

Abstract
The homeostatic oxygen sensing system (HOSS) optimizes systemic oxygen delivery. Specialized tissues utilize a conserved mitochondrial sensor, often involving NDUFS2 in complex I of the mitochondrial electron transport chain, as a site of pO2-responsive production of reactive oxygen species (ROS). These ROS are converted to a diffusible signaling molecule, hydrogen peroxide (H2O2), by superoxide dismutase (SOD2). H2O2 exits the mitochondria and regulates ion channels and enzymes, altering plasma membrane potential, intracellular Ca2+ and Ca2+-sensitization and controlling acute, adaptive, responses to hypoxia that involve changes in ventilation, vascular tone and neurotransmitter release. Subversion of this O2-sensing pathway creates a pseudohypoxic state that promotes disease progression in pulmonary arterial hypertension (PAH) and cancer. Pseudohypoxia is a state in which biochemical changes, normally associated with hypoxia, occur despite normal pO2. Epigenetic silencing of SOD2 by DNA methylation alters H2O2 production, activating hypoxia-inducible factor 1α, thereby disrupting mitochondrial metabolism and dynamics, accelerating cell proliferation and inhibiting apoptosis. Other epigenetic mechanisms, including dysregulation of microRNAs (miR), increase pyruvate dehydrogenase kinase and pyruvate kinase muscle isoform 2 expression in both diseases, favoring uncoupled aerobic glycolysis. This Warburg metabolic shift also accelerates cell proliferation and impairs apoptosis. Disordered mitochondrial dynamics, usually increased mitotic fission and impaired fusion, promotes disease progression in PAH and cancer. Epigenetic upregulation of dynamin-related protein 1 (Drp1) and its binding partners, MiD49 and MiD51, contributes to the pathogenesis of PAH and cancer. Finally, dysregulation of intramitochondrial Ca2+, resulting from impaired mitochondrial calcium uniporter complex (MCUC) function, links abnormal mitochondrial metabolism and dynamics. MiR-mediated decreases in MCUC function reduce intramitochondrial Ca2+, promoting Warburg metabolism, whilst increasing cytosolic Ca2+, promoting fission. Epigenetically disordered mitochondrial O2-sensing, metabolism, dynamics, and Ca2+ homeostasis offer new therapeutic targets for PAH and cancer. Promoting glucose oxidation, restoring the fission/fusion balance, and restoring mitochondrial calcium regulation are promising experimental therapeutic strategies.
Keywords: ABT-263 (Navitoclax), ABT-199 (Venetoclax), B-cell lymphoma 2 (BCL-2), DNA methylation, DNA methyltransferase (DNMT), dynamin-related protein 1 (Drp1), group 1 pulmonary hypertension, hypoxia-inducible factor 1α (HIF-1α), hypoxia-inducible factor 2α (HIF-2α), hypoxic pulmonary vasoconstriction, mammalian target of rapamycin (mTOR), microRNA (miRNA), miR-138, miR-25, mitochondrial calcium uniporter (MCU), mitochondrial dynamics protein of 49 kDa (MiD49), mitochondrial dynamics protein of 51 kDa (MiD51), mitofusin 2 (Mfn2), mitophagy, monocrotaline, oxygen sensing, peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), pyruvate dehydrogenase (PDH), pyruvate dehydrogenase kinase (PDK), pyruvate kinase muscle isoform 2 (PKM2), reactive oxygen species (ROS), Sugen5416, survivin, von Hippel-Lindau protein (VHL)
Graphical Abstract
1. Oxygen-sensing and experimental therapeutics in pulmonary arterial hypertension and cancer
1.1. Normal oxygen-sensing in pulmonary vs systemic arteries
Specialized tissues are specifically adapted to sense small changes in airway oxygen levels and arterial oxygen (pO2) within the physiological range [1]. These tissues, which make up the homeostatic oxygen-sensing system (HOSS), elicit changes in respiration, vascular tone and neurosecretion to adapt to changing environmental oxygen levels, or adapt to localized changes in pO2 within the body. Type 1 cells within the carotid body respond to hypoxia via exocytosis of neurotransmitters [2]. The smooth muscle cells (SMC) within the resistance pulmonary arteries (PA), fetoplacental arteries, and ductus arteriosus respond to changes in pO2 through alterations in vascular tone [1, 3]. Interestingly, hypoxia elicits vasodilation of the ductus arteriosus and systemic arteries, while stimulating vasoconstriction of the fetoplacental and resistance PA. These opposing responses are coordinated and enhance oxygen uptake and systemic oxygen delivery (Figure 1).
Figure 1. System for Homeostatic Oxygen Sensing.
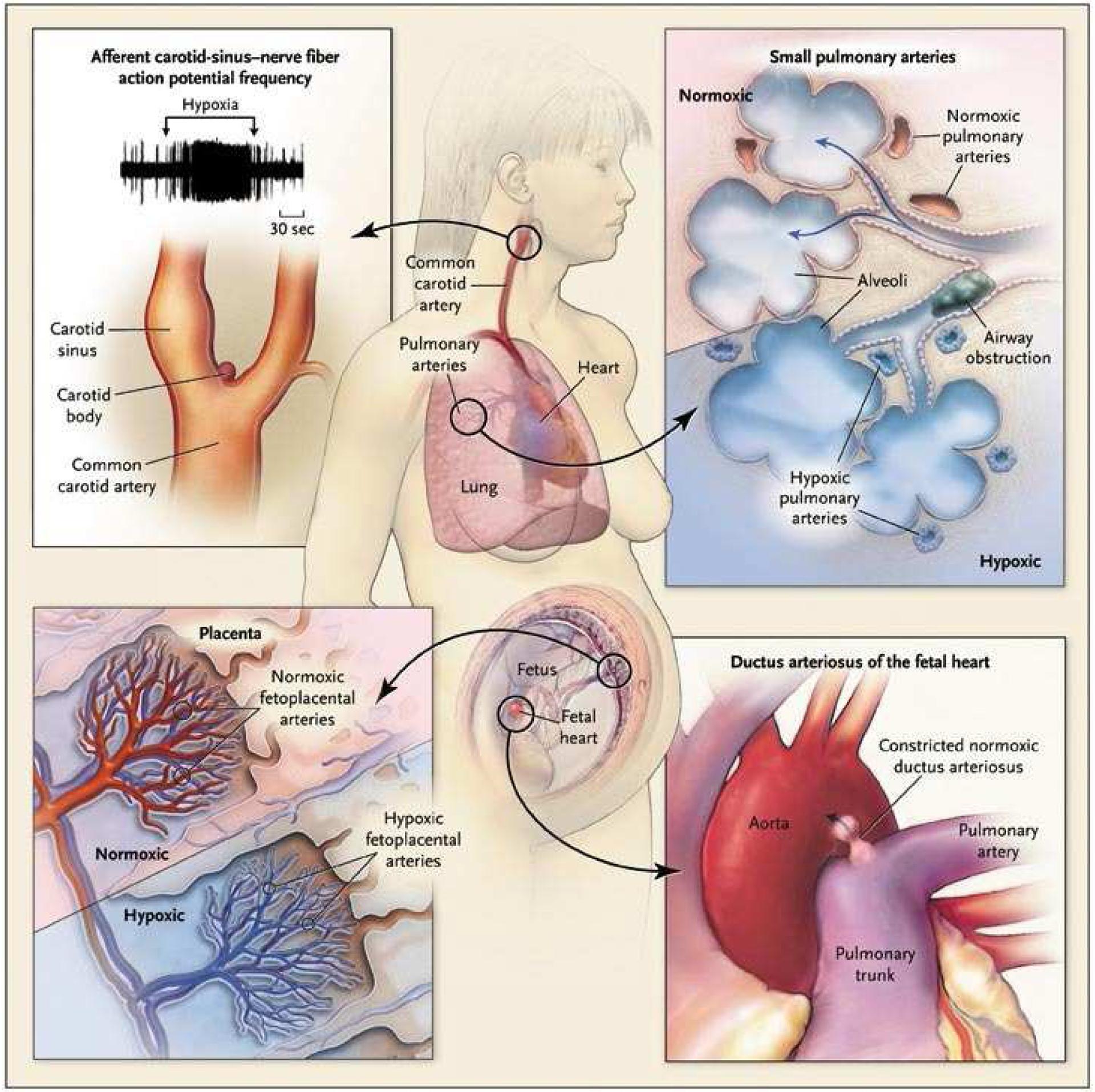
The homeostatic oxygen-sensing system (HOSS) is made up of specialized tissues specifically adapted to sense small changes in oxygen levels. These include the type 1 cells within the carotid body, smooth muscle cells (SMC) within the resistance pulmonary arteries (PA), fetoplacental arteries, and ductus arteriosus. Hypoxia increases the afferent carotid-sinus nerve fiber action potential frequency, which stimulates respiration. Hypoxia also elicits vasodilation of the ductus arteriosus and systemic arteries, while stimulates vasoconstriction of the fetoplacental and resistance PA.
Copyright © New England Journal of Medicine
Hypoxic pulmonary vasoconstriction (HPV) is a homeostatic mechanism that optimizes oxygen uptake by matching perfusion to ventilation in the lung. HPV is intrinsic to the pulmonary circulation, functioning most strongly in small, resistance level pulmonary arteries. In response to environmental hypoxia, HPV manifests ‘globally’ and all small pulmonary arteries constrict, resulting in a rise in pulmonary artery pressure (PAP). In contrast, in most lung diseases, such as pneumonia or atelectasis, segmental hypoxia results in localized HPV, which diverts blood flow to better-oxygenated lung segments [4]. This localized vasoconstrictor response improves oxygen uptake without elevating PAP. HPV occurs within seconds after the onset of modest levels of airway hypoxia, with constriction reaching a maximum intensity within minutes [5, 6]. Unless adverse vascular remodeling occurs, HPV is rapidly reversible upon restoration of normal airway oxygen levels [7, 8] (Figure 1).
1.1.1. Mechanisms of HPV
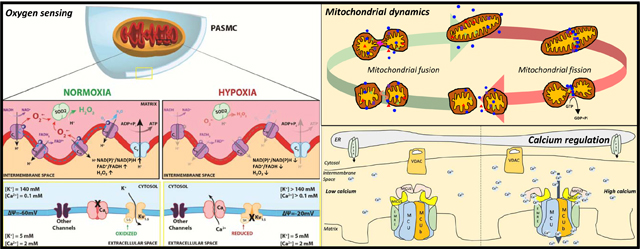
Although endothelial-derived vasoconstrictors and vasodilators do play an important role in modulating PA tone [9, 10], the core mechanism of HPV is intrinsic to the pulmonary artery smooth muscle cells (PASMC), and HPV persists in endothelium-denuded PA rings [11]. Oxygen sensing within the PASMC involves the coordination of an upstream sensor of alveolar oxygen and a downstream effector mechanism, comprised of several types of oxygen-sensitive ion channels. In response to changing oxygen tension, the sensor(s) within the mitochondria alter production of reactive oxygen species (ROS), notably superoxide anion. ROS is then locally and rapidly converted into H2O2 by mitochondrial superoxide dismutase 2 (SOD2). H2O2 diffuses from the mitochondria to the cell membrane where they modify the activity of redox-sensitive potassium (K+) and calcium (Ca2+) channels. These channels contain redox-sensitive amino acids in key positions, such as multiple cysteine residues, and are thereby susceptible to reduction and oxidation (REDOX) regulation of channel gating and open-state probability [12, 13]. Changes in the kinetics of ion channels ultimately alter membrane polarization and/or calcium influx into the oxygen-sensitive cell, leading to the physiologic response. These immediate redox changes in ion channel function are reinforced by redox regulation of enzymes, notably rho kinase, which sustains hypoxic responses.
Pharmacologic and electrophysiological studies using the patch clamp technique in isolated PASMC have identified voltage-gated potassium channels (Kv), as well as large-conductance, voltage-gated calcium channels (CaL), as major effectors of HPV. In the resistance pulmonary vessels, Kv channels are the predominant regulators of resting membrane potential and are major effectors of HPV [14]. The whole cell-patch clamp method has shown that the outward potassium current (IK) within the PASMC prevents CaL channels from opening during normoxia and that the Kv channel inhibitor 4-aminopyridine (4-AP), but not inhibitors of other channels types, such as BKCa and KATP channels, mimics the effects of hypoxia, namely inhibiting IK leading to membrane depolarization and vasoconstriction [15] (Figure 2).
Figure 2. The mechanism of acute hypoxic pulmonary vasoconstriction.
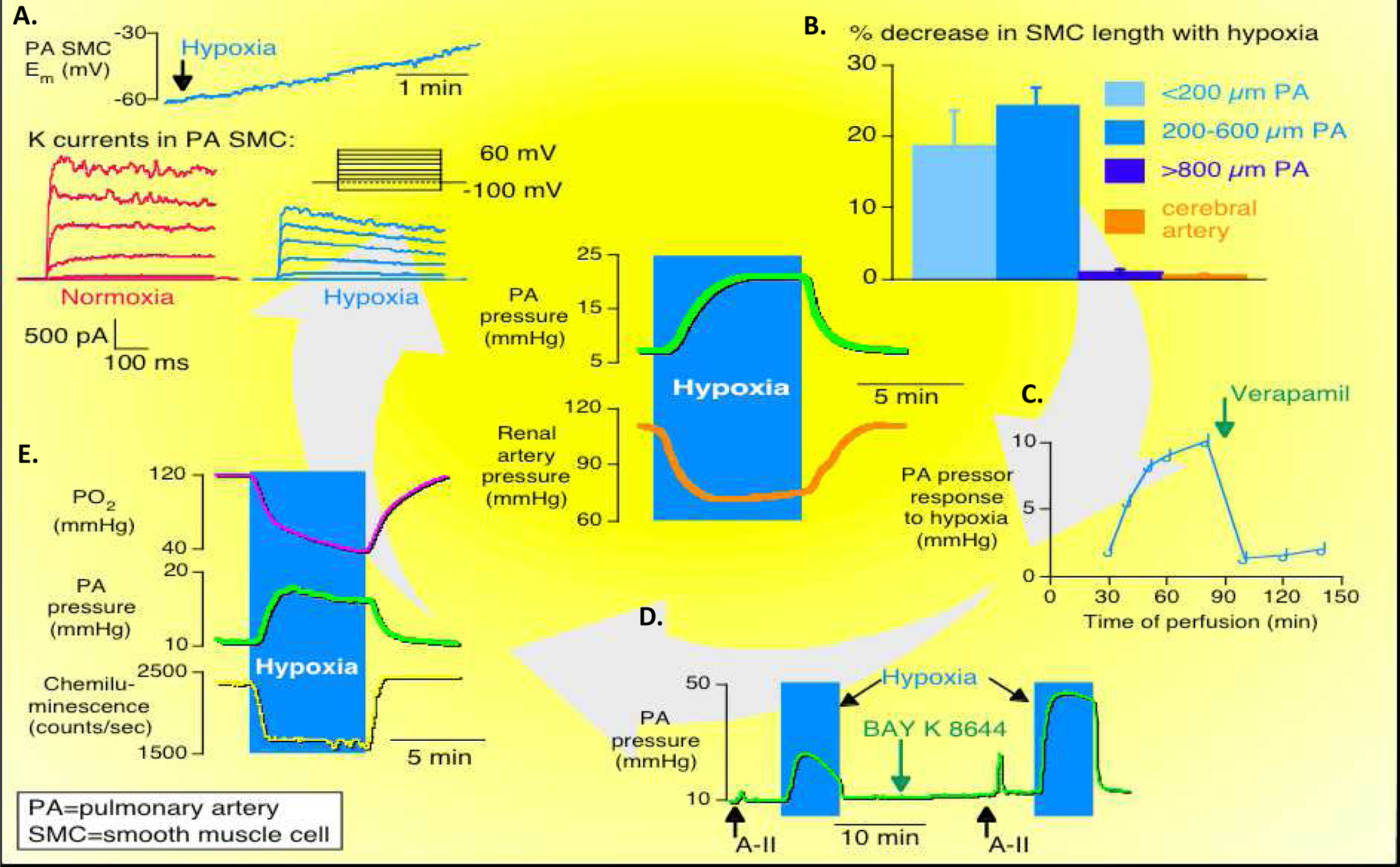
Middle: Pulmonary artery (PA) pressure is increased during hypoxia while renal artery pressure is decreased during hypoxia.
A. Potassium channel current is reduced in PA smooth muscle cells (SMC) during hypoxia.
B. Percentage decrease in SMC length with hypoxia is only observed in PAs with diameters of less than 600μm.
C. PA pressure response to hypoxia is abolished by treatment with verapamil, a calcium channel blocker.
D. PA pressure response to hypoxia is increased by BAY K8644, a calcium channel agonist.
E. PA pressure increases in response to decreased pO2.
Copyright © The FASEB Journal
In PASMC, but not in systemic arterial SMC, hypoxia inhibits Kv as well as other K+ channel types. The tonic egress of intracellular K+ down its gradient (140 mM intracellular to 5 mM extracellular) establishes a resting membrane potential of ~−60mV in normoxic PASMC. Kv channel inhibition, whether initiated by hypoxia or 4-AP, leads to accumulation of positive charges within the cytoplasm and plasma membrane depolarization (from ~−60mV to ~−20mV). This degree of membrane depolarization activates CaL channels, which have a voltage senor [16]. The resulting influx of calcium down its colossal concentration gradient (2 mM extracellular to 100 nM intracellular) initiates SMC contraction. The source of increasing cytosolic [Ca2+] that follows SMC membrane depolarization is mainly derived from the extracellular Ca2+ pool, supported by studies showing that inhibition of CaL channels by verapamil completely inhibits the hypoxic pressor response in the isolated rat lung as well as HPV in isolated PA rings [17, 18]. These electrical and ionic responses begin within seconds and account for the initiation of HPV. However, there is also an important role in the release of intracellular calcium stores and calcium sensitization in HPV, particularly with more prolonged exposure to hypoxia [19, 20].
Although several families of Kv channels exist, the Shaker family (Kv) is the most widely distributed and is implicated in HPV [21, 22]. Within this family of ion channels, Kv1.5 and Kv2.1 are central to the process of HPV. In Kv1.5 knockout mice, a significant reduction in the 4-AP- and oxygen-sensitive portions of IK as well as an impairment in HPV are observed [23]. Kv channels are tetramers and the heterotetrameric composition of a Kv channel can impact its oxygen sensitivity. For example, PASMC expressing Kv1.5/Kv1.2 heterotetrameric channels have increased sensitivity to oxygen in comparison to cells expressing homomeric channels [24]. In addition, the heterotetrametric Kv2.1/Kv9.3 channel is also oxygen-sensitive, suggesting it has a role in HPV [25] (Figure 3).
Figure 3. The Redox Theory of HPV.
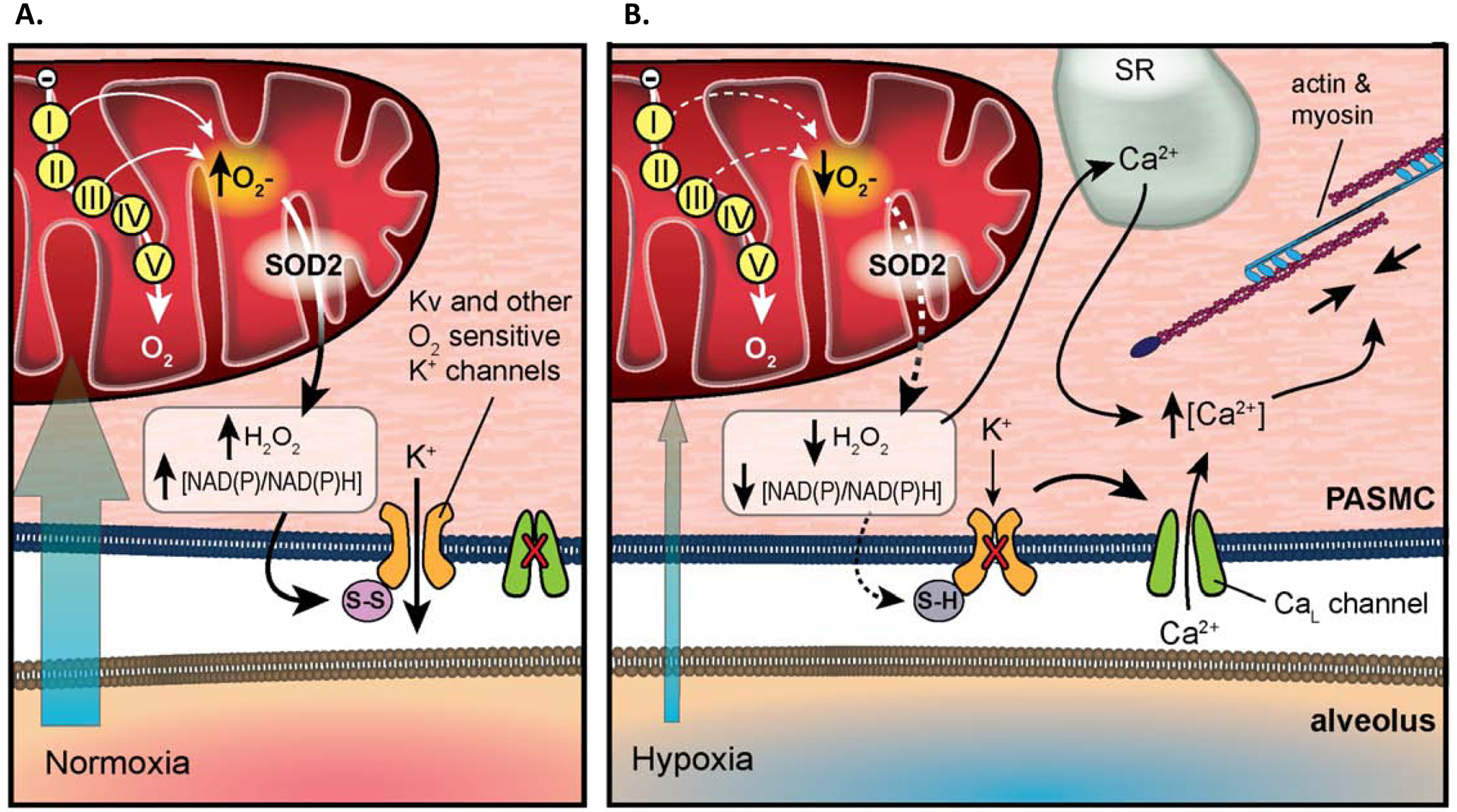
A. Under normoxic conditions, ETC produces higher level of ROS and the elevated NAD(P)/NAD(P)H ratio oxidizes the sulfhydryl groups on K+ channels, keeping them open, while the CaL channel remains closed.
B. Under hypoxic conditions, decreased ROS production and reduced NAD(P)/NAD(P)H ratio inhibits the opening of K+ channels and leads to the opening of CaL channels. Influx of Ca2+ further triggers Ca2+ release from the sarcoplasmic reticulum (SR), stimulating actin and myosin and triggering vasoconstriction.
Copyright © Pflügers Archiv: European Journal of Physiology
1.1.2. ROS as Mediators of Oxygen-Sensing
Weir and Archer et al. first demonstrated that changes in the redox status of the PA regulate PASMC membrane polarization and contraction [11]. Specifically, antioxidants cause membrane depolarization of PASMC and elicit vasoconstriction of PA rings, whereas oxidants cause membrane hyperpolarization and relaxation of constricted PA rings. These findings suggest that in the PA, reducing agents mimic hypoxia whilst oxidants mimic normoxia. Additionally, these findings demonstrated that changing the redox status of the SMC directly affects the whole-cell K+ channel current. This may reflect the impact of reducing/oxidizing key cysteine residues within Kv channels which has the effect of altering their open state probability and thereby changing membrane potential, CaL opening and vascular tone [26]. Beyond the indirect effects of redox state on Ca2+ flux mediated by changes in membrane potential, CaL channels are also themselves directly responsive to changes in pO2 [27].
The mechanism responsible for altering redox status during SMC oxygen-sensing is thought to reside within the electron transport chain (ETC) of the mitochondria. As electrons flow through the inner mitochondrial membrane, leakage of electrons occurs at several distinct sites [28]. Leaked electrons from complexes I and III combine with molecular oxygen to produce superoxide anion (O2 ·−), a toxic form of ROS [29–31]. Superoxide is quickly converted into hydrogen peroxide (H2O2) by the actions of superoxide dismutase (SOD2), a less toxic ROS with a larger diffusion radius [3, 32]. Both superoxide anion and H2O2 are capable of oxidizing different classes of cellular targets; H2O2 has an important role as a signaling molecule due to its ability to oxidize thiol moieties on cysteine or methionine residues, causing structural and functional changes in target proteins through the formation of disulfide bridges [33]. This redox signaling mechanism allows mitochondria to regulate ion channels and enzymes.
There is teleological reasoning to the ETC serving as a sensor since the ETC relies on oxygen as its terminal electron acceptor, and this upstream redox sensor function leads to changes in protein function that optimize oxygen delivery to mitochondria. During HPV, a decrease in the electron flux through the ETC leads to decreased ROS production, shifting the cell to a more reduced state and promoting reductive modification of K+ channels. These modifications reduce the open state probability of Kv channels causing membrane depolarization, leading to Ca2+ influx and subsequent vasoconstriction. Hypoxia does not have this effect on renal [34] or mesenteric arterial SMC [35], a reminder that there is mitochondrial diversity with PASMC having mitochondria that are functionally different than those in systemic arterial SMC [34].
Although the role of the mitochondria in producing ROS is well established, the vector of change in ROS production in response to physiological hypoxia (decreased vs increased) remains controversial. While several investigators, including our group, report that ROS decrease in direct proportion to falling pO2 with hypoxia [36–41], other groups found that ROS production increases in response to hypoxia [42–48]. These contrasting findings have been attributed to the degree of hypoxia used to assess HPV (hypoxia vs anoxia), the use of freshly isolated PASMC versus passaged cell lines, as well as the complexity of measuring ROS in subcellular compartments. An interesting find by Lopez-Barneo et al. suggests that ROS production during hypoxia differs depending on the mitochondrial compartment measured [49]. In the carotid body, hypoxia is found to increase ROS production in the mitochondrial intermembrane space (IMS) while decrease ROS production in the mitochondrial matrix. Similarly, Waypa et al. demonstrated that hypoxia induces compartmental effects on ROS production within vascular SMC, with the mitochondrial matrix becoming more reduced and the cytosol and mitochondrial IMS becoming more oxidized [50]. However, our group has consistently reported that in freshly isolated, resistance level PASMC or resistance level PA rings, under physiologic hypoxia (pO2 40–60mmHg) and physiologic pH (7.35–7.45), ROS decrease, both in aggregate, in the cytosol and in the mitochondria.
It is important to note that despite the controversy pertaining to the direction of ROS changes in response to hypoxia, and whether or not these changes depend on the cellular compartment measured, there is a consensus that ROS are mitochondrially-derived and act as the diffusible signaling molecules in HPV. Therefore, the signalling ROS must be able to travel out from the mitochondria towards the cytosol and cell membrane, to modulate the activity of redox-sensitive ion channels and elicit changes in vascular tone. In our recent publication, using dynamic, compartment-targeted probes (HyPer-dMito for mitochondrial H2O2 and HyPer-dCyto for cytosolic H2O2 respectively), we have demonstrated decreased levels of H2O2 in both the mitochondria and cytosol of PASMC following exposure to acute, physiologic, hypoxia [51] (Figure 3).
1.2. HPV and Pulmonary Arterial Hypertension
Pulmonary hypertension (PH) is defined by a resting mean PAP (mPAP) ≥ 20 mmHg but is further subdivided based on the aetiology and pathogenesis [52]. According to the Fifth World Symposium on Pulmonary Hypertension, PH is divided into 5 groups: PH due to pulmonary vascular disease (i.e., pulmonary arterial hypertension, PAH, Group 1), PH due to left heart disease (Group 2), PH due to lung disease or hypoxia (Group 3), PH due to chronic thromboembolic disease (CTEPH, Group 4), or PH with unclear and/or multifactorial mechanisms (Group 5) [53]. PH of any type increases mortality and can cause right ventricular failure (RVF) and death. Though most forms of PH are not driven by environmental hypoxia (except for Group 3 PH), however, epigenetic changes in the oxygen sensing pathway (which largely mimic aspects of hypoxia) are involved in all forms of PH.
A hallmark of PAH is adverse pulmonary vascular remodeling, which contributes to increased pulmonary vascular resistance (PVR). The pulmonary arterial wall is divided into three layers: the intima, media and adventitia. Pulmonary artery endothelial cells (PAEC) and PASMC are the principal components of the intimal and medial layers, respectively. The adventitial layer, whose principal resident cells are fibroblasts (PAfib), are less studied. All three layers are involved in the pulmonary remodeling in PH and this is particularly evident in Group 1 PH, where PAEC dysfunction, increased PASMC proliferation and concomitant impaired apoptosis lead to intimal hyperplasia and medial hypertrophy. Within the adventitia, fibrosis is also observed due to fibroblast proliferation and increased production of collagen, accompanied by a variable degree of perivascular inflammation [10, 54].
1.3. Abnormal oxygen-sensing in PAH – normoxic activation of Hypoxia-Inducible Factor (HIF)
In response to tissue hypoxia, the HIF family of transcription factors is activated and initiates the transcription of hundreds of genes used to combat hypoxia-induced stress [55, 56]. The HIF protein complex is comprised of two subunits, an alpha subunit (HIF-α), which exists in the cytoplasm, in three distinct isoforms (HIF-1α, HIF-2α and HIF-3α) and a beta subunit (HIF-β), which is stably expressed, independent of pO2, in the nucleus. Although both HIF subunits are persistently synthesized within the cell, the presence of HIF-α subunits is inversely proportional to the concentration of oxygen within the cell [57], reflecting a tonic process of oxygen-dependent HIF-α degradation.
Under normoxic conditions, oxygen-sensitive prolyl hydroxylases (PHD) modify proline residues within the HIF-α subunit. Proline hydroxylation allows the von Hippel-Lindau protein (VHL) to ubiquitinate HIF-α, targeting it for proteasomal degradation [58]. Under hypoxic conditions, PHD becomes inactivate. Consequently, HIF-α is not targeted for degradation by VHL, allowing HIF-α to accumulate within the cytoplasm and translocate into the cell nucleus where it dimerizes with HIF-β to form the active HIF complex [58]. HIF complexes in the nucleus are capable of binding to hypoxia response elements (HRE) located within regulatory elements of genes known to control processes related to oxygen delivery and oxygen deprivation, including pathways involved in cell metabolism, angiogenesis, erythropoiesis, cell proliferation, and apoptosis [59–61].
While activation of the HIF pathway by environmental hypoxia is often adaptive for the organism, normoxic activation of the HIF-α pathway is a hallmark of numerous pathologies. A rare but highly instructive example is Chuvash disease, which results from a homozygous missense mutation in the VHL gene. Patients with Chuvash disease manifest sustained pulmonary vasoconstriction, polycythemia and spontaneous PH, all findings that would be expected in the presence of environmental hypoxia, but which occur in the absence of tissue hypoxia [62–64]. In the absence of a functional VHL protein, HIF-α subunits become transcriptionally active. Thus, despite normoxic conditions, HIF target genes, such as erythropoietin and vascular endothelial growth factor (VEGF) are upregulated [65–67], causing these patients to display phenotypes similar to those observed during chronic hypoxia. Collectively, the role of pathologic VHL and HIF-α activation highlights one example of how subversion of oxygen-sensing pathways contributes to the pathological processes which promote disease pathogenesis in both PAH and cancer (Figure 4).
Figure 4. Role of HIF-1 under normoxia, hypoxia and pseudohypoxia.
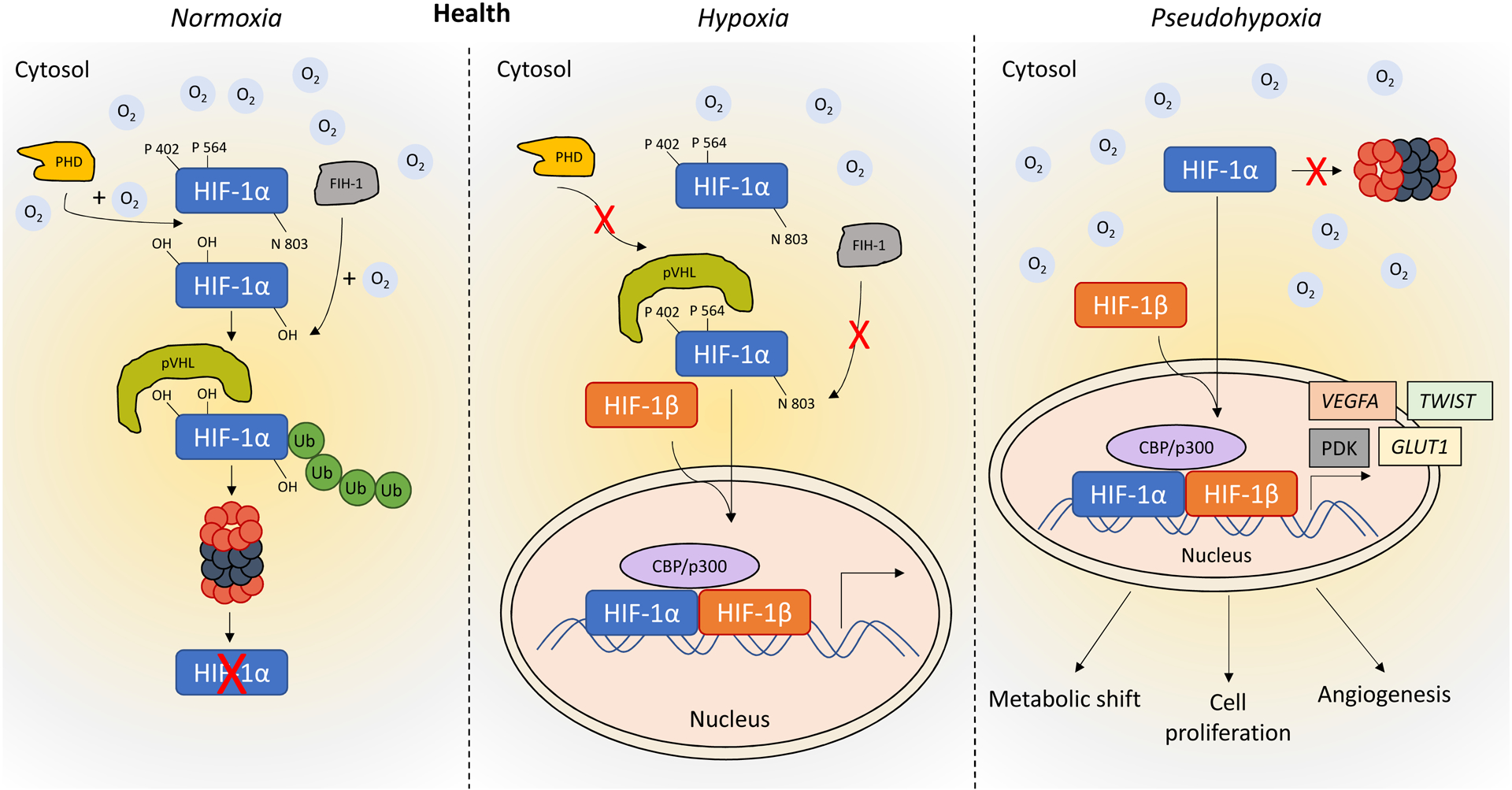
In normoxic conditions, prolyl hydroxylases (PHDs) modify proline residues within the HIF-1α subunit. This allows the von Hippel-Lindau protein (VHL) to ubiquitinate HIF-1α, targeting it for proteasomal degradation. Under hypoxic conditions, PHD is inactivate. As a consequence, HIF-α is not targeted for degradation by VHL. This allows HIF-α to accumulate within the cytoplasm and translocate into the cell nucleus where it dimerizes with HIF-1β to form the active HIF complex. HIF complexes in the nucleus are capable of binding to hypoxia response elements (HRE) located within regulatory elements of genes known to control processes related to oxygen delivery and oxygen deprivation, including pathways involved in cell metabolism, angiogenesis, erythropoiesis, cell proliferation, and apoptosis. Normoxic activation of HIF-1α (pseudohypoxia, as seen in PAH and cancer) contributes to the metabolic shift, cell proliferation and angiogenesis.
Chuvash disease demonstrates that a genetic mutation that causes HIF-1α activation results in a pseudohypoxic environment that drives a hyperproliferative, apoptosis-resistant PASMC phenotype. However, disordered oxygen sensing can also occur by epigenetic mechanisms. Fawn hooded rats (FHR) have normoxic activation of HIF-1a signaling and spontaneously develop PAH, characterized by excessive proliferation and impaired apoptosis of PASMC [68]. In contrast to Chuvash patients, normoxic activation of HIF-1α is largely a result of an epigenetic reduction in expression of mitochondrial SOD2, the main source of mitochondrial-derived H2O2. Furthermore, HIF-1α is largely regulated by the redox status. Hypoxic stabilization of HIF-1α is prevented by H2O2 and sulfhydryl oxidants where it is enhanced by reducing agents [69]. This redox regulation is similarly important to HPV and is a reminder that hypoxia is a state of reduction whilst normoxia is a state of oxidation, as originally proposed by Archer and Weir [70].
In both the FHR-PAH model and patients with PAH, SOD2 expression is decreased within the lungs and PA [71]. SOD2 deficiency in the FHR is epigenetically regulated, resulting from covalent cytosine methylation of two key CpG islands within the SOD2 gene promoter and enhancer regions [68]. These methylation events, controlled by DNA methyltransferases (DNMT), interfere with the binding of transcription factors. In FHR, upregulation of DNMT1 within the lungs, as well as DNMT3B within PASMC may explain the observed hypermethylation of the SOD2 gene. Treatment of FHR with the DNMT inhibitor, 5-aza-2’-deoxycytidine (5-AZA), restores SOD2 expression and corrects the rates of PASMC proliferation and apoptosis [68]. The therapeutic potential of 5-AZA is particularly noteworthy as it is currently approved in the treatment of myeloproliferative disorders [72]. Therapy with the SOD mimetic Mn(III)tetrakis(4-benzoic acid)porphyrin chloride (MnTBAP) also regresses PAH in the FHR, evident by its ability to reduce right ventricular (RV) hypertrophy, improve functional capacity, and decrease muscularization of pulmonary precapillary resistance vessels [68]. These results are consistent with those of 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPOL), a SOD mimetic shown to decrease HPV in rats, as well as treatment with recombinant SOD1, which reduces PVR in persistent pulmonary hypertension in newborn lambs [73, 74]. Epigenetic silencing of SOD2 by hypermethylation also enhances cell proliferation in multiple cancer types, including pancreatic cancer and myeloma [75–77]. As in PAH, overexpression of SOD2 increases H2O2 levels in cancer cells, which decreases rates of cell proliferation and tumor growth [75, 77, 78]. DNMT3B is also involved in epigenetic silencing of SOD2 within cancer cells. In human breast cancer and A549 cells (a non-small cell lung cancer cell line), depletion of DNMT3B reactivates methylation-silenced gene expression, and induces apoptosis [79].
The HIF-1α and HIF-2α subunits have similar DNA binding and dimerization domains but have distinct transactivation domains and effect unique gene targets. For example, HIF-1α regulates glycolytic genes whilst HIF-2α does not [80]. However, a number of hypoxia-regulated genes, such as VEGFA (encoding VEGF-A), are regulated by both HIFs. Activation of HIF-2α occurs in the PAEC of patients with idiopathic PAH and in rodent PAH models [81]. HIF-2α activation, secondary to PHD deficiency, contributes to obliterative vascular remodeling, leading to RVF and death [82, 83]. HIF-2α+/− mice exposed to chronic hypoxia exhibit diminished increases in RV systolic pressure and RV hypertrophy [84]. In PAEC, HIF-2α activation induces the production of the vasoconstrictor endothelin-1 and alters nitric oxide homeostasis by increasing the expression of arginase, both of which contribute to the development of HPV. Pharmacological inhibition of HIF-2α translation by the inhibitor compound 76 (C76) [85] can improve survival in rodent models of PAH by inhibiting obliterative pulmonary vascular remodelling, reducing RV hypertrophy and cardiac fibrosis and inhibiting RVF [81]. C76 treatment is found to be selective for HIF-2α, without affecting HIF-1α in human lung microvascular ECs. Selective HIF-2α inhibitors, such as PT2385 and its more potent analog, PT2977, have already been tested in clinical trials in the treatment of patients with solid tumors, including glioblastoma and renal cell carcinoma (NCT03216499, NCT02293980, NCT03401788, NCT03634540, NCT02974738). The relative importance of HIF-1α versus HIF-2α remains unclear in PAH and cancer.
1.4. HIF-activated metabolic changes in PAH and cancer
PAH is marked by metabolic abnormalities, most notable being a shift from oxidative metabolism to aerobic glycolysis, in which glycolysis rates are disproportionately increased in relation to mitochondrial pyruvate utilization and thus are considered ‘uncoupled’ [86]. This uncoupling effect is mediated in part, by the actions of HIF-1α. The two major targets of HIF-1α responsible for this effect are lactate dehydrogenase A (LDH-A) and pyruvate dehydrogenase kinase (PDK) 1 [87, 88]. LDH-A converts pyruvate into lactate, using NADH as a co-factor. This provides a means of replenishing the pool of NAD+ which is required for glycolysis, which would normally be replenished through mitochondrial metabolism. PDK1 phosphorylates and inactivates PDH, preventing the production of acetyl-CoA from pyruvate, and decreasing the net oxidative metabolic rate. In monocrotaline (MCT)-PAH rats, both PDK1 and PDK3 are upregulated and the resulting PDH inhibition contributes to the fibrogenic, hyperproliferative Warburg phenotype of RV fibroblasts (RVfib) [89]. Inhibition of either PDK1 or PDK3 reduces cell proliferation, mitochondrial fission, and collagen production within the RVfib, suggesting either of these PDK isoforms as possible therapeutic targets in treating PAH.
The HIF-1α-PDK-PDH pathway is not the only way that PAH and cancer cells develop Warburg metabolism. Pyruvate kinase (PK) is responsible for the final step of glycolysis, converting phosphoenolpyruvate into pyruvate. PK muscle isoform (PKM) 1 and 2 are isoforms of PK, formed by alternative splicing of the PKM2 RNA transcript. In cancer, a switch from PKM1 to PKM2 expression is partially responsible for increased rates of aerobic glycolysis [90]. Increased expression of PKM2, but not PKM1, increases tumor growth. Within cancer cells, PKM2 is not only a target of HIF-1α, but also acts as a transcriptional coactivator of HIF-1α, increasing HIF-1α binding to HRE and inducing expression of known HIF targets [91]. In this way, PKM2 participates in a positive feedback loop which serves to promote Warburg metabolism. PKM2 has also been observed to associate with HIF-2α, increasing its transcriptional activity [91]. In HeLa (human cervical adenocarcinoma) and Hep3B (hepatocellular carcinoma) cells, PKM2 and HIF-1α expression are associated with increased expression of metabolic genes, including LDH-A and PDK1. Although no association has been found between PKM2-HIF-2 activation and metabolic enzymes, both PKM2-induced HIF-1 and HIF-2 activation induce the expression of VEGF [91].
HIF-1α also promotes aerobic glycolysis through the regulation of a number of other proteins involved in glucose metabolism. HIF-1α activation increases the expression of glucose transporters (GLUT) 1 and 3, as well as glycolytic enzymes hexokinase (HK) 1 and 2, aldolase A, enolase, 6-phosphofructo-1-kinase, and phosphoglycerate kinase 1 [92–95]. In both cancer cells and vascular cells from PAH patients, the upregulation of glucose transporters appears to be compensatory, allowing massive increases in glucose flux to maintain ATP homeostasis. It is the reliance of these diseased cells on glucose uptake which accounts for the clinical utility of 2-[18F]-Fluoro-2-deoxy-d-glucose (FDG)-positron emission tomography (PET) in the detection of cancers and PAH [96, 97]. Using FDG as a radiotracer, PET can visualize the increase in glucose flux that is required to maintain energy homeostasis in vivo.
While originally believed to not play a role in the regulation of the glycolytic pathway, HIF-2α is capable of regulating some of the same gene targets as HIF-1α, although its role in HIF-activated Warburg metabolism is not well defined [80, 98–101] (Figure 5).
Figure 5. Mediators of Warburg metabolism in PAH and cancer.
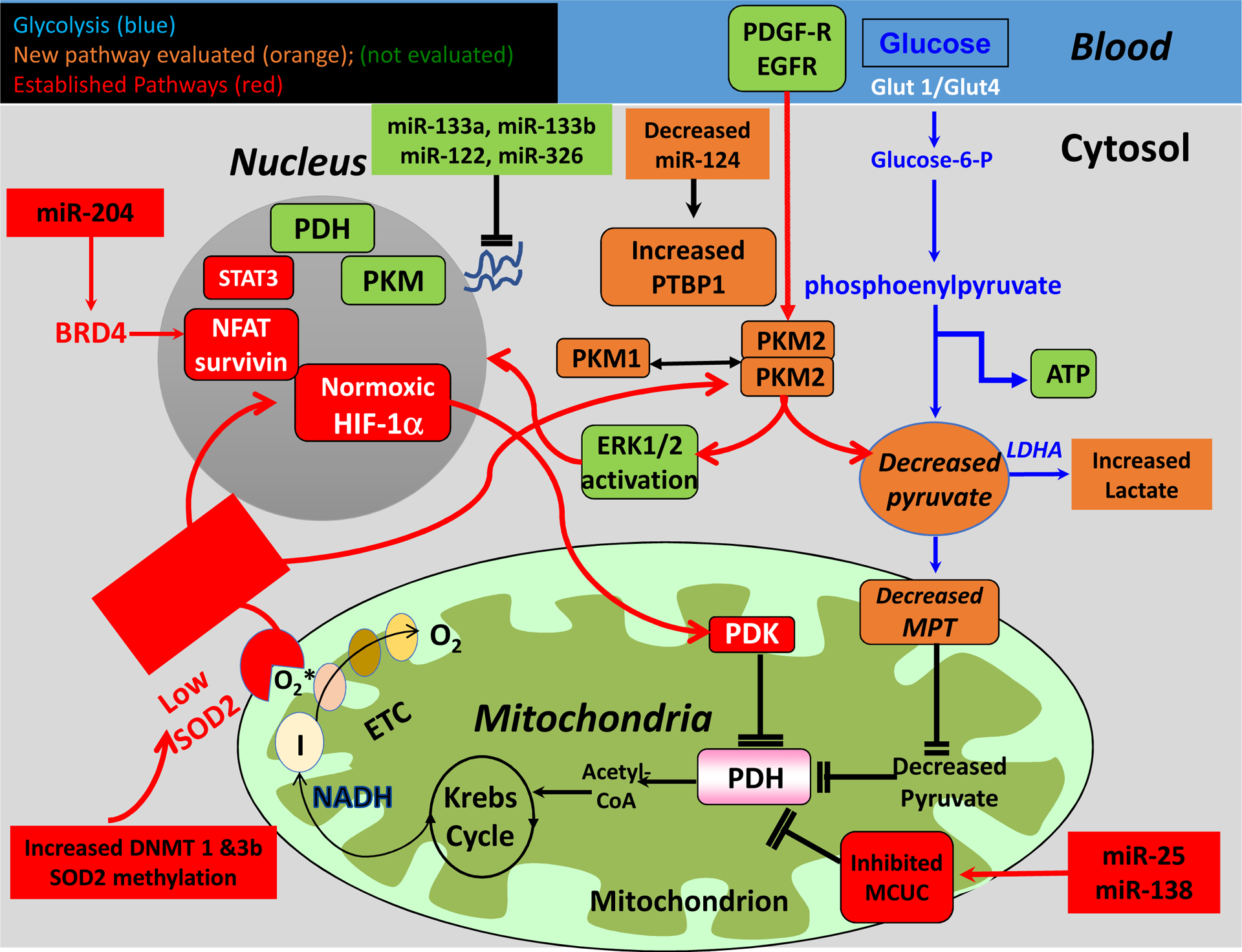
miR: microRNA; BMPR2: bone morphogenetic protein receptor type 2; PTBP1: polypyrimidine tract-binding protein; PKM: pyruvate kinase muscle isozyme; HIF-1α: hypoxia-inducible factor-1α; ERK: extracellular signal related kinase; DNMT: DNA methyltransferase; SOD2: superoxide dismutase 2; ETC: electron transport chain; PDH: pyruvate dehydrogenase; PDK: pyruvate dehydrogenase kinase; MCUC: mitochondrial calcium uniporter complex; NFAT: nuclear factor of activated T cells; EGFR: endothelial growth factor receptor; LDHA: lactate dehydrogenase A; MPT: mitochondrial pyruvate transporter; PDGF-R: platelet-derived growth factor receptor; STAT3: signal transducer and activator of transcription 3.
Copyright © Circulation
1.5. Abnormal oxygen sensing in cancer
Overexpression/activation of HIF-1α is common in cancer and can be both oxygen-dependent (due to inadequate blood supply and tumor hypoxia) and oxygen-independent (due to genetic alteration of upstream regulators).
1.5.1. HIF-activated cell proliferation in cancer
One of the major phenotypes of cancer is cell hyperproliferation and apoptosis resistance. It is common for cancer cells to undergo autocrine stimulation by the growth/survival factors they secrete. HIFs regulate a wide variety of growth factors, including transforming growth factor-α (TGF-α, encoded by TGFA), insulin-like growth factor-2 (IGF-2, encoded by IGF2), VEGF, endothelin 1, adrenomedullin and erythropoietin.
TGF-α is a mitogenic polypeptide belonging to the epidermal growth factor (EGF) family. In clear cell renal cell carcinoma (RCC), the tumor suppressor gene VHL is often mutated biallelically (VHL−/−). As a result, HIF-1α is activated regardless of the oxygen level in the tissue. Gunaratnam et al. found that in VHL−/− RCC cells, HIF-1α promotes cell proliferation by activating the TGFA/EGF receptor (EGFR) pathway [102]. In pancreatic cancer, activation of protease-activated receptor-2 (PAR-2) upregulates both HIF-1α and HIF-2α by the integrin-linked kinase (ILK) signaling pathway, which in turn increases the transcription level of TGFA [103]. This leads to the progression of human pancreatic cancer. HIF-1α-dependent TGFA activation is also observed in human non-small cell lung cancer (NSCLC) [104].
EGFR (encoded by EGFR) is a member of the ErbB family of receptors which includes four receptor tyrosine kinases. Gain-of-function mutations of EGFR is confirmed in many types of cancer. The Semenza group first found that activation of phosphoinositide 3-kinase (PI3K)/phosphatase and tensin homolog (PTEN)/protein kinase B (Akt)/FKBP-rapamycin-associated protein (FRAP) pathway contribute to the normoxic activation of HIF-1α in prostate cancer [105]. Peng et al. later reported a crosstalk between the EGFR pathway and the HIF-1α pathway contributing to the apoptosis-resistant phenotype of cancer cells [106]. In breast cancer cells, activation of EGFR by EGF stimulation increases survivin-mediated apoptosis resistance under normoxic condition. However, EGF does not increase survivin expression in normal mammary epithelial cells. EGFR upregulates normoxic HIF-1α expression through the PI3K/Akt pathway.
Human epidermal growth factor receptor-2 (HER2), another ErbB family member, is overexpressed in 20–25% of breast cancers and is correlated with both HIF-1α and HIF-2α expression [107]. Independent of oxygen availability, HER2 activation increases the levels of HIF-2α signaling in breast cancers, suggesting that HER2 activation may exacerbate tumor pathology [108]. HIF-2α-specific inhibition, through the use of siRNA or C76, decreases HER2-positive breast cancer cell growth rates, suggesting that targeting HIF-2α may be a therapeutic strategy for treatment of HER2 positive breast cancers.
IGF-2 is a protein growth factor with insulin-like structure. It stimulates mitosis and is increased in many cancers. The Semenza group first demonstrated the reciprocal positive regulation between HIF-1α and IGF-2 [109]. Both insulin and IGFs (IGF-1 and IGF-2) can induce HIF-1α activation under normoxic condition. Activated HIF-1α initiates the expression of its downstream genes, including IGF-2, IGF-binding protein (IGFBP)-2 and IGFBP-3. This suggests a role for HIF in a positive feedback loop in the autocrine regulation of tumor growth factors. Furthermore, Mohlin et al. showed that in neuroblastoma, the expression levels of HIF-1α and IGF-2 are correlated, and this correlation is strongest in high-stage tumors [110]. The expression of both IGF-2 and HIF-1α in neuroblastoma are hypoxia-dependent.
1.5.2. HIF-activated angiogenesis in cancer
In cancer cells, localized hypoxia can occur and activation of angiogenesis counteracts hypoxia by increasing the delivery of oxygen and nutrients to the tumor. Unsurprisingly, HIF is viewed as a master regulator of angiogenesis, promoting vessel growth by activating multiple pro-angiogenic pathways. Pro-angiogenic genes, including VEGFA, ANGPT2 (encoding angiopoietin 2) and TEK (encoding TEK receptor tyrosine kinase), are often used as biomarkers for tumor hypoxia, and are implicated in HIF-mediated angiogenesis.
Central to the process of angiogenesis, HIF-1α and HIF-2α regulate the expression of VEGF [56, 111–113]. VEGF increases vessel permeability and is implicated in the proliferation and migration of endothelial cells and pericytes during neovascularization. Activation of HIF-1α and HIF-2α, due to loss of VHL function, occurs in highly vascularized tumors, although the relative roles of these HIFs in angiogenesis varies depending on the cell type [113]. For example, in RCC, VHL-associated hemangioblastoma, and liver hemangioma, HIF-2α is the dominant HIF isoform controlling VEGF gene expression and vascular tumorigenesis [98, 114]. In contrast, HIF-1α plays a major role in oral cancer and breast cancer [115, 116].
HIF is also known to play a role in the activation of angiopoietin (Ang) 1 and Ang2, as well as the angiopoietin 1 receptor, Tie2. During angiogenesis, Ang1 supports the action of VEGF, acting as an agonist for the Tie2 receptor. Through Ang1/Tie2 signaling, Ang1 plays a role in vascular remodeling and stabilization of newly formed vessels by supporting interactions between endothelial cells and their surrounding environment. In contrast, Ang2 antagonizes the Tie2 receptor, destabilizing existing vessels and shifting them to a more plastic state. Ang2 upregulation is also involved in the pathogenesis of multiple cancer types, including colorectal cancer [117–121]. Ang2 is a direct target of HIF-2α and may also be indirectly targeted by HIF-1α, since VEGF activation can also stimulate Ang2 expression [122, 123]. Ang2 repression regresses tumor vascularization and tumor metastasis in mammary carcinoma as well as pancreatic insulinoma [124]. It has also been reported that Tie2 expression is partially controlled by HIF-2α in endothelial cells. Within tumor-infiltrating myeloid cells, Tie2 knockdown decreases tumor angiogenesis [124, 125].
1.5.3. HIF-activated epithelial-to-mesenchymal transition (EMT) in cancer
EMT is critical during embryonic development, tissue regeneration, organ fibrosis and wound healing [126]. This process describes the transition of a cell from an epithelial state, where cells exhibit epithelial cell-to-cell junctions and apical-basal polarity, towards a mesenchymal state, in which cells lack polarity and have heightened motility [127]. Although often viewed as a binary switch, EMT often involves only the acquisition of certain mesenchymal traits, while retaining some epithelial traits, which leads to mixed epithelial/mesenchymal phenotypes [128]. In cancer, by increasing cell motility and the ability to degrade components of the extracellular matrix, EMT activation allows cancer cells to invade the bloodstream, settle in the microvasculature, and extravasate from the vasculature to begin proliferating in secondary sites.
Adherence of epithelial cells to each other and the basement membrane is maintained by cell adhesion molecules, most notably E-cadherin, and by tight junction proteins. The transition of epithelial cells from the expression of E-cadherin to N-cadherin, as well as the increased expression of the mesenchymal markers fibronectin and vimentin, indicate EMT transition [129]. EMT is controlled largely by EMT transcription factors which bind to the promoter regions and repressing the expression of various cell adhesion genes. These transcription factors, including TWIST, Snail, SIP1, Zeb1 and Slug, have also been implicated in the progression of different cancer cell types and are controlled in part by HIF-1α [130–139]. TWIST is a master regulator of mesoderm development and embryonic morphogenesis that is also implicated in cancer metastasis [140]. HIF-1α regulates TWIST expression, promoting EMT and cancer metastasis [133]. The co-expression of HIF-1α with TWIST and Snail is associated with worsened prognosis in patients with head or neck cancers [133]. Furthermore, HIF-1α-mediated TWIST activation plays an important role in the progression of ovarian epithelial cancers and is associated with decreased survival [134]. Similar to TWIST, Upregulation of Snail induces invasion and metastasis in a variety of cancer types and promotes mammary tumor recurrence [130, 141, 142].
Although less studied than HIF-1α, HIF-2α is also implicated in EMT and cancer pathogenesis. In pancreatic cancer tissue, HIF-2α activation is associated with more aggressive cancer phenotypes and lymph node metastasis [143]. HIF-2α also increases the expression of matrix metalloproteinase (MMP) 2 and MMP9, the enzymes implicated in tumor metastasis and EMT [144]. Additionally, in gastric cancer cells, both HIF-2α and HIF-1α expression is correlated with more advanced clinical stage and are upregulated in metastasizing cancers. Small interfering RNA (siRNA) against both HIF-1 subtypes reduces the migration and invasion of both gastric and lung cancer cells, suggesting a possible role for both HIF-1 subtypes in promoting cancer metastasis [145, 146].
2. Mitochondrial biology and experimental therapeutics in PAH and cancer
2.1. Aerobic glycolysis (Warburg effect) and their experimental therapeutics
2.1.1. Glycolysis and glucose oxidation
Changes in mitochondrial metabolism vary, based on cell type, but include alterations in aerobic glycolysis, fatty acid oxidation, as well as the induction of glutaminolysis [147]. These metabolic changes are also associated with altered mitochondrial ROS production, bioenergetics and disruptions in both mitochondrial membrane potential and mitochondrial morphology [148].
Glucose is transported into the cytosol by GLUTs in the cell membrane. In the cytosol, glucose is first catalyzed to glucose-6-phosphate (G-6-P) with the enzyme, HK. After a sequence of catalytic reactions, G-6-P is converted to pyruvate by PK. Pyruvate is the final product of glycolysis and when oxygen is present, it normally enters the mitochondria via the mitochondrial pyruvate carrier (MPC) [149]. Within the mitochondrial matrix, pyruvate is oxidized by PDH to form acetyl-CoA, which feeds Krebs cycle. Krebs cycle generates the electron donors (NADP and FADH) that enter the ETC at Complex I and II respectively, leading to oxidative phosphorylation (OXPHOS) and ATP generation. Glycolysis is a cytoplasmic pathway that converts glucose into lactate and generates adenosine triphosphate (ATP). Glucose oxidation in the mitochondria generates 32 ATP molecules per molecule of glucose, while glycolysis in the cytosol only generates 2 moles of ATP per mole of glucose.
Normally, glycolysis and glucose oxidation are coupled to each other. In certain diseases, notably those characterized by excess rates of cell proliferation and impaired apoptosis, pyruvate is primarily converted to lactate by the enzyme LDH and glucose oxidation is depressed, even in the presence of oxygen. Such uncoupled aerobic glycolysis, was first discovered in cancer cells by Warburg about one century ago [150]. Warburg metabolism confers a competitive advantage to affected cells since it largely eliminates cell death by mitochondria-mediated apoptosis.
Uncoupled aerobic glycolysis contributes to the hyperproliferative and apoptosis-resistant phenotype in many cancer cell types, promoting tumor growth [151]. In PAH, uncoupled aerobic glycolysis also promotes a cancer-like phenotype in PA vascular cells [68, 152, 153] and RVfib [89]. These acquired metabolic abnormalities contribute to adverse PA modeling and lead to key pathologic features of PAH, including increased PVR. For RV myocytes, which are not proliferative, uncoupled aerobic glycolysis results in mitochondrial metabolic dysfunction, which leads to RV myocyte hypertrophy, reduced contractility and even apoptosis [154, 155]. The elevated glucose flux required to support energy homeostasis in a state of uncoupled glycolysis is also pathologic. Increased glucose levels in the PAH RV contribute to RV dysfunction by a form of posttranslational modification of proteins called O-GlcNAcylation [156]. Increased O-GlcNAcylation of mitochondrial proteins in MCT RVs leads to mitochondrial dysfunction and RVF in MCT-PAH and this glucose-related dysfunction can be reversed by colchicine [156].
Abnormalities in the many upstream regulators that affect the mitochondrial OXPHOS pathway can promote or facilitate the Warburg effect (See recent review in [148]). However, since PKM2 and PDH are rate-limiting enzymes for pyruvate production and the conversion of pyruvate to acetyl-CoA, respectively (which are essential steps for glucose oxidation), we focus on the canonical regulation, and nuclear translocation of PKM2 and PDH as key regulators of Warburg metabolism (Figure 5).
2.1.2. PKM2 In the Nucleus
2.1.2.1. Nuclear Translocation of PKM2 and its Regulation
PKM2 is predominately found in the cytosol. However, under various conditions, PKM2 can translocate into the nucleus. In the nucleus, PKM2 functions as a coactivator for several transcriptional factors to promote the Warburg effect. Factors that promote PKM2 nuclear translocation include inflammatory and proapoptotic factors, including interleukin-3, UV irradiation, H2O2, and somatostatin analog TT-232 [157, 158]. Additionally, phosphorylation of PKM2 at serine 202 by Akt, under IGF-1 stimulation, induces PKM2 nuclear translocation [159].
PKM2 nuclear translocation involves the EGF-mitogen-activated protein kinase 1 (ERK2)-PKM2 axis. ERK2 activation by mitogenic growth factors, such as platelet-derived growth factor (PDGF) and EGF, exposes its docking groove to PKM2. ERK2 phosphorylates PKM2 at serine 37 (S37), after which never in mitosis A-1 (NIMA-1, also known as PIN1)-dependent cis–trans isomerization and conversion of PKM2 S37 from a tetramer to a monomer occurs. Monomeric PKM2 then binds to importin α5 and is translocated into the nucleus [160]. This is most likely only one of several mechanisms because nuclear translocation by phosphorylating PKM2 at serine 202 by Akt under IGF-1 stimulation is not dependent on S37 phosphorylation [159]. Jumonji C domain containing dioxygenase 5 can also directly interact with PKM2 and promote PKM2 translocation into the nucleus [161]. Conversely, sirtuin 6 reduces PKM2 translocation into the nucleus by deacetylating PKM2 at lysine 433, resulting in reduced cell proliferation [162].
2.1.2.2. Nuclear PKM2 as a Coactivator
In cancer cells and under hypoxic conditions, nuclear PKM2 functions as a coactivator of HIF-1α. PKM2 interacts directly with HIF-1α and recruits the coactivator p300-acetyltransferase to enhance HIF-1α binding to the HRE of HIF-1α target genes [91]. PKM2 is also a target gene of HIF-1α, creating a positive feedback loop that maintains and continuously promotes the proglycolytic shift seen in cancer cells [91].
Nuclear PKM2 also directly regulates the Warburg effect through transcriptional regulation of c-Myc. Once bound to β-catenin phosphorylated at tyrosine 333 (Y333), the PKM2-β-catenin complex phosphorylates histone H3 at threonine 11, causing the dissociation of histone deacetylase 3 from the c-Myc promoter region [163]. PKM2-induced c-Myc expression creates a positive feedback loop since c-Myc increases the expression of heterogeneous nuclear ribonucleoproteins (hnRNP) A1, hnRNPA2, and polypyrimidine tract-binding protein (PTB), which promotes the alternative splicing of PKM2 over PKM1 [164]. Furthermore, c-Myc promotes the Warburg effect by increasing the transcription of HK2, LDHA, and PDK1 [165]. Thus, PKM2 promotes Warburg metabolism though diverse and mutually reinforcing mechanism.
2.1.3. PDH in the Nucleus
The pyruvate dehydrogenase complex (PDC) consists of three enzymes: pyruvate dehydrogenase (E1), dihydrolipoamide acetyltranseferase (E2), and dihydrolipoamide dehydrogenase (E3) [166]. Together, PDC irreversibly catalyzes the conversion of pyruvate to acetyl-CoA, thus linking glycolysis to Krebs cycle. Sutendra et al. demonstrated the nuclear presence of PDC in primary fibroblasts from human lungs, normal small airway epithelial cells, and A549 cells [167]. Nuclear PDC-E1 levels increase in parallel to decreasing mitochondrial PDC-E1 levels under serum stimulation without change in total PDC. The N-terminus of newly translated PDC contains a mitochondrial localization sequence (MLS), which is cleaved by mitochondrial-processing peptidase when entering the mitochondrial matrix [168]. Nuclear PDC does not contain a mitochondrial localization sequence, demonstrating that newly translated PDC is first processed in the mitochondria prior to translocating to the nucleus.
PDC-E1 translocation from the mitochondria to the nucleus occurs during the S phase and is undetected within the nucleus of quiescent cells. Nuclear PDC-E1 is most likely constitutively active, given the absence of nuclear PDK. This also suggests that nuclear PDC-E1 is regulated differently than mitochondrial PDC-E1. Nuclear PDC-E1 can generate acetyl-CoA from pyruvate. Cells treated with siPDC-E1 produce significantly less acetyl-CoA, while increasing levels of pyruvate restore levels of acetyl-CoA, indicating the presence of nonlimiting amounts of functional nuclear PDC-E1. Acetyl-CoA, produced by nuclear PDH-E1, is utilized for histone acetylation of histones H2B, H3, and H4 and for S phase entry. However, nuclear PDC inhibition does not alter acetylation of its other targets, such as p53 and FOXO1, indicating that nuclear PDC’s acetylation activity is tightly regulated [167]. Interestingly, nuclear PDC cannot be subject to phosphorylation regulation because PDK is absent from the nucleus. Instead, nuclear PDC may be regulated by the availability or proximity of acetyltransferases or deacetylases [167].
The mechanism by which PDC translocation occurs remains unelucidated. Although the nuclear pore complex can allow the translocation of large protein complexes, it remains unclear how a large complex like PDC be exported across the mitochondrial membrane. The authors indicate that the process may be dependent on heat shock protein 70 (Hsp70). Hsp70 is involved in the nuclear translocation of several proteins [169]. Nuclear Hsp70 levels increase with serum stimulation in parallel with nuclear PDC-E1. PDC’s PDK-binding site contains a putative Hsp70-binding motif. Hsp70 may allow PDC to remain active after translocation to the nucleus by competing with PDK for PDC’s PDK-binding site. Decreasing Hsp70 levels, using heat shock protein inhibitor I (KNK437), or using siRNA against Hsp70, decreases levels of nuclear PDC-E1.
2.1.4. Epigenetic Regulation of PKM2
PKM2 expression can be regulated at multiple levels. hnRNPs are a group of c-Myc regulated splicing factors (hnRNPA1, hnRNPA2, and PTBP1) that regulate PKM2 expression by altering PKM pre-mRNA. By binding to splicing signals that flank PKM exon 9, hnRNPs promote PKM2 expression [164].
MiRNAs provide another way in which PKM2 expression is regulated. MiRNAs are a class of small, endogenous, non-coding RNAs that serve to regulate post-transcriptional gene expression by targeting specific mRNAs for degradation or inhibiting their translation by binding to the 3’-untranslated region [170]. miRNAs regulate PKM2 activity in a tissue-specific manner either directly by targeting the PKM2 mRNA or indirectly through PTBP1. A summary of studies investigating miRNAs that target PKM2 in pulmonary hypertension and cancer cells along with their associated mechanism of action is provided in Table 1.
Table 1.
Summary of studies on miRNAs that target PKM2 in pulmonary hypertension and cancer cells and associated mechanistic pathway.
| MiRNA | Species | Cell Type | Mechanism |
|---|---|---|---|
| miR-1, miR-133b [381], miR-124, miR-137 & miR-340 [382] | Human | Colorectal cancer | PTBP1/PKM2 pathway |
| Bovine | Colorectal cancer | ||
| miR-133a & −133b [384] | Human | Tongue squamous cell carcinoma | Directly targets PKM mRNA |
| miR-122 [385] | Human | Hepatocellular carcinoma | Directly targets PKM2 mRNA |
| miR-148a & miR-326 [386] | Human | Thyroid cancer | Directly targets PKM2 mRNA |
| miR-148a & miR-152 [387] | Human | Breast cancer | Directly targets PKM2 mRNA |
| miR-152 [388] | Human | Breast cancer | miR-152/β-catenin/PKM2 pathway |
| miR-372 [389] | Human | Liver cancer | miR-372/YB-1/β-catenin/LEF/T CF 4-PKM2 pathway |
| miR-491–5p [390], miR-1294 [203] | Human | Osteosarcoma | Directly targets PKM2 mRNA |
| miR-361–5p [391] | Human | Prostate cancer | miR-361–5p/Sp1/PKM2 pathway |
| miR-let-7a [392] | Human | Cervical cancer | Directly targets PKM2 mRNA |
DNA methylation is another method through which PKM2 expression is regulated. Desai et al. discovered that intron 1 of the PKM gene is hypermethylated, resulting in reduced PKM expression. Conversely, hypomethylation of intron 1 of the PKM gene is associated with elevated PKM2 expression in various cancer types. This suggests that epigenetic regulation through DNA methylation of intron 1 may be a key mechanism through which PKM expression is increased in cancer [171]. Interestingly, Singh et al. observed higher DNA methylation at PKM exon 10 in breast cancer cells compared to normal human mammary epithelial cells, which correlates with the inclusion of the exon 10 of the PKM gene and increased PKM2 expression. DNA methylation recruits Brother of Regulator of Imprinted Sites (BORIS), a CCCTC-binding factor paralog, to bind to exon 10. BORIS increases RNA polymerase II activity at exon 10, which favours increased inclusion of exon 10 and thus increases PKM2 expression. Depletion of DNMT3B, but not DNMT1 or 3A, decreases exon 10 methylation, which correlates with decreased RNA polymerase II occupancy, and reduced exon 10 inclusion and PKM2 expression [172]. This DNA methylation-mediated recruitment of BORIS at exon 10 of PKM gene is also observed in head and neck cancer, which leads to the alternative splicing and generation of the PKM2 splice isoform [173]. These results suggest that DNA methylation is critical in regulating PKM2 expression in various types of cancers (Figure 6).
Figure 6. Mechanisms involved in imbalanced PKM2/PKM1 and therapeutic targets.
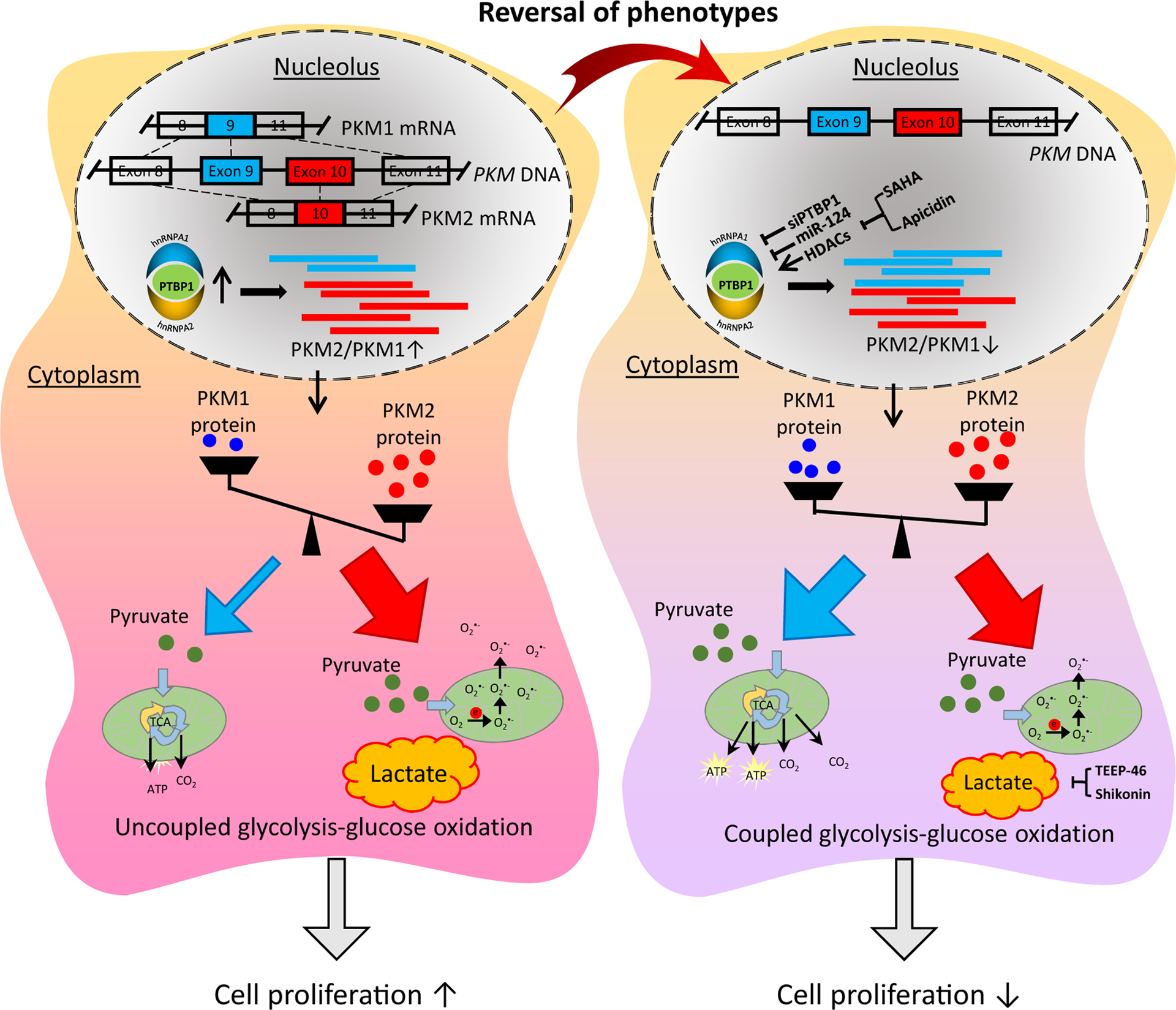
PKM: pyruvate kinase muscle isozyme isoform; PTBP1: polypyrimidine tract binding protein 1; hnRNP: heterogeneous nuclear ribonucleoprotein; TCA: tricarboxylic acid; ROS: reactive oxygen species; HDAC: histone deacetylase; SAHA: suberoylanilide hydroxamic acid.
Copyright © Comprehensive Physiology
In summary, increased expression of PKM2 is epigenetically regulated, mainly by decreased expression of its regulatory miRNAs and altered DNA methylation status of the PKM gene.
2.1.5. Epigenetic Regulation of PDH
PDH is also epigenetically regulated, mainly by DNA methylation of its upstream regulator. In PAH PASMCs, hypermethylation of the CpG islands in the SOD2 promoter, mediated by upregulation in DNMT1 and DNMT3B, reduces gene expression [174]. The resulting decrease in H2O2 activates HIF-1α, which upregulates the transcription of PDK (among other key enzymes that inhibit oxidative metabolism) [68]. Inhibition of PDH through PDK-mediated phosphorylation inhibits acetyl-CoA production and promotes the Warburg effect. This pathway has been termed the DNMT-SOD2-HIF-1α/PDK/PDH pathway [149].
2.1.6. Therapeutics targeting of PDH and PKM2 in PAH
Many in vitro, preclinical and clinical therapeutic studies have been performed to restore glucose oxidation to normalize mitochondrial metabolic function and thus cellular function in PAH. Here we focus on these two targets, i.e., PDH and PKM2, since their activity is directly involved in the glucose oxidation pathway. Additional information on clinical trials on mitochondrial metabolism in PAH can be found in previous reviews [148, 175]. A summary of therapeutics targeting PDK/PDH and PKM2 in PAH along with their associated mechanism of action is provided in Table 2.
Table 2.
Therapeutics targeting aerobic glycolysis in PAH and cancer.
| Therapy | Category | Mechanism | Tested disease(s) |
|---|---|---|---|
| DCA | Small compound | PDK inhibitor | PAH [97, 176] [89, 154, 180, 181] [182] Lung cancer, glioblastoma, breast cancer, prostate cancer [187–190], glioblastoma [192], non-Hodgkin’s lymphoma [193] |
| siPTBP1 | siRNA | Decrease PKM2/PKM1 ratio by inhibition of PTBP1 | PAH [152, 153] |
| miR-124 | miRNA | Decrease PKM2/PKM1 ratio by inhibition of PTBP1 | PAH [152, 153] |
| shikonin | Small compound | Decrease PKM2/PKM1 ratio | PAH [152, 153], Group 2 PH [183] |
| TEPP-46 | Small compound | Stabilize PKM2 tetramer form | PAH [153] Lung cancer [204, 205] |
| echinomycin | Small compound | Inhibits PDK1 by inhibiting HIF |
Colon cancer, pancreatic cancer [191], lung cancer [188], pancreatic cancer [190] |
| miR-1294 | miRNA | Inhibits PKM2 expression | Osteosarcoma [203] |
2.1.6.1. Inhibition of PDK
DCA is a metabolic modulator that inhibits all four PDK isoforms. Inhibiting PDK enhances PDH activity and OXPHOS. In vitro, DCA has been shown to restore glucose oxidation and inhibit cell proliferation and apoptosis resistance in PASMC [176, 177], PAfib [178, 179] and RVfib [89]. DCA also restores glucose oxidation in PAH RV myocytes in vitro [180] and enhances RV contractility ex vivo [181] in animal models of PAH. In vivo, DCA has been shown to improve the hemodynamic function of lungs and RV in animal models of PH. DCA reverses PA remodeling in MCT-PAH rats [97, 176], reduces RV fibrosis and improves RV systolic and diastolic function in MCT rats and FHR [89, 154, 180, 181].
Though many studies have demonstrated the benefits of DCA in animal models of PAH, clinical trials are limited. We searched on Clinicaltrials.gov using “hypertension, pulmonary” in “Condition or disease” and “DCA or glycolysis” in “Other terms” and only found 1 clinical study using DCA to treat patients with idiopathic PAH (NCT01083524; Phase I, completed). In this small clinical trial, 16 patients out of 20 completed the trial. The data from this study suggest that DCA is safe to use in patients with PAH and demonstrate that DCA leads to a reduction in mPAP and PVR and improvement in functional capacity [182]. However, DCA is not effective if patients have a certain loss-of-function SNPs in UCP2 (encoding mitochondrial uncoupling protein 2, UCP2) and SIRT3 (encoding sirtuin 3) [182]. Further clinical studies on larger populations of PAH patients (both Phases II and III) are required to assess the efficacy and safety of DCA.
2.1.6.2. Inhibition of PKM2
Decreasing PKM2/PKM1 ratio reduces glycolysis and restores glucose oxidation, resulting in reduced cell proliferation in in vitro studies in both PAEC and PAfib from PAH patients, whether achieved by PTBP1 inhibition, administration of a miR-124 mimic, or administration of shikonin, a Chinese herbal remedy [152, 153]. In a group 2 model of PH, 2 mg/kg/d of shikonin, for 2 weeks resulted in improved RV systolic pressure and regressed RV hypertrophy by normalizing PKM2/PKM1 expression [183]. Stabilizing the PKM2 tetramer formation, using a small molecule TEPP-46 (a member of the thieno[3,2-b]pyrrole[3,2-d]pyridazinones class) decreases lactate production and normalize glucose metabolism in human PAH PAfib [153]. However, preclinical studies are required to test the efficacy, effectiveness and safety of PKM2-targeted drugs before clinical trials in PAH patients.
2.1.7. Therapeutics targeting of PDH and PKM2 in cancer
The concept of aerobic glycolysis was established much earlier in cancer than in PAH, and many experimental therapeutics have focused on targeting glycolysis (see reviews [151, 184–186]). Here we focus on the therapeutics that target PDK (or PDH) and PKM2. A summary of therapeutics targeting PDK/PDH and PKM2 in cancer along with their associated mechanism of action is provided in Table 2.
2.1.7.1. Inhibition of PDK
Archer and Michelakis hold a patent for the use of PDK inhibitors to treat cancer (US11/911,299). In vitro, DCA has no effect on non-cancerous 293T cells [187], but reduces cell proliferation and/or increases apoptosis in many cancer cell lines including A549 (NSCLC), M059K (glioblastoma), MCF-7 (breast cancer), human prostate cancer cells, pancreatic cancer cell lines (PANC-1, BXPC-3), endometrial cancer cell lines (Ishikawa, RL95–2, KLE, AN3CA, and SKUT1B) [187–190]. The HIF inhibitor echinomycin, which inhibits PDK1, restores glucose oxidation in RKO (colon cancer) and Su.86 (pancreatic cancer) in vitro [191]. In vivo, DCA decreases xenograft tumor growth of A549 cells in rats [188] and in a xenograft pancreatic cancer mouse model [190]. Echinomycin also delays tumor growth from RKO xenograft in mice [191].
There are a few clinical trials using DCA to treat cancer. The first clinical report by Michelakis et al. found that 4 out of 5 patients with glioblastoma demonstrated evidence of radiologic regression by magnetic resonance imaging and were clinically stable after 15 months of oral DCA administration [192]. In a single case report, a middle-aged man with non-Hodgkin’s lymphoma who had relapsed after standard chemotherapy showed complete remission after over 4 years’ treatment with oral 1 g/day DCA as monotherapy [193]. However, ~1 week of oral DCA treatment (6.25 mg/kg/12h) does not show any benefits for 6 patients with stage IV metastatic breast cancer or advanced stage (IIIB/IV) NSCLC in an open-label Phase II trial [194]. Regarding the dose, oral DCA (~8 mg/kg/12h) treatment for 4 weeks in a Phase I trial is well-tolerated in 15 adult patients with recurrent malignant gliomas or other tumors metastatic to the brain [195]. However, 6.25 mg/kg/12h is the dose recommended for Phase II trials, based on a Phase I trial [196]. The effect of DCA may depend on the type and stage of tumors. The toxicity (a reversible peripheral neuropathy in genetically susceptible patients) [197] and the benefits of DCA will require careful monitoring in future trials.
2.1.7.2. Inhibition of PKM2
In vitro, some drugs found in plants such as resveratrol, apigenin, shikonin (and shikonin’s enantiomeric isomer, alkannin) can reduce PKM2 expression or activity, resulting in inhibition of proliferation and glycolysis and induction of apoptosis in several cancer cell lines including DLD1, HeLa, MCF-7, HCT116, HT29, MCF-7, MCF-7/Adr, MCF-7/Bcl-2, MCF-7/Bcl-xL, A549, and HCC [198–201]. The non-specific effect of these drugs is unknown. PKM2 synthetic peptide aptamers and small molecules targeting PKM2 have also been shown to reduce the growth of tumor cells [174, 202], indicating the pathologic role of PKM2 isoform in cancer cells. Overexpression of miR-1294, which inhibits PKM2 expression, inhibits cell proliferation, migration, and invasion, and induces apoptosis in osteosarcoma cells [203]; however, miRs have many targets and this miR intervention would not selectively target PKM2. On the other hand, increased expression of the PKM2 tetramer, a more active form of PKM2, inhibits glycolysis and promotes glucose oxidation [204]. PKM2 activators, 3-(trifluoromethyl)-1H-pyrazole-5-carboxamide and TEPP-46 can effectively promote PKM2 tetramerization and inhibit cell proliferation in lung cancer cell lines including NCI-H1975, A549 and NCI-H1299 [204, 205]. Currently, no clinical trial has been conducted to use PKM2-targeted drugs to treat cancer.
2.2. Mitochondrial dynamics as a target for experimental therapeutics in PAH and cancer
While the primary role of mitochondria is to generate ATP through OXPHOS, mitochondria also play a number of noncanonical roles which are important for cell survival and death. These include mitochondrial fission and fusion [206], mitochondrial quality control (via mitophagy) [207], mitochondrial biogenesis [208], calcium regulation [209], and mitochondrial apoptosis pathway [210].
Mitochondria are highly dynamic organelles which continuously undergo division (fission) and joining (fusion) [206]. In normal physiological conditions, mitochondrial dynamics are tightly regulated by two groups of proteins (fission mediators and fusion mediators), most of which are large GTPases. Dysregulation of mitochondrial fission and fusion has been identified in PAH and cancers and the most common imbalance is fission exceeding fusion, resulting in excessive mitochondrial fragmentation. This fission/fusion imbalance contributes to the excess rate of cell proliferation, apoptosis resistance and increased migration seen in both syndromes [209, 211–214]. In cancer, a fragmented mitochondrial network is also associated with an alteration of bioenergetic and biosynthetic needs that support tumor initiation [215].
2.2.1. Mitochondrial fission and its experimental therapeutics
Increased mitochondrial fission is mainly due to increased expression level/activity of Drp1 and/or its binding partners [206]. Epigenetic regulation [212], transcriptional regulation [216] and post-translational modifications [211] are each involved in the dysregulation of fission and fusion mediators in PAH and cancer.
Fission proteins include Drp1 and its four binding partners: Mff (mitochondrial fission factor), FIS1 (mitochondrial fission 1 protein), MiD49 (mitochondrial dynamics protein of 49 kDa) and MiD51 (mitochondrial dynamics protein 51 kDa) [217–221]. Since Drp1 does not have an outer mitochondrial membrane (OMM) binding domain, it requires its binding partners/membrane receptors to facilitate membrane attachment [217].
During mitochondrial fission, endoplasmic reticulum (ER) first wraps around the mitochondria and marks the fission site [222]. Next, cytosolic Drp1 is activated by post-translational modifications, and recruited to the OMM where it interacts with one or more binding partners [217]. On the OMM, Drp1 polymerizes into a ring-like structure, at a site demarcated by the ER and binding partners. This ring constricts the mitochondrial membrane, utilizing the energy generated from GTP hydrolysis [223]. There are two major phosphorylation sites in human Drp1: serine 616 (p-Drp1S616) and serine 637 (p-Drp1S637). Phosphorylation at S616 promotes mitochondrial fission [224–226] while phosphorylation at S637 decreases mitochondrial fragmentation [227, 228]. Drp1’s binding partners also likely focus the constriction apparatus, guiding division of the mitochondrial membranes.
Since Drp1 can only constrict mitochondria down to a diameter of ~30nm [229], a model of sequential constriction by multiple fission mediators was recently proposed. Lee et al. demonstrated that dynamin 2 (Dnm2), a mediator in endocytosis [230] and vesicular trafficking [231], completes the final step of mitochondrial fission [232]. However, whether Dnm2 is essential for mitochondrial fission is controversial. Fonseca et al. found that dynamin triple-knock out does not defect mitochondrial fission in mouse fibroblasts [233]. Clarification of the roles of Drp1 and Dnm2 requires additional research.
2.2.1.1. Increased mitochondrial fission in PAH
Hyperfragmentation of the mitochondrial network, whether due to increased mitochondrial fission [214] or decreased fusion [216], is observed in both PAH patients and experimental models of PAH. This pathological mitochondrial phenotype contributes not only to hyperproliferation and aerobic glycolysis of PASMC [212, 214] and RVfib [89, 234], but also to RV dysfunction in PAH [235]. The consequences of a fragmented mitochondrial network are highly contextual, ranging from metabolic inefficiency with ROS production, to impending apoptosis or increasing cell division.
Mitochondrial fission is coordinated with mitosis ensuring equitable distribution of mitochondria to daughter cells. In rapidly dividing cells, fragmented mitochondria often signify increased rates of mitotic fission, as is the case of PAH PASMC. The Archer lab first identified increased expression level of both total and activated form of Drp1 (p-Drp1S616) in PASMC from patients and rodent preclinical models of PAH [214]. In both animals and patients, HIF-dependent and cyclin B1/CDK1 activation phosphorylation of Drp1 at S616 lead to Drp1 activation. The activation of Drp1 in PAH PASMC is pathologically relevant as inhibiting Drp1 reduces rates of cell proliferation and causes cell cycle arrest [214]. Activation of Drp1 in PAH PASMC is also caused by decreased level of the inactivated form of Drp1 (p-Drp1S637), which can be reversed by treprostinil, a synthetic analog of prostacyclin [236].
Elevation of Drp1 binding partners is also observed in PAH, notably including FIS1 [214], MiD49 and MiD51 [212]. MiD49 and MiD51 are epigenetically upregulated in PAH PASMC, which eventually leads to mitotic fission and cell cycle progression via an ERK1/2 and CDK4-dependent manner [212] (Figure 7). Furthermore, a SNP (exm1300952) in the MiD49 gene (SMCR7) is associated with a higher risk of PAH [237]. Elevated MiD51 expression is also reported in the left ventricle (LV) and RV of a rodent SAB model of group 2 PH [238].
Figure 7. Pathways involved in increased MiD49/51 induced mitochondrial fission, cell proliferation and apoptosis resistance in A) PAH and B) cancer.
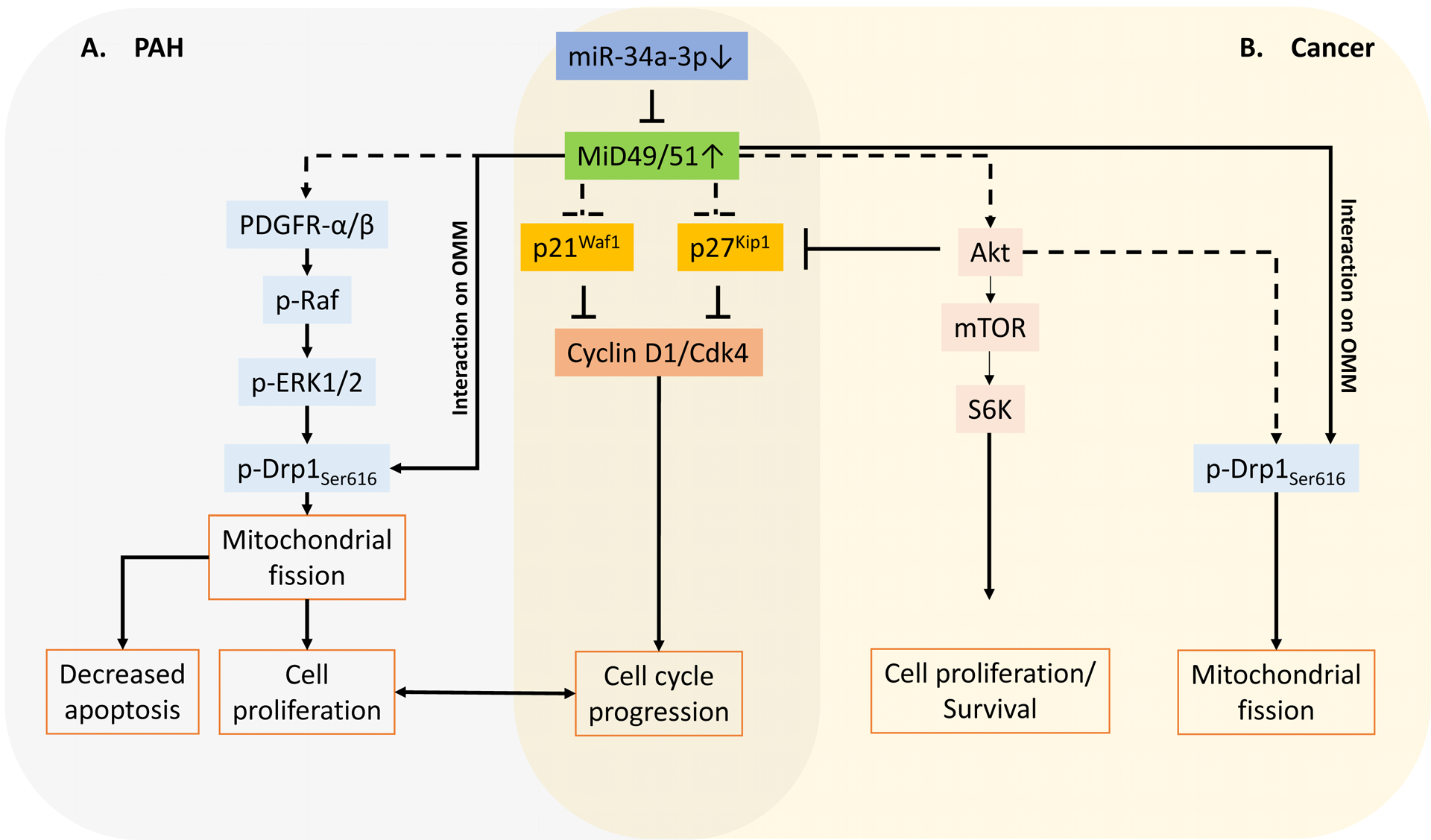
A. The expression of MiD49 and MiD51, are increased in PAH due to the downregulation of their regulatory microRNA, miR-34a-3p. This increase in MiD expression drives pathological mitochondrial fission by sequestering activated Drp1 to the outer mitochondrial membrane (OMM), cell proliferation and apoptosis resistance via PDGFR-α/β-Ras-Raf-Erk pathway.
B. In cancer, downregulation of miR-34a-3p leads to MiD upregulation which in turn drives mitochondrial fission by sequestering activated Drp1 to the outer mitochondrial membrane (OMM), cell proliferation and apoptosis resistance via Akt-mTOR-S6K axis.
The heart is intriguing because it has both dividing cells (RVfib) and nondividing cells (cardiomyocytes). While fission is seen in both cell types, the consequences are quite different. In RVfib, increased Drp1-mediated mitochondrial fission is associated with hyperproliferation and increased collagen production, which ultimately results in RV stiffness and RV failure [234]. However, increased mitochondrial fission in RV myocytes, likely also due to increased Drp1 activity and Drp1-FIS1 interaction, is associated with increase in mitochondrial ROS production and a spontaneous ischemia-reperfusion (IR) injury [235].
2.2.1.2. Increased mitochondrial fission in cancer
Excessive mitochondrial fission has been confirmed in various cancer tissues/cells, including lung cancer [211], breast cancer [239], brain cancer [240], pancreatic cancer [241], melanoma [242] and this increase in fission is associated with accelerated cell cycle progression, hyperproliferation, apoptosis resistance, invasion, and migration [243].
Excessive fission in cancer cells is due to increased expression levels/post translational activation of the fission protein, Drp1 and dysregulation of Drp1 binding partners MiD49 and MiD51. Our group demonstrated that in lung cancer, increased p-Drp1S616 and decreased p-Drp1S637, along with decreased mitofusin (Mfn) 2, contribute to the net increase in mitochondrial fragmentation in NSCLC cell lines (A549 and H1993), as well as in tumors from patients [211]. Upregulation of Drp1 also occurs in invasive breast carcinoma, in which case it is accompanied by downregulation of Mfn1 [239]. Normalization of Drp1 or Mfn1 expression in this study inhibited lamellipodia formation by breast cancer cells, a key step for cancer metastasis [239]. Brain tumor initiating cells (BTICs) are stem-cell like cells present in brain tumors. The mitochondria in BTICs are more fragmented compared to those in non-BTICs [240]. Cyclin-dependent kinase (CDK) 5 activates Drp1 in BTICs by phosphorylation at S616 while Ca2+/calmodulin-dependent protein kinase (CAMK) inhibits Drp1 in non-BTICs by phosphorylation at S637. P-Drp1S616 inhibits the downstream target, AMP-activated protein kinase (AMPK), a central cellular sensor of energy stress. Increased activity of the CDK5/CAMK-Drp1-AMPK pathway also has prognostic value in patients with primary glioblastomas [240]. Furthermore, Drp1 is also upregulated in pancreatic cancer [244] and thyroid tumors [245].
Although there is much evidence supporting the pro-tumorigenesis function of Drp1, the role of Drp1 binding partners in cancer, especially MiD49 and MiD51, is less clearly understood. We recently discovered a parallel upregulation of MiD49 and MiD51 in NSCLC and invasive breast carcinoma [213]. Similar to our observation in PAH cells, in cancer, this pathway contributes to increased mitotic fission, apoptosis resistance and accelerated cell cycle progression [213]. Different from Drp1, MiDs are mainly epigenetically regulated. In these cancers and PAH, a shared mutual upstream regulator is downregulation of miR-34a-3p (Figure 7). However, in pancreatic cancer, MiD49 is downregulated and is thought to play a role of tumor suppressor [246]. Downregulation of MiD49 in pancreatic cancer is associated with tumor growth and metastasis [246]. The reason for these opposing observations on the role of MiDs is unclear.
2.2.1.3. Therapeutic targeting of fission mediators to reduce mitochondrial hyperfragmentation
Due to the pivotal role of Drp1 in mitochondrial fission and its related pathological conditions, the GTPase domain of Drp1 has become a target domain for drug development. In 2008, the Nunnari lab discovered the first Drp1 inhibitor, mitochondrial division inhibitor (mdivi-1) [247]. They first screened ~23,000 chemical compounds in yeast using a growth assay. Candidate compounds which inhibit yeast mitochondrial fission suppressed the growth defect of mitochondrial fusion mutants (fzo1–1 and mgm1–5). This was followed by a screen based on steady-state mitochondrial morphology. Only 3 potential mitochondrial division inhibitors were identified after two rounds of screening. Among them, mdivi-1 was the most efficacious. Mdivi-1 selectively inhibited the GTPase activity of Dnm1 (the yeast dynamin-related GTPase) and its self-assembly. It also inhibited mitochondrial division [247]. Although this original study demonstrated that mdivi-1 inhibited mitochondrial apoptosis in mammalian cells by blockage of Bid-activated Bax/Bak-dependent cytochrome c (Cyt c) release, in the hyperproliferative cells in PAH and cancer, mdivi-1 is widely used as an antiproliferative and pro-apoptotic agent due to its inhibitory effect on mitotic fission. In human PAH PASMC, inhibition of Drp1 by mdivi-1 prevents mitotic fission and arrests the cell cycle at the G2/M interphase [214]. Furthermore, mdivi-1 also improves pulmonary vascular hemodynamics, RV function and exercise capacity in rodents with experimental PAH induced by a HIF-1α activator, cobalt. Rehman in our group first employed mdivi-1 as a treatment in a preclinical model of cancer [211]. Mdivi-1 significantly regressed tumor growth in a mouse xenograft model of human NSCLC. Subsequently, the efficacy of mdivi-1 has been validated by many other groups in various solid tumors, including brain tumors [240], breast cancer [248, 249] and melanoma [250].
In 2013, Qi et al. took a different approach to inhibiting Drp1-mediated fission. Rather than target the GTPase domain, they created a selective peptide P110, which inhibits Drp1 activity by blocking the interaction of Drp1 and FIS1 [251]. P110 has been successfully used to protect the RV in an ex vivo, preclinical treatment of RV-IR injury [235]. Since Drp1-mediated mitochondrial hyperfragmentation is also associated with increased apoptosis in neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease and Huntington’s disease [252–257], the therapeutic efficacies of mdivi-1 and P110 have also been tested and shown to be beneficial in these non-hyperproliferative, neurological conditions [258–260]. However, P110 would only be expected to be therapeutic in conditions in which pathologic fission requires FIS1 binding, which does not appear to be a major mechanism of fission in PAH or most cancers.
Although the therapeutic efficacy of mdivi-1 on PAH and cancer is supported by an increasing number of literatures, its lack of specificity on Drp1 [261] is a concerning fact that may lead to off-target effects. Recently, our lab discovered a novel Drp1 inhibitor, Drpitor1a [262] (patent pending), which is more specific and 50X more potent than mdivi-1 (Figure 8). Like mdivi-1, Drpitor1a inhibits lung cancer tumor growth and RV-IR injury in rodent disease models [262]. A summary of therapeutics targeting mitochondrial fusion/fission mediators in PAH and cancer is provided in Table 3.
Figure 8. Drpitor1a, a novel Drp1 GTPase inhibitor.
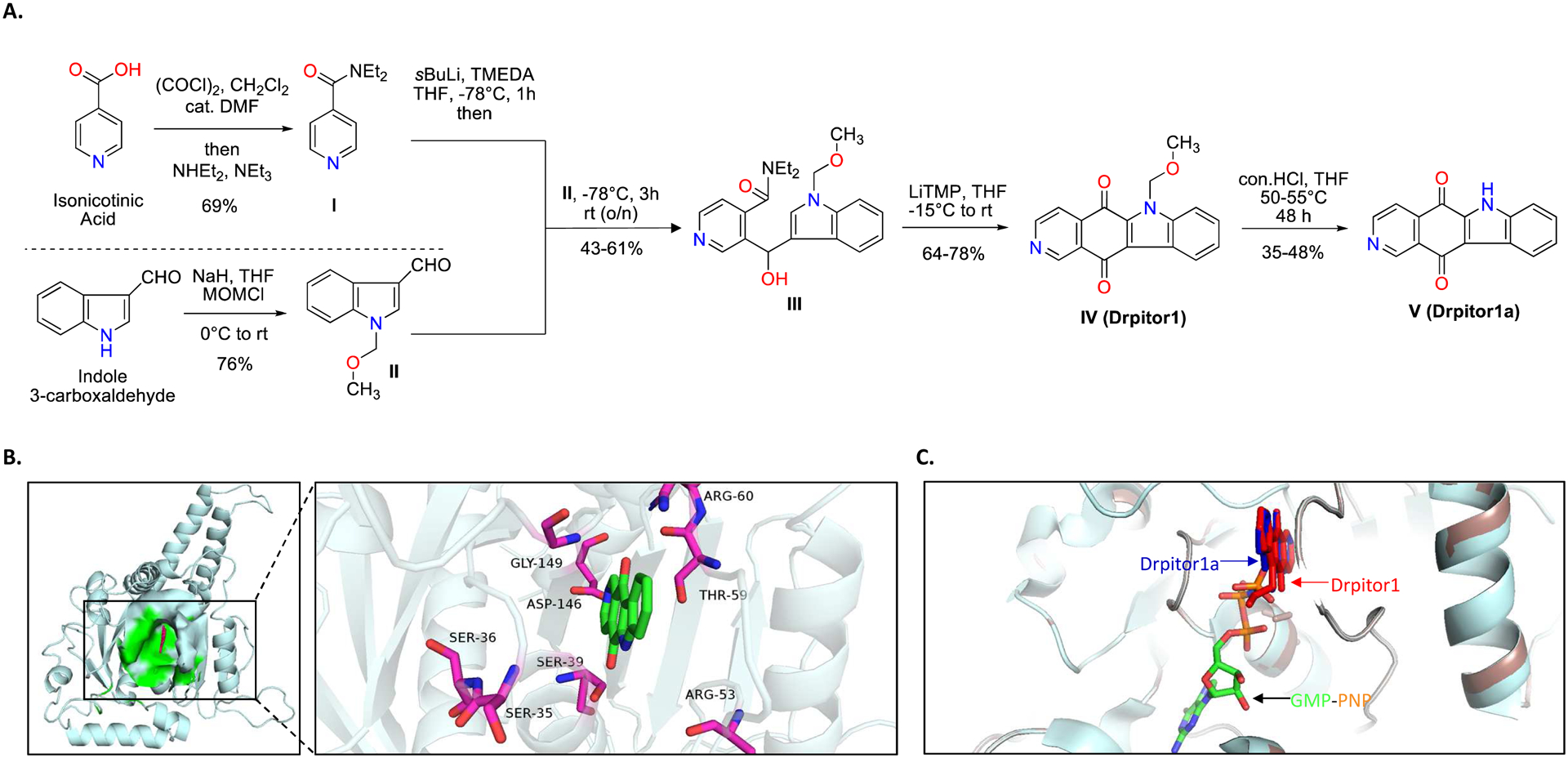
A. Synthesis of Drpitor1a.
B. Predicted interaction between Drpitor1a and the GTPase domain of Drp1.
C. The binding locations of Drpitor1 (red), Drpitor1a (blue), and guanylyl-imidodiphosphate (GMP-PNP) (orange and green) in the GTPase domain of Drp1.
Copyright © The FASEB Journal
Table 3.
Therapeutic agents targeting mitochondrial fission/fusion mediators in PAH and cancer.
| Therapy | Category | Target | Tested disease(s) |
|---|---|---|---|
| siDrp1 | siRNA | Drp1 | PAH [214], lung cancer |
| mdivi-1 | Small compound | Drp1 GTPase domain | PAH [214], lung cancer [211], brain tumors [240], breast cancer [248, 249], melanoma [250] |
| P110 | Peptide | Drp1-FIS1 interaction | RV-IR injury [235] |
| Drpitor1a | Small compound | Drp1 GTPase domain | Lung cancer [262], RV-IR injury [262] |
| siMiD49/51 | siRNA | MiD49, MiD51 | PAH [212], lung cancer [213], breast cancer [213] |
| miR-34a-3p | miRNA | MiD49, MiD51 | PAH [212], lung cancer [213], breast cancer [213] |
| Adv-Mfn2 | Adenovirus | Mfn2 | PAH [216], lung cancer [211] |
| leflunomide | Small compound | Mfn2 | Mouse spontaneous tumors [279] |
| miR-125a | miRNA | Mfn2 | PANC-1 pancreatic cancer cell [287] |
2.2.2. Therapeutic targeting of fusion mediators to reduce mitochondrial hyperfragmentation
In mammalian cells, mitochondrial fusion is primarily mediated by three large GTPases: Mfn1, Mfn2 on the OMM, and OPA1 (optic atrophy 1) on the inner mitochondrial membrane (IMM) [263, 264]. OPA1 functionally requires Mfn1, but not Mfn2, to regulate mitochondrial fusion, indicating functional differences between Mfn1 and Mfn2 [265]. In addition, there is a Ras-binding domain at the N-terminus of Mfn2 which is absent in Mfn1, suggesting specific roles of Mfn2. Previous studies have indicated that overexpression of Mfn2 (initially called the hyperplasia suppressor gene, HSG) can suppress proliferation and induce apoptosis of vascular SMCs [266, 267].
2.2.2.1. Regulation of Mfn2
Expression of Mfn2 is regulated both at transcriptional and post-translational levels. The transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), transcriptional regulator of Mfn2, is downregulated in human and rodent PAH PASMCs [216]. Mfn2 expression is also regulated at the posttranslational level by the ubiquitin-proteasome system [268]. In response to cellular stress, Mfn2 is phosphorylated at serine 27 by c-Jun N-terminal kinase (JNK), leading to the recruitment of the ubiquitin ligase (E3) Huwe1 and resulting in ubiquitin-mediated proteasomal degradation of Mfn2. This degradation of Mfn2 can result in mitochondrial fragmentation and apoptosis [269]. Mfn2 is also phosphorylated by PTEN-induced putative kinase (PINK1) at serine 442, leading to Parkin-mediated ubiquitination and proteasomal degradation [270]. In addition, Mfn2 expression is controlled by the PI3K-Akt-mTOR pathway, which plays an important role in activation-induced, proteasomal downregulation of Mfn2 in human peripheral blood T cells [271].
2.2.2.2. Dysregulation of Mfn2 in PAH and its experimental therapeutics
Mfn2 expression is downregulated in the medial layer (PASMC) of pulmonary vasculature in PAH patients and in the PAs of experimental rodent models of PAH (MCT-PAH and Su/Hx-PAH) [216]. Augmenting Mfn2 expression by adenovirus-mediated gene transfer increases mitochondrial fusion, inhibits cell proliferation and induces apoptosis in PAH PASMC, suggesting potential therapeutic benefit of Mfn2 therapy in human and experimental PAH [216]. Furthermore, augmenting Mfn2 by airway nebulization of adenovirus containing the Mfn2 gene increases lung vascularity and decreases PVR and PA medial thickness in the Su/Hx-PAH model [216] (Figure 9).
Figure 9. Therapeutic effects of Mfn2 in PAH.
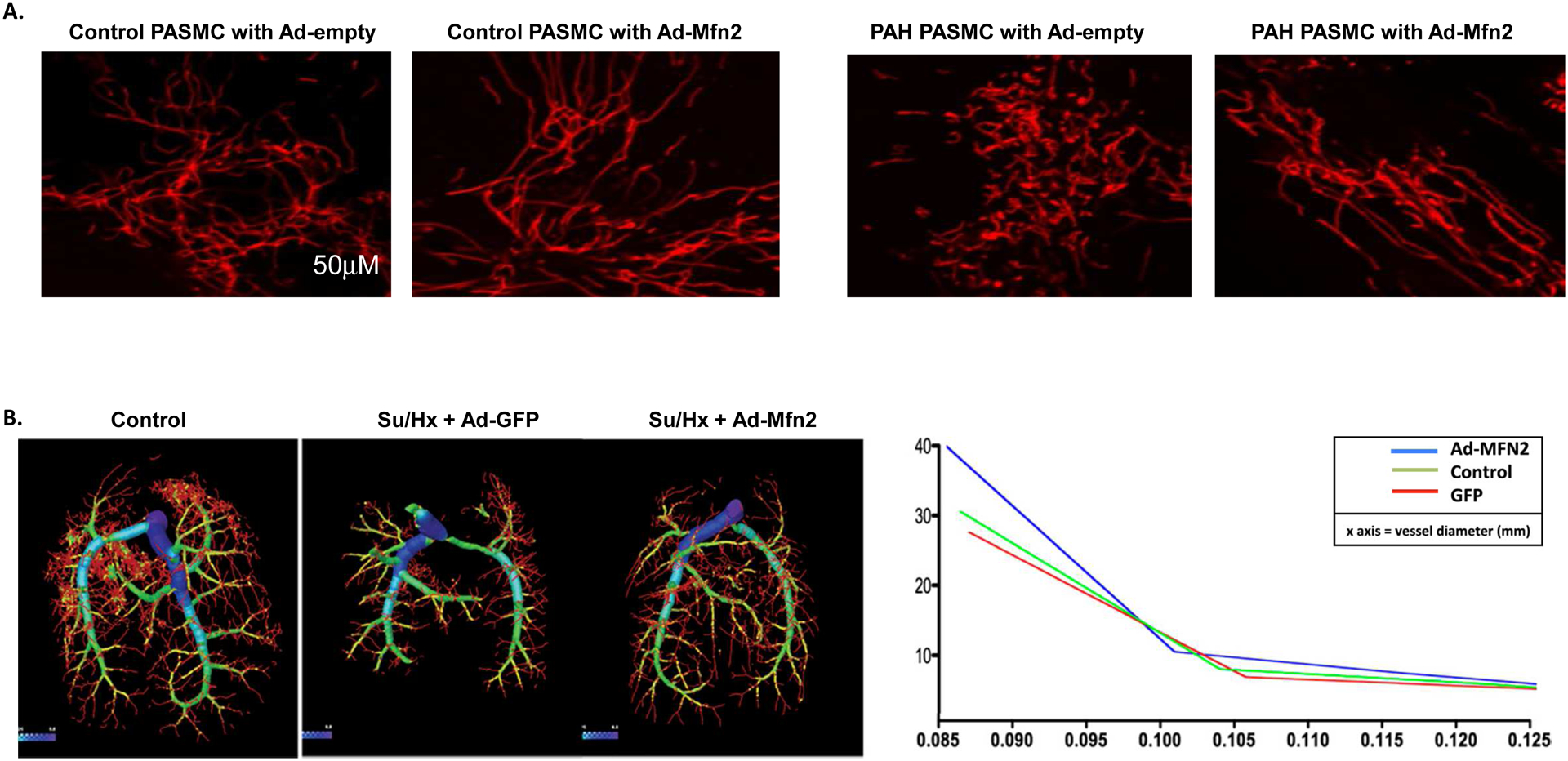
A. Overexpression of Mfn2 by adenoviral mediated gene transfer (Adv-Mfn2) induces mitochondrial fusion in control PASMC and PAH PASMC. Cells were the loaded with the potentiometric dye TMRM (red) and imaged with confocal microscope to assess the mitochondrial network structure. Scale bar: 50mm.
B. Representative CT angiogram showing pruning of small pulmonary arteries in Su/Hx rats, which can be reversed by augmenting Mfn2. Su/Hx rats were treated either with Ad-GFP or Ad-Mfn2. Compared to the control, Su/Hx rats exhibit decreased percentage of small pulmonary arteries whilst Ad-Mfn2 treatment increased the percentage of small pulmonary arteries.
Copyright © American Journal of Respiratory and Critical Care Medicine
2.2.2.3. Role of Mfn2 in the pathogenesis and therapy of cancer
Downregulation of Mfn2 in cancer was first identified in urinary bladder cancers [272] and lung cancer [211], and later, this phenomenon is confirmed in various types of cancer, including breast cancer [273], hepatocellular carcinoma [274], colorectal cancer [275] and gastric cancer [276]. Augmenting Mfn2 level in cancer cells elongates mitochondrial network [211], arrests cell cycle transition from G1 to S phase [272], inhibits cell proliferation and colony formation, decreases mitochondrial membrane potential, triggers mitochondria-associated apoptosis, and impairs the invasion and migration abilities of cancer cells [275–278].
Consistent with these findings, Xu et al. reported that overexpression of Mfn2 suppresses cancer progression through the inhibition of the mammalian target of rapamycin complex (mTORC) 2/Akt signaling pathway. Conversely, silencing of Mfn2 in the MCF7 breast cancer cells and A549 NSCLC cells promotes cell viability, colony formation, and cancer cell invasion in vitro and this is replicated in a xenotransplant murine model. In the Mfn2-silenced cancer cells, the mTORC2/Akt signaling pathway, which is activated in most cancers, becomes further activated leading to the growth and metastasis of cancer cells. Mfn2 directly interacts with mTORC2 and suppresses its kinase activity, which inactivates Akt, thereby inhibiting tumor growth [273]. Xue et al. also reported that overexpression of Mfn2 by adenovirus-mediated gene transfer in a pancreatic cancer cell line (AsPC-1) induces autophagy by inhibiting the PI3K/Akt/mTOR signaling pathway with the net effect of inhibiting cancer cell proliferation and ROS production [277].
Mfn2 expression can also be increased pharmacologically. A recent study demonstrates that oral leflunomide, an FDA-approved disease modifying drug for rheumatoid arthritis, increases Mfn2 expression 2-fold in tumors and improves the median survival of mice with spontaneous tumors by 50% compared with the control group. The mechanism of tumor suppression by leflunomide reflects reduced mitochondrial mass and ATP production and thereby suppresses tumor growth [279].
It is worth noting that there are studies reporting increased expression of Mfn2 in lung adenocarcinoma [280]. In this study, Mfn2 knockdown inhibited cell proliferation and invasion and caused cell cycle arrest [280]. However, the authors did not investigate the effect of silencing Mfn2 on mitochondrial morphology, nor did they report the effects on the expression level of other fission/fusion mediators, including Drp1 and MiDs, leaving questions about the mechanism of action of their Mfn2 knockdown. In ovarian cancer, increased Mfn2 expression is associated with its upstream regulator, cystathionine β-synthase (CBS). Although molecular inhibition of Mfn2 decreased mitochondrial fusion in ovarian cancer cells, the inhibition of cell growth might be due to a compensatory decrease in Drp1, which was not investigated in that study [281].
Mfn2 expression is not only controlled at the transcriptional level, it is also epigenetically regulated. Pan et al. reported that the expression of Mfn2 is negatively regulated by miR-125a. miR-125a expression is downregulated in several types of human cancer, including breast cancer [282], lung cancer [283], ovarian cancer [284], medulloblastoma [285] and gastric cancer [286]. Augmenting miR-125a in PANC-1 pancreatic cancer cells downregulates Mfn2 expression, resulting in mitochondrial fission via associated activation of mitochondria-dependent apoptosis and suppression of cancer cell migration. However, Mfn2 may be not the only target of miR-125a since miR-125a mimic also inhibits the expression level of two fission mediators, Drp1 and Fis1. Therefore, the mechanism how miR-125a mediates mitochondria-dependent apoptosis and suppression of cancer cell migration requires further investigation [287].
Thus, Mfn2 has been reported to have both tumor-suppressing and tumor-promoting functions, depending on the cancer type suggesting its role in cancer is more complicated than expected. The validity and therapeutic implications of the apparent differences in the roles of Mfn2 in various tumor types require further experimental study.
2.3. Mitophagy in PAH and cancer
Mitophagy is a specialized form of autophagy that serves as a quality control mechanism. Mitophagy selectively removes dysfunctional mitochondrial without killing the cell. This is achieved by targeting damaged mitochondria for proteolytic degradation [288]. The proteolytic system of mitochondria is comprised of two AAA protease complexes in the inner membrane which degrade unfolded membrane proteins [289]. The IMM and OMM proteins are also targeted for degradation by cytosolic proteasomes [290]. Mitophagy is mediated by the coordinated activation of PINK1 and Parkin. In a damaged or dysfunctional mitochondrion, the mitochondrial membrane potential depolarizes. This damage may result from oxidative stress, with the mitochondria itself generating ROS or reflect damage caused by external stimuli, like irradiation or chemotherapeutic agents. With mitochondrial damage, inhibition of mitochondrial processing peptidase (MPP) and presenilin-associated rhomboid-like (PARL) proteases stabilize PINK1 in the OMM [291, 292]. PINK1 in turn recruits Parkin, which polyubiquitinates several OMM proteins such as voltage-dependent-anion-selective channel 1 (VDAC1), Mfn1 and Mfn2. These polyubiquitinated proteins are then marked by adaptor proteins, such as p62, optineurin (OPTN) and nuclear dot protein 52 kDa (NDP52), causing them to be recognized by light chain 3 (LC3), leading to the formation of autophagosome [291]. There is limited evidence of increased mitophagy in experimental PH. Mice with PH due to endothelial-specific knockout of UCP2 exposed to intermittent hypoxia have an increase in mitophagy which in turn inhibits mitochondrial biogenesis and induces apoptosis in PAECs [293].
Mitophagy is also dysregulated in many cancers [294]. Loss of function mutation of critical genes inhibits mitophagy resulting in the accumulation of dysfunctional mitochondria, which contributes to tumorigenesis. Loss of Parkin has been reported to suppress mitophagy and contribute to the development of various cancers [295]. In a murine model, loss of Parkin results in hyperproliferative hepatocytes, leading to microscopic hepatocellular cancer [296]. Furthermore, depletion of either PINK1 or Parkin stimulates K-Ras-driven pancreatic adenocarcinoma [297]. PINK1 and Parkin also play an important role in the stability of HIF-1α in that parkin ubiquitinates HIF-1α at lysine 477 (K477), leading to its degradation [298]. In breast cancer, high HIF-1α expression correlates with low Parkin expression and increased susceptibility to metastasis [299]. PINK1 and Parkin are also critical in maintaining metabolic homeostasis and regulation of cell cycle. Loss of PINK1 or Parkin function increases ROS production, HIF-1α expression and Warburg metabolism in pancreatic adenocarcinoma and these pathologic changes are reversed by silencing HIF-1α [297]. Thus, PINK1 or Parkin inactivation promotes Warburg metabolism in cancers through HIF-1α stabilization [297]. Parkin has also been shown to regulate the stability of protein complexes such as FBX4 Cullin-RING ligase complex and controls cell cycle progression by regulating cyclin levels and by interacting with Cdc20/Cdh1 to enforce mitotic checkpoint control and genetic stability [300].
Unlike PINK1/Parkin-mediated mitophagy, which removes damaged or dysfunctional mitochondria, healthy mitochondria can be eliminated in response to nutritional stress to allow the cells to utilize the energy stored within [301]. Stress-induced mitophagy receptors, B-cell lymphoma 2 (Bcl-2) and adenovirus E1B 19-kDa-interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L, also known as NIX) are present in the OMM and interact directly with LC3 or LC3 homologs to promote mitophagy of healthy mitochondria [302]. Chromosome 10q26.3, which contains the BNIP3 locus, is often deleted in metastatic triple-negative breast cancer and is associated with poor metastasis-free survival [303, 304]. In addition, epigenetic silencing of BNIP3 has been reported during progression of tumors’ invasiveness to metastasis in several cancers, including lung, gastric, pancreatic, liver and hematological malignancies [305–308]. Chemoresistance and poor prognosis in pancreatic cancer is associated with inactivation of BNIP3 [305, 309]. In an experimental murine model of breast cancer, loss of BNIP3 resulted in higher tumor growth, faster progression to invasive carcinoma, decreased latency of lung metastasis and decreased overall survival [303]. Furthermore, silencing of BNIP3 increased tumor growth in an orthotropic model of breast cancer [310]. Thus, loss of BNIP3 decreases mitophagy and leads to increased invasiveness, ROS and HIF-1α expression [303]. Taken together, these observations indicate that BNIP3 negatively regulates the expression of HIF-1α and conversely, BNIP3 inactivation promotes tumorigenesis and invasiveness. In contrast to BNIP3, BNIP3L promotes tumorigenesis. In KRAS-mutated pancreatic ductal adenocarcinoma, loss of BNIP3L is associated with decreased tumorigenesis, reduced mitophagy, increased oxidative metabolism and better prognosis, suggesting a divergent role of these mediators of mitophagy with one acting as a tumor suppressor and the other as a promoter of tumor growth [311].
2.4. Mitochondrial Biogenesis in PAH and cancer
Mitochondrial biogenesis is characterized by the division of pre-existing mitochondria to produce new mitochondria to meet energy requirements caused by the changes in environmental and physiological conditions [208]. This process is regulated by the transcription factor, PGC-1α which promotes mitochondrial biogenesis by activating nuclear respiratory factor (Nrf) 1 and mitochondrial transcription factor A (TFAM), which drives transcription and replication of mitochondrial DNA (mtDNA) [312–315].
In human and experimental models of PAH, mitochondrial biogenesis is inhibited due to the reduced expression of its mediators [316, 317]. In a bovine model of persistent pulmonary hypertension of the newborn (PPHN), expression of PGC-1α, ETC subunits, and mtDNA copy number is decreased [318]. In addition, mice lacking endothelial bone morphogenetic protein receptor type II (BMPR2) show impaired mitochondrial biogenesis along with increased mitochondrial ROS production, decreased mitochondrial membrane potential and a predilection to the development of PH [319]. Furthermore, in a time-course study conducted with RV, gastrocnemius, and LV in MCT-PAH rats, an early decrease in the expression of genes promoting mitochondrial biogenesis (sirtuin 1, PGC-1α and TFAM) is observed in the skeletal muscles followed by a similar decrease in RV myocytes and gastrocnemius muscle; conversely, the expression their expression in the LV remained unchanged [320].
Unlike PAH, mitochondrial biogenesis is increased in cancers. An early study by Lee et al. demonstrated that in arsenic-induced Bowen’s Disease (a form of skin cancer), tumor growth is promoted by upregulation of TFAM. In addition, the transcription factors of TFAM, Nrf1 and Nrf2, are also upregulated by arsenic [321]. Consistent with these findings, several other studies demonstrated a link between increased mitochondrial biogenesis and tumor growth. Inhibition of tumorigenesis and impaired OXPHOS are associated with the loss of mtDNA [322, 323]. Furthermore, a link between increased expression of TFAM and cancer progression is reported [324, 325] which aligns with the findings that loss of TFAM suppresses K-Ras-induced tumor formation in lungs [326].
As a transcription coactivator, PGC-1α interacts with various transcription factors. It is not only the master regulator of mitochondrial biogenesis, but also a key regulator of OXPHOS. PGC-1α has been demonstrated to enhance OXPHOS, mitochondrial biogenesis and oxygen consumption rate in invasive breast cancer cells. In human epidermal growth factor receptor 2 (ERBB2)+ breast cancer, PGC-1α and estrogen-related receptor (ERR) α positively regulate the expression of genes for mediators of glutamine metabolism [327]. The PGC-1α/ERRα axis also supports de novo lipogenesis[327]. Furthermore, a strong correlation between PGC-1α expression in invasive breast cancer cells and the formation of distant metastases has been demonstrated; conversely, silencing PGC-1α suppresses invasiveness and inhibits metastasis [328].
In melanoma, there are two subpopulations of cells expressing different levels of PGC-1α which have distinct metabolic phenotypes [329]. The PGC-1α-upregulated melanoma cells are OXPHOS-dependent and have increased ROS detoxification capacities, which allows them to survive under oxidative stress conditions [329]. A high PGC-1α level is also inversely correlated with vertical growth of human melanoma and its metastasis [330]. On the contrary, low PGC-1α expressing melanoma cells are more glycolytic and sensitive to ROS-inducing drugs [329].
In prostate cancer, the role of PGC-1α is controversial. Androgen receptor (AR) is a transcription factor directly regulated by PGC-1α [331] and PGC-1α is upregulated in various prostate cancer cell lines. Activation of PGC-1α/AR promotes cancer cell growth in AR-expressing prostate cancer cells [331]. Tennakoon et al. further demonstrated that the AR-dependent cell growth in prostate cancer is due to the activation of metabolic sensor AMPK/PGC-1α pathway [332]. In contrast, Torrano et al. reported PGC-1α as a tumor suppressor in prostate cancer [333]. The found that PGC-1α is downregulated in prostate cancer and noted that patients with lower PGC-1α in the prostate cancer tissue have worse disease-free survival [333]. Mechanistically, PGC-1α might suppress tumor progression and metastasis by activation of an ERRα-dependent transcriptional program [333]. These opposing results are a reminder of the complexity of cancer and suggest that other factors, such as AR or AMPK, may modulate the impact of PGC-1α on tumorigenesis.
In pancreatic cancer, the oncogene c-Myc binds to the promoter region of PGC-1α and inhibits its transcription [334]. Pancreatic cancer stem cells have a low c-Myc/PGC-1α ratio, high level of OXPHOS and are more sensitive to metformin. However, differentiated pancreatic tumor cells have a high c-Myc/PGC-1α ratio. When pancreatic cancer stem cells develop metformin resistance during treatment, they show an intermediate metabolic phenotype, with reduced OXPHOS but increased glycolysis, due to an increase in the c-Myc/PGC-1α ratio [334]. Therefore, pharmacological inhibition of c-Myc may be a potential therapeutic intervention for pancreatic cancer by preventing/reversing their resistance to metformin.
2.5. Mitochondrial calcium homeostasis in PAH and cancer
The hallmarks of hyperproliferative diseases include uncontrolled cell division and evasion of apoptosis. Dysregulation of intracellular Ca2+ in various compartments can modulate signaling pathways relevant to these events.
In PAH, increases in cytosolic calcium ([Ca2+]cyto) promote pulmonary vasoconstriction and proliferation of PASMC [335–337]. A multifactorial mechanism is involved in the elevation of [Ca2+]cyto, which includes influx through the L-type calcium channels, activation of transient receptor potential channels (TRPC) and activation of store-operated calcium channels [338–340]. Mitochondrial calcium ([Ca2+]mito) homeostasis plays a critical role in the regulation of intracellular calcium and metabolism. Normal physiologic levels of [Ca2+]mito are also required to maintain the activity of the 3 mitochondrial dehydrogenases, including PDH. [Ca2+]mito is suppressed in PAH PASMC [209]. Increased [Ca2+]mito causes suppression of cell proliferation and increase in spontaneous apoptosis both in PAH PASMC and in cancer cells [209, 341].
Although there are complementary routes of calcium entry into mitochondria, the primary mechanism of calcium influx occurs through a multichannel complex, mitochondrial calcium uniporter complex (MCUC), located in the inner mitochondrial membrane [342]. MCUC plays an important role in maintaining the cytosolic and mitochondrial calcium balance which is critical for activation of mitochondrial calcium-dependent dehydrogenases [343]. The MCUC is a multi-protein complex with a pore-forming subunit comprised of the mitochondrial calcium uniporter (MCU) and the essential MCU regulator (EMRE). The MCUC also includes 3 regulatory subunits including the negative regulator, mitochondrial calcium uptake 1, (MICU1). Decrease in MCU expression occurs in both cancer and PAH. In PAH, MICU1 expression is also increased [209, 344]. This decrease in MCU expression and MCUC function contributes to decreased [Ca2+]mito and increased [Ca2+]cyto, resulting in PDH inhibition and mitochondrial fission respectively [345], which in turn promote cell proliferation and resistance to apoptosis in human and experimental PAH [209]. The expression of MCU is epigenetically regulated [209, 346]. Marchi et al. first reported that miR-25 is an upstream regulator of MCU. In human colon cancers and cancer-derived cells, upregulation of miR-25 silences MCU, which increases the resistance to apoptotic stimuli. Conversely, anti-miR-25 inhibits cell proliferation and increases cell apoptosis [344]. In PAH PASMC, the decreased expression of MCU is due to the increase of two regulatory miRNAs: miR-25 and miR-138. Augmenting anti-miR-25 or miR-138 restore MCU expression and regresses experimental PAH [209] (Figure 10).
Figure 10: MCU dysregulation in PAH PASMC.
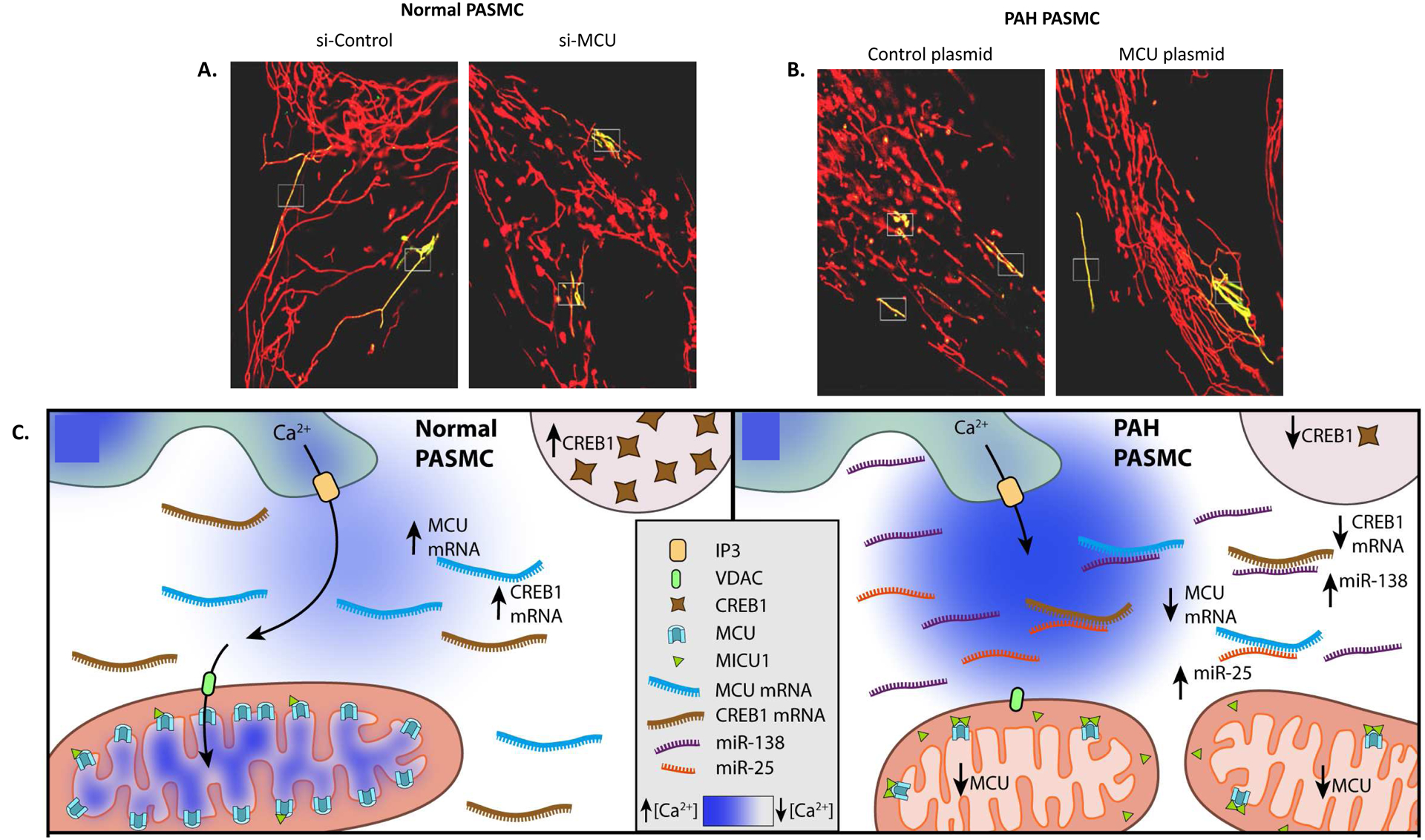
Representative images of the photoactivation experiments showing mitochondrial network in A) Normal PASMC B) PAH PASMC. The cells were co-transfected with specified siRNA or specified plasmids, then photoactivatable mitochondrial matrix targeted green fluorescent protein (mito-PA-GFP) plasmid and then loaded a with potentiometric dye TMRM (red).
A. siMCU treatment increased mitochondrial fission and restricted the diffusion of the green signal outside the white box in normal PASMC.
B. MCU augmentation inhibited mitochondrial fission as shown by the increased diffusion of the green signal outside the white box. Scale bar: 5 μm.
C. Mechanism of downregulation of MCU in PAH PASMC.
Copyright © American Journal of Respiratory and Critical Care Medicine
The role of MCU in cancer has been most investigated in breast cancer. Unlike the case in PAH and colon cancer cells [209, 344], MCU expression is increased in various types of breast cancer tissues/cells, including estrogen receptor negative and basal-like breast cancers [341] and triple negative breast cancer (TNBC) [347]. Tosatto et al. found that in TNBC, the expression of MCU is positively correlated with tumor size and lymph node infiltration [347]. Increased MCU and decreased MICU1 levels are correlated with worse patient outcomes [348]. Silencing MCU blunts cancer cell invasion, inhibits tumor growth, lymph node infiltration, and lung metastasis in a xenograft model of TNBC. In TNBC, HIF-1α is the downstream target of MCU [347]. However, MCU is not always crucial for cell biology in breast cancer cell lines. In MDA-MB-468 breast cancer cells, stimulation of EGF does not alter the expression level of MCU [349]. In MDA-MB-231 cells, manipulation (overexpression or silencing) of MCU does not change cell survival, indicating in this breast cancer cell line, MCU activity is dispensable [348]; while inhibition of MCU by ruthenium red (RuR) or siRNA abolishes serum-induced migration in the same cell line [350].
Another mechanism for regulation of mitochondrial calcium stores under physiological conditions involves UCP2 [351]. Heterologous overexpression of UCP2 in cardiomyocytes inhibits intramitochondrial calcium influx and decreases ROS production [352]. Consistent with these finding, SMCs lacking UCP2 switch to glycolysis due to dysregulation of the calcium-sensitive enzyme, PDH. Cells lacking UCP2 also exhibit impaired calcium flux from the ER to mitochondria [353]. In PASMC exposed to hypoxia, Nogo-B, a regulator of ER structure is upregulated by activating transcription factor 6 (ATF6), an ER stress-sensitive transcription factor. Induction of Nogo-B increases the distance between ER and mitochondria, resulting in the decrease of phospholipid and calcium transfer between ER and mitochondria. These changes finally result in increased cell proliferation by inhibiting mitochondria-dependent apoptosis, as seen in PAH [354].
These data suggest that calcium regulation in cancer is complex and not always MCU-dependent. The role of MCU in cellular pathology is disease- and tissue-specific. Normalizing MCU level might be a novel therapeutic target in PAH and some forms of cancer. However, attention to the basal level of pathway derangement (up or down regulated) will likely be important. As with Mfn2, one cannot make a generalization regarding the role of MCU in all tumors as there appears to be significant heterogeneity in the effects of the MCUC on tumor progression.
2.6. Apoptosis in PAH and cancer
Apoptosis is an essential physiological process by which multicellular organisms maintain quality control and eliminate abnormal cells to maintain homeostasis. At the molecular and cellular level, a series of morphological and biochemical changes are involved in apoptosis, including the formation of apoptotic bodies, shrinkage of the cells, caspase activation and DNA fragmentation [355].
In PAH, resistance to apoptosis displayed by abnormal PASMCs result in the proliferation/apoptosis imbalance, which contributes to the elevation of the PVR. In many cases, PAH patients receive a diagnosis at an advanced stage of the disease when many changes in the vessel wall layers have already taken place; therefore, cell growth inhibition may have limited value in reducing PVR at this stage. Thus, the current research is directed toward targeting these unwanted cells by induction of apoptosis in an orderly manner instead of inhibiting cell growth [356].
Likewise, in cancer, the imbalance of proliferation and apoptosis results in abnormal accumulation of cells. Apoptosis-resistance contributes to cellular multiplication and tumorigenesis; therefore, the current cancer research focuses on advancing novel therapeutic inducers to execute apoptosis [357].
Two crucial signalling pathways regulate apoptosis: the extrinsic (cell receptor) and the intrinsic (mitochondrial) pathways. Cell viability is governed at the molecular level by a balance between pro-apoptotic and anti-apoptotic signals. Within the mitochondrial pathway, anti-apoptotic signalling is mediated by several gene families, the most prominent being the Bcl-2 family (BCL-2, BCL-XL and Mcl-1), promoting the integrity of the outer mitochondrial membrane [358]. Increased expression of anti-apoptotic proteins is involved in the development and progression of PAH as well as cancer [359]. Conversely, Bax, Bak and Bcl-2 homology 3 (BH3) proteins are pro-apoptotic. Therefore, stimulating the pro-apoptotic signal or blocking anti-apoptotic function could carry a potential therapeutic benefit in reversing pulmonary vascular remodelling in PAH and cancer.
The PI3K/AKT/mTOR signalling pathway represents a key regulatory mechanism to control pro- and anti-apoptotic Bcl-2 family proteins’ activity. Small-molecule inhibitors of this signalling pathway offer new means to regulate mitochondrial apoptosis [360]. PI3K inhibitors prime neuroblastoma cells for chemotherapy by increasing apoptosis through an increased expression of pro-apoptotic Bcl-2 family proteins [361]. In this study, the use of dual-class I PI3K/mTOR inhibitor (PI103) synergistically enhanced apoptosis induced by doxorubicin in several neuroblastoma cell lines. The FDA-approved mTOR inhibitors (e.g., Rapamycin) are are antiproliferative agents that are delivered as a coating within endovascular stent and dramatically reduce local restenosis [362]. Rapamycin can trigger apoptosis in primary human ECs, human umbilical vein ECs, and aortic ECs by inhibiting mTORC2 [363, 364]. Rapamycin is a candidate pro-apoptotic drug for human pulmonary vascular cells in PAH as well. It reduced the proliferation of PASMCs from MCT rats and patients with idiopathic PAH [365, 366]. In hypoxia-induced PH mice, rapamycin decreased the thickening and proliferation of the pulmonary vasculature as well as RV hypertrophy [367]. However, McMurtry et al. reported that neither rapamycin nor rapamycin + atorvastatin has therapeutic benefits for the MCT-PAH model [368]. These discordant findings may reflect differences between the pathogenesis of preclinical models of PH (hypoxia vs MCT) as well as the methodology used (in vitro study vs in vivo study). In a clinical study, everolimus, another mTOR inhibitor, improved PVR and 6-minute walk distance (6MWD) in 8 of the 10 patients with PAH or CTEPH [369]. A Phase 1 clinical trial of albumin-bound mTOR inhibitor (ABI-009) is also now being evaluated in patients with severe PAH; the study is expected to end by December 2020 (NCT02587325).
Molecules that directly target pro- or anti-apoptotic regulator proteins, such as ABT-737 and ABT-263 (Navitoclax), have greater potential specificity than rapamycin. These BH3-mimetic small molecules activate BAX and BAK by displacing activator BH3 from BCL-2 and BCL-XL [370, 371], or selectively inhibit BCL-2, like ABT-199 (Venetoclax). In April 2016, the first inhibitor of BCL-2 (Venetoclax, Venclexta®) was approved by the FDA for the treatment of patients with chronic lymphocytic leukemia who have 17p deletion [372]. This drug has a more specific, pure, apoptotic activity compared to other cancer medications that affect both apoptosis as well as autophagy, such as anthracycline, a proteasome inhibitor, and taxane classes of drugs [373–375].
Rybka et al. assessed the pro-apoptotic effects of BH3 mimetic drugs ABT-263 (Navitoclax), ABT-199 (Venetoclax), ABT-737, and Obatoclax in experimental PAH [376]. Each of these agents promotes the death of human PASMCs in vitro. Moreover, in the rat Su/Hx-PAH model, ABT-263 promotes a beneficial death of PASMCs and reverses pulmonary vascular remodelling. This demonstration that induction of apoptosis is beneficial in PAH fits well with the benefit reported with metabolic modulators, like DCA, which inhibits cell proliferation and promotes apoptosis and also regresses experimental PAH in most preclinical models [176, 377], and even in some patients [182].
Another mitochondrial protein identified as a key signaling molecule for apoptosis is Cyt c, which is located on the IMM. During cellular stress, Cyt c is released from the mitochondria and acts as a signaling molecule. In the cytosol, Cyt c interacts with apoptosis activating factor 1 (Apaf-1), leading to apoptosome formation and caspase activation, initiating the cell death pathway. An increase in cytosolic Cyt c, in association with Apaf-1, is a trigger for the activation of caspases (e.g., caspases-3, −5, and −7), which activates cysteine proteases that are central executioners of the apoptotic pathway [378].
Inhibiting survivin, an inhibitor of apoptosis that is upregulated in cancer and PAH, has potential therapeutic benefit in decreasing adverse vascular remodeling in experimental PAH [379]. McMurtry et al. showed that the delivery of phosphorylation-deficient survivin to PASMCs from MCT-PAH rats reduces proliferation and increases apoptosis. The survivin mutant depolarizes PASMC mitochondria and initiates a leak of Cyt c and apoptosis-inducing factor (AIF) in the cytoplasm, thereby mediating mitochondria-dependent apoptosis. Gene therapy with a survivin mutant improves hemodynamics, reduces remodeling of the resistance PAs, and prolongs survival in MCT-PAH rats. Another study has shown that treating human PASMC with Puerarin, the primary active ingredient extracted from the root of a Chinese medicinal herb, kudzu, induces Cyt c release and caspase-9 activation in hypoxic but not normoxic human PASMCs in vitro. Puerarin decreases Bcl-2 and increases Bax expression [380].
Once established, PAH is characterized by apoptosis resistance, reminiscent of the apoptosis-resistant state of cancer. Induction of mitochondria-dependent apoptosis is a potential therapeutic strategy to regress PAH and cancer [379].
Conclusion
Oxygen sensing is mediated by mitochondria and their ability to transduce changes in pO2 into redox signals that regulate ion channels and enzymes. In both PAH and cancer, the oxygen sensing pathway is abnormal, and cells behave as if they were hypoxic, despite abundant ambient O2. Dysregulation of the oxygens sensing mechanisms at many levels contributes to shared features of PAH and cancer, notably increased rates of cell proliferation and impaired apoptosis. In both PAH and cancer, changes in mitochondrial metabolism, calcium handling and dynamics create a “pseudohypoxic” environment in which the normal mitochondrial mechanisms of oxygen sensing are subverted. Aberrant mitochondrial functions shared by PAH and cancer include a shift to aerobic glycolysis (the Warburg phenomenon), abnormal mitochondrial dynamics (notably increased fragmentation due to a fission/fusion imbalance), abnormal mitophagy and impaired mitochondrial biogenesis and impaired mitochondrial calcium homeostasis (due to impaired function of the MCUC). Many of these abnormalities are epigenetically regulated and/or reflect post-translational modification of mitochondrial pathways. Many of epigenetically triggered pathways are therapeutically tractable. Recent preclinical studies demonstrate the therapeutic potential of targeting mitochondrial metabolic targets, such as PDK and PKM2, mitochondrial dynamic targets (Drp1, MiD49, MiD51 and Mfn2), regulators of mitochondrial mediated apoptosis (BCL-2), and pathways that regulate intramitochondrial calcium (such as the MCUC). Mitochondrial targets are readily regulated using miRNA, siRNA and small molecule therapeutics.
Acknowledgement
This work was supported in part by U.S. National Institutes of Health (NIH) grants NIH R01HL113003 (S.L.A.), NIH R01HL071115 (S.L.A.), Canada Foundation for Innovation 33518 (S.L.A.), Tier 1 Canada Research Chair in Mitochondrial Dynamics and Translational Medicine 950-229252 (S.L.A.), the William J. Henderson Foundation 51038-13401 (S.L.A.), and the Canadian Vascular Network Scholar Award (L.T.; A.D.G.; K.D.S.; D.W.).
Abbreviations
- 4-AP
4-aminopyridine, a Kv channel inhibitor
- 5-AZA
5-aza-2’-deoxycytidine
- 6MWD
6-minute walk distance
- acetyl-CoA
acetyl coenzyme A
- AIF
apoptosis-inducing factor
- Akt
protein kinase B
- AMPK
AMP-activated protein kinase
- Ang
angiopoietin
- ANGPT2
gene encoding angiopoietin 2
- Apaf-1
apoptosis activating factor 1
- AR
androgen receptor
- ATF6
activating transcription factor 6
- ATP
adenosine triphosphate
- Bcl-2
B-cell lymphoma 2
- BH3
Bcl-2 homology 3
- BMPR2
bone morphogenetic protein receptor type II, encoded by BMPR2
- BNIP3
adenovirus E1B 19-kDa-interacting protein 3
- BNIP3L
BNIP3-like, also known as NIX
- BORIS
brother of regulator of imprinted sites
- BTIC
brain tumor initiating cell
- C76
compound 76, a HIF-2α inhibitor
- [Ca2+]cyto
cytosolic calcium
- [Ca2+]mito
mitochondrial calcium
- CaL
voltage-gated calcium channel
- CAMK
Ca2+/calmodulin-dependent protein kinase
- CBS
cystathionine β-synthase
- CDK
cyclin-dependent kinase
- Cyt c
cytochrome c
- DCA
dichloroacetate
- Dnm2
dynamin 2
- DNMT
DNA methyltransferase
- Drp1
dynamin-related protein 1, also called dynamin-1-like protein
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor, encoded by EGFR
- EMRE
essential MCU regulator
- EMT
epithelial-to-mesenchymal transition
- ER
endoplasmic reticulum
- ERBB2
human epidermal growth factor receptor 2
- ERK2
mitogen-activated protein kinase 1
- ERR
estrogen-related receptor
- ETC
electron transport chain
- FDG
2-[18F]-Fluoro-2-deoxy-d-glucose
- FHR
fawn hooded rats
- FIS1
mitochondrial fission 1 protein
- FOXO1
forkhead box protein O1
- FRAP
FKBP-rapamycin-associated protein
- G-6-P
glucose-6-phosphate
- GLUT
glucose transporter
- H2O2
hydrogen peroxide
- HER2
human epidermal growth factor receptor-2
- HIF
hypoxia-inducible factor
- HK
hexokinase
- hnRNP
heterogenous nuclear ribonucleoprotein
- HOSS
homeostatic oxygen-sensing system
- HPV
hypoxic pulmonary vasoconstriction
- HRE
hypoxia response element
- HSG
hyperplasia suppressor gene
- Hsp70
heat shock protein 70
- HyPer-dCyto
compartment-targeted probe for cytosolic H2O2
- HyPer-dMito
compartment-targeted probe for mitochondrial H2O2
- IGF
insulin-like growth factor
- IGFBP
IGF-binding protein
- Ik
potassium current
- ILK
integrin-linked kinase
- IMM
inner mitochondrial membrane
- IMS
intermembrane space
- IR
ischemia-reperfusion
- JNK
c-Jun N-terminal kinase
- KNK437
heat shock protein inhibitor I
- Kv
voltage-gated potassium channel
- LC3
light chain 3
- LDH-A
lactate dehydrogenase A, encoded by LDHA
- MCT
monocrotaline
- MCU
mitochondrial Ca2+ uniporter
- MCUC
mitochondrial Ca2+ uniporter complex
- mdivi-1
mitochondrial division inhibitor
- MET
mesenchymal-to-epithelial transition
- Mff
mitochondrial fission factor
- Mfn
mitofusin
- MICU1
mitochondrial calcium uptake 1
- MiD49
mitochondrial dynamics protein of 49 kDa
- MiD51
mitochondrial dynamics protein 51 kDa
- miRNA
microRNA
- MLS
mitochondrial localization sequence
- MMP
matrix metalloproteinase
- MnTBAP
Mn(III)tetrakis(4-benzoic acid)porphyrin chloride, a SOD mimic
- mPAP
mean pulmonary artery pressure
- MPC
mitochondrial pyruvate carrier
- MPP
mitochondrial processing peptidase
- mtDNA
mitochondrial DNA
- mTOR
the mammalian target of rapamycin
- mTORC
mammalian target of rapamycin complex
- NDP52
nuclear dot protein 52 kDa
- NIMA-1
never in mitosis A-1, also known as PIN1
- Nrf
nuclear respiratory factor
- NSCLC
non-small cell lung cancer
- O2×−
superoxide anion
- OMM
outer mitochondrial membrane
- OPA1
optic atrophy 1
- OPTN
optineurin
- OXPHOS
oxidative phosphorylation
- p-Drp1S616
phosphorylated Drp1 at serine 616
- p-Drp1S637
phosphorylated Drp1 at serine 637
- PA
pulmonary artery
- PAEC
pulmonary artery endothelial cell
- PAfib
pulmonary artery fibroblast
- PAH
pulmonary arterial hypertension
- PAP
pulmonary artery pressure
- PAR-2
protease-activated receptor-2
- PARL
presenilin-associated rhomboid-like
- PASMC
pulmonary artery smooth muscle cell
- PDC
pyruvate dehydrogenase complex
- PDGF
platelet-derived growth factor
- PDH
pyruvate dehydrogenase
- PDK
pyruvate dehydrogenase kinase
- PET
positron emission tomography
- PGC-1α
peroxisome proliferator-activated receptor γ coactivator-1 α
- PH
pulmonary hypertension
- PHD
prolyl hydroxylase
- PI3K
phosphoinositide 3-kinase
- PINK1
PTEN-induced putative kinase
- PK
pyruvate kinase
- PKM
pyruvate kinase muscle isoform
- pO2
partial pressure of oxygen
- PPHN
pulmonary hypertension of the newborn
- PTB
polypyrimidine tract-binding protein
- PTBP1
polypyrimidine-tract-binding protein 1
- PTEN
phosphatase and tensin homolog
- PVR
pulmonary vascular resistance
- RCC
renal cell carcinoma
- REDOX
reduction and oxidation
- ROS
reactive oxygen species
- RuR
ruthenium red
- RV
right ventricle
- RVF
right ventricular failure
- RVfib
right ventricular fibroblast
- SAB
supra-aortic banding
- siRNA
small interfering RNA
- SIRT3
gene encoding sirtuin 3
- SMC
smooth muscle cell
- SOD2
superoxide dismutase
- Su/Hx
Sugen5416/hypoxia, a preclinical model of PAH
- TEK
gene encoding TEK receptor tyrosine kinase (angiopoietin-1 receptor)
- TEMPOL
4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl, a SOD mimic
- TFAM
mitochondrial transcription factor A
- TGF-α
transforming growth factor-α, encoded by TGFA
- Tie2
angiopoietin-1 receptor
- TNBC
triple negative breast cancer
- TRPC
transient receptor potential channel
- UCP2
mitochondrial uncoupling protein 2, encoded by UCP2
- VDAC1
voltage-dependent-anion-selective channel 1
- VEGF
vascular endothelial growth factor
- VEGFA
gene encoding vascular endothelial growth factor A
- VHL
von Hippel-Lindau protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Weir EK, López-Barneo J, Buckler KJ, Archer SL, Acute oxygen-sensing mechanisms, The New England journal of medicine 353(19) (2005) 2042–2055. 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Weir EK, Archer SL, The role of redox changes in oxygen sensing, Respir Physiol Neurobiol 174(3) (2010) 182–91. 10.1016/j.resp.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dunham-Snary KJ, Hong ZG, Xiong PY, Del Paggio JC, Herr JE, Johri AM, Archer SL, A mitochondrial redox oxygen sensor in the pulmonary vasculature and ductus arteriosus, Pflugers Arch 468(1) (2016) 43–58. 10.1007/s00424-015-1736-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Michelakis ED, Thébaud B, Weir EK, Archer SL, Hypoxic pulmonary vasoconstriction: redox regulation of O2-sensitive K+ channels by a mitochondrial O2-sensor in resistance artery smooth muscle cells, J Mol Cell Cardiol 37(6) (2004) 1119–36. 10.1016/j.yjmcc.2004.09.007. [DOI] [PubMed] [Google Scholar]
- [5].Baraka AS, Taha SK, Yaacoub CI, Alarming hypoxemia during one-lung ventilation in a patient with respiratory bronchiolitis-associated interstitial lung disease, Canadian journal of anaesthesia = Journal canadien d’anesthesie 50(4) (2003) 411–4. 10.1007/BF03021041. [DOI] [PubMed] [Google Scholar]
- [6].Jensen KS, Micco AJ, Czartolomna J, Latham L, Voelkel NF, Rapid onset of hypoxic vasoconstriction in isolated lungs, J Appl Physiol (1985) 72(5) (1992) 2018–23. [DOI] [PubMed] [Google Scholar]
- [7].Grant JL, Naylor RW, Crandell WB, Bronchial adenoma resection with relief of hypoxic pulmonary vasoconstriction, Chest 77(3) (1980) 446–9. [DOI] [PubMed] [Google Scholar]
- [8].Shirai M, Ninomiya I, Sada K, Constrictor response of small pulmonary arteries to acute pulmonary hypertension during left atrial pressure elevation, The Japanese journal of physiology 41(1) (1991) 129–42. [DOI] [PubMed] [Google Scholar]
- [9].Lumb AB, Slinger P, Hypoxic Pulmonary Vasoconstriction: Physiology and Anesthetic Implications, Anesthesiology 122(4) (2015) 932–946. 10.1097/ALN.0000000000000569. [DOI] [PubMed] [Google Scholar]
- [10].Kylhammar D, Rådegran G, The principal pathways involved in the in vivo modulation of hypoxic pulmonary vasoconstriction, pulmonary arterial remodelling and pulmonary hypertension, Acta Physiologica 219(4) (2017) 728–756. 10.1111/apha.12749. [DOI] [PubMed] [Google Scholar]
- [11].Reeve HL, Weir EK, Nelson DP, Peterson DA, Archer SL, Opposing effects of oxidants and antioxidants on K+ channel activity and tone in rat vascular tissue, Experimental physiology 80(5) (1995) 825–34. 10.1113/expphysiol.1995.sp003890. [DOI] [PubMed] [Google Scholar]
- [12].Park MK, Bae YM, Lee SH, Ho WK, Earm YE, Modulation of voltage-dependent K+ channel by redox potential in pulmonary and ear arterial smooth muscle cells of the rabbit, Pflugers Arch 434(6) (1997) 764–71. 10.1007/s004240050463. [DOI] [PubMed] [Google Scholar]
- [13].Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M, Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation, Nature 352(6337) (1991) 711–4. 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- [14].Reeve HL, Weir EK, Archer SL, Cornfield DN, A maturational shift in pulmonary K+ channels, from Ca2+ sensitive to voltage dependent, American Journal of Physiology-Lung Cellular and Molecular Physiology 275(6) (1998) L1019–L1025. 10.1152/ajplung.1998.275.6.L1019. [DOI] [PubMed] [Google Scholar]
- [15].Hasunuma K, Rodman DM, McMurtry IF, Effects of K+ channel blockers on vascular tone in the perfused rat lung, Am Rev Respir Dis 144(4) (1991) 884–7. 10.1164/ajrccm/144.4.884. [DOI] [PubMed] [Google Scholar]
- [16].Weir EK, Archer SL, The mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channels, FASEB J 9(2) (1995) 183–9. [DOI] [PubMed] [Google Scholar]
- [17].McMurtry IF, Davidson AB, Reeves JT, Grover RF, Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs, Circ Res 38(2) (1976) 99–104. [DOI] [PubMed] [Google Scholar]
- [18].Harder DR, Madden JA, Dawson C, Hypoxic induction of Ca2+-dependent action potentials in small pulmonary arteries of the cat, J Appl Physiol (1985) 59(5) (1985) 1389–93. 10.1152/jappl.1985.59.5.1389. [DOI] [PubMed] [Google Scholar]
- [19].Robertson TP, Aaronson PI, Ward JP, Ca2+ sensitization during sustained hypoxic pulmonary vasoconstriction is endothelium dependent, Am J Physiol Lung Cell Mol Physiol 284(6) (2003) L1121–6. 10.1152/ajplung.00422.2002. [DOI] [PubMed] [Google Scholar]
- [20].Kizub IV, Strielkov IV, Shaifta Y, Becker S, Prieto-Lloret J, Snetkov VA, Soloviev AI, Aaronson PI, Ward JP, Gap junctions support the sustained phase of hypoxic pulmonary vasoconstriction by facilitating calcium sensitization, Cardiovasc Res 99(3) (2013) 404–11. 10.1093/cvr/cvt129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tipparaju SM, Li X-P, Kilfoil PJ, Xue B, Uversky VN, Bhatnagar A, Barski OA, Interactions between the C-terminus of Kv1.5 and Kvβ regulate pyridine nucleotide-dependent changes in channel gating, Pflugers Arch 463(6) (2012) 799–818. 10.1007/s00424-012-1093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen-Huu L, Reeve HL, Hampl V, Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes, J Clin Invest 101(11) (1998) 2319–30. 10.1172/jci333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Archer SL, London B, Hampl V, Wu X, Nsair A, Puttagunta L, Hashimoto K, Waite RE, Michelakis ED, Impairment of hypoxic pulmonary vasoconstriction in mice lacking the voltage-gated potassium channel Kv1.5, FASEB J 15(10) (2001) 1801–3. [DOI] [PubMed] [Google Scholar]
- [24].Hulme JT, Coppock EA, Felipe A, Martens JR, Tamkun MM, Oxygen sensitivity of cloned voltage-gated K(+) channels expressed in the pulmonary vasculature, Circ Res 85(6) (1999) 489–97. 10.1161/01.res.85.6.489. [DOI] [PubMed] [Google Scholar]
- [25].Patel AJ, Lazdunski M, Honoré E, Kv2.1/Kv9.3, a novel ATP-dependent delayed-rectifier K+ channel in oxygen-sensitive pulmonary artery myocytes, Embo j 16(22) (1997) 6615–25. 10.1093/emboj/16.22.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Oliver Dolly J, Pongs O, Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit, Nature 369(6478) (1994) 289–294. 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- [27].Franco-Obregon A, Lopez-Barneo J, Differential oxygen sensitivity of calcium channels in rabbit smooth muscle cells of conduit and resistance pulmonary arteries, J Physiol 491 (Pt 2) (1996) 511–8. 10.1113/jphysiol.1996.sp021235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wong H-S, Dighe PA, Mezera V, Monternier P-A, Brand MD, Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions, The Journal of biological chemistry 292(41) (2017) 16804–16809. 10.1074/jbc.R117.789271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Turrens JF, Boveris A, Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria, The Biochemical journal 191(2) (1980) 421–427. 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sugioka K, Nakano M, Totsune-Nakano H, Minakami H, Tero-Kubota S, Ikegami Y, Mechanism of O2- generation in reduction and oxidation cycle of ubiquinones in a model of mitochondrial electron transport systems, Biochim Biophys Acta 936(3) (1988) 377–85. 10.1016/0005-2728(88)90014-x. [DOI] [PubMed] [Google Scholar]
- [31].Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD, Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions, J Biol Chem 287(32) (2012) 27255–64. 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kaewpila S, Venkataraman S, Buettner GR, Oberley LW, Manganese superoxide dismutase modulates hypoxia-inducible factor-1 alpha induction via superoxide, Cancer Res 68(8) (2008) 2781–8. 10.1158/0008-5472.CAN-07-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Burgoyne JR, Oka S.-i., Ale-Agha N, Eaton P, Hydrogen peroxide sensing and signaling by protein kinases in the cardiovascular system, Antioxidants & redox signaling 18(9) (2013) 1042–1052. 10.1089/ars.2012.4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Michelakis ED, Hampl V, Nsair A, Wu X, Harry G, Haromy A, Gurtu R, Archer SL, Diversity in mitochondrial function explains differences in vascular oxygen sensing, Circ Res 90(12) (2002) 1307–15. [DOI] [PubMed] [Google Scholar]
- [35].Yuan XJ, Tod ML, Rubin LJ, Blaustein MP, Contrasting effects of hypoxia on tension in rat pulmonary and mesenteric arteries, Am J Physiol 259(2 Pt 2) (1990) H281–9. 10.1152/ajpheart.1990.259.2.H281. [DOI] [PubMed] [Google Scholar]
- [36].Freeman BA, Crapo JD, Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria, J Biol Chem 256(21) (1981) 10986–92. [PubMed] [Google Scholar]
- [37].Freeman BA, Topolosky MK, Crapo JD, Hyperoxia increases oxygen radical production in rat lung homogenates, Arch Biochem Biophys 216(2) (1982) 477–84. 10.1016/0003-9861(82)90236-3. [DOI] [PubMed] [Google Scholar]
- [38].Oshino N, Jamieson D, Chance B, The properties of hydrogen peroxide production under hyperoxic and hypoxic conditions of perfused rat liver, Biochem J 146(1) (1975) 53–65. 10.1042/bj1460053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Berkelhamer SK, Kim GA, Radder JE, Wedgwood S, Czech L, Steinhorn RH, Schumacker PT, Developmental differences in hyperoxia-induced oxidative stress and cellular responses in the murine lung, Free Radic Biol Med 61 (2013) 51–60. 10.1016/j.freeradbiomed.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Michelakis ED, Archer SL, Weir EK, Acute hypoxic pulmonary vasoconstriction: a model of oxygen sensing, Physiol Res 44(6) (1995) 361–7. [PubMed] [Google Scholar]
- [41].Archer SL, Nelson DP, Weir EK, Simultaneous measurement of O2 radicals and pulmonary vascular reactivity in rat lung, J Appl Physiol (1985) 67(5) (1989) 1903–11. [DOI] [PubMed] [Google Scholar]
- [42].Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT, Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes, J Biol Chem 273(19) (1998) 11619–24. 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- [43].Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT, Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes, J Biol Chem 273(29) (1998) 18092–8. 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- [44].Chandel NS, Trzyna WC, McClintock DS, Schumacker PT, Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin, Journal of immunology (Baltimore, Md. : 1950) 165(2) (2000) 1013–21. 10.4049/jimmunol.165.2.1013. [DOI] [PubMed] [Google Scholar]
- [45].Waypa GB, Marks JD, Mack MM, Boriboun C, Mungai PT, Schumacker PT, Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes, Circ Res 91(8) (2002) 719–26. [DOI] [PubMed] [Google Scholar]
- [46].Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT, Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing, Cell Metab 1(6) (2005) 401–8. 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- [47].Gao L, González-Rodríguez P, Ortega-Sáenz P, López-Barneo J, Redox signaling in acute oxygen sensing, Redox biology 12 (2017) 908–915. 10.1016/j.redox.2017.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sommer N, Huttemann M, Pak O, Scheibe S, Knoepp F, Sinkler C, Malczyk M, Gierhardt M, Esfandiary A, Kraut S, Jonas F, Veith C, Aras S, Sydykov A, Alebrahimdehkordi N, Giehl K, Hecker M, Brandes RP, Seeger W, Grimminger F, Ghofrani HA, Schermuly RT, Grossman LI, Weissmann N, Mitochondrial Complex IV Subunit 4 Isoform 2 Is Essential for Acute Pulmonary Oxygen Sensing, Circ Res 121(4) (2017) 424–438. 10.1161/circresaha.116.310482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Arias-Mayenco I, Gonzalez-Rodriguez P, Torres-Torrelo H, Gao L, Fernandez-Aguera MC, Bonilla-Henao V, Ortega-Saenz P, Lopez-Barneo J, Acute O2 Sensing: Role of Coenzyme QH2/Q Ratio and Mitochondrial ROS Compartmentalization, Cell Metab 28(1) (2018) 145–158 e4. 10.1016/j.cmet.2018.05.009. [DOI] [PubMed] [Google Scholar]
- [50].Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, Dokic D, Schumacker PT, Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells, Circ Res 106(3) (2010) 526–35. 10.1161/CIRCRESAHA.109.206334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dunham-Snary Kimberly J, Wu D, Potus F, Sykes Edward A, Mewburn Jeffrey D, Charles Rebecca L, Eaton P, Sultanian Richard A, Archer Stephen L, Ndufs2, a Core Subunit of Mitochondrial Complex I, Is Essential for Acute Oxygen-Sensing and Hypoxic Pulmonary Vasoconstriction, Circulation research 124(12) (2019) 1727–1746. 10.1161/CIRCRESAHA.118.314284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R, Haemodynamic definitions and updated clinical classification of pulmonary hypertension, Eur Respir J 53(1) (2019). 10.1183/13993003.01913-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Thenappan T, Ormiston ML, Ryan JJ, Archer SL, Pulmonary arterial hypertension: pathogenesis and clinical management, Bmj 360 (2018) j5492. 10.1136/bmj.j5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Humbert M, Lau EM, Montani D, Jaïs X, Sitbon O, Simonneau G, Advances in therapeutic interventions for patients with pulmonary arterial hypertension, Circulation 130(24) (2014) 2189–208. 10.1161/circulationaha.114.006974. [DOI] [PubMed] [Google Scholar]
- [55].Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL, Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1, Blood 105(2) (2005) 659–69. 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- [56].Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL, Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1, Mol Cell Biol 16(9) (1996) 4604–13. 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fallah J, Rini BI, HIF Inhibitors: Status of Current Clinical Development, Current Oncology Reports 21(1) (2019) 6. 10.1007/s11912-019-0752-z. [DOI] [PubMed] [Google Scholar]
- [58].Semenza GL, Hypoxia-inducible factor 1 (HIF-1) pathway, Sci STKE 2007(407) (2007) cm8. 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- [59].Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E, Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis, Nature 394(6692) (1998) 485–90. 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- [60].Pugh CW, Ratcliffe PJ, Regulation of angiogenesis by hypoxia: role of the HIF system, Nature medicine 9(6) (2003) 677–84. 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- [61].Wenger RH, Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression, Faseb j 16(10) (2002) 1151–62. 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- [62].Smith TG, Brooks JT, Balanos GM, Lappin TR, Layton DM, Leedham DL, Liu C, Maxwell PH, McMullin MF, McNamara CJ, Percy MJ, Pugh CW, Ratcliffe PJ, Talbot NP, Treacy M, Robbins PA, Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology, PLoS medicine 3(7) (2006) e290. 10.1371/journal.pmed.0030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bushuev VI, Miasnikova GY, Sergueeva AI, Polyakova LA, Okhotin D, Gaskin PR, Debebe Z, Nekhai S, Castro OL, Prchal JT, Gordeuk VR, Endothelin-1, vascular endothelial growth factor and systolic pulmonary artery pressure in patients with Chuvash polycythemia, Haematologica 91(6) (2006) 744–9. [PubMed] [Google Scholar]
- [64].Hickey MM, Richardson T, Wang T, Mosqueira M, Arguiri E, Yu H, Yu QC, Solomides CC, Morrisey EE, Khurana TS, Christofidou-Solomidou M, Simon MC, The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice, J Clin Invest 120(3) (2010) 827–39. 10.1172/JCI36362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ang SO, Chen H, Gordeuk VR, Sergueeva AI, Polyakova LA, Miasnikova GY, Kralovics R, Stockton DW, Prchal JT, Endemic polycythemia in Russia: mutation in the VHL gene, Blood Cells Mol Dis 28(1) (2002) 57–62. 10.1006/bcmd.2002.0488. [DOI] [PubMed] [Google Scholar]
- [66].Hickey MM, Lam JC, Bezman NA, Rathmell WK, Simon MC, von Hippel-Lindau mutation in mice recapitulates Chuvash polycythemia via hypoxia-inducible factor-2alpha signaling and splenic erythropoiesis, J Clin Invest 117(12) (2007) 3879–89. 10.1172/jci32614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gordeuk VR, Sergueeva AI, Miasnikova GY, Okhotin D, Voloshin Y, Choyke PL, Butman JA, Jedlickova K, Prchal JT, Polyakova LA, Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors, Blood 103(10) (2004) 3924–3932. 10.1182/blood-2003-07-2535. [DOI] [PubMed] [Google Scholar]
- [68].Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thebaud B, Husain AN, Cipriani N, Rehman J, Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target, Circulation 121(24) (2010) 2661–71. 10.1161/CIRCULATIONAHA.109.916098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Huang LE, Arany Z, Livingston DM, Bunn HF, Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit, J Biol Chem 271(50) (1996) 32253–9. 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- [70].Archer SL, Huang J, Henry T, Peterson D, Weir EK, A redox-based O2 sensor in rat pulmonary vasculature, Circ Res 73(6) (1993) 1100–12. 10.1161/01.res.73.6.1100. [DOI] [PubMed] [Google Scholar]
- [71].Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF, Oxidative stress in severe pulmonary hypertension, Am J Respir Crit Care Med 169(6) (2004) 764–9. 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- [72].Khan C, Pathe N, Fazal S, Lister J, Rossetti JM, Azacitidine in the management of patients with myelodysplastic syndromes, Ther Adv Hematol 3(6) (2012) 355–73. 10.1177/2040620712464882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Steinhorn RH, Albert G, Swartz DD, Russell JA, Levine CR, Davis JM, Recombinant human superoxide dismutase enhances the effect of inhaled nitric oxide in persistent pulmonary hypertension, Am J Respir Crit Care Med 164(5) (2001) 834–9. 10.1164/ajrccm.164.5.2010104. [DOI] [PubMed] [Google Scholar]
- [74].Elmedal B, de Dam MY, Mulvany MJ, Simonsen U, The superoxide dismutase mimetic, tempol, blunts right ventricular hypertrophy in chronic hypoxic rats, Br J Pharmacol 141(1) (2004) 105–13. 10.1038/sj.bjp.0705580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hurt EM, Thomas SB, Peng B, Farrar WL, Molecular consequences of SOD2 expression in epigenetically silenced pancreatic carcinoma cell lines, British journal of cancer 97(8) (2007) 1116–23. 10.1038/sj.bjc.6604000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hurt EM, Thomas SB, Peng B, Farrar WL, Integrated molecular profiling of SOD2 expression in multiple myeloma, Blood 109(9) (2007) 3953–62. 10.1182/blood-2006-07-035162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hodge DR, Xiao W, Peng B, Cherry JC, Munroe DJ, Farrar WL, Enforced expression of superoxide dismutase 2/manganese superoxide dismutase disrupts autocrine interleukin-6 stimulation in human multiple myeloma cells and enhances dexamethasone-induced apoptosis, Cancer Res 65(14) (2005) 6255–63. 10.1158/0008-5472.CAN-04-4482. [DOI] [PubMed] [Google Scholar]
- [78].Hitchler MJ, Oberley LW, Domann FE, Epigenetic silencing of SOD2 by histone modifications in human breast cancer cells, Free Radic Biol Med 45(11) (2008) 1573–80. 10.1016/j.freeradbiomed.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Beaulieu N, Morin S, Chute IC, Robert MF, Nguyen H, MacLeod AR, An essential role for DNA methyltransferase DNMT3B in cancer cell survival, J Biol Chem 277(31) (2002) 28176–81. 10.1074/jbc.M204734200. [DOI] [PubMed] [Google Scholar]
- [80].Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC, Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation, Mol Cell Biol 23(24) (2003) 9361–74. 10.1128/mcb.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Dai Z, Zhu MM, Peng Y, Machireddy N, Evans CE, Machado R, Zhang X, Zhao YY, Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in Pulmonary Arterial Hypertension with a HIF-2α Inhibitor, Am J Respir Crit Care Med 198(11) (2018) 1423–1434. 10.1164/rccm.201710-2079OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Dai Z, Li M, Wharton J, Zhu MM, Zhao YY, Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2α, Circulation 133(24) (2016) 2447–58. 10.1161/circulationaha.116.021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].McLaughlin VV, Shah SJ, Souza R, Humbert M, Management of pulmonary arterial hypertension, J Am Coll Cardiol 65(18) (2015) 1976–97. 10.1016/j.jacc.2015.03.540. [DOI] [PubMed] [Google Scholar]
- [84].Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P, Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia, J Clin Invest 111(10) (2003) 1519–27. 10.1172/jci15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A, Tolliday N, Lamb J, Pantopoulos K, Golub T, Iliopoulos O, Small-molecule inhibitors of HIF-2a translation link its 5’UTR iron-responsive element to oxygen sensing, Mol Cell 32(6) (2008) 838–48. 10.1016/j.molcel.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Archer SL, Fang YH, Ryan JJ, Piao L, Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension, Pulm Circ 3(1) (2013) 144–52. 10.4103/2045-8932.109960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kim JW, Tchernyshyov I, Semenza GL, Dang CV, HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia, Cell Metab 3(3) (2006) 177–85. 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- [88].Firth JD, Ebert BL, Ratcliffe PJ, Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements, J Biol Chem 270(36) (1995) 21021–7. 10.1074/jbc.270.36.21021. [DOI] [PubMed] [Google Scholar]
- [89].Tian L, Wu D, Dasgupta A, Chen KH, Mewburn J, Potus F, Lima PDA, Hong Z, Zhao YY, Hindmarch CCT, Kutty S, Provencher S, Bonnet S, Sutendra G, Archer SL, Epigenetic Metabolic Reprogramming of Right Ventricular Fibroblasts in Pulmonary Arterial Hypertension: A Pyruvate Dehydrogenase Kinase-Dependent Shift in Mitochondrial Metabolism Promotes Right Ventricular Fibrosis, Circ Res 126(12) (2020) 1723–1745. 10.1161/CIRCRESAHA.120.316443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC, The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth, Nature 452(7184) (2008) 230–3. 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- [91].Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL, Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1, Cell 145(5) (2011) 732–44. 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Robey IF, Stephen RM, Brown KS, Baggett BK, Gatenby RA, Gillies RJ, Regulation of the Warburg effect in early-passage breast cancer cells, Neoplasia 10(8) (2008) 745–56. 10.1593/neo.07724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Black EJ, Clair T, Delrow J, Neiman P, Gillespie DA, Microarray analysis identifies Autotaxin, a tumour cell motility and angiogenic factor with lysophospholipase D activity, as a specific target of cell transformation by v-Jun, Oncogene 23(13) (2004) 2357–66. 10.1038/sj.onc.1207377. [DOI] [PubMed] [Google Scholar]
- [94].Bartrons R, Caro J, Hypoxia, glucose metabolism and the Warburg’s effect, J Bioenerg Biomembr 39(3) (2007) 223–9. 10.1007/s10863-007-9080-3. [DOI] [PubMed] [Google Scholar]
- [95].Semenza GL, Regulation of metabolism by hypoxia-inducible factor 1, Cold Spring Harb Symp Quant Biol 76 (2011) 347–53. 10.1101/sqb.2011.76.010678. [DOI] [PubMed] [Google Scholar]
- [96].Almuhaideb A, Papathanasiou N, Bomanji J, 18F-FDG PET/CT imaging in oncology, Ann Saudi Med 31(1) (2011) 3–13. 10.4103/0256-4947.75771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, Thenappan T, Bache-Wiig P, Piao L, Paul J, Chen CT, Archer SL, Lung (1)(8)F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension, Am J Respir Crit Care Med 185(6) (2012) 670–9. 10.1164/rccm.201108-1562OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ, Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma, Mol Cell Biol 25(13) (2005) 5675–86. 10.1128/mcb.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR, High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq, Blood 117(23) (2011) e207–17. 10.1182/blood-2010-10-314427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, Ratcliffe PJ, Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts, J Biol Chem 284(25) (2009) 16767–75. 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wierenga ATJ, Cunningham A, Erdem A, Lopera NV, Brouwers-Vos AZ, Pruis M, Mulder AB, Günther UL, Martens JHA, Vellenga E, Schuringa JJ, HIF1/2-exerted control over glycolytic gene expression is not functionally relevant for glycolysis in human leukemic stem/progenitor cells, Cancer & Metabolism 7(1) (2019) 11. 10.1186/s40170-019-0206-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Gunaratnam L, Morley M, Franovic A, de Paulsen N, Mekhail K, Parolin DA, Nakamura E, Lorimer IA, Lee S, Hypoxia inducible factor activates the transforming growth factor-alpha/epidermal growth factor receptor growth stimulatory pathway in VHL(−/−) renal cell carcinoma cells, J Biol Chem 278(45) (2003) 44966–74. 10.1074/jbc.M305502200. [DOI] [PubMed] [Google Scholar]
- [103].Chang LH, Pan SL, Lai CY, Tsai AC, Teng CM, Activated PAR-2 regulates pancreatic cancer progression through ILK/HIF-alpha-induced TGF-alpha expression and MEK/VEGF-A-mediated angiogenesis, Am J Pathol 183(2) (2013) 566–75. 10.1016/j.ajpath.2013.04.022. [DOI] [PubMed] [Google Scholar]
- [104].Kurtipek E, Koçak N, Esme H, Düzgün N, Akin SE, Ünlü Y, Bekçi TT, The role of HIF-1 pathway in non-small-cell lung cancer, European Respiratory Journal 48(suppl 60) (2016) PA2855. 10.1183/13993003.congress-2016.PA2855. [DOI] [Google Scholar]
- [105].Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL, Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics, Cancer Res 60(6) (2000) 1541–5. [PubMed] [Google Scholar]
- [106].Peng XH, Karna P, Cao Z, Jiang BH, Zhou M, Yang L, Cross-talk between epidermal growth factor receptor and hypoxia-inducible factor-1alpha signal pathways increases resistance to apoptosis by up-regulating survivin gene expression, J Biol Chem 281(36) (2006) 25903–14. 10.1074/jbc.M603414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L, Treatment of HER2-positive breast cancer: current status and future perspectives, Nat Rev Clin Oncol 9(1) (2011) 16–32. 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- [108].Jarman EJ, Ward C, Turnbull AK, Martinez-Perez C, Meehan J, Xintaropoulou C, Sims AH, Langdon SP, HER2 regulates HIF-2α and drives an increased hypoxic response in breast cancer, Breast Cancer Res 21(1) (2019) 10. 10.1186/s13058-019-1097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL, Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2, Cancer Res 59(16) (1999) 3915–8. [PubMed] [Google Scholar]
- [110].Mohlin S, Hamidian A, Pahlman S, HIF2A and IGF2 expression correlates in human neuroblastoma cells and normal immature sympathetic neuroblasts, Neoplasia 15(3) (2013) 328–34. 10.1593/neo.121706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Levy AP, Levy NS, Wegner S, Goldberg MA, Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia, J Biol Chem 270(22) (1995) 13333–40. 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
- [112].Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D, Carmeliet P, Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice, Nature medicine 8(7) (2002) 702–10. 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- [113].Rankin EB, Rha J, Unger TL, Wu CH, Shutt HP, Johnson RS, Simon MC, Keith B, Haase VH, Hypoxia-inducible factor-2 regulates vascular tumorigenesis in mice, Oncogene 27(40) (2008) 5354–8. 10.1038/onc.2008.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Flamme I, Krieg M, Plate KH, Up-regulation of vascular endothelial growth factor in stromal cells of hemangioblastomas is correlated with up-regulation of the transcription factor HRF/HIF-2alpha, Am J Pathol 153(1) (1998) 25–9. 10.1016/s0002-9440(10)65541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lin YW, Huang ST, Wu JC, Chu TH, Huang SC, Lee CC, Tai MH, Novel HDGF/HIF-1α/VEGF axis in oral cancer impacts disease prognosis, BMC Cancer 19(1) (2019) 1083. 10.1186/s12885-019-6229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].De Francesco EM, Sims AH, Maggiolini M, Sotgia F, Lisanti MP, Clarke RB, GPER mediates the angiocrine actions induced by IGF1 through the HIF-1α/VEGF pathway in the breast tumor microenvironment, Breast Cancer Res 19(1) (2017) 129. 10.1186/s13058-017-0923-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Koga K, Todaka T, Morioka M, Hamada J, Kai Y, Yano S, Okamura A, Takakura N, Suda T, Ushio Y, Expression of angiopoietin-2 in human glioma cells and its role for angiogenesis, Cancer Res 61(16) (2001) 6248–54. [PubMed] [Google Scholar]
- [118].Sfiligoi C, de Luca A, Cascone I, Sorbello V, Fuso L, Ponzone R, Biglia N, Audero E, Arisio R, Bussolino F, Sismondi P, De Bortoli M, Angiopoietin-2 expression in breast cancer correlates with lymph node invasion and short survival, Int J Cancer 103(4) (2003) 466–74. 10.1002/ijc.10851. [DOI] [PubMed] [Google Scholar]
- [119].Caine GJ, Blann AD, Stonelake PS, Ryan P, Lip GY, Plasma angiopoietin-1, angiopoietin-2 and Tie-2 in breast and prostate cancer: a comparison with VEGF and Flt-1, Eur J Clin Invest 33(10) (2003) 883–90. 10.1046/j.1365-2362.2003.01243.x. [DOI] [PubMed] [Google Scholar]
- [120].Yoshida Y, Oshika Y, Fukushima Y, Tokunaga T, Hatanaka H, Kijima H, Yamazaki H, Ueyama Y, Tamaoki N, Miura S, Nakamura M, Expression of angiostatic factors in colorectal cancer, Int J Oncol 15(6) (1999) 1221–5. 10.3892/ijo.15.6.1221. [DOI] [PubMed] [Google Scholar]
- [121].Ahmad SA, Liu W, Jung YD, Fan F, Reinmuth N, Bucana CD, Ellis LM, Differential expression of angiopoietin-1 and angiopoietin-2 in colon carcinoma. A possible mechanism for the initiation of angiogenesis, Cancer 92(5) (2001) 1138–43.. [DOI] [PubMed] [Google Scholar]
- [122].Skuli N, Liu L, Runge A, Wang T, Yuan L, Patel S, Iruela-Arispe L, Simon MC, Keith B, Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis, Blood 114(2) (2009) 469–77. 10.1182/blood-2008-12-193581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Oh H, Takagi H, Suzuma K, Otani A, Matsumura M, Honda Y, Hypoxia and vascular endothelial growth factor selectively up-regulate angiopoietin-2 in bovine microvascular endothelial cells, J Biol Chem 274(22) (1999) 15732–9. 10.1074/jbc.274.22.15732. [DOI] [PubMed] [Google Scholar]
- [124].Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L, De Palma M, Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells, Cancer Cell 19(4) (2011) 512–26. 10.1016/j.ccr.2011.02.005. [DOI] [PubMed] [Google Scholar]
- [125].Duan LJ, Zhang-Benoit Y, Fong GH, Endothelium-intrinsic requirement for Hif-2alpha during vascular development, Circulation 111(17) (2005) 2227–32. 10.1161/01.Cir.0000163580.98098.A3. [DOI] [PubMed] [Google Scholar]
- [126].Stone RC, Pastar I, Ojeh N, Chen V, Liu S, Garzon KI, Tomic-Canic M, Epithelial-mesenchymal transition in tissue repair and fibrosis, Cell Tissue Res 365(3) (2016) 495–506. 10.1007/s00441-016-2464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Zhang Y, Weinberg RA, Epithelial-to-mesenchymal transition in cancer: complexity and opportunities, Front Med 12(4) (2018) 361–373. 10.1007/s11684-018-0656-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Lambert AW, Pattabiraman DR, Weinberg RA, Emerging Biological Principles of Metastasis, Cell 168(4) (2017) 670–691. 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Gonzalez DM, Medici D, Signaling mechanisms of the epithelial-mesenchymal transition, Science Signaling 7(344) (2014) re8–re8. 10.1126/scisignal.2005189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Peinado H, Olmeda D, Cano A, Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype?, Nature Reviews Cancer 7(6) (2007) 415–428. 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- [131].Imai T, Horiuchi A, Wang C, Oka K, Ohira S, Nikaido T, Konishi I, Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells, Am J Pathol 163(4) (2003) 1437–47. 10.1016/s0002-9440(10)63501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Evans AJ, Russell RC, Roche O, Burry TN, Fish JE, Chow VW, Kim WY, Saravanan A, Maynard MA, Gervais ML, Sufan RI, Roberts AM, Wilson LA, Betten M, Vandewalle C, Berx G, Marsden PA, Irwin MS, Teh BT, Jewett MA, Ohh M, VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail, Mol Cell Biol 27(1) (2007) 157–69. 10.1128/mcb.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Yang M-H, Wu M-Z, Chiou S-H, Chen P-M, Chang S-Y, Liu C-J, Teng S-C, Wu K-J, Direct regulation of TWIST by HIF-1α promotes metastasis, Nature Cell Biology 10(3) (2008) 295–305. 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- [134].Kim K, Park EY, Yoon MS, Suh DS, Kim KH, Lee JH, Shin DH, Kim JY, Sol MY, Choi KU, The Role of TWIST in Ovarian Epithelial Cancers, Korean J Pathol 48(4) (2014) 283–91. 10.4132/KoreanJPathol.2014.48.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Zhang J, Cheng Q, Zhou Y, Wang Y, Chen X, Slug is a key mediator of hypoxia induced cadherin switch in HNSCC: correlations with poor prognosis, Oral Oncol 49(11) (2013) 1043–50. 10.1016/j.oraloncology.2013.08.003. [DOI] [PubMed] [Google Scholar]
- [136].Iwasaki K, Ninomiya R, Shin T, Nomura T, Kajiwara T, Hijiya N, Moriyama M, Mimata H, Hamada F, Chronic hypoxia-induced slug promotes invasive behavior of prostate cancer cells by activating expression of ephrin-B1, Cancer Sci 109(10) (2018) 3159–3170. 10.1111/cas.13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Zhang W, Shi X, Peng Y, Wu M, Zhang P, Xie R, Wu Y, Yan Q, Liu S, Wang J, HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer, PloS one 10(6) (2015) e0129603. 10.1371/journal.pone.0129603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Zhu J, Huang Z, Zhang M, Wang W, Liang H, Zeng J, Wu K, Wang X, Hsieh JT, Guo P, Fan J, HIF-1α promotes ZEB1 expression and EMT in a human bladder cancer lung metastasis animal model, Oncol Lett 15(3) (2018) 3482–3489. 10.3892/ol.2018.7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Joseph JV, Conroy S, Pavlov K, Sontakke P, Tomar T, Eggens-Meijer E, Balasubramaniyan V, Wagemakers M, den Dunnen WF, Kruyt FA, Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α-ZEB1 axis, Cancer Lett 359(1) (2015) 107–16. 10.1016/j.canlet.2015.01.010. [DOI] [PubMed] [Google Scholar]
- [140].Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA, Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis, Cell 117(7) (2004) 927–939. 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- [141].Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA, The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression, Nat Cell Biol 2(2) (2000) 76–83. 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- [142].Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD, Chodosh LA, The transcriptional repressor Snail promotes mammary tumor recurrence, Cancer Cell 8(3) (2005) 197–209. 10.1016/j.ccr.2005.07.009. [DOI] [PubMed] [Google Scholar]
- [143].Yang J, Zhang X, Zhang Y, Zhu D, Zhang L, Li Y, Zhu Y, Li D, Zhou J, HIF-2α promotes epithelial-mesenchymal transition through regulating Twist2 binding to the promoter of E-cadherin in pancreatic cancer, Journal of experimental & clinical cancer research : CR 35 (2016) 26. 10.1186/s13046-016-0298-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Cockett MI, Murphy G, Birch ML, O’Connell JP, Crabbe T, Millican AT, Hart IR, Docherty AJ, Matrix metalloproteinases and metastatic cancer, Biochem Soc Symp 63 (1998) 295–313. [PubMed] [Google Scholar]
- [145].Wang Y, Li Z, Zhang H, Jin H, Sun L, Dong H, Xu M, Zhao P, Zhang B, Wang J, Pan Y, Liu L, HIF-1α and HIF-2α correlate with migration and invasion in gastric cancer, Cancer Biol Ther 10(4) (2010) 376–82. 10.4161/cbt.10.4.12441. [DOI] [PubMed] [Google Scholar]
- [146].Li Y, Qiu X, Zhang S, Zhang Q, Wang E, Hypoxia induced CCR7 expression via HIF-1alpha and HIF-2alpha correlates with migration and invasion in lung cancer cells, Cancer Biol Ther 8(4) (2009) 322–30. 10.4161/cbt.8.4.7332. [DOI] [PubMed] [Google Scholar]
- [147].Ryan JJ, Archer SL, Emerging concepts in the molecular basis of pulmonary arterial hypertension: part I: metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension, Circulation 131(19) (2015) 1691–702. 10.1161/CIRCULATIONAHA.114.006979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Dasgupta A, Wu D, Tian L, Xiong PY, Dunham-Snary KJ, Chen KH, Alizadeh E, Motamed M, Potus F, Hindmarch CCT, Archer SL, Mitochondria in the Pulmonary Vasculature in Health and Disease: Oxygen-Sensing, Metabolism, and Dynamics, Compr Physiol 10(2) (2020) 713–765. 10.1002/cphy.c190027. [DOI] [PubMed] [Google Scholar]
- [149].Archer SL, Pyruvate Kinase and Warburg Metabolism in Pulmonary Arterial Hypertension: Uncoupled Glycolysis and the Cancer-Like Phenotype of Pulmonary Arterial Hypertension, Circulation 136(25) (2017) 2486–2490. 10.1161/CIRCULATIONAHA.117.031655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Warburg O, Minami S, Versuche an Überlebendem Carcinom-gewebe, Klinische Wochenschrift 2(17) (1923) 776–777. 10.1007/BF01712130. [DOI] [Google Scholar]
- [151].Akins NS, Nielson TC, Le HV, Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer, Curr Top Med Chem 18(6) (2018) 494–504. 10.2174/1568026618666180523111351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Caruso P, Dunmore BJ, Schlosser K, Schoors S, Dos Santos C, Perez-Iratxeta C, Lavoie JR, Zhang H, Long L, Flockton AR, Frid MG, Upton PD, D’Alessandro A, Hadinnapola C, Kiskin FN, Taha M, Hurst LA, Ormiston ML, Hata A, Stenmark KR, Carmeliet P, Stewart DJ, Morrell NW, Identification of MicroRNA-124 as a Major Regulator of Enhanced Endothelial Cell Glycolysis in Pulmonary Arterial Hypertension via PTBP1 (Polypyrimidine Tract Binding Protein) and Pyruvate Kinase M2, Circulation 136(25) (2017) 2451–2467. 10.1161/CIRCULATIONAHA.117.028034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Zhang H, Wang D, Li M, Plecita-Hlavata L, D’Alessandro A, Tauber J, Riddle S, Kumar S, Flockton A, McKeon BA, Frid MG, Reisz JA, Caruso P, El Kasmi KC, Jezek P, Morrell NW, Hu CJ, Stenmark KR, Metabolic and Proliferative State of Vascular Adventitial Fibroblasts in Pulmonary Hypertension Is Regulated Through a MicroRNA-124/PTBP1 (Polypyrimidine Tract Binding Protein 1)/Pyruvate Kinase Muscle Axis, Circulation 136(25) (2017) 2468–2485. 10.1161/CIRCULATIONAHA.117.028069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Sun XQ, Zhang R, Zhang HD, Yuan P, Wang XJ, Zhao QH, Wang L, Jiang R, Jan Bogaard H, Jing ZC, Reversal of right ventricular remodeling by dichloroacetate is related to inhibition of mitochondria-dependent apoptosis, Hypertens Res 39(5) (2016) 302–11. 10.1038/hr.2015.153. [DOI] [PubMed] [Google Scholar]
- [155].Ryan JJ, Archer SL, The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure, Circ Res 115(1) (2014) 176–88. 10.1161/CIRCRESAHA.113.301129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Prisco SZ, Rose L, Potus F, Tian L, Wu D, Hartweck L, Al-Qazazi R, Neuber-Hess M, Eklund M, Hsu S, Thenappan T, Archer SL, Prins KW, Excess Protein O-GlcNAcylation Links Metabolic Derangements to Right Ventricular Dysfunction in Pulmonary Arterial Hypertension, Int J Mol Sci 21(19) (2020). 10.3390/ijms21197278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Stetak A, Veress R, Ovadi J, Csermely P, Keri G, Ullrich A, Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death, Cancer Res 67(4) (2007) 1602–8. 10.1158/0008-5472.CAN-06-2870. [DOI] [PubMed] [Google Scholar]
- [158].Hoshino A, Hirst JA, Fujii H, Regulation of cell proliferation by interleukin-3-induced nuclear translocation of pyruvate kinase, J Biol Chem 282(24) (2007) 17706–11. 10.1074/jbc.M700094200. [DOI] [PubMed] [Google Scholar]
- [159].Park YS, Kim DJ, Koo H, Jang SH, You YM, Cho JH, Yang SJ, Yu ES, Jung Y, Lee DC, Kim JA, Park ZY, Park KC, Yeom YI, AKT-induced PKM2 phosphorylation signals for IGF-1-stimulated cancer cell growth, Oncotarget 7(30) (2016) 48155–48167. 10.18632/oncotarget.10179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z, ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect, Nat Cell Biol 14(12) (2012) 1295–304. 10.1038/ncb2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Wang HJ, Hsieh YJ, Cheng WC, Lin CP, Lin YS, Yang SF, Chen CC, Izumiya Y, Yu JS, Kung HJ, Wang WC, JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1α-mediated glucose metabolism, Proc Natl Acad Sci U S A 111(1) (2014) 279–84. 10.1073/pnas.1311249111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Bhardwaj A, Das S, SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions, Proc Natl Acad Sci U S A 113(5) (2016) E538–47. 10.1073/pnas.1520045113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z, PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis, Cell 150(4) (2012) 685–96. 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].David CJ, Chen M, Assanah M, Canoll P, Manley JL, HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer, Nature 463(7279) (2010) 364–8. 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Yang W, Lu Z, Nuclear PKM2 regulates the Warburg effect, Cell Cycle 12(19) (2013) 3154–8. 10.4161/cc.26182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Patel MS, Nemeria NS, Furey W, Jordan F, The pyruvate dehydrogenase complexes: structure-based function and regulation, J Biol Chem 289(24) (2014) 16615–23. 10.1074/jbc.R114.563148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED, A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation, Cell 158(1) (2014) 84–97. 10.1016/j.cell.2014.04.046. [DOI] [PubMed] [Google Scholar]
- [168].Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N, Importing mitochondrial proteins: machineries and mechanisms, Cell 138(4) (2009) 628–44. 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Shi Y, Thomas JO, The transport of proteins into the nucleus requires the 70-kilodalton heat shock protein or its cytosolic cognate, Mol Cell Biol 12(5) (1992) 2186–92. 10.1128/mcb.12.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Liu B, Li J, Cairns MJ, Identifying miRNAs, targets and functions, Brief Bioinform 15(1) (2014) 1–19. 10.1093/bib/bbs075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Desai S, Ding M, Wang B, Lu Z, Zhao Q, Shaw K, Yung WK, Weinstein JN, Tan M, Yao J, Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers, Oncotarget 5(18) (2014) 8202–10. 10.18632/oncotarget.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Singh S, Narayanan SP, Biswas K, Gupta A, Ahuja N, Yadav S, Panday RK, Samaiya A, Sharan SK, Shukla S, Intragenic DNA methylation and BORIS-mediated cancer-specific splicing contribute to the Warburg effect, Proc Natl Acad Sci U S A 114(43) (2017) 11440–11445. 10.1073/pnas.1708447114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Yadav S, Bhagat SD, Gupta A, Samaiya A, Srivastava A, Shukla S, Dietary-phytochemical mediated reversion of cancer-specific splicing inhibits Warburg effect in head and neck cancer, BMC Cancer 19(1) (2019) 1031. 10.1186/s12885-019-6257-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Spoden GA, Mazurek S, Morandell D, Bacher N, Ausserlechner MJ, Jansen-Durr P, Eigenbrodt E, Zwerschke W, Isotype-specific inhibitors of the glycolytic key regulator pyruvate kinase subtype M2 moderately decelerate tumor cell proliferation, Int J Cancer 123(2) (2008) 312–321. 10.1002/ijc.23512. [DOI] [PubMed] [Google Scholar]
- [175].Sitbon O, Gomberg-Maitland M, Granton J, Lewis MI, Mathai SC, Rainisio M, Stockbridge NL, Wilkins MR, Zamanian RT, Rubin LJ, Clinical trial design and new therapies for pulmonary arterial hypertension, Eur Respir J 53(1) (2019). 10.1183/13993003.01908-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED, Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis, Circ Res 95(8) (2004) 830–40. 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- [177].Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL, An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension, Circulation 113(22) (2006) 2630–41. 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- [178].Plecita-Hlavata L, Tauber J, Li M, Zhang H, Flockton AR, Pullamsetti SS, Chelladurai P, D’Alessandro A, El Kasmi KC, Jezek P, Stenmark KR, Constitutive Reprogramming of Fibroblast Mitochondrial Metabolism in Pulmonary Hypertension, Am J Respir Cell Mol Biol 55(1) (2016) 47–57. 10.1165/rcmb.2015-0142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Zhao L, Ashek A, Wang L, Fang W, Dabral S, Dubois O, Cupitt J, Pullamsetti SS, Cotroneo E, Jones H, Tomasi G, Nguyen QD, Aboagye EO, El-Bahrawy MA, Barnes G, Howard LS, Gibbs JS, Gsell W, He JG, Wilkins MR, Heterogeneity in lung (18)FDG uptake in pulmonary arterial hypertension: potential of dynamic (18)FDG positron emission tomography with kinetic analysis as a bridging biomarker for pulmonary vascular remodeling targeted treatments, Circulation 128(11) (2013) 1214–24. 10.1161/CIRCULATIONAHA.113.004136. [DOI] [PubMed] [Google Scholar]
- [180].Piao L, Sidhu VK, Fang YH, Ryan JJ, Parikh KS, Hong Z, Toth PT, Morrow E, Kutty S, Lopaschuk GD, Archer SL, FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: therapeutic benefits of dichloroacetate, J Mol Med (Berl) 91(3) (2013) 333–46. 10.1007/s00109-012-0982-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang HJ, Haber I, Rehman J, Lopaschuk GD, Archer SL, The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle, J Mol Med (Berl) 88(1) (2010) 47–60. 10.1007/s00109-009-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L, Cupitt J, Paterson I, Thompson RB, Chow K, O’Regan DP, Zhao L, Wharton J, Kiely DG, Kinnaird A, Boukouris AE, White C, Nagendran J, Freed DH, Wort SJ, Gibbs JSR, Wilkins MR, Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients, Sci Transl Med 9(413) (2017). 10.1126/scitranslmed.aao4583. [DOI] [PubMed] [Google Scholar]
- [183].Xiong PY, Motamed M, Chen KH, Dasgupta A, Martin A, Neuber-Hess M, Tian L, Archer SL, Inhibiting Pyruvate Kinase Muscle Isoform 2 with Shikonin Regresses Supra-Coronary Aortic Banding Induced Group 2 Pulmonary Hypertension, C105. WITH ALL MY HEART: SEX, ESTROGEN, AND RIGHT VENTRICLE IN PULMONARY VASCULAR DISEASE AND BEYOND, American Thoracic Society 2020, pp. A6073–A6073. [Google Scholar]
- [184].Kankotia S, Stacpoole PW, Dichloroacetate and cancer: new home for an orphan drug?, Biochim Biophys Acta 1846(2) (2014) 617–29. 10.1016/j.bbcan.2014.08.005. [DOI] [PubMed] [Google Scholar]
- [185].Tataranni T, Piccoli C, Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications, Oxid Med Cell Longev 2019 (2019) 8201079. 10.1155/2019/8201079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Yadav S, Pandey SK, Goel Y, Temre MK, Singh SM, Diverse Stakeholders of Tumor Metabolism: An Appraisal of the Emerging Approach of Multifaceted Metabolic Targeting by 3-Bromopyruvate, Front Pharmacol 10 (2019) 728. 10.3389/fphar.2019.00728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I, Dichloroacetate induces apoptosis in endometrial cancer cells, Gynecol Oncol 109(3) (2008) 394–402. 10.1016/j.ygyno.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED, A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth, Cancer Cell 11(1) (2007) 37–51. 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- [189].Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ, Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation, Prostate 68(11) (2008) 1223–31. 10.1002/pros.20788. [DOI] [PubMed] [Google Scholar]
- [190].Tataranni T, Agriesti F, Pacelli C, Ruggieri V, Laurenzana I, Mazzoccoli C, Sala GD, Panebianco C, Pazienza V, Capitanio N, Piccoli C, Dichloroacetate Affects Mitochondrial Function and Stemness-Associated Properties in Pancreatic Cancer Cell Lines, Cells 8(5) (2019). 10.3390/cells8050478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Cairns RA, Papandreou I, Sutphin PD, Denko NC, Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy, Proc Natl Acad Sci U S A 104(22) (2007) 9445–50. 10.1073/pnas.0611662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC, Metabolic modulation of glioblastoma with dichloroacetate, Sci Transl Med 2(31) (2010) 31ra34. 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- [193].Strum SB, Adalsteinsson O, Black RR, Segal D, Peress NL, Waldenfels J, Case report: Sodium dichloroacetate (DCA) inhibition of the “Warburg Effect” in a human cancer patient: complete response in non-Hodgkin’s lymphoma after disease progression with rituximab-CHOP, J Bioenerg Biomembr 45(3) (2013) 307–15. 10.1007/s10863-012-9496-2. [DOI] [PubMed] [Google Scholar]
- [194].Garon EB, Christofk HR, Hosmer W, Britten CD, Bahng A, Crabtree MJ, Hong CS, Kamranpour N, Pitts S, Kabbinavar F, Patel C, von Euw E, Black A, Michelakis ED, Dubinett SM, Slamon DJ, Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer, J Cancer Res Clin Oncol 140(3) (2014) 443–52. 10.1007/s00432-014-1583-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW, Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors, Invest New Drugs 32(3) (2014) 452–64. 10.1007/s10637-013-0047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Chu QS, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJ, Venner P, Michelakis ED, A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors, Invest New Drugs 33(3) (2015) 603–10. 10.1007/s10637-015-0221-y. [DOI] [PubMed] [Google Scholar]
- [197].Langaee T, Wagner R, Horne LP, Lawson LA, Becker C, Shahin M, Starostik P, Stacpoole PW, Personalized Dosing of Dichloroacetate Using GSTZ1 Clinical Genotyping Assay, Genet Test Mol Biomarkers 22(4) (2018) 266–269. 10.1089/gtmb.2017.0261. [DOI] [PubMed] [Google Scholar]
- [198].Wu H, Wang Y, Wu C, Yang P, Li H, Li Z, Resveratrol Induces Cancer Cell Apoptosis through MiR-326/PKM2-Mediated ER Stress and Mitochondrial Fission, J Agric Food Chem 64(49) (2016) 9356–9367. 10.1021/acs.jafc.6b04549. [DOI] [PubMed] [Google Scholar]
- [199].Shan S, Shi J, Yang P, Jia B, Wu H, Zhang X, Li Z, Apigenin Restrains Colon Cancer Cell Proliferation via Targeted Blocking of Pyruvate Kinase M2-Dependent Glycolysis, J Agric Food Chem 65(37) (2017) 8136–8144. 10.1021/acs.jafc.7b02757. [DOI] [PubMed] [Google Scholar]
- [200].Liu T, Li S, Wu L, Yu Q, Li J, Feng J, Zhang J, Chen J, Zhou Y, Ji J, Chen K, Mao Y, Wang F, Dai W, Fan X, Wu J, Guo C, Experimental Study of Hepatocellular Carcinoma Treatment by Shikonin Through Regulating PKM2, J Hepatocell Carcinoma 7 (2020) 19–31. 10.2147/JHC.S237614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X, Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2, Oncogene 30(42) (2011) 4297–306. 10.1038/onc.2011.137. [DOI] [PubMed] [Google Scholar]
- [202].Vander Heiden MG, Christofk HR, Schuman E, Subtelny AO, Sharfi H, Harlow EE, Xian J, Cantley LC, Identification of small molecule inhibitors of pyruvate kinase M2, Biochem Pharmacol 79(8) (2010) 1118–24. 10.1016/j.bcp.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Yuan Q, Yu H, Chen J, Song X, Sun L, Antitumor Effect of miR-1294/Pyruvate Kinase M2 Signaling Cascade in Osteosarcoma Cells, Onco Targets Ther 13 (2020) 1637–1647. 10.2147/OTT.S232718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, Yang H, Mattaini KR, Metallo CM, Fiske BP, Courtney KD, Malstrom S, Khan TM, Kung C, Skoumbourdis AP, Veith H, Southall N, Walsh MJ, Brimacombe KR, Leister W, Lunt SY, Johnson ZR, Yen KE, Kunii K, Davidson SM, Christofk HR, Austin CP, Inglese J, Harris MH, Asara JM, Stephanopoulos G, Salituro FG, Jin S, Dang L, Auld DS, Park HW, Cantley LC, Thomas CJ, Vander Heiden MG, Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis, Nat Chem Biol 8(10) (2012) 839–47. 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Xu Y, Liu XH, Saunders M, Pearce S, Foulks JM, Parnell KM, Clifford A, Nix RN, Bullough J, Hendrickson TF, Wright K, McCullar MV, Kanner SB, Ho KK, Discovery of 3-(trifluoromethyl)-1H-pyrazole-5-carboxamide activators of the M2 isoform of pyruvate kinase (PKM2), Bioorg Med Chem Lett 24(2) (2014) 515–9. 10.1016/j.bmcl.2013.12.028. [DOI] [PubMed] [Google Scholar]
- [206].Archer SL, Mitochondrial dynamics--mitochondrial fission and fusion in human diseases, N Engl J Med 369(23) (2013) 2236–51. 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- [207].Mao K, Klionsky DJ, Participation of mitochondrial fission during mitophagy, Cell Cycle 12(19) (2013) 3131–2. 10.4161/cc.26352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Jornayvaz FR, Shulman GI, Regulation of mitochondrial biogenesis, Essays Biochem 47 (2010) 69–84. 10.1042/bse0470069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Hong Z, Chen KH, DasGupta A, Potus F, Dunham-Snary K, Bonnet S, Tian L, Fu J, Breuils-Bonnet S, Provencher S, Wu D, Mewburn J, Ormiston ML, Archer SL, MicroRNA-138 and MicroRNA-25 Down-regulate Mitochondrial Calcium Uniporter, Causing the Pulmonary Arterial Hypertension Cancer Phenotype, Am J Respir Crit Care Med 195(4) (2017) 515–529. 10.1164/rccm.201604-0814OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Wang C, Youle RJ, The role of mitochondria in apoptosis*, Annu Rev Genet 43 (2009) 95–118. 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL, Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer, FASEB J 26(5) (2012) 2175–86. 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Chen KH, Dasgupta A, Lin J, Potus F, Bonnet S, Iremonger J, Fu J, Mewburn J, Wu D, Dunham-Snary K, Theilmann AL, Jing ZC, Hindmarch C, Ormiston ML, Lawrie A, Archer SL, Epigenetic Dysregulation of the Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension: Mechanistic and Therapeutic Implications, Circulation 138(3) (2018) 287–304. 10.1161/CIRCULATIONAHA.117.031258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Dasgupta A, Chen KH, Wu D, Hoskin V, Mewburn J, Lima PDA, Parlow LRG, Hindmarch CCT, Martin A, Sykes EA, Tayade C, Lightbody ED, Madarnas Y, SenGupta SK, Elliott BE, Nicol CJB, Archer SL, An epigenetic increase in mitochondrial fission by MiD49 and MiD51 regulates the cell cycle in cancer: Diagnostic and therapeutic implications, FASEB J 34(4) (2020) 5106–5127. 10.1096/fj.201903117R. [DOI] [PubMed] [Google Scholar]
- [214].Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL, Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension, Circ Res 110(11) (2012) 1484–97. 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Senft D, Ronai ZA, Regulators of mitochondrial dynamics in cancer, Curr Opin Cell Biol 39 (2016) 43–52. 10.1016/j.ceb.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Ryan JJ, Marsboom G, Fang YH, Toth PT, Morrow E, Luo N, Piao L, Hong Z, Ericson K, Zhang HJ, Han M, Haney CR, Chen CT, Sharp WW, Archer SL, PGC1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension, Am J Respir Crit Care Med 187(8) (2013) 865–78. 10.1164/rccm.201209-1687OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Atkins K, Dasgupta A, Chen KH, Mewburn J, Archer SL, The role of Drp1 adaptor proteins MiD49 and MiD51 in mitochondrial fission: implications for human disease, Clin Sci (Lond) 130(21) (2016) 1861–74. 10.1042/CS20160030. [DOI] [PubMed] [Google Scholar]
- [218].Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT, Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology, J Cell Sci 117(Pt 7) (2004) 1201–10. 10.1242/jcs.01058. [DOI] [PubMed] [Google Scholar]
- [219].Gandre-Babbe S, van der Bliek AM, The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells, Mol Biol Cell 19(6) (2008) 2402–12. 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K, Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells, J Cell Biol 191(6) (2010) 1141–58. 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT, MiD49 and MiD51, new components of the mitochondrial fission machinery, EMBO Rep 12(6) (2011) 565–73. 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK, ER tubules mark sites of mitochondrial division, Science 334(6054) (2011) 358–62. 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Smirnova E, Griparic L, Shurland DL, van der Bliek AM, Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells, Mol Biol Cell 12(8) (2001) 2245–56. 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Qi X, Disatnik MH, Shen N, Sobel RA, Mochly-Rosen D, Aberrant mitochondrial fission in neurons induced by protein kinase C{delta} under oxidative stress conditions in vivo, Mol Biol Cell 22(2) (2011) 256–65. 10.1091/mbc.E10-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K, Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission, J Biol Chem 282(15) (2007) 11521–9. 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- [226].Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M, CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology, J Cell Biol 182(3) (2008) 573–85. 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Cribbs JT, Strack S, Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death, EMBO Rep 8(10) (2007) 939–44. 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Banes-Berceli AK, Ketsawatsomkron P, Ogbi S, Patel B, Pollock DM, Marrero MB, Angiotensin II and endothelin-1 augment the vascular complications of diabetes via JAK2 activation, Am J Physiol Heart Circ Physiol 293(2) (2007) H1291–9. 10.1152/ajpheart.00181.2007. [DOI] [PubMed] [Google Scholar]
- [229].Yoon Y, Pitts KR, McNiven MA, Mammalian dynamin-like protein DLP1 tubulates membranes, Mol Biol Cell 12(9) (2001) 2894–905. 10.1091/mbc.12.9.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Kumari S, Mg S, Mayor S, Endocytosis unplugged: multiple ways to enter the cell, Cell research 20(3) (2010) 256–75. 10.1038/cr.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Jones SM, Howell KE, Henley JR, Cao H, McNiven MA, Role of dynamin in the formation of transport vesicles from the trans-Golgi network, Science 279(5350) (1998) 573–7. 10.1126/science.279.5350.573. [DOI] [PubMed] [Google Scholar]
- [232].Lee JE, Westrate LM, Wu H, Page C, Voeltz GK, Multiple dynamin family members collaborate to drive mitochondrial division, Nature 540(7631) (2016) 139–143. 10.1038/nature20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Fonseca TB, Sanchez-Guerrero A, Milosevic I, Raimundo N, Mitochondrial fission requires DRP1 but not dynamins, Nature 570(7761) (2019) E34–E42. 10.1038/s41586-019-1296-y. [DOI] [PubMed] [Google Scholar]
- [234].Tian L, Potus F, Wu D, Dasgupta A, Chen KH, Mewburn J, Lima P, Archer SL, Increased Drp1-Mediated Mitochondrial Fission Promotes Proliferation and Collagen Production by Right Ventricular Fibroblasts in Experimental Pulmonary Arterial Hypertension, Front Physiol 9 (2018) 828. 10.3389/fphys.2018.00828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Tian L, Neuber-Hess M, Mewburn J, Dasgupta A, Dunham-Snary K, Wu D, Chen KH, Hong Z, Sharp WW, Kutty S, Archer SL, Ischemia-induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension, J Mol Med (Berl) 95(4) (2017) 381–393. 10.1007/s00109-017-1522-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Abu-Hanna J, Taanman J-W, Abraham D, Clapp L, Impact of treprostinil on dynamin-related protein 1 (DRP1) and mitochondrial fragmentation in pulmonary arterial hypertension (PAH), European Respiratory Journal 52(suppl 62) (2018) PA3059. 10.1183/13993003.congress-2018.PA3059. [DOI] [Google Scholar]
- [237].Assad TR, Hemnes AR, Larkin EK, Glazer AM, Xu M, Wells QS, Farber-Eger EH, Sheng Q, Shyr Y, Harrell FE, Newman JH, Brittain EL, Clinical and Biological Insights Into Combined Post- and Pre-Capillary Pulmonary Hypertension, J Am Coll Cardiol 68(23) (2016) 2525–2536. 10.1016/j.jacc.2016.09.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Xiong PY, Tian L, Dunham-Snary KJ, Chen KH, Mewburn JD, Neuber-Hess M, Martin A, Dasgupta A, Potus F, Archer SL, Biventricular Increases in Mitochondrial Fission Mediator (MiD51) and Proglycolytic Pyruvate Kinase (PKM2) Isoform in Experimental Group 2 Pulmonary Hypertension-Novel Mitochondrial Abnormalities, Front Cardiovasc Med 5 (2018) 195. 10.3389/fcvm.2018.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y, Mitochondrial dynamics regulates migration and invasion of breast cancer cells, Oncogene 32(40) (2013) 4814–24. 10.1038/onc.2012.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, Ferguson AN, Kashatus DF, Bao S, Rich JN, Mitochondrial control by DRP1 in brain tumor initiating cells, Nat Neurosci 18(4) (2015) 501–10. 10.1038/nn.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF, Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth, Mol Cell 57(3) (2015) 537–51. 10.1016/j.molcel.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, Jabado O, Hoehn K, Kageyama Y, Sesaki H, Chipuk JE, Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors, Mol Cell 57(3) (2015) 521–36. 10.1016/j.molcel.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Lima AR, Santos L, Correia M, Soares P, Sobrinho-Simoes M, Melo M, Maximo V, Dynamin-Related Protein 1 at the Crossroads of Cancer, Genes (Basel) 9(2) (2018). 10.3390/genes9020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Liang J, Yang Y, Bai L, Li F, Li E, DRP1 upregulation promotes pancreatic cancer growth and metastasis through increased aerobic glycolysis, J Gastroenterol Hepatol 35(5) (2020) 885–895. 10.1111/jgh.14912. [DOI] [PubMed] [Google Scholar]
- [245].Ferreira-da-Silva A, Valacca C, Rios E, Populo H, Soares P, Sobrinho-Simoes M, Scorrano L, Maximo V, Campello S, Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration, PloS one 10(3) (2015) e0122308. 10.1371/journal.pone.0122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Bai L, Liang J, Li L, Li E, Downregulation of MiD49 contributes to tumor growth and metastasis of human pancreatic cancer, Oncol Rep 43(4) (2020) 1208–1220. 10.3892/or.2020.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J, Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization, Dev Cell 14(2) (2008) 193–204. 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Han XJ, Yang ZJ, Jiang LP, Wei YF, Liao MF, Qian Y, Li Y, Huang X, Wang JB, Xin HB, Wan YY, Mitochondrial dynamics regulates hypoxia-induced migration and antineoplastic activity of cisplatin in breast cancer cells, Int J Oncol 46(2) (2015) 691–700. 10.3892/ijo.2014.2781. [DOI] [PubMed] [Google Scholar]
- [249].Lucantoni F, Dussmann H, Prehn JHM, Metabolic Targeting of Breast Cancer Cells With the 2-Deoxy-D-Glucose and the Mitochondrial Bioenergetics Inhibitor MDIVI-1, Front Cell Dev Biol 6 (2018) 113. 10.3389/fcell.2018.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Dal Yontem F, Kim SH, Ding Z, Grimm E, Ekmekcioglu S, Akcakaya H, Mitochondrial dynamic alterations regulate melanoma cell progression, J Cell Biochem (2018). 10.1002/jcb.27518. [DOI] [PubMed] [Google Scholar]
- [251].Qi X, Qvit N, Su YC, Mochly-Rosen D, A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity, J Cell Sci 126(Pt 3) (2013) 789–802. 10.1242/jcs.114439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Manczak M, Reddy PH, Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage, Hum Mol Genet 21(11) (2012) 2538–47. 10.1093/hmg/dds072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Manczak M, Calkins MJ, Reddy PH, Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage, Hum Mol Genet 20(13) (2011) 2495–509. 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Zhang Z, Liu L, Jiang X, Zhai S, Xing D, The Essential Role of Drp1 and Its Regulation by S-Nitrosylation of Parkin in Dopaminergic Neurodegeneration: Implications for Parkinson’s Disease, Antioxid Redox Signal 25(11) (2016) 609–622. 10.1089/ars.2016.6634. [DOI] [PubMed] [Google Scholar]
- [255].Zhang Q, Hu C, Huang J, Liu W, Lai W, Leng F, Tang Q, Liu Y, Wang Q, Zhou M, Sheng F, Li G, Zhang R, ROCK1 induces dopaminergic nerve cell apoptosis via the activation of Drp1-mediated aberrant mitochondrial fission in Parkinson’s disease, Exp Mol Med 51(10) (2019) 1–13. 10.1038/s12276-019-0318-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Reddy PH, Increased mitochondrial fission and neuronal dysfunction in Huntington’s disease: implications for molecular inhibitors of excessive mitochondrial fission, Drug Discov Today 19(7) (2014) 951–5. 10.1016/j.drudis.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Oliver D, Reddy PH, Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases, Cells 8(9) (2019). 10.3390/cells8090961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Reddy PH, Manczak M, Yin X, Mitochondria-Division Inhibitor 1 Protects Against Amyloid-beta induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease, J Alzheimers Dis 58(1) (2017) 147–162. 10.3233/JAD-170051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Filichia E, Hoffer B, Qi X, Luo Y, Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP, Sci Rep 6 (2016) 32656. 10.1038/srep32656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X, Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration, J Clin Invest 123(12) (2013) 5371–88. 10.1172/JCI70911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, Francis TC, Kennedy NW, Picton LK, Kumar T, Uppuluri S, Miller AM, Itoh K, Karbowski M, Sesaki H, Hill RB, Polster BM, The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species, Dev Cell 40(6) (2017) 583–594 e6. 10.1016/j.devcel.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Wu D, Dasgupta A, Chen KH, Neuber-Hess M, Patel J, Hurst TE, Mewburn JD, Lima PDA, Alizadeh E, Martin A, Wells M, Snieckus V, Archer SL, Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: Therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury, FASEB J 34(1) (2020) 1447–1464. 10.1096/fj.201901467R. [DOI] [PubMed] [Google Scholar]
- [263].Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC, Mitochondrial regulation of cell cycle and proliferation, Antioxid Redox Signal 16(10) (2012) 1150–80. 10.1089/ars.2011.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Mishra P, Carelli V, Manfredi G, Chan DC, Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation, Cell Metab 19(4) (2014) 630–41. 10.1016/j.cmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L, OPA1 requires mitofusin 1 to promote mitochondrial fusion, Proc Natl Acad Sci U S A 101(45) (2004) 15927–32. 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, Li P, Qiu X, Wen S, Xiao RP, Tang J, Dysregulation of HSG triggers vascular proliferative disorders, Nat Cell Biol 6(9) (2004) 872–83. 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- [267].Guo X, Chen KH, Guo Y, Liao H, Tang J, Xiao RP, Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway, Circ Res 101(11) (2007) 1113–22. 10.1161/CIRCRESAHA.107.157644. [DOI] [PubMed] [Google Scholar]
- [268].Cohen MM, Leboucher GP, Livnat-Levanon N, Glickman MH, Weissman AM, Ubiquitin-proteasome-dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion, Mol Biol Cell 19(6) (2008) 2457–64. 10.1091/mbc.E08-02-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Leboucher GP, Tsai YC, Yang M, Shaw KC, Zhou M, Veenstra TD, Glickman MH, Weissman AM, Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis, Mol Cell 47(4) (2012) 547–57. 10.1016/j.molcel.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Chen Y, Dorn GW, 2nd, PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria, Science 340(6131) (2013) 471–5. 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Dasgupta A, Chen KH, Munk RB, Sasaki CY, Curtis J, Longo DL, Ghosh P, Mechanism of Activation-Induced Downregulation of Mitofusin 2 in Human Peripheral Blood T Cells, Journal of immunology (Baltimore, Md. : 1950) 195(12) (2015) 5780–6. 10.4049/jimmunol.1501023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Jin B, Fu G, Pan H, Cheng X, Zhou L, Lv J, Chen G, Zheng S, Anti-tumour efficacy of mitofusin-2 in urinary bladder carcinoma, Med Oncol 28 Suppl 1 (2011) S373–80. 10.1007/s12032-010-9662-5. [DOI] [PubMed] [Google Scholar]
- [273].Xu K, Chen G, Li X, Wu X, Chang Z, Xu J, Zhu Y, Yin P, Liang X, Dong L, MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling, Sci Rep 7 (2017) 41718. 10.1038/srep41718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Wu Y, Zhou D, Xu X, Zhao X, Huang P, Zhou X, Song W, Guo H, Wang W, Zheng S, Clinical significance of mitofusin-2 and its signaling pathways in hepatocellular carcinoma, World J Surg Oncol 14(1) (2016) 179. 10.1186/s12957-016-0922-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Cheng X, Zhou D, Wei J, Lin J, Cell-cycle arrest at G2/M and proliferation inhibition by adenovirus-expressed mitofusin-2 gene in human colorectal cancer cell lines, Neoplasma 60(6) (2013) 620–6. 10.4149/neo_2013_080. [DOI] [PubMed] [Google Scholar]
- [276].Zhang GE, Jin HL, Lin XK, Chen C, Liu XS, Zhang Q, Yu JR, Anti-tumor effects of Mfn2 in gastric cancer, Int J Mol Sci 14(7) (2013) 13005–21. 10.3390/ijms140713005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Xue R, Meng Q, Lu D, Liu X, Wang Y, Hao J, Mitofusin2 Induces Cell Autophagy of Pancreatic Cancer through Inhibiting the PI3K/Akt/mTOR Signaling Pathway, Oxid Med Cell Longev 2018 (2018) 2798070. 10.1155/2018/2798070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Wang W, Liu X, Guo X, Quan H, Mitofusin-2 Triggers Cervical Carcinoma Cell Hela Apoptosis via Mitochondrial Pathway in Mouse Model, Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 46(1) (2018) 69–81. 10.1159/000488410. [DOI] [PubMed] [Google Scholar]
- [279].Yu M, Nguyen ND, Huang Y, Lin D, Fujimoto TN, Molkentine JM, Deorukhkar A, Kang Y, San Lucas FA, Fernandes CJ, Koay EJ, Gupta S, Ying H, Koong AC, Herman JM, Fleming JB, Maitra A, Taniguchi CM, Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer, JCI Insight 5 (2019). 10.1172/jci.insight.126915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Lou Y, Li R, Liu J, Zhang Y, Zhang X, Jin B, Liu Y, Wang Z, Zhong H, Wen S, Han B, Mitofusin-2 over-expresses and leads to dysregulation of cell cycle and cell invasion in lung adenocarcinoma, Med Oncol 32(4) (2015) 132. 10.1007/s12032-015-0515-0. [DOI] [PubMed] [Google Scholar]
- [281].Cheng CT, Kuo CY, Ouyang C, Li CF, Chung Y, Chan DC, Kung HJ, Ann DK, Metabolic Stress-Induced Phosphorylation of KAP1 Ser473 Blocks Mitochondrial Fusion in Breast Cancer Cells, Cancer Res 76(17) (2016) 5006–18. 10.1158/0008-5472.CAN-15-2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Shi C, Cai Y, Li Y, Li Y, Hu N, Ma S, Hu S, Zhu P, Wang W, Zhou H, Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways, Redox Biol 14 (2018) 59–71. 10.1016/j.redox.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Jiang L, Huang Q, Zhang S, Zhang Q, Chang J, Qiu X, Wang E, Hsa-miR-125a-3p and hsa-miR-125a-5p are downregulated in non-small cell lung cancer and have inverse effects on invasion and migration of lung cancer cells, BMC Cancer 10 (2010) 318. 10.1186/1471-2407-10-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Nam EJ, Yoon H, Kim SW, Kim H, Kim YT, Kim JH, Kim JW, Kim S, MicroRNA expression profiles in serous ovarian carcinoma, Clin Cancer Res 14(9) (2008) 2690–5. 10.1158/1078-0432.CCR-07-1731. [DOI] [PubMed] [Google Scholar]
- [285].Ferretti E, De Smaele E, Po A, Di Marcotullio L, Tosi E, Espinola MS, Di Rocco C, Riccardi R, Giangaspero F, Farcomeni A, Nofroni I, Laneve P, Gioia U, Caffarelli E, Bozzoni I, Screpanti I, Gulino A, MicroRNA profiling in human medulloblastoma, Int J Cancer 124(3) (2009) 568–77. 10.1002/ijc.23948. [DOI] [PubMed] [Google Scholar]
- [286].Hashiguchi Y, Nishida N, Mimori K, Sudo T, Tanaka F, Shibata K, Ishii H, Mochizuki H, Hase K, Doki Y, Mori M, Down-regulation of miR-125a-3p in human gastric cancer and its clinicopathological significance, Int J Oncol 40(5) (2012) 1477–82. 10.3892/ijo.2012.1363. [DOI] [PubMed] [Google Scholar]
- [287].Pan L, Zhou L, Yin W, Bai J, Liu R, miR-125a induces apoptosis, metabolism disorder and migrationimpairment in pancreatic cancer cells by targeting Mfn2-related mitochondrial fission, Int J Oncol 53(1) (2018) 124–136. 10.3892/ijo.2018.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Ashrafi G, Schwarz TL, The pathways of mitophagy for quality control and clearance of mitochondria, Cell Death Differ 20(1) (2013) 31–42. 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Langer T, Kaser M, Klanner C, Leonhard K, AAA proteases of mitochondria: quality control of membrane proteins and regulatory functions during mitochondrial biogenesis, Biochem Soc Trans 29(Pt 4) (2001) 431–6. 10.1042/bst0290431. [DOI] [PubMed] [Google Scholar]
- [290].Karbowski M, Youle RJ, Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation, Curr Opin Cell Biol 23(4) (2011) 476–82. 10.1016/j.ceb.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Gkikas I, Palikaras K, Tavernarakis N, The Role of Mitophagy in Innate Immunity, Front Immunol 9 (2018) 1283. 10.3389/fimmu.2018.01283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].Aggarwal S, Mannam P, Zhang J, Differential regulation of autophagy and mitophagy in pulmonary diseases, Am J Physiol Lung Cell Mol Physiol 311(2) (2016) L433–52. 10.1152/ajplung.00128.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Haslip M, Dostanic I, Huang Y, Zhang Y, Russell KS, Jurczak MJ, Mannam P, Giordano F, Erzurum SC, Lee PJ, Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia, Arterioscler Thromb Vasc Biol 35(5) (2015) 1166–78. 10.1161/ATVBAHA.114.304865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Chang JY, Yi HS, Kim HW, Shong M, Dysregulation of mitophagy in carcinogenesis and tumor progression, Biochim Biophys Acta Bioenerg 1858(8) (2017) 633–640. 10.1016/j.bbabio.2016.12.008. [DOI] [PubMed] [Google Scholar]
- [295].Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H, Trapasso F, Drusco A, Shimizu M, Masciullo V, D’Andrilli G, Scambia G, Picchio MC, Alder H, Godwin AK, Croce CM, Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27, Proc Natl Acad Sci U S A 100(10) (2003) 5956–61. 10.1073/pnas.0931262100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Fujiwara M, Marusawa H, Wang HQ, Iwai A, Ikeuchi K, Imai Y, Kataoka A, Nukina N, Takahashi R, Chiba T, Parkin as a tumor suppressor gene for hepatocellular carcinoma, Oncogene 27(46) (2008) 6002–11. 10.1038/onc.2008.199. [DOI] [PubMed] [Google Scholar]
- [297].Li C, Zhang Y, Cheng X, Yuan H, Zhu S, Liu J, Wen Q, Xie Y, Liu J, Kroemer G, Klionsky DJ, Lotze MT, Zeh HJ, Kang R, Tang D, PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism, Dev Cell 46(4) (2018) 441–455 e8. 10.1016/j.devcel.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Agnihotri S, Golbourn B, Huang X, Remke M, Younger S, Cairns RA, Chalil A, Smith CA, Krumholtz SL, Mackenzie D, Rakopoulos P, Ramaswamy V, Taccone MS, Mischel PS, Fuller GN, Hawkins C, Stanford WL, Taylor MD, Zadeh G, Rutka JT, PINK1 Is a Negative Regulator of Growth and the Warburg Effect in Glioblastoma, Cancer Res 76(16) (2016) 4708–19. 10.1158/0008-5472.CAN-15-3079. [DOI] [PubMed] [Google Scholar]
- [299].Liu J, Zhang C, Zhao Y, Yue X, Wu H, Huang S, Chen J, Tomsky K, Xie H, Khella CA, Gatza ML, Xia D, Gao J, White E, Haffty BG, Hu W, Feng Z, Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression, Nat Commun 8(1) (2017) 1823. 10.1038/s41467-017-01947-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Lee SB, Kim JJ, Nam HJ, Gao B, Yin P, Qin B, Yi SY, Ham H, Evans D, Kim SH, Zhang J, Deng M, Liu T, Zhang H, Billadeau DD, Wang L, Giaime E, Shen J, Pang YP, Jen J, van Deursen JM, Lou Z, Parkin Regulates Mitosis and Genomic Stability through Cdc20/Cdh1, Mol Cell 60(1) (2015) 21–34. 10.1016/j.molcel.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Webster BR, Scott I, Traba J, Han K, Sack MN, Regulation of autophagy and mitophagy by nutrient availability and acetylation, Biochim Biophys Acta 1841(4) (2014) 525–34. 10.1016/j.bbalip.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Chourasia AH, Boland ML, Macleod KF, Mitophagy and cancer, Cancer & Metabolism 3(1) (2015) 4. 10.1186/s40170-015-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS, Macleod KF, Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis, EMBO Rep 16(9) (2015) 1145–63. 10.15252/embr.201540759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Koop EA, van Laar T, van Wichen DF, de Weger RA, Wall E, van Diest PJ, Expression of BNIP3 in invasive breast cancer: correlations with the hypoxic response and clinicopathological features, BMC Cancer 9 (2009) 175. 10.1186/1471-2407-9-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Okami J, Simeone DM, Logsdon CD, Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer, Cancer Res 64(15) (2004) 5338–46. 10.1158/0008-5472.CAN-04-0089. [DOI] [PubMed] [Google Scholar]
- [306].Murai M, Toyota M, Satoh A, Suzuki H, Akino K, Mita H, Sasaki Y, Ishida T, Shen L, Garcia-Manero G, Issa JP, Hinoda Y, Tokino T, Imai K, Aberrant DNA methylation associated with silencing BNIP3 gene expression in haematopoietic tumours, British journal of cancer 92(6) (2005) 1165–72. 10.1038/sj.bjc.6602422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Murai M, Toyota M, Suzuki H, Satoh A, Sasaki Y, Akino K, Ueno M, Takahashi F, Kusano M, Mita H, Yanagihara K, Endo T, Hinoda Y, Tokino T, Imai K, Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer, Clin Cancer Res 11(3) (2005) 1021–7. [PubMed] [Google Scholar]
- [308].Calvisi DF, Ladu S, Gorden A, Farina M, Lee JS, Conner EA, Schroeder I, Factor VM, Thorgeirsson SS, Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma, J Clin Invest 117(9) (2007) 2713–22. 10.1172/JCI31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Erkan M, Kleeff J, Esposito I, Giese T, Ketterer K, Buchler MW, Giese NA, Friess H, Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis, Oncogene 24(27) (2005) 4421–32. 10.1038/sj.onc.1208642. [DOI] [PubMed] [Google Scholar]
- [310].Manka D, Spicer Z, Millhorn DE, Bcl-2/adenovirus E1B 19 kDa interacting protein-3 knockdown enables growth of breast cancer metastases in the lung, liver, and bone, Cancer Res 65(24) (2005) 11689–93. 10.1158/0008-5472.CAN-05-3091. [DOI] [PubMed] [Google Scholar]
- [311].Humpton TJ, Alagesan B, DeNicola GM, Lu D, Yordanov GN, Leonhardt CS, Yao MA, Alagesan P, Zaatari MN, Park Y, Skepper JN, Macleod KF, Perez-Mancera PA, Murphy MP, Evan GI, Vousden KH, Tuveson DA, Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer, Cancer Discov 9(9) (2019) 1268–1287. 10.1158/2159-8290.CD-18-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Puigserver P, Spiegelman BM, Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator, Endocr Rev 24(1) (2003) 78–90. 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- [313].Ryoo IG, Kwak MK, Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria, Toxicol Appl Pharmacol 359 (2018) 24–33. 10.1016/j.taap.2018.09.014. [DOI] [PubMed] [Google Scholar]
- [314].Virbasius JV, Scarpulla RC, Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis, Proc Natl Acad Sci U S A 91(4) (1994) 1309–13. 10.1073/pnas.91.4.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [315].Vega RB, Horton JL, Kelly DP, Maintaining ancient organelles: mitochondrial biogenesis and maturation, Circ Res 116(11) (2015) 1820–34. 10.1161/CIRCRESAHA.116.305420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Gomez-Arroyo J, Mizuno S, Szczepanek K, Van Tassell B, Natarajan R, dos Remedios CG, Drake JI, Farkas L, Kraskauskas D, Wijesinghe DS, Chalfant CE, Bigbee J, Abbate A, Lesnefsky EJ, Bogaard HJ, Voelkel NF, Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension, Circ Heart Fail 6(1) (2013) 136–44. 10.1161/CIRCHEARTFAILURE.111.966127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Ye JX, Wang SS, Ge M, Wang DJ, Suppression of endothelial PGC-1alpha is associated with hypoxia-induced endothelial dysfunction and provides a new therapeutic target in pulmonary arterial hypertension, Am J Physiol Lung Cell Mol Physiol 310(11) (2016) L1233–42. 10.1152/ajplung.00356.2015. [DOI] [PubMed] [Google Scholar]
- [318].Afolayan AJ, Eis A, Alexander M, Michalkiewicz T, Teng RJ, Lakshminrusimha S, Konduri GG, Decreased endothelial nitric oxide synthase expression and function contribute to impaired mitochondrial biogenesis and oxidative stress in fetal lambs with persistent pulmonary hypertension, Am J Physiol Lung Cell Mol Physiol 310(1) (2016) L40–9. 10.1152/ajplung.00392.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [319].Diebold I, Hennigs JK, Miyagawa K, Li CG, Nickel NP, Kaschwich M, Cao A, Wang L, Reddy S, Chen PI, Nakahira K, Alcazar MA, Hopper RK, Ji L, Feldman BJ, Rabinovitch M, BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension, Cell Metab 21(4) (2015) 596–608. 10.1016/j.cmet.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Enache I, Charles AL, Bouitbir J, Favret F, Zoll J, Metzger D, Oswald-Mammosser M, Geny B, Charloux A, Skeletal muscle mitochondrial dysfunction precedes right ventricular impairment in experimental pulmonary hypertension, Mol Cell Biochem 373(1–2) (2013) 161–70. 10.1007/s11010-012-1485-6. [DOI] [PubMed] [Google Scholar]
- [321].Martinez-Outschoorn UE, Pavlides S, Sotgia F, Lisanti MP, Mitochondrial biogenesis drives tumor cell proliferation, Am J Pathol 178(5) (2011) 1949–52. 10.1016/j.ajpath.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Berridge MV, Tan AS, Effects of mitochondrial gene deletion on tumorigenicity of metastatic melanoma: reassessing the Warburg effect, Rejuvenation Res 13(2–3) (2010) 139–41. 10.1089/rej.2009.0948. [DOI] [PubMed] [Google Scholar]
- [323].Yu M, Shi Y, Wei X, Yang Y, Zhou Y, Hao X, Zhang N, Niu R, Depletion of mitochondrial DNA by ethidium bromide treatment inhibits the proliferation and tumorigenesis of T47D human breast cancer cells, Toxicol Lett 170(1) (2007) 83–93. 10.1016/j.toxlet.2007.02.013. [DOI] [PubMed] [Google Scholar]
- [324].Toki N, Kagami S, Kurita T, Kawagoe T, Matsuura Y, Hachisuga T, Matsuyama A, Hashimoto H, Izumi H, Kohno K, Expression of mitochondrial transcription factor A in endometrial carcinomas: clinicopathologic correlations and prognostic significance, Virchows Arch 456(4) (2010) 387–93. 10.1007/s00428-010-0895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [325].Singh KP, Kumari R, Treas J, DuMond JW, Chronic exposure to arsenic causes increased cell survival, DNA damage, and increased expression of mitochondrial transcription factor A (mtTFA) in human prostate epithelial cells, Chem Res Toxicol 24(3) (2011) 340–9. 10.1021/tx1003112. [DOI] [PubMed] [Google Scholar]
- [326].Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS, Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity, Proc Natl Acad Sci U S A 107(19) (2010) 8788–93. 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].McGuirk S, Gravel SP, Deblois G, Papadopoli DJ, Faubert B, Wegner A, Hiller K, Avizonis D, Akavia UD, Jones RG, Giguere V, St-Pierre J, PGC-1alpha supports glutamine metabolism in breast cancer, Cancer Metab 1(1) (2013) 22. 10.1186/2049-3002-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [328].LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R, PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis, Nat Cell Biol 16(10) (2014) 992–1003, 1–15. 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [329].Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P, PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress, Cancer Cell 23(3) (2013) 287–301. 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Luo C, Lim JH, Lee Y, Granter SR, Thomas A, Vazquez F, Widlund HR, Puigserver P, A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis, Nature 537(7620) (2016) 422–426. 10.1038/nature19347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [331].Shiota M, Yokomizo A, Tada Y, Inokuchi J, Tatsugami K, Kuroiwa K, Uchiumi T, Fujimoto N, Seki N, Naito S, Peroxisome proliferator-activated receptor gamma coactivator-1alpha interacts with the androgen receptor (AR) and promotes prostate cancer cell growth by activating the AR, Mol Endocrinol 24(1) (2010) 114–27. 10.1210/me.2009-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [332].Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, Zhang A, Xia X, Ilkayeva OR, Xin L, Ittmann MM, Rick FG, Schally AV, Frigo DE, Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch, Oncogene 33(45) (2014) 5251–61. 10.1038/onc.2013.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [333].Torrano V, Valcarcel-Jimenez L, Cortazar AR, Liu X, Urosevic J, Castillo-Martin M, Fernandez-Ruiz S, Morciano G, Caro-Maldonado A, Guiu M, Zuniga-Garcia P, Graupera M, Bellmunt A, Pandya P, Lorente M, Martin-Martin N, Sutherland JD, Sanchez-Mosquera P, Bozal-Basterra L, Zabala-Letona A, Arruabarrena-Aristorena A, Berenguer A, Embade N, Ugalde-Olano A, Lacasa-Viscasillas I, Loizaga-Iriarte A, Unda-Urzaiz M, Schultz N, Aransay AM, Sanz-Moreno V, Barrio R, Velasco G, Pinton P, Cordon-Cardo C, Locasale JW, Gomis RR, Carracedo A, The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis, Nat Cell Biol 18(6) (2016) 645–656. 10.1038/ncb3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [334].Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Grana O, Viera CR, Yuneva M, Sainz B Jr., Heeschen C, MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells, Cell Metab 22(4) (2015) 590–605. 10.1016/j.cmet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- [335].Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX, New mechanisms of pulmonary arterial hypertension: role of Ca(2)(+) signaling, Am J Physiol Heart Circ Physiol 302(8) (2012) H1546–62. 10.1152/ajpheart.00944.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [336].Wang YX, Zheng YM, ROS-dependent signaling mechanisms for hypoxic Ca(2+) responses in pulmonary artery myocytes, Antioxid Redox Signal 12(5) (2010) 611–23. 10.1089/ars.2009.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Shimoda LA, Wang J, Sylvester JT, Ca2+ channels and chronic hypoxia, Microcirculation 13(8) (2006) 657–70. 10.1080/10739680600930305. [DOI] [PubMed] [Google Scholar]
- [338].Zamponi GW, Striessnig J, Koschak A, Dolphin AC, The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential, Pharmacol Rev 67(4) (2015) 821–70. 10.1124/pr.114.009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [339].Nilius B, Owsianik G, The transient receptor potential family of ion channels, Genome Biol 12(3) (2011) 218. 10.1186/gb-2011-12-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [340].Prakriya M, Lewis RS, Store-Operated Calcium Channels, Physiol Rev 95(4) (2015) 1383–436. 10.1152/physrev.00020.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [341].Curry MC, Peters AA, Kenny PA, Roberts-Thomson SJ, Monteith GR, Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells, Biochem Biophys Res Commun 434(3) (2013) 695–700. 10.1016/j.bbrc.2013.04.015. [DOI] [PubMed] [Google Scholar]
- [342].Foskett JK, Philipson B, The mitochondrial Ca(2+) uniporter complex, J Mol Cell Cardiol 78 (2015) 3–8. 10.1016/j.yjmcc.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [343].Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK, Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter, Nature 476(7360) (2011) 341–5. 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [344].Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corra F, Giorgi C, De Marchi E, Poletti F, Gafa R, Lanza G, Negrini M, Rizzuto R, Pinton P, Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25, Curr Biol 23(1) (2013) 58–63. 10.1016/j.cub.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [345].Denton RM, Randle PJ, Martin BR, Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase, Biochem J 128(1) (1972) 161–3. 10.1042/bj1280161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [346].Vultur A, Gibhardt CS, Stanisz H, Bogeski I, The role of the mitochondrial calcium uniporter (MCU) complex in cancer, Pflugers Arch (2018). 10.1007/s00424-018-2162-8. [DOI] [PubMed] [Google Scholar]
- [347].Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, Duchen MR, Rosato A, Bogeski I, Szabadkai G, Rizzuto R, Mammucari C, The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha, EMBO Mol Med 8(5) (2016) 569–85. 10.15252/emmm.201606255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [348].Hall DD, Wu Y, Domann FE, Spitz DR, Anderson ME, Mitochondrial calcium uniporter activity is dispensable for MDA-MB-231 breast carcinoma cell survival, PloS one 9(5) (2014) e96866. 10.1371/journal.pone.0096866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [349].Davis FM, Parsonage MT, Cabot PJ, Parat MO, Thompson EW, Roberts-Thomson SJ, Monteith GR, Assessment of gene expression of intracellular calcium channels, pumps and exchangers with epidermal growth factor-induced epithelial-mesenchymal transition in a breast cancer cell line, Cancer Cell Int 13(1) (2013) 76. 10.1186/1475-2867-13-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [350].Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, Cai G, Meng X, Zou F, Mitochondrial Ca(2)(+) uniporter is critical for store-operated Ca(2)(+) entry-dependent breast cancer cell migration, Biochem Biophys Res Commun 458(1) (2015) 186–93. 10.1016/j.bbrc.2015.01.092. [DOI] [PubMed] [Google Scholar]
- [351].Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF, Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport, Nat Cell Biol 9(4) (2007) 445–452. 10.1038/ncb1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [352].Teshima Y, Akao M, Jones SP, Marban E, Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes, Circ Res 93(3) (2003) 192–200. 10.1161/01.RES.0000085581.60197.4D. [DOI] [PubMed] [Google Scholar]
- [353].Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, Michelakis ED, Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension, Circ Res 113(2) (2013) 126–36. 10.1161/CIRCRESAHA.112.300699. [DOI] [PubMed] [Google Scholar]
- [354].Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC, Michelakis ED, The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension, Sci Transl Med 3(88) (2011) 88ra55. 10.1126/scitranslmed.3002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [355].Elmore S, Apoptosis: a review of programmed cell death, Toxicol Pathol 35(4) (2007) 495–516. 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [356].Suzuki YJ, Ibrahim YF, Shults NV, Apoptosis-based therapy to treat pulmonary arterial hypertension, J Rare Dis Res Treat 1(2) (2016) 17–24. [PMC free article] [PubMed] [Google Scholar]
- [357].Onyeagucha B, Subbarayalu P, Abdelfattah N, Rajamanickam S, Timilsina S, Guzman R, Zeballos C, Eedunuri V, Bansal S, Mohammad T, Chen Y, Vadlamudi RK, Rao MK, Novel post-transcriptional and post-translational regulation of pro-apoptotic protein BOK and anti-apoptotic protein Mcl-1 determine the fate of breast cancer cells to survive or die, Oncotarget 8(49) (2017) 85984–85996. 10.18632/oncotarget.20841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [358].Green DR, Walczak H, Apoptosis therapy: driving cancers down the road to ruin, Nature medicine 19(2) (2013) 131–3. 10.1038/nm.3076. [DOI] [PubMed] [Google Scholar]
- [359].Gautschi O, Tschopp S, Olie RA, Leech SH, Simoes-Wust AP, Ziegler A, Baumann B, Odermatt B, Hall J, Stahel RA, Zangemeister-Wittke U, Activity of a novel bcl-2/bcl-xL-bispecific antisense oligonucleotide against tumors of diverse histologic origins, J Natl Cancer Inst 93(6) (2001) 463–71. 10.1093/jnci/93.6.463. [DOI] [PubMed] [Google Scholar]
- [360].Fulda S, Shifting the balance of mitochondrial apoptosis: therapeutic perspectives, Front Oncol 2 (2012) 121. 10.3389/fonc.2012.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [361].Bender A, Opel D, Naumann I, Kappler R, Friedman L, von Schweinitz D, Debatin KM, Fulda S, PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2 proteins and enhanced mitochondrial apoptosis, Oncogene 30(4) (2011) 494–503. 10.1038/onc.2010.429. [DOI] [PubMed] [Google Scholar]
- [362].Morice MC, Serruys PW, Sousa JE, Fajadet J, Ban Hayashi E, Perin M, Colombo A, Schuler G, Barragan P, Guagliumi G, Molnar F, Falotico R, R.S.G.R.S.w.t.S.-C.B.V.B.-E.S.i.t.T.o.P.w.d.N.N.C.A. Lesions, A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization, N Engl J Med 346(23) (2002) 1773–80. 10.1056/NEJMoa012843. [DOI] [PubMed] [Google Scholar]
- [363].Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM, Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB, Mol Cell 22(2) (2006) 159–68. 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- [364].Barilli A, Visigalli R, Sala R, Gazzola GC, Parolari A, Tremoli E, Bonomini S, Simon A, Closs EI, Dall’Asta V, Bussolati O, In human endothelial cells rapamycin causes mTORC2 inhibition and impairs cell viability and function, Cardiovasc Res 78(3) (2008) 563–71. 10.1093/cvr/cvn024. [DOI] [PubMed] [Google Scholar]
- [365].Houssaini A, Abid S, Mouraret N, Wan F, Rideau D, Saker M, Marcos E, Tissot CM, Dubois-Rande JL, Amsellem V, Adnot S, Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension, Am J Respir Cell Mol Biol 48(5) (2013) 568–77. 10.1165/rcmb.2012-0429OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [366].Pena A, Kobir A, Goncharov D, Goda A, Kudryashova TV, Ray A, Vanderpool R, Baust J, Chang B, Mora AL, Gorcsan J 3rd, Goncharova EA, Pharmacological Inhibition of mTOR Kinase Reverses Right Ventricle Remodeling and Improves Right Ventricle Structure and Function in Rats, Am J Respir Cell Mol Biol 57(5) (2017) 615–625. 10.1165/rcmb.2016-0364OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [367].Paddenberg R, Stieger P, von Lilien AL, Faulhammer P, Goldenberg A, Tillmanns HH, Kummer W, Braun-Dullaeus RC, Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice, Respir Res 8 (2007) 15. 10.1186/1465-9921-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [368].McMurtry MS, Bonnet S, Michelakis ED, Bonnet S, Haromy A, Archer SL, Statin therapy, alone or with rapamycin, does not reverse monocrotaline pulmonary arterial hypertension: the rapamcyin-atorvastatin-simvastatin study, Am J Physiol Lung Cell Mol Physiol 293(4) (2007) L933–40. 10.1152/ajplung.00310.2006. [DOI] [PubMed] [Google Scholar]
- [369].Seyfarth HJ, Hammerschmidt S, Halank M, Neuhaus P, Wirtz HR, Everolimus in patients with severe pulmonary hypertension: a safety and efficacy pilot trial, Pulm Circ 3(3) (2013) 632–8. 10.1086/674311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [370].Stamelos VA, Redman CW, Richardson A, Understanding sensitivity to BH3 mimetics: ABT-737 as a case study to foresee the complexities of personalized medicine, J Mol Signal 7(1) (2012) 12. 10.1186/1750-2187-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [371].Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A, BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents, Cancer Cell 12(2) (2007) 171–85. 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- [372].Juarez-Salcedo LM, Desai V, Dalia S, Venetoclax: evidence to date and clinical potential, Drugs Context 8 (2019) 212574. 10.7573/dic.212574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [373].Ibrahim YF, Wong CM, Pavlickova L, Liu L, Trasar L, Bansal G, Suzuki YJ, Mechanism of the susceptibility of remodeled pulmonary vessels to drug-induced cell killing, J Am Heart Assoc 3(1) (2014) e000520. 10.1161/JAHA.113.000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [374].Wang X, Ibrahim YF, Das D, Zungu-Edmondson M, Shults NV, Suzuki YJ, Carfilzomib reverses pulmonary arterial hypertension, Cardiovasc Res 110(2) (2016) 188–99. 10.1093/cvr/cvw047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [375].Ibrahim YF, Shults NV, Rybka V, Suzuki YJ, Docetaxel Reverses Pulmonary Vascular Remodeling by Decreasing Autophagy and Resolves Right Ventricular Fibrosis, J Pharmacol Exp Ther 363(1) (2017) 20–34. 10.1124/jpet.117.239921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [376].Rybka V, Suzuki YJ, Shults NV, Effects of Bcl-2/Bcl-xL Inhibitors on Pulmonary Artery Smooth Muscle Cells, Antioxidants (Basel) 7(11) (2018). 10.3390/antiox7110150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [377].Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL, Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels, Circulation 105(2) (2002) 244–50. 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]
- [378].Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC, Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol, J Cell Biol 160(7) (2003) 1115–27. 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [379].McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, Bonnet S, Puttagunta L, Michelakis ED, Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension, J Clin Invest 115(6) (2005) 1479–91. 10.1172/JCI23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [380].Chen C, Chen C, Wang Z, Wang L, Yang L, Ding M, Ding C, Sun Y, Lin Q, Huang X, Du X, Zhao X, Wang C, Puerarin induces mitochondria-dependent apoptosis in hypoxic human pulmonary arterial smooth muscle cells, PloS one 7(3) (2012) e34181. 10.1371/journal.pone.0034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [381].Taniguchi K, Sakai M, Sugito N, Kumazaki M, Shinohara H, Yamada N, Nakayama T, Ueda H, Nakagawa Y, Ito Y, Futamura M, Uno B, Otsuki Y, Yoshida K, Uchiyama K, Akao Y, PTBP1-associated microRNA-1 and −133b suppress the Warburg effect in colorectal tumors, Oncotarget 7(14) (2016) 18940–52. 10.18632/oncotarget.8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [382].Sun Y, Zhao X, Zhou Y, Hu Y, miR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect, Oncol Rep 28(4) (2012) 1346–52. 10.3892/or.2012.1958. [DOI] [PubMed] [Google Scholar]
- [383].Taniguchi K, Sugito N, Kumazaki M, Shinohara H, Yamada N, Nakagawa Y, Ito Y, Otsuki Y, Uno B, Uchiyama K, Akao Y, MicroRNA-124 inhibits cancer cell growth through PTB1/PKM1/PKM2 feedback cascade in colorectal cancer, Cancer Lett 363(1) (2015) 17–27. 10.1016/j.canlet.2015.03.026. [DOI] [PubMed] [Google Scholar]
- [384].Wong TS, Liu XB, Chung-Wai Ho A, Po-Wing Yuen A, Wai-Man Ng R, Ignace Wei W, Identification of pyruvate kinase type M2 as potential oncoprotein in squamous cell carcinoma of tongue through microRNA profiling, Int J Cancer 123(2) (2008) 251–257. 10.1002/ijc.23583. [DOI] [PubMed] [Google Scholar]
- [385].Liu AM, Xu Z, Shek FH, Wong KF, Lee NP, Poon RT, Chen J, Luk JM, miR-122 targets pyruvate kinase M2 and affects metabolism of hepatocellular carcinoma, PloS one 9(1) (2014) e86872. 10.1371/journal.pone.0086872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [386].Yu G, Sun W, Shen Y, Hu Y, Liu H, Li W, Wang Y, PKM2 functions as a potential oncogene and is a crucial target of miR-148a and miR-326 in thyroid tumorigenesis, Am J Transl Res 10(6) (2018) 1793–1801. [PMC free article] [PubMed] [Google Scholar]
- [387].Xu Q, Liu LZ, Yin Y, He J, Li Q, Qian X, You Y, Lu Z, Peiper SC, Shu Y, Jiang BH, Regulatory circuit of PKM2/NF-κB/miR-148a/152-modulated tumor angiogenesis and cancer progression, Oncogene 34(43) (2015) 5482–93. 10.1038/onc.2015.6. [DOI] [PubMed] [Google Scholar]
- [388].Wen YY, Liu WT, Sun HR, Ge X, Shi ZM, Wang M, Li W, Zhang JY, Liu LZ, Jiang BH, IGF-1-mediated PKM2/beta-catenin/miR-152 regulatory circuit in breast cancer, Sci Rep 7(1) (2017) 15897. 10.1038/s41598-017-15607-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [389].Cheng X, Chen J, Huang Z, miR-372 promotes breast cancer cell proliferation by directly targeting LATS2, Exp Ther Med 15(3) (2018) 2812–2817. 10.3892/etm.2018.5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [390].Chen T, Li Y, Cao W, Liu Y, miR-491–5p inhibits osteosarcoma cell proliferation by targeting PKM2, Oncol Lett 16(5) (2018) 6472–6478. 10.3892/ol.2018.9451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [391].Ling Z, Liu D, Zhang G, Liang Q, Xiang P, Xu Y, Han C, Tao T, miR-361–5p modulates metabolism and autophagy via the Sp1-mediated regulation of PKM2 in prostate cancer, Oncol Rep 38(3) (2017) 1621–1628. 10.3892/or.2017.5852. [DOI] [PubMed] [Google Scholar]
- [392].Guo M, Zhao X, Yuan X, Jiang J, Li P, MiR-let-7a inhibits cell proliferation, migration, and invasion by down-regulating PKM2 in cervical cancer, Oncotarget 8(17) (2017) 28226–28236. 10.18632/oncotarget.15999. [DOI] [PMC free article] [PubMed] [Google Scholar]