

Abstract
Genome-wide association studies (GWAS) have identified several common genetic variants influencing major depression and general cognitive abilities, but little is known about whether the two share any of their genetic etiology. Here we investigate shared genomic architectures between major depression (MD) and general intelligence (INT) with MiXeR and their overlapping susceptibility loci with conjunctional false discovery rate (conjFDR), which evaluate the level of overlap in genetic variants and improve the power for gene discovery between two phenotypes. We analyzed GWAS data on MD (n=480,359) and INT (n=269,867) to characterize polygenic architecture and identify genetic loci shared between these phenotypes. Despite non-significant genetic correlation (rg=−0.0148, p=0.50), we observed large polygenic overlap and identified 92 loci shared between MD and INT at conjFDR<0.05. Among the shared loci, 69 and 64 are novel for MD and INT, respectively. Our study demonstrates polygenic overlap between these phenotypes with a balanced mixture of effect.
Introduction
Major depressive disorder is a debilitating mental disorder with a lifetime prevalence of 16%1. Major depressive disorder ranks among one of the major contributors to the global burden of disease2, imposing severe suffering as well as large societal costs3. In addition to the characteristic mood symptoms and aberrant thought, cognition and bodily functions, the disorder is associated with both mental and somatic comorbidities4,5 indicating complex underlying mechanisms. However, despite clinical heterogeneity, heritability estimates of major depressive disorder amounts to 35 %6, implying sizeable genetic effects.
Cognitive impairment is a central feature in major depressive disorder, and is a mediator of the impaired social and occupational functioning observed in the disorder7. In addition to being part of the diagnostic criteria of major depressive episodes, cognitive symptoms are also observed during euthymia8, suggesting mechanisms distinct from those of mood symptoms. However, the underlying pathophysiology of the cognitive impairments remains largely unknown, although some neurobiological correlates have been proposed9. Importantly, cognitive genetic effects are indicated, as unaffected relatives of patients with major depressive disorder display cognitive deficits10, and family and twin studies find that the genetic liabilities of major depressive disorder and cognitive abilities covary10,11.
Like most complex conditions, major depressive disorder is a polygenic disorder12,13, and genome-wide association studies (GWAS) have not until recently identified genetic susceptibility loci14. The most recent GWAS of more than n=480,359 participants with an estimated single nucleotide polymorphism (SNP) heritability of 8.7% identified 44 genetic loci associated with major depression (MD)15. General intelligence (INT) is also regarded as a polygenic trait, with estimated SNP heritability of 22%16. It has also been difficult to identify genetic loci associated with INT until the recent well powered GWASs of INT with n=300,48617 and n=269,86716 participants, which identified 148 and 205 genetic loci, respectively. Despite the phenotypic overlap indicating genetic pleiotropy, common genetic mechanisms for INT and major depression remain elusive and recent genetic correlation estimates are low (rg=−0.1448) and nonsignificant16. However, a significant negative correlation (rg=−0.2675, p=6.20×10−10) between depressive symptoms and INT has been reported previously16.
In the present study, we aimed to identify single SNPs shared between major depression and INT. Despite the assembly of very large GWAS cohorts, the identified genome-wide significant loci only explain a small portion of the variance of these complex phenotypes15,16. The lack of power of the traditional GWAS statistical approach can be attributed to the polygenic architecture of complex traits which is largely expected to be accounted for by numerous loci with very small effects18. Shared genetic background is expected for any pair of highly polygenic traits, in line with the omnigenic model19. It is now possible to use the MiXeR model20 to quantify the level of overlap and thus address how large the overlap is between polygenic phenotypes.
To improve the discovery of loci with small effects and to identify genomic loci shared between polygenic traits, we applied the condFDR/conjFDR method. This method employs the entire original set of p-values from the GWAS summary statistics for two traits of interest and intrinsically controls for the type 1 error rate using Benjamini-Hochberg-like FDR correction21. The condFDR/conjFDR method builds on an empirical Bayesian statistical framework and increases the power to detect novel phenotype-specific and shared loci by leveraging the combined power of two GWAS22. Specifically, we condition the false discovery rate (FDR) for one trait on association with another, even in the presence low genetic correlation between the two, to prioritize candidate susceptibility loci for the former22,23. Furthermore, SNPs enriched for cross-trait associations are more likely to replicate24. Like standard GWAS analysis, the condFDR/conjFDR method does not operate on a causal level, but identifies LD proxies of the underlying causal variants, which remain unknown. The method has successfully uncovered shared loci in various complex traits and disorders, even in the absence of genetic correlation25. Here, we used this statistical approach to analyze the recent large GWAS datasets on MD15 and INT16. We aimed to characterize the polygenic architecture shared between INT and major depression, and to discover shared genetic loci with small effects with both the same and opposite directions of effect.
Results
Shared genomic architectures (MiXeR)
MiXeR20 analysis provided evidence of large polygenic overlap between major depression and intelligence. Of 13.9K causal variants linked to MD, 11K (SE=0.4) were shared with intelligence, while the overall measure of polygenic overlap, on 0–100% scale, was 88.3% (as quantified by the Dice coefficient) and 0.2K variants associated with intelligence were not shared with major depression, despite no genetic correlation (Figure 1). MiXeR also estimated a much lower number of SNPs shared between height and MD (1.1K, SE=0.5) and height and intelligence (1.6K, SE=0.2 ), with Dice coefficient 12.2% and 21%, respectively (Supplementary Figures 2 and 3).
Figure 1.
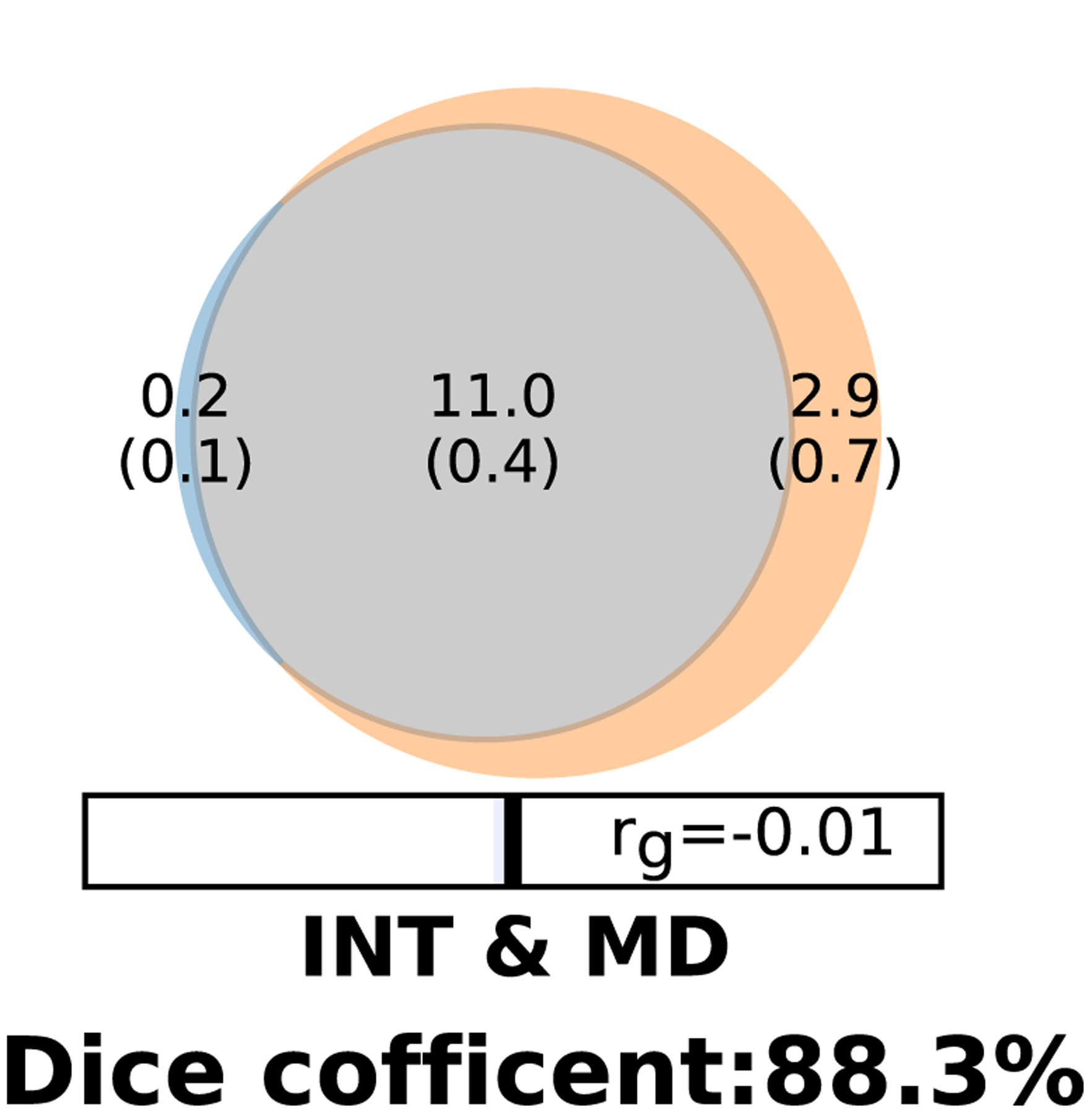
Venn diagram shows the estimated number of causal variants shared (grey) between intelligence (INT) and major depression (MD) and unique (colors) to either of them. The number of causal variants in thousands is shown, with the standard error in thousands provided in parentheses. The Dice coefficient, which is the percentage of shared causal variants between two phenotypes, is 88.3. The genetic correlation (rg) is estimated between the two phenotypes.
Enrichment pattern
To provide a visual pattern of pleiotropic enrichment between the two phenotypes, we generated conditional Q-Q plots26 conditioning INT on MD and vice versa. The conditional Q-Q plots showed a strong enrichment pattern for MD given INT (Figure 2A) and for INT given MD (Figure 2B). The blue line shows association p-values in the main trait of interest (major depression) for all SNPs irrespective of their association p-values in INT. Red, yellow and purple lines show association p-values in the main trait for successively nested subsets of SNPs with increasing significance in the conditional trait. The consistently increasing degree of leftward deflection for subsets of variants with higher significance in the conditional trait in both directions indicates substantial polygenic overlap between major depression and INT (Figure 2A & B).
Figure 2. Conditional Quantile-Quantile Plots.
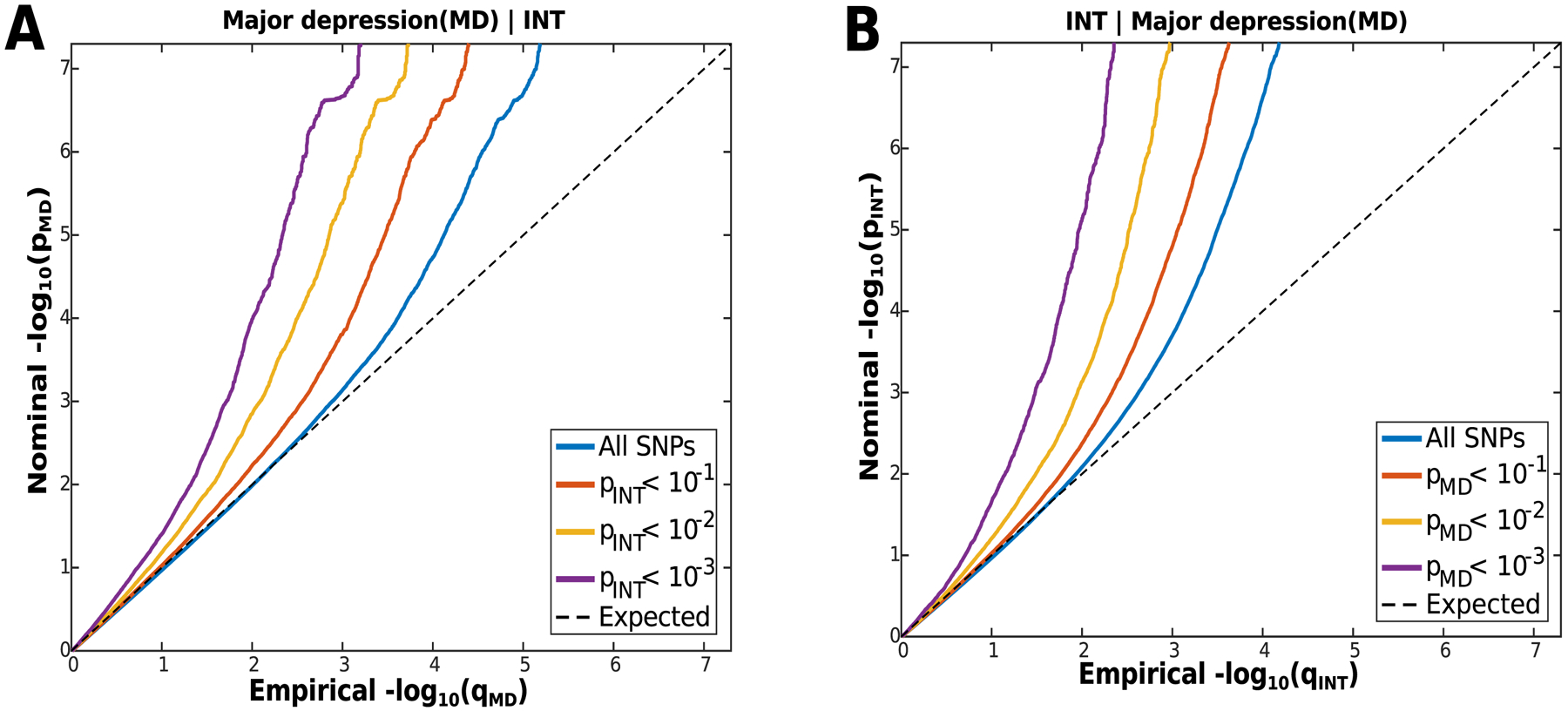
A) Conditional Q-Q plots of nominal versus empirical major depression (MD) −log10 p-values (corrected for inflation) below the standard GWAS threshold of p<5×10−8 as a function of the significance of the association with INT, at the level of p≤0.1, p≤0.01, p≤0.001, respectively. The blue line indicates all SNPs. The dashed line indicates the null hypothesis. B) Conditional Q-Q plots of nominal versus empirical INT −log10 p-values (corrected for inflation) below the standard GWAS threshold of p<5×10−8 as a function of significance of association with major depression (MD), at the level of p≤0.1, p≤0.01, p≤0.001, respectively. The blue line indicates all SNPs. The dashed line indicates the null hypothesis.
To test the statistical significance of the enrichment for the Q-Q plot strata, we used the partitioned LD-score regression approach13. The results showed a significant increase in the enrichment parameter for major depression given INT, Further details are provided in the Supplementary Methods.
Shared genetic loci (cond/conj FDR)
To improve the discovery of genetic variants associated with major depression and INT, we applied the condFDR statistical analysis on the association of major depression with INT, and vice versa. At condFDR<0.01, we identified 68 loci associated with major depression conditionally on INT (Supplementary Table 1). The reversed condFDR analysis identified 298 loci associated with INT conditional on major depression at condFDR<0.01 (Supplementary Table 2)15,16. The LD-score regression13 resulted in a non-significant genetic correlation (rg=−0.0148 with p-value=0.5008).
At conjFDR<0.05, we identified 92 distinct genomic loci jointly associated with major depression and INT (Figure 3; Supplementary Table 3): 69 of these loci were not identified in the original major depression GWAS15, and 64 were not reported in the original INT GWAS16. In total, 53 shared loci are novel for both major depression and INT (Supplementary Table 3).
Figure 3. Manhattan Plot.
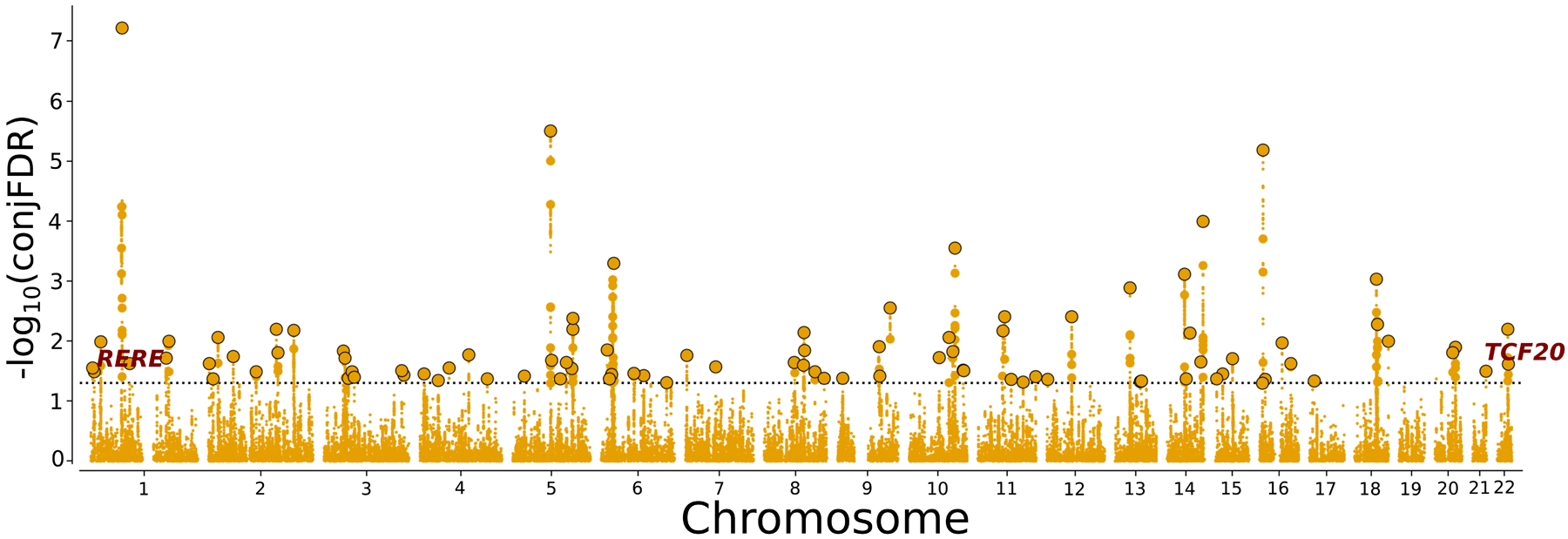
Common genetic variants jointly associated with major depression and intelligence at conjunctional false discovery rate (conjFDR)<0.05. Manhattan plot showing the −log10 transformed conjFDR values for each SNP on the y-axis and chromosomal positions along the x-axis. The dotted horizontal line represents the threshold for significant shared associations (conjFDR<0.05). Independent lead SNPs are encircled in black. The significant shared signal in the major histocompatibility complex region (chr6:25119106–33854733) is represented by one independent lead SNP. Further details are provided in Supplementary Table 3.
By comparing the effect directions of the lead SNPs at the shared loci (conjFDR<0.05), we found that 48 lead SNPs have consistent effect directions in major depression and INT and 44 have opposite effect directions. This is in line with the observed absence of significant genetic correlation and supports the hypothesis of mixed effect directions between major depression and INT.
Functional annotations
The functional annotation of all SNPs in the loci shared between major depression and INT (n=5,234; Figures 4 A–C) at conjFDR value<0.05 indicates that these are mostly intronic and intergenic, 54.7% and 30.6% respectively, while 1.4% were exonic (Figure 4 A, Supplementary Table 4). Of the 92 top lead SNPs in the loci shared between major depression and INT, 42 were located inside a protein-coding gene and 16 inside a non-coding RNA (Supplementary Table 3).
Figure 4. Distribution of the Annotation for All SNPs Jointly Associated With between major depression and intelligence at Conjunctional False Discovery Rate Less Than 0.10 Including Functional Consequences of SNPs.
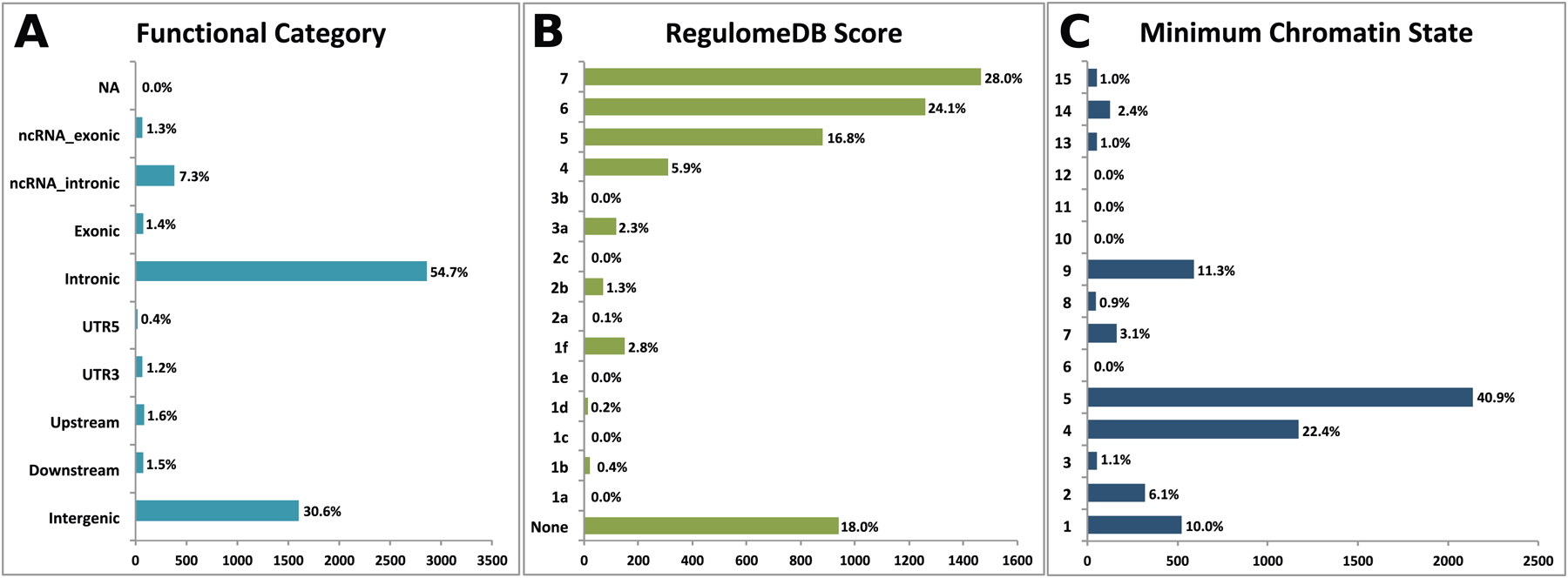
A) Distribution of functional consequences of SNPs in the shared genomic risk loci. B) Distribution of RegulomeDB score for SNPs in shared genomic loci, with a low score indicating a higher likelihood of having a regulatory function. C) The minimum chromatin state across 127 tissue and cell types for SNPs in shared genomic loci, with lower states indicating higher accessibility and states 1–7 referring to open chromatin states.
Seven of the 92 top lead SNPs (rs301807, rs62255971, rs12204714, rs13276452, rs4740502, rs3896224 and rs1952042) had CADD scores27 above 12.37 suggesting high deleteriousness. Four of these SNPs, rs62255971, rs12204714, rs3896224 and rs1952042, were intronic variants within CCDC66, ESR1, SORCS3 and RTN1, respectively (Supplementary Table 3). The distribution of minimum chromatin state showed that 80.5% of the top SNPs are located in open chromatin states regions. (Figure 4 C; Supplementary Table 3). Two of the top SNPs (rs301807 and rs9620039) had RegulomeDB scores28 of 1f and 1b, respectively, indicating that they may affect transcription factor binding (Supplementary Table 3); rs301807 had a CADD score of 16.18; these SNPs annotated to RERE and TCF20 as nearest genes, respectively (Supplementary Table 3). We further investigated the gene regulatory effects of rs301807 and rs9620039 using the GTEx and Braineac eQTLs datasets. These databases showed that rs301807 was significantly associated with altered expression of RP5–1115A15.1, RERE, GPR157 and SLC2A5 in several human tissues, including frontal cortex, hippocampus, putamen, temporal cortexand intralobular white matter (Figure 5; Supplementary Tables 5 and 6).
Figure 5. Gene expression level in human brain tissues.
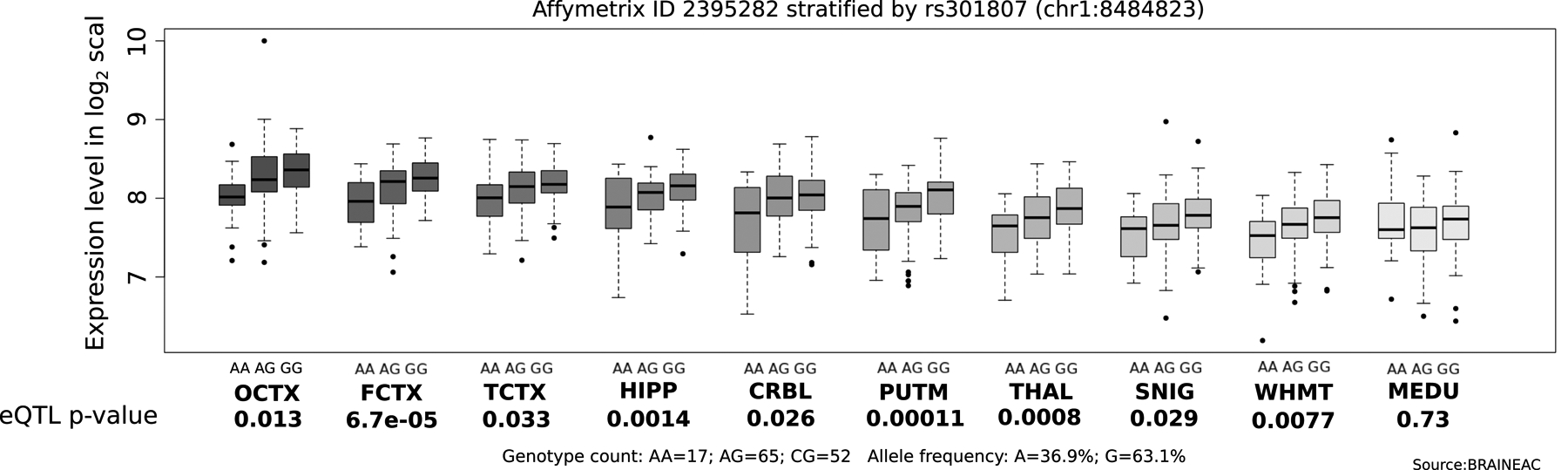
The rs301807 variant shared between major depression and intelligence is significantly associated with brain grene expression of RERE, with the major depression risk allele associated with lower/higher intelligence and lower/higher RERE expression. Figure was generated with Brianeac35.
We then found that rs9620039 was significantly associated with the expression levels of multiple genes in human brain tissues (Supplementary Tables 5 and 7), including TCF20 in the cerebellar cortex (Supplementary Figure 5). These findings provide complementary information about the potential gene regulatory mechanisms of rs301807 and rs9620039 that may influence major depression and INT.
Using GTEx data29, we found that loci having concordant effect directions in major depression and INT were annotated to genes significantly overexpressed in cerebellum, hypothalamus and frontal cortex BA9 (Supplementary Figures 3A, 4 and 5). GO gene-set analysis for the genes annotated to the loci with concordant effect directions revealed nine significantly associated biological processes, including “regulation of short term neuronal synaptic plasticity” “homophilic cell adhesion via plasma membrane adhesion molecules”, and “cellcell adhesion via plasma membrane adhesion molecules” (Supplementary Table 8). These genes were also significantly associated with six cellular component gene-sets, including “AMPA glutamate receptor complex”, “ionotropic glutamate receptor complex” and “endoplasmic reticulum”, as well as one molecular function gene set, i.e., “calcium ion binding” (Supplementary Table 8). On the other hand, the expression of genes annotated to the loci with opposite effect directions was not higher in brain than in other tissues (Supplementary Figures 3B, 6 and 7). GO gene-set analysis showed 18 significantly associated biological processes for the genes annotated to the loci with opposite effect directions; “central nervous system projection neuron axonogenesis”, “negative regulation of dendrite development” and “central nervous system neuron axonogenesis” were among the most strongly associated processes (Supplementary Table 9). Further, these genes were significantly associated with eight cellular component gene-sets, including “t_tubule”, “synapse”, and “excitatory synapse” and with six molecular function gene sets, where the most strongly associated were “alpha actinin binding”, “transcription coactivator activity” and “voltage gated cation channel activity” (Supplementary Table 9).
The pathway over-representation analysis indicated “MECP2 and Associated Rett Syndrome” and “GABAergic synapse” pathways as significantly overrepresented (p-value=0.00466, p-value=0.00892, respectively) based on the genes annotated to the loci with consistent effect directions in MD and INT, and “Validated nuclear estrogen receptor alpha network”, “Regulation of nuclear SMAD2/3 signaling” and “Phenytoin (Antiarrhythmic) Action” pathways as significantly overrepresented based on the genes annotated to the loci with opposite effect directions (Supplementary Table 10).
Discussion
The main finding of the present study is the substantial polygenic overlap between INT and MD identified with a condFDR/conjFDR approach. We singled out multiple genomic loci shared by the two phenotypes with a mixture of allelic effect directions (48 lead SNPs having concordant effect directions and 44 SNPs with discordant effect directions) despite their low genetic correlation. Half of the 92 genetic loci shared between major depression and INT have a consistent direction of allelic effect, while the other half has an opposite direction, which is in line with the observed non-significant genetic correlation. Furthermore, the MiXeR results address the question of levels of polygenic overlap. The shared number of causal variants was 88.3% between MD and INT; which is a substantial part of the polygenic architecture. In comparison, the percentage of shared causal variants between MD and INT was estimated to be 12.2% and 21% with height, respectively. The overlap with height, can inform the proportion of overlap potentially driven by omnigenic effects, and suggest that a large fraction of overlap between MD and INT is not driven by omnigenic factors.
Gene-set analysis of the identified loci shared by INT and major depression with concordant effects implicated functional pathways related to cell adhesion and metabolism, while the loci shared with opposite effects were associated with regulation of gene silencing, central nervous system projection neuron axonogenesis and dendrite development. This provides neurobiological insights that can be tested in experimental studies. Further, the extensive polygenic overlap between major depression and INT is likely to lead to hypotheses about the neurobiological underpinnings of cognitive functioning and major depressive disorder both related to depressive episodes as well as in euthymia8.
A recent study reported a small but non-significant genetic correlation between INT and major depressive disorder (using a smaller sample of n=18,75916,30), while the genetic correlation between INT and the larger major depression sample15 analyzed here was close to zero n (rg=−0.0148, p=0.5008). It is worth noting that genetic correlation can be significant only if the bulk of variants associated with both phenotypes reveals consistent direction of effects (same or opposite but not mixed). In contrast, both conditional QQ plots and LD score based partitioned heritability are agnostic to effect directions and capture shared genetic basis even when the effects are mixed and so cancel each other out in correlation analyses. Consistent with the absence of genetic correlation between INT and major depression, we identified an equal distribution of concordant and discordant directions among the shared loci. The conjFDR statistical approach, which enables the identification of individual loci jointly affecting both phenotypes, singled out 92 loci shared between INT and major depression, further dissecting their polygenic overlap. Further leveraging the increased power from the combined samples, condFDR identified 68 loci in major depression conditioned on INT, and 298 loci in INT conditioned on major depression. These discovered loci can help explain more of the heritability underlying major depression and INT traits. The present study proves the usefulness and cost-effectiveness of applying the cond/conjFDR statistical tools to increase gain from available GWAS data26,31.
Interestingly, the balanced mixture of directional effects among the loci shared between major depression and INT is similar to that observed between bipolar disorder and INT25. This contrasts findings for other psychiatric disorders such as schizophrenia25,31, where the majority of risk variants shared with INT have a detrimental effect on cognitive performance, and is in line with the more severe cognitive impairments seen in schizophrenia relative to affective disorders32. Many of the loci identified here are also found in GWAS on various complex traits and disorders related to brain function, corroborating that pleiotropy is ubiquitous among brain phenotypes (Supplementary Tables 8 and 9). The genetic effects on INT identified here are determined in a sample representative of the general population, who were not depressed during their cognitive examination, although it is possible that participants in the INT study may develop depression later in life16. Hence, our results indicate that genetic risk variants for major depression contribute to the variance in INT, even among those who are not depressed. This provides further evidence for the strong relationship between INT and psychiatric disorders, implicating a substantial shared genetic basis33 and thus neurobiological mechanisms.. This may inform cognitive theories of psychopathology of depression9 where cognitive processes are involved in generating negative thoughts characteristic of MDD. The large number of risk loci associated with higher cognitive performance may provide genetic clues into the increased inclination to many cultural and artistic professions among people with mood disorders34.
An interesting shared signal with the same direction of effect in major depression and INT was detected on chromosome 1p36.23, at a locus that contains many genes (nearest gene RERE; Supplementary Table 3). The lead SNP at this locus (rs301807) was identified as an eQTL for RERE in the GTEx portal and Braineac database and significantly regulates the expression of the gene in several human brain regions29,35. The G allele of this SNP was associated with major depression risk and increased brain expression of RERE. The variant had a RegulomeDB score of 1f and a CADD score of 16.18 showing that it may affect transcription factor binding and be deleterious27. This locus was genome-wide significant in the primary GWASs on MDD15, but is a novel finding for INT. RERE is a widely-expressed nuclear receptor coregulator that positively regulates retinoic acid signaling. Mutations in RERE cause a genetic syndrome and haploinsufficiency of RERE might be sufficient to cause many of the phenotypes associated with proximal 1p36 deletions36. Another interesting locus singled out by our analyses is located on chromosome 22 in the vicinity of several genes (nearest gene TCF20; Supplementary Table 3) and has a RegulomeDB score of 1b. This locus had a strong joint effect on schizophrenia and cognitive abilities31, and was genome-wide significant in the primary GWASs on INT, but is a novel finding for major depression. TCF20 encodes a widely expressed transcriptional co-regulator, and TCF20 mutations are associated with autism and intellectual disability. Using Braineac eQTLs datasets, we determined that the variant could significantly regulate the expression of TCF20 in the cerebellar cortex.
The genes annotated to the loci with concordant effect directions in major depression and INT were significantly overexpressed in multiple brain regions (Supplementary Figures 3A, 4 and 5). Although the phenotypic effect doesn’t mainly depend on the genetic variants of the nearest gene, the findings support the importance of brain expressed genes in the shared genetic etiology underlying major depression and INT. Gene-set analysis implicated nine significantly associated biological processes for these genes, converging on processes related to cell adhesion and neuronal synapses (Supplementary Table 8). Such processes have been previously linked to major depressive disorder37 and INT16,38. In line with these results, the cellular component gene-set analysis revealed significant associations for the AMPA glutamate receptor complex, and the endoplasmic reticulum39. Interestingly, the over-represented pathways for the genes annotated to these loci were GABAergic synapses, and MECP2 which is associated with Rett Syndrome, indicating potential involvement of neurodevelopmental processes40. The genes annotated to the loci with opposite effect direction were not significantly overexpressed in brain tissue (Supplementary Figures 3B, 6 and 7). Gene-set analysis showed 18 significantly associated biological processes, related to regulation of gene silencing and central nervous system projection neuron axonogenesis (Supplementary Table 9). In line with these results, these genes were over-represented in pathways for validated nuclear estrogen receptor alpha network, regulation of nuclear SMAD2/3 signaling and phenytoin action (Supplementary Table 10). Estrogens are synthesized in various tissues throughout the body, including the brain and have a number of effects on cognition and brain function41.
To conclude, our study extends the current understanding of the common genetic variation influencing MD and INT. Our findings provide evidence for an extensive polygenic overlap between MD and INT, and identify a series of specific gene loci shared between MD and INT. The results provide insights into the shared genetic architecture between two important human traits, suggesting a shared neurobiological basis.
Methods
Participant Samples
For this study we used GWAS summary statistics results for major depression and INT15,16. Data on major depression were obtained from the Psychiatric Genomics Consortium (PGC) and 23andMe which also included cases with self-reported depression. The major depression sample consisted of 135,458 cases and 344,901 controls including seven cohorts15. To avoid sample overlap, we excluded the UK Biobank cohort (n=29,740) contribution to the MD GWAS15 from all analyses. Any systematic sample overlap between major depression and INT GWASs was thereby prevented. Data on general cognitive ability were based on 269,867 individuals drawn from 14 cohorts, primarily consisting of data from the UK Biobank (n=195,653)16. (For details, see Supplementary Methods and the original publications)15,16.
Statistical analysis
To quantify polygenic overlap between major depression and intelligence, we employed the MiXeR statistical tool20. MiXeR uses GWAS summary statistics of each phenotype to provide univariate estimates of the proportion of non-null SNPs (polygenicity) and the variance of effect size of non-null SNPs in each phenotype. Then it constructs a bivariate causal mixture model to estimate the total number of shared and phenotype-specific causal variants. MiXeR results are presented as a Venn diagram of shared and unique polygenic components across traits. The Dice coefficient score (polygenic overlap measure in 0–100% scale), and genetic correlation between two phenotypes are also computed. We also applied MiXeR to quantify the polygenic overlap between major depression/intelligence and height, to demonstrate non-specific genetic overlap with a trait not association with behavioral, mental or cognitive traits.
Conditional quantile-quantile (Q-Q) plots26 give a visual impression of the overlap in associations with two traits. Conditional Q-Q plots compare the association with one trait within SNP strata determined by the significance of their association with the second trait. Specifically, we computed the empirical cumulative distribution of nominal p-values in one phenotype for all SNPs and for subsets of SNPs with significance levels in another phenotype below the indicated cut-offs (p≤1, p≤0.1, p≤0.01, p≤0.001, respectively)26. In this analysis we focused on polygenic effects below the standard GWAS significance threshold (p>5×10−8)26. Enrichment for overlapping signals exists for a given phenotype if the degree of leftward deflection from the expected null line increases with increasing association significance in the second phenotype26.
The condFDR statistical method26 improves the detection of genetic variants associated with major depression and INT via re-ranking SNPs compared to their nominal p-value−based ranking26 (for details, see Supplementary Methods). To identify shared genetic variants associated with both traits and make a comprehensive list of overlapping signals, we used a conjunctional FDR (conjFDR) procedure26. The conjFDR statistic is based on condFDR and is defined as the maximum of the two mutual condFDR for a specific SNP. The conjFDR approach estimates a posterior probability that an SNP is null for either phenotype or both, given that the p-values for both phenotypes are lower than the observed p-values26 (for details, see Supplementary Methods). Two genomic regions, the extended major histocompatibility complex genes region (hg19 location Chr 6: 25119106–33854733) and chromosome 8p23.1 (hg19 location Chr 8: 7242715–12483982), were excluded from the FDR fitting procedures because complex correlations in regions with intricate LD can bias FDR estimation.
As the empirical null distribution in GWASs is affected by global variance inflation due to factors such as population stratification and cryptic relatedness, all p-values were corrected for inflation using a genomic inflation control procedure26. To minimize inflation resulting from LD dependency between variants we performed random pruning of all SNPs across 100 iterations both in conditional Q-Q plots and condFDR/conjFDR analyses. For each random pruning iteration, only one random SNP representative was retained for each LD-independent block (cluster of SNPs with r2>0.1). The results were then averaged across all iterations. LD-score based significance testing of enrichment and genetic correlation analyses were also performed42.
Genomic loci definition and Functional annotation
To detail the LD-independent genomic regions surrounding the identified signals we used the Functional Mapping and Annotation (FUMA) protocol43. SNPs having a condFDR<0.01 and at r2<0.6 with each other were considered as independent significant SNPs and a fraction of the independent significant SNPs in approximate linkage equilibrium with each other at r2<0.1 were considered as lead SNPs. To determine the distinct genomic loci and their borders we used the default parameters from FUMA43.
For the functional annotation we used all candidate SNPs in the genomic loci with a condFDR or conjFDR value<0.10 having an r2≥0.6 with one of the independent significant SNPs. We annotated SNPs based on functional Categories, Combined Annotation Dependent Depletion (CADD) scores, RegulomeDB scores and chromatin states using FUMA43. The CADD score is the score of deleteriousness of SNPs predicted by 63 functional annotations. CADD=12.37 is the score for the most deleterious variants27. The RegulomeDB score is a categorical score showing the regulatory functionality of SNPs based on the overlap of existing functional data including annotation to cis-expression quantitative trait loci (cis-eQTLs) and evidence for transcription factor binding28. Chromatin state shows the accessibility of genomic regions with 15 categorical states predicted by ChromHMM based on 5 chromatin marks for 127 epigenomes28,44,45.
We performed gene-set enrichment analysis based on the gene ontology classification system using FUMA43. As an input for this step, we took the closest gene to the lead SNP of each LD-independent locus identified in our conjFDR analysis. To determine gene expression and assess eQTL functionality of likely regulatory lead SNPs, we also applied data from the genotype tissue expression (GTEx) resource and the Braineac eQTLs dataset29,35. Pathway over-representation analyses were performed for the genes nearest the identified shared loci using ConsensusPathDB for the loci having consistent effect directions in major depression and INT and the loci having opposite effect direction separately46. All analyses were corrected for multiple comparisons. For details, see Supplementary Methods.
Supplementary Material
Acknowledgements
We would like to thank the research participants and the Major Depression Working Group of the Psychiatric Genomics Consortium, 23andMe and Intelligence cohorts for making their available their GWAS summary statistics available. We gratefully acknowledge support from the American National Institutes of Health (NS057198, EB00790), European Union’s Horizon2020 Research and Innovation Action Grant #847776 CoMorMent, the Research Council of Norway (229129, 213837, 248778, 273291, 223273); the South-East Norway Regional Health Authority (2017-112) and KG Jebsen Stiftelsen (SKGJ-MED-008).
Footnotes
Competing interests
Dr. Andreassen has received speaker’s honorarium from Lundbeck, and is a consultant to HealthLytix. Dr. Dale is a Founder of and holds equity in CorTechs Labs, Inc, and serves on its Scientific Advisory Board. He is a member of the Scientific Advisory Board of Human Longevity, Inc. and receives funding through research agreements with General Electric Healthcare and Medtronic, Inc. The terms of these arrangements have been reviewed and approved by UCSD in accordance with its conflict of interest policies. The other authors declare no competing interests.
Data availability
Data used in this article were obtained from the UK Biobank [https://www.ukbiobank.ac.uk/], from the Psychiatric Genomics Consortium [https://www.med.unc.edu/pgc/] and 23andMe [https://www.23andme.com/].
Code availability
The codes for MiXeR and conditional and conjunctional FDR analyses are publicly available on [https://github.com/precimed/mixer] and [https://github.com/precimed/pleiofdr/], respectively.
References:
- 1.Kupfer DJ, Frank E & Phillips ML Major depressive disorder: new clinical, neurobiological, and treatment perspectives. Lancet 379, 1045–1055, doi: 10.1016/S0140-6736(11)60602-8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferrari AJ et al. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med 10, e1001547, doi: 10.1371/journal.pmed.1001547 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gustavsson A et al. Cost of disorders of the brain in Europe 2010. Eur Neuropsychopharmacol 21, 718–779, doi: 10.1016/j.euroneuro.2011.08.008 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Kessler RC et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 289, 3095–3105, doi: 10.1001/jama.289.23.3095 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Vancampfort D et al. Metabolic syndrome and metabolic abnormalities in patients with major depressive disorder: a meta-analysis of prevalences and moderating variables. Psychol Med 44, 2017–2028, doi: 10.1017/S0033291713002778 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Geschwind DH & Flint J Genetics and genomics of psychiatric disease. Science 349, 1489–1494, doi: 10.1126/science.aaa8954 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan Z et al. Cognitive impairment in major depressive disorder. CNS Spectr 24, 22–29, doi: 10.1017/S1092852918001207 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Szmulewicz AG et al. Neuropsychological profiles of major depressive disorder and bipolar disorder during euthymia. A systematic literature review of comparative studies. Psychiatry Res 248, 127–133, doi: 10.1016/j.psychres.2016.12.031 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Beck AT The evolution of the cognitive model of depression and its neurobiological correlates. Am J Psychiatry 165, 969–977, doi: 10.1176/appi.ajp.2008.08050721 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Otte C et al. Major depressive disorder. Nat Rev Dis Primers 2, 16065, doi: 10.1038/nrdp.2016.65 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Navrady LB et al. Intelligence and neuroticism in relation to depression and psychological distress: Evidence from two large population cohorts. Eur Psychiatry 43, 58–65, doi: 10.1016/j.eurpsy.2016.12.012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cross-Disorder Group of the Psychiatric Genomics, C. et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 45, 984–994, doi: 10.1038/ng.2711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47, 291–295, doi: 10.1038/ng.3211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyde CL et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet 48, 1031–1036, doi: 10.1038/ng.3623 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wray NR et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 50, 668–681, doi: 10.1038/s41588-018-0090-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savage JE et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet 50, 912–919, doi: 10.1038/s41588-018-0152-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies G et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun 9, 2098, doi: 10.1038/s41467-018-04362-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glazier AM, Nadeau JH & Aitman TJ Finding genes that underlie complex traits. Science 298, 2345–2349, doi: 10.1126/science.1076641 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Boyle EA, Li YI & Pritchard JK An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186, doi: 10.1016/j.cell.2017.05.038 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frei O et al. Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat Commun 10, 2417, doi: 10.1038/s41467-019-10310-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Efron B Large-scale inference : empirical Bayes methods for estimation, testing, and prediction. Vol. 1 (2010). [Google Scholar]
- 22.Andreassen OA, Thompson WK & Dale AM Boosting the power of schizophrenia genetics by leveraging new statistical tools. Schizophr Bull 40, 13–17, doi: 10.1093/schbul/sbt168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smeland OB et al. Discovery of shared genomic loci using the conditional false discovery rate approach. Hum Genet, doi: 10.1007/s00439-019-02060-2 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Wang Y et al. Leveraging Genomic Annotations and Pleiotropic Enrichment for Improved Replication Rates in Schizophrenia GWAS. PLoS Genet 12, e1005803, doi: 10.1371/journal.pgen.1005803 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smeland OB et al. Genome-wide analysis reveals extensive genetic overlap between schizophrenia, bipolar disorder and general cognitive ability. Mol. Psychiatry 25 844–853 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andreassen OA et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet 92, 197–209, doi: 10.1016/j.ajhg.2013.01.001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kircher M et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–315, doi: 10.1038/ng.2892 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyle AP et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22, 1790–1797, doi: 10.1101/gr.137323.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Consortium GT et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213, doi: 10.1038/nature24277 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Major Depressive Disorder Working Group of the Psychiatric, G. C. et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 18, 497–511, doi: 10.1038/mp.2012.21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smeland OB et al. Identification of Genetic Loci Jointly Influencing Schizophrenia Risk and the Cognitive Traits of Verbal-Numerical Reasoning, Reaction Time, and General Cognitive Function. JAMA Psychiatry 74, 1065–1075, doi: 10.1001/jamapsychiatry.2017.1986 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barch DM Neuropsychological abnormalities in schizophrenia and major mood disorders: similarities and differences. Curr Psychiatry Rep 11, 313–319 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill WD, Harris SE & Deary IJ What genome-wide association studies reveal about the association between intelligence and mental health. Curr Opin Psychol 27, 25–30, doi: 10.1016/j.copsyc.2018.07.007 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Taylor CL Creativity and Mood Disorder: A Systematic Review and Meta-Analysis. Perspect Psychol Sci 12, 1040–1076, doi: 10.1177/1745691617699653 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Ramasamy A et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci 17, 1418–1428, doi: 10.1038/nn.3801 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fregeau B et al. De Novo Mutations of RERE Cause a Genetic Syndrome with Features that Overlap Those Associated with Proximal 1p36 Deletions. Am J Hum Genet 98, 963–970, doi: 10.1016/j.ajhg.2016.03.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hidese S et al. Cerebrospinal fluid neural cell adhesion molecule levels and their correlation with clinical variables in patients with schizophrenia, bipolar disorder, and major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry 76, 12–18, doi: 10.1016/j.pnpbp.2017.02.016 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Deary IJ, Penke L & Johnson W The neuroscience of human intelligence differences. Nat Rev Neurosci 11, 201–211, doi: 10.1038/nrn2793 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Bannerman DM et al. Somatic Accumulation of GluA1-AMPA Receptors Leads to Selective Cognitive Impairments in Mice. Front Mol Neurosci 11, 199, doi: 10.3389/fnmol.2018.00199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Armstrong DD, Deguchi K & Antallfy B Survey of MeCP2 in the Rett syndrome and the non-Rett syndrome brain. J Child Neurol 18, 683–687, doi: 10.1177/08830738030180100601 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Hara Y, Waters EM, McEwen BS & Morrison JH Estrogen Effects on Cognitive and Synaptic Health Over the Lifecourse. Physiol Rev 95, 785–807, doi: 10.1152/physrev.00036.2014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 47, 1236–1241, doi: 10.1038/ng.3406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8, 1826, doi: 10.1038/s41467-017-01261-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roadmap Epigenomics C et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330, doi: 10.1038/nature14248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Z et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48, 481–487, doi: 10.1038/ng.3538 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Kamburov A, Stelzl U, Lehrach H & Herwig R The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res 41, D793–800, doi: 10.1093/nar/gks1055 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.