic relevance of pathogenic genetic variants did not differ in isolated LV dysfunction compared with DCM in a recent cohort study,33 suggesting that genetic testing should not depend on the degree of cardiac systolic dysfunction or dilatation.
The reticence to recommend universal genetic testing in DCM stems from a number of factors. The sensitivity for genetic testing in DCM has been variable (25–40% for familial DCM, 10–25% for isolated DCM31 , 33) and there have been limited genetic-prognostic associations that would directly change the management for the proband. However, we propose that genetic testing for DCM has a higher yield than many of the other screening investigations that are recommended for the work-up of a patient with a new diagnosis of DCM (e.g. checking thyroid function tests and ferritin levels). We also propose that genetic testing should be strongly considered in non-familial DCM as 10–25% may have pathogenic variants and that genetic testing should be considered in ‘acquired’ DCM such as exposure to alcohol or pregnancy as these conditions have been demonstrated to have a similar genetic basis to ‘non-acquired’ DCM.7 , 34 Conversely, in patients with a proven genetic basis, other ‘acquired’ triggers, such as auto-immunity, alcohol, and chemotherapy, should be considered. We outline our suggested approach in Figure 3.
Figure 3.

Approach to genetic testing in dilated cardiomyopathy. CMR, cardiac magnetic resonance; ICD, implantable cardioverter-defibrillator; LP, likely pathogenic; P, pathogenic.
How should the patient be counselled before genetic testing?
It is very important that all individuals at risk of a genetic disease undergo genetic counselling, and certainly prior to genetic testing. This can be with a genetic counsellor or other clinician (doctor—cardiologist or geneticist, specialist nurse) with appropriate expertise, in line with local regulations and care systems. Genetic counselling is a communication process and requires both the provision of information as well as psychosocial support as the patient adjusts to their genetic status.35 Genetic counselling will involve taking a detailed medical and family history, discussion of genetic testing, informed consent if genetic testing is performed, result disclosure, and psychosocial support. Key concepts to be discussed include the limitations of genetic testing, including the possibility of a negative test or the identification of a variant of uncertain significance (VUS; when a variant cannot be confidently reported as either pathogenic or benign, explained further in next section). In these scenarios, it does not mean that their disease does not have a genetic aetiology, rather than a monogenic cause has not been identified on current testing of genes known to be linked to DCM. It is important to highlight that the results may change over time as other genes are identified or curated and that testing may need to be repeated. It should also be highlighted that genetic testing will not always change the patient’s management but may be undertaken predominantly to benefit the patient’s relatives in facilitating family screening (so that unaffected relatives who do not carry the pathogenic variant identified in the proband can be discharged from ongoing follow-up). Patients should be made aware of the EU charter of fundamental rights (Lisbon treaty), article 21, which prohibits discrimination on the basis of ‘genetic features’, and relevant national legislation or protections, e.g. pertaining to health insurance, life assurance, etc. Patients should also be aware that their rights may differ in different geographic regions if they travel or relocate and their rights may change in the future. For example, in the USA, under a federal law called the Genetic Information Nondiscrimination Act (GINA), it is illegal for health insurance providers to use genetic information to determine health insurance eligibility or coverage. However, life insurance is not covered under GINA; therefore, life insurance companies can use genetic testing results to determine eligibility and/or cost of life insurance. Therefore, in the USA, it is recommended that unaffected family members obtain life insurance prior to genetic testing.
Side Boxes 1 and 2—How to take a family history and how to draw a pedigree (see end of the article).
Side Box 1 How to take a family history
Always take at least a three-generation family history
Ask about a family history of cardiomyopathy or heart muscle disease
Establish if family members have undergone clinical screening and the outcome of the screening
In affected living family members, ask if they have undergone genetic testing and if the results are available
Ask about a family history of early pacemakers (<55 years old) or heart transplantation
Ask about a family history of sudden cardiac death
Ask about a history of unexplained deaths under the age of 50 years, unexplained accidents or drownings
- If there is a history of sudden death, ask if post-mortem reports are available or if they can be obtained
- Cardiomyopathy specialists will want to (i) see the post-mortem report, (ii) establish whether a specialist cardiac exam was undertaken, and (iii) establish whether DNA has been retained.
Many patients do not understand the difference between heart attack, stroke, and cardiac arrest, so spend some time asking what they mean by each term.
Side Box 2 How to draw a pedigree
Example pedigree:
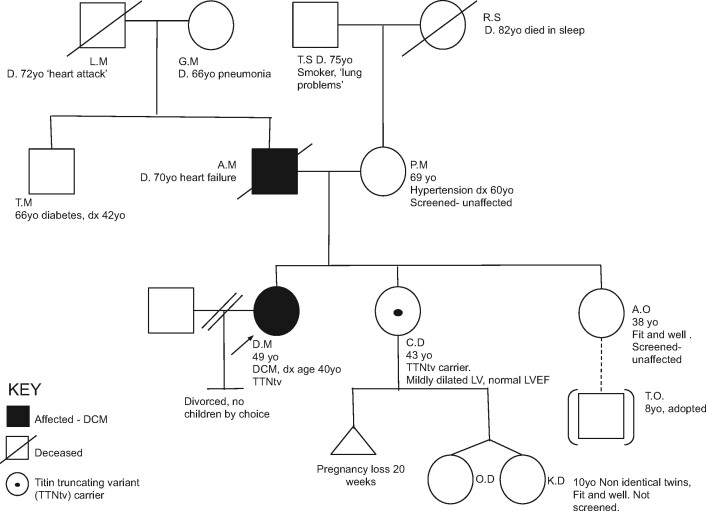
Start with a solid square (male) or circle (female) for the first person with disease who presented to medical attention (the proband). Indicate that they are the proband with an arrow in the lower-left corner.
Draw a key on your pedigree to indicate what the shading means. This is particularly helpful if you are attempting to indicate multiple pathologies.
Add the person’s name/initials, current age or date of birth, disease, and age at disease onset below the symbol.
Then draw the proband’s parents. Consanguinity (reproductive relationship between related individuals) is indicated by a double horizontal line instead of a single horizontal line between parents. Annotate the parents’ names, current age or age at death, and any medical diagnoses.
If an individual is deceased, put a line through their symbol.
Add siblings on the same line as the proband and connect using lines as illustrated. Annotate age and medical information as before.
Continue for children, aunts, uncles, and grandparents for at least a three-generation pedigree.
What are the possible outcomes from genetic testing?
Variant classification and the identification of an informative variant
Genetic variants are classified as pathogenic, likely pathogenic, of uncertain significance, likely benign, or benign.36 Following a genetic test, there are two broad categories of result; an informative variant is identified (pathogenic or likely pathogenic), or no informative variant is identified (no variant, or benign, likely benign, or VUS). Laboratories should also indicate if a VUS is identified that may become informative with further investigation.
Variant classification is based on integration of data from multiple sources including population data (presence of the variant in people with and without disease), segregation data for variants previously found in families, computational annotations, and functional data from experimental systems. A detailed description of variant identification and classification is beyond the scope of this review but we refer the reader to other resources.31 , 36
What does no genetic variant mean?
The absence of an identifiable genetic variant does not necessarily mean that there is no genetic aetiology to the DCM. The possibilities include (i) gene linked to DCM not on tested gene panel—this could occur with recently reported disease genes; (ii) genetic aetiology of monogenic DCM not fully characterized, so gene not yet identified for inclusion on testing panel; or (iii) genuinely not monogenic aetiology of DCM (though polygenic disease may still cluster in families). However, there is emerging evidence from the study of hypertrophic cardiomyopathy that the absence of an identifiable causative variant substantially increases the likelihood that there is no underlying monogenic/Mendelian disease,37 and that ongoing surveillance of family members may not be needed if initial evaluation is reassuring.
How to deal with a variant of uncertain significance?
A variant is defined a VUS when it cannot be confidently reported as either pathogenic or benign. A VUS can arise following the identification of a rare genetic variant in either a gene not known to be linked to DCM or if there is insufficient evidence to assign pathogenicity as per the criteria listed above (<90% chance of pathogenicity). It is also established that genetic testing can be less informative for the interpretation of variants in non-European ancestry populations, due to less complete understanding of background genetic variation in these populations.38
A VUS cannot be used for cascade screening and it is unlikely to change management for the proband. In clinical practice, the diagnostic laboratory should highlight if the variant might be interpretable with additional information and we may try to gain further evidence for possible pathogenicity including whether it segregates with disease in affected family members.
A VUS may be redefined as pathogenic or likely pathogenic in light of such segregation data or following the publication of reports confirming pathogenicity of that variant in another population/family. As such patients should be counselled about this possibility. Publicly available datasets can be used to revisit pathogenicity such as ClinVar (a free resource of variant-phenotype information, to enable ongoing re-evaluation of variants),39 , 40 population datasets such as gnomAD,41 or gene-specific resources, e.g. atlas of cardiac genetic variation (https://www.cardiodb.org/acgv/; last accessed 18 May 2021). As a community, we encourage data sharing of these rare variants and phenotypes so that we may better understand how to interpret genetic variation in cardiomyopathy genes, though we recognize that uncertainties around data protection have hindered routine sharing of these data.42
Genotype-phenotype associations and implications for clinical practice
As outlined, there are a limited number of genes in DCM that are informative for the direct management of the proband. We focus on lamin in this main text but refer the reader to the Supplementary material online, which provide a comprehensive review of titin, other sarcomere-encoding genes, and genes associated with arrhythmogenic cardiomyopathies. We highlight the genes in which variants are most likely to be encountered by a practising cardiologist looking after patients with DCM and genes in which identified variants may change clinical care of patients with DCM.
LMNA variants
The lamin A/C gene (LMNA) encodes the nuclear envelope proteins lamin A and lamin C. In addition to DCM, variants in LMNA cause a diverse range of phenotypes including Emery-Dreifuss muscular dystrophy, limb-girdle muscular dystrophy, lipodystrophy, progeria, and restrictive dermopathy.
LMNA protein-altering variants are found in ∼4–6% of patients with DCM.12 , 43–46
LMNA-associated DCM is frequently associated with conduction disease or atrial and ventricular arrhythmias and is often associated with skeletal involvement, early-onset cardiomyopathy and a higher risk of sudden cardiac death.47–49 LMNA-associated DCM is also typically highly penetrant, with development of the phenotype between 20 and 39 years of age in two-thirds of cases and complete penetrance by 60 years.46 , 48
LMNA variant carriers have a poor prognosis compared with the broader cohort of DCM patients, because of a high rate of progression to malignant arrhythmias (∼20% over 5 years50 , 51) and pump failure (∼19% heart transplantation, ∼8% mortality over 8 years45). The clinician should be aware of the risk factors that are associated with a higher risk of adverse outcome in LMNA-associated DCM. In a landmark multicentre cohort of 269 LMNA variant carriers, non-sustained ventricular tachycardia, LVEF <45%, male sex, and non-missense variants, were independent and cumulative risk factors for malignant ventricular arrythmias.49 Separating predictors of adverse arrhythmic and non-arrhythmic outcomes, in a study of 122 LMNA variant carriers, male sex, non-missense variants, and LV dysfunction at index presentation were associated with the development of ventricular arrhythmias, whereas LV dysfunction at presentation was associated with end-stage heart failure or death.51 Index presentation LVEF is a key predictor of end-stage heart failure.45 Patients should be referred to a specialized heart failure centre once clinical heart failure or recurrent ventricular tachycardia are present.
The key implication for clinical care in light of these adverse outcomes is a lower threshold for implantation of an ICD. Guidelines recommend that an ICD is considered in patients with LMNA-associated DCM where a pacemaker is indicated.32 As yet, there is no absolute recommendation for a primary prevention ICD in LMNA-associated DCM patients without an indication for pacing or a conventional ICD indication. However, in a multicentre cohort of 589 LMNA variant carriers, a risk score has been developed and validated for the prediction of life-threatening ventricular arrhythmias (https://lmna-risk-vta.fr).50 Predictor variables were male sex, non-missense LMNA variant, first-degree and higher atrioventricular block, non-sustained ventricular tachycardia, and reduced LVEF. A 5-year estimated risk threshold ≥7% predicted 96.2% of ventricular arrhythmias and led to the net reclassification of 29% of patients with ventricular arrhythmias compared with guideline-based management. These data support broader criteria for placement of a primary prevention ICD in LMNA variant carriers, though there is no randomized controlled trial evidence to support this recommendation. At present, according to the 2019 Heart Rhythm Society recommendations, in individuals with lamin A/C arrhythmogenic cardiomyopathy and two or more risk factors (LVEF <45%, non-sustained ventricular tachycardia, or male sex) an ICD is reasonable (class IIa recommendation).52 Due to the high rates of heart block and need for pacing, a transvenous ICD, not a subcutaneous device, is recommended for patients with LMNA cardiomyopathy.
The adverse prognosis also has implications for the management of asymptomatic variant carriers. In a Norwegian series of LMNA carriers, asymptomatic gene-positive family members had a 9% annual incidence of a newly documented cardiac phenotype and a 61% cardiac penetrance during 4.4 years of follow-up.45
As yet, gene-specific targeted therapies for LMNA-associated DCM are lacking, but a Phase 3 randomized double-blind clinical trial is currently underway evaluating ARRY-371797, an oral, selective p38 mitogen-activated protein kinase inhibitor (ClinicalTrials.gov Identifier: NCT03439514). This has been shown to prevent LV dilatation and deterioration in a mouse model of LMNA cardiomyopathy53 and was associated with improved functional capacity in patients in a Phase 2 clinical study.54
Areas for future research
The last 10 years have seen strong progress in the characterization and refinement of the genetic basis of DCM. Harnessing this knowledge to provide targeted therapies should be the goal for the next 10 years.
Understand biological basis of disease and identify therapeutic targets
Discovery of disease associated molecular pathways and targeted molecular therapies, such as the p53-mediated fibrosis in lamin A/C DCM, will be facilitated through RNA and protein profiling of cardiac samples, preferably of diagnostic and not end-stage ventricular samples. Titin-truncating variant (TTNtv) in DCM is characterized by pronounced alterations in mitochondrial energetics, with up-regulation of all components of the metabolic mitochondrial electron transport chain in human and experimental TTNtv hearts55 , 56 and this opens the path for metabolic interventions.
Improving diagnostic yield
With regard to genetic testing itself, there is potential for the refinement of potential DCM disease-gene associations to improve diagnosis and cascade screening. New gene discovery will increase the sensitivity of genetic testing in DCM but as previously outlined, the incremental gain in Mendelian gene discovery is likely to be limited for DCM. In our opinion, the greatest opportunity is likely through investment to improve variant interpretation—that is to improve discrimination of pathogenic variants from rare but benign bystanders. This is likely to arise from a combination of in silico methods and high-throughput in vitro functional perturbation assays, though defining high-throughput assay endpoints for many DCM genes is far from trivial.
Many DCM genes show variable penetrance but we still need to better define penetrance for many of these genes in order to be able to counsel gene carriers effectively. Understanding the reasons for the variable penetrance will also be a major focus of future research. One of the most promising avenues will be exploring the collective additive or interactive contributions of common variants with individually smaller effects as contributors to polygenic disease and modifiers of penetrance and expressivity. Polygenic risk scores will capture some of the common variant contribution to disease risk and may improve our ability to discriminate which family members will manifest disease.57 , 58 Polygenic risk scores for incident disease as well as risk stratification/outcomes have been developed for many common diseases59 , 60 and often the risk of disease cannot be predicted from conventional (non-genetic) risk factors; the same principles may apply to cardiomyopathy.
Risk and therapeutic stratification
Precision risk stratification and therapeutic stratification to improve clinical outcomes will also be a key focus of future research. More pressingly, both rare variants and genomic risk scores need to be placed in the context of conventional DCM risk markers such as age, New York Heart Association class, biomarkers, and phenotypic variables including LVEF and mid-wall fibrosis to provide multi-modality risk stratification. Regarding therapeutic stratification, much of the evidence base surrounding device implantation in genetic DCM comes from registry and observational data. We would welcome the development of trials stratified by genetic status to evaluate medical and device therapy further.
Targeted therapies
In the last decade, several strategies have been developed to remove, correct, or silence genetic defects, including genome editing, exon skipping, allele-specific silencing, spliceosome-mediated RNA trans-splicing, and gene replacement. Most of these technologies have already been tested for efficacy and efficiency in animal- or human-induced pluripotent stem cell models of hypertrophic cardiomyopathy, DCM or other cardiomyopathy with promising results61–63 and some have been tested in humans for other conditions.64 For example, nonsense variants in LMNA are anticipated to cause disease through haploinsufficiency. Therapies that target read-through of the premature stop codon may allow normal expression and cellular function. Application of genetic therapy will require extensive efficacy and safety studies.
Conclusion
We have outlined the genetic basis of DCM as relevant to clinical practice, highlighting genes which are informative for management and how genetic testing can underpin evaluation of family members, and increasingly inform the management of affected individuals. We have also addressed some of the challenges in interpreting genetic testing in DCM patients which we hope will be useful for the practicing clinician. The landscape of genetic DCM is continuously evolving and we expect the next 10 years to be characterized by greater understanding of disease beyond the monogenic model, refinement of variant interpretation, further target identification, and the application of genetic biomarkers to patient stratification for improved outcomes in DCM.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This work was supported by the National Heart Lung Institute, Imperial College London; the Wellcome Trust [107469/Z/15/Z]; Medical Research Council (UK); NIHR Royal Brompton Biomedical Research Unit; and the NIHR Imperial Biomedical Research Centre. S.K.P. is supported by the Rosetrees Trust, Alexander Jansons Foundation, and CORDA. J.S.W. acknowledges additional support from the British Heart Foundation. S.H. acknowledges support from the Netherlands Cardiovascular Research Initiative, an initiative with support of the Dutch Heart Foundation, CVON Early HFPEF, 2015-10, She-PREDICTS, 2017-21, Arena-PRIME, 2017-18.
Conflict of interest: S.H. reports personal fees from Astra Zeneca, Cell Prothera, and Merck, grants from Dutch Heart Foundation, outside the submitted work. N.K.L. reports personal fees from MyoKardia Inc and Array BioPharma, outside the submitted work. J.S.W. reports grants and non-financial support from the Wellcome Trust [107469/Z/15/Z], Medical Research Council (UK), British Heart Foundation, NIHR Royal Brompton Cardiovascular Biomedical Research Unit, and the NIHR Imperial College Biomedical Research Centre, during the conduct of the study; grants and personal fees from Myokardia, outside the submitted work. The remaining authors have nothing to disclose.
Supplementary Material
Contributor Information
Upasana Tayal, National Heart Lung Institute, Imperial College London, UK; Cardiovascular Research Centre, Royal Brompton & Harefield Hospitals, London, UK.
James S Ware, National Heart Lung Institute, Imperial College London, UK; Cardiovascular Research Centre, Royal Brompton & Harefield Hospitals, London, UK; MRC London Institute of Medical Sciences, London, UK.
Neal K Lakdawala, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA.
Stephane Heymans, Department of Cardiology, CARIM School for Cardiovascular Diseases Faculty of Health, Medicine and Life Sciences, Maastricht University, The Netherlands; Department of Cardiovascular Sciences, Centre for Molecular and Vascular Biology, Leuven, KU, Belgium; The Netherlands Heart Institute, Nl-HI, Utrecht, The Netherlands.
Sanjay K Prasad, National Heart Lung Institute, Imperial College London, UK; Cardiovascular Research Centre, Royal Brompton & Harefield Hospitals, London, UK.
References
- 1. Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M, Duboc D, Gimeno J, de Groote P, Imazio M, Heymans S, Klingel K, Komajda M, Limongelli G, Linhart A, Mogensen J, Moon J, Pieper PG, Seferovic PM, Schueler S, Zamorano JL, Caforio ALP, Charron P. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 2016;37:1850–1858. [DOI] [PubMed] [Google Scholar]
- 2. Codd MB, Sugrue DD, Gersh BJ, Melton LJ 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975-1984. Circulation 1989;80:564–572. [DOI] [PubMed] [Google Scholar]
- 3. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013;10:531–547. [DOI] [PubMed] [Google Scholar]
- 4. Petretta M, Pirozzi F, Sasso L, Paglia A, Bonaduce D. Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am J Cardiol 2011;108:1171–1176. [DOI] [PubMed] [Google Scholar]
- 5. Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Müller S, Kayvanpour E, Vogel B, Sedaghat-Hamedani F, Lim W-K, Zhao X, Fradkin D, Köhler D, Fischer S, Franke J, Marquart S, Barb I, Li DT, Amr A, Ehlermann P, Mereles D, Weis T, Hassel S, Kremer A, King V, Wirsz E, Isnard R, Komajda M, Serio A, Grasso M, Syrris P, Wicks E, Plagnol V, Lopes L, Gadgaard T, Eiskjær H, Jørgensen M, Garcia-Giustiniani D, Ortiz-Genga M, Crespo-Leiro MG, Deprez RHLD, Christiaans I, van Rijsingen IA, Wilde AA, Waldenstrom A, Bolognesi M, Bellazzi R, Mörner S, Bermejo JL, Monserrat L, Villard E, Mogensen J, Pinto YM, Charron P, Elliott P, Arbustini E, Katus HA, Meder B. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J 2015;36:1123–1135a. [DOI] [PubMed] [Google Scholar]
- 6. Jansweijer JA, Spaendonck-Zwarts KV, Tanck MWT, Tintelen JV, Christiaans I, Smagt JVD, Vermeer A, Martijn Bos J, Moss AJ, Swan H, Priori S, Rydberg A, Tfelt-Hansen J, Ackerman M, Olivotto I, Charron P, Gimeno JR, Berg MVD, Wilde A, Pinto YM. Heritability in genetic heart disease: the role of genetic background. Open Heart 2019;6:e000929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ware JS, Amor-Salamanca A, Tayal U, Govind R, Serrano I, Salazar-Mendiguchía J, García-Pinilla JM, Pascual-Figal DA, Nuñez J, Guzzo-Merello G, Gonzalez-Vioque E, Bardaji A, Manito N, López-Garrido MA, Padron-Barthe L, Edwards E, Whiffin N, Walsh R, Buchan RJ, Midwinter W, Wilk A, Prasad S, Pantazis A, Baski J, O’Regan DP, Alonso-Pulpon L, Cook SA, Lara-Pezzi E, Barton PJ, Garcia-Pavia P. Genetic etiology for alcohol-induced cardiac toxicity. J Am Coll Cardiol 2018;71:2293–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ware SM. Genetics of paediatric cardiomyopathies. Curr Opin Pediatr 2017;29:534–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kindel SJ, Miller EM, Gupta R, Cripe LH, Hinton RB, Spicer RL, Towbin JA, Ware SM. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail 2012;18:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choudhry S, Puri K, Denfield SW. An update on pediatric cardiomyopathy. Curr Treat Options Cardiovasc Med 2019;21:36. [DOI] [PubMed] [Google Scholar]
- 11. Mestroni L, Brun F, Spezzacatene A, Sinagra G, Taylor MRG. Genetic causes of dilated cardiomyopathy. Prog Pediatr Cardiol 2014;37:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hershberger RE, Morales A, Dilated cardiomyopathy overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds. GeneReviews®. Seattle, WA: University of Washington; 2007. pp 1993–2021. [Google Scholar]
- 13. Tayal U, Prasad S, Cook SA. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med 2017;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, Govind R, Mazaika E, de Marvao A, Dawes TJW, Felkin LE, Ahmad M, Theotokis PI, Edwards E, Ing AY, Thomson KL, Chan LLH, Sim D, Baksi AJ, Pantazis A, Roberts AM, Watkins H, Funke B, O'Regan DP, Olivotto I, Barton PJR, Prasad SK, Cook SA, Ware JS, Walsh R. Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation 2020;141:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, Celeghin R, Edwards M, Fan J, Ingles J. An evidence-based assessment of genes in dilated cardiomyopathy. medRxiv 2020. doi: 10.1101/2020.12.10.20247197. [DOI] [PMC free article] [PubMed]
- 16. Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, Crowley SB, Dougherty K, Harrison SM, McGlaughon J, Milko LV, Morales A, Seifert BA, Strande N, Thomson K, Peter van Tintelen J, Wallace K, Walsh R, Wells Q, Whiffin N, Witkowski L, Semsarian C, Ware JS, Hershberger RE, Funke B. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med 2019;12:e002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thomson KL, Ormondroyd E, Harper AR, Dent T, McGuire K, Baksi J, Blair E, Brennan P, Buchan R, Bueser T, Campbell C, Carr-White G, Cook S, Daniels M, Deevi SVV, Goodship J, Hayesmoore JBG, Henderson A, Lamb T, Prasad S, Rayner-Matthews P, Robert L, Sneddon L, Stark H, Walsh R, Ware JS, Farrall M, Watkins HC, NIHR BioResource – Rare Diseases Consortium. Analysis of 51 proposed hypertrophic cardiomyopathy genes from genome sequencing data in sarcomere negative cases has negligible diagnostic yield. Genet Med 2019;21:1576–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alamo L, Ware JS, Pinto A, Gillilan RE, Seidman JG, Seidman CE, Padrón R. Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife 2017;6:e24634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deacon DC, Happe CL, Chen C, Tedeschi N, Manso AM, Li T, Dalton ND, Peng Q, Farah EN, Gu Y, Tenerelli KP, Tran VD, Chen J, Peterson KL, Schork NJ, Adler ED, Engler AJ, Ross RS, Chi NC. Combinatorial interactions of genetic variants in human cardiomyopathy. Nat Biomed Eng 2019;3:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roncarati R, Viviani Anselmi C, Krawitz P, Lattanzi G, Kodolitsch YV, Perrot A, Pasquale ED, Papa L, Portararo P, Columbaro M, Forni A, Faggian G, Condorelli G, Robinson PN. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur J Hum Genet 2013;21:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hazebroek MR, Moors S, Dennert R, Wijngaard A V D, Krapels I, Hoos M, Verdonschot J, Merken JJ, Vries BD, Wolffs PF, Crijns HJGM, Brunner-La Rocca H-P, Heymans S. Prognostic relevance of gene-environment interactions in patients with dilated cardiomyopathy: applying the MOGE(S) classification. J Am Coll Cardiol 2015;66:1313–1323. [DOI] [PubMed] [Google Scholar]
- 22. Haas J, Mester S, Lai A, Frese KS, Sedaghat‐Hamedani F, Kayvanpour E, Rausch T, Nietsch R, Boeckel J, Carstensen A, Völkers M, Dietrich C, Pils D, Amr A, Holzer DB, Bordalo DM, Oehler D, Weis T, Mereles D, Buss S, Riechert E, Wirsz E, Wuerstle M, Korbel JO, Keller A, Katus HA, Posch AE, Meder B. Genomic structural variations lead to dysregulation of important coding and non‐coding RNA species in dilated cardiomyopathy. EMBO Mol Med 2018;10:107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Minoche AE, Horvat C, Johnson R, Gayevskiy V, Morton SU, Drew AP, Woo K, Statham AL, Lundie B, Bagnall RD, Ingles J, Semsarian C, Seidman JG, Seidman CE, Dinger ME, Cowley MJ, Fatkin D. Genome sequencing as a first-line genetic test in familial dilated cardiomyopathy. Genet Med 2019;21:650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nair NU, Das A, Amit U, Robinson W, Park SG, Basu M, Lugo A, Leor J, Ruppin E, Hannenhalli S. Putative functional genes in idiopathic dilated cardiomyopathy. Sci Rep 2018;8:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Evans JP, Skrzynia C, Burke W. The complexities of predictive genetic testing. BMJ 2001;322:1052–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lakdawala NK, Funke BH, Baxter S, Cirino AL, Roberts AE, Judge DP, Johnson N, Mendelsohn NJ, Morel C, Care M, Chung WK, Jones C, Psychogios A, Duffy E, Rehm HL, White E, Seidman JG, Seidman CE, Ho CY. Genetic testing for dilated cardiomyopathy in clinical practice. J Card Fail 2012;18:296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, Helio T, Keren A, McKenna WJ, Monserrat L, Pankuweit S, Perrot A, Rapezzi C, Ristic A, Seggewiss H, van Langen I, Tavazzi L, European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2010;31:2715–2726. [DOI] [PubMed] [Google Scholar]
- 28. Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, Fonarow GC, Francis GS, Lenihan D, Lewis EF, McNamara DM, Pahl E, Vasan RS, Ramasubbu K, Rasmusson K, Towbin JA, Yancy C, American Heart Association Committee on Heart Failure and Transplantation of the Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Epidemiology and Prevention; and Council on Quality of Care and Outcomes Research Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 2016;134:e579–e646. [DOI] [PubMed] [Google Scholar]
- 29. Baxi DS, Kuliev A, Pakhalchuk T, Prokhorovich M, Rechitsky S. Preimplantation genetic diagnosis (PGD) for borderline indications including human leukocyte antigens (HLA) matching, cancer predisposition, cardiac disease and their proportion in overall PGD cases for single gene disorders. Fertil Steril 2017;108:e268–e269. [Google Scholar]
- 30. Kuliev A, Pomerantseva E, Polling D, Verlinsky O, Rechitsky S. PGD for inherited cardiac diseases. Reprod Biomed Online 2012;24:443–453. [DOI] [PubMed] [Google Scholar]
- 31. Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, Morales A, Taylor MRG, Vatta M, Ware SM, ACMG Professional Practice and Guidelines Committee. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2018;20:899–909. [DOI] [PubMed] [Google Scholar]
- 32. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze-Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP, Heart Rhythm Society (HRS), European Heart Rhythm Association (EHRA). HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011;13:1077–1109. [DOI] [PubMed] [Google Scholar]
- 33. Hazebroek MR, Krapels I, Verdonschot J, van den Wijngaard A, Vanhoutte E, Hoos M, Snijders L, van Montfort L, Witjens M, Dennert R, Crijns HJGM, Brunner-La Rocca H-P, Brunner HG, Heymans S. Prevalence of pathogenic gene mutations and prognosis do not differ in isolated left ventricular dysfunction compared with dilated cardiomyopathy. Circ Heart Fail 2018;11:e004682. [DOI] [PubMed] [Google Scholar]
- 34. Ware JS, Li J, Mazaika E, Yasso CM, DeSouza T, Cappola TP, Tsai EJ, Hilfiker-Kleiner D, Kamiya CA, Mazzarotto F, Cook SA, Halder I, Prasad SK, Pisarcik J, Hanley-Yanez K, Alharethi R, Damp J, Hsich E, Elkayam U, Sheppard R, Kealey A, Alexis J, Ramani G, Safirstein J, Boehmer J, Pauly DF, Wittstein IS, Thohan V, Zucker MJ, Liu P, Gorcsan J, McNamara DM, Seidman CE, Seidman JG, Arany Z, IMAC-2 and IPAC Investigators. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med 2016;374:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Skirton H, Cordier C, Ingvoldstad C, Taris N, Benjamin C. The role of the genetic counsellor: a systematic review of research evidence. Eur J Hum Genet 2015;23:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Walsh R, Buchan R, Wilk A, John S, Felkin LE, Thomson KL, Chiaw TH, Loong CCW, Pua CJ, Raphael C, Prasad S, Barton PJ, Funke B, Watkins H, Ware JS, Cook SA. Defining the genetic architecture of hypertrophic cardiomyopathy: re-evaluating the role of non-sarcomeric genes. Eur Heart J 2017;38:3461–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Landry LG, Rehm HL. Association of racial/ethnic categories with the ability of genetic tests to detect a cause of cardiomyopathy. JAMA Cardiol 2018;3:341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Neale BM, Daly MJ, MacArthur DG, Genome Aggregation Database Consortium. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wright CF, Ware JS, Lucassen AM, Hall A, Middleton A, Rahman N, Ellard S, Firth HV. Genomic variant sharing: a position statement. Wellcome Open Res 2019;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J 2008;156:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, Minikel EV, MacArthur DG, Farrall M, Cook SA, Watkins H, Exome Aggregation Consortium. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med 2017;19:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hasselberg NE, Haland TF, Saberniak J, Brekke PH, Berge KE, Leren TP, Edvardsen T, Haugaa KH. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur Heart J 2018;39:853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Crasto S, My I, Di PE. The broad spectrum of LMNA cardiac diseases: from molecular mechanisms to clinical phenotype. Front Physiol 2020;11:761. doi: 10.3389/fphys.2020.00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Müehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 1999;341:1715–1724. [DOI] [PubMed] [Google Scholar]
- 48. Arbustini E, Pilotto A, Repetto A, Grasso M, Negri A, Diegoli M, Campana C, Scelsi L, Baldini E, Gavazzi A, Tavazzi L. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J Am Coll Cardiol 2002;39:981–990. [DOI] [PubMed] [Google Scholar]
- 49. van Rijsingen IAW, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Pilotto A, Pasotti M, Jenkins S, Rowland C, Aslam U, Wilde AAM, Perrot A, Pankuweit S, Zwinderman AH, Charron P, Pinto YM. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers: a European cohort study. J Am Coll Cardiol 2012;59:493–500. [DOI] [PubMed] [Google Scholar]
- 50. Wahbi K, Ben Yaou R, Gandjbakhch E, Anselme F, Gossios T, Lakdawala NK, Stalens C, Sacher F, Babuty D, Trochu J-N, Moubarak G, Savvatis K, Porcher R, Laforêt P, Fayssoil A, Marijon E, Stojkovic T, Béhin A, Leonard-Louis S, Sole G, Labombarda F, Richard P, Metay C, Quijano-Roy S, Dabaj I, Klug D, Vantyghem M-C, Chevalier P, Ambrosi P, Salort E, Sadoul N, Waintraub X, Chikhaoui K, Mabo P, Combes N, Maury P, Sellal J-M, Tedrow UB, Kalman JM, Vohra J, Androulakis AFA, Zeppenfeld K, Thompson T, Barnerias C, Bécane H-M, Bieth E, Boccara F, Bonnet D, Bouhour F, Boulé S, Brehin A-C, Chapon F, Cintas P, Cuisset J-M, Davy J-M, De Sandre-Giovannoli A, Demurger F, Desguerre I, Dieterich K, Durigneux J, Echaniz-Laguna A, Eschalier R, Ferreiro A, Ferrer X, Francannet C, Fradin M, Gaborit B, Gay A, Hagège A, Isapof A, Jeru I, Juntas Morales R, Lagrue E, Lamblin N, Lascols O, Laugel V, Lazarus A, Leturcq F, Levy N, Magot A, Manel V, Martins R, Mayer M, Mercier S, Meune C, Michaud M, Minot-Myhié M-C, Muchir A, Nadaj-Pakleza A, Péréon Y, Petiot P, Petit F, Praline J, Rollin A, Sabouraud P, Sarret C, Schaeffer S, Taithe F, Tard C, Tiffreau V, Toutain A, Vatier C, Walther-Louvier U, Eymard B, Charron P, Vigouroux C, Bonne G, Kumar S, Elliott P, Duboc D. Development and validation of a new risk prediction score for life-threatening ventricular tachyarrhythmias in laminopathies. Circulation 2019;140:293–302. [DOI] [PubMed] [Google Scholar]
- 51. Kumar S, Baldinger SH, Gandjbakhch E, Maury P, Sellal J-M, Androulakis AFA, Waintraub X, Charron P, Rollin A, Richard P, Stevenson WG, Macintyre CJ, Ho CY, Thompson T, Vohra JK, Kalman JM, Zeppenfeld K, Sacher F, Tedrow UB, Lakdawala NK. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J Am Coll Cardiol 2016;68:2299–2307. [DOI] [PubMed] [Google Scholar]
- 52. Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, Chillou C. D, DePasquale EC, Desai MY, Estes NAM 3rd, Hua W, Indik JH, Ingles J, James CA, John RM, Judge DP, Keegan R, Krahn AD, Link MS, Marcus FI, McLeod CJ, Mestroni L, Priori SG, Saffitz JE, Sanatani S, Shimizu W, Peter van Tintelen J, Wilde AAM, Zareba W. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019;16:e301–e372. [DOI] [PubMed] [Google Scholar]
- 53. Muchir A, Wu W, Choi JC, Iwata S, Morrow J, Homma S, Worman HJ. Abnormal p38 mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum Mol Genet 2012;21:4325–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. MacRae C, Taylor MRG, Mestroni L, Moses JR, Ashley EA, Wheeler MT, Lakdawala NK, Hershberger RE, Ptaszynski M, Sandor V. Phase 2 study of a797, an oral, selective p38 mitogen-activated protein kinase inhibitor, in patients with lamin a/c-related dilated cardiomyopathy. Eur Heart J 2016;37:P4981. [Google Scholar]
- 55. Verdonschot JAJ, Hazebroek MR, Derks KWJ, Barandiarán Aizpurua A, Merken JJ, Wang P, Bierau J, van den Wijngaard A, Schalla SM, Abdul Hamid MA, van Bilsen M, van Empel VPM, Knackstedt C, Brunner-La Rocca H-P, Brunner HG, Krapels IPC, Heymans SRB. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur Heart J 2018;39:864–873. [DOI] [PubMed] [Google Scholar]
- 56. Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, Rackham OJL, van Heesch S, Pua CJ, Kui M, Walsh R, Tayal U, Prasad SK, Dawes TJW, Ko NSJ, Sim D, Chan LLH, Chin CWL, Mazzarotto F, Barton PJ, Kreuchwig F, de Kleijn DPV, Totman T, Biffi C, Tee N, Rueckert D, Schneider V, Faber A, Regitz-Zagrosek V, Seidman JG, Seidman CE, Linke WA, Kovalik J-P, O'Regan D, Ware JS, Hubner N, Cook SA. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet 2017;49:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Harper AR, Goel A, Grace C, Thomson KL, Petersen SE, Xu X, Waring A, Ormondroyd E, Kramer CM, Ho CY, Neubauer S, Tadros R, Ware JS, Bezzina CR, Farrall M, Watkins H, HCMR Investigators. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet 2021;53:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tadros R, Francis C, Xu X, Vermeer AMC, Harper AR, Huurman R, Kelu Bisabu K, Walsh R, Hoorntje ET, Te Rijdt WP, Buchan RJ, van Velzen HG, van Slegtenhorst MA, Vermeulen JM, Offerhaus JA, Bai W, de Marvao A, Lahrouchi N, Beekman L, Karper JC, Veldink JH, Kayvanpour E, Pantazis A, Baksi AJ, Whiffin N, Mazzarotto F, Sloane G, Suzuki H, Schneider-Luftman D, Elliott P, Richard P, Ader F, Villard E, Lichtner P, Meitinger T, Tanck MWT, van Tintelen JP, Thain A, McCarty D, Hegele RA, Roberts JD, Amyot J, Dubé M-P, Cadrin-Tourigny J, Giraldeau G, L'Allier PL, Garceau P, Tardif J-C, Boekholdt SM, Lumbers RT, Asselbergs FW, Barton PJR, Cook SA, Prasad SK, O'Regan DP, van der Velden J, Verweij KJH, Talajic M, Lettre G, Pinto YM, Meder B, Charron P, de Boer RA, Christiaans I, Michels M, Wilde AAM, Watkins H, Matthews PM, Ware JS, Bezzina CR. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet 2021;53:128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rao AS, Knowles JW. Polygenic risk scores in coronary artery disease. Curr Opin Cardiol 2019;34:435–440. [DOI] [PubMed] [Google Scholar]
- 60. Khera AV, Chaffin M, Wade KH, Zahid S, Brancale J, Xia R, Distefano M, Senol-Cosar O, Haas ME, Bick A, Aragam KG, Lander ES, Smith GD, Mason-Suares H, Fornage M, Lebo M, Timpson NJ, Kaplan LM, Kathiresan S. Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell 2019;177:587–596.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bondue A, Arbustini E, Bianco A, Ciccarelli M, Dawson D, De Rosa M, Hamdani N, Hilfiker-Kleiner D, Meder B, Leite-Moreira AF, Thum T, Tocchetti CG, Varricchi G, Van der Velden J, Walsh R, Heymans S. Complex roads from genotype to phenotype in dilated cardiomyopathy: scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc Res 2018;114:1287–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Prondzynski M, Mearini G, Carrier L. Gene therapy strategies in the treatment of hypertrophic cardiomyopathy. Pflugers Arch 2019;471:807–815. [DOI] [PubMed] [Google Scholar]
- 63. Zaleta-Rivera K, Dainis A, Ribeiro AJS, Cordero P, Rubio G, Shang C, Liu J, Finsterbach T, Parikh VN, Sutton S, Seo K, Sinha N, Jain N, Huang Y, Hajjar RJ, Kay MA, Szczesna-Cordary D, Pruitt BL, Wheeler MT, Ashley EA. Allele-specific silencing ameliorates restrictive cardiomyopathy attributable to a human myosin regulatory light chain mutation. Circulation 2019;140:765–778. [DOI] [PubMed] [Google Scholar]
- 64. Brenner D, Ludolph AC, Weishaupt JH. Gene specific therapies—the next therapeutic milestone in neurology. Neurol Res Pract 2020;2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.