

Abstract
Aberrant activation of the Wnt/β-catenin signaling circuit is associated with cancer recurrence and relapse, cancer invasion and metastasis, and cancer immune evasion. Direct targeting of β-catenin, the central hub in this signaling pathway, is a promising strategy to suppress hyperactive β-catenin signaling but has proven to be highly challening. Substantial efforts have been made to discover compounds that bind with β-catenin, block β-catenin-mediated protein–protein interactions (PPIs), and suppress β-catenin signaling. Herein, we characterize potential small-molecule binding sites in β-catenin, summarize bioactive small molecules that directly target β-catenin, and review structure-based inhibitor optimization, structure-activity relationship (SAR), and biological activities of reported inhibitors. This knowledge will benefit future inhibitor development and β-catenin-related drug discovery.
Keywords: Wnt/β-catenin signaling, Protein–protein interaction, β-Catenin, TCF, BCL9, Binding site analysis, Small-molecule inhibitor
Introduction
The Wnt/β-catenin signaling pathway plays important roles in regulating embryogenesis, stem cell renewal, and tissue maintenance.1–3 β-Catenin is the central mediator of this pathway, and Wnt/β-catenin signaling is balanced through a precise control of β-catenin levels in the cytosol (Figure 1).4 Without a Wnt signal, cytosolic β-catenin is actively phosphorylated by a destruction complex comprising adenomatous polyposis coli (APC), Axin, glycogen synthase kinase 3β (GSK3β), casein kinase 1α (CK1α), and protein phosphatase 2A (PP2A).5 Within this destruction complex APC and Axin serve as the scaffold that facilitates phosphorylation of β-catenin at residue S45 by CK1α and phosphorylation at residues S33, S37, and T41 by GSK3β. These phosphorylation events drive β-catenin to undergo ubiquitination and proteasome degradation. As a result, only a minimal amount of β-catenin are maintained in unstimulated cells.6 Upon binding of a Wnt ligand to a member of the Frizzled (Fzd) family of G-protein-coupled receptors (GPCRs) and one of two low density lipoprotein receptor-related proteins 5 and 6 (Lrp5/6), Axin and the adaptor protein Dishevelled (Dvl) will be recruited to this membrane-anchored protein complex, resulting in disassembly of the destruction complex. This allows β-catenin to be stabilized into the dephosphorylated state, accumulated in the cytoplasm, and translocated into the cell nucleus, where the active, unphosphorylated β-catenin displaces co-repressor Groucho/transducin-like enhancer of split (TLE) from the T-cell factor (TCF) and lymphoid enhancer-binding factor (LEF) family of transcriptional factors, and recruits co-activators B-cell lymphoma 9 (BCL9) or BCL9-like (BCL9L), Pygopus (Pygo 1 or Pygo 2), CREB-binding protein (CBP)/p300, and among others to transcribe Wnt/β-catenin target genes (Figure 1).7–13 In addition to its involvement in Wnt signaling, β-catenin acts as a structural component of adherens junctions, where it binds to the cytoplasmic domain of E-cadherin to recruit and organize actin filaments.14 This pool of β-catenin in most case is highly stable and not involved in the Wnt pathway-related machinery.15
Figure 1.
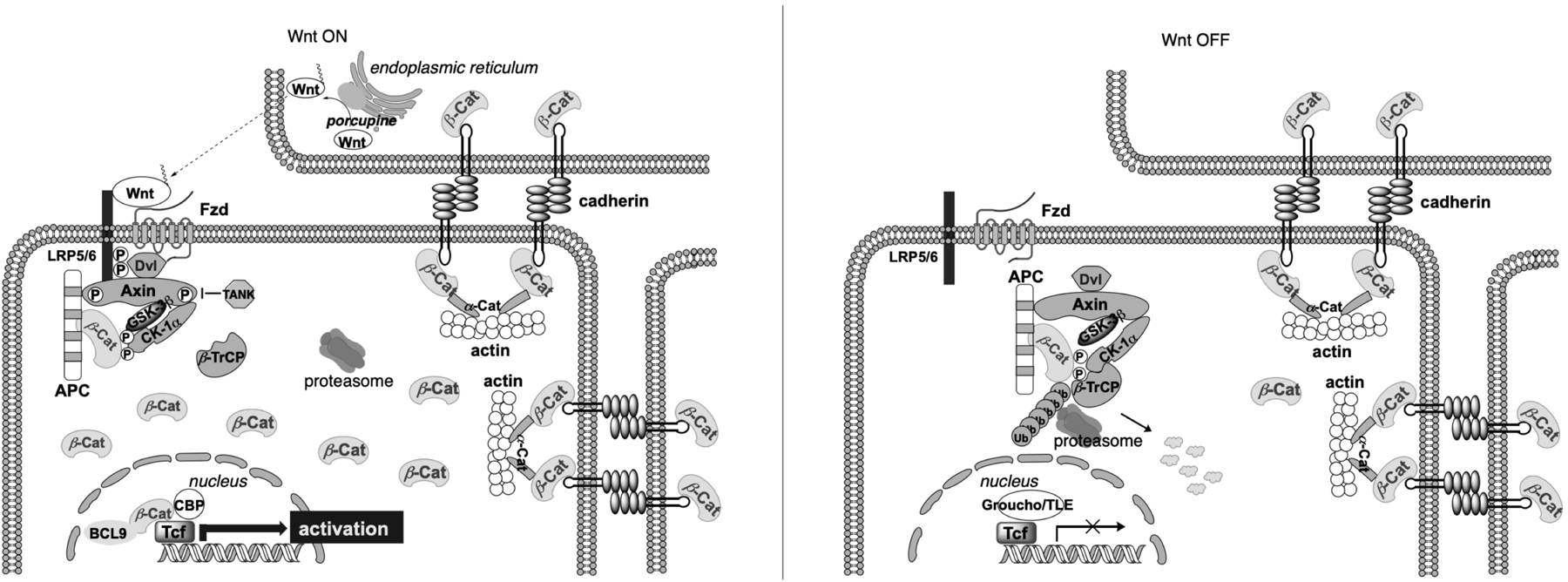
Overview of the Wnt/β-catenin signaling pathway in its on and off state. β-cat: β-catenin.
Hyperactive Wnt/β-catenin signaling is implicated in initiation and progression of various types of cancer, such as colorectal cancer (CRC), noncolorectal gastrointestinal cancer, lung cancer, prostate cancer, leukemia, and breast cancer. In most cases, oncogenic activation of Wnt/β-catenin signaling is triggered by inactivation mutations of the components of the destruction complex (e.g., APC and Axin), or activation mutations at the N-terminal phosphorylation domain of the β-catenin gene CTNNB1,16–26 driving transcription of Wnt/β-catenin target genes that induce cancer cell epithelial-to-mesenchymal transition (EMT), activate self-renewal of stem-like cancer cells, promote tumor cell invasion and metastasis, confer treatment resistance, and foster tumor immune evasion. Autocrine/paracrine activation of the upstream effectors (Fzd, Dvl, and Wnt ligands) of the Wnt/β-catenin pathway and epigenetic silence of Wnt antagonist genes also causes hyperactivation of this signaling pathway and has been recorded in many types of cancers.27–34 Several proof-of-concept studies targeting this pathway for potential treatment of cancer have been conducted. Restoration of APC was shown to promote cellular differentiation and reestablish crypt homeostasis in CRC.35 Enforced expression of Axin suppressed proliferation and self-renewal capacity of chronic myelogenous leukemia (CML) cells that bear activated β-catenin.36 Dicer-substrate siRNAs-mediated β-catenin knockdown reduces liver tumor burden in vivo.37 In addition, collective research data have indicated that β-catenin signaling mediates immune escape of cancer cells and resistance to checkpoint blockade immunotherapies.38–44 These studies have demonstrated that suppression of Wnt/β-catenin signaling offers a new approach toward anticancer therapies.
Substantial efforts have been made to target this pathway by developing therapeutic agents against the upstream effectors. OMP-18R5 (Vantictumab),45 a monoclonal antibody against Fzd receptors, has completed phase I clinical trials (Clinicaltrials.gov IDs: NCT01345201, NCT02005315, NCT01957007, and NCT01973309). OMP-54F28,46 a recombinant protein of the Fzd 8 cysteine-rich domain fused to the Fc part of immunoglobulin, acts as a decoy receptor and binds with all Wnt ligands to suppress Wnt/β-catenin signaling. OMP-54F28 is also ready for phase II clinical trials (Clinicaltrials.gov IDs: NCT02069145, NCT02092363, NCT02050178, and NCT01608867). Porcupine is a membrane-bound O-acyltransferase47, and the inhibition of this enzyme prevents production of bioactive Wnt ligands to suppress Wnt/β-catenin signaling. LGK974 and ETC-1922159 are two representative porcupine inhibitors that are in phase I clinical trials (LGK974, Clinicaltrials.gov ID: NCT01351103; and ETC-1922159, Clinicaltrials.gov ID: NCT02521844).48, 49 Other proteins such as tankyrases 1/2, CK1α, CK1ε, GSK3β, etc. which play important roles in Wnt/β-catenin signaling are also actively being targeted, but neither the inhibitors of tankyrase, CK1ε, and GSK3β nor the activators of CK1α have entered into clinical trials.50–58 Despite the progress, targeting of the upstream effectors might have a limited application due to the following concerns: 1) cancer cells with more downstream genetic or epigenetic mutations, such as loss-of-function mutations of the destructive complex or β-catenin activation mutations, and upregulation of downstream effectors such as β-catenin by crosstalk with the other signaling pathways are anticipated to be resistant to such agents; and 2) the upstream targets such as tankyrase, GSK3β and CK1α are involved in multiple cellular processes. The intervention of these targets increases risks of undesired off-pathway toxicities.51, 59 As a consequence, β-catenin-containing transcriptional complex emerges as the most promising target for the development of inhibitors, because this nuclear transcriptional complex is formed at the very late stage and determines the transcriptional activities of the Wnt/β-catenin signaling cascacde.60 To date, the only reported drug-like inhibitors targeting this transcriptional complex are ICG-00161 and its second-generation prodrug derivatives PRI-724,62 CWP232228,63, 64 and CWP232291.65 This series of compounds binds with the general coactivator CBP for the disruption of the CBP/β-catenin interaction. The mechanism, hence, may less likely be Wnt pathway-specific, because CBP has been reported to be involved in many transcriptional processes.66 On the other hand, direct targeting of β-catenin is expected to result in specific inhibition of the pathway, but has been proven challenging. Over years, dozens of β-catenin-containing protein complex structures have been reported, and the potential binding sites of β-catenin for native protein binding partners and inhibitors have been analyzed by different research groups using different structural and biochemical methods. Herein, for the first time we provide a review to characterize possible small-molecule binding sites in β-catenin, summarize the efforts on development of bioactive small molecules that directly bind with β-catenin, and assess their biological characteristics. Structure-based inhibitor design and structure-activity relationship (SAR) of the reported inhibitors are discussed in the context to reveal new directions for future inhibitor development and drug discovery.
Potential small-molecule binding sites in β-catenin
Human β-catenin is comprised of 781 amino acids with a central structural core of 12 armadillo repeats (residues 138–664) and the intrinsically disordered N- and C-terminal regions.67 Structural biology studies reveal that β-catenin utilizes its armadillo repeat domain to interact with most of its known ligand proteins. These structures include β-catenin in complex with Wnt signaling suppressors APC,68–70 Axin,71 and ICAT,72, 73 transcription activators LEF1,74 TCF3,75 and TCF4,76–78 transcription coactivator BCL9,78 and Wnt signaling-releveant proteins E-cadherin14 and LRH1,79 among which LEF/TCF and BCL9 interact with β-catenin in the Wnt transcription complex. Disruption of either of these two types of protein-protein interactions (PPIs) (i.e., β-catenin/TCF PPI and β-catenin/BCL9 PPI) would disassemble the transcriptional complex, resulting in inactivation of Wnt/β-catenin signaling. These PPIs are well-characterized, and the distinct binding sites on β-catenin can be extracted by analyses of the crystallographic data of protein complex structures. Specifically, the β-catenin/TCF PPI interface covers ~4800 Å2, and this PPI interface structure offers four key binding areas.80–84 The first key binding area is around residues N426, K435, R469, H470, and K508 of β-catenin where these residues form a concave pocket to interact with residues D16, E17, L18, and I19 of TCF4 or the equivalent residues in TCF1, LEF1 and TCF3. Mutations of these β-catenin residues to alanine strongly reduced the binding affinity with LEF1 and TCF4 in co-immunoprecipitation (co-IP) experiments.80 Residue D16 of TCF4 forms salt bridge interactions with residue K435 of β-catenin and a hydrogen bond with β-catenin residue H470, as well as hydrogen bond networks with structural water molecules. This aspartate residue is the most important protruding hot spot of the TCF/LEF family of transcriptional factors by site-directed mutagenesis studies (Box 2 shows the definition of protruding hot spot, hot spot pocket, and hot spot interactions of protein–protein interactions).81–84 For example, the D16A mutation of TCF4 led to a significant decrease in binding affinity with β-catenin in enzyme-linked immunosorbent assays (ELISA) and induced a ΔΔG of +2.4 kcal/mol in isothermal titration calorimetry (ITC) experiments. The double mutation (D16A and E17A) of TCF4 further greatly reduced the TCF4 binding affinity, while the binding affinity of the single E17A mutation was weakened by 4-fold when compared to that of wild-type TCF4.81 The second key binding area is around β-catenin residues K312 and K345 where TCF4 residues E24 and E29 bind. The dissociation constant (KD) of the PPI between β-catenin and TCF4 E24A/E29A double mutant was six-fold higher than that of the β-catenin/wild type TCF4 PPI in surface plasmon resonance (SPR) studies, and the IC50 value of the TCF4 E24A/E29A double mutant for disrupting β-catenin/TCF4 PPI was increased by 62-fold compared with wild type TCF4 in fluorescence polarization (FP) competitive inhibition assays.85 The co-IP experiments further demonstrated that mutations of all four glutamate in this region (E24A, E26A, E28A, and E29A) abolished the binding with β-catenin.77 However, no obvious binding pocket was observed in this region of β-catenin. The third binding area is around residues H578 and R582 of β-catenin where D11 of TCF4 interacts. Mutagenesis and ITC studies showed ~30-fold increase in KD between wild type TCF4 and β-catenin mutation H578A or R582A. The D11A mutation had a large effect on TCF4 binding than the deletion of the entire TCF4 N-terminal 9 or 12 residues.83 The fourth key binding site is a hydrophobic area around β-catenin residues F253, F293, and I296, where TCF4 residues L41, V44, and L48 adopted an α-helical structure to bind with these residues through hydrophobic interactions. Mutation of the hydrophobic residues, L41A, V44A, and L48A, of TCF4 significantly reduced their binding affinity with β-catenin.81–84
Box 2. Hot spot interactions between proteins.
For the protein–protein interaction a small subset of residues at the protein-protein interface contributes to the majority of the binding free energy. These residues are called hot spots.131 A hot spot is defined as a residue which substitution by an alanine leads to a significant decrease in the free energy of binding (ΔΔGbinding > 1.5 kcal/mol). These hot spots tend to cluster and contact with each other to form an area of conserved interactions called hot regions.132 The protruding hot spot of one protein (called the ligand protein) packs against the concave hot region of the other protein (called the target protein). The protruding hot spot is also often called the projecting hot spots. The concave hot region is often called the hot spot pocket.133
The contacting surface area of β-catenin/BCL9 PPI is much smaller (~1450 Å2), where BCL9 adopts an α-helix to bind with β-catenin. Crystallographic and biochemical experiments identified two key binding areas at this PPI interface. One is a hydrophobic pocket formed by residues L156, L159, V167, A171, M174, and L178 of β-catenin, which interacts with BCL9 residues L366, I369, and L373. The β-catenin L156A/L159A double mutant loses its binding affinity with BCL9 in pulldown experiments.78 Other double mutants of β-catenin including L156S/L159S and L156S/L178S also block their binding with BCL9 in both fluorescence anisotropy binding experiments and AlphaScreen competitive binding assays.86 The protein pulldown experiments demonstrate the binding of BCL9 with β-catenin was abrogated by BCL9 mutations L366K, L373A, and L366A/I369A.78, 87 BCL9 mutations L366A, I369A, and L373A showed no inhibition of the β-catenin/wild-type BCL9 PPI in FP competitive inhibition assays.88 The second key binding area is an acidic knob formed by β-catenin residues D162, E163, and D164, which form salt bridge interactions with BCL9 residues H358 and R359. The β-catenin D162A mutant decreased its binding affinity with BCL989 and the β-catenin D164A mutant abolished its interaction with BCL9 or BCL9L90. BCL9 H358A and R359A mutants significantly reduced their binding affinity with β-catenin.78 The binding of BCL9 with β-catenin was fully abrogated by the BCL9358HRE360/358AKQ360 mutant.87
Inhibitors of the β-catenin/TCF PPI
Most efforts targeting β-catenin have been focused on discovering the agents that antagonize the β-catenin/TCF PPI, and to date various inhibitors have been reported. Peptide-based inhibitors including hydrocarbon-stapled peptides and peptoid-peptide macrocycles have been reported.91, 92 Based on the observation that the β-catenin binding domain (CBD) of Axin adopts an α-helical structure to bind with β-catenin and the binding site of β-catenin for Axin is overlapped with that for the TCF4 α-helical domain, hydrocarbon-stapled peptides were designed by modifying the Axin sequence with an i/i+4 side-chain staple and then optimizing the sequence using phage display. These efforts led to two stapled peptides, StAx-35 (KD = 13 nM) and StAx-35R (KD = 53 nM) (Figure 2a). The direct binding of StAx-35 and StAx-35R with β-catenin was demonstrated by the protein pulldown assays using the cell lysates. N-terminally acetylated StAX-35 (aStAx-35) was successfully co-crystalized with β-catenin (residues 134–665) (PDB id, 4DJS), offering the first and the only co-crystal structure of β-catenin with its inhibitor (Figure 2b).91 Cell-based studies indicated these stapled peptides can penetrate the cell membrane, selectively suppress TOPFlash luciferase reporter activity while sparing FOPFlash luciferase reporter activity, and downregulate Wnt target genes LEF1, LGR5, and Axin2. The proliferation of Wnt-hyperactive cancer cells was blocked at the micromole levels after 5-day incubation. Further structural optimization was performed to increase the cellular uptake and cell-based potency. The nuclear localization sequence (NLS) of the SV40 large T-antigen that was previously shown increasing both cellular uptake and nuclear localization was introduced to the N-terminal end of the stapled peptide StAx-h, in which all R residues of StAx-35R were replaced by homoarginine, resulting in NLS-StAx-h. The fluorescently labeled version of NLS-StAx-h (f-NLS-StAx-h) exhibits 7-fold improvement in cellular uptake at 5 mM and considerable cytosolic distribution with respect to the fluorescently labeled StAx-35R (f-StAx). The core sequence of NLS-StAx-h (StAx-h) was shown to bind with β-catenin in pulldown experiments using the lysate of DLD-1 cells. Further studies reveal that NLS-StAx-h suppresses Wnt target gene expression and inhibits proliferation and migration of CRC cells.93 It should be noted that disruption of the β-catenin/Axin PPI can potentially activate Wnt/β-catenin signaling. Indeed, two Axin-mimicking stapled peptides, SAHPA1 and SAHPA2, were reported.94 SAHPA1 disrupted the interaction between β-catenin and Axin, and activated rather than suppressed Wnt/β-catenin signaling. Similarly, a small molecule SKL2001 that disrupts the β-catenin/Axin PPI was demonstrated to be an agonist of the Wnt/β-catenin signaling pathway.95 Using the Rosetta suite of protein design algorithms, a series of cyclized peptide-peptoid macrocycles was designed to bind with β-catenin. The most active macrocycle that potently disrupts the β-catenin/TCF PPI also markedly decreases growth of prostate cancer cells and inhibits Wnt signaling in the in vivo zebrafish model.92
Figure 2.
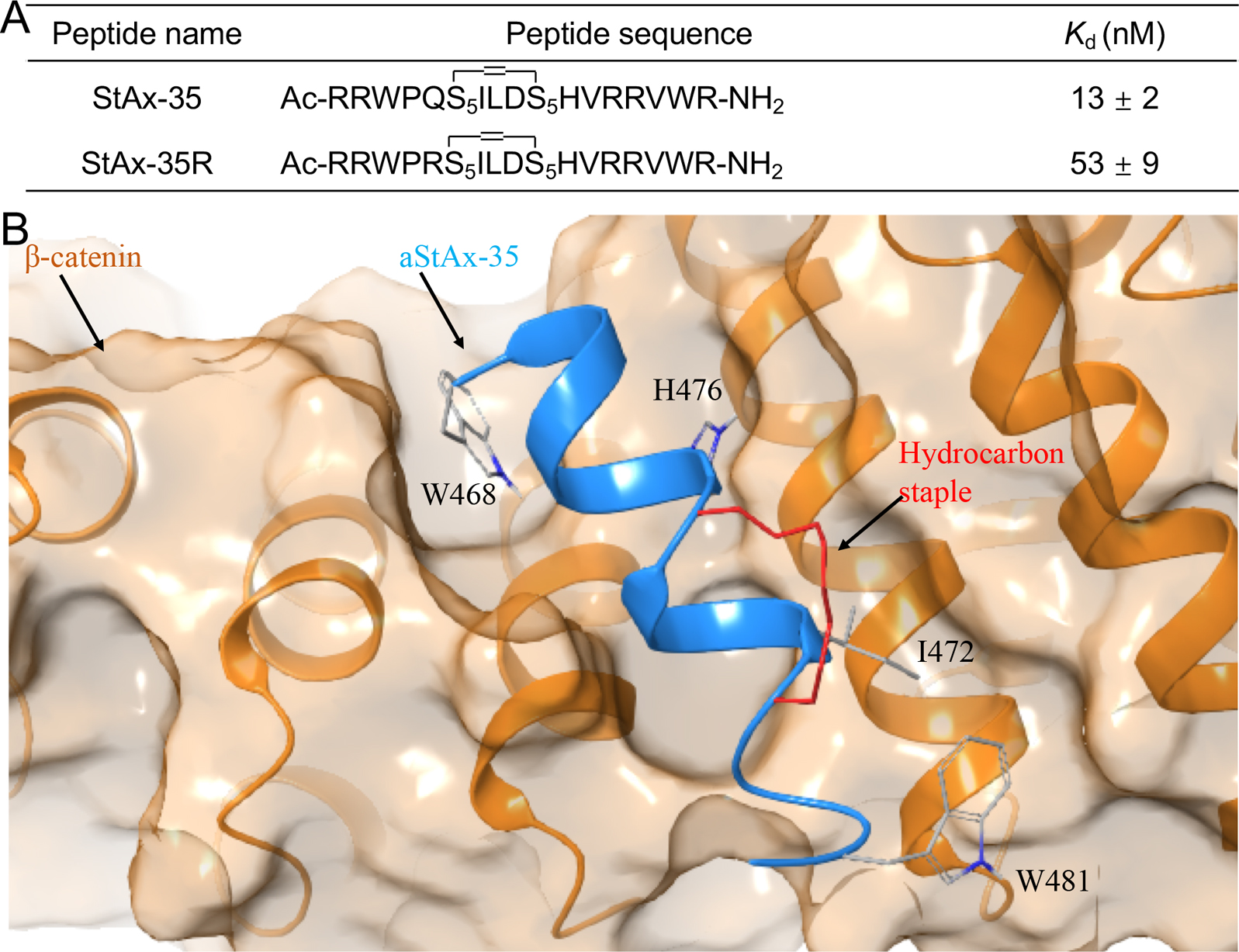
(A) Structures and the binding affinity of StAx-35 and StAx-35R. (B). Crystal structure of β-catenin in complex with aStAx-35.
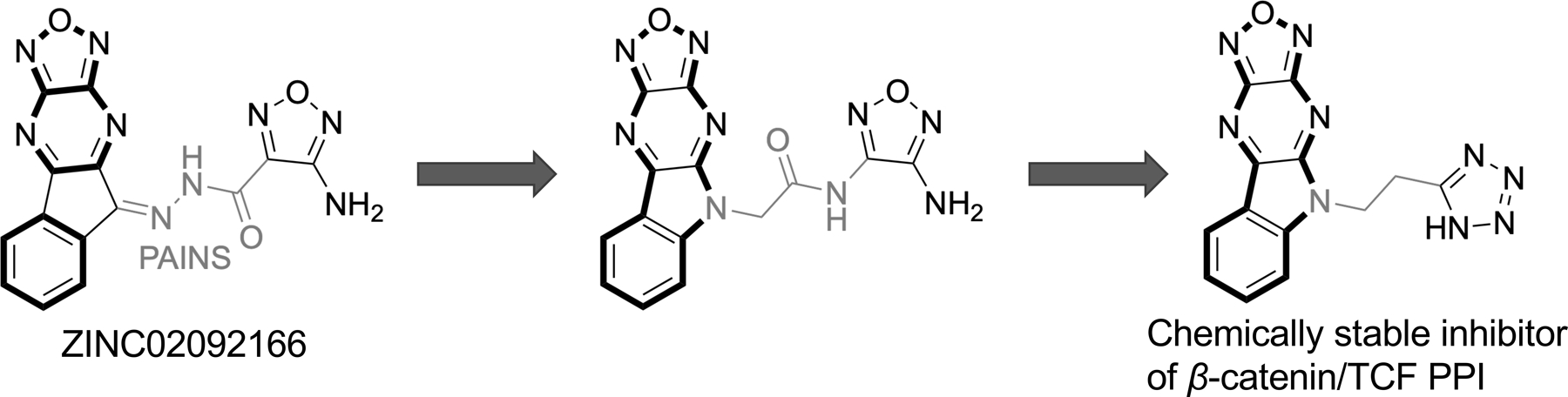
Some natural products including PKF115–584,96, 97 GCP049090,96 Henryin,98 organocopper compound BC2199, iCRT series (iCRT3, iCRT5, iCRT14),100 ZINC02092166 and its chemically stable derivatives,101 and compound LF3102 were reported to disrupt the β-catenin/TCF PPI and showed inhibitory activities in cell-based studies. Compounds LF3 and iCRT3 have been extensively characterized. LF3 was identified from 16,000 compounds via compound screening using AlphaScreen and ELISA assays. LF3 suppressed Wnt/β-catenin signaling in cells with high Wnt activity, and reduced expression of Wnt target genes. LF3 also showed potent inhibitory activities against cancer cells related to Wnt signaling in vitro and in a mouse xenograft model. Co-IP assays using HCT116 colon cancer cells indicated that LF3 dose-dependently disrupted the β-catenin/TCF4 interaction but spared the β-catenin/E-cadherin interaction, indicating that this compound does not disturb β-catenin-mediated cell adhesion. iCRT3 was identified from 14,997 compounds through an Axin RNAi-based chemical genetic screen. iCRT3 suppressed not only β-catenin but also androgen receptor (AR) signaling pathways. Both pathways are hyperactive in prostate cancer. Correspondingly, iCRT3 inhibited growth of prostate cancer cells in vitro and in an in vivo xenograft model.103 However, the other compounds (PKF115–584, GCP049090, iCRT5, iCRT14, and ZINC02092166) contain pan-assay interference compounds (PAINS) substructures that can cause frequent hits in biochemical assays.104–106 More importantly, all of these compounds have not been reported to directly bind with β-catenin by biochemical or biophysical experiments. Therefore, we only review in details the following small-molecule inhibitors, which were reported to bind directly with β-catenin.
Inhibitor PNU-74654 (Year 2006)
PNU-74654 in Table 1 was initially discovered by in silico docking of 17,700 compounds to a well-defined binding pocket around β-catenin residues K435 and R469, followed by evaluation of 22 compounds with the best docking scores using WaterLOGSY NMR and competitive ITC studies.107 The direct binding of PNU-74654 with β-catenin was assessed by the ITC experiment (KD = 450 nM). The binding mode between PNU-74654 and β-catenin (around hot spot residues K435/R469) was investigated by extensive docking studies (Figure 5). The results predicted that the methyl group on the furan ring and the phenyl moiety bound with two small pockets on both side of the hot spot pair, respectively. The great contribution of these two groups to the binding affinity of PNU-74654 was supported by the experimental data of its two analogs, in which the methyl group on the furan ring and the distal phenyl moiety were replaced by a proton and a piperidine ring, respectively, and both show an apparent decrease in binding affinity. Neither selectivity nor cellular activity data were reported for PNU-74654 although this compound was claimed to specifically inhibit TCF4 transactivation in the luciferase reporter experiment. It is worth noting that PNU-74654 has a reactive functional group, acyl hydrozone, which has been recognized as a PAINS moiety.104–106 This PAINS substructure could be problematic when further optimizing this compound to improve potency, selectivity, and physicochemical properties. One example for the modification of this linker group was the optimization of ZINC02092166,101 in which the acyl hydrozone group of ZINC02092166 was substituted by a new substructure, resulting in a series of chemically stable derivatives of ZINC02092166 as the inhibitors of the β-catenin/TCF PPI. The tetraheterocyclic ring of ZINC02092166 in Figure 3 contains 17 electrons and is not aromatic. The substitution of the imine carbon atom with a nitrogen atom resulted in a new tetraheterocyclic ring with 18 electrons, which constitutes a stable large aromatic system. These new derivatives not only show selectivity for β-catenin/TCF PPI over β-catenin/cadherin and β-catenin/APC PPIs, but also suppress transactivation of Wnt/β-catenin signaling, downregulate Wnt target genes Axin2, cyclin D1, and c-myc, and inhibit growth of cancer cells. More importantly, their cell-based inhibitory activities are in agreement with that from the biochemical assays. On the contrary, ZINC02092166 displayed higher inhibitory activities in cellular experiments than in biochemical studies.
Table 1.
Small-molecule inhibitors that were reported to directly bind with β-catenin.
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | M.W. | Discovery method | Direct binding | KD, μM | Binding sites | TOP-Flash, IC50, μM | In vivo efficacy |
| PNU-74654 | 320 | virtual screening | NMR, ITC | 0.450 | K435, R469 | N.D. | N.D. |
| UU-T01 | 230 | hot spot-based design | ITC, mutagenesis | 0.531 | K435, K469 K508 | N.D. | N.D. |
| UU-T02 | 665 | peptidomimetic design | ITC, mutagenesis | 0.418 | K435, R469 R474, K508 R515 | SW480: 232 | N.D. |
| MSAB | 305 | luciferase-reporter screening | SPR, NMR | N.D. | residues 301–670 | HCT116: 0.58 | 20 mg/kg p.o., q.2d. |
| HI-B1 | 255 | rational design based on a hit | protein pull-down | N.D. | K312 | DLD1: 13 CACO2: 13 | 50mg/kg i.p., q.d. |
| carnosic acid | 332 | screening by in vitro ELISA | NMR | 5–20 | ARD N-terminus | SW480: ~25 | diet containing 0.1% or 1% carnosate |
| PNPB-22 | 646 | hot spot-based design | ITC, mutagenesis | 0.330 | L156, L159, V167, A171, M174, L178 | SW480: 13 | N.D. |
N.D.: not determined. ARD: Armadillo repeat domain.
Figure 5.
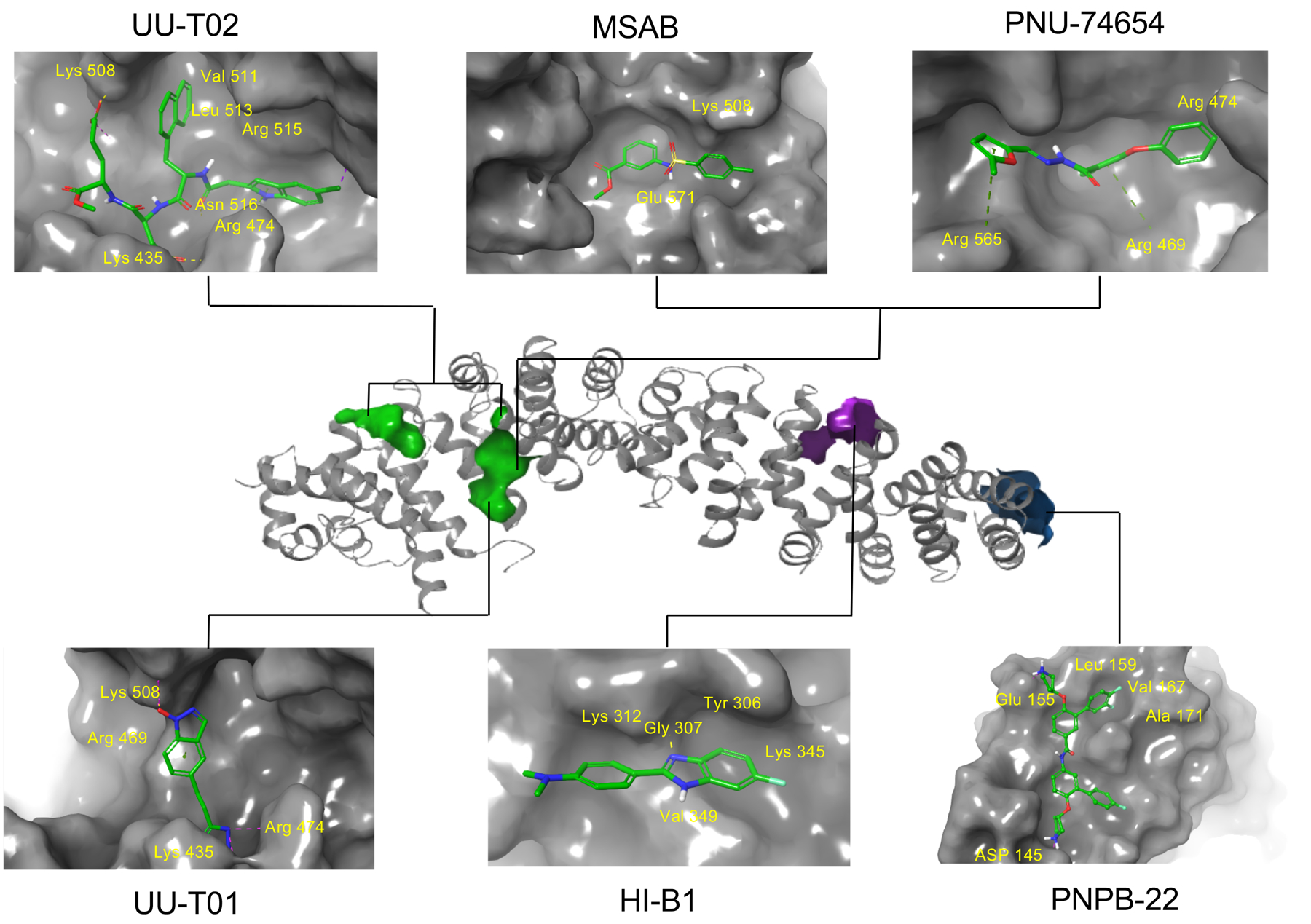
Predicted binding sites of the reported small-molecule inhibitors with β-catenin.
Figure 3.
Optimization of ZINC02092166 as inhibitors of β-catenin/TCF PPI.
Inhibitor UU-T01 (2013)
Compound UU-T01 in Table 1 was designed by a hot spot-based bioisostere replacement strategy.85 Specifically, different bioisosteres were used to mimic the carboxylic acid groups of TCF4 hot spots D16 and E17, and the selected fragments were merged with the assistance of a linker library. UU-T01 was obtained by combining the optimal fragments and linkers. The direct binding between β-catenin and UU-T01 was evaluated in the ITC experiment (KD = 531 nM). The inhibition of β-catenin/TCF4 PPI was demonstrated by FP (Ki = 3.1 μM) and AlphaScreen (Ki = 7.6 μM) competitive inhibition assays. The binding mode was assessed using Autodock docking (Figure 5). The indazole-1-ol group mimics the carboxylate moiety of TCF4 E17, makes salt bridge interactions with residue K508 of β-catenin, and forms cation–π interactions with the positively charged guanidino group of β-catenin R469. The tetrazole ring mimics the carboxylate moiety of TCF4 D16, and forms salt bridge and hydrogen bond interactions with β-catenin residues K435 and N430, respectively. The binding mode was supported by site-directed mutagenesis and SAR studies. For example, UU-T01 shows 3- to 7.6-fold loss in activity against β-catenin K435A, R469A, and K508A mutations in ITC studies. Moreover, compounds containing the indazole-1-ol moiety are more potent than those with the benzotriazole-1-ol ring in FP competitive inhibition assays. This observation is consistent with the hypothesis that the cation–π interaction plays an important role for this series of compounds to bind with β-catenin residue R469. The indazole-1-ol ring has a higher π-electron density than the benzotriazole-1-ol ring due to the different electronegativities between the nitrogen and carbon atoms, and is more favorable for forming the cation–π interaction. The other carboxyl acid bioisosteres, 5-oxo-1, 2, 4-oxadiazole and 5-thioxo-1, 2, 4-oxadiazole, are inferior to tetrazole for binding. The length of linker between indazole-1-ol and tetrazole is also critical for inhibitor binding affinity, and the CH2CH2 linker is superior to the CH2 and CH2OCH2 linkers. This study not only presents a new hot spot-based bioisosteric replacement approach to designing small-molecule PPI modulators, but also provides evidence demonstrating the ligandability of this binding area (around residues K435 and K508), which is in consistent with the results of binding site analyses. It should be noted that the selectivity and cell-based activity data were not collected for UU-T01.
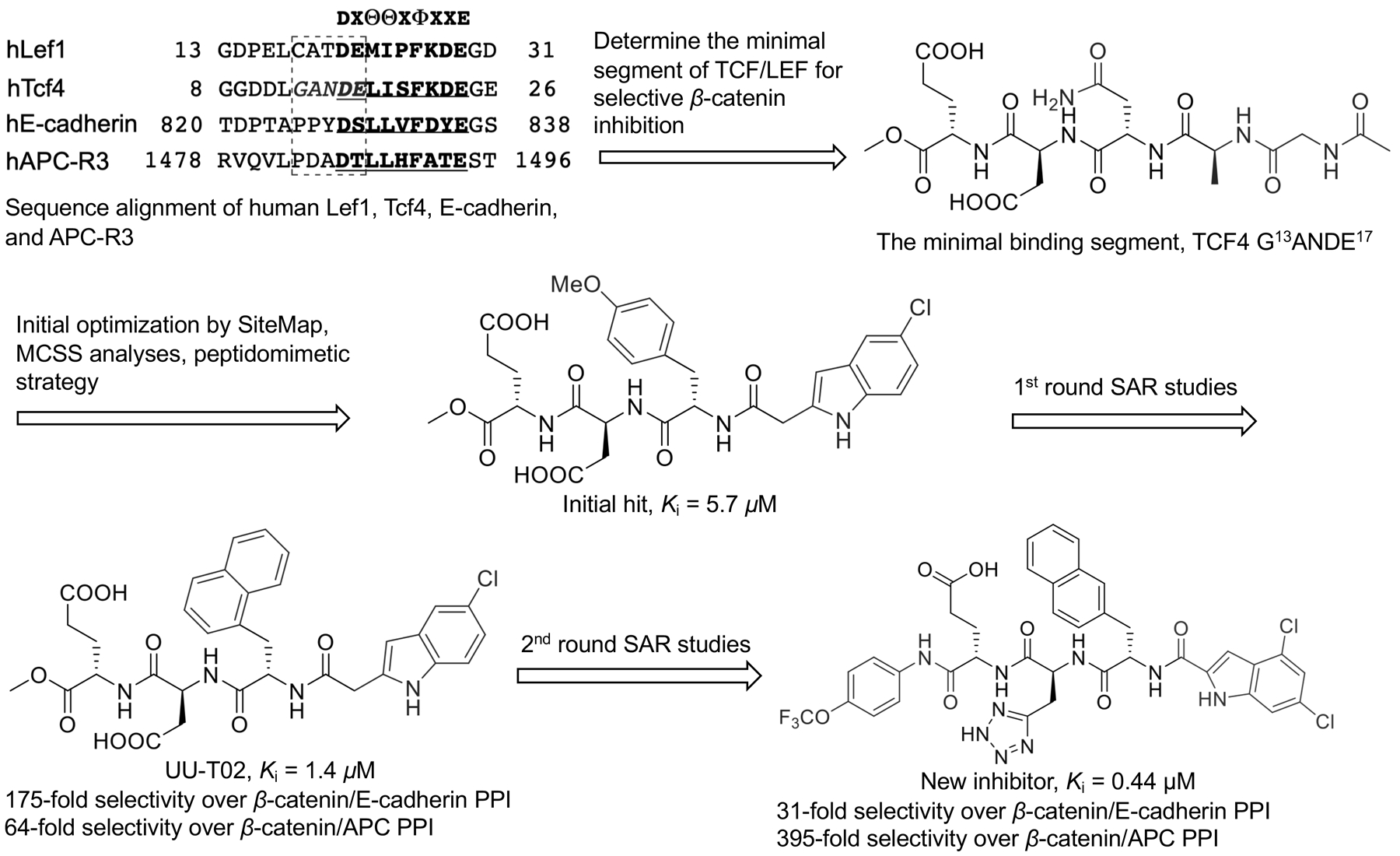
Inhibitor UU-T02 (2014)
UU-T02 in Table 1 was also designed to target the potential binding pockets surrounding β-catenin residues K435 and R469.108 As described above, crystallographic studies indicated that β-catenin utilized its armadillo repeat domain to bind with a diverse group of protein partners such as TCF/LEF, BCL9, E-cadherin, APC, Axin, and ICAT. Available crystal structures14, 69, 70, 74–78 and biochemical studies82, 109–111 have indicated that the same surface area of β-catenin is used to bind with TCF, APC, and regions III and IV of E-cadherin. Both in vitro and in vivo studies indicate the binding mode of β-catenin with TCF, APC and E-cadherin is mutually exclusive. Therefore, inhibitor selectivity turns out to be the main concern when designing compounds to disrupt the β-catenin/TCF PPI. UU-T02 was designed to approach this problem (Figure 4). Specifically, although the most important protruding hot spot D16 of TCF4 is also conserved in E-cadherin (D830) and APC (D1486 of the APC-R3 repeat), the binding features adjacent to this critical residue are different between TCF, E-cadherin, and APC when binding with β-catenin.75, 80, 84 For example, as described above, TCF4 E17 is relatively important for TCF4 binding with β-catenin while the residues at the same position in E-cadherin and APC-R3 (S831 and T1487, respectively) do not make direct interactions with β-catenin. Additionally, deletion experiments showed that the TCF4 G8-A14 sequence adjacent to hot spot D16 is important for TCF4 binding with β-catenin.77, 83, 84 The E-cadherin and APC-R3 residues at the same position of TCF4 G13AN15 do not bind with β-catenin directly. The D16A/E17A TCF4 peptide cannot disrupt the wild-type β-catenin/wild-type TCF4 PPI in FP competitive inhibition assays, while the alanine mutations of E-cadherin P826YDS829 and APC-R3 D1484ADT1487 sequences only leads to 10- and 30-fold increase of IC50 values for disrupting the wild-type β-catenin/wild-type E-cadherin PPI and the wild-type β-catenin/wild-type APC-R3 PPI, respectively.108 On the basis of these analyses, it was hypothesized that occupying the TCF4 G13ANDE17 binding area by capturing the binding features of G13ANDE17 would produce selective inhibitors for the β-catenin/TCF PPI. Careful analyses of the co-crystal structures of β-catenin with TCF4 reveal that this TCF4 G13ANDE17 binding area can be divided into four pockets: pocket A is formed by residues C429, N430, K435, H470, S473, and R474 of β-catenin; pocket B is lined with β-catenin residues I507, K508, V511, L539, I569, C573, and this pocket is relatively deep; pockets C and D are shallow surface pockets with pocket C surrounded by residues L519 and I579, and pocket D defined by residues E462, C466, L506, K508 (the side chain carbon atoms), and A509. Pockets A and B are connected to pocket C through an arginine channel formed by residues R474 and R515. Pockets B and D are connected through residues R469 and K508. With the assistance of multiple-copy simultaneous search (MCSS) and SiteMap analyses, new β-catenin/TCF inhibitors were designed by the peptidomimetic approach based on TCF4 G13ANDE17 sequence. The critical residues D16 and E17 were kept in new inhibitors to bind with pockets A and K508, while the 4-OMe benzyl and indole moieties were initially selected to occupy pocket B and the arginine channel toward pocket C, respectively. The first designed compound disrupts the β-catenin/TCF4 PPI with a Ki of 5.7 μM in FP competitive inhibition assays. SAR studies on this scaffold reveal that the 5-Cl indole moiety is the optimal substituent for the arginine channel and the larger hydrophobic group is preferred for pocket B. The representative compound, UU-T02, inhibits the β-catenin/TCF4 interaction with a Ki of 1.32 μM in FP assays and is 175- and 64-fold selective against β-catenin/E-cadherin and β-catenin/APC interactions, respectively. The direct binding between UU-T02 and β-catenin was examined by ITC experiments (KD = 418 nM). The binding mode was assessed by Autodock and Glide docking (Figure 5). Two carboxylate groups of UU-T02 were predicted to form salt bridge interactions with β-catenin residues K435 and K508. The naphthyl group binds deeply with the hydrophobic pocket B, and the indole N-H could form a hydrogen bond with C=O group of N516 side chain, and the indole ring is designed to form cation–π interactions with R474 and R515. This predicted binding model was supported by SAR and site-directed mutagenesis studies. UU-T02 exhibits low cell-based activity probably due to its poor cell membrane permeability which is caused by its two carboxylate groups. Further optimization of UU-T02 was performed to increase its activities in both biochemical and cell-based studies.112 Extensive SAR studies not only demonstrate that the naphthyl group, two carboxylate groups, and the 5-Cl indole moiety are critical for maintaining the inhibitory activity, but also reveal that the methyl ester of UU-T02 can be replaced by the larger hydrophobic groups, and the carboxylate groups can be substituted by its bioisosteres to improve activity. These efforts have led to new inhibitors with improved activity (Ki = 0.44 μM).112 Cell-based studies showed that the last inhibitor in Figure 4 selectively disrupted β-catenin/TCF over β-catenin/E-cadherin and β-catenin/APC interactions, dose-dependently suppressed transactivation of Wnt/β-catenin signaling, inhibited growth of Wnt/β-catenin signaling-hyperactive cancer cells, and blocked migration and invasiveness of Wnt/β-catenin-dependent cancer cells.
Figure 4.
Discovery and optimization of UU-T02.
Inhibitor MSAB (2016)
Compound MSAB in Table 1 was identified from a library of 22,000 compounds using a TCF-dependent luciferase reporter system as the pilot assay.113 This compound selectively suppresses transactivation of Wnt/β-catenin signaling and inhibits growth of cancer cells with hyperactive Wnt/β-catenin signaling. It also displays anti-tumor effects in Wnt-dependent xenograft tumor models in vivo. Mechanistic analyses indicated this compound bound with β-catenin, promoted its degradation, and selectively downregulated Wnt target genes expression. The binding of MSAB with β-catenin was first demonstrated by biotin-based affinity purification combined with tandem mass spectrometry (MS/MS) showing that β-catenin was the leading candidate among MSAB-binding Wnt effector proteins, and further confirmed by ligand-observed NMR and SPR studies. Pull-down and NMR experiments suggested that MSAB likely bound to the second armadillo repeat of β-catenin (residues K301-Y670). The SAR analysis reveals that the para substitution of the phenyl ring and the ester group of phenylsulfonamidobenzoates are important for maintaining the inhibitory activity, which is in consistent with the predicted binding mode in Figure 5. Specifically, the methyl and methoxy groups are favored for the para substitution of the phenyl ring while the fluoro or chloro substituent at this position results in the complete loss of activity. The replacement of the ester group of phenylsulfonamidobenzoates with amides also abolishes the inhibitory activity.
Inhibitor HI-B1 (2017)
HI-B1 in Table 1 was designed by cyclization of resveratrol, a previously discovered Wnt inhibitor.114 This compound inhibited the β-catenin/TCF4 luciferase reporter activity, downregulated transcription and expression Wnt target genes cyclin D1 and Axin2, and selectively suppressed growth of Wnt-dependent cancer cells in vitro and in vivo. The direct binding of HI-B1 with β-catenin was demonstrated by incubation of HI-B1-sepharose 4B beads with DLD cell lysates and then Western blotting of the proteins that were pulled down. The binding of HI-B1 with β-catenin led to disruption of the β-catenin/TCF4 PPI, which was confirmed by co-IP experiments at both biochemical and cellular levels. The binding mode was assessed based on the results of computer modeling (Figure 5) and preliminary SAR studies, which suggested that the nitrogen atom in imidazole ring of HI-B1 may form a hydrogen bond with β-catenin K312. The importance of this nitrogen atom in the imidazole ring of HI-B1 for binding with β-catenin was further evaluated by SAR studies.
Inhibitors of β-catenin/BCL9 PPI
Compared with β-catenin/TCF PPI, β-catenin/BCL9 PPI has the smaller contacting area (1450 Å2) and the moderate binding affinity (KD = 470 nM), representing a promising alternative binding site for inhibitor design to suppress Wnt/β-catenin signaling. The roles of the β-catenin-BCL9-Pygo axis in Wnt target gene transcription is described in Box 1. Peptides-based inhibitors including triazole- and hydrocarbon-stapled peptides and sulfono-γ-AApeptide have been designed to disrupt this PPI interface.115–118 By mimicking the binding features of the α-helical HD2 domain of BCL9, a stabilized α-helix of BCL9 (SAH-BCL9B) with the i, i+4 side-chain stapling was designed and synthesized. SAH-BCL9B can dissociate the β-catenin/BCL9 complex in cells, selectively suppress Wnt transcriptional activity, and inhibit proliferation, angiogenesis and migration of β-catenin-hyperactive cancer cells. Moreover, in INA-6 multiple myeloma and Colo320 colorectal carcinoma mouse models, SAH-BCL9B was found to suppress tumor growth, angiogenesis, invasion, and metastasis. The helical foldamer scaffold based on the unnatural sulfono-γ-AApeptide scaffold was used to design and synthesize derivatives to disrupt β-catenin/BCL9 PPI. Although sulfono-γ-AApeptides do not faithfully mimic α-helix, these sulfono-γ-AApeptide derivatives can capture key structural features of BCL9 for binding with β-catenin, disrupt β-catenin/BCL9 PPI, and exhibit cell-based activity. The merit of using these sulfono-γ-AApeptides is that they are absolutely resistant against proteases and proteinases when compared to peptide-based inhibitors. A small-molecule natural product, carnosic acid in Table 1, was reported as an inhibitor of the β-catenin/BCL9 PPI after screening two libraries containing 1,250-compounds by in vitro ELISA assay to monitor the interaction between His6-tagged BCL9 HD2 (His-HD2) and the glutathione S-transferase (GST) tagged β-catenin armadillo repeat domain that was immobilized on glutathione-coated microplates.119 NMR saturation transfer difference (STD) studies and heteronuclear single-quantum correlation (HSQC) experiments between 1H and 15N demonstrated that carnosic acid bound with β-catenin at the first four armadillo repeat. NMR and analytical ultracentrifugation analyses in combination with crystallographic analysis reveal an intrinsically labile α-helix adjacent to the BCL9-binding site in β-catenin. This labile α-helix is responsive to the addition of carnosic acid. The binding of carnosic acid promoted degradation of active β-catenin, suppressed Wnt/β-catenin signaling transactivation, and regulated expression of Wnt target genes. It is noted that the catechol moiety of carnosic acid is liable for oxidization and reactions with protein nucleophiles, and has been identified as a PAINS moiety.104, 105 This compound is also associated with many biological activities. The inhibitor optimization based on carnosic acid is yet to be reported.
Box.1. How does β-catenin regulate Wnt target gene expression?
Upon activation of the Wnt/β-catenin signaling pathway, β-catenin is translocated into the cell nucleus and activates transcription of Wnt/β-catenin target genes. Due to lack of the DNA binding domains in β-catenin, the DNA-binding partners are necessary to bring β-catenin to the promoter of its specific DNA sequences.67 The TCF/LEF family of transcription factors is the main nuclear partners that guide β-catenin to its specific DNA loci. The core interaction region for β-catenin binding with TCF/LEF transcriptional factors is armadillo repeats 3–10 of β-catenin.75 Meanwhile, the proteins of the chromatin remodeling complex including CBP, p300, Tip60, SWI/SNF, ISWI, etc. are recruited to the C-terminal domain of β-catenin. These proteins occupy the similar binding site in β-catenin. The C-terminal domain of β-catenin serves as a platform for the recruitment and sequential exchange of these transcriptional co-activators.129 The armadillo repeat 1 of β-catenin interacts with the homology domain 1 (HD1) of β-catenin co-activator, BCL9 or BCL9 paralog, BCL9L. The homology domain 2 (HD2) of BCL9 or BCL9L binds with Pygo which will localize chromatin-binding proteins to the modified nucleosomes.130
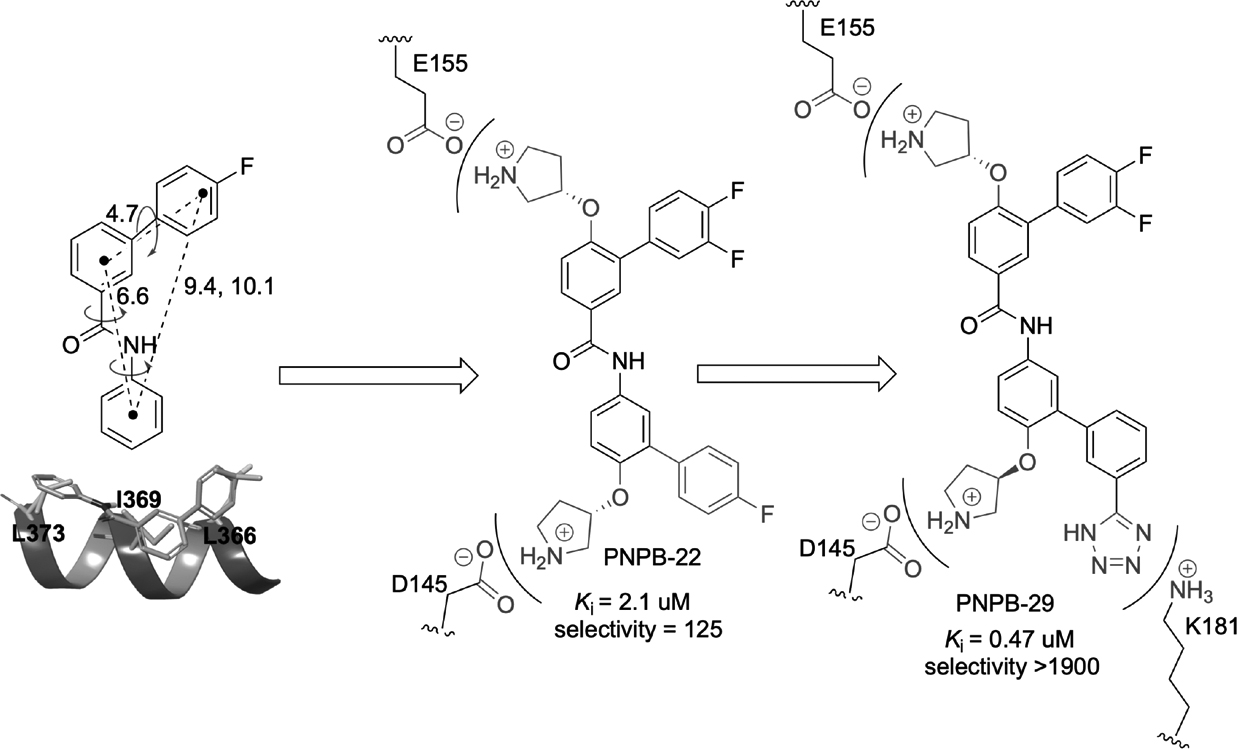
Based on the observation that the HD2 domain (residues S352-F374) of BCL9 adopts an α-helical structure to bind with β-catenin first armadillo repeat, and BCL9 residues L366 (i), I369 (i + 3), and L373 (i + 7) serve as the protruding hot spots, a fragment-size scaffold 3-(4-fluorophenyl)-N-phenylbenzamide (PNPB) was designed as a generalizable scaffold to mimic the binding features of these three protruding α-helical hot spots at positions i, i + 3, and i + 7 and was used as the starting point to design inhibitors for disruption of the β-catenin/BCL9 PPI (Figure 6).86 The potency of the inhibitor was improved by introducing substituents to this scaffold. Specifically, two positively charged pyrrolidino groups were introduced to make salt bridge interactions with β-catenin D145 and E155, respectively. One F atom was added to the 4-fluorophenyl moiety to enhance hydrophobic interactions in the pocket lined with β-catenin residues L159, L160, V167, and A171. An additional 4-phenyl ring was introduced to occupy the pocket lined with residues Q177, L178, and K181 of β-catenin. This design strategy was assessed by SAR and site-directed mutagenesis studies. The direct binding of the representative compound (PNPB-22, Ki = 2.1 μM, Table 1) with β-catenin was demonstrated by ITC experiments (Kd = 330 nM). PNPB-22 selectively disrupted the β-catenin/BCL9 interaction over the β-catenin/E-cadherin interaction in biochemical and cell-based experiments, inhibited Wnt/β-catenin signaling transactivation, regulated transcription and expression of Wnt target genes, and suppressed growth of Wnt-dependent cancer cells. Further modification was carried out to improve the activity and drug-like properties of this series of inhibitors. By the analysis of the predicted binding mode between PNPB-22 and β-catenin (Figures 5 and 6), the tetrazole group was introduced to the PNPB scaffold to capture the additional interaction with β-catenin K181, resulting in a compound with improved activity (Ki = 0.47 μM).120 The success of this strategy was again demonstrated by SAR and mutagenesis analyses. To improve drug-like properties of the PNPB series, a separate effort was made by introducing the piperazine linker to replace one phenyl ring in PNPB-22, followed by SAR studies.121
Figure 6.
Discovery and optimization of the PNPB series of small-molecule β-catenin/B-cell lymphoma 9 (BCL9) inhibitors. PNPB, 3-(4-fluorophenyl)-N-phenylbenzamide.
Concluding Remarks and Future Perspectives
Aberrant activation of β-catenin signaling has strongly been implicated in initiation and progression of many types of cancer. Collective evidence has suggested suppression of this signaling pathway circuit would offer a novel strategy for treatment of metastatic cancers and addressing cancer dormancy. Rather than regulating upstream effectors, direct targeting of oncogenic β-catenin, a key signal hub of this pathway, is a biologically compelling approach to suppress hyperactive β-catenin signaling, but has proven to be a formidable challenge. Peptide-based inhibitors have been designed to bind with β-catenin, but they commonly suffer from low protease and proteinase stability and poor cell permeability. Small molecules are also reported to bind with β-catenin and suppress Wnt/β-catenin signaling. The major lessons that we have learned from these discovery programs are listed below.
Firstly, although these small-molecule inhibitors have been demonstrated to directly bind with β-catenin using various biophysical and/or biochemical experiments, none of them have been moved to the latter stages of inhibitor development. One question to be addressed is how to optimize the compounds to obtain more potent β-catenin inhibitors. The co-crystal structure of small molecules with its target protein often plays a crucial role in inhibitor optimization, especially for the challenging therapeutic targets such as PPIs.122 To date, only the co-crystal structure of N-terminally acetylated 17-mer stapled peptide aStAx-35 in complex with β-catenin (residues 134–665) was resolved (PDB id, 4DJS, resolution = 3.03 Å, Rfree = 0.291, Figure 2b), highlighting the challenge in obtaining the co-crystal structure of small molecules with β-catenin. One of the reasons behind might be suboptimal crystallization conditions. The second question to be addressed is the inferior physiochemical properties of some reported β-catenin inhibitors. For example, some reported inhibitors contain PAINS substructures, which were not identified in the early hit triage and characterization processes and have caused problems for optimizing these compounds in the β-catenin inhibitor campaigns. These undesired structures could cause severe off-target effects that contribute to the exaggerated pharmacological readouts, further complicating the inhibitor optimization efforts. On one hand, medicinal chemistry efforts should continue to improve the potency and physiochemical properties of the inhibitors. On the other hand, new technologies such as cryo-electron microscopy (cryo-EM) might be a powerful additional tool in determining β-catenin-small molecule complex structures.123, 124 Cryo-EM was reported to be advantageous for determining the structures of ‘intractable’ targets for which X-ray and NMR do not work.
Secondly, in silico docking, NMR, and mutagenesis studies revealed that most of these inhibitors bind to a region around hot spot residues K435, N426, H470, R469, and K508 of β-catenin where residues D16, E17, L18, and I19 of TCF4 interact, implying this hot-spot region might be a ligandable site. Future screening and structure-based design efforts could focus on this site. Alternatively, the hot-spot regions identified from the BCL9 binding site in β-catenin emerge as new potential sites for small-molecule binding, given that the contacting area between β-catenin and BCL9 is much smaller (1450 Å2 for β-catenin/BCL9 vs. 4800 Å2 for β-catenin/TCF) and this PPI displays moderate binding affinity (KD = 470 nM for β-catenin/BCL9 PPI vs. 7–10 nM for β-catenin/TCF PPI). Encouragingly, carnosic acid and PNPB series were reported to occupy the BCL9 binding site of β-catenin, and the residues of β-catenin for binding with these two series of compounds were determined by NMR and mutagenesis studies, respectively. These results provide preliminary evidence supporting that the BCL9-binding site in β-catenin can be ligandable. More efforts need to be made to diversify the chemical spaces of the inhibitors, identify more potent inhibitors, and determine the druggability of this binding site.
Thirdly, β-catenin not only uses the same surface area for binding with TCF, E-cadherin (regions III and IV), and APC, but also uses the same area for binding with BCL9 and region V of E-cadherin. Therefore, selectivity is a major concern when designing small molecules to disrupt β-catenin/TCF or β-catenin/BCL9 interaction. The selectivity data have been obtained for some of the inhibitors discussed above. UU-T02 and PNPB series represent two examples of designing selective inhibitors for β-catenin/TCF and β-catenin/BCL9 interactions, respectively. By analyzing the binding mode of the sequences flanking the hot spot residue D16 of TCF, a binding area at the β-catenin/TCF interface that is selective for β-catenin/TCF over β-catenin/E-cadherin and β-catenin/APC PPIs was revealed by site-directed mutagenesis studies. To occupy this selective binding site, small-molecule inhibitors were designed by a peptidomimetic strategy that includes SiteMap and MCSS technologies. As a result, the representative compound, UU-T02, displayed good selectivity for β-catenin/TCF4 over β-catenin/E-cadherin and β-catenin/APC interactions. A generalizable PNPB scaffold was designed to mimic the hydrophobic side chains of α-helical hot spots at positions i, i + 3, and i + 7. This scaffold was used to mimic the binding mode of BCL9 hot spots L366, I369, and L373. Optimization of this scaffold was conducted by introducing two positively charged pyrrolidino groups to interact with β-catenin residues E155 and D145. These initial efforts led to a collection of selective inhibitors of β-catenin/BCL9 PPI with compound PNPB-22 (the selectivity for β-catenin/BCL9 over β-catenin/E-cadherin PPIs = 125-fold) being the representative. Further optimization was conducted by introducing the tetrazole group to interact with another adjacent K181 of β-catenin, resulting in a compound with improved activity and selectivity. In addition to the selectivities between β-catenin binding partners, the specificities of β-catenin inhibitors over off-targets should also be assessed by designing appropriate recuse experiments and using appropriate control compounds.125 The off-target effects from non-β-catenin targets could cause various adverse effects and hamper further development of these inhibitors as cancer therapeutics.
Last but not least, human β-catenin has 781 amino acids while only residues 140–664 (central armadillo domain) have been targeted. The N-terminal and C-terminal domains are essential for β-catenin phosphorylation, stabilization, and target gene transcription. The binding of small molecules with these two intrinsically disordered domains is expected to regulate Wnt/β-catenin signaling, especially given that the areas in the central armadillo domain occupied by TCF/LEF is very large.126 The challenge is how make these two intrinsically disordered regions to form binding pockets for small-molecule binding. In addition, proteolysis targeting chimeras (PROTAC) technology can particularly be useful for targeting the abnormally accumulated β-catenin in cancer cells.127 This approach may be more interesting than disruption of the challenging PPIs. A PROTAC peptide (xStAx-VHLL) has been reported by connecting stapled peptide StAx-35R with the von Hippel-Lindau (VHL) ligand.128 xStAx-VHLL achieved durable β-catenin degradation and impaired Wnt/β-catenin signaling in cancer cells. Moreover, xStAx-VHLL decreased colorectal cancer cell proliferation and tumor formation in nude mice, and reduced the existing tumors in mouse model with APC gene mutation. xStAx-VHLL also inhibited survival of organoids derived from colorectal cancer patients. These results indicate the PROTAC might have potential for developing therapeutic agents to treat β-catenin-related cancer. On the other hand, it is worth noting that small molecules like MSAB and carnosic acid promote the proteasomal degradation of β-catenin.
Taken together, β-catenin is a highly promising target for cancer drug discovery because it is a downstream effector of the Wnt/β-catenin signaling pathway and all upstream Wnt signaling pathways are convergent onto this transcription coactivator. However, targeting this protein has proven challenging. A number of small-molecule inhibitors have been reported to bind with β-catenin, disrupt β-catenin-mediated PPIs, and suppress Wnt/β-catenin signaling. Although none of these inhibitors go beyond preclinical studies, they serve as good starting-points for further inhibitor discovery.
Acknowledgments
This work was supported by the Department of Defense CDMRP BCRP breakthrough award W81XWH-14-1-0083, the Susan G. Komen Career Catalyst Research Grant CCR16380693, Floridian Breast Cancer Foundation Scientific Grant (19012901), and the 2020 Moffitt Team Science Award. The H. Lee Moffitt Cancer Center & Research Institute is a NCI-designated Comprehensive Cancer Center, supported by the Cancer Center Support Grant P30 CA076292.
Author Biosketches
Zhen Wang obtained his Ph.D. degree in Medicinal Chemistry from Guangzhou Institute of Biomedicine and Health (GIBH), University of Chinese Academy of Sciences. His Ph.D. project was design and synthesis of small-molecule inhibitors targeting receptor kinase discoidin domain receptors (DDRs). Currently, he is a postdoctoral fellow at H. Lee Moffitt Cancer Center and Research Institute. He is working on design and synthesis of small-molecule inhibitors for β-catenin/TCF and β-catenin/BCL9 protein–protein interactions.
Zilu Li received his B.S. degree in Pharmaceutical Science from Shandong University. He is currently a Ph.D. student at the University of South Florida and a research associate at H. Lee Moffitt Cancer Center and Research Institute. He is working on design and synthesis of small-molecule inhibitors to disrupt the β-catenin/BCL9 protein-protein interaction.
Haitao Ji is Associate Professor and Associate Member at H. Lee Moffitt Cancer Center and Research Institute. He received his B.S. degree in Pharmacy and Ph.D. degree in Medicinal Chemistry from the Second Military Medical University. He received the postdoctoral training with Prof. Richard B. Silverman at Northwestern University. Dr. Ji’s research has been focused on developing new technologies to design and characterize inhibitors for PPIs, applying newly developed tools/techniques to design and synthesize potent and selective inhibitors of important PPI targets, and developing new PPI inhibitors into novel targeted therapies.
Footnotes
Conflict of Interest Statement
The authors declare no competing financial interest.
Data Availability Statement
Data sharing not applicable as no datasets were generated or analyzed during the current study.
References.
- 1.Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31(1):99–109. [DOI] [PubMed] [Google Scholar]
- 2.Nusse R, Brown A, Papkoff J, et al. A new nomenclature for int-1 and related genes: the Wnt gene family. Cell. 1991;64(2):231. [DOI] [PubMed] [Google Scholar]
- 3.Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–999. [DOI] [PubMed] [Google Scholar]
- 4.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26. [DOI] [PubMed] [Google Scholar]
- 5.Gumbiner BM. Carcinogenesis: a balance between β-catenin and APC. Curr Biol. 1997;7(7):R443–446. [DOI] [PubMed] [Google Scholar]
- 6.Rubinfeld B, Albert I, Porfiri E, et al. Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science. 1996;272(5264):1023–1026. [DOI] [PubMed] [Google Scholar]
- 7.Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst. 2014;106(1):djt356. [DOI] [PubMed] [Google Scholar]
- 8.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. [DOI] [PubMed] [Google Scholar]
- 9.Polakis P Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunner E, Peter O, Schweizer L, et al. Pangolin encodes a Lef-1 homologue that acts downstream of Armadillo to transduce the Wingless signal in Drosophila. Nature. 1997;385(6619):829–833. [DOI] [PubMed] [Google Scholar]
- 11.Van De Wetering M, Cavallo R, Dooijes D, et al. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell. 1997;88(6):789–799. [DOI] [PubMed] [Google Scholar]
- 12.Takemaru KI, Moon RT. The transcriptional coactivator CBP interacts with β-catenin to activate gene expression. J Cell Biol. 2000;149(2):249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hecht A, Vleminckx K, Stemmler MP, et al. The p300/CBP acetyltransferases function as transcriptional coactivators of β-catenin in vertebrates. EMBO J. 2000;19(8):1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber AH, Weis WI. The structure of the β-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by β-catenin. Cell. 2001;105(3):391–402. [DOI] [PubMed] [Google Scholar]
- 15.Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol. 2010;2(2):a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103(2):311–320. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi K, Nagatoishi S, Tsumoto K, et al. Discovery of chemical probes that suppress Wnt/β-catenin signaling through high-throughput screening. Cancer Sci. 2020;111(3):783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakajima M, Fukuchi M, Miyazaki T, et al. Reduced expression of Axin correlates with tumour progression of oesophageal squamous cell carcinoma. Br J Cancer. 2003;88(11):1734–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu W, Dong X, Mai M, et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat Genet. 2000;26(2):146–147. [DOI] [PubMed] [Google Scholar]
- 20.Lammi L, Arte S, Somer M, et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74(5):1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kahn M Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13(7):513–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubinfeld B, Robbins P, El-Gamil M, et al. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 1997;275(5307):1790–1792. [DOI] [PubMed] [Google Scholar]
- 23.Han ZG. Functional genomic studies: insights into the pathogenesis of liver cancer. Annu Rev Genomics Hum Genet. 2012;13:171–205. [DOI] [PubMed] [Google Scholar]
- 24.Sastre-Perona A, Santisteban P. Role of the wnt pathway in thyroid cancer. Front Endocrinol (Lausanne). 2012;3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teeuwssen M, Fodde R. Wnt Signaling in Ovarian Cancer Stemness, EMT, and Therapy Resistance. J Clin Med. 2019;8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herr P, Hausmann G, Basler K. WNT secretion and signalling in human disease. Trends Mol Med. 2012;18(8):483–493. [DOI] [PubMed] [Google Scholar]
- 27.Bafico A, Liu G, Goldin L, et al. An autocrine mechanism for constitutive Wnt pathway activation in human cancer cells. Cancer Cell. 2004;6(5):497–506. [DOI] [PubMed] [Google Scholar]
- 28.Caldwell GM, Jones C, Gensberg K, et al. The Wnt antagonist sFRP1 in colorectal tumorigenesis. Cancer Res. 2004;64(3):883–888. [DOI] [PubMed] [Google Scholar]
- 29.Lee AY, He B, You L, et al. Expression of the secreted frizzled-related protein gene family is downregulated in human mesothelioma. Oncogene. 2004;23(39):6672–6676. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki H, Watkins DN, Jair KW, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36(4):417–422. [DOI] [PubMed] [Google Scholar]
- 31.Fukui T, Kondo M, Ito G, et al. Transcriptional silencing of secreted frizzled related protein 1 (SFRP 1) by promoter hypermethylation in non-small-cell lung cancer. Oncogene. 2005;24(41):6323–6327. [DOI] [PubMed] [Google Scholar]
- 32.Zou H, Molina JR, Harrington JJ, et al. Aberrant methylation of secreted frizzled-related protein genes in esophageal adenocarcinoma and Barrett’s esophagus. Int J Cancer. 2005;116(4):584–591. [DOI] [PubMed] [Google Scholar]
- 33.Milovanovic T, Planutis K, Nguyen A, et al. Expression of Wnt genes and frizzled 1 and 2 receptors in normal breast epithelium and infiltrating breast carcinoma. Int J Oncol. 2004;25(5):1337–1342. [PubMed] [Google Scholar]
- 34.Uematsu K, Kanazawa S, You L, et al. Wnt pathway activation in mesothelioma: evidence of Dishevelled overexpression and transcriptional activity of β-catenin. Cancer Res. 2003;63(15):4547–4551. [PubMed] [Google Scholar]
- 35.Dow LE, O’rourke KP, Simon J, et al. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell. 2015;161(7):1539–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–667. [DOI] [PubMed] [Google Scholar]
- 37.Dudek H, Wong DH, Arvan R, et al. Knockdown of β-catenin with dicer-substrate siRNAs reduces liver tumor burden in vivo. Mol Ther. 2014;22(1):92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–235. [DOI] [PubMed] [Google Scholar]
- 39.Spranger S, Dai D, Horton B, et al. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31(5):711–723.e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luke JJ, Bao R, Sweis RF, et al. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res. 2019;25(10):3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiménez-Sánchez A, Memon D, Pourpe S, et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell. 2017;170(5):927–938.e920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grasso CS, Giannakis M, Wells DK, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 2018;8(6):730–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruiz De Galarreta M, Bresnahan E, Molina-Sanchez P, et al. β-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019;9(8):1124–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waaler J, Mygland L, Tveita A, et al. Tankyrase inhibition sensitizes melanoma to PD-1 immune checkpoint blockade in syngeneic mouse models. Commun Biol. 2020;3(1):196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gurney A, Axelrod F, Bond CJ, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. 2012;109(29):11717–11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dealmeida VI, Miao L, Ernst JA, et al. The soluble wnt receptor Frizzled8CRD-hFc inhibits the growth of teratocarcinomas in vivo. Cancer Res. 2007;67(11):5371–5379. [DOI] [PubMed] [Google Scholar]
- 47.Takada R, Satomi Y, Kurata T, et al. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev Cell. 2006;11(6):791–801. [DOI] [PubMed] [Google Scholar]
- 48.Chen B, Dodge ME, Tang W, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5(2):100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu J, Pan S, Hsieh MH, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110(50):20224–20229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hsiao SJ, Smith S. Tankyrase function at telomeres, spindle poles, and beyond. Biochimie. 2008;90(1):83–92. [DOI] [PubMed] [Google Scholar]
- 51.Huang SM, Mishina YM, Liu S, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461(7264):614–620. [DOI] [PubMed] [Google Scholar]
- 52.Waaler J, Machon O, Von Kries JP, et al. Novel synthetic antagonists of canonical Wnt signaling inhibit colorectal cancer cell growth. Cancer Res. 2011;71(1):197–205. [DOI] [PubMed] [Google Scholar]
- 53.Buchstaller HP, Anlauf U, Dorsch D, et al. Discovery and optimization of 2-arylquinazolin-4-ones into a potent and selective tankyrase inhibitor modulating Wnt pathway activity. J Med Chem. 2019;62(17):7897–7909. [DOI] [PubMed] [Google Scholar]
- 54.Rodriguez-Blanco J, Li B, Long J, et al. A CK1α activator penetrates the brain and shows efficacy against drug-resistant metastatic medulloblastoma. Clin Cancer Res. 2019;25(4):1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen C, Li B, Astudillo L, et al. The CK1α activator pyrvinium enhances the catalytic efficiency (kcat/Km) of CK1α. Biochemistry. 2019;58(51):5102–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheong JK, Nguyen TH, Wang H, et al. IC261 induces cell cycle arrest and apoptosis of human cancer cells via CK1δ/ɛ and Wnt/β-catenin independent inhibition of mitotic spindle formation. Oncogene. 2011;30(22):2558–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monastyrskyi A, Nilchan N, Quereda V, et al. Development of dual casein kinase 1δ/1ɛ (CK1δ/ɛ) inhibitors for treatment of breast cancer. Bioorg Med Chem. 2018;26(3):590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shirai F, Mizutani A, Yashiroda Y, et al. Design and discovery of an orally efficacious spiroindolinone-based tankyrase inhibitor for the treatment of colon cancer. J Med Chem. 2020;63(8):4183–4204. [DOI] [PubMed] [Google Scholar]
- 59.Thorne CA, Hanson AJ, Schneider J, et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat Chem Biol. 2010;6(11):829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kramps T, Peter O, Brunner E, et al. Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear β-catenin-TCF complex. Cell. 2002;109(1):47–60. [DOI] [PubMed] [Google Scholar]
- 61.Emami KH, Nguyen C, Ma H, et al. A small molecule inhibitor of β-catenin/CREB-binding protein transcription. Proc Natl Acad Sci U S A. 2004;101(34):12682–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kimura K, Ikoma A, Shibakawa M, et al. Safety, tolerability, and preliminary efficacy of the anti-fibrotic small molecule PRI-724, a CBP/β-catenin inhibitor, in patients with hepatitis C virus-related cirrhosis: A single-center, open-label, dose escalation phase 1 trial. EBioMedicine. 2017;23:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Benoit YD, Mitchell RR, Risueño RM, et al. Sam68 allows selective targeting of human cancer stem cells. Cell Chem Biol. 2017;24(7):833–844.e839. [DOI] [PubMed] [Google Scholar]
- 64.Jang G-B, Hong I-S, Kim R-J, et al. Wnt/β-catenin small-molecule inhibitor CWP232228 preferentially inhibits the growth of breast cancer stem-like cells. Cancer Res. 2015;75(8):1691–1702. [DOI] [PubMed] [Google Scholar]
- 65.Lee J-H, Faderl S, Pagel JM, et al. Phase 1 study of CWP232291 in patients with relapsed or refractory acute myeloid leukemia and myelodysplastic syndrome. Blood Adv. 2020;4(9):2032–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim YM, Ma H, Oehler VG, et al. The gamma catenin/CBP complex maintains survivin transcription in β-catenin deficient/depleted cancer cells. Curr Cancer Drug Targets. 2011;11(2):213–225. [DOI] [PubMed] [Google Scholar]
- 67.Xing Y, Takemaru K, Liu J, et al. Crystal structure of a full-length β-catenin. Structure. 2008;16(3):478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eklof Spink K, Fridman SG, Weis WI. Molecular mechanisms of β-catenin recognition by adenomatous polyposis coli revealed by the structure of an APC-β-catenin complex. EMBO J. 2001;20(22):6203–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ha NC, Tonozuka T, Stamos JL, et al. Mechanism of phosphorylation-dependent binding of APC to β-catenin and its role in β-catenin degradation. Mol Cell. 2004;15(4):511–521. [DOI] [PubMed] [Google Scholar]
- 70.Xing Y, Clements WK, Le Trong I, et al. Crystal structure of a β-catenin/APC complex reveals a critical role for APC phosphorylation in APC function. Mol Cell. 2004;15(4):523–533. [DOI] [PubMed] [Google Scholar]
- 71.Xing Y, Clements WK, Kimelman D, et al. Crystal structure of a β-catenin/axin complex suggests a mechanism for the β-catenin destruction complex. Genes Dev. 2003;17(22):2753–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Graham TA, Clements WK, Kimelman D, et al. The crystal structure of the β-catenin/ICAT complex reveals the inhibitory mechanism of ICAT. Mol Cell. 2002;10(3):563–571. [DOI] [PubMed] [Google Scholar]
- 73.Daniels DL, Weis WI. ICAT inhibits β-catenin binding to Tcf/Lef-family transcription factors and the general coactivator p300 using independent structural modules. Mol Cell. 2002;10(3):573–584. [DOI] [PubMed] [Google Scholar]
- 74.Sun J, Weis WI. Biochemical and structural characterization of β-catenin interactions with nonphosphorylated and CK2-phosphorylated Lef-1. J Mol Biol. 2011;405(2):519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Graham TA, Weaver C, Mao F, et al. Crystal structure of a β-catenin/Tcf complex. Cell. 2000;103(6):885–896. [DOI] [PubMed] [Google Scholar]
- 76.Poy F, Lepourcelet M, Shivdasani RA, et al. Structure of a human Tcf4-β-catenin complex. Nat Struct Biol. 2001;8(12):1053–1057. [DOI] [PubMed] [Google Scholar]
- 77.Graham TA, Ferkey DM, Mao F, et al. Tcf4 can specifically recognize beta-catenin using alternative conformations. Nat Struct Biol. 2001;8(12):1048–1052. [DOI] [PubMed] [Google Scholar]
- 78.Sampietro J, Dahlberg CL, Cho US, et al. Crystal structure of a β-catenin/BCL9/Tcf4 complex. Mol Cell. 2006;24(2):293–300. [DOI] [PubMed] [Google Scholar]
- 79.Yumoto F, Nguyen P, Sablin EP, et al. Structural basis of coactivation of liver receptor homolog-1 by β-catenin. Proc Natl Acad Sci U S A. 2012;109(1):143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Von Kries JP, Winbeck G, Asbrand C, et al. Hot spots in β-catenin for interactions with LEF-1, conductin and APC. Nat Struct Biol. 2000;7(9):800–807. [DOI] [PubMed] [Google Scholar]
- 81.Knapp S, Zamai M, Volpi D, et al. Thermodynamics of the high-affinity interaction of TCF4 with β-catenin. J Mol Biol. 2001;306(5):1179–1189. [DOI] [PubMed] [Google Scholar]
- 82.Omer CA, Miller PJ, Diehl RE, et al. Identification of Tcf4 residues involved in high-affinity β-catenin binding. Biochem Biophys Res Commun. 1999;256(3):584–590. [DOI] [PubMed] [Google Scholar]
- 83.Fasolini M, Wu X, Flocco M, et al. Hot spots in Tcf4 for the interaction with β-catenin. J Biol Chem. 2003;278(23):21092–21098. [DOI] [PubMed] [Google Scholar]
- 84.Gail R, Frank R, Wittinghofer A. Systematic peptide array-based delineation of the differential β-catenin interaction with Tcf4, E-cadherin, and adenomatous polyposis coli. J Biol Chem. 2005;280(8):7107–7117. [DOI] [PubMed] [Google Scholar]
- 85.Yu B, Huang Z, Zhang M, et al. Rational design of small-molecule inhibitors for β-catenin/T-cell factor protein-protein interactions by bioisostere replacement. ACS Chem Biol. 2013;8(3):524–529. [DOI] [PubMed] [Google Scholar]
- 86.Hoggard LR, Zhang Y, Zhang M, et al. Rational design of selective small-molecule inhibitors for β-catenin/B-cell lymphoma 9 protein-protein interactions. J Am Chem Soc. 2015;137(38):12249–12260. [DOI] [PubMed] [Google Scholar]
- 87.De La Roche M, Worm J, Bienz M. The function of BCL9 in Wnt/β-catenin signaling and colorectal cancer cells. BMC Cancer. 2008;8:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kawamoto SA, Thompson AD, Coleska A, et al. Analysis of the interaction of BCL9 with β-catenin and development of fluorescence polarization and surface plasmon resonance binding assays for this interaction. Biochemistry. 2009;48(40):9534–9541. [DOI] [PubMed] [Google Scholar]
- 89.Hoffmans R, Basler K. Identification and in vivo role of the Armadillo-Legless interaction. Development. 2004;131(17):4393–4400. [DOI] [PubMed] [Google Scholar]
- 90.Hoffmans R, Basler K. BCL9–2 binds Arm/β-catenin in a Tyr142-independent manner and requires Pygopus for its function in Wg/Wnt signaling. Mech Dev. 2007;124(1):59–67. [DOI] [PubMed] [Google Scholar]
- 91.Grossmann TN, Yeh JT, Bowman BR, et al. Inhibition of oncogenic Wnt signaling through direct targeting of β-catenin. Proc Natl Acad Sci U S A. 2012;109(44):17942–17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schneider JA, Craven TW, Kasper AC, et al. Design of peptoid-peptide macrocycles to inhibit the β-catenin TCF interaction in prostate cancer. Nat Commun. 2018;9(1):4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dietrich L, Rathmer B, Ewan K, et al. Cell permeable stapled peptide inhibitor of Wnt signaling that targets β-catenin protein-protein interactions. Cell Chem Biol. 2017;24(8):958–968.e955. [DOI] [PubMed] [Google Scholar]
- 94.Cui HK, Zhao B, Li Y, et al. Design of stapled α-helical peptides to specifically activate Wnt/β-catenin signaling. Cell Res. 2013;23(4):581–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gwak J, Hwang SG, Park HS, et al. Small molecule-based disruption of the Axin/β-catenin protein complex regulates mesenchymal stem cell differentiation. Cell Res. 2012;22(1):237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lepourcelet M, Chen YN, France DS, et al. Small-molecule antagonists of the oncogenic Tcf/β-catenin protein complex. Cancer Cell. 2004;5(1):91–102. [DOI] [PubMed] [Google Scholar]
- 97.Sukhdeo K, Mani M, Zhang Y, et al. Targeting the β-catenin/TCF transcriptional complex in the treatment of multiple myeloma. Proc Natl Acad Sci U S A. 2007;104(18):7516–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li X, Pu J, Jiang S, et al. Henryin, an ent-kaurane diterpenoid, inhibits Wnt signaling through interference with β-catenin/TCF4 interaction in colorectal cancer cells. PLoS One. 2013;8(7):e68525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tian W, Han X, Yan M, et al. Structure-based discovery of a novel inhibitor targeting the β-catenin/Tcf4 interaction. Biochemistry. 2012;51(2):724–731. [DOI] [PubMed] [Google Scholar]
- 100.Gonsalves FC, Klein K, Carson BB, et al. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc Natl Acad Sci U S A. 2011;108(15):5954–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Catrow JL, Zhang Y, Zhang M, et al. Discovery of selective small-molecule inhibitors for the β-catenin/T-cell factor protein-protein interaction through the optimization of the acyl hydrazone moiety. J Med Chem. 2015;58(11):4678–4692. [DOI] [PubMed] [Google Scholar]
- 102.Fang L, Zhu Q, Neuenschwander M, et al. A small-molecule antagonist of the β-catenin/TCF4 interaction blocks the self-renewal of cancer stem cells and suppresses tumorigenesis. Cancer Res. 2016;76(4):891–901. [DOI] [PubMed] [Google Scholar]
- 103.Lee E, Madar A, David G, et al. Inhibition of androgen receptor and β-catenin activity in prostate cancer. Proc Natl Acad Sci U S A. 2013;110(39):15710–15715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53(7):2719–2740. [DOI] [PubMed] [Google Scholar]
- 105.Baell J, Walters MA. Chemistry: Chemical con artists foil drug discovery. Nature. 2014;513(7519):481–483. [DOI] [PubMed] [Google Scholar]
- 106.Pouliot M, Jeanmart S. Pan assay interference compounds (PAINS) and other promiscuous compounds in antifungal research. J Med Chem. 2016;59(2):497–503. [DOI] [PubMed] [Google Scholar]
- 107.Trosset JY, Dalvit C, Knapp S, et al. Inhibition of protein-protein interactions: the discovery of druglike β-catenin inhibitors by combining virtual and biophysical screening. Proteins. 2006;64(1):60–67. [DOI] [PubMed] [Google Scholar]
- 108.Huang Z, Zhang M, Burton SD, et al. Targeting the Tcf4 G13ANDE17 binding site to selectively disrupt β-catenin/T-cell factor protein-protein interactions. ACS Chem Biol. 2014;9(1):193–201. [DOI] [PubMed] [Google Scholar]
- 109.Hülsken J, Birchmeier W, Behrens J. E-cadherin and APC compete for the interaction with β-catenin and the cytoskeleton. J Cell Biol. 1994;127(6 Pt 2):2061–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Orsulic S, Huber O, Aberle H, et al. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci. 1999;112 (Pt 8):1237–1245. [DOI] [PubMed] [Google Scholar]
- 111.Choi HJ, Huber AH, Weis WI. Thermodynamics of β-catenin-ligand interactions: the roles of the N- and C-terminal tails in modulating binding affinity. J Biol Chem. 2006;281(2):1027–1038. [DOI] [PubMed] [Google Scholar]
- 112.Wang Z, Zhang M, Wang J, et al. Optimization of peptidomimetics as selective inhibitors for the β-catenin/T-cell factor protein-protein interaction. J Med Chem. 2019;62(7):3617–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hwang SY, Deng X, Byun S, et al. Direct targeting of β-catenin by a small molecule stimulates proteasomal degradation and suppresses oncogenic Wnt/β-catenin signaling. Cell Rep. 2016;16(1):28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shin SH, Lim DY, Reddy K, et al. A small molecule inhibitor of the β-catenin-TCF4 interaction suppresses colorectal cancer growth in vitro and in vivo. EBioMedicine. 2017;25:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sang P, Zhang M, Shi Y, et al. Inhibition of β-catenin/B cell lymphoma 9 protein-protein interaction using α-helix-mimicking sulfono-γ-AApeptide inhibitors. Proc Natl Acad Sci U S A. 2019;116(22):10757–10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kawamoto SA, Coleska A, Ran X, et al. Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. J Med Chem. 2012;55(3):1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Takada K, Zhu D, Bird GH, et al. Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci Transl Med. 2012;4(148):148ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Feng M, Jin JQ, Xia L, et al. Pharmacological inhibition of β-catenin/BCL9 interaction overcomes resistance to immune checkpoint blockades by modulating Treg cells. Sci Adv. 2019;5(5):eaau5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.De La Roche M, Rutherford TJ, Gupta D, et al. An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nat Commun. 2012;3:680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang M, Wang Z, Zhang Y, et al. Structure-based optimization of small-molecule inhibitors for the β-catenin/B-cell lymphoma 9 protein-protein interaction. J Med Chem. 2018;61(7):2989–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wisniewski JA, Yin J, Teuscher KB, et al. Structure-based design of 1,4-dibenzoylpiperazines as β-catenin/B-cell lymphoma 9 protein-protein interaction inhibitors. ACS Med Chem Lett. 2016;7(5):508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cierpicki T, Grembecka J. Targeting protein-protein interactions in hematologic malignancies: still a challenge or a great opportunity for future therapies? Immunol Rev. 2015;263(1):279–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jones CG, Martynowycz MW, Hattne J, et al. The CryoEM method MicroED as a powerful tool for small molecule structure determination. ACS Cent Sci. 2018;4(11):1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Renaud JP, Chari A, Ciferri C, et al. Cryo-EM in drug discovery: achievements, limitations and prospects. Nat Rev Drug Discov. 2018;17(7):471–492. [DOI] [PubMed] [Google Scholar]
- 125.Kaelin WG, Jr. Common pitfalls in preclinical cancer target validation. Nat Rev Cancer. 2017;17(7):425–440. [DOI] [PubMed] [Google Scholar]
- 126.Riascos-Bernal DF, Chinnasamy P, Cao LL, et al. β-Catenin C-terminal signals suppress p53 and are essential for artery formation. Nat Commun. 2016;7:12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Salami J, Crews CM. Waste disposal-An attractive strategy for cancer therapy. Science. 2017;355(6330):1163–1167. [DOI] [PubMed] [Google Scholar]
- 128.Liao H, Li X, Zhao L, et al. A PROTAC peptide induces durable β-catenin degradation and suppresses Wnt-dependent intestinal cancer. Cell Discov. 2020;6:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mosimann C, Hausmann G, Basler K. β-Catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol. 2009;10(4):276–286. [DOI] [PubMed] [Google Scholar]
- 130.Mellor J It takes a PHD to read the histone code. Cell. 2006;126(1):22–24. [DOI] [PubMed] [Google Scholar]
- 131.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267(5196):383–386. [DOI] [PubMed] [Google Scholar]
- 132.Keskin O, Ma B, Nussinov R. Hot regions in protein--protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005;345(5):1281–1294. [DOI] [PubMed] [Google Scholar]
- 133.Guo W, Wisniewski JA, Ji H. Hot spot-based design of small-molecule inhibitors for protein-protein interactions. Bioorg Med Chem Lett. 2014;24(11):2546–2554. [DOI] [PubMed] [Google Scholar]