

Abstract
Spinocerebellar ataxia type 1 (SCA1) is a fatal neurodegenerative disease caused by an abnormal expansion of CAG repeats in the Ataxin1 (ATXN1) gene. SCA1 is characterized by motor deficits, cerebellar neurodegeneration, gliosis and gene expression changes. Expression of brain derived neurotrophic factor (BDNF), growth factor important for the survival and function of cerebellar neurons is decreased in ATXN1[82Q] mice, the Purkinje neuron specific transgenic mouse model of SCA1. As this decrease in BDNF expression may contribute to cerebellar neurodegeneration we tested whether delivery of extrinsic human BDNF via osmotic Alzet pumps has a beneficial effect on disease severity in this mouse model of SCA1. Additionally, to test the effects of BDNF on established and progressing cerebellar pathogenesis and motor deficits we delivered BDNF post-symptomatically.
We have found that post-symptomatic delivery of extrinsic BDNF ameliorated motor deficits, and cerebellar pathology (i.e. dendritic atrophy of Purkinje cells, and astrogliosis) indicating therapeutic potential of BDNF even after the onset of symptoms in SCA1.However, BDNF did not alter Purkinje cell gene expression changes indicting that certain aspects of disease pathogenesis cannot be ameliorated/slowed down with BDNF and that combinational therapies may be needed.
Keywords: ATAXIN-1, BDNF, gliosis, Purkinje cells, cerebellar ataxia, neurodegeneration
Introduction
Spinocerebellar ataxia type 1 (SCA1) is a dominantly inherited and fatal neurodegenerative disease resulting from an overexpansion of glutamine(Q)-encoding CAG repeats within the Ataxin1 (ATXN1) gene [1,2]. SCA1 belongs to a group of polyglutamine (polyQ) disorders that also includes SCA2, 3, 6, 7, 17, spinobulbar muscular atrophy, Huntington’s disease (HD), and dentatorubropallidoluysian atrophy [3–5]. Clinical onset of SCA1 is characterized by progressive ataxia, or loss of motor coordination and balance, which typically starts during patients’ mid-thirties [5–8]. In addition, patients with SCA1 exhibit cognitive deficits, depression, and premature lethality 10–20 years after disease onset [6]. Currently, there are no disease modifying treatments for SCA1 [9].
Treatment with neuroprotective factors, such as brain derived neurotrophic factor (BDNF), has been demonstrated to ameliorate neural toxicity in several neurodegenerative diseases, including mouse models of Alzheimer’s disease and Huntington’s disease. This is thought to occur because BDNF, as a neurotrophin, supports neuronal health and function by promoting cell survival and synapse plasticity in adult tissue [10–12]. Moreover, previous studies show that BDNF plays an important role in both cerebellar development and cerebellar neuronal function in adult mice [13–15].
We have previously reported a reduction in cerebellar BDNF in ATXN1[82Q] mice, a Purkinje neuron specific transgenic mouse model of SCA1, that may contribute to the pathological degeneration of the cerebellar cortex [16]. In addition, administration of extrinsic BDNF intracereberoventricularly (ICV) to early stage ATXN1[82Q] mice delays the onset of motor deficits and ameliorates several markers of SCA1 pathology[16]. While the dominantly inherited nature of SCA1 allows for pre-symptomatic treatment, prolonged chronic BDNF treatment may not be affordable and may have side effects. Therefore, we examined the therapeutic efficacy of BDNF treatment after the onset of motor symptoms. We observed that post-symptomatic BDNF delivery ameliorates SCA1 disease severity through improvement of rotarod performance and decrease in gliosis and neuronal atrophy.
Materials and Methods
Mice
The creation of the ATXN1[82Q] mice was previously described [17]. Equal number of male and female ATXN1[82Q] and wild-type mice were randomly allocated to BDNF or control artificial cerebrospinal fluid (aCSF) groups (N =16, 12, 12, 15 respectively). We surgically implanted ALZET pumps (Alzet Model 1004) into 12-week-old mice in a subcutaneous pocket in their back. Delivery cannula was placed into the right lateral ventricle using stereotaxic surgery (A/P, 1.1 mm; M/L, 0.5 mm D/V, −2.5 mm from Bregma) as previously described [16]. ALZET pumps delivered BDNF or aCSF at a steady flow rate for 4 weeks following implantation (20 μg of human recombinant BDNF (R&D Systems Cat. 248-BD-250/CF) in 100μl per micropump, resulting in a delivery rate of 0.71 μg/day).
In all experiments, investigators were blinded to the genotype/treatment. While we started with the same number of animals in each group, the final number of animals per condition varied depending on the success of surgery, including survival from surgery and correct placement of cannula. All mice in which postmortem examination revealed that cannula was misplaced or obstructed were eliminated from the analysis.
Animal experimentation was approved by University of Minnesota and was conducted in accordance with the National Institutes of Health’s (NIH) Principles of Laboratory Animal Care (86–23, revised 1985), and the American Physiological Society’s Guiding Principles in the Use of Animals.
Rotarod analysis
Mice were tested on rotarod (#47600; Ugo Basile) to evaluate motor deficits as described previously [16,18,19] prior to BDNF delivery, 2 and 5 weeks after the BDNF delivery started. Rotarod paradigm consisted of four trials per day over four days with acceleration from 5 to 40 rotations per minute (rpm) over minutes 0 to 5, followed by 40 rpm constant speed from 5 to 10 min. Latency to fall was recorded.
Immunofluorescent (IF) staining
IF was performed on minimum of six different floating 45 μm thick brain slices from each mouse (six technical replicates per mouse). We used primary antibodies against Purkinje cell marker calbindin (mouse, Sigma-Aldrich) and astrocytic marker glial fibrillary acidic protein (GFAP) (rabbit, DAKO) as previously described [19,20]. Confocal images were acquired using a confocal microscope (Olympus FV1000) using a 20X oil objective. Z-stacks consisting of twenty non-overlapping 1 μm thick slices were taken of each stained brain slice (i.e. six z-stacks per each mouse, each taken from a different brain slice). The laser power and detector gain were standardized and fixed between mice within a surgery cohort, and all images for mice within a cohort were acquired in a single imaging session to allow for quantitative comparison.
Quantitative analysis was performed using ImageJ (NIH) as described previously [19]. We quantified a minimum of six different images from each mouse, each taken from a different brain slice. Measurements of molecular layer thickness were taken on calbindin immunostained sections by measuring the distance from the base of the Purkinje cell body to the end of the dendrite at the pial surface. Six measurements at the primary fissure were taken from each cerebellar section per animal. These measurements were averaged to represent the molecular thickness of each biological replicate. GFAP intensity was quantified by measuring the mean gray value of GFAP staining in the molecular and Purkinje cell layers of the primary fissure. At least six measurements were taken from images of different slices and averaged. Relative GFAP intensity of each mouse was normalized to the WT aCSF treated mouse within the surgery cohort.
Enzyme-linked immunosorbent assay (ELISA)
Tissue was extracted from each mouse, and a quarter of the cerebellum and quarter of the cerebrum separated and weighed to get total tissue weight of each. Tissue protein was extracted from mouse cerebellum and cerebrum using 300uL and 600uL of Tris-Triton Lysis Buffer [150nM sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS (sodium dodecyl sulfate), and 50mM Tris (pH 8.0)] respectively, as has been described previously [20–22]. Following lysate preparation, total BDNF was quantified in duplicates using the Total BDNF Quantikine ELISA kit (Biotechne-R&D Systems).
Reverse transcription and quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from mouse cerebella using TRIzol (Life Technologies) and RT-qPCR was performed as described previously [16,23].
Western Blot
Cerebellar protein lysates prepared for ELISA were used for Western blotting. Protein concentration was measured using a Bradford Assay as previously described [21,22]. Western blot (N=3 samples per group) was performed using 50ug protein per well with two negative controls (cerebellar lysate from an Atxn1 null mouse[24] and water loading control). To detect ATXN1 we used an anti-ATXN1 antibody (11NQ, rabbit, gift from Dr. Harry Orr) and normalized samples using α-tubulin (mouse, Sigma) as a loading control.
Statistical analysis
Wherever possible, sample sizes were calculated using power analyses based on the standard deviations from our previous studies, significance level of 5%, and power of 90%. Statistical tests were performed with GraphPad Prism. Data was analyzed using unpaired Student’s t-test (to test statistical significance of differences between two groups), or one way ANOVA followed by the Sidak’s multiple comparisons post-hoc test. Outliers were determined using GraphPad PRISM’s Robust regression and Outlier removal (ROUT) with a Q=1% for non-biased selection.
Data availability
All the data from this study are available from the authors.
Results
Delivery of BDNF late in disease ameliorates motor deficits in SCA1
ATXN1[82Q] mice demonstrate reproducible motor deficits and cerebellar pathology by 12 weeks[21,23,25–28]. Thus, to determine whether ICV BDNF delivery starting after the onset of motor deficits has therapeutic benefits we choose 14 weeks as treatment starting time point (Figure 1A). Pre-surgery rotarod was performed at 12 weeks of age to confirm presence of motor deficits in ATXN1[82Q] mice. As expected, ATXN1[82Q] mice performed significantly worse on the rotarod compared to their wild-type (WT) littermate controls (latency to fall for WT mice was 347±30.56 s, N=24, while for ATXN1[82Q] mice it was 188.9±12.82 s, N=28, two-tailed unpaired t-test P<0.0001, Figure 1B) confirming that ATXN1[82Q] mice exhibit motor deficits at 12 weeks. At 14 weeks we randomly divided mice into four experimental groups (WT and ATXN1[82Q] mice treated with BDNF or aCSF) and performed stereotaxic surgeries to implant cannulas into the right lateral ventricle. Cannulas were connected to Osmotic ALZET pumps to deliver recombinant BDNF (at a rate of ~ 0.71 μg/day) or control artificial cerebrospinal fluid (aCSF) for a total of 4 weeks. Importantly, there was no difference in the pre-surgery rotarod performance of ATXN1[82Q] mice that underwent BDNF or aCSF treatments (latency to fall of ATXN1[82Q] mice chosen for future aCSF delivery was 187.5±16.82 s, N=13, and for ATXN1[82Q] mice chosen for future BDNF delivery latency was 183.5±18.07 s, N=15, two-tailed t-test P=0.8839, Figure 1C).
Figure 1. Delivery of BDNF after onset of motor symptoms in SCA1 mice.
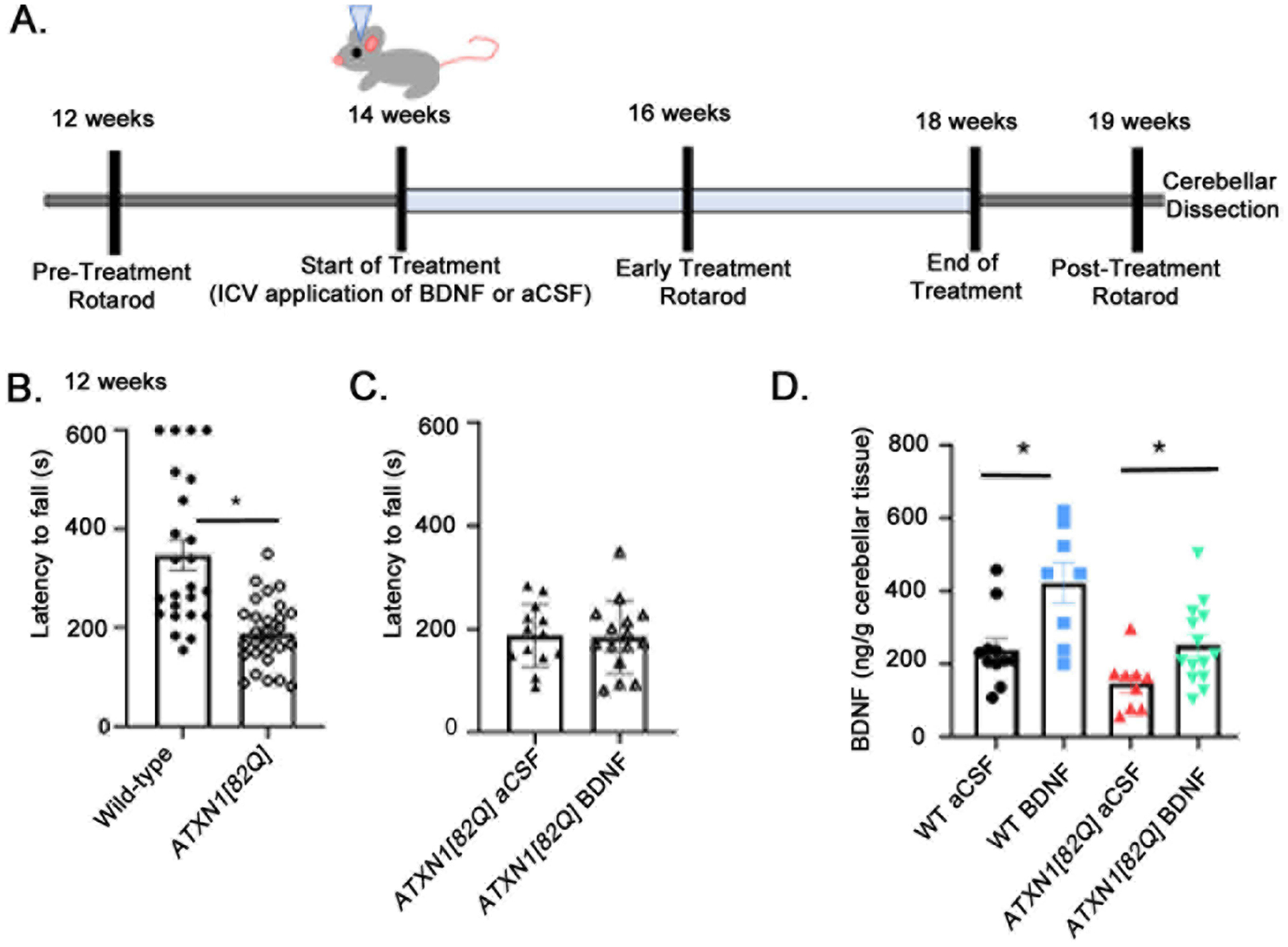
A. Schematic of intracerebroventricular BDNF treatment and evaluation. In brief, ATXN1[82Q] mice and their WT littermates were treated with either ICV BDNF or aCSF starting at 14 weeks of age. Rotorod testing to assess relative motor function was done at 12 weeks (pre-treatment), 16 weeks (mid-treatment), and 19 weeks (post-treatment). Following the final rotarod test, cerebellar tissue was dissected and processed for analysis. B. Rotorod at 12 weeks of age (WT, N=24, ATXN1[82Q], N=28). C. Rotarod performance of ATXN1[82Q] mice randomly assigned into two groups (ATXN1[82Q] mice chosen for future aCSF delivery, N=13, or future BDNF delivery, N=15). * two-tailed, unpaired t-test P<0.05. D.BDNF enzyme-linked immunosorbent assay (ELISA) of the cerebella of experimental mice (WT aCSF (N=12), WT BDNF (N=8), ATXN1[82Q] aCSF (N=10), and ATXN1[82Q] BDNF (N=14) . *P< 0.05 one way ANOVA with posthoc Sidak’s test. Each data point represent individual mouse, bars show the average with error bars = SEM.
We confirmed extrinsic BDNF delivery using an enzyme-linked immunosorbent assay (ELISA). We found a significant increase in cerebellar BDNF levels in mice receiving extrinsic BDNF compared to mice receiving aCSF both in wild-type and in ATXN1[82Q] mice (P=0.0001 one way ANOVA F (3, 38)=8.836). There was 76.7% increase in cerebellar BDNF levels in WT mice (WT aCSF: 234.5±28.4 ng/g cerebella, N=12, and WT BDNF: 422.4±55.7 ng/g, N=8, P = 0.0011, one way ANOVA with posthoc Sidak’s test). BDNF in ATXN1[82Q] mice similarly increased (71.9%, ATXN1[82Q] aCSF: 146.1±26.4 ng/g, N=9, ATXN1[82Q] BDNF: 251.1±29.5 ng/g, N=14, P=0.0347 one way ANOVA with post-hoc Sidak’s tests Figure 1D). Moreover WT-aCSF and ATXN1[82Q]-BDNF cerebella had very similar BDNF levels (P=0.791 one way ANOVA with post-hoc Sidak’s test). These results indicate that ICV delivery of exogenous BDNF can increase cerebellar BDNF levels to similar % in both WT and ATXN1[82Q] mice.
Next, to determine whether two weeks of BDNF treatment are sufficient to ameliorate SCA1 motor deficits, we tested mice on rotarod at 16 weeks of age. BDNF-treated ATXN1[82Q] mice performed significantly better on rotarod at 16 weeks compared to control aCSF-treated ATXN1[82Q] mice (e.g. day 4 average latency to fall of BDNF-treated ATXN1[82Q] mice was 329.4±23.7 sec, N=16; compared to aCSF-treated ATXN1[82Q] mice 239±22.97 sec, N=12, P<0.0001, one-way ANOVA F(3, 49)=21, followed by posthoc Sidak’s test P=0.042, Figure 2A). Intriguingly, BDNF-treated WT mice also performed better on rotarod (latency for aCSF- and BDNF- treated WT mice was 469.1±45.16 sec, N=15, and 594.7±3.193, N=10 respectively, post-hoc Sidak’s test P=0.0094). Both aCSF- and BDNF-treated ATXN1[82Q] mice performed worse compared to wild-type mice (post-hoc Sidak’s tests comparing to WT mice P<0.05). These results suggest that two weeks of BDNF treatment are sufficient to ameliorate, but not fully restore motor deficits in SCA1 mice even when treatment was started after the onset of motor symptoms.
Figure 2. Post-symptomatic BDNF delivery ameliorates motor deficits.
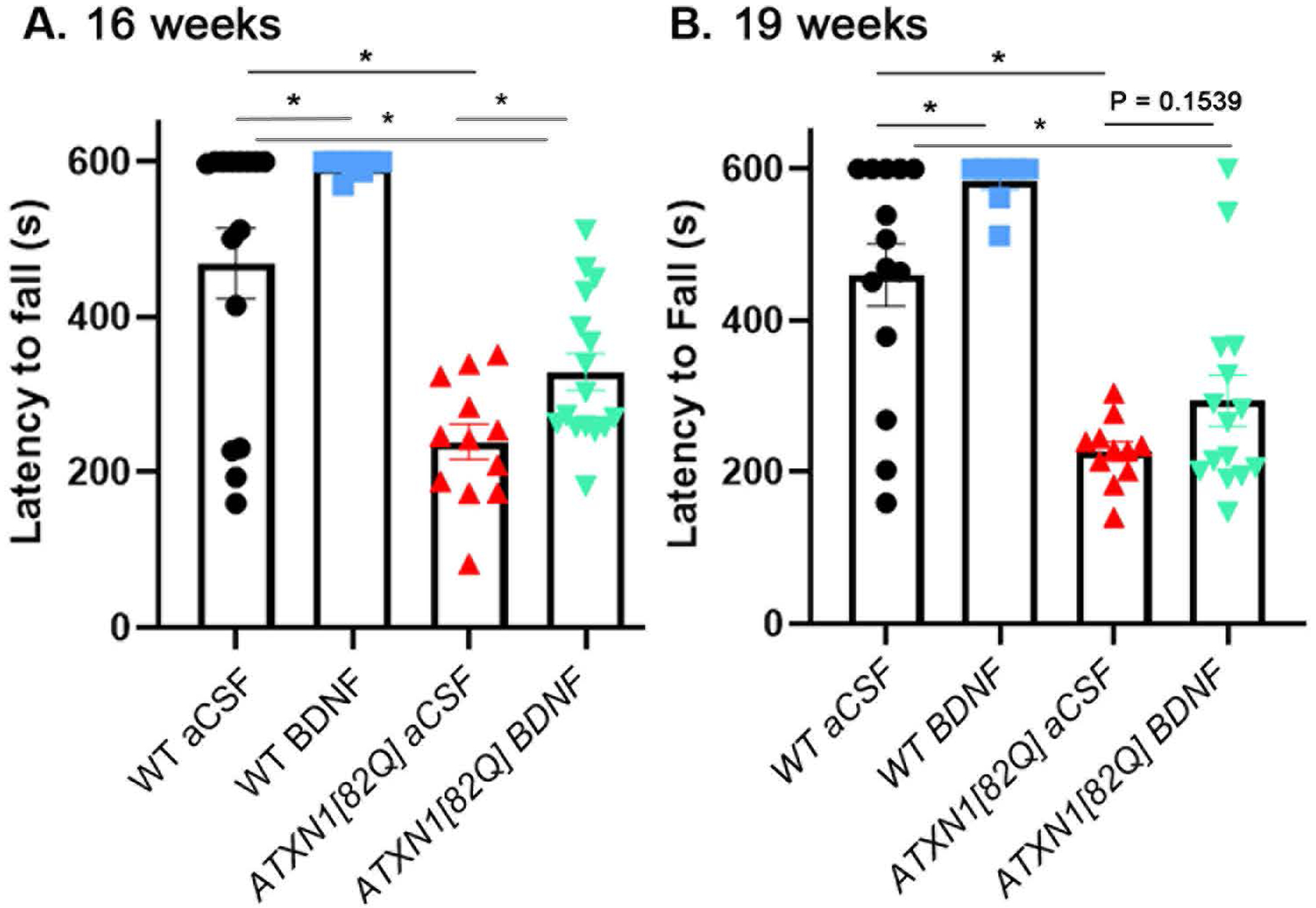
A. Rotarod performance of mice at 16 weeks, two weeks after the start of treatment (WT aCSF (N=15), WT BDNF (N=10), ATXN1[82Q] aCSF (N=12), and ATXN1[82Q] BDNF (N=16). B. Rotorod performance of mice at 19 weeks, one week after the cessation of treatment (ATXN1[82Q] aCSF, N=11, ATXN1[82Q] BDNF, N=15). Each data point represent individual mouse, bars show the average with error bars = SEM. * P<0.05, one-way ANOVA with post hoc Sidak’s test.
We then tested how long the positive effect of BDNF administration on motor function lasts. The ALZET pumps delivered BDNF for four weeks, thus we chose to test mice on the rotarod one week after BDNF delivery ended at 19 weeks. Extrinsic BDNF delivery again improved motor performance of WT mice (Figure 2B, latency for aCSF- and BDNF-treated WT mice was 460.1±41 sec, N=14, and 560.1±21, N=9 respectively, one way ANOVA F (3, 44)=20.1, post-hoc Sidak’s test P=0.017). Both aCSF- and BDNF-treated ATXN1[82Q] mice performed worse than WT mice (P<0.05 posthoc Sidak’s tests comparing aCSF- and BNF-treated ATXN1[82Q] mice with aCSF- and BDNF-treated WT mice). While BDNF-treated ATXN1[82Q] mice demonstrate a trend towards improved latency to fall on rotarod compared to control aCSF-treated ATXN1[82Q] mice, this was not statistically significant (day 4 average latency to fall of BDNF-treated ATXN1[82Q] mice=294.4±33.6 sec, N=15; compared to aCSF-treated ATXN1[82Q] mice =227.3±13.24sec, N=11, P=0.1539 one way ANOVA with post-hoc Sidak’s test). This suggests that post-symptomatic administration of BDNF may have a time-limited effect on SCA1-associated motor deficits after cessation of delivery.
Delivery of BDNF late in disease improves SCA1 Purkinje neuron pathology
We then investigated whether BDNF ameliorates cerebellar pathology characteristic for SCA1 including Purkinje cell dendritic atrophy and gene expression changes [21,23,26,29–33]. Dendritic atrophy can be quantified as a decrease in the width of molecular layer in cerebellar slices stained with the Purkinje cell marker calbindin (Figure 3A). There was a significant reduction in molecular layer width in 20 week old ATXN1[82Q] mice (average width of the molecular layer for aCSF-treated WT mice was 209.8±1.51 μm compared to 136.6±3.1 μm in aCSF-treated ATXN1[82Q] mice, N=5 of each, P<0.05 one-way ANOVA F(3, 16)=71.72 followed by Sidak’s post-hoc test), suggesting that Purkinje cell dendritic arbors are degenerating. There is notable amelioration of molecular layer degeneration in ATXN1[82Q] BDNF-treated mice (Figure 3B, average width of the molecular layer for aCSF-treated ATXN1[82Q] mice was 136.6±1.51 μm compared to 157.6±5.06 μm in BDNF-treated ATXN1[82Q] mice, N=5 of each, P=0.0021, one-way ANOVA followed by post-hoc Sidak’s test).
Figure 3. Delivery of BDNF late in disease improves SCA1 cerebellar Purkinje neuron dendritic atrophy.
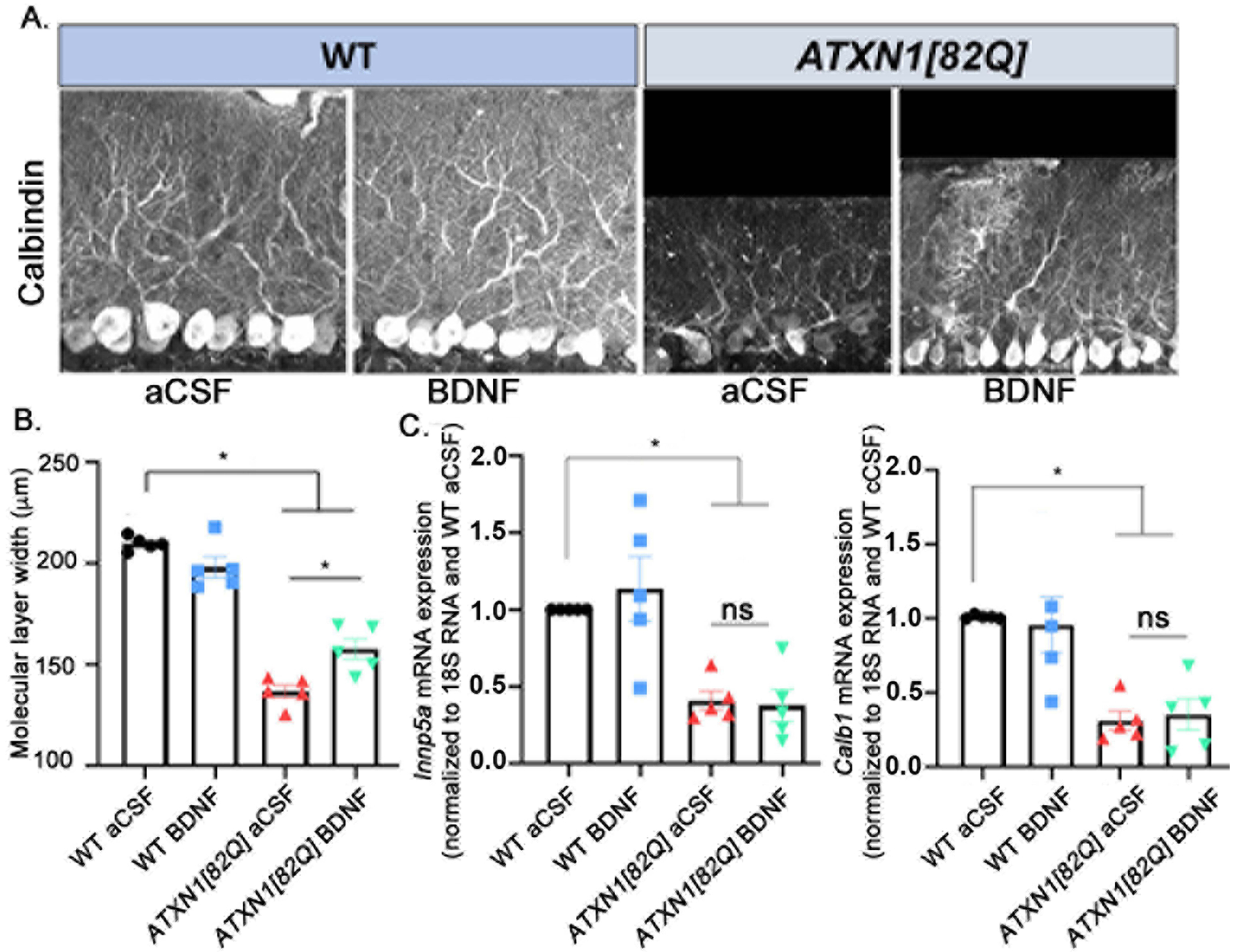
A. Representative images of cerebella stained for Calbindin. B. Width of the molecular layer quantification. N=5 in each group. C. Relative expression of two representative Magenta Module genes associated with SCA1 pathology in the cerebellum- Calbindin 1 (Calb1) and Inpp5a, mRNA in total cerebellar extracts, measured with RTqPCR (normalized to 18S RNA using WT aCSF as reference). N=5 per group. Each data point represent individual mouse, bars show the average with error bars = SEM. * P<0.05, one-way ANOVA followed by post hoc Sidak’s test.
We also examined molecular Purkinje cell pathology by quantifying SCA1-associated gene expression changes. The Magenta Module is a hub of genes whose expression is reduced and correlates with disease progression in ATXN1[82Q] Purkinje cells [33]. We used RT-qPCR to determine whether BDNF treatment rescued expression of Purkinje neuron genes belonging to magenta module (Calbindin, Inpp51, Garnl3, ITPR, Homer 3, Rgs8, Pcp4). Notably, none of the genes tested were rescued with BDNF treatment (representative data included for Calbindin and Inpp5a; all qPCR normalized to WT aCSF littermates, N=5 per group; for Calbindin relative mRNA levels for ATXN1[82Q] aCSF=0.3100±0.0639, ATXN1[82Q] BDNF=0.3520±0.1049, and WT BDNF=0.9580±0.1894; for Inpp5a, relative mRNA levels for ATXN1[82Q] aCSF=0.4100±0.0616, ATXN1[82Q] BDNF=0.3780±0.1042, and WT BDNF=1.136±0.2105; one-way ANOVA F(3, 16)=10.49, P<0.05 post-hoc Sidak’s test comparing WT to either ATXN1[82Q] aCSF or ATXN1[82Q] BDNF, and P >0.05 comparing ATXN1[82Q] aCSF and ATXN1[82Q] BDNF, additional magenta genes exhibit similar results, Figure 3C).
Finally, because expression of ATXN1 is tightly controlled [34] and correlates with SCA1 pathology [33], we assessed whether application of exogenous BDNF affected ATXN1 expression. Western blot of cerebellar protein lysates demonstrated no significant change in the expression of either wild type mouse ATXN1[2Q] or human mutant ATXN1[82Q] proteins expression upon BDNF treatment (Supplemental Figure 1).
Together, these data suggest that exogenous BDNF supports the health of Purkinje cell dendritic arbors, though Purkinje cell disease-associated gene expression changes are not rescued.
Cerebellar gliosis is altered with BDNF treatment
We have previously shown that cerebellar glia become reactive early in SCA1 mouse models and contribute to disease pathogenesis [19,20]. As Bergmann glia, a cerebellum specific type of astroglia, are intimately associated with Purkinje cells and also express BDNF receptors, we investigated whether BDNF treatment affects their activation [35–37].
Astrogliosis in the molecular and Purkinje cell layer where Bergmann glia reside was examined using immunohistochemistry of parasagittal cerebellar slices with the antibody against astroglial marker, glial fibrillary acidic protein (GFAP), which is increased during their activation (Figure 4A). In accordance with our previous findings, we noted an increase in the intensity of GFAP staining in the molecular and Purkinje cells layers of the cerebella of ATXN1[82Q] mice compared to their wild-type littermates (1.557±0.106 fold increase over wild type levels, one-way ANOVA F(3, 12)=16.27, P= 0.002, followed by post-hoc Sidak’s test, P=0.0002) [38]. This increase in GFAP intensity was significantly reduced upon BDNF treatment (normalized intensity for aCSF ATXN1[82Q] mice was 1.557±0.106 compared to 1.223±0.085 in BDNF treated mice, one-way ANOVA followed by post-hoc Sidak’s test P=0.0073; Figure 4B), indicating reduction in Bergmann gliosis with BDNF treatment.
Figure 4. Astrogliosis is reduced with BDNF treatment.
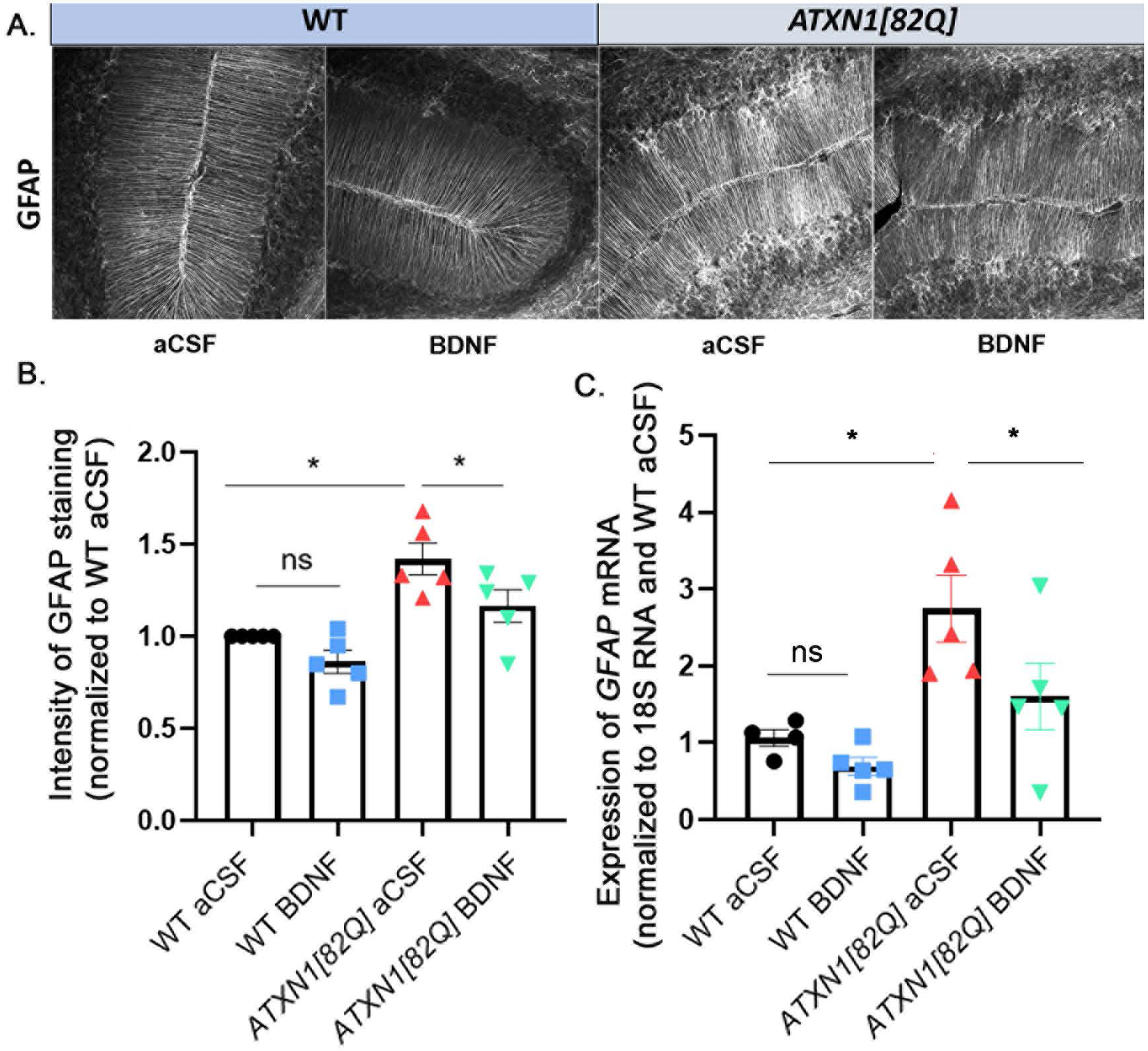
A. Representative images of GFAP staining in the cerebellar molecular and Purkinje cell layers. B. Quantification of GFAP intensity in Bergmann glia. N=5 per group. C. Relative expression of GFAP mRNA in total cerebellar extracts, measured with RTqPCR (normalized to 18S RNA using WT aCSF as reference) N=5 per group. Each data point represent individual mouse, bars show the average with error bars = SEM. * P<0.05, one-way ANOVA followed by Sidak’s post hoc test.
We obtained similar results at the level of total cerebellar GFAP mRNA. First, expression of total cerebellar GFAP mRNA was significantly increased in ATXN1[82Q] mice compared to wild type mice (Figure 4C, normalized to 18S loading control and WT aCSF treated littermates relative mRNA expression levels in ATXN1[82Q] aCSF was 2.746 ± 0.43, P=0.0035, one way ANOVA F(3,15)=7.48 followed by the post-hoc test). Moreover, we observed a decrease in total cerebellar GFAP mRNA levels in BDNF-treated ATXN1[82Q] mice (ATXN1[82Q] BDNF mice 1.604 ± 0.43, P=0.007, one way ANOVA followed by the post-hoc Sidak’s test P = 0.0249). Interestingly, there was a slight decrease in total cerebellar GFAP mRNA levels in BDNF treated WT mice that was not statistically significant (WT BDNF mice 0.694 ± 0.11, one way ANOVA followed by the post-hoc Sidak’s test P=0.4601). This result may indicate that the BDNF-associated decrease in Bergmann glia GFAP is at the transcriptional or mRNA stability level.
Discussion
We report here that post-symptomatic delivery of extrinsic BDNF ameliorates motor deficits and aspects of cerebellar pathology in the Purkinje cell specific transgenic mouse model of SCA1, including shrinkage of the Purkinje cell dendrites and astrogliosis. Based on these results, we propose BDNF as a potential candidate for post-symptomatic therapeutic treatments targeting motor deficits and cerebellar degeneration in SCA1.
The ATXN1[82Q] mouse model recapitulates the cerebellar aspects of SCA1, including ataxia, motor dysfunction, Purkinje cell molecular layer atrophy, and gliosis. To assess the efficacy of BDNF treatment over time, we measured motor function using the rotarod at three key points: before, during, and after end of ICV BDNF application. At 12 weeks of age, ATXN1[82Q] mice demonstrate reduced motor function as compared to WT littermate controls. ICV application of BDNF lead to ~ 70% increase in BDNF levels and reduced the severity of motor dysfunction with two weeks of treatment (16 weeks of age). However, this beneficial effect was not statistically significant one week after BDNF application had ceased, suggesting that BDNF may need to be continuously delivered for sustained beneficial effects. In addition, it is possible that BDNF stability was declining in the implanted Alzet pumps during the four weeks of treatment.
Accordingly, four weeks of extrinsic BDNF treatment ameliorated key pathological changes in cerebellar tissue, reducing molecular layer degeneration and lessening SCA1-associated astrogliosis. While extrinsic BDNF ameliorated shrinking of the PC dendritic arbor, it did not rescue expression of Purkinje neuron genes associated with disease progression. As BDNF treatment in early stage SCA1 did ameliorate expression of these genes [16], this may indicate that some pathological changes, such as altered PC gene expression cannot be ameliorated after the onset of symptoms. This is reminiscent of previous reports that timing of the treatment is a critical determinant of degree of the therapeutic rescue. For instance, in the conditional SCA1 transgenic mice stopping mutant ATXN1 expression in the early disease stage (6 weeks of age) was much more beneficial compared to stopping mutant ATXN1 expression in the mid stage (12 weeks), while stopping mutant ATXN1 at the late stage (32 weeks) had no beneficial effect [28]. Similarly, anti-sense oligonucleotides (ASOs) targeting Atxn1 were much more efficient if injected early (5 weeks) than if injected at a later disease stage (8–9 weeks) [22]. Our results build on this, proposing that after the onset of motor symptoms, BDNF may be beneficial in maintaining the Purkinje cell dendrites without altering the expression of genes that belong to the disease-associated Magenta Module.
BDNF belongs to the family of neurotrophins [37], which are largely responsible for neuron proliferation, survival, and synaptic plasticity [11,39]. Dysregulation of BDNF levels has been noted in several neurodegenerative disorders including proteinopathies such as Alzheimer’s and Parkinson’s diseases [12,40,41] as well as a number of polyglutamine repeat disorders (HD[10], SCA6[42]). Correction of aberrant BDNF levels in models of neurodegenerative disease has been shown to ameliorate different aspects of disease symptomology, from preventing the loss of vulnerable neuron populations to improving disease-associated cognitive and motor deficits [43–45].
BDNF is expressed in cerebellum from early development [46], and its cognate receptor, Tyrosine kinase B receptor (TrkB), is highly expressed on Purkinje neurons. Recent work has shown that BDNF dysregulation in cerebellum may be a common characteristic of ataxias, including SCA1 [16], SCA6 [6,42], and Friedreich’s Ataxia [47]. Notably, BDNF has been identified as one of the top four upstream regulators of gene expression changes in SCA1 [33]. Takahashi et al. described reduced BDNF mRNA levels in the cerebellar extract from patients with SCA6 [42], and BDNF was also decreased in patients with Friedreich’s Ataxia [47]. In addition, BDNF expression is reduced in several mouse models of cerebellar degeneration – Lurcher, Purkinje cell degeneration (pcd), and stargazer mice [48,49]. As BDNF-TrkB signaling regulates Purkinje cell dendritic branching and synaptic strength [11,13,15,39,50], reduced BDNF signaling may contribute to cerebellar Purkinje cell pathology described in SCA1 and these other ataxias, including shrinkage of the dendritic arbor [20,23,27]. In support of this theory, we have previously found that treatment with recombinant BDNF delivered intracerebroventricularly (ICV) early in SCA1- prior to the onset of motor symptoms- ameliorated both motor deficits and dendritic atrophy in a mouse model of SCA1 [16].
We have found a similar relative increase in cerebellar BDNF levels in both WT and SCA1 mice with Alzet pumps ICV (72 and 77% increase compared to aCSF treated mice respectively). However, since the pumps were set to deliver equivalent BDNF amounts, due to the lower BDNF in SCA1 mice we expected a higher percent of increase. There are several mutually non-excluding possibilities: due to lower BDNF in the mutant mice there might be more available BDNF receptors to sequester extrinsic BDNF, neuroinflammation in the cerebella of mutant mice may reduce the ability of BDNF to be extracted from CSF as well as stability of BDNF in the extracellular space, and BDNF may be less efficient in upregulating its production in neurodegenerative cerebella [51].
Our results suggest that BDNF treatment even after the onset of symptoms can improve motor function in transgenic ATXN1[82Q] mice. In this mouse model, ATXN1 is expressed only in Purkinje cells, which recreates many of the cerebellar aspects of the disease. However, in patients with SCA1, ATXN1 is expressed throughout the brain. Knock-in SCA1 mice, Atxn1154Q/2Q , which express mutant ATXN1 globally exhibit additional SCA1 features not present in transgenic ATXN1[82Q] mice, such as premature lethality, reduced adult hippocampal neurogenesis, cognitive deficits, and kyphosis [52,53]. Interestingly, BDNF treatment in neurodegenerative proteinopathies like Alzheimer’s and Parkinson’s diseases improves cognitive function through improving adult hippocampal neurogenesis [54,55]. In addition, we have also found that BDNF treatment improves motor performance of wild-type mice, but did not alter PC morphology or expression of PC genes. This may indicate that extrinsic BDNF has a beneficial effect in healthy aging brains, but it is unclear whether this effect is due to the changes in cerebellar function or outside of the cerebellum. Indeed, using BDNF ELISA of cerebrum we have found that ICV application, in addition to increasing BDNF levels in cerebellum (Figure 1D), also causes a significant increase in a brain BDNF level in BDNF treated mice (Supplemental Figure 2). Given this, it would be of interest to assess the effect of BDNF treatment on a range of SCA1 phenotypes that may be caused by non-cerebellar pathologies using the Atxn1154Q/2Q knock in mouse model that expresses mutant ATXN1 throughout the brain.
Summary
To date, there is no cure for SCA1. However, administration of exogenous BDNF seems to improve key behavioral and pathological phenotypes associated with cerebellar aspects of SCA1. To continue to unearth how this treatment strategy could be extended to patients, the next step would be to assess how BDNF administration affects broader aspects of SCA1, including cognitive deficits and premature lethality. In addition, this treatment strategy could be considered for combination therapy with drugs currently under review targeting ATXN1 expression, allowing for a two-pronged approach for improving neuron health and delaying onset of disease symptoms.
Supplementary Material
Supplemental Figure 1. Protein levels of ATXN1 are not altered as a result of BDNF treatment
A. Representative image of western blot of cerebellar lysates probed with anti-ATXN1 antibody. B. Quantification of the lower, 110kB band, representing the wild-type, unexpanded ATXN1[2Q] protein. C. Quantification of the higher band, which represents the mutant human ATXN1[82Q] protein. N=3 per group. Each data point represent individual mouse, bars show the average with error bars = SEM.
Supplemental Figure 2. ICV-Application of Exogenous BDNF increases levels of BDNF in Cerebrum.
ELISA of cerebral tissue. BDNF levels were normalized to WT aCSF treated littermate controls. Each data point represent individual mouse, bars show the average with error bars = SEM. * P<0.05 one way ANOVA followed by posthoc Sidak’s test.
Acknowledgements
We are grateful to Drs. Harry Orr and Huda Zoghbi for the generous gift of mice and to all the members of Cvetanovic and Orr laboratories for suggestions. This study is funded by the Regenerative Medicine Minnesota (RMM 101617 TR 001) and National Institutes of Health (NS107387-02) grants.
Footnotes
Availability of data and material
All data will be freely available from authors upon request.
Competing interests
The authors have no conflict of interest or competing interests to declare.
References:
- 1.Zoghbi HY, Orr HT. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, Spinocerebellar ataxia type 1. J. Biol. Chem 2009;284:7425–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banfi S, Servadio A, Chung MY, Capozzoli F, Duvick LA, Elde R, et al. Cloning and developmental expression analysis of the murine homolog of the spinocerebellar ataxia type 1 gene (Sca1). Hum. Mol. Genet 1996; [DOI] [PubMed] [Google Scholar]
- 3.Gusella JF, Macdonald ME. MOLECULAR GENETICS : UNMASKING POLYGLUTAMINE TRIGGERS IN NEURODEGENERATIVE DISEASE. Nat. Rev. Neurosci 2000;1:109–15. [DOI] [PubMed] [Google Scholar]
- 4.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat. Rev. Genet. [Internet] Nature Publishing Group; 2010;11:247–58. Available from: 10.1038/nrg2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Genis D, Matilla T, Volpini V, Rosell J, Dávalos A, Ferrer I, et al. Clinical, neuropathologic, and genetic studies of a large spinocerebellar ataxia type 1 (SCA1) kindred: (CAG)n expansion and early premonitory signs and symptoms. Neurology [Internet]. 1995;45:24–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7824128 [DOI] [PubMed] [Google Scholar]
- 6.Rüb U, Schöls L, Paulson H, Auburger G, Kermer P, Jen JC, et al. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol 2013. [DOI] [PubMed] [Google Scholar]
- 7.Matilla-Dueñas A Ashizawa T, Brice A, Magri S, McFarland KN, Pandolfo M, Pulst, et al. Consensus Paper: Pathological Mechanisms Underlying Neurodegeneration in Spinocerebellar Ataxias. Cerebellum. 2014;13:269–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orr HT, Zoghbi HY. Trinucleotide Repeat Disorders. Annu. Rev. Neurosci. [Internet] 2007;30:575–621. Available from: http://www.annualreviews.org/doi/abs/10.1146/annurev.neuro.29.051605.113042 [DOI] [PubMed] [Google Scholar]
- 9.Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias-from genes to potential treatments. Nat. Rev. Neurosci 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martín-Ibañez R, Muñoz MT, et al. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J. Neurosci 2004;24:7727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vigers AJ, Amin DS, Talley-Farnham T, Gorski JA, Xu B, Jones KR. SUSTAINED EXPRESSION OF BDNF IS REQUIRED FOR MAINTENANCE OF DENDRITIC SPINES AND NORMAL BEHAVIOR HHS Public Access. Neuroscience. 2012;212:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldberg NRS, Caesar J, Park A, Sedgh S, Finogenov G, Masliah E, et al. Neural stem cells rescue cognitive and motor dysfunction in a transgenic model of dementia with lewy bodies through a BDNF-dependent mechanism. Stem Cell Reports. The Authors; 2015;5:791–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, Segal RA. Abnormal Cerebellar Development and Foliation in BDNF Ϫ / Ϫ Mice Reveals a Role for Neurotrophins in CNS Patterning. Neuron. 1997;19:269–81. [DOI] [PubMed] [Google Scholar]
- 14.Bao S, Chen L, Qiao X, Knusel B, Thompson RF. Impaired Eye-Blink Conditioning in waggler, a Mutant Mouse With Cerebellar BDNF Deficiency. Learn. Mem 1998;5:355–64. [PMC free article] [PubMed] [Google Scholar]
- 15.Carter AR, Chen C, Schwartz PM, Segal RA. Brain-Derived Neurotrophic Factor Modulates Cerebellar Plasticity and Synaptic Ultrastructure. J Neurosci. 2002;22:1316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mellesmoen A, Sheeler C, Ferro A, Rainwater O, Cvetanovic M. Brain derived neurotrophic factor (BDNF) delays onset of pathogenesis in transgenic mouse model of spinocerebellar ataxia type 1 (SCA1). Front. Cell. Neurosci 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burright EN, Clark BH, Servadio A, Matilla T, Feddersen RM, Yunis WS, et al. SCA1 transgenic mice: A model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82:937–48. [DOI] [PubMed] [Google Scholar]
- 18.Cvetanovic M, Patel JM, Marti HH, Kini AR, Opal P. Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1. Nat. Med. [Internet] 2011;17:1445–7. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3287040&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JH, Lukowicz A, Qu W, Johnson A, Cvetanovic M. Astroglia contribute to the pathogenesis of spinocerebellar ataxia Type 1 (SCA1) in a biphasic, stage-of-disease specific manner. Glia. 2018;66:1972–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qu W, Johnson A, Kim JH, Lukowicz A, Svedberg D, Cvetanovic M. Inhibition of colony-stimulating factor 1 receptor early in disease ameliorates motor deficits in SCA1 mice. J. Neuroinflammation. Journal of Neuroinflammation; 2017;14:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duvick L, Barnes J, Ebner B, Agrawal S, Andresen M, Lim J, et al. SCA1-like disease in mice expressing wild-type Ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron [Internet]. Elsevier Inc.; 2010;67:929–35. Available from: 10.1016/j.neuron.2010.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedrich J, Kordasiewicz HB, O’Callaghan B, Handler HP, Wagener C, Duvick L, et al. Antisense oligonucleotide–mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruegsegger C, Stucki DM, Steiner S, Angliker N, Radecke J, Keller E, et al. Impaired mTORC1-Dependent Expression of Homer-3 Influences SCA1 Pathophysiology. Neuron. 2016;89:129–46. [DOI] [PubMed] [Google Scholar]
- 24.Matilla A, Roberson ED, Banfi S, Morales J, Armstrong DL, Burright EN, et al. Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J. Neurosci 1998; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, et al. Purkinje Cell Expression of a Mutant Allele of SCA1 in Transgenic Mice Leads to Disparate Effects on Motor Behaviors , Followed by a Progressive Cerebellar Dysfunction and Histological Alterations. J. Neurosci 1997;17:7385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ibrahim MF, Power EM, Potapov K, Empson RM. Motor and Cerebellar Architectural Abnormalities during the Early Progression of Ataxia in a Mouse Model of SCA1 and How Early Prevention Leads to a Better Outcome Later in Life. Front. Cell. Neurosci 2017;11:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hourez R, Servais L, Orduz D, Gall D, Millard I, de Kerchove d’Exaerde A, et al. Aminopyridines Correct Early Dysfunction and Delay Neurodegeneration in a Mouse Model of Spinocerebellar Ataxia Type 1. J. Neurosci 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zu T Recovery from Polyglutamine-Induced Neurodegeneration in Conditional SCA1 Transgenic Mice. J. Neurosci 2004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, et al. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J. Neurosci 2004;24:8853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ebner B, Ingram MA, Barnes JA, Duvick LA, Frisch JL, Clark HB, et al. Purkinje cell ataxin-1 modulates climbing fiber synaptic input in developing and adult mouse cerebellum. J. Neurosci. [Internet] 2013;33:5806–20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23536093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnes JA, Ebner BA, Duvick LA, Gao W, Chen G, Orr HT, et al. Abnormalities in the Climbing Fiber-Purkinje Cell Circuitry Contribute to Neuronal Dysfunction in ATXN1[82Q] Mice. J. Neurosci 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Serra HG, Duvick L, Zu T, Carlson K, Stevens S, Jorgensen N, et al. ROR a -Mediated Purkinje Cell Development Determines Disease Severity in Adult SCA1 Mice. Cell. 2006;127:697–708. [DOI] [PubMed] [Google Scholar]
- 33.Ingram M, Wozniak EAL, Duvick L, Yang R, Bergmann P, Carson R, et al. Cerebellar Transcriptome Profiles of ATXN1 Transgenic Mice Reveal SCA1 Disease Progression and Protection Pathways. Neuron [Internet]. Elsevier Inc.; 2016;89:1194–207. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0896627316001045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ju H, Kokubu H, Lim J. Beyond the Glutamine Expansion: Influence of Posttranslational Modifications of Ataxin-1 in the Pathogenesis of Spinocerebellar Ataxia Type 1. Mol. Neurobiol 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poblete-Naredo I, Guillem AM, Juárez C, Zepeda RC, Ramírez L, Caba M, et al. Brain-derived neurotrophic factor and its receptors in Bergmann glia cells. Neurochem. Int 2011; [DOI] [PubMed] [Google Scholar]
- 36.Pöyhönen S, Er S, Domanskyi A, Airavaara M. Effects of Neurotrophic Factors in Glial Cells in the Central Nervous System : Expression and Properties in Neurodegeneration and Injury. Front. Physiol 2019;10:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellamy TC. Interactions between Purkinje neurones and Bergmann glia. The cerebellum. 2006;5:116–26. [DOI] [PubMed] [Google Scholar]
- 38.Cvetanovic M, Ingram M, Orr H, Opal P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience [Internet]. IBRO; 2015;289:289–99. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0306452215000159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rico B, Xu B, Reichardt LF. TrkB receptor signaling is required for establishment of GABAergic synapses in the cerebellum. Nat. Neurosci 2002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ventriglia M, Zanardini R, Bonomini C, Zanetti O, Volpe D, Pasqualetti P, et al. Serum brain-derived neurotrophic factor levels in different neurological diseases. Biomed Res. Int 2013;2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sadanand A, Janardhanan A, Vanisree AJ, Pavai T. Neurotrophin Expression in Lymphocytes: a Powerful Indicator of Degeneration in Parkinson’s Disease, Amyotrophic Lateral Sclerosis and Ataxia. J. Mol. Neurosci. Journal of Molecular Neuroscience; 2018;64:224–32. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi M, Ishikawa K, Sato N, Obayashi M, Niimi Y, Ishiguro T, et al. Reduced brain-derived neurotrophic factor (BDNF) mRNA expression and presence of BDNF-immunoreactive granules in the spinocerebellar ataxia type 6 (SCA6) cerebellum. Neuropathology. 2012;32:595–603. [DOI] [PubMed] [Google Scholar]
- 43.Vidal-Martinez G, Najera K, Miranda JD, Gil-Tommee C, Yang B, Vargas-Medrano J, et al. FTY720 Improves Behavior, Increases Brain Derived Neurotrophic Factor Levels and Reduces α-Synuclein Pathology in Parkinsonian GM2 +/− Mice. Neuroscience. The Authors; 2019;411:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Razgado-Hernandez LF, Espadas-Alvarez AJ, Reyna-Velazquez P, Sierra-Sanchez A, Anaya-Martinez V, Jimenez-Estrada I, et al. The transfection of BDNF to dopamine neurons potentiates the effect of dopamine D3 receptor agonist recovering the striatal innervation, dendritic spines and motor behavior in an aged rat model of Parkinson’s disease. PLoS One. 2015;10:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Pins B, Cifuentes-Díaz C, Thamila Farah A, López-Molina L, Montalban E, Sancho-Balsells A, et al. Conditional BDNF delivery from astrocytes rescues memory deficits, spine density, and synaptic properties in the 5xFAD mouse model of alzheimer disease. J. Neurosci 2019;39:2441–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dieni S, Rees S. Distribution of brain-derived neurotrophic factor and TrkB receptor proteins in the fetal and postnatal hippocampus and cerebellum of the guinea pig. J. Comp. Neurol 2002;454:229–40. [DOI] [PubMed] [Google Scholar]
- 47.Misiorek JO, Schreiber AM, Urbanek-Trzeciak MO, Jazurek-Ciesiołka M, Hauser LA, Lynch DR, et al. A Comprehensive Transcriptome Analysis Identifies FXN and BDNF as Novel Targets of miRNAs in Friedreich’s Ataxia Patients. Mol. Neurobiol 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salomova M, Tichanek F, Jelinkova D, Cendelin J. Abnormalities in the cerebellar levels of trophic factors BDNF and GDNF in pcd and lurcher cerebellar mutant mice. Neurosci. Lett Elsevier; 2020;725:134870. [DOI] [PubMed] [Google Scholar]
- 49.Meng H, Larson SK, Gao R, Qiao X. BDNF transgene improves ataxic and motor behaviors in stargazer mice. Brain Res. 2007;1160:47–57. [DOI] [PubMed] [Google Scholar]
- 50.Bamji SX, Rico B, Kimes N, Reichardt LF. BDNF mobilizes synaptic vesicles and enhances synapse formation by disrupting cadherin-β-catenin interactions. J. Cell Biol 2006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minichiello L TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci Nature Publishing Group; 2009;10:850–60. [DOI] [PubMed] [Google Scholar]
- 52.Watase K, Weeber EJ, Xu B, Antalffy B, Yuva-Paylor L, Hashimoto K, et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron. 2002;34:905–19. [DOI] [PubMed] [Google Scholar]
- 53.Asher M, Johnson A, Zecevic B, Pease D, Cvetanovic M. Ataxin-1 regulates proliferation of hippocampal neural precursors. Neuroscience [Internet]. IBRO; 2016;322:54–65. Available from: 10.1016/j.neuroscience.2016.02.011 [DOI] [PubMed] [Google Scholar]
- 54.Palasz E, Wysocka A, Gasiorowska A, Chalimoniuk M, Niewiadomski W, Niewiadomska G. BDNF as a promising therapeutic agent in parkinson’s disease. Int. J. Mol. Sci 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi SH, Bylykbashi E, Chatila ZK, Lee SW, Pulli B, Clemenson GD, et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science (80-. ) 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Protein levels of ATXN1 are not altered as a result of BDNF treatment
A. Representative image of western blot of cerebellar lysates probed with anti-ATXN1 antibody. B. Quantification of the lower, 110kB band, representing the wild-type, unexpanded ATXN1[2Q] protein. C. Quantification of the higher band, which represents the mutant human ATXN1[82Q] protein. N=3 per group. Each data point represent individual mouse, bars show the average with error bars = SEM.
Supplemental Figure 2. ICV-Application of Exogenous BDNF increases levels of BDNF in Cerebrum.
ELISA of cerebral tissue. BDNF levels were normalized to WT aCSF treated littermate controls. Each data point represent individual mouse, bars show the average with error bars = SEM. * P<0.05 one way ANOVA followed by posthoc Sidak’s test.
Data Availability Statement
All the data from this study are available from the authors.