

Abstract
Iron is an essential micronutrient metal for cellular functions but can generate highly reactive oxygen species resulting in oxidative damage. For these reasons its uptake and metabolism is highly regulated. A small but dynamic fraction of ferrous iron inside the cell, termed intracellular labile iron, is redox-reactive and ready to participate multiples reactions of intracellular enzymes. Due to its nature its determination and precise quantification has been a roadblock. However, recent progress in the development of intracellular labile iron probes are allowing the reevaluation of our current understanding and unmasking new functions. The role of intracellular labile iron in regulating the epigenome was recently discovered. This chapter examine how intracellular labile iron can modulate histone and DNA demethylation and how its pool can mediate a signaling pathway from cAMP serving as a sensor of the metabolic needs of the cells.
Keywords: Iron, intracellular labile Fe(II), reactive oxygen species, DNA methylation, histone methylation, TET methylcytosine dioxygenases, JmjC domain-containing demethylases, G-protein coupled receptor, cAMP, RapGEF2
Graphical Abstract
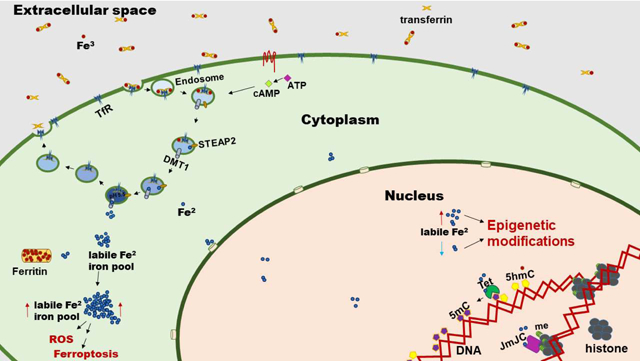
1. Introduction
Iron is an essential metal needed by cells to properly function. The reduced form of iron, Fe(II), is a cofactor in numerous enzymatic reactions in the cell and is therefore required for many vital physiological functions. However, Fe(II) is highly reactive and must therefore be tightly controlled in order to reduce the risk of generating reactive oxygen species (ROS) and eliciting profound cellular damage. Therefore, several cellular pathways have evolved to tightly regulate iron uptake, sequestering, and metabolism to safely utilize iron for vital physiological processes. Furthermore, cells have evolved mechanisms to sense and utilize labile Fe(II) via epigenetic pathways that allow them to adapt and fine tune their responses to the dynamic nature of their environment. Recent insights and technological developments have shed light on the dynamic nature of labile Fe(II) and its novel role in epigenetic regulation of both DNA and histone methylation. In this chapter, we will briefly describe how cells generate a pool of intracellular Fe(II) and how this pool changes due to metabolic needs and environmental pressures. We will focus on the novel role of the labile Fe(II) pool in modulating transcription via epigenomic events, such as DNA and histone methylation, and how these events may serve as therapeutic targets for various diseases including cancer.
2. Overview of iron uptake and metabolism
Iron is one of the most abundant metals in cells. It is an important cofactor and a vital nutrient required for proper cellular function. It is tightly regulated, the demand can be localized inside the cell, and its excess can generate free radicals that cause cellular damage. For these reasons, a delicate balance of iron uptake, storage, utilization, and export must be struck for the maintenance of cellular functions.
Dietary iron is first absorbed in the duodenum by enterocytes1. Most of the unbound iron in the intestinal lumen is oxidized and present as ferric iron, Fe(III). Prior to absorption, Fe(III) is reduced to ferrous iron, Fe(II), by the duodenal cytochrome B (Dcytb) enzyme presented in the brush border of the cells1. Ferrous iron is able to enter the duodenal cells through the divalent metal transporter 1 (DMT1)1–3. Iron can also enter the duodenal cells when bound to hemeproteins1 and is transported inside cells by the heme carrier protein 1 (HCP1)4. Iron within the enterocyte can go into the bloodstream by two sequential steps: 1) ferrous iron crosses the basolateral membrane via the iron exporter ferroportin; 2) ferrous iron is oxidized to ferric iron by hephaestin ferroxidase5. Once in circulation, iron is largely bound to transferrin5,6. Diferric transferrin can bind to the membrane-bound transferrin receptors (TFR1 and TFR2) on target cells and is subsequently internalized via clathrin-mediated endocytosis1. It is likely that most iron enters the cells using this mechanism, since transferrin is only 30% saturated under physiological conditions1,7. The acidification of the internalized endosomes will result in the release of the ferric iron from transferrin7. Subsequently, the endosomal ferrireductase STEAP3 converts ferric into ferrous iron7. DMT1 can then transport the ferrous iron into the cytosol7. Once in the cytoplasm, ferrous iron can be transported to other organelles or stored within ferritin via Fe(II) oxidation by the intrinsic ferroxidase activity of ferritin. The transferrin receptor and transferrin without iron (apo-transferrin) are sorted to recycle endosomes back to the cell surface where apo-transferrin dissociates6.
Iron bound to transferrin is not the only mechanism that cells use to internalize iron. Mutant mice lacking transferrin receptors die in utero from severe anemia and have defects in the development of erythrocytes, muscle cells and neurons6,8,9. However, other types of cells are still able to internalize iron without major dysfunction in the knockout animals, suggesting that there must be other mechanisms for iron internalization. An additional mechanism for iron internalization is through metal transporters. The ferrous iron not bound to transferrin can enter cells using transporters like DMT1, natural resistance-associated macrophage protein 1 (NRAMP1), or ZRT/IRT-like proteins (ZIPs)7,10. Heme-bound iron can provide a source of iron for certain cell populations. Macrophages can obtain iron from heme after phagocytosis10. Moreover, other cell types could obtain heme-iron through various effectors such as using the heme importer Heme-Responsive Gene 1 (HGR1), the heme-carrier protein 1 (HCP1), the feline leukemia virus group C cellular receptor 1 (FLVCR1) , the heme-hemopexin complex via the LDL receptor related protein (LRP1), or the haptoglobin hemoglobin complex via the CD163 receptor6,10,11. Lipocalin 2 bound to catechol-ferric complexes can also enter the cell via the solute carrier protein SLC22A1712. In addition, ferritin released to the serum and bound to iron can enter cells via TFR1 in humans, via the T cell immunoglobulin and mucin domain containing 2 receptor or the scavenger receptor class A member 5 in mice13,14–16.
Cytoplasmic iron can be transported to different organelles like mitochondria, stored with ferritin, exported out of cells by ferroportin 1 (FPN1), or can be part of the labile iron pool and used in different metabolic processes. Within the cytoplasm, iron is transported by the iron chaperone poly C binding protein (PCBP) family. The PCBP family of proteins delivers iron to specific locations like ferritin for storage and can also deliver iron to enzymes which require iron as a cofactor for enzymatic activity, such as ribonucleotide reductase, deoxyhypusine hydroxylase, and iron-dependent and 2-oxoglutarate (2OG, also known as α-ketoglutarate)-dependent dioxygenase family proteins, like prolyl hydroxylase 2 (PHD2) and the asparaginyl hydroxylase factor inhibiting HIF-1α (FIH)17,18. Ultimately, a large portion of internalized iron is localized to the mitochondria where it is incorporated into heme and iron-sulfur proteins that are part of the electron transfer chain17,19.
3. Detection and characterization of labile iron
Labile iron is hard to define because of its dynamic nature and difficulty to detect. The terminology was initially used to describe the iron release from choleglobin and hemoglobin after an acid treatment20,21. Later it was discovered with experimental transfusion of radioactive iron that a portion of it remained in the blood as an intermediate stage before it is incorporated into hemoglobin22. This iron was first termed the labile iron pool and later the extracellular labile iron pool.
A pool of free intracellular iron was also suspected after initial cellular experiments with iron chelators23 and was later named by Allan Jacobs as the intracellular labile iron pool24. While chelatable iron suggested that there was a pool of free intracellular iron, a direct quantification method for free intracellular iron was missing. The chelator itself can affect the metabolism of iron by inducing intracellular shuffling of iron and by sequestering iron bound with low affinity to other proteins or lipids25,26. Initial experiments to determine the intracellular iron pool were done with chelators having a high affinity to iron (transferrin and deferoxamine), or with a low affinity and specificity (EDTA, phenanthroline, and DTPA). Most of these chelators have higher affinity to Fe(III) or are bound to Fe(III) because the Fe(II) is oxidized once they are formed in a complex27,28. For example, radiolabeled iron was supplied to cells and after cellular fractionation, iron not bound to ferritin, or bound to other molecules, were identified25,29,30. Another method was to chelate iron and determine the relative amount of free iron using electron paramagnetic resonance31,32. Additional studies with chelators also determined the functional role of “intracellular labile iron”28. These methods shed light on intracellular iron dynamics and definitively indicate the existence of several intracellular iron pools. However, these methods lack specificity and sensitivity, since chelators change the iron balance and composition within the cell and fractionation techniques damage cellular compartments that result in leakage of its milieu. Moreover, probable redistribution of iron binding molecules, changes in the relative amount of Fe(II) and Fe(III), and changes in endocytosis dynamics that influence the rate of iron uptake from the extracellular space affect these interpretations.
For these reasons, fluorescence cellular probes were recently developed with the goal of specifically measuring iron without affecting cellular function or viability. The first set of probes contained phenanthroline moiety, or calcein fluorescence molecules, with EDTA-like moiety28,33–39. These probes are very convenient because they can be used to measure the abundance of iron in living cells and determine its localization. However, most of them are unable to distinguish clearly between Fe(II) or Fe(III) and other metals like Cu or Mg. Additionally, they exhibit unequal cellular distribution, are based on “turn off” mechanisms, and are unable to function in very low pH environments, such as lysosomes, due to their pH-dependent sensitivity40. Nevertheless these fluorescence probes successfully steered research into new directions and next-generation probes were subsequently developed that can target specific cellular compartments like lysosomes and mitochondria or utilize a “turn on” mechanism41–45.
Only recently were fluorescence-based probes developed that are specific to either Fe(II) or Fe(III)42,46–50. This means that we have to be very cautious in interpreting the extensive previous literature that claims to detect “label intracellular iron” based on techniques that result in a disruption of cells or probes that are not specific to iron or its ionic states. For example, calcein-AM, one of the most widely used probes for iron, was initially believed to be a good sensor for Fe(II) but was later found to be a sensor for Fe(III) and not especially specific to iron40,51. Interpretations about the increased level of labile iron due to ROS in cancer cells, based on calcein or PhenGreen experiments, should therefore be carefully reevaluated or replicated using next-generation probes.
One of the biggest challenges was to develop probes that could sense labile Fe(II) without chelation and sequestration of iron. To overcome this obstacle, activity-based probes were developed to be able to emit fluorescence by reacting with Fe(II). These types of probes are very sensitive to nM concentrations of intracellular labile Fe(II)46,52–58. For example, an Fe(II)-dependent probe was designed to be able to modulate the fluorescence resonance energy transfer (FRET) between two dyes linked by an Fe(II)-responsive trigger . These types of probes allowed a more accurate quantification, localization, and detection of labile ferrous iron in living cells59. This activity-based approach was used in synthetic biology to design artificial molecules to control gene activation, signal transduction, and cytoskeletal remodeling in response to Fe(II) and as a specific delivery method of medication to cells with high concentration of Fe(II)60,61. Additionally, efforts to develop intracellular labile iron probes that could be used in living organism are under way62–65.
The intracellular labile iron pool is very dynamic in its size and availability, which affects downstream functions of cells that are iron-dependent. A complete picture of its role inside the cell is still lacking due to difficulties in detection and quantification. However, it is clear that it performs essential functions in modulating the kinetics of iron-dependent enzymes, such as Fe(II) and 2OG-dependent dioxygenases, and iron-sulfur center proteins66.
4. Labile iron as a determinant of epigenomic control.
Labile iron as a modulator of DNA and histone demethylation
The epigenome comprises all the modifications to the chromatin that regulates the expression of the genome without altering the DNA sequence. This control of gene expression needs to be fine-tuned to the environment so that cells can appropriately, and sometimes relatively rapidly under certain situations, respond to environmental challenges. Intracellular iron plays a central role as one of the many mechanisms that inform cells of environmental changes. The function of many epigenetic effectors which regulate the epigenome requires, or is influenced by, intracellular iron levels. Changes in the environment or in the metabolism of the cell could therefore result in fluctuations of the iron pool, and consequently in the epigenome. Therefore, labile iron can be a determining factor that controls the epigenome.
Ten-eleven translocation (TET) enzymes and JmjC domain-containing demethylases are two important classes of epigenetic regulators which require Fe(II). TET enzymes (TET1/2/3) catalyze the cascade oxidation of 5-methylcytosine (5mC, the classical mark of silenced transcription) into 5-hydroxymethylcytosine (5hmC) and furthermore into transient intermediates which are excised by DNA base excision repair and are ultimately replaced by unmodified cytosine, thus completing the active DNA demethylation. Besides being a DNA demethylation intermediate, 5hmC is also a stable epigenetic mark which itself can influence transcription. Conversely, JmjC domain-containing demethylases (~20 members) are the chief enzymes which demethylate histone lysine residues and subsequently regulate chromatin accessibility and transcription. Both TETs and JmjC domain-containing demethylases belong to the Fe(II) and 2-oxoglutarate-dependent dioxygenase superfamily, a large class of enzymes which require Fe(II) for proper enzymatic activity. This suggests that changes in the intracellular labile Fe(II) pool can influence the activity of these enzymes and subsequently mediate downstream epigenetic changes.
Experiments with iron chelators were the first indication that iron is involved in the epigenetic landscape. For example, the transcription of a gene termed serpina3g, which encodes serine protease inhibitor A3G to be involved in the differentiation of memory T cells, was found to be downregulated by exposure to iron chelator deferoxamine (DFO) and by nickel exposure67,68. Chromatin immunoprecipitation (ChIP) assays following the treatment demonstrated a change in the methylation pattern of the serpina3g promoter which affects Serpina3g transcription. DFO chelates iron while nickel displaces iron from the active site of enzymes, like the Fe(II) and 2OG-dependent dioxygenases, thus resulting in reduced activity of these enzymes68,69. The interpretation of these experiments were hard to make at the time since TETs and JmjC domain-containing demethylases were discovered later70,71. A similar effect of iron chelators in changing histone methylation patterns was observed later in human breast cancer cell lines treated with DFO72. Moreover, in vivo depletion of ferrous iron with ferrous chelator thiosemicarbazone-24 reduces 5hmC expression and blastocyst formation during embryo development73. This effect in 5hmC expression and blastocyst formation can be explained by a lower TET enzyme activity, as the opposite is seen with increased TET activity with vitamin C74–76. A reduction of intracellular labile iron could be responsible for the alteration of DNA and histone hippocampal methylation in neonatal iron deficiency77–79. Vitamin C is considered a cofactor for Fe(II) and 2OG-dependent dioxygenases, such as collagen hydroxylases, TETs and JmjC domain-containing demethylases, by converting catalytically inactive Fe(III) to catalytically active Fe(III)80. Thus, the bioavailability of vitamin C, via Fe(II), regulates the epigenome. Moreover, quinones, a large class of cyclic organic compounds, were also found to increase the labile iron pool and consequently increase global levels of genomic 5hmC81. Quinone-induced 5hmC elevation was found to be mediated by TET enzymes and can be blocked by iron chelator treatment81. These findings gave the first indications that cellular determinants which regulate the labile iron pool result in epigenetic consequences.
The correlation of 5hmC and intracellular labile Fe(II) levels suggests that both events might be directly connected. Labile iron could act as a propagating signal that regulates the Fe(II)-dependent oxidase activity of TETs, subsequently affecting the epigenetic landscape. This previously unrecognized function for iron is persuasive since an essential characteristic of cellular iron metabolism is its entry into the cytosolic Fe(II) labile pool from cellular iron uptake or from ferritin that is able to store large quantities of Fe(III) and release it as Fe(II). Even though this process is clearly involved in maintaining iron homeostasis and the biosynthesis of Fe(II) cofactors, it appears to have been appropriated for cellular signaling with enzymes that employ labile Fe(II), like TETs and JmjC domain-containing histone demethylases.
A mouse model of hemochromatosis that resulted in an increase of labile iron in the brain was found to result in a reduced global brain DNA methylation82. This reduction was correlated with a reduction of DNA methyltransferase activity by iron in vitro82. It won’t be a surprise to find out that in this model TET activity could be increased, which would contribute to the global DNA demethylation83–85.
cAMP signaling and labile iron
Growing evidence suggests that the labile iron pool may be part of a global signaling pathway for epigenomic regulation of DNA and histone methylation. Recent work found that changes in intracellular cAMP, the second messenger for numerous signaling pathways, increases global 5hmC levels in primary Schwann cells and other cell types86. This increase correlates with cAMP-induced elevation of the intracellular labile Fe(II) pool and was abolished by treatment with iron chelators. cAMP was found to propagate this effect through RapGEF2 which causes enhanced acidification of endosomes that resulted in increased Fe(II) release to the labile Fe(II) pool86. As a result of the changes in global 5hmC levels, the transcriptional profile of these cells subsequently changed. The effect of cAMP could also be mimicked by treatment with adenylate cyclase activators, phosphodiesterase inhibitors, and by GPCR ligands which increased intracellular cAMP86, suggesting that changes in cAMP signaling alter the labile Fe(II) pool which subsequently regulates DNA demethylation. These changes in cAMP and iron could be modulated by a non-canonical GPCR pathway connecting the dynamic changes in the cellular environment with an epigenetic response.
Like TET enzymes, JmjC domain-containing histone demethylases which modulate histone methylation, also require Fe(II) for proper enzymatic activity. Therefore, cellular determinants which alter labile Fe(II) are also expected to influence histone methylation dynamics. Recent work found that cAMP signaling and subsequent Fe(II) elevation resulted in increased demethylation of H3K4me3, a histone mark of actively transcribed genes87. Interestingly, while prolonged stimulation maintained decreased H3K4me3, brief stimulation resulted in only a transient increase of Fe(II) and decreased H3K4me3, both of which returned to baseline levels shortly after removal of the stimulus87. This suggests that the labile Fe(II) pool is sensitive to fast changes in cAMP signaling and thus allows for cells to dynamically respond to environmental stimuli through rapid alterations of histone methylation. cAMP-induced Fe(II) elevation was found to be mediated by RapGEF2 which causes an increase in endosome acidification by vacuolar H+-ATPase assembly, thus resulting in endosomal iron release and increased cytosolic iron86,87. Removal of iron from the media or treatment with iron chelators abolished cAMP-induced demethylation of H3K4me387. Moreover, inhibition of KDM5 demethylase, which antagonizes H3K4me3, also abolished cAMP-induced demethylation87. Overall, these data highlight a growing body of evidence showing that labile Fe(II) is part of a broad signaling pathway which regulates DNA and histone methylation in response to environmental stimuli, thus allowing cells to rapidly alter transcription and dynamically respond to extracellular challenges.
5. Labile iron and disease pathogenesis.
It is clear that the intracellular ferrous iron pool isn’t static, but is instead dynamically needed for the activity of downstream enzymes and reactions, including those that could affect the epigenetic program of cells. While deficiency diminishes these vital functions, its excess could result in the generation of damaging ROS or cell death via ferroptosis. Therefore, it should be no surprise that the amount of labile iron has been implicated in several cellular pathological states. It also implies that therapeutic approaches could be improved or conceived by modulating the levels of labile ferrous iron. For example, the levels of labile iron has been implicated in numerous diseases beyond the scope of this chapter, including aging88–92 , the immune response93,94, Parkinsons disease95–97, age related macular degeneration98–100, retinitis pigmentosa101, kidney disease102, neurodegeneration103–105, malaria106,107, viral infections108,109, and inflammation110. How labile iron flux results in epigenetic changes to drive pathogenesis of these diseases or can be a therapeutic target has yet to be thoroughly explored.
The best studied role of labile iron in a pathology is cancer. For example, downregulation of ferritin by H-Ras results in a rise of the labile iron pool that leads to cell growth stimulation111,112. Increase of the labile iron pool is probably needed for the high metabolic requirements of cancer cells. This correlation of decreased ferritin levels and increased labile iron was also observed in cancer cells like adenocarcinoma and breast cancer113,114
In adenocarcinoma the increased labile iron pool also increases ROS and contributes to the epithelial mesenchymal transition (EMT) observed in this cancer between the early stage and the later most invasive stage113. The EMT and changes in cell growth and proliferation implicate a change in the transcription program which may be driven by epigenetic signatures of these cells. It will be important to elucidate the role of increased iron in cancer pathogenesis and the contribution of iron-induced epigenetic changes in disease progression. Also, it will be important to interrogate these questions using the newly-developed iron probes, since some measurements of labile iron were previously done using probes like calcein that detect Fe(III) rather than Fe(II). If the ferrous iron pool is involved in the epigenetic signature of cancer cells, then any approach to change the pool could result in changes to the epigenome and may be a therapeutic target for cancer treatment. Interestingly, iron chelation with deferoxamine in colorectal cancer results in significant inhibition of cell growth and an increase in global histone methylation115. Aberrant histone methylation has been observed in numerous cancers and has long been a therapeutic target in pre-clinical studies. The labile iron pool may therefore be targeted to modulate aberrant histone methylation, restore transcriptional programs, and treat cancer progression. Another potential therapeutic approach is to use the increase labile iron to trigger ferroptosis which has been used in glioma, renal cell carcinoma, and in neuroblastoma116,117. Future work should continue exploring the therapeutic value of labile iron modulation in cancer treatment and in the numerous diseases where disrupted iron metabolism has been observed.
6. Concluding Remarks
Intracellular labile Fe(II) is like a “fire” that cells require and must maintain to sustain a plethora of intracellular enzymatic reactions. But this fire in cells can result in the production of damaging reactive oxygen species (ROS). Intracellular labile Fe(II) can burn the cells via a Fenton reaction that results in the generation of hydroxyl radicals which damage the cell. Yet enzymes such as the Fe(II) and 2OG-dependent dioxygenase superfamily require Fe(II) as do many critical cellular functions. These contrasting pressures require cells to sustain a delicate balance: Cells must tightly regulate the uptake and storage of iron to maintain a pool of intracellular labile iron large enough to perform vital cellular functions while minimizing the subsequent damage resulting from ROS. This pool of iron must shrink or swell depending on the cell’s metabolic need and its dynamic environmental pressures. For these reasons, it is probable that cells developed these mechanisms for sensing intracellular iron fluctuations and utilizing labile Fe(II) to rapidly respond to environmental stimuli. Epigenetic enzymes which utilize Fe(II) can rapidly alter transcription in response to cellular stress or environmental challenges. Within this group of enzymes, JmjC domain-containing histone demethylases and TET methylcytosine dioxygenases, which depend on Fe(II), are essential in modulating the balance of methylation-demethylation of histones and DNA, respectively. The methylation changes in the DNA or histones can result in profound alterations in gene expression and cellular identity. Additionally, these enzymes are highly sensitive to changes in the intracellular labile Fe(II) pool which can be modulated by extracellular signaling from GPCRs. Therefore, by regulating the intracellular labile Fe(II) pool, GPCRs via cAMP can change the methylation status of both DNA and histones and subsequently alter transcription in response to environmental stimuli. Signaling oscillations and rapid changes in the extracellular environment results in dynamic changes to the intracellular labile Fe(II) pool, a vital “fire” that keeps the epigenetic pot set at the right temperature for proper cellular function. Dysregulation in the intracellular labile Fe(II) pool is present in several diseases and might be an important contributing factor that alter the epigenetic landscape of those diseases.
Highlights.
Labile Fe(II) is redox-reactive, dynamic and required for enzymatic reactions.
Labile Fe(II), as an essential cofactor, is involved in the demethylation of DNA and histones.
Diseases that alter the intracellular labile Fe(II) pool can affect the epigenome.
cAMP signaling promotes DNA and histone demethylation by increasing intracellular labile Fe(II) pool.
Acknowledgements
We thank Ms. Erna Stoddart for her administrative assistance. We apologize to our colleagues whose work we were not able to cite in this review due to the space limitation. The work on the regulation of DNA and histone demethylation by cAMP signaling and vitamin C in the Wang lab was supported by the Sylvester Comprehensive Cancer Center at the University of Miami, and NIH grant R01NS089525.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson GJ & Frazer DM Current understanding of iron homeostasis. The American Journal of Clinical Nutrition 106, 1559S–1566S (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gunshin H, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388, 482–488 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Fleming MD, et al. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nature genetics 16, 383–386 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Rouault TA The intestinal heme transporter revealed. Cell 122, 649–651 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Fleming RE & Bacon BR Orchestration of Iron Homeostasis. New England Journal of Medicine 352, 1741–1744 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Muckenthaler MU, Rivella S, Hentze MW & Galy B A Red Carpet for Iron Metabolism. Cell 168, 344–361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lane DJR, et al. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1853, 1130–1144 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Levy JE, Jin O, Fujiwara Y, Kuo F & Andrews N Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nature Genetics 21, 396–399 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Barrientos T, et al. Metabolic Catastrophe in Mice Lacking Transferrin Receptor in Muscle. EBioMedicine 2, 1705–1717 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sukhbaatar N & Weichhart T Iron Regulation: Macrophages in Control. Pharmaceuticals 11, 137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajagopal A, et al. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 453, 1127–1131 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bao G, et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nature chemical biology 6, 602–609 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li L, et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proceedings of the National Academy of Sciences 107, 3505–3510 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen TT, et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. The Journal of Experimental Medicine 202, 955–965 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li JY, et al. Scara5 Is a Ferritin Receptor Mediating Non-Transferrin Iron Delivery. Developmental Cell 16, 35–46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leimberg MJ, Prus E, Konijn AM & Fibach E Macrophages function as a ferritin iron source for cultured human erythroid precursors. Journal of cellular biochemistry 103, 1211–1218 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Philpott CC & Ryu M-S Special delivery: distributing iron in the cytosol of mammalian cells. Frontiers in pharmacology 5(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nandal A, et al. Activation of the HIF Prolyl Hydroxylase by the Iron Chaperones PCBP1 and PCBP2. Cell Metabolism 14, 647–657 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shvartsman M & Ioav Cabantchik Z Intracellular iron trafficking: role of cytosolic ligands. Biometals 25, 711–723 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Legge JW & Lemberg R Coupled oxidation of ascorbic acid and haemoglobin: The ;labile iron’ in blood and its increase in choleglobin formation. The Biochemical journal 35, 353–362 (1941). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O B.G.a.S. Watersoluble c-Hemin from Blood. Hoppe-Seyler‚Äôs Zeitschrift fur Physiologische Chemie 246, 181–193 (1937). [Google Scholar]
- 22.Greenberg GR & Wintrobe MM A labile iron pool. The Journal of biological chemistry 165, 397 (1946). [PubMed] [Google Scholar]
- 23.White GP, Bailey-Wood R & Jacobs A The effect of chelating agents on cellular iron metabolism. Clinical science and molecular medicine 50, 145–152 (1976). [DOI] [PubMed] [Google Scholar]
- 24.Jacobs A An intracellular transit iron pool. Ciba Foundation symposium, 91–106 (1976). [DOI] [PubMed] [Google Scholar]
- 25.Vyoral D & Petrák J Iron transport in K562 cells: a kinetic study using native gel electrophoresis and 59Fe autoradiography. Biochimica et biophysica acta 1403, 179–188 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Konijn AM, et al. The cellular labile iron pool and intracellular ferritin in K562 cells. Blood 94, 2128–2134 (1999). [PubMed] [Google Scholar]
- 27.Espósito BP, Breuer W & Cabantchik ZI Design and applications of methods for fluorescence detection of iron in biological systems. Biochemical Society transactions 30, 729–732 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Kakhlon O & Cabantchik ZI The labile iron pool: characterization, measurement, and participation in cellular processes(1). Free radical biology & medicine 33, 1037–1046 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Rothman RJ, Serroni A & Farber JL Cellular pool of transient ferric iron, chelatable by deferoxamine and distinct from ferritin, that is involved in oxidative cell injury. Molecular pharmacology 42, 703–710 (1992). [PubMed] [Google Scholar]
- 30.Weaver J & Pollack S Low-Mr iron isolated from guinea pig reticulocytes as AMP-Fe and ATP-Fe complexes. Biochem J 261, 787–792 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozlov AV, Yegorov D, Vladimirov YA & Azizova OA Intracellular free iron in liver tissue and liver homogenate: studies with electron paramagnetic resonance on the formation of paramagnetic complexes with desferal and nitric oxide. Free radical biology & medicine 13, 9–16 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Cairo G, et al. Induction of ferritin synthesis by oxidative stress. Transcriptional and post-transcriptional regulation by expansion of the “free” iron pool. The Journal of biological chemistry 270, 700–703 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Breuer W, Epsztejn S & Cabantchik ZI Dynamics of the cytosolic chelatable iron pool of K562 cells. FEBS letters 382, 304–308 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Breuer W, Epsztejn S & Cabantchik ZI Iron acquired from transferrin by K562 cells is delivered into a cytoplasmic pool of chelatable iron(II). The Journal of biological chemistry 270, 24209–24215 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Epsztejn S, Kakhlon O, Glickstein H, Breuer W & Cabantchik I Fluorescence analysis of the labile iron pool of mammalian cells. Analytical biochemistry 248, 31–40 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Petrat F, de Groot H & Rauen U Subcellular distribution of chelatable iron: a laser scanning microscopic study in isolated hepatocytes and liver endothelial cells. Biochem J 356, 61–69 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrat F, Rauen U & de Groot H Determination of the chelatable iron pool of isolated rat hepatocytes by digital fluorescence microscopy using the fluorescent probe, phen green SK. Hepatology 29, 1171–1179 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Picard V, Epsztejn S, Santambrogio P, Cabantchik ZI & Beaumont C Role of ferritin in the control of the labile iron pool in murine erythroleukemia cells. The Journal of biological chemistry 273, 15382–15386 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Ma Y, Liu Z, Hider RC & Petrat F Determination of the labile iron pool of human lymphocytes using the fluorescent probe, CP655. Analytical chemistry insights 2, 61–67 (2007). [PMC free article] [PubMed] [Google Scholar]
- 40.Tenopoulou M, Kurz T, Doulias PT, Galaris D & Brunk UT Does the calcein-AM method assay the total cellular ‘labile iron pool’ or only a fraction of it? Biochem J 403, 261–266 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glickstein H, El RB, Shvartsman M & Cabantchik ZI Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood 106, 3242–3250 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Fakih S, et al. Targeting the lysosome: fluorescent iron(III) chelators to selectively monitor endosomal/lysosomal labile iron pools. Journal of medicinal chemistry 51, 4539–4552 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Abbate V, Reelfs O, Hider RC & Pourzand C Design of novel fluorescent mitochondria-targeted peptides with iron-selective sensing activity. Biochem J 469, 357–366 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Abbate V, Reelfs O, Kong X, Pourzand C & Hider RC Dual selective iron chelating probes with a potential to monitor mitochondrial labile iron pools. Chemical communications (Cambridge, England) 52, 784–787 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Carney IJ, et al. A ratiometric iron probe enables investigation of iron distribution within tumour spheroids. Metallomics 10, 553–556 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Au-Yeung HY, Chan J, Chantarojsiri T & Chang CJ Molecular imaging of labile iron(II) pools in living cells with a turn-on fluorescent probe. Journal of the American Chemical Society 135, 15165–15173 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niwa M, Hirayama T, Okuda K & Nagasawa H A new class of high-contrast Fe(II) selective fluorescent probes based on spirocyclized scaffolds for visualization of intracellular labile iron delivered by transferrin. Organic & biomolecular chemistry 12, 6590–6597 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Liu Z, Wang S, Li W & Tian Y Bioimaging and Biosensing of Ferrous Ion in Neurons and HepG2 Cells upon Oxidative Stress. Analytical chemistry 90, 2816–2825 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Long L, et al. A coumarin-based fluorescent probe for monitoring labile ferrous iron in living systems. Analyst 143, 2555–2562 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Sreedevi P, et al. Calix[4]arene Based Redox Sensitive Molecular Probe for SERS Guided Recognition of Labile Iron Pool in Tumor Cells. Analytical Chemistry 90, 7148–7153 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Thomas F, et al. Calcein as a fluorescent probe for ferric iron. Application to iron nutrition in plant cells. The Journal of biological chemistry 274, 13375–13383 (1999). [DOI] [PubMed] [Google Scholar]
- 52.Hirayama T, et al. A universal fluorogenic switch for Fe(ii) ion based on N-oxide chemistry permits the visualization of intracellular redox equilibrium shift towards labile iron in hypoxic tumor cells. Chemical science 8, 4858–4866 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirayama T, Okuda K & Nagasawa H A highly selective turn-on fluorescent probe for iron(ii) to visualize labile iron in living cells. Chemical Science 4, 1250–1256 (2013). [Google Scholar]
- 54.Spangler B, et al. A reactivity-based probe of the intracellular labile ferrous iron pool. Nature Chemical Biology 12, 680–685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aron AT, Reeves AG & Chang CJ Activity-based sensing fluorescent probes for iron in biological systems. Current opinion in chemical biology 43, 113–118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aron AT, Ramos-Torres KM, Cotruvo JA Jr. & Chang CJ Recognition- and reactivity-based fluorescent probes for studying transition metal signaling in living systems. Accounts of chemical research 48, 2434–2442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niwa M, Hirayama T, Oomoto I, Wang DO & Nagasawa H Fe(II) Ion Release during Endocytotic Uptake of Iron Visualized by a Membrane-Anchoring Fe(II) Fluorescent Probe. ACS chemical biology 13, 1853–1861 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Gao G, et al. A simple and effective dansyl acid based “turn-on” fluorescent probe for detecting labile ferrous iron in physiological saline and live cells. Talanta 215, 120908 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Aron AT, Loehr MO, Bogena J & Chang CJ An Endoperoxide Reactivity-Based FRET Probe for Ratiometric Fluorescence Imaging of Labile Iron Pools in Living Cells. Journal of the American Chemical Society 138, 14338–14346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zeng G, et al. Engineering Iron Responses in Mammalian Cells by Signal-Induced Protein Proximity. ACS synthetic biology 6, 921–927 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Spangler B, et al. Toward a Ferrous Iron-Cleavable Linker for Antibody-Drug Conjugates. Molecular pharmaceutics 15, 2054–2059 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Goel A, et al. A dual colorimetric-ratiometric fluorescent probe NAP-3 for selective detection and imaging of endogenous labile iron(III) pools in C. elegans. Chemical communications (Cambridge, England) 51, 5001–5004 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Aron AT, et al. In vivo bioluminescence imaging of labile iron accumulation in a murine model of Acinetobacter baumannii infection. Proceedings of the National Academy of Sciences of the United States of America 114, 12669–12674 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muir RK, et al. Measuring Dynamic Changes in the Labile Iron Pool in Vivo with a Reactivity-Based Probe for Positron Emission Tomography. ACS central science 5, 727–736 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu L, et al. Visualization of Dynamic Changes in Labile Iron(II) Pools in Endoplasmic Reticulum Stress-Mediated Drug-Induced Liver Injury. Analytical chemistry 92, 1245–1251 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Cyr AR & Domann FE The Redox Basis of Epigenetic Modifications: From Mechanisms to Functional Consequences. Antioxidants & Redox Signaling 15, 551–589 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao J, Yan Y, Salnikow K, Kluz T & Costa M Nickel-induced downregulation of serpin by hypoxic signaling. Toxicology and applied pharmacology 194, 60–68 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Costa M, et al. Nickel carcinogenesis: epigenetics and hypoxia signaling. Mutation research 592, 79–88 (2005). [DOI] [PubMed] [Google Scholar]
- 69.Macomber L & Hausinger RP Mechanisms of nickel toxicity in microorganisms. Metallomics 3, 1153–1162 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shi YG & Tsukada Y The discovery of histone demethylases. Cold Spring Harbor perspectives in biology 5(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pogribny IP, et al. Modulation of intracellular iron metabolism by iron chelation affects chromatin remodeling proteins and corresponding epigenetic modifications in breast cancer cells and increases their sensitivity to chemotherapeutic agents. International journal of oncology 42, 1822–1832 (2013). [DOI] [PubMed] [Google Scholar]
- 73.Zhao MH, et al. Analysis of Ferrous on Ten-Eleven Translocation Activity and Epigenetic Modifications of Early Mouse Embryos by Fluorescence Microscopy. Microscopy and microanalysis 22, 342–348 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Minor EA, Court BL, Young JI & Wang G Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. The Journal of biological chemistry 288, 13669–13674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blaschke K, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500, 222–226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yin R, et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. Journal of the American Chemical Society 135, 10396–10403 (2013). [DOI] [PubMed] [Google Scholar]
- 77.Chen J, et al. Vitamin C modulates TET1 function during somatic cell reprogramming. Nature Genetics 45, 1504–1509 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Schachtschneider KM, et al. Impact of neonatal iron deficiency on hippocampal DNA methylation and gene transcription in a porcine biomedical model of cognitive development. BMC Genomics 17, 856 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tran PV, Kennedy BC, Lien YC, Simmons RA & Georgieff MK Fetal iron deficiency induces chromatin remodeling at the Bdnf locus in adult rat hippocampus. American journal of physiology. Regulatory, integrative and comparative physiology 308, R276–282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Young JI, Züchner S & Wang G Regulation of the Epigenome by Vitamin C. Annual review of nutrition 35, 545–564 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao B, et al. Redox-active quinones induces genome-wide DNA methylation changes by an iron-mediated and Tet-dependent mechanism. Nucleic acids research 42, 1593–1605 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ye Q, et al. Brain iron loading impairs DNA methylation and alters GABAergic function in mice. FASEB journal 33, 2460–2471 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maiti A & Drohat AC Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. The Journal of biological chemistry 286, 35334–35338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ito S, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Camarena V, et al. cAMP signaling regulates DNA hydroxymethylation by augmenting the intracellular labile ferrous iron pool. eLife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huff TC, et al. Oscillatory cAMP signaling rapidly alters H3K4 methylation. Life science alliance 3(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Galaris D, Mantzaris M & Amorgianiotis C Oxidative stress and aging: the potential role of iron. Hormones (Athens) 7, 114–122 (2008). [DOI] [PubMed] [Google Scholar]
- 89.Kaur D, Rajagopalan S & Andersen JK Chronic expression of H-ferritin in dopaminergic midbrain neurons results in an age-related expansion of the labile iron pool and subsequent neurodegeneration: implications for Parkinson’s disease. Brain Res 1297, 17–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee JH, Jang H, Cho EJ & Youn HD Ferritin binds and activates p53 under oxidative stress. Biochem Biophys Res Commun 389, 399–404 (2009). [DOI] [PubMed] [Google Scholar]
- 91.Nakamura T, Naguro I & Ichijo H Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim Biophys Acta Gen Subj 1863, 1398–1409 (2019). [DOI] [PubMed] [Google Scholar]
- 92.Picca A, et al. Altered Expression of Mitoferrin and Frataxin, Larger Labile Iron Pool and Greater Mitochondrial DNA Damage in the Skeletal Muscle of Older Adults. Cells 9(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cronin SJF, Woolf CJ, Weiss G & Penninger JM The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front Mol Biosci 6, 116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nairz M & Weiss G Iron in infection and immunity. Mol Aspects Med 75, 100864 (2020). [DOI] [PubMed] [Google Scholar]
- 95.Kaur D, Lee D, Ragapolan S & Andersen JK Glutathione depletion in immortalized midbrain-derived dopaminergic neurons results in increases in the labile iron pool: implications for Parkinson’s disease. Free Radic Biol Med 46, 593–598 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wypijewska A, et al. Iron and reactive oxygen species activity in parkinsonian substantia nigra. Parkinsonism Relat Disord 16, 329–333 (2010). [DOI] [PubMed] [Google Scholar]
- 97.Devos D, et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid Redox Signal 21, 195–210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lukinova N, et al. Iron chelation protects the retinal pigment epithelial cell line ARPE-19 against cell death triggered by diverse stimuli. Invest Ophthalmol Vis Sci 50, 1440–1447 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hadziahmetovic M, et al. Bmp6 regulates retinal iron homeostasis and has altered expression in age-related macular degeneration. Am J Pathol 179, 335–348 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mandala A, et al. Fenofibrate prevents iron induced activation of canonical Wnt/β-catenin and oxidative stress signaling in the retina. NPJ Aging Mech Dis 6, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Obolensky A, et al. Zinc-desferrioxamine attenuates retinal degeneration in the rd10 mouse model of retinitis pigmentosa. Free Radic Biol Med 51, 1482–1491 (2011). [DOI] [PubMed] [Google Scholar]
- 102.Shah SV & Rajapurkar MM The role of labile iron in kidney disease and treatment with chelation. Hemoglobin 33, 378–385 (2009). [DOI] [PubMed] [Google Scholar]
- 103.Campanella A, et al. Skin fibroblasts from pantothenate kinase-associated neurodegeneration patients show altered cellular oxidative status and have defective iron-handling properties. Hum Mol Genet 21, 4049–4059 (2012). [DOI] [PubMed] [Google Scholar]
- 104.Martelli A, et al. Iron regulatory protein 1 sustains mitochondrial iron loading and function in frataxin deficiency. Cell Metab 21, 311–323 (2015). [DOI] [PubMed] [Google Scholar]
- 105.Danielpur L, et al. GLP-1-RA Corrects Mitochondrial Labile Iron Accumulation and Improves β-Cell Function in Type 2 Wolfram Syndrome. J Clin Endocrinol Metab 101, 3592–3599 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Slavic K, et al. A vacuolar iron-transporter homologue acts as a detoxifier in Plasmodium. Nat Commun 7, 10403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Clark M, Fisher NC, Kasthuri R & Cerami Hand C Parasite maturation and host serum iron influence the labile iron pool of erythrocyte stage Plasmodium falciparum. Br J Haematol 161, 262–269 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Crowe WE, Maglova LM, Ponka P & Russell JM Human cytomegalovirus-induced host cell enlargement is iron dependent. Am J Physiol Cell Physiol 287, C1023–1030 (2004). [DOI] [PubMed] [Google Scholar]
- 109.Lin SJ, et al. White spot syndrome virus protein kinase 1 defeats the host cell’s iron-withholding defense mechanism by interacting with host ferritin. J Virol 89, 1083–1093 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nakamura K, et al. Activation of the NLRP3 inflammasome by cellular labile iron. Exp Hematol 44, 116–124 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Kakhlon O, Gruenbaum Y & Cabantchik ZI Ferritin expression modulates cell cycle dynamics and cell responsiveness to H-ras-induced growth via expansion of the labile iron pool. Biochem J 363, 431–436 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kakhlon O, Gruenbaum Y & Cabantchik ZI Repression of ferritin expression modulates cell responsiveness to H-ras-induced growth. Biochem Soc Trans 30, 777–780 (2002). [DOI] [PubMed] [Google Scholar]
- 113.Zhang KH, et al. Ferritin heavy chain-mediated iron homeostasis and subsequent increased reactive oxygen species production are essential for epithelial-mesenchymal transition. Cancer Res 69, 5340–5348 (2009). [DOI] [PubMed] [Google Scholar]
- 114.Wang W, et al. IRP2 regulates breast tumor growth. Cancer Res 74, 497–507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cao LL, et al. Iron chelation inhibits cancer cell growth and modulates global histone methylation status in colorectal cancer. Biometals 31, 797–805 (2018). [DOI] [PubMed] [Google Scholar]
- 116.Zhao N, et al. Ferronostics: Measuring Tumoral Ferrous Iron with PET to Predict Sensitivity to Iron-Targeted Cancer Therapies. J Nucl Med (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hassannia B, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest 128, 3341–3355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]