

Abstract
Intracellular protein trafficking via the endosomes plays a key role in the maintenance of normal neuronal function. Although many diseases of the central nervous system exhibit specific pathological hallmarks, abnormalities of the endosome system are common traits for several of them, including Alzheimer disease (AD). Three main routes originate from the endosomes: the recycling, degradation, and retrograde pathways. Studies have shown that the majority of Down syndrome subjects develop AD pathology and manifest altered morphology and number of endosomes, and abnormalities in lysosome acidification and exosome secretion, suggesting that dysfunction of one of these pathways could play a functional role in the AD‐like phenotype of the syndrome. Two of the major endosomal routes are mediated by the retromer complex, a multimeric system responsible for transport of cargo from the endosome to the trans‐Golgi network or to the cell membrane. Recently, a new endosome system structurally related to the retromer, called “retriever,” has been reported. Whereas we know a great deal about the neuropathophysiology of the retromer complex, no precise pathogenic role for the retriever has yet been identified. Here, we will review the neurobiology of the endosome system and its role as key player in the development of AD‐like pathology in Down syndrome. Additionally, we will discuss current knowledge on these two main endosome systems, retromer and retriever, and their potential as novel therapeutic targets. ANN NEUROL 2021;90:4–14
Down syndrome (DS), or trisomy 21, is the most common genetic abnormality resulting from a partial or complete triplication of chromosome 21 (HSA21). It affects one of every 700 births in the United States and is the leading cause of genetically defined intellectual disability. 1 Individuals with DS have a significant increased risk of developing Alzheimer disease (AD), and clinical studies estimated that by the age of 40 to 50 years more than 60% of all individuals with DS will display classical AD‐like neuropathology and cognitive decline, and for this reason they are referred as AD‐DS cases. 2 Interestingly, DS and AD share some pathophysiological aspects, as supported by genetic evidence and neuronal mechanisms. Thus, genetic alteration in DS might somewhat parallel that in AD, because an additional copy of amyloid‐β precursor protein (APP) is present on HSA21. Furthermore, the polymorphism of the presenilin‐1 and apolipoprotein E genes, two well‐known risk factors for AD, occur more frequently among mothers of DS children versus controls. 3 Regarding the neuronal mechanisms, recent evidence has clearly shown altered protein homeostasis, as reflected by protein trafficking defects, in both AD and DS. In particular, it has been reported that in brains from individuals with DS, like in AD, the endosome system is the first site of amyloid‐β (Aβ) accumulation, suggesting that the subcellular localization of APP more than its absolute levels may be critical during the early stages of AD‐DS pathogenesis. 4 Under physiological conditions, early endosomes mature into late endosomes, and the invagination of late endosomal membrane forms intraluminal vesicles (ILVs), which will then mature into multivesicular bodies (MVBs). MVBs are delivered to the lysosomes for degradation of the cargo, or to the plasma membrane and released as exosomes. 5 In DS and AD, the higher level of ILVs released as exosomes in the extracellular space is generally ascribed to the higher level of MVB production (Fig 1), 6 because early exosome blood biomarkers are found in patients with sporadic AD and DS. 7 Like in AD, DS also manifests lysosomal dysfunction, which can be characterized by acidification insufficiency, altered enzymatic activity, and decreased turnover (see Fig 1). 8 Considering that the endosomal system is responsible for protein recycling to the cell membrane or degradation to the lysosomes, in recent years its dysfunction has gained considerable attention as the potential biologic link between AD and DS. 9 A key regulator of endosome protein sorting is the retromer complex, a multimeric system responsible for retrograde transport of cargo from the endosome to the trans‐Golgi network (TGN). Initially, dysfunction of the retromer complex has been primarily involved with Aβ processing and brain amyloidosis; more recently, new evidence demonstrated that the complex is downregulated in primary tauopathy and tau transgenic mice independently from Aβ, suggesting for the first time that the system is also an active player in the development of tau pathology. 10 Similar to AD, retromer complex anomalies have also been recently reported in postmortem brain tissues and fibroblasts derived from DS subjects when compared with matched controls. 11 Although the retromer complex is a major controller of protein sorting, there are numerous cargoes, including α5β1 integrin, which do not depend on it. 12 This observation indirectly indicated for the first time the existence of an additional system/mechanism working in parallel with the classical retromer. Thus, a homologous endosome trafficking system known as the “retriever complex,” comprised of some subunits structurally equivalent to the retromer complex, has recently been identified. 13 In mammals, the retromer and retriever complexes can bind to different and common cargo subunits, which are members of the “membrane‐associated sorting nexins” (SNXs). Typically, nexins form dimers and are composed of different combinations of SNX1, SNX2, SNX5, SNX6, and SNX27, which then directly control the formation and stabilization of tubules that protrude from the endosomes. 14 Given that, similarly to AD, protein trafficking dysfunction is considered an upstream event in DS pathophysiology, it is obviously important to elucidate the mechanisms whereby the two systems, retromer and retriever, could contribute to aberrant protein trafficking and ultimately the neuropathology of the syndrome. In this review, we will summarize some of the aspects of the complex neurobiology of the endosome system, and will focus specifically on these two complexes and their biologic link(s) to the pathophysiology of DS.
FIGURE 1.
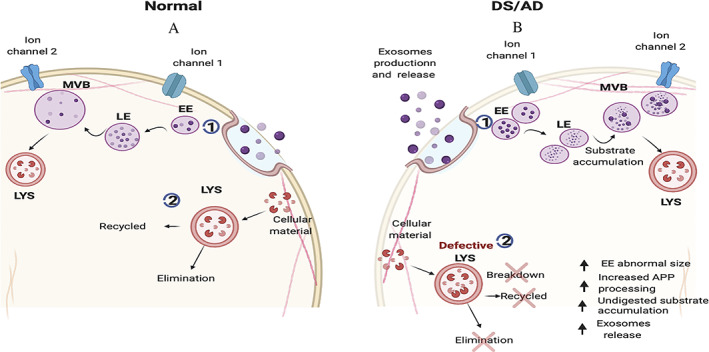
Common cellular changes detected in the central nervous system of Down syndrome (DS)/Alzheimer disease (AD) subjects compared to controls. (A) 1. Endosomal body/multivesicular body (MVB) processing in normal individuals. 2. Uncorrupted lysosome (LYS)‐mediated degradative system. (B) 1. Altered endosome pathway. Individuals with DS/AD manifest changes and impairments in cargo sorting and metabolism from early endosomes (EEs) or late endosome (LEs) to MVBs toward the degradation pathway, resulting in increased number and enlargement of EEs, LEs, and MVB, and increased exosome production and release. 2. Degradative pathway defects. Proteolysis and recycling deficit in DS/AD result from selectively impaired lysosomal degradation of cellular material. APP = amyloid‐β precursor protein. [Color figure can be viewed at www.annalsofneurology.org]
Protein Trafficking: Overview of the Endosome System
Membrane trafficking is a highly regulated process that facilitates intracellular sorting and degradation of proteins, small vesicles, lipids, and other organelles first into specific membrane carriers, and then into specific compartments to their final destination. In the central nervous system (CNS), these processes are important especially for protein turnover at the pre‐ and postsynaptic terminals, because they preserve the continuity of synthesis and degradation of different materials that ultimately ensure neuronal health and synaptic plasticity. 15
The first subcellular organelle charged with internalizing of cargoes is the early endosome (EE), a dynamic compartment enriched in phosphoinositide phosphatidylinositol 3‐phosphate. 16 The entry into the EE network begins with the endocytosis of integral membrane proteins from the cell surface through clathrin‐dependent or independent mechanisms, as well as through phagocytosis and pinocytosis. 17 A simplified view of this phenomenon shows 3 possible sorting pathways originating from EE: recycling route to the plasma membrane (PM), degradation route through the lysosome, and retrograde transport to the TGN. 18 First, the recycling route allows the transport of cargo protein from the EE to the endosome recycling compartment (ERC), composed by tubulovesicular membranes, and from the ERC to the PM. This route is divided into 2 steps: a “fast” or direct step that occurs from EEs, and a “slow” or indirect step that occurs from the ERC, both controlled by various guanosine triphosphatases (GTPases) and utilized for instance during transferrin recycling by interacting directly with Rab11 protein, a member of the GTPase family and member of the recycling pathway. In addition to Rab11, Rab35 is also present in sorting endosomes and endosomal tubules and controls trafficking between the PM and endocytic compartments (Fig 2, route I). 19 Second, cargoes targeted for lysosomal degradation are organized into the ILVs, which once formed allow endosome biogenesis from early to late forms through the endosome sorting complex required for transport. Then, the late endosome (LE) fuses with the lysosome, generating the endolysosome, which then follows a unidirectional fate of end‐dead. 20 Rab GTPases, especially the switch between Rab5 and Rab7, control diverse signaling and cell functions of the endosomes. Rab5 regulates EEs uptake and fusion, and the switch to Rab7 is required for initiation and maturation of early‐to‐late endosome; both have been shown to actively participate in cargo trafficking and lysosomal degradation (see Fig 2, route II). 21 Third, cargoes could take a different route to reach the TGN via the retrograde pathway. In the endosome‐to‐TGN trafficking, cargoes emerge from the endosomes, travel along cytoskeleton tracks, tether at, and then fuse with the TGN (see Fig 2, route III). Similar to other endosome trafficking routes, this retrograde path is highly conserved from yeast to humans and involves small GTPases, coat proteins, SNX, and SNARE proteins.
FIGURE 2.
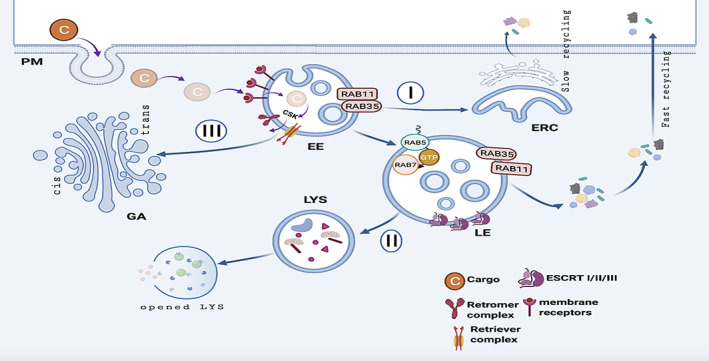
Protein trafficking routes. Extracellular proteins are targeted (as cargoes; C) to allow their internalization through plasma membrane (PM) invagination. Within the cell, the targeted cargo enters into the early endosome (EE) through membrane receptors, and is further redirected to the proper destination via 3 different routes. (I) The recycling route allows the transport of the cargo protein to the endosome recycling compartment (ERC), and from the ERC to the PM. Rab guanosine triphosphatases, such as Rab11 and Rab35, participate in the recycling. (II) Late endosome (LE) formation initiates cargo protein transport to the lysosome (LYS), where they are degraded by proteases. The process is mediated by the Rab5 to Rab7 switch, and by the endosomal sorting complex required for transport (ESCRT). (III) In the retrograde pathway, the cargo is released from the EE. The retromer and retriever complexes facilitate cargo transport to the Golgi apparatus (GA; trans network), along the cytoskeleton (CSK). GTP = guanosine triphosphate. [Color figure can be viewed at www.annalsofneurology.org]
The major player that orchestrates retrograde pathway is the retromer system (Fig 3), a heteromeric complex, consisting of 2 subcomplexes: a constitutive trimer of vacuolar protein sorting (VPS) VPS26–VPS29–VPS35, also known as the retromer recognition core, and the SNX dimers. Although VPS recognition core is thought to operate together with the SNXs within the same complex, a recent study supports that the two major components can also operate independently from each other. 22
FIGURE 3.
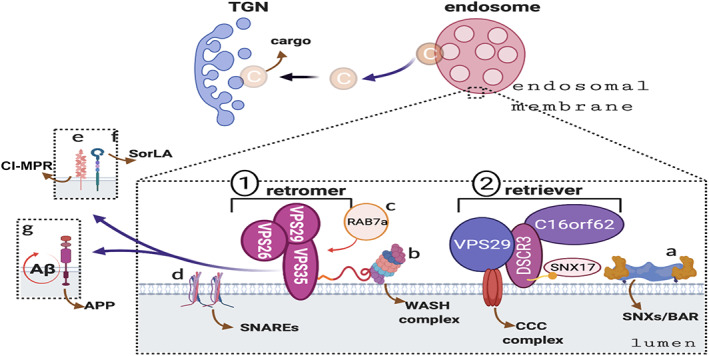
The retromer and retriever complexes. The retromer recognition core Vps35–Vps29–Vps26 trimer (1) recruits subcomplexes, such as the SNXs‐BAR (a) and WASH (b), and Rab7a (c) and SNARE (d) proteins to promote cargo (C) sorting. Targeted cargoes, including cation independent mannose 6‐phosphate receptor (CI‐MPR; e), sortilin‐related receptor (SorLa; f), and amyloid‐β (Aβ) precursor protein (APP; g), are transported from the endosome to the trans‐Golgi network (TGN) by the retromer complex. The retriever complex (DSCR3–C16orf62–VPS29) (2), associated with sorting nexin 17 (SNX17) protein, WASH, and CCC complexes, cooperates with the retromer complex to mediate the endosomal protein sorting process. [Color figure can be viewed at www.annalsofneurology.org]
As the retromer, the retriever is a functional and evolutionarily conserved multimeric complex composed of 3 main units: C16orf62, containing a HEAT‐repeat protein fold and considered the analog to the VPS35; DSCR3, a paralogue of VPS26; and VPS29, which is identical to the one present in the retromer recognition core (see Fig 3). 23 The retriever system was first identified following proteomic studies in search of proteins that potentially cooperate with the sorting nexin SNX17 in the endosome to cell surface recycling pathway. These experiments revealed that SNX17 bind and forms a stable trimer with the retriever as well as the subunits of the CCC complex, the coiled‐coil domain‐containing proteins 22 and 93, and the copper metabolism MURR1 (mouse U2af1‐rs1 region 1) domain proteins. 23 Moreover, the CCC complex requires the association of the WASH complex to orchestrate the SNX17‐retriever–dependent retrieval and recycling process. 24
Endosome System Dysfunction in DS
The endosome system is a vital component for the cell biology, because it acts as an extension of the PM in regulating cell signaling and function. Because of this important physiologic aspect, any dysfunction will have a significant impact at the cellular level and potentially influence the pathophysiology of numerous disorders. Thus, progressive and persistent endosome dysregulation leads to a corrupted and failed system of protein sorting and trafficking, which ultimately results in the accumulation of abnormal proteins and disrupted cellular proteostasis.
One of the earliest reports associating endosome dysfunction with DS is a paper in which compared with matched controls, brains from DS subjects had an abnormal accumulation of endosomes. 25 Another study demonstrated that the enlargement of endosomes could serve as an early marker for DS neuropathology, because it is frequently already detected at the fetal stage (28 weeks of gestation). 26 Although morphological changes in the endosomes have been found in postmortem brain tissues of DS patients and in the CNS of Ts65Dn mice (a transgenic mouse model of DS), 27 enlarged endosomes are also observed in the periphery, such as blood mononuclear cells and lymphoblastoid cells from DS individuals. 4 Another protein that has been associated with endosomal abnormalities in DS is synaptojanin 1 (SYNJ1), a brain‐enriched lipid phosphatase. 28 The biologic link between SYNJ1 and DS has been supported by studies showing a significant increase of enlarged endosomes in a neuroblastoma cell line (SH‐SY5Y cells) and human DS fibroblasts overexpressing this protein. 4 Additionally, SYNJ1 can be phosphorylated by a serine/threonine kinase encoded by the dual‐specificity tyrosine (Y)‐phosphorylation regulated kinase 1A (DYRK1A) gene, which is located in the critical region of HSA21. 29 Currently, major efforts are being made in developing selective DYRK1A inhibitors for therapies of many neurodegenerative disorders, including DS. To this end, a recent study showed that pharmacological inhibition of DYRK1A ameliorates behavioral deficits in Ts65Dn mice. 30 Intersectin 1 (ITSN1), another gene mapping to HSA21, has been reported to be potentially involved in endosomal abnormalities found in DS. Its product is a scaffolding protein known to regulate endocytosis, neurotransmitter release, and cell signaling. 31 , 32 ITSN1 knockout in mice, in Caenorhabditis elegans, and in Drosophila results in endosomal vesicle trafficking defects in neurons. 33 , 34 , 35 , 36 The active involvement of ITSN1 in regulating endocytosis would predict that any deregulation of its level could be deleterious for this pathway. Interestingly, a study has demonstrated that ITSN1 is upregulated in the CNS of DS patients when compared with controls. 37 Besides the enlargement of endosomes, the total number of endosomes is also considered to be a marker of its altered function in DS. The number of endosomes under physiological condition is proportional to the rate of endocytic and endosome uptake; however, because during neurodegenerative processes the number of endosomes is typically increased, this proportion is lost. 25 Interestingly, by morphometric analysis, the number of early endosome‐associated protein‐1–positive endosomes of all sizes is increased in primary fibroblast from DS patients compared to diploid controls, and the majority of them were identified particularly as large‐sized endosomes. 38 In DS retromer complex function can be also indirectly affected by changes in the expression level of the microRNA‐155 (miR155), a chromosome 21‐encoded microRNA that by negatively regulating C/EBPβ reduces levels of SNX27, an important component of the sorting nexin dimer. Interestingly, upregulating SNX27 in the hippocampus of DS mice rescues synaptic and cognitive deficits, whereas in the CNS of DS patients miR155 upregulation is strongly correlated with dementia (Table1). 39 , 40
TABLE 1.
Key Findings on the Retromer System Biology in DS
| Retromer Correlated Function | Pathological Processing | Finding | |
|---|---|---|---|
| VPS35/26/29 | Core recognition component |
Reduced expression |
Human brain from DS individuals 11 |
| SNX27 | Endosome‐associated cargo adaptor |
Reduced expressions Axonal demyelination |
Hippocampus of DS mice 39 Ts65Dn mice 41 |
| Rab5 and Rab7 | VPS subcomplex regulators |
Rab7 colocalization with p62/LC3B Large rab5‐positive endosomes |
DS fibroblast cell lines 42 Fibroblast from DS individuals 25 |
| Sorl1 | Retromer complex cargo |
Various genetic variants Decreased transport of NGF |
AD individuals with DS 43 Ts65Dn DS mice 44 |
| Lysosome | Component of cellular homeostasis |
Decreased lysosomal turnover Increased lysosomal hydrolases |
Ts2 mice and fibroblast from DS subjects 45 DS fibroblasts 25 |
| Exosome | Endosomal pathway process |
Upregulated secretion Compromised exosome release |
Ts2 mice 6 Human and murine DS brains 46 |
AD = Alzheimer disease; DS = Down syndrome; NGF: nerve growth factor; VPS = vacuolar protein sorting.
Moreover, recent data have suggested a potential link between endosome dysfunction and abnormalities in exosome secretion, a phenomenon that can be easily assessed by measuring the expression levels of CD63, a marker associated with membranes of intracellular vesicles, and the exosome‐related protein Rab35. Thus, it has been shown that frontal cortices of DS patients and brains from 12‐month‐old Ts2 mice express much higher levels of CD63 and Rab35 compared with controls (see Table1). 46
Endosomal–Lysosomal System in DS
The delivery of ubiquitinated cargoes to lysosomal acid hydrolases for degradation occurs after transient or complete fusion events occur between LE and the lysosomes, which ultimately form hybrid organelles, called endolysosomes. This is considered the main intracellular site of the endolysosomal system, and comprises of a series of dynamic membranous organelles specialized for regulating intracellular trafficking but most importantly cellular proteostasis. 47 Although decades of research in non‐neuronal cells have revealed important regulatory mechanisms for the endolysosomal trafficking system, 48 only recently studies have begun to unveil remarkably complex and spatially compartmentalized mechanisms regulating this system within the CNS. 49 Interestingly, upregulated endocytosis and progressive endolysosomal disruption have been considered to be emerging characteristics of DS neuropathology. 50 Some evidence suggests that an early dysfunction in the lysosomal degradative capacity could be dependent on the third copy of the APP gene on HSA21, because the excessive amount of endogenous APP would favor the conversion of normal lysosomal function into lysosomal disruption. However, defects of lysosomal lumen such as its insufficient acidification, cathepsin D (CTSD) inactivation, and altered hydrolases activity could also contribute to the deregulated endolysosomal system in DS independently from the extra copy of APP. The maintenance of acidic pH (4.2–5.3) is essential for regulating many functions of lysosomes as well as for CTSD to operate optimally. However, both parameters are altered in human DS fibroblasts and primary cortical neurons from Ts2 mice, reflecting an abnormal turnover of endocytic substrates (see Table1). 45 , 51
Retromer and Retriever in DS: In Vivo Evidence
As mentioned earlier, endocytic trafficking involves a dynamic and continuous maturation of specific endosomal compartments, with some cargos predominantly sorted at the EE and others at the LE level. Regarding protein trafficking processes, the retromer complex is the most appreciated regulator that drives the endosome‐to‐TGN protein sorting pathway.
Interestingly, the complex is directly involved in the transport of several important cargo proteins that have been linked and considered relevant to the risk of developing AD in DS subjects. Among them SorLa (sortilin‐related receptor), the endocytic receptor for APP, and the cation‐independent mannose 6‐phosphate receptor (CI‐MPR), which transports the lysosomal protease CTSD, were found downregulated in cortices from young DS individuals compared to controls (see Table1). 11 Moreover, a variant of this receptor has been found in association with AD pathology in adults with DS.48 Another protein cargo, the beta‐secretase 1 (BACE1) enzyme, a transmembrane protein involved in APP cleavage rapidly internalized from the cell surface and transported to EE where it can be recycled by the retromer complex, has also been reported to be upregulated in DS. 52 Interestingly, in addition to the retromer, a clathrin adaptor Golgi‐localized γ‐ear‐containing ARF binding protein 3 (GGA3) has recently been shown to play a key role in the sorting and trafficking of BACE1 to the lysosomes, where it is normally degraded. GGA3 depletion results in BACE1 accumulation both in vitro and in vivo, its levels are reduced and inversely related to BACE1 levels in postmortem brains of AD patients, and a GGA3 rare variant cosegregates with increased risk for late onset AD. 53
Low‐density lipoprotein receptor‐related protein 1 (LRP1) is a type I transmembrane glycoprotein. It has been demonstrated that LRP1 can affect APP trafficking and processing through APP binding interactions with its extracellular and intracellular domains. 20 , 21 , 22
Finally, the α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptors (AMPARs), a group of cation‐permeable ionotropic glutamate receptors involved in synaptic transmission, are internalized at the level of the EE in hippocampal neurons. 54 These receptors use the retromer complex for basal dendritic trafficking 55 , 56 and have been reported to be significantly altered in the hippocampus of Ts65Dn mice. 57
The retromer is expressed in multiple tissues but plays a noticeable role in the CNS, where its dysfunction and/or altered expression of its components have been linked to several neurodegenerative diseases. Thus, studies showed that expression of the retromer recognition core is downregulated in AD brain, and contributes to amyloidogenic APP processing and Aβ generation, whereas in vitro studies showed that its downregulation increases Aβ formation. Additionally, genetic reduction of VPS35 in Tg2576 mice, a model of AD‐like brain amyloidosis, results in higher levels of Aβ peptides and plaques, cognitive impairments, and synaptic dysfunction. 58 Levels of VPS35 and the other 2 components of the retromer recognition core module (i.e., VPS26 and VPS29) are decreased in an age‐ and region‐specific manner in the same mouse model, suggesting early involvement of the system in the onset and development of the AD‐like phenotype. 59 Importantly, a gain of function of VPS35 in the CNS of triple transgenic AD mice rescued their phenotype, including amelioration of long‐term memory loss, lower Aβ levels, and decrease in pathological tau. On the other hand, downregulation of VPS35 results in worsening of the phenotype in a transgenic mouse model of tauopathy, P301S transgenic mice. 6
Interestingly, dysregulation of VPS35 has also been linked to early and late onset Parkinson disease (PD), the most common movement disorder, and similar to AD, the consensus is that a loss of function in the retromer complex is responsible for it. PD was first linked to the retromer complex in 2011, when exome sequencing revealed a mutation in residue 620 (D620N) in the VPS35 orthologue. 60 Additionally, postmortem analysis of brain tissues revealed decreased protein levels of VPS35 in the substantia nigra of PD patients. 61 Using a Drosophila model, one study found that RNAi‐mediated Vps35 knockdown led to aberrant alpha‐synuclein degradation in the lysosome, specifically, via impaired delivery of CTSD to the lysosome.38 In cellular models, studies indicate that D620N VPS35 mutation resulted in defective CI‐MPR sorting as well as altered localization of the AMPARs, indicating a link between retromer and postsynaptic transmission. 62
Given that EE enlargement is a key feature of DS, in a recent study we examined the status of the retromer complex system in DS using both postmortem brain tissues and patient‐derived fibroblasts from individuals with DS and unaffected controls. To start establishing a temporal profile of retromer dysregulation within the context of the evolution of DS phenotype, we investigated the retromer core and associated proteins in young patients prior to significant Aβ deposition (age 15–30 years), and older subjects (age 40–65 years) at a time when virtually all the individuals with DS display Aβ plaques. VPS35 and VPS29 were significantly decreased in the cortex, and all 3 recognition core proteins were reduced in the hippocampus of the oldest subgroup of DS patients when compared to controls (see Table1). Surprisingly, we found that retromer deficiency was not primarily age‐related in DS subjects. Dysregulation of the retromer complex was also observed in younger patient subgroups, suggesting that the dysfunction may begin prior to full development of AD‐like pathology and cognitive symptoms in DS. Interestingly, in both young and aged subjects, retromer depletion was most evident in the hippocampus. Additionally, we examined the relationship between retromer proteins and both soluble and insoluble Aβ 1–40 and Aβ 1–42 peptide levels for all subjects. Although reduction of retromer proteins seems to precede Aβ deposition, we found that the degree of retromer depletion for all retromer core proteins significantly and inversely correlates with the accumulation of both Aβ peptides in cortex and hippocampus. Although we recognize that the observed inverse correlation between retromer and Aβ does not necessarily indicate causation, we believe that our finding further supports the evidence that in DS retromer dysfunction and neurodegeneration are closely associated. 7 Although many SNX components have been shown to regulate intracellular trafficking, SNX27 is the only one comprising a cargo‐binding domain, involved in endosomal trafficking, highly expressed in the CNS and colocalized with EE and recycling endosomes. Interestingly, it has been shown that in DS upregulation of the miR155, which is encoded in HSA21, results in a significant reduction of SNX27 expression. This observation supports the hypothesis of a possible involvement of this nexin in DS pathogenesis, because SNX27 normally interacts with the PS‐1 of the γ‐secretase complex and inhibits APP cleavage and Aβ generation. 39 Moreover, lower levels of SNX27 were reported in the brains of Ts65Dn mice (see Table1). 63 In addition to SNX27, some other retromer components may also be associated with DS. 10 DS critical region 3 (DSCR3), a gene located in HSA21, is upregulated in adult DS brain. 64 DSCR3 is considered to be a VPS26 analog, as it adopts an arrestinlike fold as found in the VPS26 family, and is a major component of the retriever complex. The retriever, together with the WASH complex, can also regulate SNX17‐mediated sorting of different proteins, 23 some of which are involved in synaptic plasticity and Aβ clearance. 65 , 66 Although it is known that the retriever and the retromer share similarities in their structure, evidence of direct interaction between them is currently lacking, and further investigation is required to study a potential biologic link between the two systems, and the role of their concurrent dysfunction in DS pathogenesis.
Retromer and Retriever as Therapeutic Targets
From the data presented, it is evident that overall CNS health is particularly dependent on the normal function of this complex endosomal system and highly sensitive to its dysfunction. Interestingly, so far, all the pathological processes secondary to these abnormalities have been linked to a loss rather than a gain in retromer complex system function. 67 , 68 The balance between cargo degradation and recycling involves the integrity of the whole retromer complex as a functional unit, in which VPS35 has emerged as the central scaffold responsible for the structural assembly and stability of the entire complex. Thus, VPS35 could be considered a novel therapeutic target, given that its loss would induce pathology, and restoring defects by increasing its level would normalize protein sorting and trafficking processes and ultimately halt or delay disease progression. 69 , 70 , 71 Similarly, the upregulation of the retriever complex system could be achieved not only by exogenous molecules as pharmacological targets but also through reinforcement of endogenous proteins directly interacting with the retriever complex. 72
Taking into consideration all the available data, particularly on the retromer complex (i.e., VPS35) in diseases such as AD, PD, and DS it is obvious that although we do not know in their entirety the precise molecular mechanisms involved in each of them, we know that at least so far, they are all characterized by a loss rather than a gain of function. With this concept in mind, it is easy to envision a condition in which a recovery of the retromer function will potentially have a therapeutic effect for these diseases. To this end, early work showed that in a Drosophila model of PD, mutant LRRK2 flies, overexpression of the VPS35 or VPS26 ameliorates the eye phenotype, significantly protects from the locomotor deficits, and rescues their shortened lifespan. 68 Moreover, in an animal model of AD with amyloid beta plaques and tau tangles, the gain of function of the retromer complex achieved by genetically overexpressing VPS35 in the CNS rescues the entire AD‐like phenotype. 73 Interestingly, in both cases, no evidence of any cytotoxic effect was reported.
Recently, pharmacological chaperones, small molecules that selectively target, bind, and stabilize a protein and increase its cellular steady‐state concentration, have emerged as potential facilitators of cargoes trafficking. The possible application of pharmacological chaperones to target a multiprotein complex such as the retromer represents a new idea, because originally it was not considered that this type of drug could bind to a complex that does not have an allosteric site. A recent paper has identified and then validated 2 pharmacological chaperones named R55 and R33 (both thiophene thiourea derivates), which were able to stabilize the retromer recognition core against thermal denaturation. 74 Subsequent in vitro studies showed that these compounds upregulate endogenous levels of VPS35 and other major components of the recognition core, resulting in an increase of APP trafficking out of the endosomes and reduced Aβ formation. 59 Interestingly, other reports have also demonstrated that these drugs can modulate levels of tau protein and its phosphorylated isoforms in human induced pluripotent stem cell (iPSC)‐derived neurons 75 and neuronal cells stably expressing all 6 tau isoforms (N2A‐Htau cells) 10 independently from any effect on APP and Aβ. On the other hand, a recent preclinical study using the 3xTg AD mouse model has provided evidence for the efficacy of chronic treatment with a pharmacologic chaperone against the onset of the AD‐like phenotype (Aβ, tau, and behavioral impairments). 76
The specific mechanism of action of many pharmacological chaperones could represent an advantage from a therapeutic point of view for several diseases including DS if one considers also their efficacy at low concentrations, lack of off‐target effects, and good bioavailability. 77 , 78
Therefore, the possibility of targeting the retromer and retriever complex systems by implementing a pharmacological chaperone approach in DS is now becoming not only an interesting but also a feasible opportunity. This approach will be a significant departure from previous ones that have been implemented in DS as well as other neurologic diseases. Whereas most of the therapeutic strategies adopted so far have aimed at a specific protein or enzyme that is often considered part of a metabolic pathway or regulatory feedback, the pharmacological chaperone approach will, by contrast, target a cellular mechanism that is universal and at the base of cell health and homeostasis.
Conclusions and Future Directions
Based on the available literature, consistent evidence clearly points out that, like in AD, one of the earliest neuropathologic features of DS are abnormalities of protein sorting and trafficking, a process in which the endosomal system functions as a master regulator. Although in recent years significant progress in this field has been made, and important basic new knowledge on the physiopathology of these processes has been gained, several areas remain to be investigated; this underscores an incomplete understanding of the neurobiology of the endosomal system, its regulatory mechanisms, and changes that occur during the disease pathogenesis. Identification of some of the mechanisms involved in retromer system dysfunction and DS has helped us to move the field forward. However, the recent discovery of the retriever, an additional system central to sorting and trafficking of proteins and of which we know very little, has added an extra layer of complexity and challenge for this area of research.
Although useful model systems such as murine models of DS (Ts2 and Ts65Dn mice), human fibroblasts, and neurons derived from DS iPSCs are widely available and are allowing the elucidation and exploration of unchartered endosomal mechanisms and pathways, we have to recognize that all of them have some limitations, and there is an urgent need for better ones.
Future investigation of the endosomal system in DS should focus not only on the cell biology of these two major systems, but also on the accurate characterization of other aspects such as cell type contribution and brain region distribution and relative roles for each of them. Moreover, work should focus on some open questions, such as the following: can one system (ie, retromer) compensate for the deficit of the other (ie, retriever)? Is the effect of their simultaneous dysfunction synergistic/additive or independent? Because they share a subunit (ie, VPS29), will targeting one have an effect on the other?
We believe that addressing these questions not only will shed important new light on these two systems and their interplay as early and active contributors to DS pathogenesis, but most importantly will provide a new therapeutic framework for DS patients.
Author Contributions
Both authors contributed to the conception and design of the review, the interpretation of studies included in the review, and drafting the text: A.F. contributed to preparing the figures and the table.
Potential Conflicts of Interest
Nothing to report.
Acknowledgments
D.P. is the Scott Richards North Star Charitable Foundation Chair for Alzheimer's Research. Some of the work described in the article and performed in the laboratory of the corresponding author was supported by grants from the NIH, National Instititute on Aging (NIA) (AG055707 and AG056689) and the Pennsylvania Department of Health Commonwealth Universal Research Enhancement program (4100083099).
References
- 1. Parker SE, Mai CT, Canfield MA, et al. Updated national birth prevalence estimates for selected birth defects in the United States, 2004–2006. Birth Defects Res A Clin Mol Teratol 2010;88:1008–1688. [DOI] [PubMed] [Google Scholar]
- 2. Hithersay R, Startin CM, Hamburg S, et al. Association of dementia with mortality among adults with Down syndrome older than 35 years. JAMA Neurol 2019;76:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhaumik P, Ghosh P, Ghosh P, et al. Combined association of presenilin‐1 and apolipoprotein E polymorphisms with maternal meiosis II error in Down syndrome births. Genet Mol Biol 2017;40:577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wiseman FK, Pulford L, Barkus C, et al. Trisomy of human chromosome 21 enhances amyloid‐b deposition independently of an extra copy of APP. Brain 2018;141:2457–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Perez‐Gonzalez R, Gauthier SA, Kumar A, Levy E. The exosome secretory pathway transports amyloid precursor protein carboxyl‐terminal fragments from the cell into the brain extracellular space. J Biol Chem 2012;287:43108–43115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. D'Acunzo P, Hargash T, Pawlik M, et al. Enhanced generation of intraluminal vesicles in neuronal late endosomes in the brain of a Down syndrome mouse model with endosomal dysfunction. Dev Neurobiol 2019;79:656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hamlett E, Ledreux A, Potter H, et al. Exosomal biomarkers in Down syndrome and Alzheimer's disease. Free Radic Biol Med 2018;114:110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lie PP, Nixon RA. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol Dis 2019;122:94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choy RW‐Y, Chen Z, Schekman R. Amyloid precursor protein traffics from the cell surface via endosomes for amyloid beta production in the trans‐Golgi network. Proc Natl Acad Sci U S A 2012;109:E2077–E2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vagnozzi A, Li JG, Chiu J, et al. VPS35 regulates tau phosphorylation and neuropathology in tauopathy. Mol Psychiatry (in press). 10.1038/s41380-019-0453-x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11. Curtis ME, Yu D, Praticò D. Dysregulation of the retromer complex system in Down syndrome. Ann Neurol 2020;88:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McNally KE, Faulkner R, Steinberg F, et al. Retriever, a multiprotein complex for retromer‐independent endosomal cargo recycling. Nat Cell Biol 2017;19:1214–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang J, Fedoseienko A, Chen B, et al. Endosomal receptor trafficking: retromer and beyond. Traffic 2018;19:578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang H, Huang T, Hong Y, et al. The retromer complex and sorting nexins in neurodegenerative diseases. Front Aging Neurosci 2018;10:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO J 2017;36:1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dickson EJ, Hille B. Understanding phosphoinositides: rare, dynamic, and essential membrane phospholipids. Biochem J 2019;476:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mayor S, Parton RG, Donaldson JG. Clathrin‐independent pathways of endocytosis. Cold Spring Harb Perspect Biol 2014;6:a016758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martínez‐Núñez L, Munson M. Retro is cool: structure of the versatile retromer complex. Structure 2020;28:387–389. [DOI] [PubMed] [Google Scholar]
- 19. Kouranti I, Sachse M, Arouche N, et al. Rab35 regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis. Curr Biol 2006;16:1719–1725. [DOI] [PubMed] [Google Scholar]
- 20. Huotari J, Heleniusa A. Endosome maturation. EMBO J 2011;30:3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chavrier P, Parton RG, Hauri HP, et al. Comparative study cell localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell 1990;62:317–329. [DOI] [PubMed] [Google Scholar]
- 22. Kvainickas A, Jimenez‐Orgaz A, Nägele H, et al. Cargo‐selective SNX‐BAR proteins mediate retromer trimer independent retrograde transport. J Cell Biol 2017;216:3677–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McNally KE, Cullen PJ. Endosomal retrieval of cargo: retromer is not alone. Trends Cell Biol 2018;28:807–822. [DOI] [PubMed] [Google Scholar]
- 24. Phillips‐Krawczak CA, Singla A, Starokadomskyy P, et al. COMMD1 is linked to the WASH complex and regulates endosomal trafficking of the copper transporter ATP7A. Mol Biol Cell 2015;26:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cataldo A, Mathews PM, Boiteau AB, et al. Down syndrome fibroblast model of Alzheimer‐related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am J Pathol 2008;173:370–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nixon RA. Amyloid precursor protein and endosomal‐lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease. FASEB J 2017;31:2729–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marland JRK, Smillie KJ, Cousin MA. Synaptic vesicle recycling is unaffected in the Ts65Dn mouse model of Down syndrome. PLoS One 2016;11:e0147974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cossec J, Lavaur J, Berman D, et al. Trisomy for synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum Mol Genet 2012;21:3156–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Papoulidis I, Papageorgiou E, Siomou E, et al. A patient with partial trisomy 21 and 7q deletion expresses mild Down syndrome phenotype. Gene 2014;536:441–443. [DOI] [PubMed] [Google Scholar]
- 30. Nguyen TL, Duchon A, Manousopoulou A, et al. Correction of cognitive deficits in mouse models of Down syndrome by a pharmacological inhibitor of DYRK1A. Dis Model Mech 2018;11::dmm035634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hunter MP, Russo A, O'Bryan JP. Emerging roles for intersectin (ITSN) in regulating signaling and disease pathways. Int J Mol Sci 2013;14:7829–7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Bryan JP. Intersecting pathways in cell biology. Sci Signal 2010;3:re10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu Y, Chu P, Bowser D, et al. Mice deficient for the chromosome 21 ortholog Itsn1 exhibit vesicle‐trafficking abnormalities. Hum Mol Genet 2008;17:3281–3290. [DOI] [PubMed] [Google Scholar]
- 34. Wang W, Bouhours M, Gracheva EO, et al. ITSN‐1 controls vesicle recycling at the neuromuscular junction and functions in parallel with DAB‐1. Traffic 2008;9:742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rose S, Malabarba MG, Krag C, et al. Caenorhabditis elegans intersectin: a synaptic protein regulating neurotransmission. Mol Biol Cell 2007;18:5091–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koh TW, Verstreken P, Bellen HJ. Dap160/intersectin acts as a stabilizing scaffold required for synaptic development and vesicle endocytosis. Neuron 2004;43:193–205. [DOI] [PubMed] [Google Scholar]
- 37. Herrero‐Garcia E, O'Bryan J. Intersectin scaffold proteins and their role in cell signaling and endocytosis. Biochim Biophys Acta Mol Cell Res 2017;1864:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cataldo A, Petanceska S, Peterhoff CM, et al. App gene dosage modulates endosomal abnormalities of Alzheimer's disease in a segmental trisomy 16 mouse model of Down syndrome. J Neurosci 2003;23:6788–6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang X, Zhao Y, Zhang X, et al. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat Med 2013;19:473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tili E, Mezache L, Michaille JJ, et al. MicroRNA 155 up regulation in the CNS is strongly correlated to Down's syndrome dementia. Ann Diagn Pathol 2018;34:103–109. [DOI] [PubMed] [Google Scholar]
- 41. Olmos‐Serrano JL, Kang HJ, Tyler WA, et al. Down syndrome developmental brain transcriptome reveals defective oligodendrocyte differentiation and myelination. Neuron 2016;89:1208–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aivazidis S, Jain A, Anderson C, et al. Down syndrome fibroblasts exhibit diminished autophagic clearance and endosomal dysfunction after serum starvation. bioRxiv 2018. 10.1101/436782. [DOI] [Google Scholar]
- 43. Lee JH, Chulikavit M, Pang D, et al. Association between genetic variants in sortilin‐related receptor 1 (SORL1) and Alzheimer's disease in adults with Down syndrome. Neurosci Lett 2007;425:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cooper JD, Salehi A, Delcroix JD, et al. Failed retrograde transport of NGF in a mouse model of Down's syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc Natl Acad Sci U S A 2001;98:10439–10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ying J, Sato Y, Im E, et al. Lysosomal dysfunction in Down syndrome is APP‐dependent and mediated by APP‐βCTF (C99). J Neurosci 2019;39:5255–5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gauthier S, Pérez‐González R, Sharma A, et al. Enhanced exosome secretion in Down syndrome brain—a protective mechanism to alleviate neuronal endosomal abnormalities. Acta Neuropathol Commun 2017;5:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Winckler B, Faundez V, Maday S, et al. The endolysosomal system and proteostasis: from development to degeneration. J Neurosci 2018;38:9364–9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cui Y, Carosi JM, Yang Z, et al. Retromer has a selective function in cargo sorting via endosome transport carriers. J Cell Biol 2019;218:615–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kiral FR, Kohrs FE, Jin EJ, Hiesinger PR. Rab GTPases and membrane trafficking in neurodegeneration. Curr Biol 2018;28:R471–R486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gomez W, Morales R, Maracaja‐Coutinho V, et al. Down syndrome and Alzheimer's disease: common molecular traits beyond the amyloid precursor protein. Aging 2020;12:1011–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perluigi M, Pupo G, Tramutola A, et al. Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim Biophys Acta 2014;1842:1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nistor M, Don M, Parekh F, et al. Alpha‐ and beta‐secretase activity as a function of age and beta‐amyloid in Down syndrome and normal brain. Neurobiol Aging 2007;28:1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lomoio S, Willen R, Kim W, et al. Gga3 deletion and a GGA3 rare variant associated with late onset Alzheimer's disease trigger BACE1 accumulation in axonal swellings. Sci Transl Med 2020;12:eaba1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hanley JG. Endosomal sorting of AMPA receptors in hippocampal neurons. Biochem Soc Trans 2010;38:460–465. [DOI] [PubMed] [Google Scholar]
- 55. Tian Y, Tang FL, Sun X, et al. VPS35‐deficiency results in an impaired AMPA receptor trafficking and decreased dendritic spine maturation. Mol Brain 2015;8:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Temkin P, Morishita W, Goswami D, et al. The retromer supports AMPA receptor trafficking during LTP. Neuron 2017;94:74–82. [DOI] [PubMed] [Google Scholar]
- 57. Kameyama K, Lee HK, Bear MF, Huganir RL. Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long‐term depression. Neuron 1998;21:1163–1175. [DOI] [PubMed] [Google Scholar]
- 58. Wen L, Tang FL, Hong W, et al. VPS35 haploinsufficiency increases Alzheimer's disease neuropathology. J Cell Biol 2011;195:765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chu J, Praticò D. The retromer complex system in a transgenic mouse model of AD: influence of age. Neurobiol Aging 2017;52:32–38. [DOI] [PubMed] [Google Scholar]
- 60. Williams ET, Chen X, Moore DJ. VPS35, the retromer complex and Parkinson's disease. J Parkinsons Dis 2017;7:219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vilarino‐Guell C, Wider C, Ross O, et al. VPS35 mutations in Parkinson's disease. Am J Hum Genet 2011;89:162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. MacLeod DA, Rhinn H, Kuwahara T, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron 2013;77:425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Meraviglia V, Ulivi AF, Boccazzi M, et al. SNX27, a protein involved in Down syndrome, regulates GPR17 trafficking and oligodendrocyte differentiation. Glia 2016;64:1437–1460. [DOI] [PubMed] [Google Scholar]
- 64. Lockstone HE, Harris LW, Swatton JE, et al. Gene expression profiling in the adult Down syndrome brain. Genomics 2007;90:647–660. [DOI] [PubMed] [Google Scholar]
- 65. Matter ML, Zhang Z, Nordstedt C, Ruoslahti E. The alpha5beta1 integrin mediates elimination of amyloid‐beta peptide and protects against apoptosis. J Cell Biol 1998;141:1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jongewaard IA, Lauer R, Behrendt D, et al. Beta 1 integrin activation mediates adhesive differences between trisomy 21 and non‐trisomic fibroblasts on type VI collagen. Am J Med Genet 2002;109:298–305. [DOI] [PubMed] [Google Scholar]
- 67. Seaman MN, Marcusson EG, Cereghino JL, Emr SD. Endosome to Golgi retrieval of the vacuolar protein sorting receptor, Vps10p, requires the function of the VPS29, VPS30, and VPS35 gene products. J Cell Biol 1997;137:79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Linhart R, Wong SA, Cao J, et al. Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson's disease mutant of leucine‐rich repeat kinase 2 (LRRK2). Mol Neurodegener 2014;9:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tang F, Zhao L, Zhao Y, et al. Coupling of terminal differentiation deficit with neurodegenerative pathology in Vps35‐deficient pyramidal neurons. Cell Death Differ 2020;27:2099–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Small S, Simoes‐Spassov S, Mayeux R, Petsko G. Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer's disease. Trends Neurosci 2017;40:592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Berman DE, Ringe D, Petsko G, Small S. The use of pharmacological retromer chaperones in Alzheimer's disease and other endosomal‐related disorders. Neurotherapeutics 2015;12:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Baños‐Mateos S, Adriana L, Hierro RA. VPS29, a tweak tool of endosomal recycling. Curr Opinion Cell Biol 2019;59:81–87. [DOI] [PubMed] [Google Scholar]
- 73. Li JG, Chiu J, Praticò D. Full recovery of the Alzheimer's disease phenotype by gain of function of vacuolar protein sorting 35. Mol Psychiatry 2020;25:2630–2640. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74. Mecozzi V, Berman D, Simoes S, et al. Pharmacological chaperones stabilize retromer to limit APP. Nat Chem Biol 2014;10:443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Young JE, Fong LK, Frankowski H, et al. Stabilizing the retromer complex in a human stem cell model of Alzheimer's disease reduces tau phosphorylation independently of amyloid precursor protein. Stem Cell Rep 2018;10:1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li JG, Chiu J, Ramanjulu R, et al. A pharmacological chaperone improves memory by reducing Aβ and tau neuropathology in a mouse model with plaques and tangles. Mol Neurodegener 2020;15:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bernier V, Lagacé M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab 2004;15:222–228. [DOI] [PubMed] [Google Scholar]
- 78. Syed Haneef SA, Priya Doss CG. Personalized pharmacoperones for lysosomal storage disorder: approach for next‐generation treatment. Adv Protein Chem Struct Biol 2016;102:225–265. [DOI] [PubMed] [Google Scholar]