

Abstract
Background & aims:
Gain of function (GOF) mutation of CTNNB1 gene is one of the most frequent genetic events in hepatocellular carcinoma (HCC). T-box transcription factor 3 (TBX3) is a liver-specific target of the Wnt/β-Catenin pathway and thought to be an oncogene mediating activated β-Catenin driven HCC formation.
Methods:
We evaluated TBX3 expression pattern in human HCC specimens. Tbx3 was conditionally knocked out in murine HCC models by hydrodynamic tail vein injection of Cre together with c-Met and ΔN90-β-Catenin (c-Met/β-Catenin) in Tbx3flox/flox mice. TBX3 was overexpressed in human HCC cell lines to investigate the functions of TBX3 in vitro.
Results:
A bimodal expression pattern of TBX3 in human HCC samples was detected: high expression of TBX3 in GOF CTNNB1 HCC and downregulation of TBX3 in non-CTNNB1 mutant tumors. High expression of TBX3 was associated with increased differentiation and decreased tumor growth gene expression signatures. Using Tbx3flox/flox mice, we found that ablation of Tbx3 significantly accelerates c-Met/β-Catenin driven HCC formation. Moreover, Tbx3(−) HCC demonstrated increased YAP/TAZ activity. The accelerated tumor growth induced by loss of TBX3 in c-Met/β-Catenin mouse HCC was successfully prevented by overexpression of LATS2, which inhibited YAP/TAZ activity. In human HCC cell lines, overexpression of TBX3 inhibited HCC cell growth as well as YAP/TAZ activation. A negative correlation between TBX3 and YAP/TAZ target genes was observed in human HCC samples. Mechanistically, phospholipase D1 (PLD1), a known positive regulator of YAP/TAZ, was identified as a novel transcriptional target repressed by TBX3.
Conclusion:
Our study suggests that TBX3 is induced by GOF CTNNB1 mutants and suppresses HCC growth by inactivating PLD1, thus leading to the inhibition of YAP/TAZ oncogenes.
Keywords: Hepatocellular carcinoma, T-Box Transcription Factor 3, β-Catenin, HIPPO cascade
Graphical Abstract
Lay Summary
TBX3 is a liver-specific target of the Wnt/β-Catenin pathway and thought to be an oncogene in promoting liver development. Here, we demonstrate that TBX3 is in fact a tumor suppressor gene, and it restricts liver tumor growth induced by activated CTNNB1 mutants. Mechanistically, we discover that TBX functions via regulating YAP/TAZ pathway. TBX3 therefore likely contributes to the specific pathological phenotypes observed in CTNNB1 mutant human HCCs. Strategies which increase TBX3 expression and/or activities may be effective for HCC treatment.
Introduction
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related death in the world.[1] Several multi-kinase inhibitors have been used for advanced HCC with limited survival benefits.[2] Immune checkpoint inhibitors show promise for HCC, however, most patients still progress with the regimen.[3] Thus, there is a continued need to elucidate the molecular mechanisms underlying HCC pathogenesis in order to develop novel biomarker-based precision medicine for HCC treatment.
The Wnt/β-Catenin pathway is a prominent signaling cascade controlling cell proliferation, differentiation, and tumor development in many organs.[4] In the liver, it has been implicated in the regulation of homeostasis, regeneration, and hepatocarcinogenesis.[5] Once activated, β-Catenin promotes target gene expression in association with T cell factor (TCF) family members and induces target gene expression. Mutations in CTNNB1, which encodes β-Catenin, result in the stabilization, nuclear translocation, and activation of the Wnt/β-Catenin cascade. Importantly, gain of function (GOF) mutations of CTNNB1 gene could be found in ~15 to 30% of human HCCs.[5] Studies have shown that HCCs with GOF CTNNB1 mutations display a particular phenotype, characterized by well-differentiated phenotype, low serum α-fetoprotein (AFP) levels, infrequent microvascular invasion, and better outcome.[6] However, the mechanisms underlying these observations remain unknown.
Yes-associated protein (YAP) is the major transcriptional co-activator downstream of Hippo tumor-suppressing pathway. Once activated, YAP translocates into the nucleus and interacts with TEA Domain (TEAD) DNA binding proteins to activate the expression of downstream targets, such as CTGF and CYR61.[7] YAP activation has been detected in multiple tumor types, including HCC, where YAP nuclear localization has been observed in ~60% of cases.[8] Recently, the biochemical crosstalk between Hippo/YAP/TAZ and Wnt/β-Catenin cascades has been unraveled.[9] However, whether and how these two pathways interact during hepatocarcinogenesis remains unclear.
TBX3 is a transcription repressor of the T-box gene family, whose members share a common DNA-binding domain, the T-box.[10] TBX3 has been found to be overexpressed in multiple tumor types and it contributes to the oncogenic process.[10] Intriguingly, some other studies suggest that TBX3 can also function as a tumor suppressor in specific tumor types, suggesting a context- and/or tissue-dependent function of TBX3 in cancer.[11] TBX3 has been identified as a downstream target gene of the Wnt/β-Catenin pathway in the liver,[12] and its expression is upregulated in human and mouse HCCs with GOF CTNNB1 mutations.[12] However, the overall expression pattern of TBX3 in human HCCs, and its functional role in hepatocarcinogenesis remain to be determined.
Here, we investigated the expression and functional contribution of TBX3 as a downstream effector of Wnt/β-Catenin during hepatocarcinogenesis.
Materials and Methods
Constructs and reagents
The plasmids used in this study, including pT3-EF1α, pT3-EF1α-c-Met (human), pT3-EF1α-ΔN90-β-Catenin (human), pT3-EF1α-YapS127A (human), pT3-EF1α-Lats2 (mouse), pCMV, pCMV/Cre, and pCMV/sleeping beauty (SB) transposase have been described previously.[13] Human PLD1 was purchased from Addgene (#45268) and cloned into the pT3-EF1α vector via the Gateway cloning technology (Invitrogen, Carlsbad, CA). pLenti-puro-TBX3 and pT3-EF1α-TBX3 (with C-terminal HA-tag) were constructed by inserting the corresponding human TBX3 cDNA (BD5458526) (Dharmacon, Lafayette, CO, USA) into pLenti-puro or pT3-EF1α vector via the Gateway cloning strategy. pLenti-puro-EGFP or pT3-EF1α-EGFP were used as control. All plasmids were purified by using the GenElute™ HP Endotoxin-Free Plasmid Maxiprep Kit (Cat# NA0410, Sigma-Aldrich, St. Louis, MO, USA) before used.
Mice and hydrodynamic tail vein injection
Tbx3flox/flox mice were kindly provided by Dr. Anne M. Moon from Weis Center for Research, Geisinger Clinic, USA. FVB/N mice were obtained from Jackson Laboratory (Bar Harbor, ME). Hydrodynamic tail vein injection (HTVi) was performed in 5.5- to 6.5-week-old mice as described previously.[13] Detailed information is available in Table S1. Mice were monitored by abdominal palpation and euthanized when they developed a high burden of liver tumors, i.e. large abdominal masses. Mice were housed, fed, and monitored in accordance with protocols approved by the Committee for Animal Research at the University of California, San Francisco.
Statistical analysis
The Prism 8.0 software (GraphPad, San Diego, CA, USA) was used to analyze the data. The data were presented as Mean ± SD. Comparisons between two groups were performed with the two-tailed unpaired t-test. Comparisons among three or more groups were performed with analysis of variance (ANOVA). Unpaired t-test with Welch correction, paired t-test, Mann-Whitney test, Chi-square test, or Fisher's exact test was applied when necessary. Survival curves were estimated using the Kaplan-Meier method and compared using the log-rank test. P-value < 0.05 was considered statistically significant.
Additional information is available in online Supplementary Materials and Methods.
Results
TBX3 expression in human HCCs
We analyzed the expression level of TBX3 mRNA using the TCGA-LIHC human HCC dataset.[14] We discovered that TBX3 expression was variable among HCCs, and the overall expression of TBX3 was significantly lower in HCCs when compared with surrounding liver tissues (Fig. 1A). We examined the correlation between TBX3 mRNA levels and CTNNB1 mutations as well as other mutation status in the TCGA-LIHC dataset._Consistent with TBX3 as a Wnt/β-Catenin target, [12] HCCs with high TBX3 expression were mainly characterized by CTNNB1 mutations (Fig. 1B and Fig. S1A, B). Subsequently, we divided HCCs into two groups: GOF CTNNB1 mutant HCCs and non-CTNNB1 mutant HCCs. The analysis clearly showed a bimodal expression pattern of TBX3 in human HCC samples: while TBX3 was highly expressed in GOF CTNNB1 mutant HCCs, its expression was downregulated in non-CTNNB1 mutant tumors (Fig. 1C). Similar results were obtained when comparing the levels of TBX3 in HCC tissues with paired non-tumorous livers (Fig. 1D). Consistent with previous reports that mutations of TP53 and CTNNB1 are mutually exclusive in HCCs, [15] TBX3 expression was found to be negatively correlated with TP53 mutations (Fig. 1B and Fig. S1A, C). No significant correlation between TBX3 expression levels and other genetic events in human HCCs was observed (Fig. S1A).
Fig. 1. Expression pattern of TBX3 mRNA in human HCC samples based on the TCGA-LIHC dataset.

(A) The overall expression of TBX3 mRNA. (B) Heatmap of TBX3 mRNA levels with common gene mutations involved in HCCs. (C) TBX3 mRNA expression is upregulated in GOF CTNNB1 mutation HCCs and downregulated in non-CTNNB1 mutant HCCs when compared to ST. (D) TBX3 mRNA expression in paired samples. (E) KEGG pathway analysis of TBX3 co-expressed genes. HCC, hepatocellular carcinoma; ST, surrounding liver tissues; GOF, gain of function. **P < 0.01; ***P < 0.001; ****P < 0.0001. (A) Mean ± SD; Welch's t-test. (C) Mean ± SD; One-way ANOVA test. (D) Paired t-test.
To investigate the mechanisms whereby TBX3 regulates hepatocarcinogenesis, we analyzed the genes whose mRNA expression was correlated with TBX3 levels in human HCCs using the TCGA-LIHC dataset. A total of 1,596 genes positively correlated with TBX3 expression, and 3,632 genes negatively correlated with TBX3 expression were identified (Table S5, 6). Noticeably, the top positive correlated genes were all canonical Wnt/β-Catenin targets, including GLUL, CYP2E1, LGR5, and SP5, providing further evidence that TBX3 is a main Wnt/β-Catenin target (Fig. S2A). In addition, KEGG pathway analysis revealed that TBX3 positively co-expressed genes were enriched in metabolic pathways (Fig. 1E and Fig. S2B). For instance, TBX3 expression was positively correlated with the expression of cytochrome P450 enzymes, such as CYP1A1, CYP1A2, CYP2E1, and CYP3A4 (Fig. S3). TBX3 negatively co-expressed genes were implicated in oncogenic pathways and cellular mechanisms, such as “PI3K-Akt signaling pathway” and “cell cycle” (Fig. 1E and Fig. S2B).
Altogether, we show a bimodal expression pattern of TBX3 in human HCC samples, with high TBX3 expression in GOF CTNNB1 HCCs and low TBX3 expression in non-CTNNB1 mutant HCCs.
Ablation of Tbx3 accelerates tumor growth in c-Met/β-Catenin driven HCC in mice
First, we analyzed TBX3 protein levels in nine human HCC cell lines and one hepatoblastoma (HB) cell line. TBX3 was found to be expressed at low levels in most HCC cell lines, whereas HepG2 cells, a HB cell line with GOF CTNNB1 mutation, showed the highest TBX3 protein expression (Fig. S4A). Subsequently, three HCC cell lines with low TBX3 expression, namely Li-7, MHCC-97H, and SNU182, were selected and transfected with TBX3 (Fig. S4B). Overexpression of TBX3 significantly inhibited HCC cell growth in vitro (Fig. S4C).
Next, we characterized the functional role of TBX3 during hepatocarcinogenesis in vivo. For this purpose, TBX3 protein expression was analyzed in six oncogene-induced mouse liver tumor models (Fig. S5). Consistent with human HCC data,[12] TBX3 protein was expressed at high levels in c-Met/β-Catenin and Yap/β-Catenin driven mouse HB tumors. To determine whether the loss of Tbx3 affects c-Met/β-Catenin HCC development, we hydrodynamically co-injected the c-Met and ΔN90-β-Catenin constructs together with the pCMV/Cre plasmid in Tbx3flox/flox conditional knockout (KO) mice (c-Met/β-Catenin/Cre). Additional Tbx3flox/flox mice were injected with c-Met/β-Catenin and pCMV empty vector as control (c-Met/β-Catenin/pCMV). Mice were monitored and euthanized when they developed large abdominal masses and became moribund (Fig. 2A). Importantly, we found that ablation of Tbx3 significantly accelerated tumor growth in c-Met/β-Catenin mice. Indeed, while all c-Met/β-Catenin/pCMV injected Tbx3flox/flox mice had to be harvested between 8- and 10-weeks post-injection, all c-Met/β-Catenin/Cre injected mice had to be sacrificed by 6 weeks post-injection (Fig. 2B). This was accompanied by increased liver weight and liver to body weight ratio in c-Met/β-Catenin/Cre mice (Fig. 2C). Overexpression of c-MET and ΔN90-β-Catenin oncogenes as well as deletion of TBX3 were validated by Western blot analysis (Fig. 2D). Microscopically, c-Met/β-Catenin/Cre tumors showed slight morphological/cytological differences when compared with corresponding c-Met/β-Catenin/pCMV tumors, although they were not very pronounced. Tumors in c-Met/β-Catenin/Cre mice displayed a lesser degree of differentiation; indeed, the neoplastic hepatocytes showed a cytoplasmic basophilia, higher nuclear/cytoplasmic ratio owing to the smaller size of the cytoplasm, increased lipid storage, numerous areas of necrosis of different size, and an increase in hematopoiesis. However, no remarkable differences such as lineage shift (no cholangiocarcinoma/mixed tumors) were detected (Fig. S6).
Fig. 2. Ablation of Tbx3 accelerates tumor growth in c-Met/β-Catenin driven hepatocarcinogenesis in mice.

(A) Study design. (B) Survival curves of c-Met/β-Catenin/pCMV and c-Met/β-Catenin/Cre mice. (C) The liver weight and liver to body weight ratio data. (D) Western blot analysis of lysates from normal livers (NL) and HCC tissues. GAPDH was used as a loading control. (E) Gross images, H&E staining, and immunohistochemical staining of Ki67 and SOX9 in mouse HCCs. (F) Western blot analysis of nuclear lysates from mouse HCCs. β-Tubulin and Histone H3 were used as loading controls. (G) Quantification of Ki67 positive cells in mouse HCCs. (H) Relative mRNA levels of YAP/TAZ signaling targets (Ctgf, and Cyr61), Notch signaling targets (Hes1, Hey1, and HeyL) in mouse HCCs. HTVi, hydrodynamic tail vein injection; Scale bar: 200 μm for 100x; 100 μm for 200x. *P < 0.05; **P < 0.01; ***P < 0.001. (C, G, H) Mean ± SD; Unpaired t-test, Welch's t-test or Mann-Whitney test.
We investigated the signaling pathways that might mediate the tumor-suppressing activity of TBX3. Tbx3 KO tumors displayed significantly higher Ki67 immunoreactivity (Fig. 2E, G), suggesting that increased tumor cell proliferation might be the major cellular mechanism leading to accelerated HCC growth in the Tbx3 KO genetic background. We analyzed the major pathways regulating tumor proliferation, including ERK, AKT, and STAT3 pathways, but found no difference between control and Tbx3 KO tumors (Fig. S7). Immunohistochemical (IHC) staining revealed that the tumor cells in both groups were positive for the epithelial marker E-Cadherin, the hepatocellular marker Hepatocyte nuclear factor 4α (HNF4α), Wnt/β-Catenin target Glutamine synthetase (GS), and negative for the mesenchymal marker Vimentin (Fig. S8). Human HCC studies suggest that high TBX3 is associated with increased hepatocyte differentiation (Fig. S2B). Accordingly, in Tbx3 KO tumors, markers of HCC and hepatoblasts, such as Afp and epithelial cell adhesion molecule (Epcam), were significantly upregulated (Fig. S9A), whereas mature hepatocyte genes such as metabolic pathway related genes Cyp1a1, Cyp1a2, and Cyp2e1 were significantly downregulated (Fig. S9B). Intriguingly, the nuclear expression of SOX9 was prominent in Tbx3 KO tumor cells (Fig. 2D, E). As the Hippo cascade has been demonstrated to regulate hepatocyte lineage commitment,[13, 16] as well as HCC cell proliferation,[17] we hypothesized that YAP/TAZ might be activated in Tbx3 KO HCC samples. We found that the phosphorylated forms of YAP (p-YAPSer127 and p-YAPSer397) and YAP/TAZ negative regulator LATS2 were downregulated in Tbx3 KO tumors, whereas no changes were observed in total YAP/TAZ proteins and other Hippo pathway members, including MST1/2, SAV1, MOB1, and p-MOB1 (Fig. 2D). Nuclear YAP/TAZ levels were upregulated (Fig. 2F) and their downstream targets, including Ctgf and Cyr61, were significantly increased in Tbx3 KO tumors (Fig. 2H). Notch is a key pathway regulated by YAP/TAZ.[16] Accordingly, we found augmented NOTCH2 protein levels in Tbx3 KO tumors (Fig. 2D). Notch2 and other canonical Notch target genes were also upregulated at mRNA level (Fig. 2H and Fig. S9C).
In summary, our study demonstrates that TBX3 could act as a tumor suppressor during hepatocarcinogenesis both in vitro and in vivo. Mechanistically, deletion of Tbx3 led to increased YAP/TAZ activity, resulting in augmented Notch activation, decreased tumor differentiation status as well as increased tumor proliferation.
Activation of Hippo prevents accelerated hepatocarcinogenesis induced by Tbx3 loss in c-Met/β-Catenin mice
Since our results suggest that TBX3 might inhibit HCC formation via the Hippo/YAP/TAZ cascade, we hypothesized that activation of Hippo via overexpression of LATS2 would prevent accelerated c-Met/β-Catenin driven hepatocarcinogenesis induced by loss of Tbx3. To test this hypothesis, we injected c-Met, ΔN90-β-Catenin, and Cre plasmids together with Lats2 construct (c-Met/β-Catenin/Cre/Lats2) into Tbx3flox/flox mice (Fig. 3A). This allowed the expression of c-Met/β-Catenin oncogenes together with Lats2 in Tbx3 KO hepatocytes. As a control, additional Tbx3flox/flox mice were injected with c-Met/β-Catenin/Cre together with pT3-EF1α empty vector (c-Met/β-Catenin/Cre/pT3) (Fig. 3A). We found that consistent with our previous data, c-Met/β-Catenin/Cre/pT3 injected Tbx3flox/flox mice rapidly developed lethal burden of liver tumors, and all mice had to be euthanized ~5 weeks post-injection. In contrast, c-Met/β-Catenin/Cre/Lats2 injected Tbx3flox/flox mice were harvested around 8 weeks post-injection due to high tumor burden (Fig. 3B). Indeed, c-Met/β-Catenin/Cre/Lats2 Tbx3flox/flox mice developed liver tumors at the similar latency as c-Met/β-Catenin/pCMV injected Tbx3flox/flox mice (Fig. 3B, C). Histologically, similar tumor lesions were observed in c-Met/β-Catenin/Cre/Lats2 Tbx3flox/flox and c-Met/β-Catenin/pCMV injected Tbx3flox/flox mice (Fig. S10). Overexpression of c-MET, ΔN90-β-Catenin as well as deletion of TBX3 were validated by Western blotting in both mouse tumor cohorts (Fig. 3E). Overexpression of Lats2 was validated by qRT-PCR analysis in c-Met/β-Catenin/Cre/Lats2 tumors (Fig. 3G). As expected, LATS2 overexpression led to decreased total YAP/TAZ as well as nuclear YAP/TAZ protein levels (Fig. 3E, F), as well as decline of their downstream targets Ctgf and Cyr61 mRNA (Fig. 3G), as well as decreased SOX9 expression (Fig. 3D, E). Tumor cell proliferation was significantly inhibited (Fig. 3D, H). No significant changes in the expressions of E-Cadherin, HNF4α, GS, and Vimentin were observed (Fig. S11).
Fig. 3. Activation of Hippo by overexpression of LATS2 prevents loss of Tbx3 induced accelerated HCC growth in c-Met/β-Catenin HCC model.

(A) Study design. (B) Survival curves among c-Met/β-Catenin/pCMV, c-Met/β-Catenin/Cre/pT3, and c-Met/β-Catenin/Cre/Lats2 mice. (C) The liver weight and liver to body weight ratio among mouse cohorts. (D) Gross images, H&E staining, and immunohistochemical staining of Ki67 and SOX9 in mouse HCCs. (E) Western blot analysis of lysates from normal mouse livers (NL), mouse HCCs. GAPDH was used as a loading control. (F) Western blot analysis of nuclear lysates from mouse HCC tissues. β-Tubulin and Histone H3 were used as loading controls. (G) Relative mRNA levels of Lats2 and YAP/TAZ signaling targets (Ctgf, and Cyr61) in mouse HCCs. (H) Quantification of Ki67 positive cells in mouse HCCs. HTVi, hydrodynamic tail vein injection; Scale bar: 200 μm for 100x; 100 μm for 200x. n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (C, G) Mean ± SD; One-way ANOVA test. (H) Mean ± SD; Unpaired t-test.
In summary, our data demonstrate that activation of Hippo, which leads to the inactivation of the YAP/TAZ pathway, prevents loss of Tbx3 induced accelerated liver tumorigenesis in c-Met/β-Catenin mice.
Ablation of Tbx3 does not accelerate tumor growth in Yap/β-Catenin driven tumorigenesis in mice
Previously, we established a murine HB model via the co-expression of activated YAP (YapS127A) and ΔN90-β-Catenin (Yap/β-Catenin).[18] YAPSer127 is the major phosphorylation site through which YAP is inactivated by the LATS kinases, while mutant YapS127A is the constitutively activated form of this oncogene that escapes LATS mediated inactivation. TBX3 was found to be upregulated in Yap/β-Catenin liver tumor samples (Fig. S5A). We hypothesized that, if TBX3 delays tumor growth by inactivation of YAP (via induction of the LATS/Hippo axis), it is conceivable that ablation of Tbx3 would have limited impact on Yap/β-Catenin induced liver tumors. We co-injected Yap/β-Catenin/Cre or control Yap/β-Catenin/pCMV plasmids into Tbx3flox/flox mice (Fig. 4A). We found that all injected mice developed liver tumors and were euthanized between 9 and 17 weeks after injection, with no statistical difference in survival length between the two cohorts (Fig. 4B). Overexpression of YAP and ΔN90-β-Catenin as well as deletion of TBX3 were validated by Western blotting (Fig. 4C). Histologically, HB lesions from Yap/β-Catenin/Cre and control Yap/β-Catenin/pCMV appeared identical. Indeed, HBs of pure epithelial type, dominated by fetal-like HBs cells, with a minor component of lesser mature embryonal-like cells were detected in the two mouse cohorts (Fig. S12). No significant changes in the expressions of E-Cadherin, HNF4α, GS, and Vimentin were observed (Fig. S13). Tumor cells were highly proliferative, showing similar proliferation rates as indicated by Ki67 index (Fig. 4D, F). No difference in the levels of ERK, AKT, and STAT3 cascades was noted (Fig. S14). As concerns the Hippo/YAP/TAZ pathway, similar expression levels for most of the proteins (Fig. 4C), including nuclear localized YAP and TAZ (Fig. 4E) were detected in Tbx3(+) and Tbx3(−) Yap/β-Catenin tumors. However, LATS2 and p-YAPSer397 levels were consistently lower in Tbx3(−) tumors (Fig. 4C). qRT-PCR analysis suggested a similar expression of Yap/Notch pathway genes, including Ctgf, Cyr61, and Hes1, in tumor tissues in the two mouse cohorts (Fig. 4G and Fig. S15A). No difference in hepatocyte differentiation markers (Cyp1a1, Cyp1a2, and Cyp2e1), as well as markers of HCC and hepatoblasts (Afp and Epcam), was found between Tbx3(+) and Tbx3(−) tumor samples (Fig. S15B, C).
Fig. 4. Ablation of Tbx3 does not accelerate tumor growth in Yap/β-Catenin mice.

(A) Study design. (B) Survival curves of Yap/β-Catenin/pCMV and Yap/β-Catenin/Cre mice. (C) Western blot analysis of lysates from normal livers (NL) and mouse HCCs. GAPDH was used as a loading control. (D) Gross images, H&E staining, and immunohistochemical staining of Ki67 in mouse HCCs. (E) Western blot analysis of nuclear lysates from mouse HCCs. β-Tubulin and Histone H3 were used as loading controls. (F) Quantification of Ki67 positive cells in mouse HCCs. (G) Relative mRNA levels of YAP/TAZ signaling targets (Ctgf, and Cyr61), Notch signaling targets (Hes1, and Hey1) in mouse HCCs. HTVi, hydrodynamic tail vein injection; Scale bar: 200 μm for 100x; 100 μm for 200x. n.s., not significant; **P < 0.01; ***P < 0.001; ****P < 0.0001. (F) Mean ± SD; Unpaired t-test. (G) Mean ± SD; One-way ANOVA test.
In summary, the present data indicate that ablation of Tbx3 has a limited impact on liver tumor development driven by constitutively activated YAP.
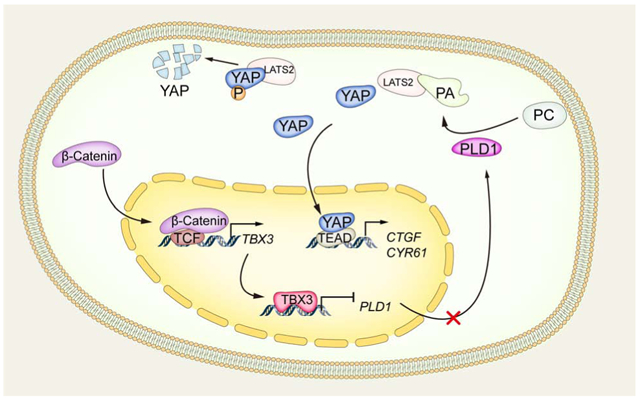
TBX3 directly represses PLD1 expression at transcriptional level leading to inhibition of YAP/TAZ activity
The data from the mouse models suggested Hippo/YAP/TAZ as the major signaling cascade regulated by TBX3 during hepatocarcinogenesis. To test whether a similar phenotype could be observed in human HCCs, we evaluated the correlation between TBX3 and YAP/TAZ downstream targets in human HCC samples using the TCGA-LIHC dataset. We found that TBX3 mRNA expression was negatively correlated with the mRNA levels of YAP/TAZ downstream targets, including CCN2, AXL, and JAG1, as well as the canonical Notch signaling targets (NOTCH1, NOTCH2, JAG2, HES1, HEYL, and NRARP) in human HCCs (Fig. 5). We overexpressed TBX3 in three human HCC cell lines. TBX3 significantly reduced the protein levels of total YAP/TAZ as well as nuclear YAP/TAZ (Fig. 6A, B). YAP/TAZ targets CTGF and CYR61 mRNA expression were significantly downregulated (Fig. 6C). However, the mRNA levels of LATS2, YAP, and TAZ were not regulated by TBX3 (Fig. S16), suggesting that TBX3 likely modulates YAP/TAZ activity at the post-transcriptional level.
Fig. 5. TBX3 mRNA expression is negatively correlated with YAP/TAZ and Notch signaling cascades in human HCC samples based on the TCGA-LIHC dataset.

(A) TBX3 mRNA levels are negatively correlated with the mRNA levels of YAP/TAZ signaling targets. (B) TBX3 mRNA levels are negatively correlated with the mRNA levels of Notch signaling targets. r, Spearman’s correlation coefficient.
Fig. 6. Overexpression of TBX3 inactivates YAP/TAZ signaling in human HCC cell lines.

(A) Western blotting analysis of HCC cell lines with low TBX3 expression (Li-7, MHCC-97H, and SNU182) transfected with either HA-tagged TBX3 or EGFP (Control). GAPDH was used as a loading control. (B) Overexpression of TBX3 significantly reduces nuclear YAP/TAZ protein levels, as determined by Western blotting. β-Tubulin and Histone H3 were used as loading controls. (C) Overexpression of TBX3 significantly reduces mRNA levels of YAP/TAZ downstream targets (CTGF, and CYR61) in HCC cell lines analyzed by qRT-PCR. qRT-PCR, quantitative reverse transcription PCR. ****P < 0.0001. (C) Mean ± SD; Unpaired t-test.
As TBX3 is a transcriptional repressor, we searched for its potential target in human HCCs. For this purpose, we searched and identified the top 50 genes whose expression levels were negatively correlated with TBX3 in the TCGA-LIHC dataset (Table S6). Each of the genes was searched using PubMed for any possible link to the Hippo cascade. Among these genes was phospholipase D1 (PLD1), a possible regulator of the YAP.[19] Specifically, PLD-mediated production of phosphatidic acid (PA) inhibits LATS kinases, leading to YAP nuclear localization and activation.[19] In the TCGA-LIHC database, a strong negative correlation between PLD1 and TBX3 mRNA expression was noted (Fig. 7A). In addition, forced overexpression of TBX3 in human HCC cell lines significantly reduced PLD1 mRNA expression (Fig. 7B), whereas Pld1 mRNA levels were significantly elevated in Tbx3(−) c-Met/β-Catenin tumors when compared to Tbx3(+) liver tumors (Fig. 7C). Importantly, overexpression of TBX3 led to decreased cellular PA levels in Li-7 human HCC cells (Fig. 7D).
Fig. 7. TBX3 directly represses PLD1 expression at transcriptional level leading to inhibition of YAP/TAZ activity.

(A) TBX3 mRNA expression is negatively correlated with PLD1 mRNA expression in the TCGA-LIHC dataset. (B) Overexpression of TBX3 significantly reduces PLD1 mRNA expression in human HCC cell lines. (C) Ablation of Tbx3 significantly increases Pld1 mRNA expression in c-Met/β-Catenin mouse HCCs. (D) Overexpression of TBX3 leads to reduced cellular PA levels in Li-7 HCC cells. No serum was used as a control. (E) ChIP-PCR assay shows that exogenous TBX3 protein directly binds to the PLD1 promoter in Li-7 and MHCC-97H cells. (F) ChIP-PCR assay shows that endogenous TBX3 protein directly binds to the PLD1 promoter in HepG2 cells. (G) qRT-PCR shows that exogenous PA rescues TBX3 induced downregulation of YAP/TAZ targets (CTGF and CYR61). (H) qRT-PCR shows that overexpression of PLD1 prevents TBX3 induced downregulation of CTGF and CYR61. (I) Study design. (J) The liver weight and liver to body weight ratio of c-Met/β-Catenin/PLD1 and c-Met/β-Catenin/pT3 mice. r, Spearman’s correlation coefficient; IP, immunoprecipitation; ChIP-PCR: Chromatin immunoprecipitation-PCR; qRT-PCR, quantitative reverse transcription PCR; HTVi, hydrodynamic tail vein injection. n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (B, C, J) Mean ± SD; Unpaired t-test or Welch's t-test. (D, G, H) Mean ± SD; One-way ANOVA test.
Next, we investigated whether PLD1 is a direct target of TBX3 transcriptional repressor. We searched for putative TBX3 binding sites in the PLD1 promoter region. JASPAR database analysis revealed TBX3 as a candidate transcription regulator of PLD1 (Fig. S17A), and one putative TBX3 binding site was identified along the PLD1 promoter region (Fig. S17B). To investigate whether TBX3 binds to PLD1 promoter region, Chromatin immunoprecipitation (ChIP) assay was performed in human HCC cells. HA-tagged TBX3 was transfected into the cells, and ChIP-PCR assay confirmed the binding of TBX3 to the PLD1 promoter sequence (Fig. 7E). Furthermore, in HepG2 cells, characterized by high TBX3 expression, ChIP with anti-TBX3 antibody demonstrated the binding of endogenous TBX3 to the PLD1 promoter (Fig. 7F).
We investigated whether TBX3 inhibits YAP/TAZ via PLD1/PA pathway. TBX3 was overexpressed in HCC cells and exogenous PA was added. We found that overexpression of TBX3 significantly reduced mRNA levels of YAP/TAZ downstream targets, including CTGF and CYR61. Adding exogenous PA to the culture medium rescued this phenotype (Fig. 7G). Furthermore, overexpression of PLD1 together TBX3 prevented TBX3 induced downregulation of CTGF and CYR61 in HCC cell lines (Fig. 7H).
Finally, we tested the hypothesis that overexpression of PLD1 would accelerate c-Met/β-Catenin driven HCC pathogenesis, recapitulating the phenotypes when Tbx3 was deleted in mice. We hydrodynamically co-injected c-Met and ΔN90-β-Catenin together with the PLD1 plasmid in FVB/N mice (c-Met/β-Catenin/PLD1). Additional mice were injected with c-Met/β-Catenin and pT3-EF1α empty vector as control (c-Met/β-Catenin/pT3) (Fig. 7I). Mice were euthanized around 7 weeks post injection. We found a significantly higher tumor burden in c-Met/β-Catenin/PLD1 than that of c-Met/β-Catenin/pT3 mice (Fig. 7J).
In summary, our study indicates that TBX3 directly binds to the PLD1 promoter region, inhibiting its transcription. This leads to decreased YAP/TAZ activity, and eventually increasing HCC differentiation and decreasing proliferation.
Discussion
TBX3 is a well-characterized target of the Wnt/β-Catenin cascade in HCC.[11, 12] However, the specific role of TBX3 in human HCC remains poorly defined. In the current study, we found that TBX3 mRNA expression is high in HCCs with CTNNB1 GOF mutations, but low in most non-CTNNB1 mutant HCCs. Further analysis of genes whose expression levels correlated with that of TBX3 revealed that high TBX3 expression was associated with increased differentiation status and less malignancy. Functionally, we found that overexpression of TBX3 inhibits HCC growth in vitro, whereas ablation of Tbx3 accelerates tumor growth in c-Met/β-Catenin mice. GOF CTNNB1 mutant HCCs have been shown to display distinctive features, such as a relatively well-differentiated phenotype and genomic stability.[6] Our data suggest TBX3 may function downstream of activated Wnt/β-Catenin signaling in maintaining these unique phenotypes. These findings are also consistent with the data from Seehawer M et al, suggesting that TBX3 is a key microenvironment-dependent lineage-commitment factor in promoting HCC formation.[20]
It has been reported TBX3 can either promote tumor development or suppress tumor progression, depending on the tumor type.[12, 21-24] Consistently, pan-cancer gene expression analysis revealed a variable TBX3 mRNA expression in different tumor types (Fig. S18). While some tumor types, such as rectal adenocarcinoma and esophageal carcinoma, demonstrate elevated TBX3 expression, others, such as renal clear cell carcinoma, cholangiocarcinoma, and lung adenocarcinoma, display decreased TBX3 expression. In the analysis of murine HCC models, we noticed that in addition to HCCs with overexpression of GOF β-Catenin, TBX3 is upregulated in c-Myc induced HCCs. Thus, we analyzed Wnt/β-Catenin pathway status in c-Myc mouse HCCs. We found no evidence of Wnt/β-Catenin activation, as only membranous β-Catenin was observed and tumors demonstrated no GS immunoreactivity (Fig. S19A). Gene sequencing revealed no mutations in exon 3 of the mouse Ctnnb1 allele (Fig. S19B). The data suggest that c-Myc may induce TBX3 expression independent of the Wnt/β-Catenin pathway. Indeed, analysis of the TCGA-LIHC dataset revealed that almost 50% of HCCs with elevated TBX3 do not show CTNNB1 mutations (Fig. S1A, B). Clearly, TBX3 may have distinct expression patterns and functional roles under different oncogenic stimuli, which require further studies.
It should be also noted that in the TCGA-LIHC dataset, among 96 human HCCs with CTNNB1 mutations, 83 samples showed high TBX3 expression, whereas 13 samples exhibited low TBX3 levels. Further analysis revealed the presence of additional genetic events in the 13 CTNNB1 mutant HCC samples with low TBX3 expression, such as TP53 or ARID1A mutations (Fig. S20). We cannot exclude that these genetic events could also regulate TBX3 expression, and additional studies are needed to fully unravel the genes and pathways regulating TBX3 in HCC.
Mechanistically, we identified PLD1 as a novel transcriptional target of TBX3. The phospholipase D (PLD) genes encode the intracellular PLDs enzymes, which are activated due to extracellular signaling events and generate the lipid second-messenger PA via hydrolysis of phosphatidylcholine (PC). The two best-characterized PLD isoforms, PLD1 and PLD2, are involved in the regulation of multiple key signaling pathways that control a variety of cellular functions such as cell proliferation and death. PLD1 is frequently upregulated in various types of cancer including HCC.[25] For instance, Xiao and colleagues reported that PLD1, but not PLD2, is upregulated in human HCC tissues and HCC cell lines. PLD1 activation contributes to HCC development via regulation of proliferation, migration, and invasion of HCC cells, as well as by promoting the EMT process.[26] In the current study, we discovered that PLD1 is a direct target of TBX3 and a regulator of the Hippo cascade. Based on the TCGA-LIHC dataset, PLD1 mRNA negatively correlates with TBX3 mRNA expression (Fig. 7A). In addition, CTNNB1 mutant HCCs exhibit high TBX3 and low PLD1 expression (Fig. S21). Consistent with previous studies, we found that PLD1 levels positively correlate with those of YAP/TAZ target genes, including CCN2, JAG1, NOTCH2, and AXL in human HCCs (Fig. S22). These data envisage the existence of a β-Catenin/TBX3/Hippo cascade in hepatocarcinogenesis. Specifically, GOF CTNNB1 mutations activate β-Catenin and induce TBX3 expression that, in turn, inhibits PLD1. The consequent decrease of PLD1 expression triggers the activation of LATS kinases, leading to inactivation of YAP and TAZ. This might contribute to maintaining a relatively less aggressive status of HCC. It is also worth noting that activation of YAP has been shown to induce chromosomal instability in HCC,[27] whereas GOF CTNNB1 mutant human and mouse HCCs are known to be chromosomally stable.[28] In addition, activation of β-Catenin has been linked to a better prognosis in some human HCC cohorts,[29] whereas YAP levels predict poor prognosis in this tumor type.[8] Our study might provide mechanistic insights into these clinical observations.
Supplementary Material
Highlights.
TBX3 is upregulated in CTNNB1 mutant HCC and downregulated in non-CTNNB1 mutant HCC.
TBX3 overexpression increases HCC differentiation and decreases tumor proliferation.
TBX3 deceases YAP/TAZ pathway activation.
PLD1, a positive regulator of YAP/TAZ, is a novel transcriptional target repressed by TBX3.
TBX3 is a tumor suppressor downstream of activated Wnt/β-Catenin in HCC.
Acknowledgments:
The authors would like to thank Dr. Anne M. Moon (Department of Molecular and Functional Genomics, Weis Center for Research, Geisinger Clinic, USA) for the Tbx3flox/flox mice; and Dr. Binbin Liu (Liver Cancer Institute and Zhongshan Hospital of Fudan University, China) for the MHCC-97H cell line. We thank Dr. Wenqi Wang and Dr. Han Han (Department of Developmental and Cell Biology, University of California, Irvine, USA) for the advice on PA experiments.
Financial support statement: This study is supported by NIH grants R01CA239251 to XC; R01CA204586 and R01CA250227 to XC and SPM; P30DK026743 to UCSF Liver Center; K08DK101603 to BW; Burroughs Wellcome Fund Career Award for Medical Scientists to BW.
List of abbreviations:
- AFP
α-fetoprotein
- ANOVA
analysis of variance
- ChIP
Chromatin immunoprecipitation
- CTNNB1
β-Catenin (cadherin-associated protein) beta 1
- Epcam
epithelial cell adhesion molecule
- GOF
Gain of function
- GS
Glutamine synthetase
- HB
Hepatoblastoma
- HCC
Hepatocellular carcinoma
- H&E
Hematoxylin-Eosin staining
- HNF4α
Hepatocyte nuclear factor 4α
- HTVi
Hydrodynamic tail vein injection
- IHC
Immunohistochemistry
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KO
Knockout
- LATS 2
Large tumor suppressor kinase 2
- PA
Phosphatidic acid
- PLD1
Phospholipase D1
- qRT-PCR
quantitative reverse transcription PCR
- SB
Sleeping beauty
- TAZ
PDZ-binding motif
- TBX3
T-box transcription factor 3
- TCF4
T cell factor 4
- TCGA
The Cancer Genome Atlas
- TEAD
TEA domain transcription factor
- TFBS
Transcription factor binding site
- wt
wild-type
- YAP
Yes-associated protein
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability:
The data that support the findings of this study are included within the article and its supplementary materials.
References
- [1].Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- [2].Faivre S, Rimassa L, Finn RS. Molecular therapies for HCC: Looking outside the box. J Hepatol 2020;72:342–352. [DOI] [PubMed] [Google Scholar]
- [3].Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med 2020;382:1894–1905. [DOI] [PubMed] [Google Scholar]
- [4].Nusse R, Clevers H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017;169:985–999. [DOI] [PubMed] [Google Scholar]
- [5].Perugorria MJ, Olaizola P, Labiano I, Esparza-Baquer A, Marzioni M, Marin JJG, et al. Wnt-beta-catenin signalling in liver development, health and disease. Nat Rev Gastroenterol Hepatol 2019;16:121–136. [DOI] [PubMed] [Google Scholar]
- [6].Rebouissou S, Nault JC. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J Hepatol 2020;72:215–229. [DOI] [PubMed] [Google Scholar]
- [7].Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015;163:811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xu MZ, Yao TJ, Lee NP, Ng IO, Chan YT, Zender L, et al. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer 2009;115:4576–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li N, Lu N, Xie C. The Hippo and Wnt signalling pathways: crosstalk during neoplastic progression in gastrointestinal tissue. FEBS J 2019;286:3745–3756. [DOI] [PubMed] [Google Scholar]
- [10].Khan SF, Damerell V, Omar R, Du Toit M, Khan M, Maranyane HM, et al. The roles and regulation of TBX3 in development and disease. Gene 2020;726:144223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Willmer T, Cooper A, Peres J, Omar R, Prince S. The T-Box transcription factor 3 in development and cancer. Biosci Trends 2017;11:254–266. [DOI] [PubMed] [Google Scholar]
- [12].Renard CA, Labalette C, Armengol C, Cougot D, Wei Y, Cairo S, et al. Tbx3 is a downstream target of the Wnt/beta-catenin pathway and a critical mediator of beta-catenin survival functions in liver cancer. Cancer Res 2007;67:901–910. [DOI] [PubMed] [Google Scholar]
- [13].Zhang S, Wang J, Wang H, Fan L, Fan B, Zeng B, et al. Hippo Cascade Controls Lineage Commitment of Liver Tumors in Mice and Humans. Am J Pathol 2018;188:995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–1341 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM, Sung CO, et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology 2014;60:1972–1982. [DOI] [PubMed] [Google Scholar]
- [16].Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell 2014;157:1324–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Manmadhan S, Ehmer U. Hippo Signaling in the Liver - A Long and Ever-Expanding Story. Front Cell Dev Biol 2019;7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tao J, Calvisi DF, Ranganathan S, Cigliano A, Zhou L, Singh S, et al. Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 2014;147:690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Han H, Qi R, Zhou JJ, Ta AP, Yang B, Nakaoka HJ, et al. Regulation of the Hippo Pathway by Phosphatidic Acid-Mediated Lipid-Protein Interaction. Mol Cell 2018;72:328–340 e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Seehawer M, Heinzmann F, D'Artista L, Harbig J, Roux PF, Hoenicke L, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 2018;562:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hansel DE, Rahman A, House M, Ashfaq R, Berg K, Yeo CJ, et al. Met proto-oncogene and insulin-like growth factor binding protein 3 overexpression correlates with metastatic ability in well-differentiated pancreatic endocrine neoplasms. Clin Cancer Res 2004;10:6152–6158. [DOI] [PubMed] [Google Scholar]
- [22].Perkhofer L, Walter K, Costa IG, Carrasco MC, Eiseler T, Hafner S, et al. Tbx3 fosters pancreatic cancer growth by increased angiogenesis and activin/nodal-dependent induction of stemness. Stem Cell Res 2016;17:367–378. [DOI] [PubMed] [Google Scholar]
- [23].Li Z, Wang Y, Duan S, Shi Y, Li S, Zhang X, et al. Expression of TBX3 in Hepatocellular Carcinoma and Its Clinical Implication. Med Sci Monit 2018;24:9324–9333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Krstic M, Kolendowski B, Cecchini MJ, Postenka CO, Hassan HM, Andrews J, et al. TBX3 promotes progression of pre-invasive breast cancer cells by inducing EMT and directly up-regulating SLUG. J Pathol 2019;248:191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Brown HA, Thomas PG, Lindsley CW. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat Rev Drug Discov 2017;16:351–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Xiao J, Sun Q, Bei Y, Zhang L, Dimitrova-Shumkovska J, Lv D, et al. Therapeutic inhibition of phospholipase D1 suppresses hepatocellular carcinoma. Clin Sci (Lond) 2016;130:1125–1136. [DOI] [PubMed] [Google Scholar]
- [27].Weiler SME, Pinna F, Wolf T, Lutz T, Geldiyev A, Sticht C, et al. Induction of Chromosome Instability by Activation of Yes-Associated Protein and Forkhead Box M1 in Liver Cancer. Gastroenterology 2017;152:2037–2051 e2022. [DOI] [PubMed] [Google Scholar]
- [28].Calvisi DF, Factor VM, Ladu S, Conner EA, Thorgeirsson SS. Disruption of beta-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterology 2004;126:1374–1386. [DOI] [PubMed] [Google Scholar]
- [29].Mao TL, Chu JS, Jeng YM, Lai PL, Hsu HC. Expression of mutant nuclear beta-catenin correlates with non-invasive hepatocellular carcinoma, absence of portal vein spread, and good prognosis. J Pathol 2001;193:95–101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are included within the article and its supplementary materials.