

Abstract
Background:
Mature primary neuronal cultures are an important model of the nervous system, but limited scalability has been a major challenge in their use for drug discovery of neurodegenerative diseases. This work describes a method for improving scalability through the use of larger format microtiter plates while preserving culture quality.
New Method:
Here we describe a method and quality control procedures for growing embryonic day 18 rat hippocampal/cortical neuronal cultures in 384-well microtiter plates for three weeks in vitro.
Results:
We use these cultures in two assays measuring intracellular lipid vesicle trafficking and synapse density for routine screening of small molecule libraries. Together this culture system and screening platform have successfully identified therapeutics capable of improving cognitive function in transgenic models of Alzheimer’s disease that have advanced to clinical trials, validating their translational applicability.
Comparison with Existing Methods:
Our method enables the growth of healthy, mature neurons in larger format microtiter plates than in traditional primary neuronal culturing protocols, making it ideal for drug screening and mechanism of action studies.
Conclusion:
The predictive capacity of this culture system and screening platform provides a method for rapidly identifying novel disease-modifying neurodegenerative therapeutics.
Keywords: Primary neurons, Tissue culture, Alzheimer’s disease, Aβ, Amyloid beta, Oligomers, Neurons, Therapeutics discovery, Drug discovery, Screening methods
1. Introduction
Rodent primary neuronal cultures are a robust tool for uncovering the molecular and cellular mechanisms underlying brain function in both health and disease (Fath et al., 2009). Beginning with the pioneering studies of Gary Banker and others, the use of these cultures in neurobiology is widespread (Dotti et al., 1988; Kaech and Banker, 2006; Banker and Goslin, 1998; Benson et al., 1994; Banker and Cowan, 1977; Westbrook and Nelson, 1983). For example, hippocampal and cortical cultures are an important model of synaptic dysfunction in neurodegenerative diseases such as Alzheimer’s disease (AD), for which there are currently no disease-modifying therapies.
In order to model the mature nervous system, maturation of primary neurons in vitro is necessary. Primary neuronal cultures are typically derived from embryonic vs. postnatal brain tissue because of the high cellular mortality observed in postnatal tissue following dissociation that likely arises from truncation of the more developed dendritic and axonal arbors in postnatal tissue (Sciarretta and Minichiello, 2010). Characterization of the in vitro development and maturation of mixed hippocampal and cortical neurons in culture has been particularly extensive (Fath et al., 2009; Kaech and Banker, 2006). Following culturing of embryonic day 18 (E18) hippocampal and cortical tissue, neuronal polarity develops within two days in vitro (DIV2) and dendritic spines first emerge around DIV14 (Kaech and Banker, 2006). By three weeks in culture, neurons have formed a dense axonal network (Fath et al., 2009) and extensive dendritic branching (Sahu et al., 2019; Ramakers et al., 1998; Yang et al., 2010; Roppongi et al., 2017) with large numbers of synapse-containing spines (Kaech and Banker, 2006; Lu et al., 2016). Comparable to development of the brain in situ, synaptic density increases steeply during the third week (Grabrucker et al., 2009) and peaks at DIV21 (Ramakers et al., 1998). By three weeks in vitro cultures also exhibit spontaneous oscillatory activity (Banker and Goslin, 1998; Opitz et al., 2002) and express the full complement of synaptic proteins found in the adult (Torre and Nicholls, 1998). Therefore, mixed cultures containing both hippocampal and cortical primary neurons grown to DIV21 are an appropriate model of the mature nervous system for use in in vitro drug discovery experiments.
Identification of brain-penetrant small molecule therapeutics for neurodegenerative diseases usually begins with translational in vitro systems that can test many compounds simultaneously in multi-point dose response. Alternatives to dissociated primary neurons include acute slices, organotypic cultures, organoids, tumor cell lines of neuronal origin, and differentiated human stem cells. However, each of these has drawbacks that make them less than ideal for primary screening. Acute slices, organoids and organotypic cultures have intact brain circuitry but are difficult and costly to scale up to the throughput typically required for primary screens. Differentiated tumor cell lines can be scaled up since they are capable of the cell division that primary neurons are not, but they lack several of the features of primary neurons (Al-Ali et al., 2013; Kaech and Banker, 2006; Banker and Goslin, 1998). For example, continuous neuronal cell lines do not exhibit well-defined axons, dendrites, and synapses (Kaech and Banker, 2006; Banker and Goslin, 1998). Neural progenitor cells derived from human patients, rodent embryonic stem cells, or induced pluripotent stem cells require extended periods of time to differentiate and still do not fully replicate all of the network properties of primary neurons (Gazina et al., 2018). Dissociated primary neurons and glia derived from embryonic rodent brain and do not suffer from the drawbacks of these surrogate systems (Al-Ali et al., 2013).
Larger numbers of wells per microtiter plate provide opportunity for more replicate data points per dose and are therefore typically favored in screening operations. However, there are few reports of dissociated neuronal cultures (DIV14) in 384-well microtiter plates being used in primary compound screening (Spicer et al., 2017), and only one other report (Crowe et al., 2020) of cultures being grown all the way to maturity (DIV21) on these plates for the purpose of drug discovery. As we reported previously (Izzo et al., 2014a, b), we have successfully used DIV21 hippocampal and cortical cultures at DIV21 for routine compound screening; here we provide both more detailed methods and additional information on the culture quality control process that has been optimized from more than 10 years of weekly use.
The Aβ oligomer hypothesis of Alzheimer’s disease etiology is based on the observation that the protein Aβ builds up with age in the brain and self-associates to form oligomers; this structural state is the most toxic shape of the protein, because published evidence demonstrates that it binds saturably to a single receptor site at neuronal synapses and disrupts synaptic plasticity (Jin and Selkoe, 2015; Selkoe and Hardy, 2016; Lacor et al., 2004; Cline et al., 2018; Haas and Strittmatter, 2016; Brody and Strittmatter, 2018; Smith and Strittmatter, 2017; Kim et al., 2013), resulting in synapse loss and cognitive dysfunction. We model Alzheimer’s disease by treating our DIV21 culture model of the mature nervous system with Aβ oligomers, and search for drug candidates that block the impact of Aβ oligomers on cell biology functions related to synaptic plasticity (Izzo et al., 2014a, b). Here we provide additional detail on the performance parameters of two assays used in the drug discovery process: the primary screening Trafficking Assay, which measures the negative impact of Aβ oligomers on intracellular lipid vesicle trafficking rate, and the secondary screening Immunofluorescence Assay, capable of measuring cell type, Aβ oligomer binding, and the negative impact of Aβ oligomer binding on synapse density and synaptic protein and receptor expression.
We previously reported that compounds that block the negative impact of Aβ oligomers on intracellular lipid vesicle trafficking rate and synapse number (whether added before or after Aβ oligomer addition to cultures, defined as a “hit phenotype”) were forwarded to behavioral efficacy testing in transgenic Alzheimer’s mouse models. This hit phenotype successfully identified seven compounds from two structurally distinct chemical series capable of significantly improving cognitive function in transgenic mouse models of AD (Izzo et al., 2014a). The most advanced of these compounds, CT1812, is currently in four Phase 2 trials in Alzheimer’s patients. To our knowledge this is the first report of cultures in 384-well microtiter plates being used in conjunction with a screening assays to reliably predict compounds that will be effective in behavioral models. The predictive capacity of this culture system and screening platform allows for the rapid identification of novel disease-modifying neurodegenerative therapeutics.
2. Materials and methods
The methods for generating and maintaining a healthy hippocampal/cortical mixed neuronal culture are outlined below. These steps have been optimized for health and performance in our assays.
2.1. Culturing
All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Cognition Therapeutics and are in compliance with the Office of Laboratory and Animal Welfare (OLAW), the Animal Veterinary Medical Association (AVMA) and the Guide for the Care and Use of Laboratory Animals, Eighth Edition. Pregnant Sprague Dawley rats were obtained on embryonic day 14 (E14) from Taconic Biosciences (Rensselaer, NY, USA). Pregnant rats were housed under pathogen-free conditions with iso-BLOX enrichment material (Envigo, Indianapolis, Indiana, USA), food, and water ad libitum until reaching embryonic day 18 (E18) to acclimate to their new environment. Embryos from a single rat were pooled to produce a unique dissociated neuronal culture. All analyses described below were performed on at least 3 unique cultures. E18 pregnant rats were anesthetized using CO2 asphyxiation per AVMA guidelines. After euthanasia was verified, the embryonic sac was removed and placed into a 150 mm × 25 mm petri dish containing Hank’s Balanced Salt Solution (HBSS). The petri dish containing the embryos was transferred to the culturing area (e.g. clean room or hood), using a sterilized plastic transport container to contain spills. Table 1 lists all reagents and tools that are needed for the culturing of hippocampal/cortical neurons. Using Delicate Operating Scissors, the embryo was removed from the amniotic sac. The embryo was placed in a sterile petri dish containing HBSS and decapitated with scissors, rinsed in another petri dish containing HBSS and then placed into a silgar tray filled with HBSS. Following overlying bone removal using Castroviejo Angled Scissors, the brain was dissected using a Freer Elevator starting from under the frontal lobe. The leading edge of the elevator was kept along the bottom of the skull to eliminate damage to the brain. Once the brain was removed, it was placed in a clean 100 mm × 15 mm petri dish containing HBSS under a dissection microscope.
Table 1.
List of Reagents and Tools for Neuronal Culture.
| Culturing Reagents | Company | Catalogue Number |
|---|---|---|
| Plating Media (see Table 3 for recipe) | Thermo Fisher | various |
| Feeding Media (see Table 3 for recipe) | Thermo Fisher | various |
| Hank’s Balanced Salt Solution (HBSS) | Thermo Fisher | 14,175,103 |
| Trypsin, 2.5 % (10X) | Thermo Fisher | 15090–046 |
| 1x Phosphate Buffer Saline | Thermo Fisher | 14040–182 |
| Trypan Blue 0.4% | Thermo Fisher | 45000–717 |
| Tools for Hippocampal/Cortical Isolation | Company | Catalogue Number |
| NO. 3c Dumont | Roboz | RS-5043 |
| NO. PP Heavy Duty Dumont | Roboz | RS-4950 |
| Micro Diss FCPS 4 in. 1 × 2 teeth | Roboz | RS-5152 |
| Adson FCPS | Roboz | RS-5234 |
| Micro Diss Needle Holder | Roboz | RS-6060 |
| Castroviejo Angled Scissors | Roboz | RS-5668 |
| Delicate Operating Scissors | Roboz | RS-6700 |
| Freer Elevator Double Ended | Roboz | RS-8820 |
| 0.25 mm Tungsten wire | Roboz | RS-6065 |
The meninges (membrane containing red blood vessels) were removed using two sharp forceps (No. 3c Dumont), beginning at the ventral side of the brain, and working toward the dorsal side. Care was taken not to distort or poke the brain tissue itself during this step. With the dorsal side of the brain facing up, the two hemispheres were separated through the telencephalon using a 0.25 mm Tungsten wire. Working on one hemisphere at a time, the excess meninges were removed, and the hippocampus and cortex isolated from the striatum and ventral midline by means of the ventricular opening using two distinct cuts. The hippocampus was visible as a slightly thicker portion lining the curved medial edge of the cortex. Fig. 1 illustrates a diagram for isolation of hippocampal and cortical sections from the embryonic brain. The isolated tissue was carefully transferred to a conical tube containing HBSS. The brain dissection and hippocampal/cortical isolation were repeated until the required number of embryos were dissected. Pooling 10 brains yielded between 5.0–6.0 × 106 cells/mL or about 5.0–6.0 × 105 cells/mL per brain (5–6 million cells per culture). Hippocampal and cortical tissue from the embryo brains were digested in 2.5 % Trypsin (Life Technologies, Carlsbad, California, USA) for 10 min at room temperature (21 °C) and then aspirated and dispensed between 30–40 times using a glass 10 mL pipette to dissociate cells. Isolated cells were plated at a density of 4.6 × 104 cells per cm2 in 384-well poly-D Lysine coated plates (Greiner, Monroe, North Carolina, USA) in Neurobasal Media (Life Technologies) supplemented with B27 (Life Technologies), Glutamax (Life Technologies) and antibiotics (penicillin, 50 units/mL and streptomycin 50 mg/mL, Life Technologies) (see Table 2 for plating media recipe). Cultures were maintained at 37 °C in 5% CO2 with weekly media changes of 50 % of culture media for 3 weeks prior to Quality Control evaluation for experimentation (see Table 2 for feeding media recipe).
Fig. 1.
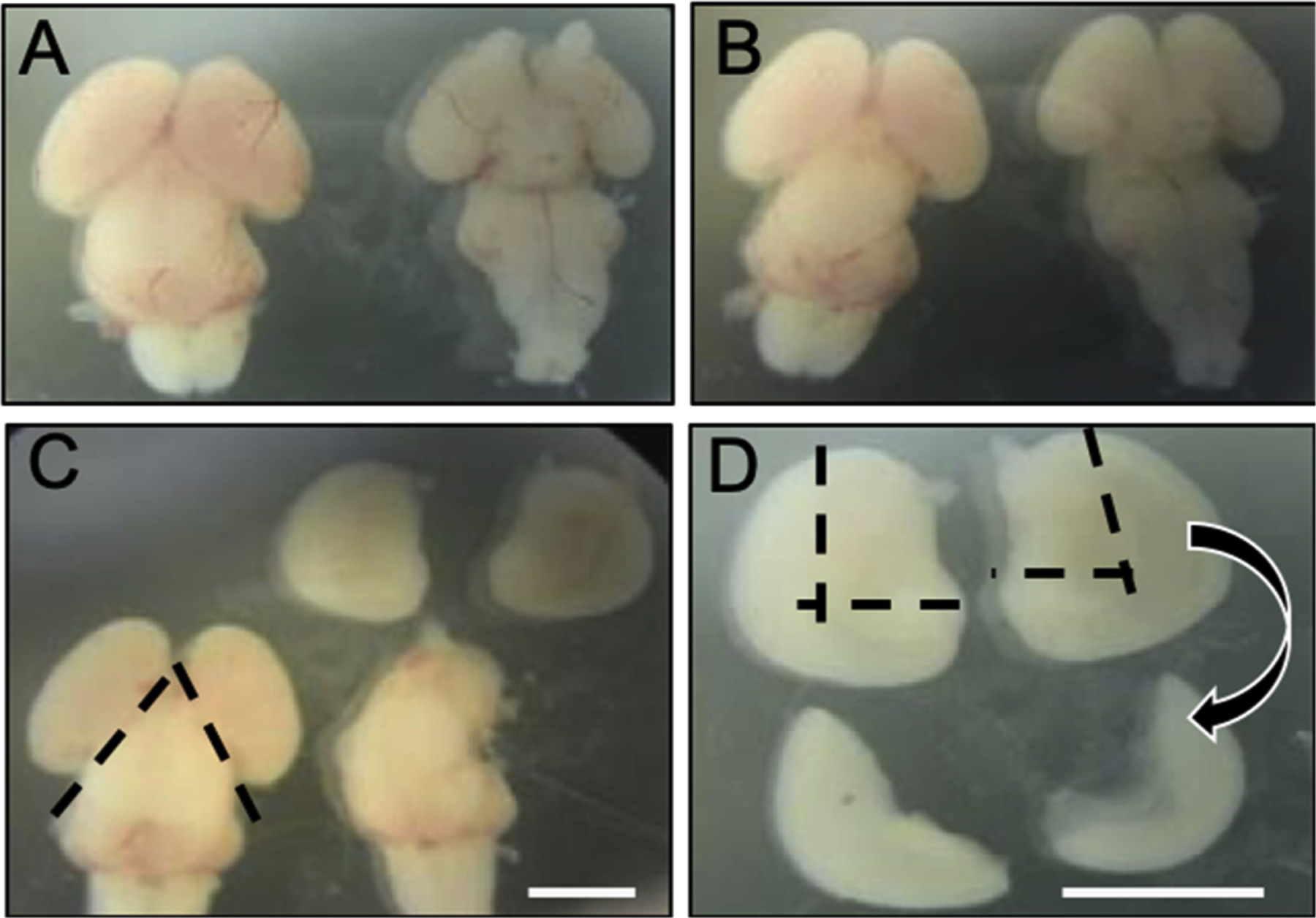
Isolation of hippocampal and cortical tissue from embryonic day 18 (E18) Sprague-Dawley rat brain. (A) Dorsal (left) and ventral (right) view of E18 rat brain with meningial membrane intact. (B) Dorsal (left) and ventral (right) view of E18 rat brain with meninges removed. (C) Following removal of meninges, left and right hemisphere were dissected along the dashed lines. (D) Finally, hippocampal and cortical brain tissue were isolated by cutting along the dashed lines. Scale bars = 3 mm.
Table 2.
Plating Media and Feeding Media Recipe.
| Plating Media Components | Company | Catalogue Number | Volume |
|---|---|---|---|
| Neurobasal | Thermo Fisher | 12348–017 | 500 mL |
| B27 | Thermo Fisher | 17504–044 | 10 mL |
| Glutamax | Thermo Fisher | 35050–061 | 5 mL |
| Glutamate* | Thermo Fisher | BP378–100 | 5 mL |
| Penicillin/Streptomycin | Thermo Fisher | 15070–063 | 0.5 mL |
| Total Volume of Liquid | 520.5 mL | ||
| Feeding Media Components | Company | Catalogue Number | Volume |
| Neurobasal | Thermo Fisher | 12348–017 | 500 mL |
| B27 | Thermo Fisher | 17504–044 | 10 mL |
| Glutamax | Thermo Fisher | 35050–061 | 5 mL |
| Penicillin/Streptomycin | Thermo Fisher | 15070–063 | 0.5 mL |
| Total Volume of Liquid | 515.5 mL |
Glutamate is made at 100X Stock (500 mL of ddH20 with 0.184 g of L-Glutamic Acid) at pH of 7.
2.2. Aβ oligomer preparations
Synthetic amyloid beta (Aβ) 1–42 was purchased from California Peptide (catalog number 641 15). An Aβ monomer film was prepared by evaporating the 1,1,1,3,3,3,hexafluoro-2-propanol (HFIP) (Sigma-Aldrich, St. Louis, Missouri, USA; catalog number 105,228) at room temperature from a solution of 253 μg Aβ(1–42) in HFIP at room temperature for 20 min using N2 gas. The film was then dissolved in 10.12 μL anhydrous DMSO (Sigma-Aldrich; catalog number D2650) to make a 5 mM working solution. This solution was diluted to 100 μM with cold Basal Media Eagle media (Life Technologies; catalog number 21,010), followed by incubation at 4 °C for 24 h to form oligomers. The resulting oligomer preparations were centrifuged at 16,000 × g to pellet any insoluble fibrils and the supernatant was diluted in Neurobasal media prior to addition to cultures.
2.3. Trafficking assay
Neurons were treated with compounds and/or Aβ 1–42 oligomer preparations (0.086 % DMSO final assay concentration in culture media) and incubated for 1–24 hours at 37 °C in 5% CO2 as previously described (Izzo et al., 2014a). Tetrazolium salts (3-(4,5-dimethylthiazol-2 yl)-2,5diphenyl tetrazolium bromide, Roche Molecular Biochemicals, Basel, Switzerland) were added at a final concentration of 0.75 mM and neurons were incubated at 37 °C for 60–90 min. Vesicular formazan remaining in cells was quantified by absorbance spectrometry (590 nm with 690 nm subtracted) following extraction with 1.6 % Tween-20.
2.4. Immunofluorescence assay
Oligomeric Aβ protein was added to neuronal cultures for 60 min at 37 °C and automated immunofluorescent imaging was then used to measure oligomerized Aβ binding to neurons and glia (microtubule-associated protein 2 [MAP2]-negative cells), as described previously (Izzo et al., 2014a). In brief, cells were fixed with 3.75 % formaldehyde (Polysciences Warrington, Pennsylvania, USA; catalog number 4018) and blocking was performed with 5% normal donkey serum (GFAP experiments only; Rockland Immunochemicals, Gilbertsville, PA, USA; catalog number D220-00-0100) or 5% normal goat serum (all other antibody experiments; Tissue Culture Biologicals, Seal Beach, California, USA; catalog number 701D) and 0.5 % Triton X-100 (Sigma-Aldrich, St. Louis, Missouri, USA; catalog number T8787). Cells were then incubated with primary antibodies for Aβ, drebrin (to identify spines, a surrogate for synapses, and MAP2 (to identify dendrites on neurons), or glial markers for oligodendrocytes (Oligo-2 antibody) or astrocytes (GFAP antibody), followed by appropriate fluorescently labeled secondary antibodies and fluorescent nuclear stain, DAPI (4′,6-dia-midino-2-phenylindole, Thermo Fisher catalog #D1306, RRID: AB_2629482) (Table 3 contains primary and secondary antibody details). Images were captured on a Cellomics VTi or CX7 (RRID: SCR_018706) automated microscope with a 20X, 0.75 numerical aperture objective and analyzed using the ThermoFisher/Cellomics Neuronal Profiling bioapplication, which measured punctate labeling of Aβ along MAP2-labeled dendrites, as well as neuron count, nuclear area, and dendritic segment length (which the bioapplication refers to as neurite length).
Table 3.
Primary and Secondary Antibodies Used in Immunofluorescence Studies.
| Antibody Name | Final Assay Concentration | Vendor and Catalog Number; RRID | Clonality | Host |
|---|---|---|---|---|
| Anti-Amyloid β (Clone 4G8) | 1 μg/mL | Covance Cat # SIG-39,200; RRID: AB_10175152 | Monoclonal | Mouse |
| Anti-Drebrin | 2 μg/mL | Millipore Cat # AB10140; RRID: AB_1977159 | Polyclonal | Rabbit |
| Anti-Glial fibrillary acidic protein (GFAP) | 2 μg/mL | R&D Systems Cat # AF2594; RRID: AB_2109656 | Polyclonal | Sheep |
| Anti-Microtubule associated protein 2 (MAP2) | 0.2 μg/mL | Millipore Sigma Cat # AB5543; RRID: AB_571049 | Polyclonal | Chicken |
| Anti-Oligo-2 | 2.7 μg/mL | Sigma-Aldrich Cat # ABN899; RRID: AB_2877641 | Polyclonal | Rabbit |
| Goat Anti-Chicken Alexa Fluor 546 | 2 μg/mL | Life Technologies Cat # A11040; RRID: AB_2534097 | Polyclonal | Goat |
| Goat Anti-Mouse Alexa Fluor 647 | 2 μg/mL | Life Technologies Cat # A21235; RRID: AB_2535804 | Polyclonal | Goat |
| Goat Anti-Rabbit Alexa Fluor 488 | 1 μg/mL | Thermo Fisher Scientific Cat # A11008; RRID: AB_143165 | Polyclonal | Goat |
| Donkey Anti-Chicken Alexa Fluor 560 | 1 μg/mL | Jackson Immuno Research Cat # 703-165-155; RRID: AB_2340363 | Polyclonal | Donkey |
| Donkey Anti-Sheep Alexa Fluor 488 | 1 μg/mL | Jackson Immuno Research Cat # 713-545-003; RRID: AB_2340744 | Polyclonal | Donkey |
| Donkey Anti-Mouse Alexa Fluor 647 | 1 μg/mL | Jackson Immuno Research Cat # 715-605-151; RRID: AB_2340863 | Polyclonal | Donkey |
| Donkey Anti-Rabbit Alexa Fluor 488 | 1 μg/mL | Jackson Immuno Research Cat # 711-545-152; RRID: AB_2313584 | Polyclonal | Donkey |
| Donkey Anti-Sheep Alexa Fluor 594 | 1 μg/mL | Jackson Immuno Research Cat # 713-585-003; RRID: AB_2340747 | Polyclonal | Donkey |
| Anti-Iba1 antibody | 2 μg/mL | Abcam Cat # AB5076 RRID: AB_2224402 | Polyclonal | Goat |
3. Results
3.1. Culture properties
Limited scalability has been a major challenge in the use of primary neuronal cultures for drug discovery of neurodegenerative diseases. Here, we describe a method and quality control procedures for growing E18 rat mixed hippocampal and neocortical neuronal cultures in 384-well microtiter plates to DIV21 for use as a model of the mature central nervous system. Photomicrographs of the dissection procedure used to isolate hippocampus and cortex from E18 rat brains are shown in Fig. 1.
The length of time that cells are grown in vitro is an important factor in the translational applicability of a culture; only cultures grown for a minimum of DIV21 replicate morphological and electrophysiological features of mature hippocampal and cortical neurons in vivo (Ramakers et al., 1998; Banker and Goslin, 1998; Opitz et al., 2002; Torre and Nicholls, 1998). Culture maturation over time is shown in Fig. 2A–C. Immunofluorescent labeling with MAP2 at DIV7, DIV14, and DIV21 demonstrates the natural growth of dendritic segments over time, reaching 529.5 +/− 73.8 μM in length by DIV21. Using MAP2 to measure length of neuronal dendritic segments and soma was an appropriate measurement of the overall health of the neurons in these cultures, a critical feature to ascertain prior to using a given cell culture in drug discovery assays. Dendritic segment length was measured using Cellomics Neuronal Profiling bioapplication (more detail in the methods section above). The maturity of neurons in vitro also affects the expression of receptors. AD is thought to be caused by toxic Aβ oligomers, which bind specifically and saturably to receptors along dendritic spines (Izzo et al., 2014b). Published evidence indicates that this oligomer receptor is composed of LilRB2, cellular prion protein and NgR1 (Smith and Strittmatter, 2017; Kim et al., 2013). At DIV18, low levels of oligomer binding are observed, however by DIV25 bright punctate binding of oligomers can be observed along dendritic shafts (Fig. 2D–E); previously published co-localization studies demonstrated that this punctate binding represents Aβ oligomer binding to synaptic receptor sites on dendritic spines (Izzo et al., 2014b; Kim et al., 2013; Smith and Strittmatter, 2017).
Fig. 2.
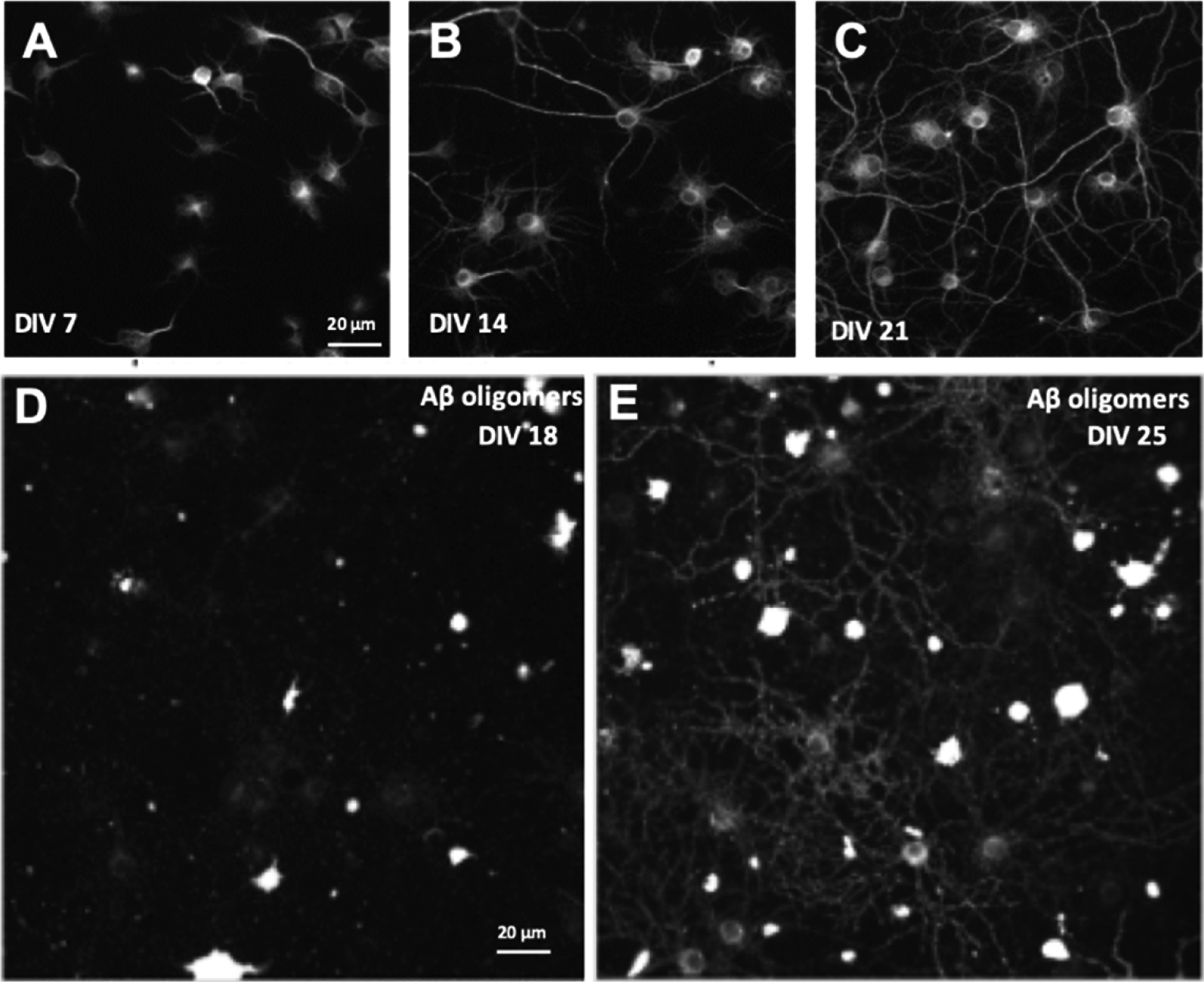
Immunofluorescent labeling of primary mixed hippocampal and neocortical neuronal cultures reveals the temporal maturation of neurons and glia in vitro. Cultures that have been maintained for increasing length of time in culture seven days in vitro (DIV7, A), DIV14 (B), and DIV21. (C) cultures were labeled with MAP2 primary antibody and the dendrite segment length quantified via automated image processing. Average dendrite segment length increases over time (DIV7 = 72.7 ± 6.2, DIV14 = 264.2 ± 30, and DIV21 = 529.5 ± 73.8 microns). Cultures were treated with Aβ 1–42 oligomers at DIV18 (D) and DIV25 (E); low binding of oligomers to synaptic receptors was observed at DIV18 but sister cultures treated with oligomers seven days later exhibited increased oligomer binding due to maturation of synaptic oligomer receptor expression (N = 3 independent cell culture preparations). Scale bar (20 μm) for B and C shown in A; scale bar (20 μm) for E shown in D.
We utilize serum-free Neurobasal media supplemented with B27, a nutritional supplement that contains essential fatty acids, hormones, vitamins and antioxidants. This defined media format is optimized for dissociated neuronal cultures and has been widely used (Chen et al., 2008), as it supports long-term viability of a variety of neuronal populations (Brewer et al., 1993; Brewer, 1995). Healthy cultures typically contain 20–35 % MAP2-positive neurons. This defined media also prevents glial overgrowth by inhibiting glial cell division (Brewer et al., 1993). We previously characterized the glial population in these cultures based on the nuclear morphology visualized by the DNA-binding dye DAPI (Izzo et al., 2014a). Approximately 27 % of MAP2-negative glial cells have a normal symmetrical nuclear morphology, with the remaining cells having an abnormal nuclear morphology and bright DAPI staining typical of fragmented and condensed chromatin, likely corresponding to unhealthy or dying glial cells (Figure S1 in Izzo et al., 2014a). Here, we further characterized the glial population by subtype based on protein expression. Out of all nucleated cells in culture (labeled with the DNA binding dye DAPI), approximately 36 % ± 7% are oligo2-positive oligodendrocytes, and 7% ± 2% GFAP-positive astrocytes (Fig. 3), with the remainder likely other glial populations. Microglial antibody Iba1 (Abcam, Cat #ab5076) labeled <1% of the cell (data not shown), therefore, it is unlikely that microglial populations represent a significant fraction of the cell population in these cultures.
Fig. 3.
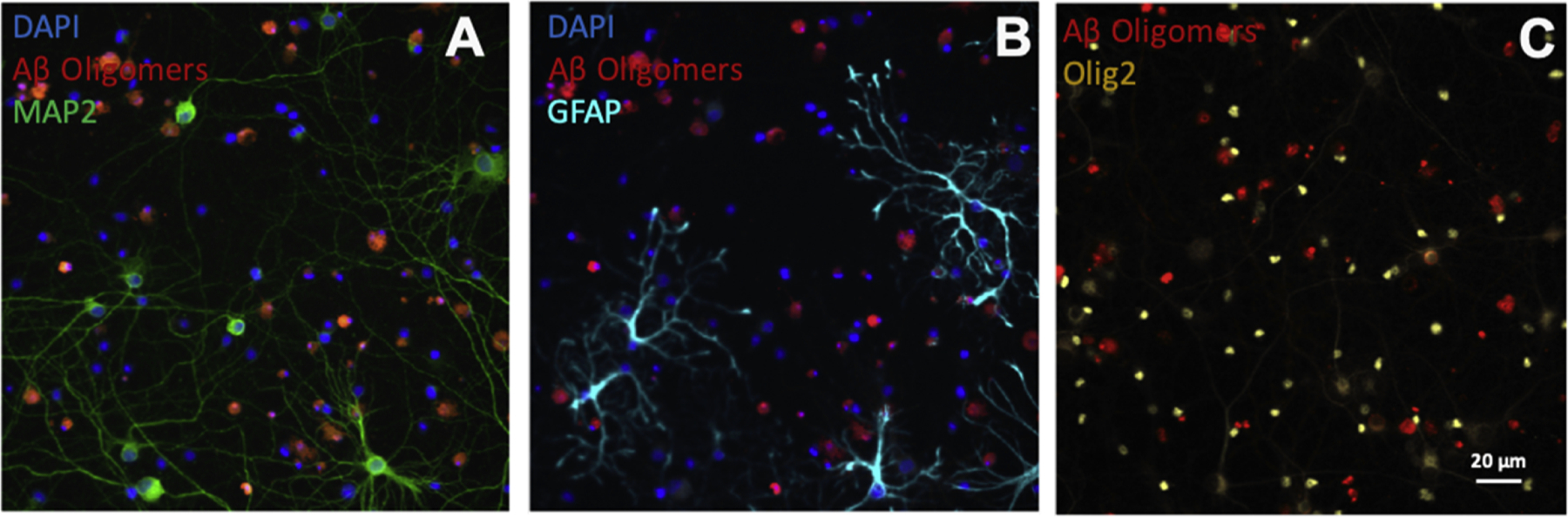
Cellular composition of 384-well DIV21 cultures. Approximately 20–35 % of the cells in these cultures are neurons (MAP2-positive cells, A), 7% ± 2% are astrocytes (GFAP-positive cells, B) and 36 % ± 7% are oligodendrocytes (oligo-2 positive cells, C). Scale bar for all images is shown in C (20 μm).
3.2. Quality control
Morphological features of neurons provide a sensitive measure of culture health, and so this is evaluated for each unique cell culture at DIV18 prior to culture use in screening assays. Immunofluorescence Assays as described in the Materials and Methods section are performed on a subset of wells from one randomly chosen plate (240 wells, roughly 2500 neurons) from each unique cell culture as a Quality Control procedure. The following parameters were quantified by automated image processing: percentage of neurons to glia (image processing parameter “Average % Neuronal Cells”, or the number of DAPI stained nuclei that are MAP2 positive), the total length of all the dendritic segments per neuron (“Neurite Length”, or the total lengths of all MAP2-positive dendritic segments in a well divided by the number of neurons in that well) and the number of drebrin-positive synaptic puncta on each neuron (“Synapse Count per Neuron”, or spot intensity per neurite length). Drebrin positive puncta correspond to spines, the vast majority of which contain a single synapse, and so are a reasonable numeric surrogate for synapse number. Historic data correlating average values for plates with performance metrics for the Trafficking Assay provided quantitative criteria for quality control based on mean +/− 2 standard deviations for each parameter. A given set of plates cultured from one set of brain tissue is deemed suitable for use in screening assays (i.e., passing Quality Control) if the quality control plate’s average values were within all of the following ranges: percentage of neurons = 20–35 %, dendritic segment length per neuron = 400–600 microns per neuron, and the number of drebrin-positive puncta = 50–150 per neuron.
Additionally, qualitative visual inspection of plates was conducted and pates were rejected if large numbers of wells (more than 30 % or 115 wells per microtiter plate) contained 1) neurons with abnormal morphologies such as dystrophic dendrites with a disorganized cytoskeleton (Fig. 4B); 2) poorly adherent cells with excessive dendritic segment fasciculation (Fig. 4C); 3) cells with no dendrite outgrowth (Fig. 4D); or 4) neurons with multiple large internal vesicles in the cell body (Fig. 4E). These features reflect poor cell health which negatively impacts assay performance. The source of poor cell health can range from microtiter plate manufacturing issues (including poor poly-D-lysine coating [Fig. 4 F–G]; fluorescent inclusions in microtiter plate plastic; non-sterile plates; and biological activity of plastic hardner leechate) to reagent quality issues (such as lot-to-lot variability in media or media supplements). Lot-to-lot reagent variability is controlled by side-by-side comparison of Quality Control assay measurement values in cultures prepared with different reagent lots for all of the culture and assay reagents described in the Methods section. For example, although B27 has been optimized by the manufacturer to minimize variability, we still observed lot-to-lot variation that must be closely monitored (see Fig. 5).
Fig. 4.
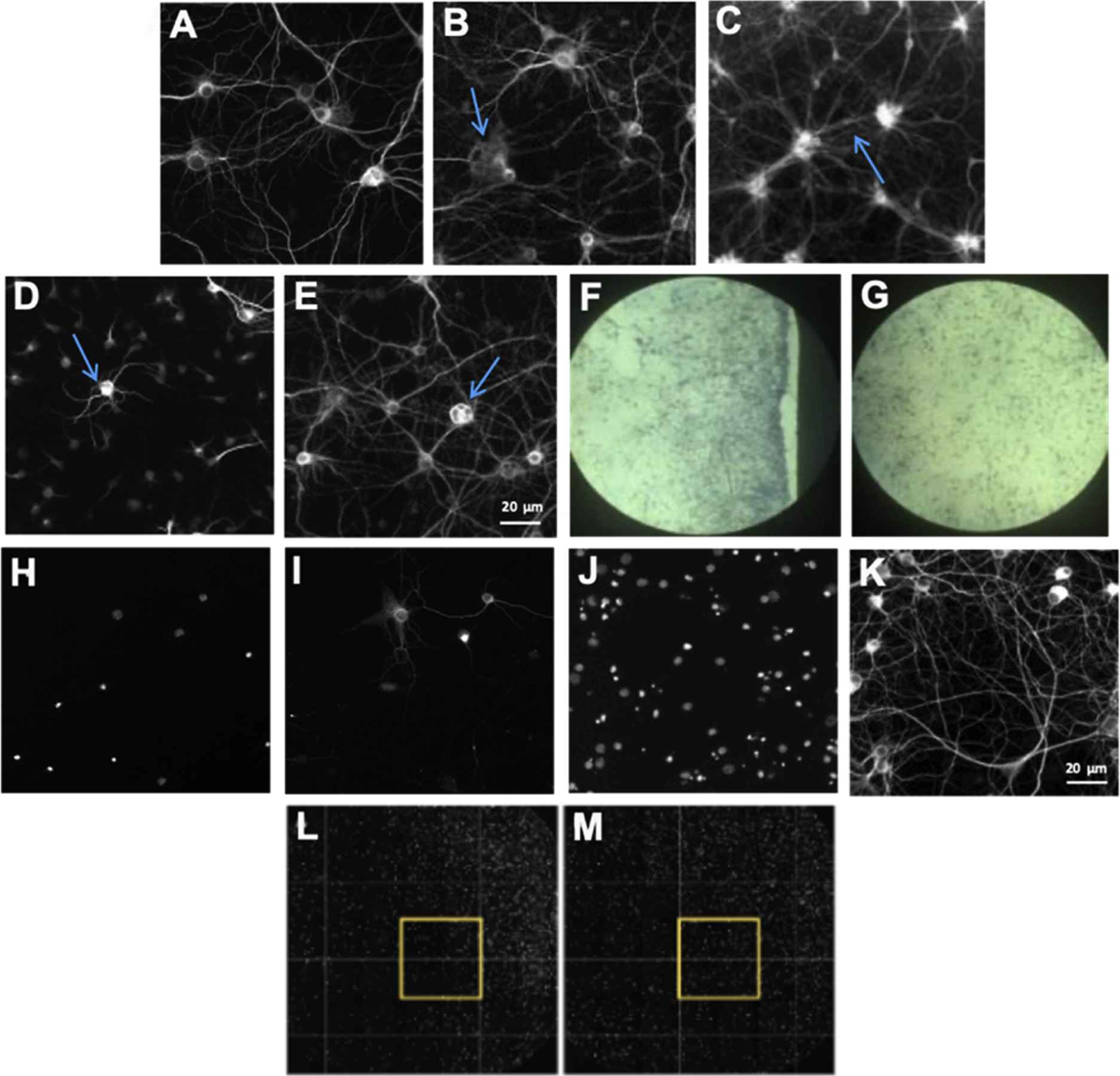
Optimization of culture health and quality. DIV21 cultures stained with MAP2 primary antibody. Healthy neurons are shown in (A). Examples of abnormal morphologies that are indicators of poor neuronal health include dystrophic dendrites with a disorganized cytoskeleton (arrow in B); poorly adherent cells with excessive dendritic segment fasciculation (arrow in C); cells with no dendritic outgrowth (arrow in D); and cells with multiple large internal vesicles in the cell body (arrow in E). Abnormal morphology can be due to a variety of causes, including incomplete poly-D-lysine coating on the microtiter plate that pulls apart at the edge of the well, leading to uneven neuronal adherence (F). With appropriate poly-D-lysine coating, neurons spread out evenly on the plate (G). Scale bar for A–D is shown in E. Additional optimization included a comparison of glass (H–I) and plastic (J–K) plates, with neurons adhering well only to plastic plates. (H) DIV21 DAPI-labeled nuclei and (I) MAP2-labeled neurons on a glass poly-D-lysine-coated 384-well plate. (J) DIV21 DAPI-labeled nuclei and (K) MAP2-labeled neurons on a plastic poly-D-lysine-coated 384-well plate. A final optimization indicates that, after plating, incubation of plates at room temperature for 30 min prior to 37 °C incubation produces nuclei that are more evenly dispersed throughout the well. (L–M) Whole well DAPI immunofluorescent labeling at DIV18. (L) Plate that was incubated at 37 °C according to the standard procedure. (M) Plate that was incubated at room temperature for 30 min before incubating at 37 °C exhibits more even distribution of cells across the well. The yellow box indicates the standard scan area of the immunofluorescence assay. Scale bar for H–J is shown in K.
Fig. 5.
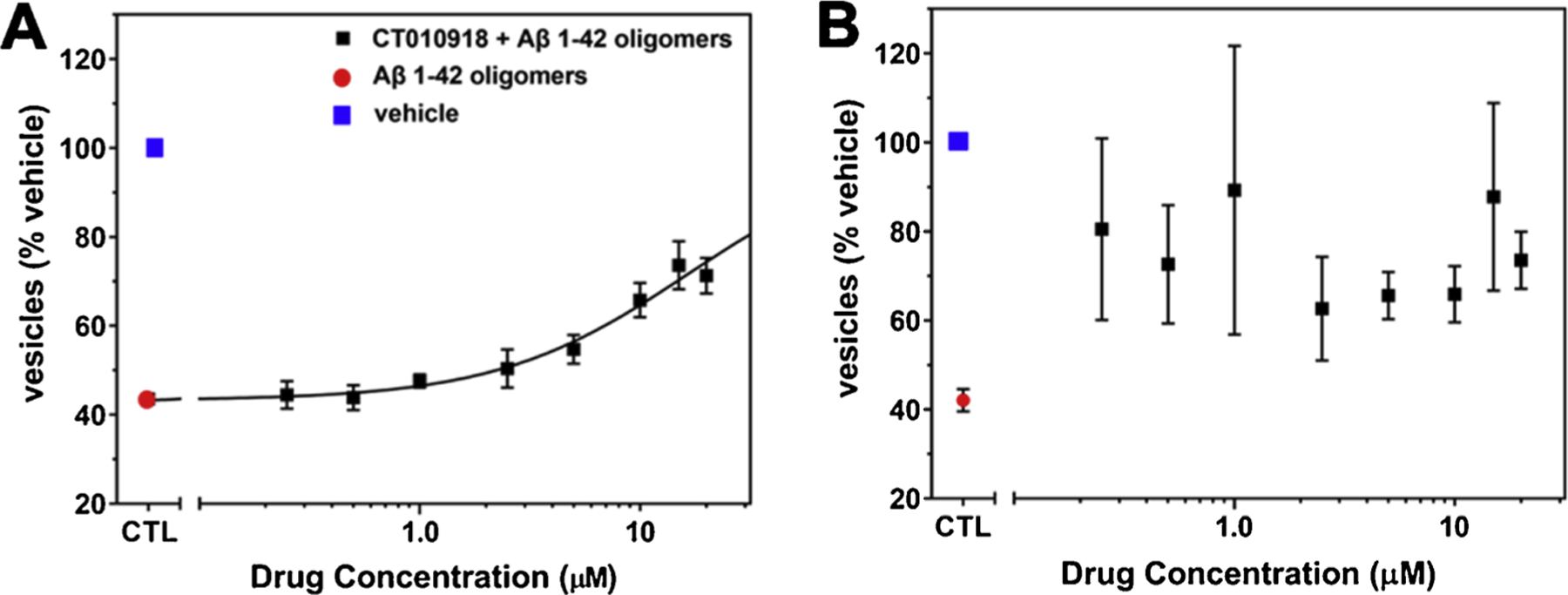
Reagent lot Quality Control. (A–B) In the Trafficking Assay (Izzo et al., 2014a) treatment of cultures with Aβ oligomers negatively impacts trafficking rate (red circles) compared to vehicle-treated cultures (blue square). (A) Cultures produced with passing lots of B27 media supplement exhibit a dose-dependent inhibition of Aβ oligomer-induced deficits by a positive control compound (CT010918) included on each assay plate. (B) Culture grown with a failed B27 lot does not exhibit dose-dependent inhibition of Aβ oligomer-induced deficits with the positive control compound. Mean and standard deviation of 3 replicate experimental plates are shown for each graph.
3.3. Optimization of cell culture performance/health
Several publications outline methods for use of primary neuronal cultures in 96-well (or smaller) format, however the smaller surface area-to-volume ratio of 384-well microtiter plates present unique challenges. In order to optimize cultures in the 384-well microtiter plate format, we compared poly-lysine-coated plates with a glass (Fig. 4H,I) or plastic (Fig. 4J,K) bottom. We found that glass-bottom plates from a variety of lots and manufacturers caused cell adherence problems, and primary hippocampal/cortical neuronal cells adhered only to the plastic surface in this 384-well footprint. Even disbursement of cells within the well is important for assay performance. Standard procedure (Fig. 4L) is to plate and immediately incubate the cells at 37 °C, however, we found that adding a 30-minute incubation at room temperature prior to placing the cells at 37 °C produced a more uniform distribution of cells across wells (Fig. 4M) in this 384-well footprint than any other mechanical factor (speed of cell dispensing, plate movement post-cell addition) and have implemented this step since our previously published papers. This procedure avoids the thermal gradients created within individual wells of multiwell plate that negatively impact cell dispersion (Lundholt et al., 2003).
3.4. Screening assays
As previously noted, plates passing Quality Control were used for screening assays. We have adapted the previously published MTT assay to measure the intracellular trafficking rate of endosomal/lysosomal lipid vesicles in neurons and glia for use with these cultures (Hong et al., 2007; Kreutzmann et al., 2010; Liu and Schubert, 1997; Izzo et al., 2014b). In this Trafficking Assay, extracellular tetrazolium salts are taken up by the cell in endocytosis and reduced to insoluble purple formazan in the endosomal/lysosomal compartment, which is then trafficked out of the cell in exocytosis. The location of this reduced formazan cargo dye within intracellular vesicles or on the surface of the cell can be measured with a variety of methods (imaging or plate-based detection). As described previously (Izzo et al., 2014a), Aβ 1–42 oligomers induce a selective acceleration of the exocytosis and not endocytosis rate vs. vehicle, such that the amount of cargo dye contained in intracellular lipid vesicles of Aβ 1–42 oligomer-treated cultures (Fig. 5A–B, red dots) is lower than that of vehicle treated cultures (Fig. 5A–B, blue dots). Positive control allosteric sigma-2 receptor antagonists (such as CT010918) can dose dependently inhibit the Aβ 1–42 oligomer-induced trafficking deficit (Fig. 5A, black dots) and are included as standards on each set of microtiter plates in routine screening. In assays performed on cultures prepared with lots of B27 that do not pass Quality Control measures, identical lots of these positive control compounds fail to produce a dose-dependent inhibition of Aβ 1–42 oligomer-induced deficits (Fig. 5B).
Performance criteria for routine screening were calculated as follows: Aβ oligomer-treated control wells divided by vehicle-treated control well values x100; values < 80 % were considered acceptable. Microtiter plates that passed control well performance criteria were accepted for subsequent compound analysis. The percentage of total microtiter plates in a one-year period that achieved this passing criteria (84 % on average) are shown in Fig. 6. By eliminating many of the variables that can hinder culture health and assay performance, historic averages of z’ = 0.285 (Zhang et al., 1999) and 15 % coefficient of variation are routinely achieved. By replicating concentration-response curves on six independent replicate plates a power of >95 % is achieved with this assay, an acceptable level of performance for reducing false positive and negative compound identification rates. It should be noted that the statistical performance of this assay does not lead to a z’-factor sufficient for single point screening. This limitation is overcome by screening in an 8-point concentration-response format in 6 replicate experiments to achieve statistical power. In practice we combine at least two replicates from three separate cell cultures to account for variability between primary preparations
Fig. 6.
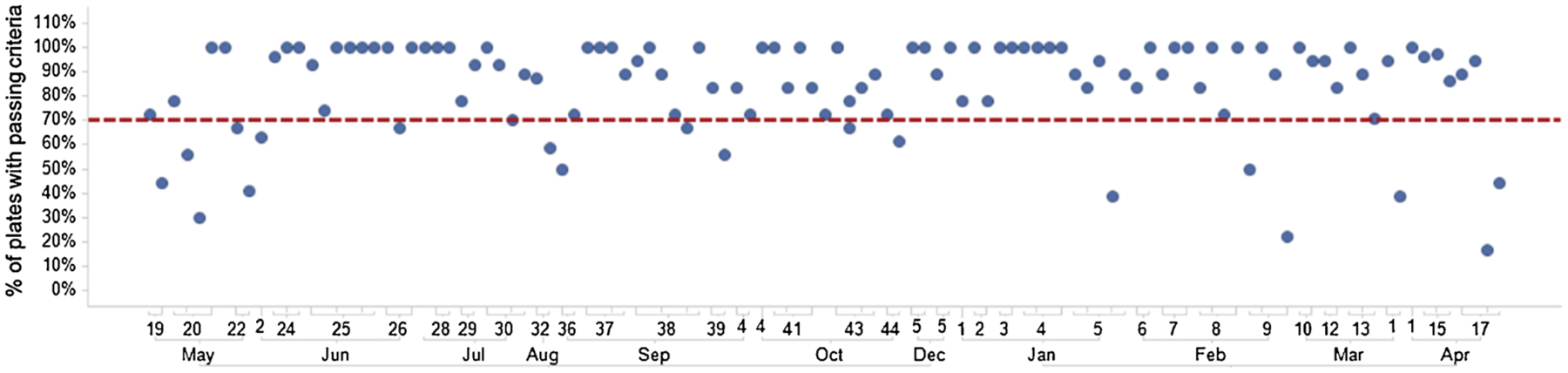
Historic data for Trafficking Assay performance over a one-year period. The percentage of total microtiter plates that passed control well performance criteria are graphed for each unique culture. The average number of plates with passing windows was 84 % ± 19 %, surpassing the benchmark of 70 % (red-dashed line).
In the screening cascade assembled to discover and optimize small molecules capable of blocking Aβ 1–42 oligomer-induced toxicity, the Trafficking Assay was used as a primary screen with a typical throughput of 50 compounds per week on a single set of liquid handling robotics for compound addition (three compounds per microtiter plate in 8-point dose response concentration curve +/− Aβ oligomers, plate layout shown in Supplemental Fig. 1). Compounds that blocked Aβ 1–42 oligomer-induced toxicity with EC50 < 1 u M were forwarded on to a secondary immunofluorescence assay that measured the ability of compounds to displace receptor-bound Aβ 1–42 oligomers and rescue Aβ 1–42 oligomer-induced synapse loss. This assay was similar to the immunofluorescence assay used for Quality Control. Automated image processing was used to quantify the number of drebrin-positive puncta (Izzo et al., 2014a). Compounds that rescued Aβ 1–42 oligomer-induced synapse loss with EC50 < 1 μM but had no effect on synapse number when dosed alone, were then forwarded on to behavioral testing in aged transgenic Alzheimer’s mouse models. The compounds tested in these assays that achieved target brain concentrations in mice, blocked Aβ 1–42 oligomer-induced trafficking deficits and synapse loss but had no effect when dosed in the absence of Aβ 1–42 oligomers were all behaviorally efficacious in the Alzheimer’s mouse model (Izzo et al., 2014a).
4. Conclusions
The present results describe an optimized culturing process for producing mature (DIV21) neuronal and glial cultures from hippocampus and neocortex. We also describe the use of these cultures with assays that model disease-relevant aspects of cellular function that are disrupted by age-related stressors like synaptotoxic Aβ 1–42 oligomers that cause neurodegenerative disorders such as Alzheimer’s disease. Together they constitute an in vitro screening platform system that models the mature central nervous system and is suitable for use in drug discovery. This screening platform has a demonstrated ability to predict efficacy in in vivo mouse studies based on compound hit phenotype in in vitro assays. The predictive ability of this screening platform to determine which compounds would be behaviorally efficacious, and which would not, accelerated the process of compound identification and optimization, and led to the discovery and development of drug candidate CT1812, a selective allosteric sigma-2 receptor complex antagonist currently in Phase 2 clinical trials.
Supplementary Material
Acknowledgements
The authors received support from the Alzheimer’s Drug Discovery Foundation (20100501), the National Institute of Neurodegenerative Disease and Stroke (NS083175), the National Institute of Aging (AG037337, AG047059, AG052252, AG052249, AG055247, AG055206, AG06212), and from Cognition Therapeutics, Inc. The authors thank Allison Marin, Ph.D., for providing assistance with the preparation of this manuscript.
Abbreviations:
- Aβ
Amyloid beta
- AD
Alzheimer’s disease
- DIV
Days in vitro
- E18
Embryonic Day 18
- GFAP
Glial fibrillary acidic protein
- HBSS
Hank’s Balanced Salt Solution
- MAP2
Microtubule-associated protein 2
Footnotes
Declaration of Competing Interest
All authors are employees of Cognition Therapeutics, Inc. and report no other conflicts of interest.
Data Availability Statement
All data are accessible upon request.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.jneumeth.2021.109180.
References
- Al-Ali H, Blackmore M, Bixby JL, Lemmon VP, 2013. High content screening with primary neurons. Assay Guid. Man 1–34. [Google Scholar]
- Banker GA, Cowan WM, 1977. Rat hippocampal neurons in dispersed cell culture. Brain Res. 126, 397–425. [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K, 1998. Types of nerve cell cultures, their advantages, and limitations. In: Banker G, Goslin K (Eds.), Cult. Nerve Cells The MIT Press, Cambridge, Massachusetts, pp. 11–36. [Google Scholar]
- Benson DL, Watkins FH, Steward O, Banker G, 1994. Characterization of GABAergic neurons in hippocampal cell cultures. J. Neurocytol 23, 279–295. [DOI] [PubMed] [Google Scholar]
- Brewer G, 1995. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J. Neurosci. Res 42, 674–683. [DOI] [PubMed] [Google Scholar]
- Brewer G, Torricelli J, Evege E, Price P, 1993. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res 35, 567–576. [DOI] [PubMed] [Google Scholar]
- Brody AH, Strittmatter SM, 2018. Synaptotoxic signaling by amyloid Beta oligomers in alzheimer’s disease through prion protein and mGluR5. Adv. Pharmacol 82, 293–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Stevens B, Chang J, Milbrandt J, Barres Ba, Hell JW, 2008. NS21: re-defined and modified supplement B27 for neuronal cultures. J. Neurosci. Methods 171, 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline EN, Bicca MA, Viola KL, Klein WL, 2018. The Amyloid-β oligomer hypothesis: beginning of the third decade. J. Alzheimers Dis 64, S567–S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe A, Henderson MJ, Anderson J, Titus SA, Zakharov A, Simeonov A, Buist A, et al. , 2020. Compound screening in cell-based models of tau inclusion formation: comparison of primary neuron and HEK293 cell assays. J. Biol. Chem 295, 4001–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA, 1988. The establishment of polarity by hippocampal neurons in culture. J. Neurosci 8, 1454–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fath T, Ke YD, Gunning P, Götz J, Ittner LM, 2009. Primary support cultures of hippocampal and substantia nigra neurons. Nat. Protoc 4, 78–85. [DOI] [PubMed] [Google Scholar]
- Gazina EV, Morrisroe E, Mendis GDC, Michalska AE, Chen J, Nefzger CM, Rollo BN, Reid CA, Pera MF, Petrou S, 2018. Method of derivation and differentiation of mouse embryonic stem cells generating synchronous neuronal networks. J. Neurosci. Methods 293, 53–58. [DOI] [PubMed] [Google Scholar]
- Grabrucker A, Vaida B, Bockmann J, Boeckers TM, 2009. Synaptogenesis of hippocampal neurons in primary cell culture. Cell Tissue Res. 338, 333–341. [DOI] [PubMed] [Google Scholar]
- Haas LT, Strittmatter SM, 2016. Targeting abeta receptors to modify alzheimer’s disease progression. In: Wolfe MS (Ed.), Dev. Ther. Alzheimer’S Dis. Prog. Challenges Academic Press, pp. 227–250. [Google Scholar]
- Hong H-S, Maezawa I, Yao N, Xu B, Diaz-Avalos R, Rana S, Hua DH, Cheng RH, Lam KS, Jin L-W, 2007. Combining the rapid MTT formazan exocytosis assay and the MC65 protection assay led to the discovery of carbazole analogs as small molecule inhibitors of Abeta oligomer-induced cytotoxicity. Brain Res. 1130, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo NJ, Staniszewski A, To L, Fa M, Teich AF, Saeed F, Wostein H, et al. , 2014a. Alzheimer’s Therapeutics Targeting Amyloid Beta 1–42 Oligomers I: Abeta 42 Oligomer Binding to Specific Neuronal Receptors Is Displaced by Drug Candidates That Improve Cognitive Deficits. PLoS One 9, e111898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo NJ, Xu J, Zeng C, Kirk MJ, Mozzoni K, Silky C, Rehak C, et al. , 2014b. Alzheimer’s therapeutics targeting amyloid Beta 1–42 oligomers II: Sigma-2/PGRMC1 receptors mediate abeta 42 oligomer binding and synaptotoxicity. PLoS One 9, e111899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Selkoe DJ, 2015. Systematic analysis of time-dependent neural effects of soluble amyloid β oligomers in culture and in vivo: prevention by scyllo-inositol. Neurobiol. Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G, 2006. Culturing hippocampal neurons. Nat. Protoc 1, 2406–2415. [DOI] [PubMed] [Google Scholar]
- Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, Hyman BT, Shatz CJ, 2013. Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 341, 1399–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzmann P, Wolf G, Kupsch K, 2010. Minocycline recovers MTT-formazan exocytosis impaired by amyloid beta peptide. Cell. Mol. Neurobiol 30, 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, et al. , 2004. Synaptic targeting by Alzheimer’s-related amyloid β oligomers. J. Neurosci 24, 10191–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Schubert D, 1997. Cytotoxic amyloid peptides inhibit cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction by enhancing MTT formazan exocytosis. J. Neurochem 69, 2285–2293. [DOI] [PubMed] [Google Scholar]
- Lu Z, Piechowicz M, Qiu S, 2016. A simplified method for ultra-low density, long-term primary hippocampal neuron culture. J. Vis. Exp 2016, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundholt BK, Scudder KM, Pagliaro L, 2003. A simple technique for reducing edge effect in cell-based assays. J. Biomol. Screen 8, 566–570. [DOI] [PubMed] [Google Scholar]
- Opitz T, De Lima A.D., Voigt T, 2002. Spontaneous development of synchronous oscillatory activity during maturation of cortical networks in vitro. J. Neurophysiol 88, 2196–2206. [DOI] [PubMed] [Google Scholar]
- Ramakers GJA, Kloosterman F, van Hulten P., van Pelt J., Corner MA, 1998. Activity-dependent regulation of neuronal network excitability. Neural Circuits and Networks. Springer, Berlin Heidelberg, Berlin, Heidelberg, pp. 141–151. [Google Scholar]
- Roppongi R, Champagne-Jorgensen K, Siddiqui T, 2017. Low-density primary hippocampal neuron culture. J. Vis. Exp 122, 55000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu MP, Nikkilä O, Lågas S, Kolehmainen S, Castrén E, 2019. Culturing primary neurons from rat hippocampus and cortex. Neuronal Signal 3. NS20180207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarretta C, Minichiello L, 2010. The preparation of primary cortical neuron cultures and a practical application using immunofluorescent cytochemistry. Methods Mol. Biol Humana Press, pp. 221–231. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J, 2016. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, Strittmatter SM, 2017. Binding sites for Amyloid-β oligomers and synaptic toxicity. Cold Spring Harb. Perspect. Med 7, a024075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer TP, Hubbs C, Vaissiere T, Collia D, Rojas C, Kilinc M, Vick K, et al. , 2017. Improved scalability of neuron-based phenotypic screening assays for therapeutic discovery in neuropsychiatric disorders. Mol. Neuropsychiatry 3, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neural circuits and networks. In: Torre V, Nicholls J (Eds.), 1998. Proc. NATO Adv. Study Inst. Neuronal Circuits Netw. Springer, Berlin Heidelberg, Heidelberg. [Google Scholar]
- Westbrook GL, Nelson PG, 1983. [7] Electrophysiological techniques in dissociated tissue culture. Methods Enzymol. 103, 111–132. [DOI] [PubMed] [Google Scholar]
- Yang H, Cong R, Na L, Ju G, You S-W, 2010. Long-term primary culture of highly-pure rat embryonic hippocampal neurons of low-density. Neurochem. Res 35, 1333–1342. [DOI] [PubMed] [Google Scholar]
- Zhang J-H, Chung T, Oldenburg K, 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.