

Abstract
We have previously demonstrated that the transcription co-factor yes-associated protein 1 (YAP1) promotes vascular smooth muscle cell (VSMC) de-differentiation. Yet, the role and underlying mechanisms of YAP1 in neointima formation in vivo remain unclear. The goal of this study was to investigate the role of VSMC-expressed YAP1 in vascular injury-induced VSMC proliferation and delineate the mechanisms underlying its action. Experiments employing gain- or loss-of-function of YAP1 demonstrated that YAP1 promotes human VSMC proliferation. Mechanistically, we identified platelet-derived growth factor receptor beta (PDGFRB) as a novel YAP1 target gene that confers the YAP1-dependent hyper-proliferative effects in VSMCs. Furthermore, we identified TEA domain transcription factor 1 (TEAD1) as a key transcription factor that mediates YAP1-dependent PDGFRβ expression. ChIP assays demonstrated that TEAD1 is enriched at a PDGFRB gene enhancer. Luciferase reporter assays further demonstrated that YAP1 and TEAD1 co-operatively activate the PDGFRB enhancer. Consistent with these observations, we found that YAP1 expression is upregulated after arterial injury and correlates with PDGFRβ expression and VSMC proliferation in vivo. Using a novel inducible SM-specific Yap1 knockout mouse model, we found that the specific deletion of Yap1 in adult VSMCs is sufficient to attenuate arterial injury-induced neointima formation, largely due to inhibited PDGFRβ expression and VSMC proliferation. Our study unravels a novel mechanism by which YAP1/TEAD1 promote VSMC proliferation via transcriptional induction of PDGFRβ, thereby enhancing PDGF-BB downstream signaling and promoting neointima formation.
Keywords: Smooth Muscle Proliferation and Differentiation, Restenosis, Vascular Biology, Gene Expression and Regulation, Genetically Altered and Transgenic Models
1. Introduction
Unlike cardiac and skeletal muscle cells, vascular smooth muscle cells (VSMCs) exhibit remarkable phenotypic plasticity, such that in response to environmental cues, they can undergo de-differentiation and proliferation [1]. Occlusive vascular diseases, such as atherosclerosis and post-angioplasty restenosis, are largely a result of phenotypically-modulated VSMCs that have switched from a contractile phenotype to a proliferative and migratory phenotype [1, 2]. However, the mechanisms responsible for VSMC proliferation after arterial injury remain incompletely understood. Delineating underlying mechanisms will not only lead us towards a better understanding of the pathology of occlusive vascular diseases, but also to ultimately developing new therapeutic strategies for their treatment.
The Hippo signaling pathway is evolutionarily conserved from Drosophila to mammals. The Hippo pathway has been shown to play a critical role in controlling tumorigenesis by regulating cell proliferation and apoptosis [3, 4]. YAP1 (yes-associated protein 1) is a Hippo signaling pathway effector, which acts as a potent oncogene [5]. Mechanistically, YAP1 is a transcriptional co-factor that lacks a DNA-binding domain and thus requires the interaction with transcription factors, mainly TEADs (transcriptional enhancer activator domain proteins) to regulate gene expression. TEADs bind to a consensus DNA sequence 5′-CATTCC-3′, named the muscle-specific cytidine-adenosine-thymidine (MCAT) element [6, 7]. Importantly, we have previously demonstrated that YAP1 can induce de-differentiation of cultured VSMCs while promoting their proliferation and migration [8]. Furthermore, we previously found that YAP1 is essential for cardiomyocyte and SMC proliferation during cardiovascular development [9]. Consistent with this, YAP1 has been demonstrated to mediate the growth-promoting effects of various stimuli in VSMCs [10–12]. While the effects of YAP1 on VSMC de-differentiation have been attributed to its ability to disrupt the interaction between SRF (serum response factor) and myocardin [13], the underlying mechanisms that mediate YAP1 regulation of VSMC proliferation and neointima formation in vivo remain unclear.
Platelet-derived growth factor-BB (PDGF-BB) is a potent VSMC mitogen that mediates its hyper-proliferative effects by binding to PDGF receptors (PDGFRβ and PDGFRα), leading to activation of multiple downstream pro-mitogenic signaling pathways, such as MAPK1/3 (also known as ERK1/2), AKT, and mTORC1 [1, 14–17]. Previous studies have demonstrated that PDGFRβ is upregulated and plays a critical role in atherosclerotic lesion development [18–20] and injury-induced neointima formation [21–25]. However, the transcriptional mechanisms regulating the expression of PDGFRβ in VSMCs are not clear.
In this study, we sought to assess the role and to understand the underlying mechanism(s) of vascular smooth muscle-expressed YAP1 in injury-induced neointima formation. Analysis of gain- and loss-of-function of YAP1 assays in human VSMCs identified PDGFRB as a novel YAP1 target gene that largely mediates the effects of YAP1 on VSMC proliferation. Furthermore, we have identified TEAD1 as a key transcription factor that mediates YAP1-dependent PDGFRβ expression. Using a novel inducible SM-specific Yap1 knockout mouse model, we found that the specific deletion of Yap1 in VSMCs is sufficient to attenuate arterial injury-induced neointima formation and VSMC proliferation, resulting from attenuated PDGFRβ expression. Our study suggests that the YAP1/TEAD1-PDGFRβ axis is a promising therapeutic target to ameliorate injury-induced neointima formation.
2. Material and methods
Inducible SM-specific Yap1 knockout (Yap1 iKO) mice were generated by crossing Yap1 flox (F) female mice [9, 26] with male mice expressing tamoxifen-inducible Cre driven by the SM-specific Myh11 gene promoter (Myh11-CreERT2) [27]. Since Myh11-CreERT2 transgene is localized on the Y chromosome [27], only male mice were used in this study. To generate control and Yap1 iKO mice, 10-week-old Myh11-CreERT2+/Yap1F/F male mice were injected intraperitoneally with either sunflower oil (control) or tamoxifen (iKO; 1 mg/mouse) for 2 rounds of 5 days each, with 2 days’ break in-between. After the last tamoxifen administration, a 2-week washout period was allowed before mice were used for experiments. All mice used in this study were maintained on a C57BL/6J background. The use of experimental mice for arterial injury procedures and BSL-2 viral work was approved by the Institutional Animal Care and Use Committee and Biosafety committees at Augusta University. A detailed, expanded Materials and Methods section is included in the Online Supplement.
3. Results
3.1. YAP1 induces PDGFRβ expression and enhances PDGF-BB-mediated signaling.
We have previously demonstrated that YAP1 can promote rat aortic SMC proliferation, migration, and de-differentiation [8]. In the current study, we found that adenoviral-mediated expression of YAP1 in human coronary artery SMCs (HCASMCs) enhances cell proliferation, as indicated by cell count analysis, WST-1 proliferation assays, and EdU incorporation assays (Figure 1A–D). In an attempt to delineate the underlying mechanism(s) that mediate YAP1’s induction of VSMC proliferation, we utilized a qPCR array that covers a subset of genes implicated in cell proliferation. This analysis demonstrated that YAP1 induces the expression of multiple growth factor receptor genes including PDGFRB, ERBB3 (human epidermal growth factor receptor 3), and FLT1 (vascular endothelial growth factor receptor 1), in addition to multiple cell-cycle regulatory genes, such as CDC25A (cell division cycle 25A), CDK5 (cyclin-dependent kinase 5), and PLK1 (polo-like kinase 1) (Figure 1E and Online Table I). Given the well-documented role of PDGF-BB and its receptor PDGFRβ in promoting VSMC proliferation and neointima formation [18–25], we focused on PDGFRB as a potential novel YAP1 target gene that may mediate its effect on VSMC proliferation. qRT-PCR and Western blot assays validated the qPCR array data and showed that over-expression of YAP1 in HCASMCs induces the expression of PDGFRβ at both the mRNA and protein levels, which is correlated with up-regulation of cell proliferation markers CCND1 and PCNA, and down-regulation of multiple SM contractile markers such as ACTA2, CNN1, and TAGLN (Figure 1F–H). Surprisingly, YAP1 over-expression did not alter the expression of bona fide YAP1 targets, such as CCN1 and MYC [11], or the glutamine transporter SLC1A5, which we recently demonstrated as a target gene of the transcription factor TEAD1 [28] (Figure 1G–H). Taken together, these data suggest that YAP1 specifically induces PDGFRB expression in VSMCs.
Figure 1. YAP1 is sufficient to promote VSMC proliferation, induce PDGFRβ expression, and enhance PDGF-BB-mediated signaling in VSMCs.
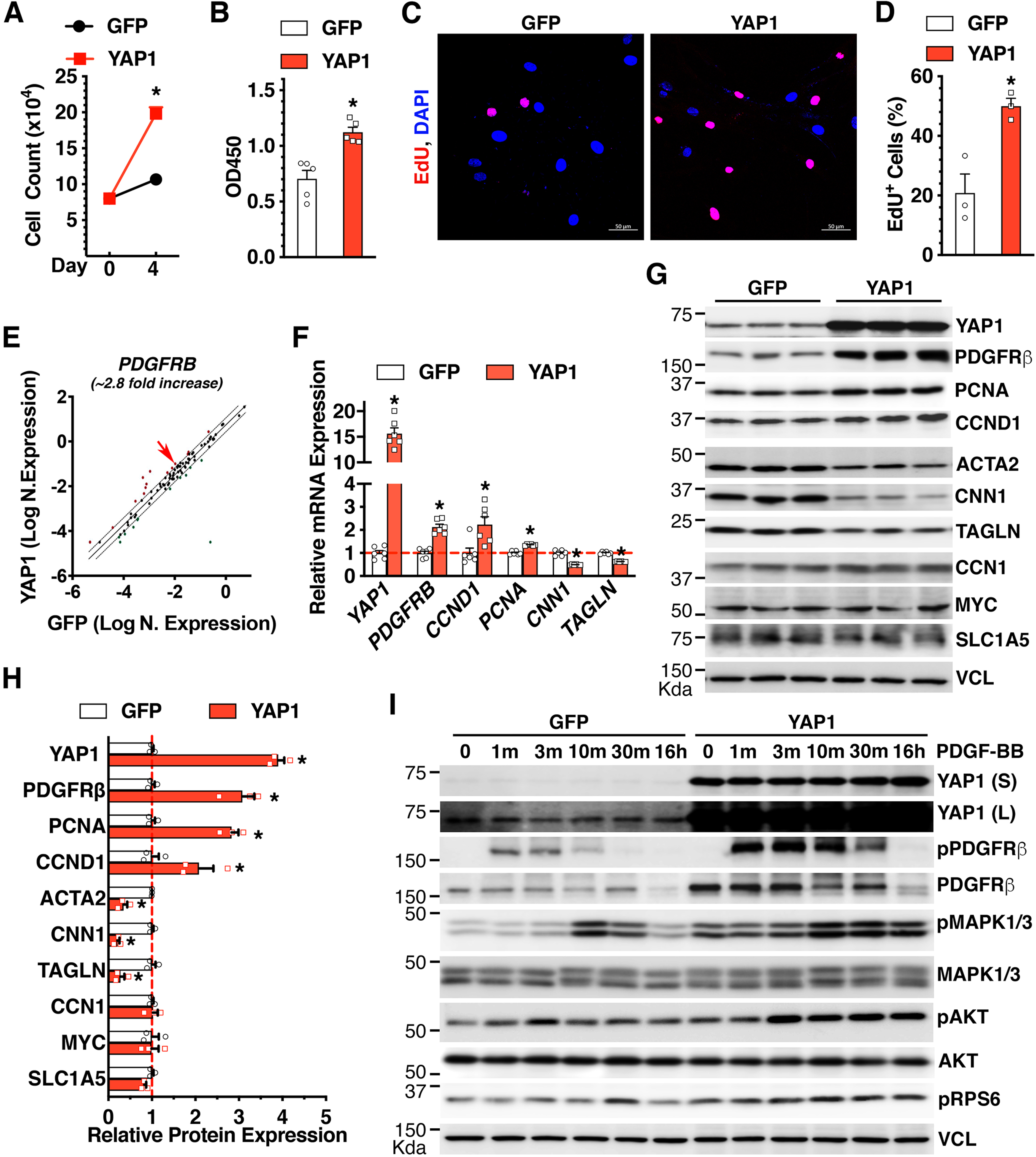
A. HCASMCs were transduced with GFP or YAP1 adenovirus and plated at equal numbers (Day 0). Cell counting analysis was performed at day 4 post-viral transduction. *p<0.05. N=3 per group. B. WST-1 proliferation assay was carried out at day 4 post-viral transduction. *p<0.05. N=5 per group. C. HCASMCs were transduced with GFP or YAP1 adenovirus and incubated with EdU for 48 hours for IF staining to visualize EdU incorporation (red). Cell nuclei were co-stained with DAPI (blue). D. The percentage of EdU+ cells to the total number of DAPI+ cells shown in “C” is plotted. *p<0.05. N=3 per group. E. HCASMCs were transduced with GFP or YAP1 adenovirus for qPCR array analysis. Over-expression of YAP1 induces 2.8-fold increase of PDGFRB (red arrow) expression compared to GFP control in HCASMCs. F-G. HCASMCs were transduced with GFP or YAP1 adenovirus for qRT-PCR analysis (F) or Western blotting (G). *p<0.05. N=6 per group in “F”. H. Quantification of relative protein expression in “G” after normalization to the loading control VCL (Vinculin). *p<0.05. N=3 per group. I. HCASMCs were transduced with GFP or YAP1 adenovirus for 48 hours and then treated with or without PDGF-BB (30 ng/ml) for different time periods. Subsequently cells were harvested for Western blotting. VCL served as the loading control. S: short exposure. L: long exposure.
In contrast to the positive effects of YAP1 on PDGFRβ expression, our qPCR array data demonstrated that YAP1 downregulates PDGFRA mRNA levels (Online Table I). qRT-PCR assays validated this array result (Online Figure IA). Interestingly, we found that the basal expression of PDGFRB at the mRNA level was ~5 fold higher than PDGFRA in HCASMCs (Online Figure IB), suggesting that PDGFRβ may play a more important role in mediating the pro-mitogenic signaling activation by PDGF-BB in human VSMCs. Consistent with this notion, we found that YAP1-mediated up-regulation of PDGFRβ correlated with enhanced PDGF-BB activation of PDGFRβ, as indicated by enhanced PDGFRβ tyrosine phosphorylation [29], and enhanced/sustained activation of downstream signaling molecules such as MAPK1/3, AKT, and mTORC1 (using pRPS6 as a biochemical readout for mTORC1 activity) (Figure 1I). Notably, consistent with previous studies showing that sustained ligand stimulation with PDGF-BB can induce PDGFRβ internalization followed by lysosomal or proteasomal degradation [30, 31], we found that treatment of HCASMCs with PDGF-BB for 16 hours led to a significant downregulation of PDGFRβ (Figure 1I).
Consistent with our YAP1 gain-of-function studies (Figure 1), depletion of endogenous YAP1 by siRNA inhibited HCASMC proliferation (Figure 2A–D) and down-regulated basal PDGFRβ expression, which was accompanied with down-regulation of PCNA and induction of SM contractile markers such as ACTA2, CNN1, and TGFβ1I1 (Figure 2E–G). Notably, we found that the inhibitory effects of silencing YAP1 on PDGFRB mRNA expression are modest, compared to its effects on PDGFRβ protein expression, suggesting that both transcriptional and post-transcriptional regulatory mechanisms underlie YAP1-mediated PDGFRB expression. In addition, silencing YAP1 inhibited PDGF-BB-mediated activation of PDGFRβ, MAPK1/3, AKT, and mTORC1 signaling (Figure 2H). Consistently, silencing YAP1 inhibited basal- and PDGF-BB-induced CCND1 expression, where the latter was only detectable after PDGF-BB stimulation for 48 hours.
Figure 2. YAP1 is required for VSMC proliferation, PDGFRβ expression and PDGFRβ-dependent signaling activation in VSMCs.
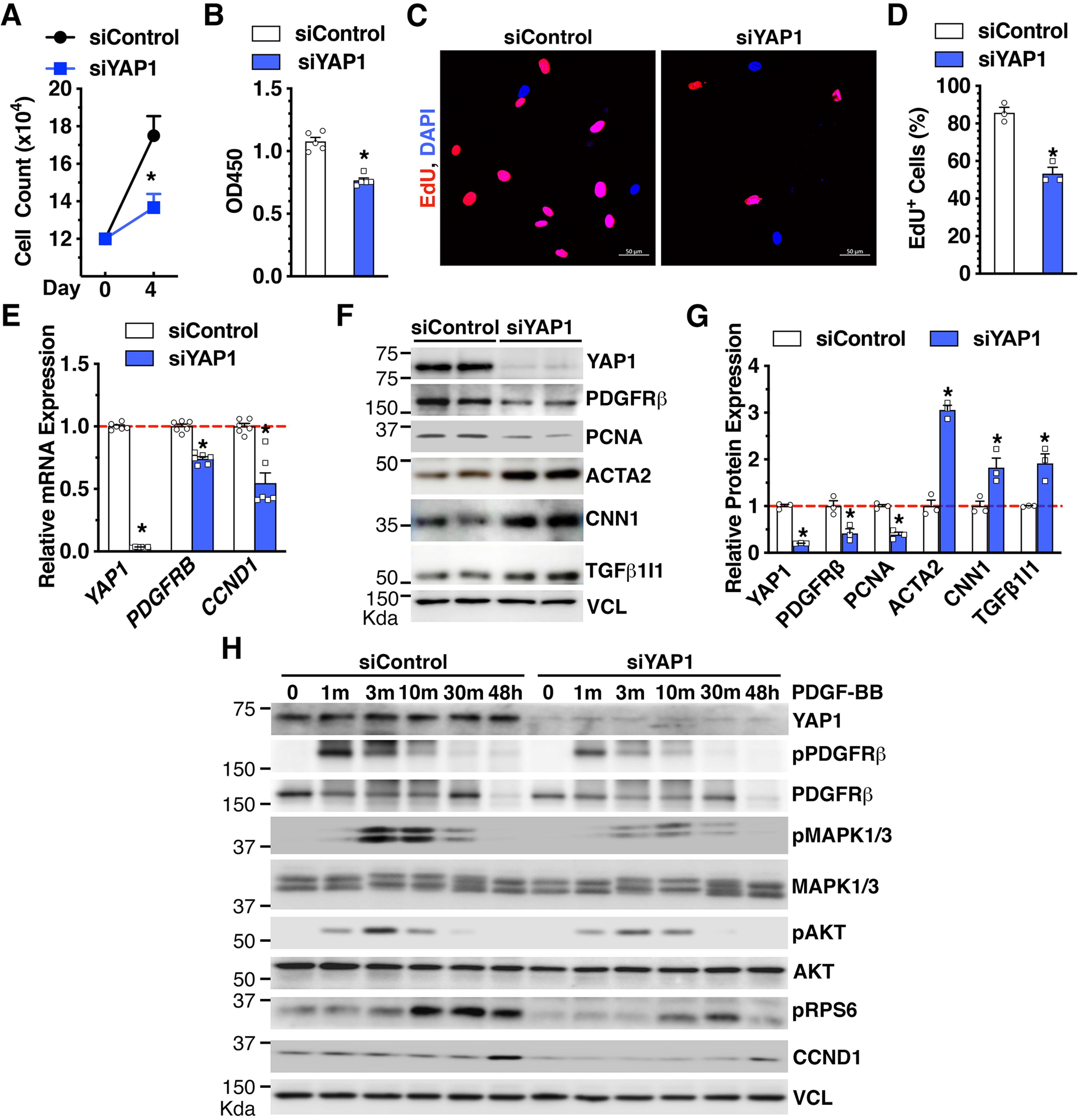
A. HCASMCs were transfected with scrambled control silencing RNA duplex (siControl) or silencing RNA duplex against YAP1 (siYAP1) and plated at equal numbers (Day 0). Cell counting analysis was performed at day 4 post-siRNA transfection *p<0.05. N=3 per group. B. WST-1 proliferation assay at day 4 post-siRNA transfection. *p<0.05. N=5 per group. C. HCASMCs were transfected with siControl or siYAP1 for 48 hours and then incubated with EdU for additional 48 hours for IF staining to visualize EdU incorporation (red). Cell nuclei were co-stained with DAPI (blue). D. The percentage of EdU+ cells to the total number of DAPI+ cells is plotted. *p<0.05. N=3 per group. E-F. HCASMCs were transfected with siControl or siYAP1 for 48 hours and then were harvested for qRT-PCR analysis (E) or Western blotting (F). *p<0.05. N=3–6 per group. G. Quantification of relative protein expression in “F” after normalization to the loading control VCL. *p<0.05. H. HCASMCs were transfected with siControl or siYAP1 in the absence or presence of PDGF-BB (50 ng/ml) for different time periods as indicated and then harvested for Western blotting.
Together, these YAP1 gain- and loss-of-function studies in HCASMCs demonstrate that YAP1 is sufficient and required to promote human VSMC proliferation, induce PDGFRβ expression, and enhance PDGF-BB-mediated activation of pro-mitogenic signaling.
3.2. PDGFRβ confers YAP1-mediated effects on VSMC proliferation.
Next, we tested the role of PDGFRβ in mediating VSMC proliferation under basal conditions or after over-expression of YAP1. We found that silencing endogenous PDGFRB markedly attenuated serum-induced VSMC proliferation and almost completely abolished YAP1-induced VSMC proliferation and CCND1 expression (Figure 3A–C). Notably, we found that silencing endogenous PDGFRB slightly downregulated both basal- and adenovirus-induced protein expression of YAP1 (Online Figure IIA and Figure 3A), suggesting a positive-feedback loop that links PDGFRβ and YAP1 expression. However, we found that treatment of HCASMCs with PDGF-BB for 2- or 5-days did not alter YAP1 expression at the protein level (Online Figure IIB). Furthermore, we found that silencing endogenous PDGFRB did not alter the expression of multiple SM contractile markers (Online Figure IIA).
Figure 3. PDGFRβ is required for YAP1-dependent VSMC proliferation.
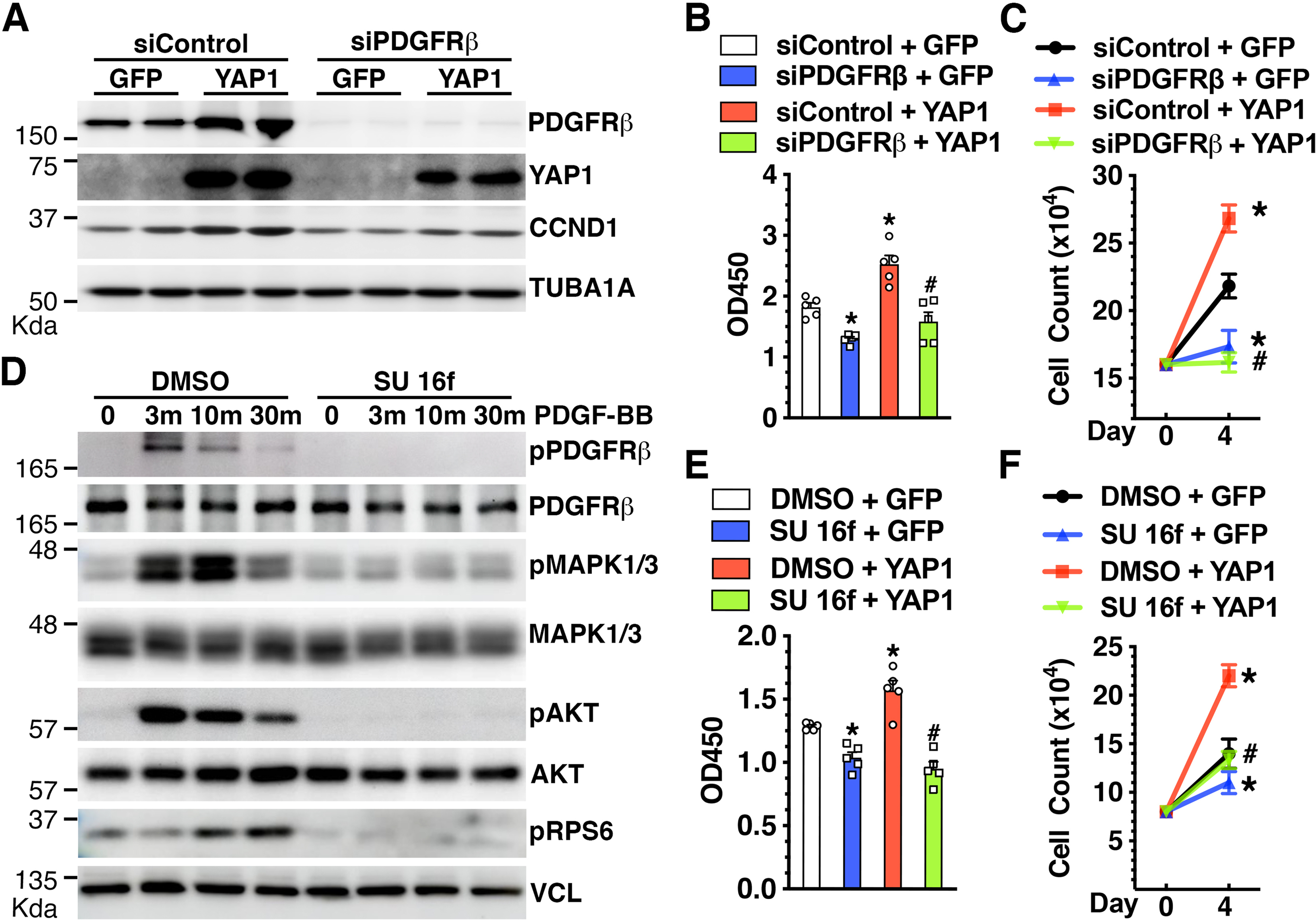
A-C. HCASMCs were transfected with siControl or siPDGFRβ for 24 hours and then transduced with control GFP or YAP1 adenovirus for additional 48 hours. Cells were then harvested for Western blot analysis (A), WST-1 proliferation assay (B), or cell count analysis (C). *p<0.05, vs siControl + GFP; #p<0.05, vs siControl + YAP1. N=3–5 per group. D. HCASMCs were incubated with DMSO (control) or PDGFRβ tyrosine kinase inhibitor SU 16f (10 nM) for 48 hours before treatment with PDGF-BB (50 ng/ml) for different time periods. Subsequently, cells were harvested for Western blot analysis. E-F. HCASMCs were plated at equal numbers (Day 0) and incubated with DMSO (control) or SU 16f (10 nM). Cells were then transduced with control GFP or YAP1 adenovirus for 2 days for WST-1 proliferation assay (E) or 4 days for cell count analysis (F). *p<0.05, vs DMSO + GFP; #p<0.05, vs DMSO + YAP1. N=3–5 per group.
Next, we used a potent and selective PDGFRβ tyrosine kinase inhibitor (SU 16f [32]) to further test the role of PDGFRβ in mediating YAP1 effects on VSMC proliferation. As shown in Figure 3D, pretreatment of VSMCs with PDGFRβ tyrosine kinase inhibitor SU 16f (10 nM) led to a slight decrease in the total protein expression of PDGFRβ, MAPK1/3, and AKT (Figure 3D). However, unlike the negligible effects on the expression level of total proteins, SU 16f completely abolished PDGF-BB-induced phosphorylation/stimulation of PDGFRβ, MAPK1/3, and AKT, suggesting the specific effects of SU16F through inhibiting PDGFRβ activity. Importantly, SU 16f treatment attenuated both basal and YAP1-induced VSMC proliferation (Figure 3E–F). Together, these studies suggest that PDGFRβ confers, at least in part, the YAP1-mediated effects on VSMC proliferation.
3.3. YAP1-induced PDGFRβ expression is dependent on TEAD1.
YAP1 primarily interacts with TEAD family transcription factors in order to regulate gene transcription [6, 7]. Recently, we have demonstrated that TEAD1 is the most abundant TEAD family protein in HCASMCs [28]. In addition, similar to YAP1, TEAD1 can promote VSMC proliferation [28]. These prior observations prompted us to test whether TEAD1 mediates YAP1-induced PDGFRβ expression in VSMCs. We found that over-expression of TEAD1 in HCASMCs induces PDGFRβ expression at both mRNA and protein levels and up-regulates the proliferative marker PCNA, without affecting the basal expression of YAP1 (Figure 4A–C). Conversely, silencing endogenous TEAD1 down-regulated basal PDGFRβ expression (Figure 4D–E) and markedly attenuated YAP1-induced PDGFRβ and CCND1 expression (Figure 4F). The slight increase in PDGFRβ expression observed in siTEAD1 + YAP1 compared to siTEAD1 + GFP is likely attributed to the functional activity of the residual TEAD1 protein (~15%) and/or other functionally redundant TEAD family proteins such as TEAD2–4 in HCASMCs. Together, these data suggest that TEAD1 mediates YAP1-induced PDGFRβ expression.
Figure 4. YAP1 requires TEAD1 to induce PDGFRβ expression.
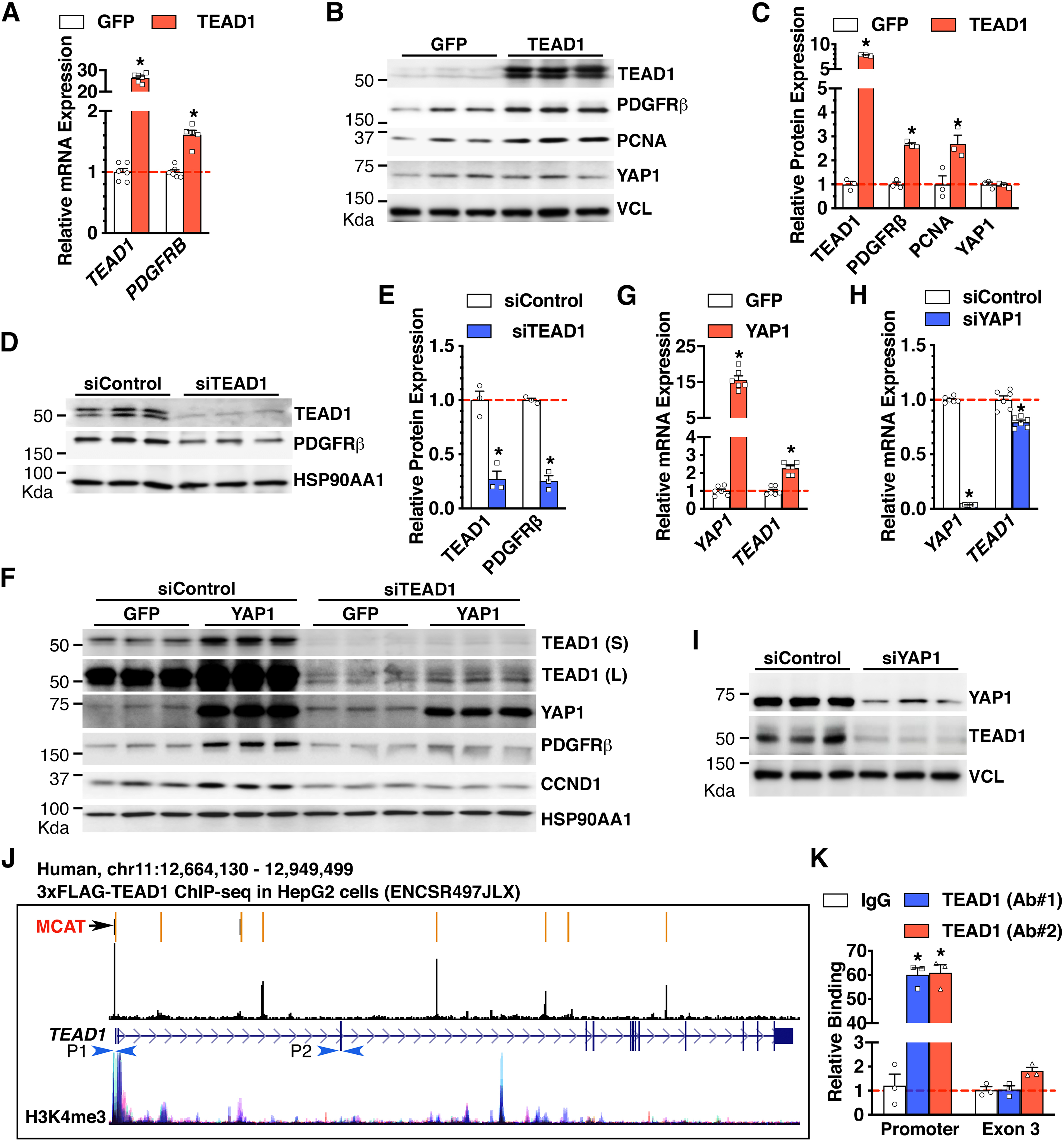
A-B. HCASMCs were transduced with GFP or TEAD1 adenovirus for 48 hours and then harvested for qRT-PCR analysis (A) or Western blotting (B). *p<0.05, N=6 per group in “A”. C. Quantification of relative protein expression in “B” after normalization to the loading control VCL. *p<0.05. N=3 per group. D. HCASMCs were transfected with siControl or siTEAD1 for 2 days and then were harvested for Western blot analysis. E. Quantification of relative protein expression in “D” after normalization to the loading control HSP90AA1. *p<0.05. N=3 per group F. HCASMCs were transfected with siControl or siTEAD1 for 2 days then transduced with control GFP or YAP1 adenovirus for 2 additional days. Cells were then harvested for Western blotting. N=3 per group. S: short exposure. L: long exposure. G. HCASMCs were transduced with GFP or YAP1 adenovirus for 2 days and then harvested for qRT-PCR analysis. *p<0.05. N=6 per group. H-I. HCASMCs were transfected with siControl or siYAP1 for 3 days and then harvested for qRT-PCR analysis (H) or for Western blotting (I). *p<0.05. N=3–6 per group. J. Schematic depicting identified MCAT elements in the human TEAD1 gene locus and TEAD1 binding peaks as revealed by chromatin immunoprecipitation (ChIP)-seq data in HepG2 cells. The H3K4me3 ChIP-seq tracks demonstrate the active transcription regions in TEAD1 gene locus across 7 cell types from the ENCODE project. P1 and P2 depict the primer sets used for ChIP-qPCR in “K”. K. Adenovirus expressing TEAD1 was transduced into HCASMCs for ChIP assay using TEAD1 antibodies from 2 different vendors (Ab #1 and Ab #2) or IgG control. The precipitated DNA was amplified by real-time PCR using TEAD1 promoter-specific primers that span the MCAT elements depicted in “J” (P1), or primers targeting TEAD1 exon 3 region (P2). *p<0.05. N=3 per group.
Notably, over-expression of YAP1 not only induced basal PDGFRβ expression, but also induced basal TEAD1 protein expression (Figure 4F). Dose response experiments further demonstrated that even a slight increase in YAP1 expression is sufficient to induce the expression of TEAD1 and PDGFRβ proteins, concomitant with upregulation of PCNA (Online Figure III). Consistently, we found that YAP1 over-expression induces, while silencing YAP1 down-regulates, TEAD1 mRNA and protein expression, respectively (Figure 4F–I). Together, these data suggest that TEAD1 is a direct transcriptional target of YAP1/TEADs complex. Furthermore, we found that the inhibitory effects of silencing YAP1 on TEAD1 mRNA expression are modest, compared to its effects on TEAD1 protein expression, suggesting both transcriptional and post-transcriptional regulatory mechanisms. De novo analysis of a TEAD1 ChIP-seq dataset generated in HepG2 cells (ENCODE) demonstrated that TEAD1 is enriched at the TEAD1 promoter by ~73 fold over IgG control and correlated with the active transcription histone mark H3K4me3 (Figure 4J) [33]. Our bioinformatic analysis further revealed multiple putative TEAD DNA binding motifs (MCAT elements) in the human TEAD1 gene including in the promoter region (Figure 4J, indicated by an arrow). To validate the binding of TEAD1 to the MCAT element identified in the TEAD1 promoter in HCASMCs, we performed ChIP-qPCR assays using two TEAD1 antibodies from two different vendors (Ab #1 and Ab #2). Data from the ChIP-qPCR assays revealed that, compared to IgG control, TEAD1 was indeed significantly enriched at the TEAD1 promoter in HCASMCs by ~60 folds (Figure 4K). The specificity of TEAD1 binding to the promoter site was validated by using primer sets flanking TEAD1 exon 3. Furthermore, re-analysis of TEAD1 ChIP-seq datasets generated in fetal or adult mouse hearts [34, 35] revealed that the Tead1 gene promoter contains a TEAD1 binding peak (Online Figure IV). Together, these data suggest that YAP1 forms a complex with TEAD1 to transcriptionally activate TEAD1 expression through a novel feed-forward mechanism in both human and mouse.
3.4. YAP1/TEAD1 transcriptionally activate a PDGFRB gene enhancer.
Since we found that both YAP1 and TEAD1 can regulate PDGFRβ gene expression (Figures 1, 2, and 4), we tested the role of PDGFRβ in mediating VSMC proliferation in response to co-transduction with YAP1 and TEAD1 adenoviruses. We found that silencing endogenous PDGFRB almost completely abolished YAP1/TEAD1-induced VSMC proliferation and the expression of the cell proliferative marker PCNA (Figure 5A–B).
Figure 5. YAP1/TEAD1 transcriptionally activate PDGFRB.
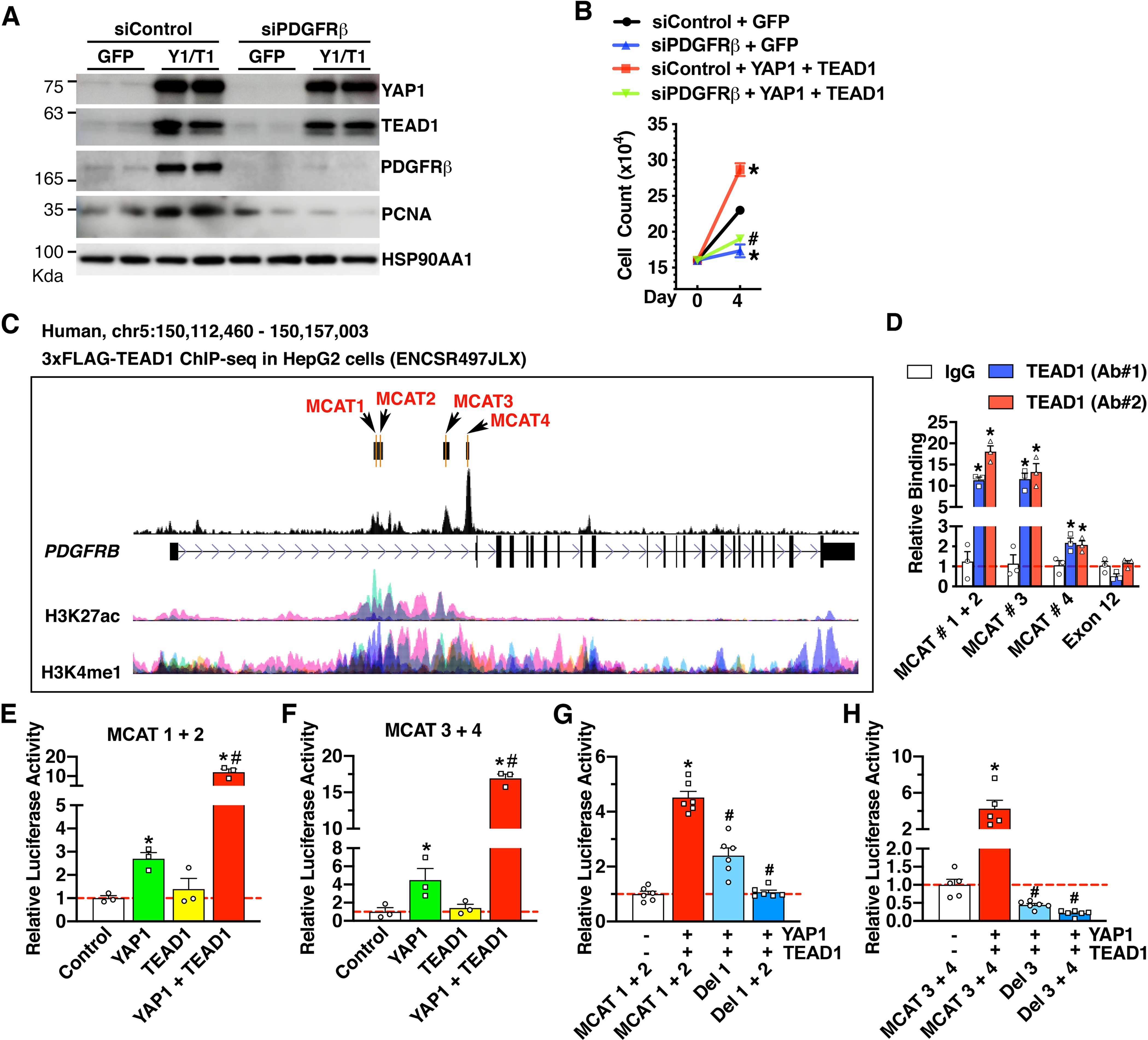
A-B. HCASMCs were transfected with siControl or siPDGFRβ for 24 hours before transduction with control GFP or a mix of YAP1 (Y1) and TEAD1 (T1) adenoviruses for additional 3 days. Cells were then harvested for Western blot analysis (A) or cell count analysis (B). *p<0.05, vs siControl + GFP; #p<0.05, vs siControl + YAP1 + TEAD1. N=3–5 per group. C. Schematic depicting the identified MCAT elements and TEAD1 binding peaks in the human PDGFRB gene locus as revealed by ChIP-seq data in HepG2 cells. H3K27ac and H3K4me1 ChIP-seq tracks show the enhancer regions within TEAD1 gene locus across 7 cell types from the ENCODE project. D. Adenovirus expressing TEAD1 was transduced into HCASMCs for ChIP assay using TEAD1 antibodies from 2 different vendors (Ab #1 and Ab #2) or IgG control. The precipitated DNA was amplified by real-time PCR using PDGFRB enhancer-specific primers that span the MCAT elements depicted in “C”, or primers targeting PDGFRB exon 12 region as a negative control. *p<0.05. N=3 per group. E-H. 10T1/2 cells were co-transfected with YAP1 or TEAD1 expression plasmids, together with luciferase reporter constructs harboring PDGFRB enhancer region spanning MCAT 1 + 2 (E), MCAT 3 + 4 (F), truncation mutants lacking MCAT1 (Del 1) or both MCAT 1 and 2 (Del 1 + 2) (G), or truncation mutants lacking MCAT3 (Del 3) or both MCAT 3 and 4 (Del 3 + 4) (H) for dual luciferase reporter assays. Reporter activity was expressed relative to the transfection with control empty plasmid (set to 1). *p<0.05, vs empty plasmid (control); #p<0.05, vs YAP1 alone (E & F); #p<0.05, vs MCAT1 + 2 (G) or MCAT 3 + 4 (H). N=3–6 per group.
Next, we sought to further examine the transcriptional mechanism by which YAP1/TEAD1 regulates PDGFRB gene expression. ChIP-seq data from the ENCODE project using HepG2 cells demonstrated that TEAD1 is enriched at multiple regions in the first intron of the human PDGFRB gene locus, which correlated with epigenetic signatures of active enhancers (H3K27ac and H3K4me1; Figure 5C) [33]. Our de novo bioinformatics analysis revealed there are 4 putative MCAT elements residing in the TEAD1 binding peaks. Data from our ChIP-qPCR assays in HCASMCs revealed that, compared to IgG control, TEAD1 was significantly enriched at MCAT sequences 1/2, 3, and 4, by ~15, ~12, and ~2 folds, respectively (Figure 5D). Together, these data demonstrate that TEAD1 is enriched at the PDGFRB enhancer across different cell types.
To test the functional role of the identified MCAT elements in driving PDGFRB enhancer activity, we generated 2 luciferase reporter constructs spanning “MCAT 1 + 2” and “MCAT 3 + 4’” sequences, respectively. Data from the luciferase reporter assays showed that YAP1 expression is sufficient to activate the enhancer activity of both reporter constructs in 10T1/2 cells (Figure 5E–F). Surprisingly, transfection with TEAD1 expression plasmid alone failed to activate the enhancer activity of either reporter. However, co-transfection with YAP1 and TEAD1 expression plasmids led to a dramatic augmentation of YAP1-mediated reporter activity of both constructs, suggesting a synergistic effect between YAP1 and TEAD1 in activating PDGFRB enhancer activity (Figure 5E–F).
We next sought to determine the relative importance of the 4 identified MCAT elements in YAP1/TEAD1-mediated PDGFRB enhancer activity. Compared to the “MCAT 1 + 2” reporter construct, the deletion of MCAT1 (Del 1) partially attenuated, whereas the deletion of both MCAT1 and 2 (Del 1 + 2) completely abolished YAP1/TEAD1-mediated PDGFRB enhancer activity to baseline levels (Figure 5G). On the other hand, compared to the “MCAT 3 + 4” reporter construct, deletion of MCAT3 only (Del 3) was sufficient to abolish YAP1/TEAD1-induced PDGFRB enhancer activity to baseline levels, while further deletion of MCAT 4 (Del 3 + 4) had no additional effects on YAP1/TEAD1-induced PDGFRB enhancer activity (Figure 5H). Together, these data suggest that MCATs 1, 2, and 3 play a critical role in mediating YAP1/TEAD1-induced PDGFRB enhancer activity, while MCAT 4 is likely dispensable in this regard.
3.5. SM-specific deletion of Yap1 attenuates neointima formation and decreases VSMC proliferation in vivo by abolishing PDGFRβ induction.
As an initial step to test the functional relevance of the novel YAP1-PDGFRβ axis in promoting VSMC proliferation and neointima formation in vivo, we examined the expression of YAP1 and PDGFRβ in left common carotid arteries (LCA) after ligation injury, a mouse model that triggers VSMC proliferation thereby resulting in neointima formation [36]. We found that ligation injury in WT C57BL/6J adult mice induces the expression of YAP1, PDGFRβ, and proliferative markers (PCNA and pHIST1H3A) at day 7 post-injury (Figure 6A–B), suggesting the activation of the YAP1-PDGFRβ axis after arterial injury. To test the role of SM-specifically expressed YAP1 in neointima formation in adult mice, we generated an inducible SM-specific Yap1 KO (iKO) mice by crossing female Yap1 flox mice with male mice expressing tamoxifen-inducible Cre recombinase under the control of the SM-specific Myh11 promotor (Online Figure VA) [27]. Characterization of the Yap1 iKO mouse model demonstrated the specific deletion of Yap1 in aortic tissues by ~80% at both mRNA and protein levels (Online Figure VB and Figure 6C–E). To our surprise, we found that Yap1 iKO does not affect the basal expression of PDGFRβ, TEAD1, or a panel of SM marker genes in the intact dorsal aorta (Figure 6C–E). To determine the effect of Yap1 ablation on vascular injury-induced PDGFRβ expression, we isolated contralateral RCAs and their respective injured LCAs from control and Yap1 iKO mice for Western blotting. Data from the Western blot assays revealed that PDGFRβ protein expression is induced in response to vascular injury while the deletion of Yap1 in VSMCs almost completely abolishes PDGFRβ and PCNA upregulation after arterial injury (Figure 6F–G). As a result, at day 21 post-injury, we found that Yap1 iKO attenuates neointimal area and neointima/media ratio in injured LCA by ~66% and 65%, respectively, without affecting the relative medial layer area (Figure 7A–D). Furthermore, Yap1 iKO markedly decreased the number of MKI67 positive cells (a marker of cell proliferation) in both neointimal and medial areas of injured LCA by ~40% and ~51%, respectively (Figure 7E–G). Together, these data demonstrate that while YAP1 is not required for basal PDGFRβ expression in intact mouse vessels where VSMCs are quiescent, YAP1 is required for PDGFRβ induction in response to arterial injury. Therefore, Yap1 deletion in VSMCs is sufficient to attenuate neointima formation, likely through inhibiting PDGFRβ-driven VSMC proliferation in vivo.
Figure 6. SM-specific deletion of Yap1 attenuates injury-induced PDGFRβ expression.
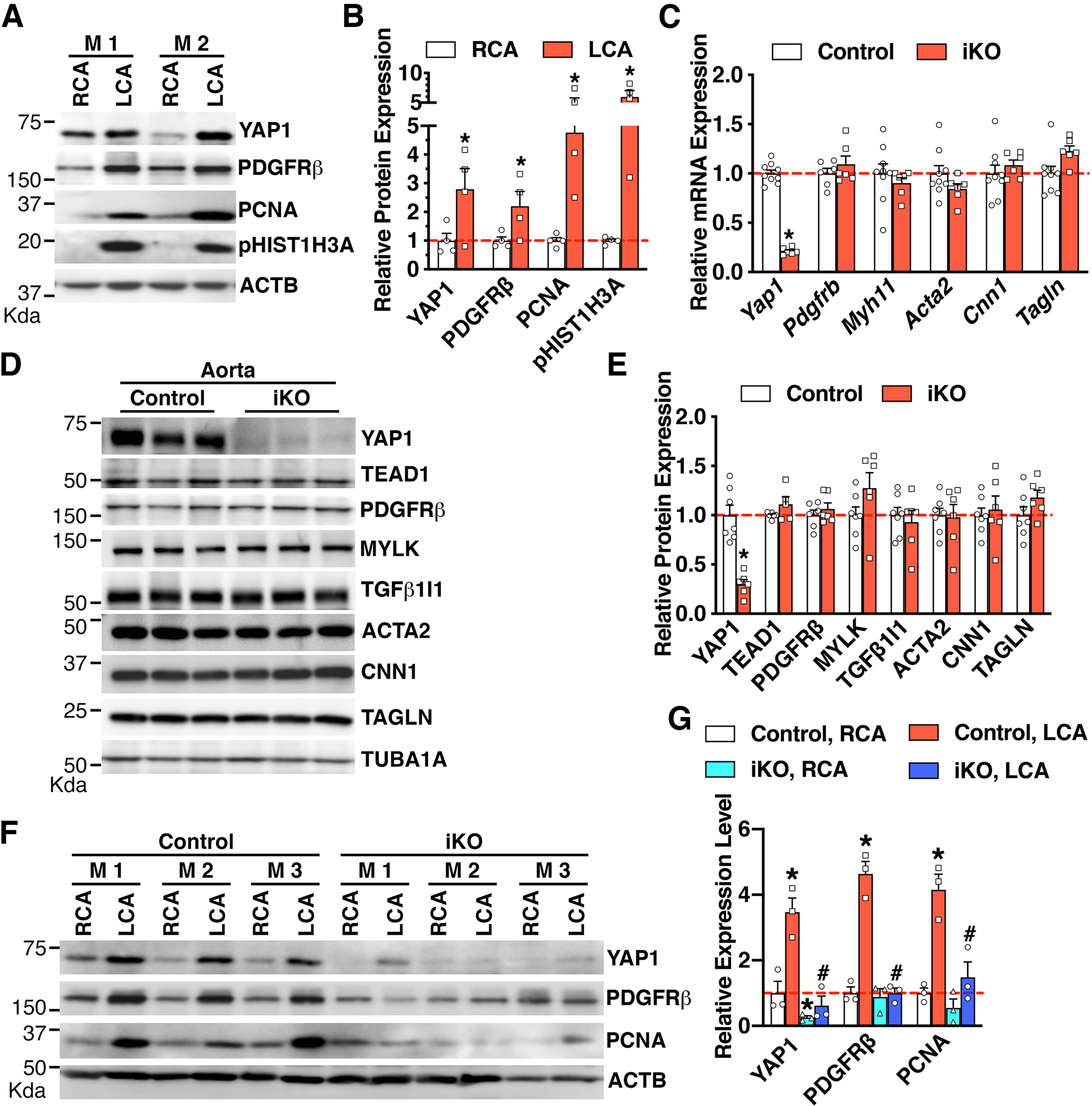
A. Control right carotid artery (RCA) and ligation-injured left carotid artery (LCA) were harvested from adult male C57BL/6J mice at day 7 post-injury for Western blotting. M: mouse. B. Quantification of protein expression in “A” relative to signals from the uninjured control RCA (set to 1, red dashed line) after normalization to the loading control ACTB. N=4. *p<0.05. C-D. Aortic tissues were harvested from control or SM-specific Yap1 iKO mice for qRT-PCR analysis (C) or Western blotting (D). *p<0.05. N=6–9 per group in “C”. E. Quantification of relative protein expression in “D” after normalization to the loading control TUBA1B. *p<0.05. N=6–7 per group. F. Control or Yap1 iKO mice were subjected to LCA ligation injury. RCAs or LCAs were harvested at day 7 post-injury for Western blotting. M: mouse. G. Quantification of relative protein expression in “F”. Relative expression of proteins in the RCA of the control group was set as 1 (red dashed line) after normalization to the loading control ACTB. *p<0.05, vs control RCA group; #p<0.05, vs control LCA group. N=3 per group.
Figure 7. SM-specific deletion of Yap1 attenuates neointima formation by inhibiting VSMC proliferation.
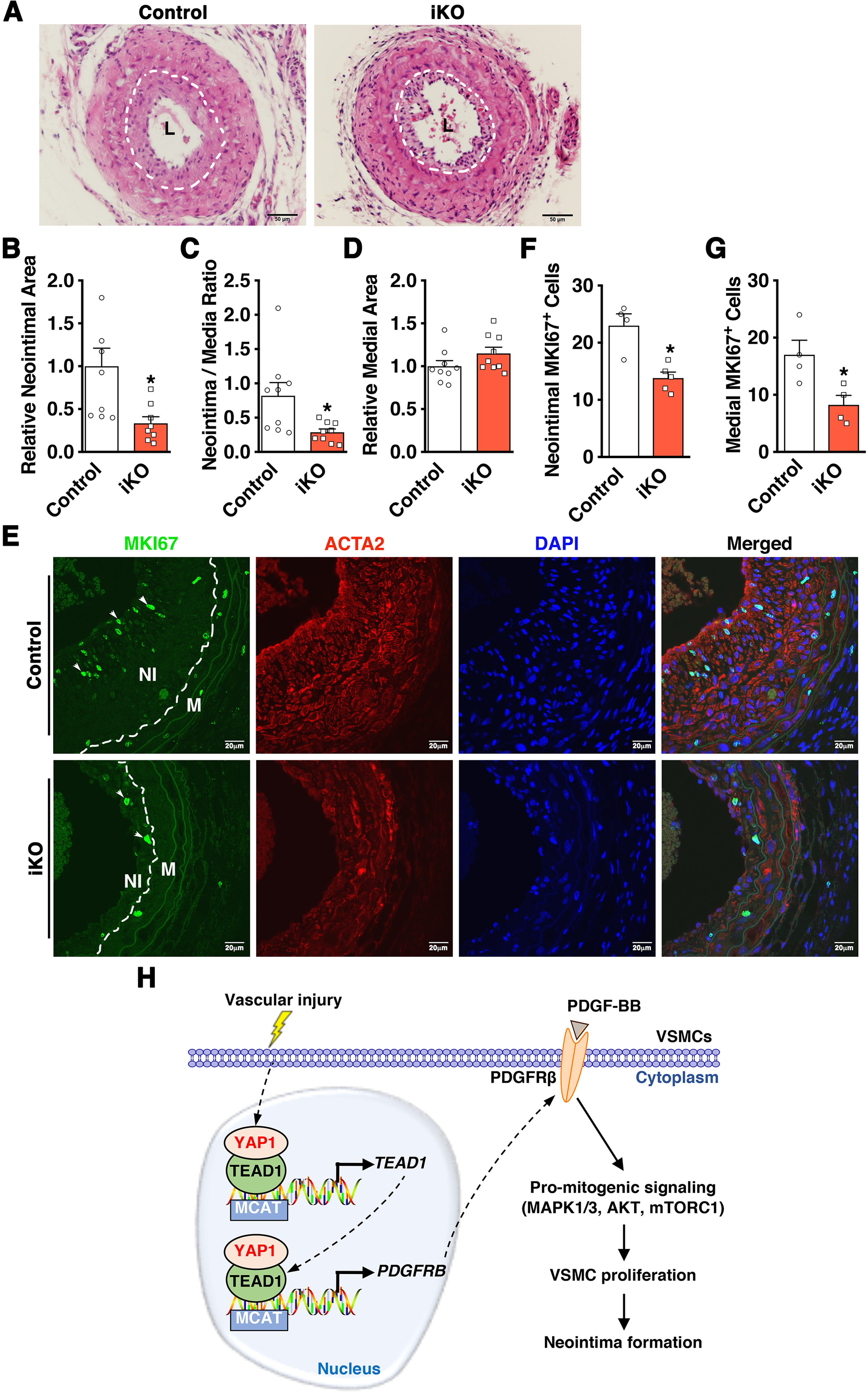
A. Control or SM-specific Yap1 iKO (iKO) mice were subjected to LCA ligation injury. At day 28 post-injury, LCAs were harvested for morphometric analysis by Hematoxylin and Eosin staining. Dashed lines depict the internal elastic laminae. L: lumen. B-D. Quantification of relative neointimal area (B), neointima-to-media ratio (C), and relative medial area (D) in LCA at day 28 post-injury in iKO versus control groups. *p<0.05. E. Control or iKO mice were subjected to LCA ligation injury for IF staining using antibodies against MKI67 (green) and ACTA2 (red). Nuclei were counter-stained with DAPI. White dashed lines in the far-right panels depict the internal elastic laminae. Arrows point to representative MKI67 positive cells. NI: neointima; M: media. F-G. Quantification of MKI67-positive cells within the neointimal area (F) or the medial area (G) of LCA in control or iKO mice. *p<0.05. H. Schematic diagram depicting the major findings of this study. We found that vascular injury induces YAP1 expression, which induces TEAD1 expression through a novel feed-forward mechanism. The induced YAP1 and TEAD1 can form a transcriptional complex to co-operatively promote PDGFRβ expression. Upregulated PDGFRβ expression, in turn, promotes PDGF-BB-dependent pro-mitogenic signaling, leading to enhanced VSMC proliferation and injury-induced neointima formation.
In summary, our study uncovers a previously unrecognized YAP1/TEAD1-PDGFRβ axis in VSMC proliferation and neointima formation. Our data demonstrate that injury-induced YAP1 expression promotes the expression of TEAD1, which forms a transcriptional complex with YAP1 to synergistically activate PDGFRβ expression. Upregulated PDGFRβ expression, in turn, promotes PDGF-BB-mediated activation of pro-mitogenic signaling, leading to enhanced VSMC proliferation that eventually contributes to injury-induced neointima formation (Figure 7H).
4. Discussion
YAP1 is activated in several vascular pathologies such as pulmonary hypertension [37], atherosclerosis [38, 39], and injury-induced neointima formation [8]. We previously demonstrated that Yap1 mRNA is upregulated in balloon-injured rat carotid arteries [8]. A recent study demonstrated that vascular injury induces PIK3CG (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit gamma) to promote the transcriptional activation of Yap1 in injured vessels via a CREB (cyclic AMP-response element-binding protein)-dependent mechanism [12]. YAP1 may also be regulated at the post-transcriptional level. For instance, we recently reported that microRNA-15b/16, which is downregulated in vessels after arterial injury, targets Yap1 by binding to Yap1 3’-UTR in VSMCs, thereby contributing to the upregulated YAP1 expression in injured vessels [40]. In this study, we found that silencing endogenous PDGFRB inhibits basal YAP1 expression in VSMCs, suggesting that injury-induced PDGFRB expression can regulate YAP1 expression in a positive feedback loop. However, treatment of VSMCs with PDGF-BB for 2- or 5-days did not seem to alter basal YAP1 expression, suggesting that while PDGFRB may be required for basal YAP1 expression, sustained activation of PDGFRβ is insufficient to promote YAP1 expression and that other additional regulators may be required to mediate this response.
YAP1 has a well-established role in promoting cell proliferation and oncogenesis in different systems [3–5]. We and others have demonstrated that YAP1 can promote switching of VSMCs from a differentiated to a proliferative phenotype [8, 13]. With regards to YAP1 transcriptional target genes that may mediate its effects on VSMC proliferation, previous studies in rat aortic SMCs demonstrated that YAP1 induces the expression of the bona fide YAP1 targets CCN2, CCN1, and MYC [11]. However, the role of these genes in mediating YAP1 proliferative effects in VSMCs was not fully delineated. In the current study, we found that YAP1 does not affect the expression of CCN1 or MYC in human VSMCs (Figure 1G). Instead, we found that YAP1 induces the expression of several growth factor receptors, among which we identify PDGFRB as a novel YAP1/TEAD1 target gene that mediates, at least in part, the growth-promoting effects of YAP1 in VSMCs. Notably, recent studies in cancer cells demonstrated that YAP1/TEAD1 can also promote PDGFB gene expression, which leads to increased levels of secreted PDGF-BB and subsequent cell proliferation [41, 42]. Together, these observations suggest that YAP1 transcriptional activation of PDGFB and/or its receptor PDGFRB is a key mechanism underlying YAP1 function across different cell types. Future studies are required to test the role of the other identified potential target genes in our qPCR array in mediating YAP1 function in VSMCs.
While PDGFRB is expected to contribute to YAP1-mediated effects on SM de-differentiation, surprisingly, we found that silencing endogenous PDGFRB did not alter the expression of multiple SM contractile markers. However, this unexpected finding is in line with our recent finding that PDGF-BB requires a relatively long time (up to 5 days) to induce de-differentiation in HCASMCs [28]. To directly test the contribution of PDGFRB in YAP1-mediated effects on SM de-differentiation, future studies should utilize short-hairpin RNA targeting YAP1, or similar approaches, instead of transient knocking down using silencing RNA, to achieve a long-term depletion of YAP1.
Previous studies have demonstrated that in response to arterial injury, several growth factors and cytokines, such as PDGF-BB, FGF, and EGF, are released from vascular cells to promote the proliferation and migration of medial VSMCs, thereby facilitating neointima formation [1]. Several lines of evidence suggest that the PDGF/PDGFRβ signaling pathway plays a key role in injury-induced VSMC proliferation. For instance, PDGF-BB is regarded as the most potent mitogen promoting VSMC growth in culture [43]. Consistent with these in vitro findings, infusion of recombinant PDGF-BB or local transfection of a PDGF-B expression vector greatly enhances intimal thickening in models of arterial injury [44]. Conversely, neutralizing antibodies against PDGF-AB that suppress the activity of PDGF-AA, PDGF-BB and PDGF-AB attenuate neointima formation after arterial injury [45]. Furthermore, previous studies have demonstrated that PDGFRβ expression and activation is enhanced after arterial injury [18, 46], and inhibition of PDGFRβ, but not PDGFRα, inhibits neointima formation in animal models of neointima formation [24, 25]. In the current study, we found that YAP1 exerts opposing effects on the expression of PDGFR isoforms in VSMCs, inducing PDGFRβ while downregulating PDGFRα. Importantly, time-course experiments with PDGF-BB treatment in the presence or absence of YAP1 demonstrate that YAP1 promotes rather than inhibits PDGF-BB-elicited activation of pro-mitogenic signaling. Furthermore, we found that depletion or pharmacological inhibition of PDGFRβ is sufficient to attenuate both basal and YAP1-induced VSMC proliferation. The dominant effects of PDGF-BB on PDGFRβ in VSMCs may be attributed to the relative abundance of PDGFRβ versus PDGFRα in human VSMCs (Online Figure IB). Consistent with our in vitro findings, we also demonstrate here that YAP1 deficiency in postnatal VSMCs in mice is sufficient to abolish PDGFRβ induction after arterial injury and attenuate neointima formation and VSMC proliferation in vivo. However, we found that YAP1 deficiency does not alter the basal expression of PDGFRβ in quiescent vessels in vivo. The discrepancy between our in vitro findings, using siRNA, and in vivo findings, using iKO, on basal PDGFRβ expression in VSMCs may be attributed to the differences in proliferation/differentiation states of VSMCs in culture versus quiescent VSMCs in the intact vessels. Another possibility is that the basal expression of PDGFRβ in quiescent VSMCs may be dependent on both YAP1 and its paralogue TAZ, due to their functional redundancy [47]. To address this possibility, it will be necessary to generate and analyze SM-specific double Yap1 and Taz KO mice.
We have found that most of the MKI67-positive cells in the neointima area are ACTA2-positive, suggesting proliferating VSMCs contribute the neointima formation (Figure 7E). However, it has become increasingly clear that the cell identity/source in the neointima area cannot be determined by staining of a cell-specific marker alone [48]. Recent lineage tracing studies have reported that ligation-injury induced neointima formation in mouse carotid arteries is mainly attributed to the migration and subsequent proliferation of medial SMCs [49], which ultimately constitute up to 80% of the neointimal area [50]. Together, these observations suggest that SMC origin is the likely source of MKI67-positive cells in the neointima although we cannot preclude the possibility of other cell sources, such as proliferative macrophages, to contribute to the neointima formation in response to injury.
Current clinical approaches to ameliorate post-angioplasty restenosis are limited to drug-eluting stents that release high concentrations of mTORC1 inhibitors (rapamycin and its analogs). However, several lines of evidence argue that PDGFRβ inhibitors may be more effective than mTORC1 inhibitors. PDGFRβ activation leads to activation of multiple pro-mitogenic signaling pathways, such as AKT, mTORC1, and MAPK1/3. Accordingly, PDGFRβ inhibitors are expected to inhibit multiple downstream signaling pathways as compared to the specific inhibition of mTORC1 by rapamycin and its analogs. In support of this notion, in this study, we found that YAP1 deficiency-mediated downregulation of PDGFRβ inhibits AKT, mTORC1, and MAPK1/3 signaling in HCASMCs (Figure 2). Furthermore, inhibition of endothelial cell-expressed YAP1 was previously demonstrated to attenuate the expression of inflammatory factors and limit the development of atherosclerosis [38, 39]. Together, these observations suggest that targeting YAP1 in the vessel wall may inhibit both PDGFRβ-mediated pro-mitogenic signaling in VSMCs and pro-inflammatory factors in ECs, respectively, and thus may represent a more effective approach to ameliorate post-angioplasty restenosis.
Previous studies have demonstrated that YAP1 can interact with a multitude of transcription factors, including TEADs, SMADs, RUNX1/2, TP63/TP73, and ERBB4 [51]. However, while the oncogenic activity of YAP1 requires binding to TEAD proteins [52], the functional significance of YAP1 interactions with other transcription factors remains unclear [53]. In VSMCs, previous studies demonstrated that verteporfin, which disrupts the interaction between YAP1 and TEADs, can inhibit VSMC proliferation [11]. Consistent with this finding, we demonstrate here that YAP1-mediated induction of PDGFRβ is dependent on TEAD1 and that TEAD1 can positively regulate itself by binding to the TEAD1 gene promoter. These findings suggest that a YAP/TEAD interaction inhibitor may have clinical utility for treatment of restenosis.
5. Conclusions
In summary, our study unravels a novel mechanism of YAP1-induced VSMC proliferation and neointima formation via induction of PDGFRβ expression, and suggests that blocking this axis may represent a promising approach to ameliorate post-angioplasty restenosis.
Supplementary Material
Highlights.
Vascular injury-induces YAP1 expression to promote the expression of TEAD1
YAP1 forms a transcriptional complex with TEAD1 to activate PDGFRβ expression
Upregulated PDGFRβ expression promotes PDGF-BB-mediated pro-mitogenic signaling
PDGF-BB signaling enhances VSMC proliferation and injury-induced neointima formation
Blocking YAP1/TEAD1-PDGFRβ axis is a promising approach to ameliorate restenosis
Acknowledgements
We thank Dr. B. Paul Herring for his critical reading of this manuscript.
Funding
The work at the JZ laboratory is supported by a grant from the National Heart, Lung, and Blood Institute, NIH (R01HL149995). JZ is a recipient of Established Investigator Award (17EIA33460468) and Transformational Project Award (19TPA34910181) from the American Heart Association. IO is supported by a K99 award (K99HL153896) from the National Heart, Lung, and Blood Institute, NIH, and a postdoctoral fellowship (18POST34030400) from the American Heart Association. KD is supported by a postdoctoral fellowship (19POST34450071) from the American Heart Association.
Footnotes
Declaration of Competing Interest
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Owens GK, Kumar MS, Wamhoff BR, Molecular regulation of vascular smooth muscle cell differentiation in development and disease, Physiological reviews 84(3) (2004) 767–801. [DOI] [PubMed] [Google Scholar]
- [2].Bennett MR, Sinha S, Owens GK, Vascular Smooth Muscle Cells in Atherosclerosis, Circ.Res. 118(4) (2016) 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D, Elucidation of a universal size-control mechanism in Drosophila and mammals, Cell 130(6) (2007) 1120–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhao B, Li L, Lei Q, Guan KL, The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version, Genes & development 24(9) (2010) 862–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moroishi T, Hansen CG, Guan KL, The emerging roles of YAP and TAZ in cancer, Nature reviews. Cancer 15(2) (2015) 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL, TEAD mediates YAP-dependent gene induction and growth control, Genes & development 22(14) (2008) 1962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, Agarinis C, Schmelzle T, Bouwmeester T, Schubeler D, Bauer A, YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers, PLoS Genet 11(8) (2015) e1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang X, Hu G, Gao X, Wang Y, Zhang W, Harmon EY, Zhi X, Xu Z, Lennartz MR, Barroso M, Trebak M, Chen C, Zhou J, The induction of yes-associated protein expression after arterial injury is crucial for smooth muscle phenotypic modulation and neointima formation, Arterioscler Thromb Vasc Biol 32(11) (2012) 2662–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang Y, Hu G, Liu F, Wang X, Wu M, Schwarz JJ, Zhou J, Deletion of yes-associated protein (YAP) specifically in cardiac and vascular smooth muscle cells reveals a crucial role for YAP in mouse cardiovascular development, Circ Res 114(6) (2014) 957–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Feng X, Liu P, Zhou X, Li MT, Li FL, Wang Z, Meng Z, Sun YP, Yu Y, Xiong Y, Yuan HX, Guan KL, Thromboxane A2 Activates YAP/TAZ Protein to Induce Vascular Smooth Muscle Cell Proliferation and Migration, J Biol Chem 291(36) (2016) 18947–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kimura TE, Duggirala A, Smith MC, White S, Sala-Newby GB, Newby AC, Bond M, The Hippo pathway mediates inhibition of vascular smooth muscle cell proliferation by cAMP, J Mol Cell Cardiol 90 (2016) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yu Q, Li W, Jin R, Yu S, Xie D, Zheng X, Zhong W, Cheng X, Hu S, Li M, Zheng Q, Li G, Song Z, PI3Kgamma (Phosphoinositide 3-Kinase gamma) Regulates Vascular Smooth Muscle Cell Phenotypic Modulation and Neointimal Formation Through CREB (Cyclic AMP-Response Element Binding Protein)/YAP (Yes-Associated Protein) Signaling, Arteriosclerosis, thrombosis, and vascular biology (2019) Atvbaha118312212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xie C, Guo Y, Zhu T, Zhang J, Ma PX, Chen YE, Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin, J Biol Chem 287(18) (2012) 14598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ross R, Glomset J, Kariya B, Harker L, A platelet-dependent serum factor that stimulates the proliferation of arterial smooth muscle cells in vitro, Proceedings of the National Academy of Sciences of the United States of America 71(4) (1974) 1207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Thyberg J, Palmberg L, Nilsson J, Ksiazek T, Sjolund M, Phenotype modulation in primary cultures of arterial smooth muscle cells. On the role of platelet-derived growth factor, Differentiation; research in biological diversity 25(2) (1983) 156–67. [DOI] [PubMed] [Google Scholar]
- [16].Gomez D, Owens GK, Smooth muscle cell phenotypic switching in atherosclerosis, Cardiovascular research 95(2) (2012) 156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Holycross BJ, Blank RS, Thompson MM, Peach MJ, Owens GK, Platelet-derived growth factor-BB-induced suppression of smooth muscle cell differentiation, Circ Res 71(6) (1992) 1525–32. [DOI] [PubMed] [Google Scholar]
- [18].Rubin K, Tingstrom A, Hansson GK, Larsson E, Ronnstrand L, Klareskog L, Claesson-Welsh L, Heldin CH, Fellstrom B, Terracio L, Induction of B-type receptors for platelet-derived growth factor in vascular inflammation: possible implications for development of vascular proliferative lesions, Lancet 1(8599) (1988) 1353–6. [DOI] [PubMed] [Google Scholar]
- [19].Wilcox JN, Smith KM, Williams LT, Schwartz SM, Gordon D, Platelet-derived growth factor mRNA detection in human atherosclerotic plaques by in situ hybridization, J Clin Invest 82(3) (1988) 1134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sano H, Sudo T, Yokode M, Murayama T, Kataoka H, Takakura N, Nishikawa S, Nishikawa SI, Kita T, Functional blockade of platelet-derived growth factor receptor-beta but not of receptor-alpha prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice, Circulation 103(24) (2001) 2955–2960. [DOI] [PubMed] [Google Scholar]
- [21].Uchida K, Sasahara M, Morigami N, Hazama F, Kinoshita M, Expression of platelet-derived growth factor B-chain in neointimal smooth muscle cells of balloon injured rabbit femoral arteries, Atherosclerosis 124(1) (1996) 9–23. [DOI] [PubMed] [Google Scholar]
- [22].Panek RL, Dahring TK, Olszewski BJ, Keiser JA, PDGF receptor protein tyrosine kinase expression in the balloon-injured rat carotid artery, Arteriosclerosis, thrombosis, and vascular biology 17(7) (1997) 1283–8. [DOI] [PubMed] [Google Scholar]
- [23].Rosenkranz S, Kazlauskas A, Evidence for distinct signaling properties and biological responses induced by the PDGF receptor alpha and beta subtypes, Growth factors (Chur, Switzerland) 16(3) (1999) 201–16. [DOI] [PubMed] [Google Scholar]
- [24].Giese NA, Marijianowski MMH, McCook O, Hancock A, Ramakrishnan V, Fretto LJ, Chen CY, Kelly AB, Koziol JA, Wilcox JN, Hanson SR, The role of alpha and beta platelet-derived growth factor receptor in the vascular response to injury in nonhuman primates, Arterioscler. Thromb. Vasc. Biol. 19(4) (1999) 900–909. [DOI] [PubMed] [Google Scholar]
- [25].Sirois MG, Simons M, Edelman ER, Antisense oligonucleotide inhibition of PDGFR-beta receptor subunit expression directs suppression of intimal thickening, Circulation 95(3) (1997) 669–76. [DOI] [PubMed] [Google Scholar]
- [26].Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D, The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals, Developmental cell 19(1) (2010) 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S, G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension, Nat Med 14(1) (2008) 64–68. [DOI] [PubMed] [Google Scholar]
- [28].Osman I, He X, Liu J, Dong K, Wen T, Zhang F, Yu L, Hu G, Xin H, Zhang W, Zhou J, TEAD1 (TEA Domain Transcription Factor 1) Promotes Smooth Muscle Cell Proliferation Through Upregulating SLC1A5 (Solute Carrier Family 1 Member 5)-Mediated Glutamine Uptake, Circ Res 124(9) (2019) 1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kazlauskas A, Cooper JA, Autophosphorylation of the PDGF receptor in the kinase insert region regulates interactions with cell proteins, Cell 58(6) (1989) 1121–33. [DOI] [PubMed] [Google Scholar]
- [30].Sorkin A, Westermark B, Heldin CH, Claesson-Welsh L, Effect of receptor kinase inactivation on the rate of internalization and degradation of PDGF and the PDGF beta-receptor, The Journal of cell biology 112(3) (1991) 469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mori S, Heldin CH, Claesson-Welsh L, Ligand-induced polyubiquitination of the platelet-derived growth factor beta-receptor, The Journal of biological chemistry 267(9) (1992) 6429–34. [PubMed] [Google Scholar]
- [32].Sun L, Tran N, Liang C, Tang F, Rice A, Schreck R, Waltz K, Shawver LK, McMahon G, Tang C, Design, synthesis, and evaluations of substituted 3-[(3- or 4-carboxyethylpyrrol-2-yl)methylidenyl]indolin-2-ones as inhibitors of VEGF, FGF, and PDGF receptor tyrosine kinases, J Med Chem 42(25) (1999) 5120–30. [DOI] [PubMed] [Google Scholar]
- [33].Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R, Histone H3K27ac separates active from poised enhancers and predicts developmental state, Proceedings of the National Academy of Sciences of the United States of America 107(50) (2010) 21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Akerberg BN, Gu F, VanDusen NJ, Zhang X, Dong R, Li K, Zhang B, Zhou B, Sethi I, Ma Q, Wasson L, Wen T, Liu J, Dong K, Conlon FL, Zhou J, Yuan GC, Zhou P, Pu WT, A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers, Nature communications 10(1) (2019) 4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu J, Wen T, Dong K, He X, Zhou H, Shen J, Fu Z, Hu G, Ma W, Li J, Wang W, Wang L, Akerberg BN, Xu J, Osman I, Zheng Z, Wang W, Du Q, Pu WT, Xiang M, Chen W, Su H, Zhang W, Zhou J, TEAD1 protects against necroptosis in postmitotic cardiomyocytes through regulation of nuclear DNA-encoded mitochondrial genes, Cell death and differentiation (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kumar A, Lindner V, Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow, Arterioscler Thromb Vasc Biol 17(10) (1997) 2238–44. [DOI] [PubMed] [Google Scholar]
- [37].Kudryashova TV, Goncharov DA, Pena A, Kelly N, Vanderpool R, Baust J, Kobir A, Shufesky W, Mora AL, Morelli AE, Zhao J, Ihida-Stansbury K, Chang BJ, DeLisser H, Tuder RM, Kawut SM, Sillje HHW, Shapiro S, Zhao YT, Goncharova EA, HIPPO-Integrin-linked Kinase Cross-Talk Controls Self-Sustaining Proliferation and Survival in Pulmonary Hypertension, American Journal of Respiratory and Critical Care Medicine 194(7) (2016) 866–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan KL, Li YJ, Chien S, Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis, Proc Natl Acad Sci U S A 113(41) (2016) 11525–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, Liu J, Deng D, Lau CW, Wan S, Ai D, Mak KK, Tong KK, Kwan KM, Wang N, Chiu JJ, Zhu Y, Huang Y, Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow, Nature 540(7634) (2016) 579–582. [DOI] [PubMed] [Google Scholar]
- [40].Xu F, Ahmed AS, Kang X, Hu G, Liu F, Zhang W, Zhou J, MicroRNA-15b/16 Attenuates Vascular Neointima Formation by Promoting the Contractile Phenotype of Vascular Smooth Muscle Through Targeting YAP, Arterioscler Thromb Vasc Biol 35(10) (2015) 2145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang K-J, Wang C, Dai L-H, Yang J, Huang H, Ma X-J, Zhou Z, Yang Z-Y, Xu W-D, Hua M-M, Lu X, Zeng S-X, Wang H-Q, Zhang Z-S, Cheng Y-Q, Liu D, Tian Q-Q, Sun Y-H, Xu C-L, Targeting an Autocrine Regulatory Loop in Cancer Stem-like Cells Impairs the Progression and Chemotherapy Resistance of Bladder Cancer, Clin. Cancer Res. 25(3) (2019) 1070–1086. [DOI] [PubMed] [Google Scholar]
- [42].Liang YY, Deng XB, Lin XT, Jiang LL, Huang XT, Mo ZW, Yuan YW, Teh MT, RASSF1A inhibits PDGFB-driven malignant phenotypes of nasopharyngeal carcinoma cells in a YAP1-dependent manner, Cell death & disease 11(10) (2020) 855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Raines EW, PDGF and cardiovascular disease, Cytokine Growth Factor Rev 15(4) (2004) 237–54. [DOI] [PubMed] [Google Scholar]
- [44].Jawien A, Bowenpope DF, Lindner V, Schwartz SM, Clowes AW, Platelet-Derived Growth-Factor Promotes Smooth-Muscle Migration and Intimal Thickening in a Rat Model of Balloon Angioplasty, J. Clin. Invest. 89(2) (1992) 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lewis CD, Olson NE, Raines EW, Reidy MA, Jackson CL, Modulation of smooth muscle proliferation in rat carotid artery by platelet-derived mediators and fibroblast growth factor-2, Platelets 12(6) (2001) 352–8. [DOI] [PubMed] [Google Scholar]
- [46].Abe J, Deguchi J, Takuwa Y, Hara K, Ikari Y, Tamura T, Ohno M, Kurokawa K, Tyrosine phosphorylation of platelet derived growth factor beta receptors in coronary artery lesions: implications for vascular remodelling after directional coronary atherectomy and unstable angina pectoris, Heart 79(4) (1998) 400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN, Hippo pathway effector Yap promotes cardiac regeneration, Proc Natl Acad Sci U S A 110(34) (2013) 13839–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liu M, Gomez D, Smooth Muscle Cell Phenotypic Diversity, Arteriosclerosis, thrombosis, and vascular biology 39(9) (2019) 1715–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chappell J, Harman JL, Narasimhan VM, Yu HX, Foote K, Simons BD, Bennett MR, Jorgensen HF, Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models, Circ.Res. 119(12) (2016) 1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Herring BP, Hoggatt AM, Burlak C, Offermanns S, Previously differentiated medial vascular smooth muscle cells contribute to neointima formation following vascular injury, Vascular cell 6 (2014) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yu FX, Guan KL, The Hippo pathway: regulators and regulations, Genes & development 27(4) (2013) 355–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D, Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP, Genes & development 26(12) (2012) 1300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Varelas X, The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease, Development (Cambridge, England) 141(8) (2014) 1614–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.