

Abstract
Tumor necrosis is a common histological feature and poor prognostic predictor in various cancers. Despite its significant clinical implications, the mechanism underlying tumor necrosis remains largely unclear due to lack of appropriate pre-clinical modeling. We propose that tumor necrosis is a synergistic consequence of metabolic stress and inflammation, which lead to oxidative stress-induced cell death, such as ferroptosis. As a natural consequence of tumor expansion, tumor cells are inevitably stripped of vascular supply, resulting in deprivation of oxygen and nutrients. The resulting metabolic stress has commonly been considered the cause of tumor necrosis. Recent studies found that immune cells, such as neutrophils, when recruited to tumors, can directly trigger ferroptosis in tumor cells, suggesting that immune cells can be involved in amplifying tumor necrosis. This article will discuss potential mechanisms underlying tumor necrosis development and its impact on tumor progression as well as the immune response to tumors.
Keywords: Tumor necrosis, hypoxia, neutrophil, ferroptosis, metabolic stress, inflammation, immunosuppression
Introduction
Cancer is equally a derangement of cell proliferation and death. Large-scale unscheduled cell/tissue death commonly occurs in cancers, particularly in solid tumors, collectively and histopathologically termed tumor necrosis. In addition to being a histological hallmark of solid tumors, tumor necrosis frequently predicts poor prognoses in a variety of advanced cancers, such as metastatic breast cancer [1], non-small cell lung cancer [2], malignant mesothelioma [3], clear cell renal cell carcinoma [4], malignant gastrointestinal stromal tumors [5], Ewing’s sarcoma of the bone [6], endometrial cancer [7], and glioblastomas (GBMs) [8, 9]. Moreover, tumor necrosis often positively correlates with sizes, stages, and histological grades of tumors [6, 10, 11]. Such a strong association between necrosis and tumor aggressiveness raises a longstanding yet unresolved question as to whether necrosis is an epiphenomenon accompanying tumor progression or a direct cause of tumor aggressiveness. Resolving the uncertainty could have significant clinical implications. However, there has been little experimental testing of these notions, probably due to gaps in knowledge of the mechanisms underlying tumor necrosis and lack of experimental models which recapitulate the extent of necrosis observed in cancer patients.
In contrast to well-characterized programmed cell death, such as apoptosis and autophagy, necrosis was previously thought to be a catastrophic and disordered cell death process characterized by compromised plasma membrane integrity, swelling of cellular organelles, random DNA degradation, and uncontrolled release of pro-inflammatory molecules [12]. However, studies in the past decade have discovered that necrosis can occur in a regulated fashion and includes several cell death mechanisms known as necroptosis, parthanatos, oxytosis, ferroptosis, NETosis, pyronecrosis and pyroptosis [13]. These cell death mechanisms have been employed to explain a variety of necroses observed in vivo in various pathological situations [14]. Still, whether tumor necrosis is regulated through these mechanisms and how necrosis unfolds in tumor development remain largely unclear. As a natural consequence of tumor expansion, tumor cells are inevitably stripped of vascular supply, resulting in deprivation of oxygen and nutrients. The resulting metabolic stress of the affected cells has been commonly considered the cause of tumor necrosis. This notion suggests that the extent of tumor necrosis directly depends on tumor size and extent of intratumoral hypoxia and ischemia. While it is generally true that the extent of tumor necrosis positively correlates with tumor size [10], this correlation can be inconsistent and vague in some advanced solid-organ malignancies, such as GBMs, where small tumors can also form necrosis [15]. Pre-clinical GBM models also indicate that large tumors do not always form necrosis [16]. Moreover, although hypoxia can directly cause tumor cell injury and subsequently trigger tumor necrosis, some studies reported that the area of extensive tumor necrosis was oddly and surprisingly accompanied by enormous amounts of neo-angiogenesis and microvascular proliferation, as reflected in remarkably higher numbers of microvessels [17]. Therefore, the formation of tumor necrosis appears to be a complex process detertermined by multlple interconnected factors. Recent studies found that immune cells, such as neutrophils, when recruited to tumors, can directly trigger ferroptosis in tumor cells, suggesting that immune cells can contribute to formation and amplification of tumor necrosis [16]. This article will discuss potential mechanisms which may explain the development of tumor necrosis, with a particular focus on the interaction between tumor cells and neutrophils. The impact of tumor necrosis on tumor progression as well as the immune response to tumors will also be discussed.
Metabolic stress induces initial tumor necrosis
Convention has held that tumor necrosis is caused by rapid tumor expansion outstripping vascular supply, which results in ischemia and eventually hypoxia as well as nutrient (e.g., glucose) starvation in the tumor. This notion is supported by several observations. First, tumor necrosis is usually localized in the inner region of solid tumors, often termed the necrotic core. Second, tumor cells in this inner region of a solid tumor are more likely to undergo hypoxia as a direct consequence of diffusion limitation of oxygen. Third, these tumor cells also experience enhanced aerobic glycolysis due to their farther distance from vasculature [18–20]. Moreover, necrosis, along with hypoxia, is more commonly observed when solid tumors have expanded to more than 4 mm in diameter [21]. Along with this canonical view, several interrelated microenvironmental determinants, such as hypoxia, glucose deprivation, and reactive oxygen species (ROS) have been proposed to instigate tumor cell injury and contribute to the development of tumor necrosis.
Hypoxia-induced cell death
Hypoxia is a feature of solid tumors and is closely associated with poor prognosis in cancer [22, 23]. It has been proposed that tumor cells undergoing hypoxic insults are forced to switch from aerobic to anaerobic metabolism, which in turn leads to reduction in ATP production, overproduction of lactate, and lowering of extracellular pH [24]. Such perturbations of acid/base homeostasis in combination with ATP shortage may trigger malfunction of ATPase-dependent ion transport, leading to elevation of calcium levels both within the cytoplasm and within mitochondria, precipitating the eventual swelling and rupture of organelles and cell death [25].
Glucose deprivation-induced cell death
Glucose deprivation has been implicated as another microenvironmental instigator of tumor necrosis. Under normal physiological conditions, metabolic stress, such as glucose deprivation, can directly compromise mitochondrial membrane potential and impermeability, specifically of the inner mitochondrial membrane, resulting in leakage of mitochondrial apoptotic molecules and triggering apoptosis [26]. The uncontrolled and accelerated growth of tumor cells creates high demand for energy, which is normally supplied by aerobic glycolysis. Glucose deprivation in tumor cells may lead to metabolic stress, which, if unable to be resolved by alternative energy supplies, such as autophagy, eventually leads to metabolic catastrophe [20]. This will cause cancer cell death, either through apoptosis or, in most cases, through necrosis, as many cancer cells are resistant to apoptosis due to mutation of TP53 or other apoptotic genes.
ROS-induced cell death
Oxidative stress caused by accumulation of intracellular ROS is another trigger that can lead to tumor cell death and is closely associated with metabolic stress such as glucose deprivation. The impacts of ROS on cancer cells can drastically vary, largely depending on the local amounts of ROS, the types of ROS, and the locations where ROS are generated. For instance, while low yet tolerable amounts of hydrogen peroxide (H2O2) and superoxide (O2−) have been implicated in promotion of tumor growth in various types of cancers, high concentrations of the same types of ROS can induce cancer cell death [27]. Additionally, the type of cell death mechanism involved in ROS-instigated tumor cell death seems to depend on the concentration of intracellular ROS. Those tumor cells undergoing apoptotic cell death at low levels of ROS may be influenced to undergo necrosis when subject to high levels of ROS [26]. In the latter condition, high concentrations of intracellular ROS were reported to precipitate dysfunction of mitochondria, which are among the most ROS-sensitive organelles. This latter event in turn leads to a shortage of energy supply and ATP depletion, eventually resulting in necrosis [12, 28].
Both hypoxia and glucose deprivation have been shown to increase intracellular ROS production in cancer cells, leading to mitochondrial dysfunction, energy depletion, and eventual cellular demise [12, 29]. Hypoxia is closely associated with elevation in ROS production, specifically under the circumstances of ischemic-reperfusion injury [25]. Disrupted redox homeostasis triggered by elevated oxidative stress can further amplify hypoxia-initiated tumor cell death. In many cases, accumulation and production of mitochondrial ROS, including both H2O2 and O2−, is a direct consequence of glucose deprivation in various types of cancer cells. Pharmacological inhibition of ROS production via treatment with N-acetyl-L-cysteine (NAC) and catalase abolished tumor cell death in a study using A549 human lung cancer cells, suggesting glucose deprivation-instigated cell death is accomplished via production of intracellular ROS [30].
Overall, hypoxia and nutrient deprivation, especially of glucose, may serve as initial triggers of necrosis in certain tumors. These insults could instigate cell death by precluding oxidative phosphorylation and depleting intracellular ATP. In turn, mitochondrial strain and uncontrolled accumulation of ROS would lead to catastrophic and irreversible perturbations of a series of intracellular homeostatic processes and, eventually, to tumor cell demise. Still, the exact order of, interrelationship among, and individual contributions of the aforementioned intracellular events in tumor cell death as a result of hypoxia and nutrient deprivation remain to be clarified. While several molecular and environmental instigators of tumor cell death/necrosis have been identified, the majority of these previous studies relied on in vitro models and focused on one microenvironmental perturbation at a time. The experimental setting contrasts with the reality that tumor cells, specifically those localized in the inner regions/cores of tumors, are subjected to a combination of all of these stressors. In addition, these stressors can also induce inflammatory responses and infiltration of immune cells, which may also influence tumor cell survival. Therefore, development of tumor necrosis in vivo could be a more complex process.
Recruitment of neutrophils by tumor cells
Neutrophil genesis in normal physiological conditions
Neutrophils are the most abundant leukocytes in humans and account for more than 70% of all white blood cells. Compared to other immune cells, such as macrophages or lymphocytes, neutrophils are relatively short-lived. To maintain a steady supply available to respond to infection, inflammation, or other pathological stimuli, the physiological turnover rate of neutrophils is very high, with production of up to 2 × 1011 cells per day in humans [31]. Under normal physiological conditions, neutrophils are derived from the granulocyte-monocyte progenitors (GMPs) in the bone marrow and released into the circulation as well as several peripheral lymphoid organs, including spleen, liver, and lung, upon maturation [32]. Within the marrow, where granulocyte colony-stimulating factor (G-CSF) or granulocyte-macrophage colony-stimulating factor (GM-CSF) is continuously supplied, GMPs in turn differentiate into a granulocyte-designated lineage followed by further maturation processes [33]. During this differentiation process, the developing neutrophils alter their nuclear morphology from a round shape to a banded shape and then eventually into the segmented, multi-lobular morphology observed in terminally-differentiated neutrophils. Overall, neutrophil production and differentiation within the bone marrow are governed by the G-CSF receptor (G-CSFR)-signal transducer and activator of transcription 3 (STAT3) signaling pathway [34]. Outside of the bone marrow, a cascade of cytokine signaling, such as that from IL-23-expressing phagocytes (macrophages and dendritic cells) and IL-17-producing T lymphocytes (e.g., Tregs and natural killer T cells), intricately increases the production of G-CSF within the bone marrow so that neutrophil production and differentiation are strictly maintained and regulated [35, 36]. The presence of G-CSF is not absolutely required for the generation of neutrophils, as other molecules, such as GM-CSF, interleukin 6 (IL-6), and KIT ligand (KITL, a.k.a. KITLG) share similar and redundant roles with G-CSF, although to a lesser extent [37, 38]. Circulating mature neutrophils account for merely 1–2 % of all neutrophils throughout the body. The majority of mature cells are retained in the bone marrow via the C-X-C motif chemokine 12 - C-X-C chemokine receptor type 4 (CXCL12-CXCR4) signaling axis-mediated bone marrow retention [39–43]. Additionally, several adhesion molecules, such as integrin subunit α4 (ITGα4) and vascular cell adhesion molecule 1 (VCAM1), as well as some proteases, are also important in neutrophil retention [40, 42–45]. As neutrophils differentiate, they downregulate surface receptors required for their retention in the bone marrow while upregulating receptors for neutrophil-specific chemotactic factors, such as CXCR2. When neutrophil-specific chemotactic factors, such as CXCL1 and CXCL2, are released by endothelial cells or megakaryocytes during tissue injury or damage, CXCR2-CXCL1/2 signaling then liberates mature neutrophils into the circulation [40, 42–44].
Cancer induces distorted neutrophil genesis
In the context of cancer, both production and retention of bone marrow neutrophils are disrupted. Neutrophil production is pathologically enhanced due to overproduction of G-CSF, GM-CSF, and other granulopoiesis-promoting cytokines, such as IL-6, by tumor cells [31, 46]. These cytokines shift marrow hematopoiesis from lymphocytic to granulocytic, resulting in neutrophilia as observed in a variety of pre-clinical and clinical cancer models. Under certain contexts, tumor cells can trigger a process termed emergency granulopoiesis, shifting steady-state neutrophil production into rapid and uncontrolled production in both marrow and secondary lymphoid organs, such as the spleen, as observed in both pre-clinical and clinical models of pancreatic or colon cancers [47]. In addition to its direct contribution to the promotion of granulopoiesis, G-CSF is well-known for its counteracting action on neutrophil retention in the bone marrow. In the context of cancer, tumor-secreted G-CSF is capable of pressuring bone marrow into releasing neutrophils, both mature and immature, by various mechanisms [48–51]. These include 1) thrombopoietin (THPO)-mediated upregulation of neutrophil-specific chemotactic factors (e.g., CXCL1 and 2), which in turn leads to activation of CXCR2 signaling [51], 2) reduction of CXCL12 expression in bone marrow stromal cells[48], and 3) downregulation of CXCR4 expression in neutrophils [50]. Moreover, several factors regulating neutrophil release from the bone marrow are commonly upregulated, either locally within tumors or systemically, as a result of cancers [52–55]. These neutrophil release-promoting factors overpower retention signals within the marrow, resulting in release of neutrophils regardless of their differentiation status. For instance, interleukin-1β (IL-1β) released by tumor cells or tumor-associated macrophages within the tumor microenvironment stimulates neutrophil production and egress from the bone marrow and increases the number of circulating neutrophils, both peripherally and within tumor tissues, in breast and gastric cancer mouse models [53, 56–58]. Together, these studies suggested that dysregulated production of cytokines involved in neutrophil production and bone marrow egress by tumor cells, stromal cells, or other immune cells within the tumor microenvironment leads to pathological overproduction and release of neutrophils (Fig. 1a). In addition, such aberrant signaling also results in the emergence of immature neutrophils in the peripheral circulation. In the context of cancer, these immature neutrophils often assume an immunosuppressive phenotype, lacking typical cytotoxic neutrophilic granules and even inhibiting the production and proliferation of tumor-specific T effector cells. Such immature neutrophils are thus termed myeloid-derived suppressor cells (MDSCs), or more specifically, PMN-MDSCs [59].
Figure 1. Recruitment of neutrophils by a tumor.
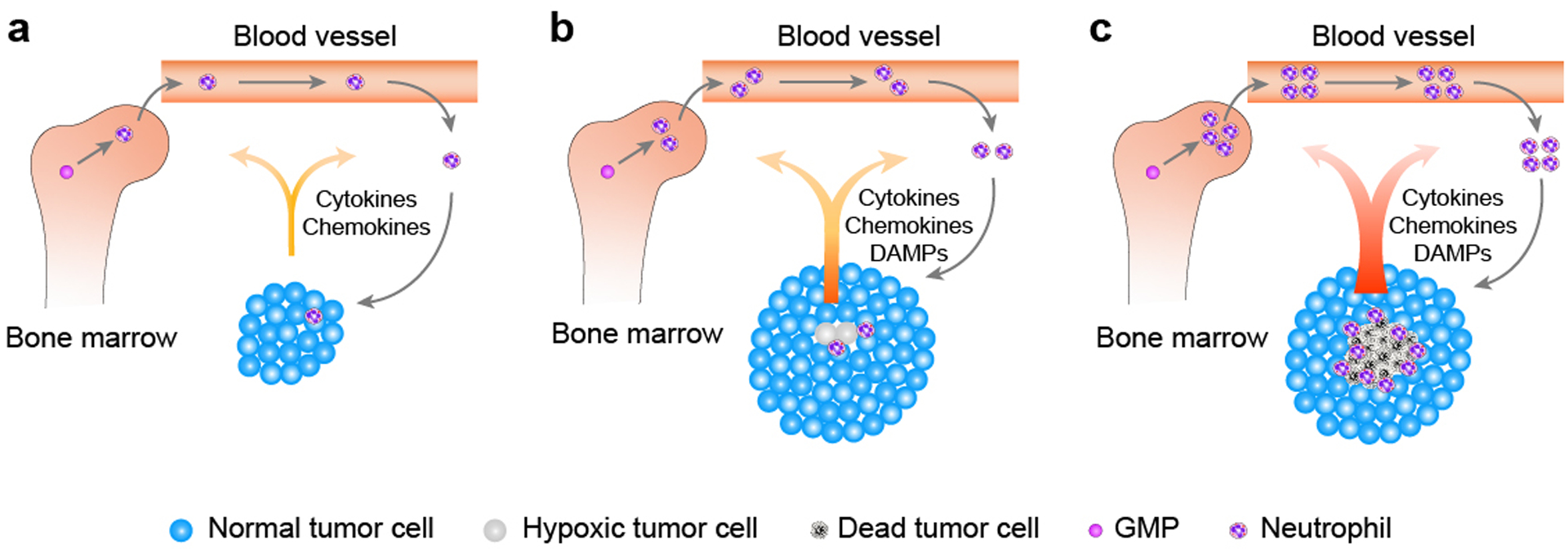
a) Under normal physiology, neutrophils arise from granulocyte-monocyte progenitors (GMPs) in bone marrow and are released upon maturation, and the whole process is tightly regulated. In the context of cancer, both production and retention of neutrophils are disrupted. Tumor cells overproduce a variety of granulopoiesis-promoting factors, which not only pathologically promote granulopoiesis in the bone marrow but also overpower retention signals within the marrow, resulting in peripheral neutrophilia. Certain tumor cells can further secrete neutrophilic chemotactic factors, forming a chemokine gradient that guides neutrophils to transmigrate and extravasate into the site of the tumor. Neutrophils that are recruited into the tumor are collectively termed tumor-associated neutrophils (TANs). b) Following natural tumor progression and expansion, cells in the center of a solid tumor are likely to undergo hypoxia and other potential stressors within the tumor microenvironment, resulting in tumor cell/tissue damage. Damaged tumor cells further release damage-associated molecular patterns (DAMPs), which can preferentially recruit neutrophils over other immune cells to tissue damage sites, further augmenting the extent of TANs infiltration. c) As tumor progression advances, oxygen- and nutrient-deprived tumor cells in the center of the solid tumor die, forming a visible tumor necrosis. Dead tumor cells aberrantly upregulate expression and release of a variety of factors, reinforcing bone marrow granulopoiesis and neutrophil release and further magnifying the extent of TANs infiltration within the tumor. Once recruited, TANs can cause additional tumor cell damage/death, augmenting inflammatory responses within the tumor, resulting in further tumor cell/tissue death. Together, these aberrations of marrow granulopoiesis and recruitment of neutrophils into the damaged tumor site facilitated by necrotic tumor cells, as well as TANs-triggered tumor cytotoxicity, form a positive feedback loop, magnifying and accelerating both TANs infiltration and tumor necrosis to their fullest extents.
Neutrophil recruitment by cancer
In most tissues, the neutrophil recruitment cascade involves several steps shared with all other leukocytes, including tethering, rolling, adhesion, crawling, and, finally, transmigration to leave vessels and go into peripheral tissues. Chemokines, such as CXCL8 (i.e., IL-8) in humans and its murine analogues, CXCL1, CXCL2, and CXCL5, establish an intravascular chemotactic gradient. The guidance provided by such a chemokine gradient allows neutrophils to transmigrate and exit the vasculature at the designated site(s) adjacent to the site of inflammation or tumors, where the concentrations of chemokines are the highest (Fig. 1a) [60, 61]. Necrotic tumor cells appear to have enhanced expression and release of these chemokines (Fig. 1c). Gene expression analysis indicated that IL-8 is the most upregulated factor (approximately 100-fold increase) released by tumor cells located in the peri-necrotic region as compared to the cellular tumor region in human GBMs [16]. In non-neoplastic tissue inflammation, damaged/dying cells are known to passively release damage-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB1), which are able to preferentially recruit neutrophils over other immune cells to tissue damage sites [62]. The temporal and spatial correlation between tumor necrosis and infiltrating neutrophils suggests that when tumors experience hypoxia/ischemic insults, initial hypoxic cellular demise or inflammation might already start releasing these neutrophil-recruiting factors (Fig. 1b). Cytokine profiling suggested that necrotic/dying GBM tumor cells secrete a variety of factors related to neutrophil recruitment, such as IL-8, IL-6, and CXCL1 [16]. Studies using a mouse model of PTEN-deficient uterine cancer showed a tight association between hypoxia and neutrophil accumulation in the tumors [63]. Relief of tumor hypoxia by housing mice in a hyperoxic environment reduces neutrophil infiltration [64]. Because these observations were made at early tumorigenic stages, the hypoxia-induced neutrophil recruitment is unlikely an indirect effect of tumor cell death. In fact, hypoxia-induced STAT3 activation followed by CXCL5 expression and release from the tumor cells is a major signaling effector in hypoxic tumor cells, and CXCR2 on neutrophils is responsible for sensing CXCL5 [63, 64]. Furthermore, hypoxia-induced activation of the master transcriptional mediator responsible for cellular response under hypoxic stress—hypoxia inducible factor 1α (HIF1α) leads to upregulation of IL-8 [65], the major chemotactic factor of neutrophils in humans. Therefore, hypoxia in tumors not only directly induces tumor cell death, but also leads to an inflammatory response by recruiting neutrophils and prolonging their survival [63, 64, 66]. In addition, hypoxia may also modulate the function of neutrophils in various circumstances. On one hand, hypoxia can enhance their degranulation-associated tissue damage capacity [67]; on the other hand, hypoxia can inhibit neutrophil respiratory burst activity and the NADPH oxidase-dependent cell killing ability [64, 68].
Neutrophils induce tumor cell death
Anti-tumor effect of neutrophil-induced cell death
Neutrophils have been known to be a heterogeneous cell population with conflicting roles in cancer [31, 46, 69]. These cells can play a pro-tumorigenic role by promoting tumor cell proliferation, angiogenesis, metastasis, and immunosuppression in the tumor microenvironment. On the other hand, neutrophils can play an anti-tumorigenic role by directly inducing tumor cell death and facilitating the anti-tumor ability of cytotoxic T cells. Previous studies have provided mechanistic insights into the neutrophil cytotoxicity (Fig. 2). Clark and Klebanoff demonstrated that the neutrophil-derived peroxidase system, including the myeloperoxidase (MPO)−H2O2−chloride (Cl−) cascade, is involved in neutrophil-triggered tumor cytotoxicity [70, 71]. Once secreted from neutrophils, MPO and H2O2 work with extracellular Cl−to generate cytotoxic reactive oxidants [72]. Slivka and Weiss further proposed that hypochlorous acid, a product of the MPO−H2O2−Cl− system, could be a mediator of such neutrophil-mediated tumor cytotoxicity [73, 74]. These early studies revealed that neutrophil-mediated cytotoxicity against tumor cells relies on an oxidative process. Later, Saito et al. [75] observed that the oxidative process occurs at the site of contact between tumor cells and neutrophils, suggesting direct contact is required for the cell-killing by neutrophils. In studying lung metastasis of breast cancer cells, Granot et al. [76] found that the NADPH oxidase-H2O2 pathway is required for neutrophil-triggered tumor cytotoxicity in killing 4T1 tumor cells and preventing their metastasis. Such tumor cell-killing appears to be independent of hypochlorous acid, because the hypochlorous acid scavenger, taurine, does not inhibit the tumor cell-killing. This study also suggested that physical contact of tumor cells is required for triggering neutrophils to secrete H2O2. Later, it was found that cathepsin G on the surface of neutrophils and the human receptor for advanced glycation end products (RAGE) on tumor cells form a bridge for the interaction of these cells, leading to the subsequent tumor cell-killing [77]. In follow-up studies, Gershkovitz et al. [78] found that transient receptor potential cation channel subfamily M member 2 (TRPM2), a H2O2-dependent Ca2+ channel, in tumor cells is required for neutrophil-mediated cell-killing by increasing Ca2+ in tumor cells. Consistently, elevated TRPM2 expression level sensitized tumor cells to neutrophil cytotoxicity [79]. In addition to direct involvement in the cell-killing signaling, TRPM2 is also involved in tumor cells’ capacities to recruit neutrophils in a CXCL2-mediated fashion [80]. Although neutrophil cytotoxicity relies on reactive oxidants in most situations, these cells can also generate other cytotoxic factors, such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), which is able to induce apoptosis in other cells [81].
Figure 2. Neutrophils can induce tumor cell death via multiple mechanisms.
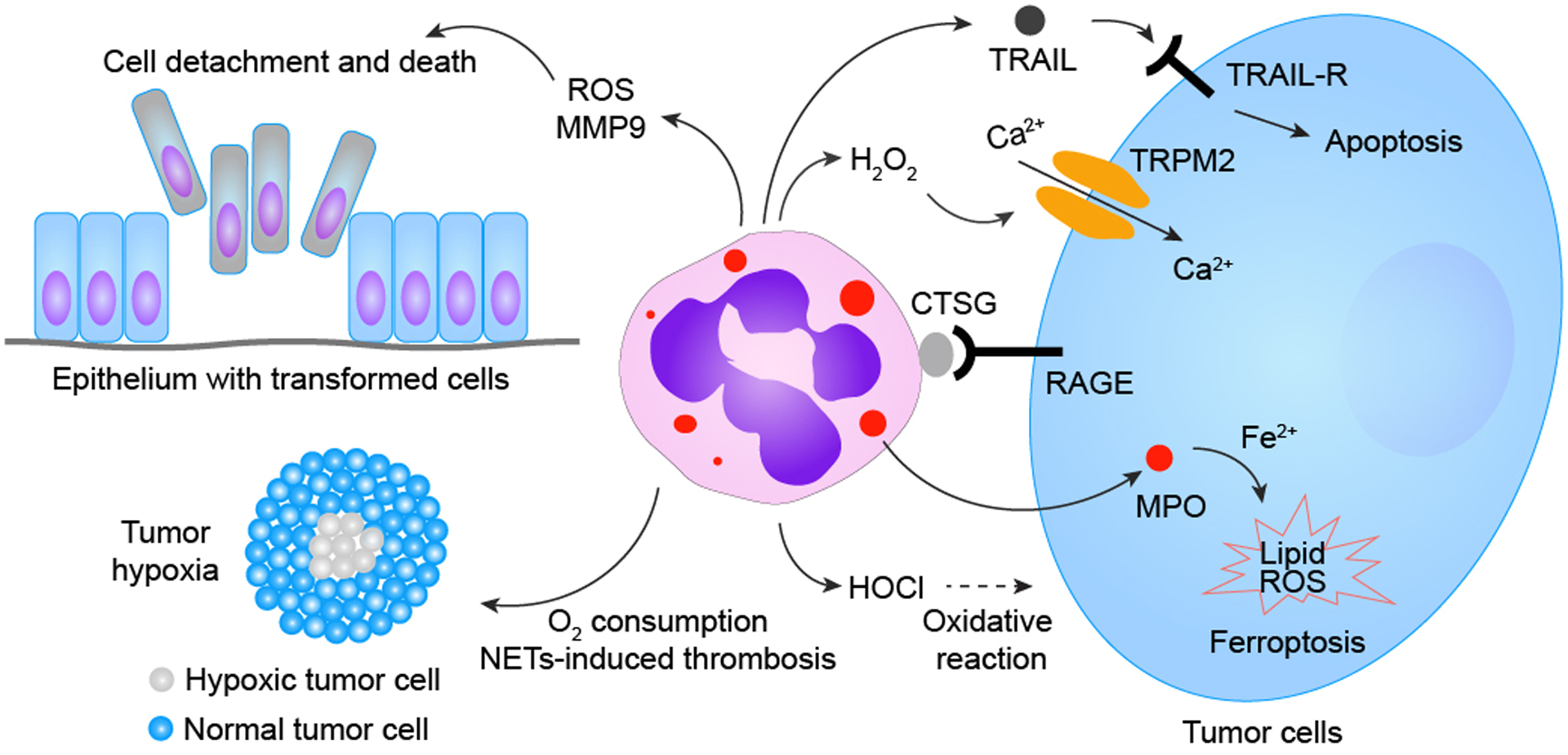
Neutrophils are well-known for their innate capacities to execute oxidative bursts, causing oxidative damage to surrounding cells/tissues. Unsurprisingly, most previously identified neutrophil-triggered tumor cell-killing primarily relies on neutrophils’ production of reactive oxidants. Several byproducts (e.g., hypochlorous acid, HOCl) generated via the neutrophilic MPO−H2O2−Cl− cascade, has been proposed to play a role in neutrophil-mediated tumor cytotoxicity. A study revealed that H2O2 generated in the neutrophilic oxidase cascade can directly instigate tumor cell death via the H2O2-activated nonselective cation channel—transient receptor potential cation channel subfamily M member 2 (TRPM2)—resulting in TRPM2-facilitated uncontrolled Ca2+ influx into tumor cells and subsequent tumor cell death. Neutrophils also execute tumor cytotoxicity via transfer of neutrophilic MPO into tumor cells, which in turn leads to aberrant intracellular accumulation of lipid ROS and eventual tumor cell ferroptosis. Moreover, neutrophils can also generate other tumor-cytotoxic factors, such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), to instigate tumor apoptotic cell death. Neutrophils’ capacities to generate tumor-cytotoxic materials depend on physical contact between neutrophils and tumor cells mediated by neutrophilic cathepsin G (CTSG) and receptor for advanced glycation end products (RAGE). Additionally, neutrophils are capable of triggering tumor cytotoxicity indirectly by causing damage or metabolic disturbances to tumor cells, such as 1) detachment of neoplastically-transformed epithelial cells in a neutrophilic NADPH oxidase-mediated manner, 2) competitive consumption of oxygen in the tumor microenvironment to augment hypoxic stress, and 3) NETs-facilitated vascular damage, leading to thrombosis and obstruction of blood flow, further magnifying hypoxia and nutrient deprivation among tumor cells.
In addition to direct cell-killing, neutrophils can eliminate tumor cells through other mechanisms (Fig. 2). For example, in the mouse model of PTEN-deficient uterine cancer, infiltrated neutrophils induce basement membrane detachment of the transformed but viable epithelial cells, which then die after the detachment [63, 64]. NADPH oxidase-derived ROS were also suggested to be involved in the basement membrane detachment induced by neutrophils. In addition, infiltrating neutrophils may also modulate the tumor microenvironment by enhancing hypoxia in tumors. This could be achieved by consuming microenvironmental O2, resulting in local tissue hypoxia, similar to what occurs when neutrophils transmigrate into intestinal epithelium in an acute colitis model [82]. Alternatively, neutrophils in cancers can generate neutrophil extracellular traps (NETs), which contribute to thrombosis and subsequent vascular obstruction [83], resulting in hypoxia and nutrient deprivation.
Pro-tumor effect of neutrophil-induced cell death
Neutrophil cytotoxicity has usually been studied as an anti-tumor property. This may work when tumors are at early developmental stages or when tumor cells colonize secondary organs in metastasis [63, 76]. In these situations, the neutrophil-triggered cytotoxic effects may be sufficient to wipe out the whole transformed cell group due to their relatively small numbers. However, in developed tumors with relatively large sizes, neutrophil-induced tumor cell death may not necessarily have an overall anti-tumor effect due to the recurrent and persistent local inflammatory response elicited by tumor cell death. More specifically, dying tumor cells (necrotic cores) may stimulate growth of surrounding tumor cells by directly secreting tumorigenic factors or initiating local inflammatory responses (see below). This wound-healing-like response may offset the loss of tumor mass caused by cell death and eventually promote tumor growth. Recently, in studying the development of tumor necrosis during glioblastoma progression, Yee et al. found that neutrophil-induced tumor cell death can counterintuitively promote tumor aggressiveness [16]. In this process, tumor cells undergo ferroptosis when encountering neutrophils infiltrating into the tumor stroma. Ferroptosis is a type of iron-dependent regulated cell death mediated by lethal accumulation of lipid peroxides [84]. When cocultured with activated neutrophils in vitro or encountering tumor-infiltrating neutrophils in an orthotopic GBM mouse model, LN229 GBM cells show increased amounts of lipid peroxides. Adding ferroptosis inhibitors or an iron chelator into the coculture rescued tumor cells from being killed by neutrophils. Ferroptosis blockade by either expressing phospholipid peroxidase glutathione peroxidase 4 (GPX4) or depleting acyl-CoA synthetase long chain family member 4 (ACSL4) in tumor cells dampened necrosis and lessened tumor aggressiveness in this tumor model. In both in vitro and in vivo settings, MPO-containing granules were found in tumor cells. Adding MPO inhibitors to the coculture or knocking down MPO from neutrophils rescued tumor cells from the neutrophil-induced tumor cell-killing in vitro. These observations suggested that neutrophils induce tumor cell ferroptosis by transferring MPO-containing granules into tumor cells (Fig. 2) [16]. This notion is consistent with the early observation that neutrophils from MPO-deficient patients lack such cytotoxicity [70].
Necrosis promotes tumor progression
Necrotic cells are able to release a broad spectrum of factors that can modulate the tumor microenvironment. Certain factors, such as HMGB1, IL-8 and IL-6, can affect tumor cells or stroma cells to enhance tumor cell proliferation, invasion, and angiogenesis [85]. Several intracellular components, when released, are able to directly or indirectly activate and recruit immune cells which then facilitate inflammation or immunosuppression. These components include HMGB1 [86–90], uric acid [91–93], nucleosomes [94–96], heat shock protein family members hsp 70, hsp 60, and gp96 [97–101], as well as fragmented chromatin/DNA [102–104]. The released cellular components have been generally called DAMPs. Because these factors could be released throughout the whole dying process, including when cells may still be metabolically active, these doomed cells may impact the tumor even before the appearance of overt necrosis.
Chronic inflammation induced by tumor necrosis
Chronic inflammation has been shown in several studies to promote initiation and progression of cancers [105, 106]. Many recent studies, undertaken to explain how and why tumor necrosis negatively impacts patients’ prognoses, have provided new insights regarding the link between tumor necrosis and inflammation. In addition to local (within the tumor microenvironment) inflammation triggered by cell death, tumor necrosis may cause systemic (within the whole body of the host) inflammation, as demonstrated by elevation of serum inflammatory markers, erythrocyte sedimentation rate (ESR), and total white blood cell count commonly observed in cancer patients [107]. The presence of tumor necrosis, through so-called necroinflammation, is capable of persistently triggering pro-inflammatory responses and thereby creating a chronically pro-inflammatory environment at both local and systemic levels, thereby shortening the host’s survival. Long-term local inflammation can escalate into chronic dysregulated inflammation, subjecting the host to constant exposure to pro-inflammatory cytokines (e.g., IL-1, IL-6, and TNFα), which in turn increases production and recruitment of a variety of pro-inflammatory immune cells (Fig. 3). While these pro-inflammatory responses may be beneficial for the host to eliminate pathogens under normal physiological circumstances, chronic activation of inflammatory reactions within the host may lead to generalized cachexia, failure to thrive, and eventual multi-organ deterioration, thereby worsening prognoses in cancer patients [108, 109]. Additionally, subjection to a chronic inflammatory state may lower the host’s tolerance threshold to cytotoxic anti-cancer chemotherapy or radiotherapy and increase chances of complications.
Figure 3. A proposed model of tumor necrosis development and its impact on tumor progression.
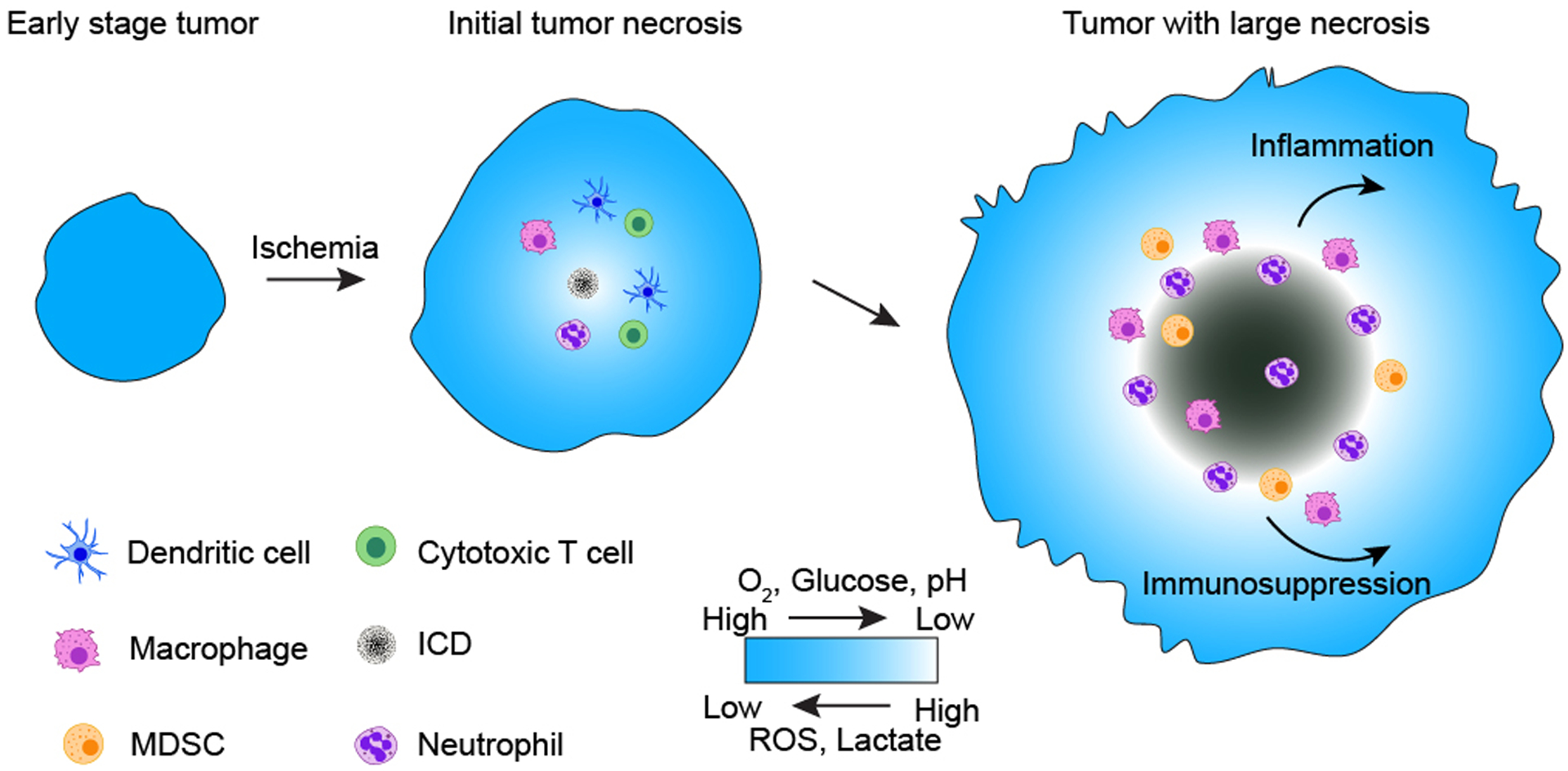
As the tumor expands during early tumorigenesis, rapid tumor expansion outstrips vascular supply and results in ischemia. Ischemic tumor cells at the center of the tumor face multiple metabolic challenges, including deprivation of oxygen (O2) and glucose as well as aberrant accumulation of ROS and lactate, resulting in pH reduction. Tumor cells residing in such a harsh microenvironment could die in a way that may be immunogenic and able to recruit immune cells involved in both innate and adaptive responses. These immune cells could further facilitate tumor cell death. As tumor cell death in this zone persists, the size of tumor necrosis expands; meanwhile, dying tumor cells release various chemotactic molecules, recruiting more macrophages and neutrophils as well as MDSCs into the tumor necrotic zone. These recruited immune cells, along with other released tumor cellular contents and metabolites, together create an immunosuppressive tumor microenvironment and dampen resolution of inflammation, leading to persistence of unresolved, so-called chronic inflammation. The chronic inflammation and immune escape could then promote tumor growth and malignant tumor progression.
Immunogenic and immunosuppressive effects by tumor necrosis
In certain conditions, tumor cell death can initiate both innate and adaptive immune responses. Such immunogenic cell death (ICD) may trigger enhancing signals that facilitate tumor-associated neoantigens generated by mutations in stimulating the adaptive immune response (Fig. 3) [110]. In this scenario, the tumor cell-derived DAMPs activate pattern recognition receptors (PRRs) that initiate the immunity. Through the DAMPs-PRRs signaling, necroptotic cancer cells can promote dendritic cell maturation, cytotoxic T cell priming, and IFN-γ production [111, 112]. Although it is still unclear whether tumor cells undergoing ferroptosis are immunogenic like necroptotic cancer cells, it was suggested that lipid metabolites and autophagy-mediated HMGB1 released from ferroptotic cancer cells can recruit antigen-presenting cells and other immune cells [113]. These ferroptosis-associated DAMPs may trigger Toll-like receptor 4 (TLR4) singling, which can then recruit and activate multiple immune cells, including dendritic cells and neutrophils [113].
The fact that certain cancers (e.g., GBMs) almost always develop remarkable necrotic cores suggests that necrotic cells, likely initially immunogenic, find a way eventually to circumvent the immune surveillance (Fig. 3). Although it is still unclear how such escape occurs in necrotic cells, some mechanisms may be similar to that used by tumor cells when escaping immune surveillance in early tumorigenesis [114]. In this latter case, the escaping process may rely on reducing release and sensing of DAMPs, interfering with the downstream immunological processes, or more immunosuppressive mechanisms as discussed below [110]. First, shortages of glucose (hypoglycemia) and oxygen (hypoxia) in tumors associated with necrosis provide a metabolic challenge for cytotoxic T cells to generate sufficient energy for proliferation and activation in tumors [115]. Second, released metabolic products may have immunosuppressive functions. For example, tumor cells can generate ROS in response to hypoxia [116]. In addition, tumor infiltrating immune cells, such as MDSCs, macrophages, regulatory T cells, and neutrophils, can also release ROS into the tumor microenvironment. While low levels of ROS are required for T cell propagation and activation, when ROS reaches a certain level, it can inhibit the antitumor function and proliferation of T cells and reduce T cell survival [117]. In addition, accumulation of lactate due to increased tumor-associated glycolysis can impede the proliferation and cell-killing ability of cytotoxic T cells [118]. This is likely due to disrupted export of lactate from T cells and reduced glycolysis in T cells for energy production. Other metabolic products or released cell contents may also contribute to immunosuppression within the tumor microenvironment. For example, while antigen-presenting dendritic cells can provide a chemically-reducing microenvironment favorable for T lymphocyte activation via the extracellular release of cysteine and thioredoxin, extracellular glutamate, which is aberrantly elevated in the GBM microenvironment, can inhibit antigen-dependent T lymphocyte proliferation [119]. In addition, because nucleosomes can induce lymphocyte necrosis [96], nucleosomes released from necrotic tumor cells may contribute to immunosuppression in necrotic tumors. Furthermore, the arachidonic acid metabolites, such as prostaglandin E2, generated under the cellular peroxide tone may allow immune evasion by tumors [113, 120]. Other inhibitory DAMPs, such as adenosine, may also suppress the immunogenic response [121]. Notably, the above immunosuppressive components in the necrotic tumor microenvironment could be directly generated by tumor cells at various stages of necrosis. However, these tumor cells may also recruit or modulate other immune cells, such as M2-polarized macrophages, neutrophils, MDSCs, or regulatory T cells, which in turn facilitate creation of the immunosuppressive microenvironment [114]. It is still unclear whether there is a point of no return beyond which the balance of immunogenic and immunosuppressive properties of necrotic cells is tipped toward immunosuppression and eventually lead to immune surveillance escape. However, amplifying the release of DAMPs may convert the non-immunogenic cell death to ICD [122]. In addition, blockade of IL-8 or its receptors may inhibit neutrophil recruitment and the formation of neutrophil extracellular traps, thereby relieving immunosuppression and benefitting immunotherapy [123]. Therefore, finding a method to counteract the ways by which necrotic tumors escape immune surveillance and revive the immunogenicity of cell death associated with necrosis may benefit cancer therapeutics.
Conclusions and Outlook
Emerging evidence suggests that the formation of tumor necrosis is a complex process orchestrated by multlple interconnected factors, including molecular aberrations within the tumor cells, the metabolic statuses of the tumors, as well as the influence of non-neoplastic (e.g., immune) cells in the tumor microenvironment (Fig. 3). Ferroptotic cell death in necrotic GBM offers a glimpse into this complex regulation. The molecular aberrations associated with activation of TAZ [124] and RAS-RAF-ERK signaling [125] could increase ferroptosis sensitivity [126–129], thereby sensitizing GBM cells to ferroptosis when intracellular ROS imbalance is triggered by metabolic insults, such as hypoxia, glucose deprivation, and extracellular glutamate accumulation [130]. The resulting tumor damage would in turn recruit neutrophils to the site of tissue damage and thereby result in a positive feedback loop, amplifying GBM necrosis development to its fullest extent [16]. Given that other immune cells, such as macrophages and microglia, are also abundant in GBMs, whether they play a role similar to that of neutrophils in necrosis formation is an open question. In addition, whether the necrosis development modality laid out in GBMs also applies to other cancers awaits future investigation. The interrelationship among tumor necrosis and tumor progression remains complex and challenging to study. Tumor cells dedicated to necrosis may impact tumor progression in various ways that are manifested by chronic inflammation as well as the immunogenic and immunosuppressive effects. The fact that inhibiting ferroptosis can prolong the survival of mice bearing xenografted GBM suggests that necrosis may enhance tumor aggressiveness and that targeting ferroptotic cell death in necrotic GBM might benefit GBM patients by curtailing tumor necrosis-triggered comorbidities [16]. On the other hand, identification of the tipping point at which ICDs transform to non-immunogenic cell death may benefit immunotherapy by stimulating the adaptive immune response and blocking immunosuppression.
Acknowledgements
We acknowledge support from the National Institute of Neurological Disorders and Stroke (R01 NS109147 and NS119547 to W.L.), the Four Diamonds Fund for Pediatric Cancer Research (to PSU), and Penn State College of Medicine Medical Scientist Training Program (5T32GM118294 to P.Y. through PSU).
Abbreviations:
- DAMP
damage-associated molecular pattern
- GBM
glioblastoma
- G-CSF
granulocyte colony-stimulating factor
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- GMP
granulocyte-monocyte progenitor
- ICD
immunogenic cell death
- MDSC
myeloid-derived suppressor cells
- PRR
pattern recognition receptor
- ROS
reactive oxygen species
- TAN
tumor-associated neutrophil
Footnotes
Competing Interests statement
The authors declare no competing interests.
References
- [1].Fisher ER, Anderson S, Redmond C, & Fisher B (1993). Pathologic findings from the National Surgical Adjuvant Breast Project protocol B-06. 10-year pathologic and clinical prognostic discriminants. Cancer, 71(8), 2507–2514. doi: [DOI] [PubMed] [Google Scholar]
- [2].Swinson DE, Jones JL, Richardson D, Cox G, Edwards JG, & O’Byrne KJ (2002). Tumour necrosis is an independent prognostic marker in non-small cell lung cancer: correlation with biological variables. Lung cancer (Amsterdam, Netherlands), 37(3), 235–240. [DOI] [PubMed] [Google Scholar]
- [3].Edwards JG, Swinson DE, Jones JL, Muller S, Waller DA, & O’Byrne KJ (2003). Tumor necrosis correlates with angiogenesis and is a predictor of poor prognosis in malignant mesothelioma. Chest, 124(5), 1916–1923. [DOI] [PubMed] [Google Scholar]
- [4].Cheville JC, Lohse CM, Zincke H, Weaver AL, & Blute ML (2003). Comparisons of Outcome and Prognostic Features Among Histologic Subtypes of Renal Cell Carcinoma. AMERICAN JOURNAL OF SURGICAL PATHOLOGY, 27, 612–624. [DOI] [PubMed] [Google Scholar]
- [5].Muro-Cacho CA, Cantor AB, & Morgan M (2000). Prognostic factors in malignant gastrointestinal stromal tumors. Ann Clin Lab Sci, 30(3), 239–247. [PubMed] [Google Scholar]
- [6].Llombart-Bosch A, Contesso G, Henry-Amar M, Lacombe MJ, Oberlin O, Dubousset J, … Sarrazin D (1986). Histopathological predictive factors in Ewing’s sarcoma of bone and clinicopathological correlations. A retrospective study of 261 cases. Virchows Arch A Pathol Anat Histopathol, 409(5), 627–640. [DOI] [PubMed] [Google Scholar]
- [7].Bredholt G, Mannelqvist M, Stefansson IM, Birkeland E, B¯ TH, ÿyan AM, … Akslen LA (2015). Tumor necrosis is an important hallmark of aggressive endometrial cancer and associates with hypoxia, angiogenesis and inflammation responses. Oncotarget Oncotarget, 6(37), 39676–39691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hammoud MA, Sawaya R, Shi W, Thall PF, & Leeds NE (1996). Prognostic significance of preoperative MRI scans in glioblastoma multiforme. Journal of Neuro-Oncology, 27(1), 65–73. doi: 10.1007/bf00146086 [DOI] [PubMed] [Google Scholar]
- [9].Shah AH, Snelling B, Bregy A, Patel PR, Tememe D, Bhatia R, … Komotar RJ (2013). Discriminating radiation necrosis from tumor progression in gliomas: a systematic review what is the best imaging modality? Journal of Neuro-Oncology, 112(2), 141–152. doi: 10.1007/s11060-013-1059-9 [DOI] [PubMed] [Google Scholar]
- [10].Gustafson P, Akerman M, Alvegard TA, Coindre JM, Fletcher CD, Rydholm A, & Willen H (2003). Prognostic information in soft tissue sarcoma using tumour size, vascular invasion and microscopic tumour necrosis-the SIN-system. Eur J Cancer, 39(11), 1568–1576. doi: 10.1016/s0959-8049(03)00369-1 [DOI] [PubMed] [Google Scholar]
- [11].Carneiro A, Bendahl PO, Engellau J, Domanski HA, Fletcher CD, Rissler P, … Nilbert M (2011). A prognostic model for soft tissue sarcoma of the extremities and trunk wall based on size, vascular invasion, necrosis, and growth pattern. Cancer, 117(6), 1279–1287. doi: 10.1002/cncr.25621 [DOI] [PubMed] [Google Scholar]
- [12].Zong WX, & Thompson CB (2006). Necrotic death as a cell fate. Genes Dev, 20(1), 1–15. doi: 10.1101/gad.1376506 [DOI] [PubMed] [Google Scholar]
- [13].Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, & Vandenabeele P (2014). Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol, 15(2), 135–147. doi: 10.1038/nrm3737 [DOI] [PubMed] [Google Scholar]
- [14].Tonnus W, & Linkermann A (2017). The in vivo evidence for regulated necrosis. Immunol Rev, 277(1), 128–149. doi: 10.1111/imr.12551 [DOI] [PubMed] [Google Scholar]
- [15].Raza SM, Lang FF, Aggarwal BB, Fuller GN, Wildrick DM, & Sawaya R (2002). Necrosis and glioblastoma: a friend or a foe? A review and a hypothesis. Neurosurgery, 51(1), 2–12; discussion 12–13. [DOI] [PubMed] [Google Scholar]
- [16].Yee PP, Wei Y, Kim SY, Lu T, Chih SY, Lawson C, … Li W (2020). Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat Commun, 11(1), 5424. doi: 10.1038/s41467-020-19193-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Leek RD, Landers RJ, Harris AL, & Lewis CE (1999). Necrosis correlates with high vascular density and focal macrophage infiltration in invasive carcinoma of the breast. Br J Cancer, 79(5–6), 991–995. doi: 10.1038/sj.bjc.6690158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tomes L, Emberley E, Niu Y, Troup S, Pastorek J, Strange K, … Watson PH (2003). Necrosis and hypoxia in invasive breast carcinoma. Breast Cancer Res Treat, 81(1), 61–69. doi: 10.1023/A:1025476722493 [DOI] [PubMed] [Google Scholar]
- [19].Gatenby RA, & Gillies RJ (2004). Why do cancers have high aerobic glycolysis? Nat Rev Cancer, 4(11), 891–899. doi: 10.1038/nrc1478 [DOI] [PubMed] [Google Scholar]
- [20].Jin S, DiPaola RS, Mathew R, & White E (2007). Metabolic catastrophe as a means to cancer cell death. J Cell Sci, 120(Pt 3), 379–383. doi: 10.1242/jcs.03349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li XF, Carlin S, Urano M, Russell J, Ling CC, & O’Donoghue JA (2007). Visualization of hypoxia in microscopic tumors by immunofluorescent microscopy. Cancer Res, 67(16), 7646–7653. doi: 10.1158/0008-5472.CAN-06-4353 [DOI] [PubMed] [Google Scholar]
- [22].Wilson WR, & Hay MP (2011). Targeting hypoxia in cancer therapy. Nat Rev Cancer, 11(6), 393–410. doi: 10.1038/nrc3064 [DOI] [PubMed] [Google Scholar]
- [23].Bristow RG, & Hill RP (2008). Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer, 8(3), 180–192. doi: 10.1038/nrc2344 [DOI] [PubMed] [Google Scholar]
- [24].Hao G, Xu ZP, & Li L (2018). Manipulating extracellular tumour pH: an effective target for cancer therapy. RSC Advances, 8(39), 22182–22192. doi: 10.1039/C8RA02095G [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kalogeris T, Baines CP, Krenz M, & Korthuis RJ (2012). Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol, 298, 229–317. doi: 10.1016/B978-0-12-394309-5.00006-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim JS, He L, & Lemasters JJ (2003). Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun, 304(3), 463–470. doi: 10.1016/s0006-291x(03)00618-1 [DOI] [PubMed] [Google Scholar]
- [27].Liou GY, & Storz P (2010). Reactive oxygen species in cancer. Free Radic Res, 44(5), 479–496. doi: 10.3109/10715761003667554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Golstein P, & Kroemer G (2007). Cell death by necrosis: towards a molecular definition. Trends Biochem Sci, 32(1), 37–43. doi: 10.1016/j.tibs.2006.11.001 [DOI] [PubMed] [Google Scholar]
- [29].Nikoletopoulou V, Markaki M, Palikaras K, & Tavernarakis N (2013). Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta, 1833(12), 3448–3459. doi: 10.1016/j.bbamcr.2013.06.001 [DOI] [PubMed] [Google Scholar]
- [30].Kim CH, Han SI, Lee SY, Youk HS, Moon JY, Duong HQ, … Kang HS (2007). Protein kinase C-ERK1/2 signal pathway switches glucose depletion-induced necrosis to apoptosis by regulating superoxide dismutases and suppressing reactive oxygen species production in A549 lung cancer cells. J Cell Physiol, 211(2), 371–385. doi: 10.1002/jcp.20941 [DOI] [PubMed] [Google Scholar]
- [31].Coffelt SB, Wellenstein MD, & de Visser KE (2016). Neutrophils in cancer: neutral no more. Nat Rev Cancer, 16(7), 431–446. doi: 10.1038/nrc.2016.52 [DOI] [PubMed] [Google Scholar]
- [32].Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, & Chilvers ER (2010). Neutrophil kinetics in health and disease. Trends Immunol, 31(8), 318–324. doi: 10.1016/j.it.2010.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kolaczkowska E, & Kubes P (2013). Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol, 13(3), 159–175. doi: 10.1038/nri3399 [DOI] [PubMed] [Google Scholar]
- [34].Panopoulos AD, Zhang L, Snow JW, Jones DM, Smith AM, El Kasmi KC, … Watowich SS (2006). STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood, 108(12), 3682–3690. doi: 10.1182/blood-2006-02-003012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schwarzenberger P, Huang W, Ye P, Oliver P, Manuel M, Zhang Z, … Kolls JK (2000). Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J Immunol, 164(9), 4783–4789. doi: 10.4049/jimmunol.164.9.4783 [DOI] [PubMed] [Google Scholar]
- [36].Forlow SB, Schurr JR, Kolls JK, Bagby GJ, Schwarzenberger PO, & Ley K (2001). Increased granulopoiesis through interleukin-17 and granulocyte colony-stimulating factor in leukocyte adhesion molecule-deficient mice. Blood, 98(12), 3309–3314. doi: 10.1182/blood.v98.12.3309 [DOI] [PubMed] [Google Scholar]
- [37].Molineux G, Migdalska A, Szmitkowski M, Zsebo K, & Dexter TM (1991). The effects on hematopoiesis of recombinant stem cell factor (ligand for c-kit) administered in vivo to mice either alone or in combination with granulocyte colony-stimulating factor. Blood, 78(4), 961–966. [PubMed] [Google Scholar]
- [38].Liu F, Wu HY, Wesselschmidt R, Kornaga T, & Link DC (1996). Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity, 5(5), 491–501. doi: 10.1016/s1074-7613(00)80504-x [DOI] [PubMed] [Google Scholar]
- [39].Ma Q, Jones D, & Springer TA (1999). The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity, 10(4), 463–471. doi: 10.1016/s1074-7613(00)80046-1 [DOI] [PubMed] [Google Scholar]
- [40].Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, & Rankin SM (2003). Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity, 19(4), 583–593. doi: 10.1016/s1074-7613(03)00263-2 [DOI] [PubMed] [Google Scholar]
- [41].Suratt BT, Petty JM, Young SK, Malcolm KC, Lieber JG, Nick JA, … Worthen GS (2004). Role of the CXCR4/SDF-1 chemokine axis in circulating neutrophil homeostasis. Blood, 104(2), 565–571. doi: 10.1182/blood-2003-10-3638 [DOI] [PubMed] [Google Scholar]
- [42].Eash KJ, Means JM, White DW, & Link DC (2009). CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood, 113(19), 4711–4719. doi: 10.1182/blood-2008-09-177287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Eash KJ, Greenbaum AM, Gopalan PK, & Link DC (2010). CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest, 120(7), 2423–2431. doi: 10.1172/JCI41649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Petty JM, Lenox CC, Weiss DJ, Poynter ME, & Suratt BT (2009). Crosstalk between CXCR4/stromal derived factor-1 and VLA-4/VCAM-1 pathways regulates neutrophil retention in the bone marrow. J Immunol, 182(1), 604–612. doi: 10.4049/jimmunol.182.1.604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Levesque JP, Liu F, Simmons PJ, Betsuyaku T, Senior RM, Pham C, & Link DC (2004). Characterization of hematopoietic progenitor mobilization in protease-deficient mice. Blood, 104(1), 65–72. doi: 10.1182/blood-2003-05-1589 [DOI] [PubMed] [Google Scholar]
- [46].Shaul ME, & Fridlender ZG (2019). Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. doi: 10.1038/s41571-019-0222-4 [DOI] [PubMed] [Google Scholar]
- [47].Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, … Pittet MJ (2012). Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A, 109(7), 2491–2496. doi: 10.1073/pnas.1113744109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, … Lapidot T (2002). G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol, 3(7), 687–694. doi: 10.1038/ni813 [DOI] [PubMed] [Google Scholar]
- [49].Semerad CL, Liu F, Gregory AD, Stumpf K, & Link DC (2002). G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity, 17(4), 413–423. doi: 10.1016/s1074-7613(02)00424-7 [DOI] [PubMed] [Google Scholar]
- [50].Kim HK, De La Luz Sierra M, Williams CK, Gulino AV, & Tosato G (2006). G-CSF down-regulation of CXCR4 expression identified as a mechanism for mobilization of myeloid cells. Blood, 108(3), 812–820. doi: 10.1182/blood-2005-10-4162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kohler A, De Filippo K, Hasenberg M, van den Brandt C, Nye E, Hosking MP, … Gunzer M (2011). G-CSF-mediated thrombopoietin release triggers neutrophil motility and mobilization from bone marrow via induction of Cxcr2 ligands. Blood, 117(16), 4349–4357. doi: 10.1182/blood-2010-09-308387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Casbon AJ, Reynaud D, Park C, Khuc E, Gan DD, Schepers K, … Werb Z (2015). Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A, 112(6), E566–575. doi: 10.1073/pnas.1424927112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, … de Visser KE (2015). IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature, 522(7556), 345–348. doi: 10.1038/nature14282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Waight JD, Hu Q, Miller A, Liu S, & Abrams SI (2011). Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS One, 6(11), e27690. doi: 10.1371/journal.pone.0027690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, … Ferrara N (2010). Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A, 107(50), 21248–21255. doi: 10.1073/pnas.1015855107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, … Apte RN (2005). CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J Immunol, 175(12), 8200–8208. doi: 10.4049/jimmunol.175.12.8200 [DOI] [PubMed] [Google Scholar]
- [57].Bunt SK, Sinha P, Clements VK, Leips J, & Ostrand-Rosenberg S (2006). Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol, 176(1), 284–290. doi: 10.4049/jimmunol.176.1.284 [DOI] [PubMed] [Google Scholar]
- [58].Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, … Wang TC (2008). Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell, 14(5), 408–419. doi: 10.1016/j.ccr.2008.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gabrilovich DI (2017). Myeloid-Derived Suppressor Cells. Cancer Immunol Res, 5(1), 3–8. doi: 10.1158/2326-6066.CIR-16-0297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Williams MR, Azcutia V, Newton G, Alcaide P, & Luscinskas FW (2011). Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol, 32(10), 461–469. doi: 10.1016/j.it.2011.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pruenster M, Mudde L, Bombosi P, Dimitrova S, Zsak M, Middleton J, … Rot A (2009). The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat Immunol, 10(1), 101–108. doi: 10.1038/ni.1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, … Schwabe RF (2015). The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest, 125(2), 539–550. doi: 10.1172/JCI76887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Blaisdell A, Crequer A, Columbus D, Daikoku T, Mittal K, Dey SK, & Erlebacher A (2015). Neutrophils Oppose Uterine Epithelial Carcinogenesis via Debridement of Hypoxic Tumor Cells. Cancer Cell, 28(6), 785–799. doi: 10.1016/j.ccell.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mahiddine K, Blaisdell A, Ma S, Crequer-Grandhomme A, Lowell CA, & Erlebacher A (2020). Relief of tumor hypoxia unleashes the tumoricidal potential of neutrophils. J Clin Invest, 130(1), 389–403. doi: 10.1172/JCI130952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Feng W, Xue T, Huang S, Shi Q, Tang C, Cui G, … Guo H (2018). HIF-1alpha promotes the migration and invasion of hepatocellular carcinoma cells via the IL-8-NF-kappaB axis. Cell Mol Biol Lett, 23, 26. doi: 10.1186/s11658-018-0077-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, … Chilvers ER (2005). Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med, 201(1), 105–115. doi: 10.1084/jem.20040624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hoenderdos K, Lodge KM, Hirst RA, Chen C, Palazzo SG, Emerenciana A, … Condliffe AM (2016). Hypoxia upregulates neutrophil degranulation and potential for tissue injury. Thorax, 71(11), 1030–1038. doi: 10.1136/thoraxjnl-2015-207604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].McGovern NN, Cowburn AS, Porter L, Walmsley SR, Summers C, Thompson AAR, … Chilvers ER (2011). Hypoxia selectively inhibits respiratory burst activity and killing of Staphylococcus aureus in human neutrophils. J Immunol, 186(1), 453–463. doi: 10.4049/jimmunol.1002213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Vols S, Sionov RV, & Granot Z (2017). Always Look On the Bright Side: Anti-Tumor Functions of Neutrophils. Curr Pharm Des, 23(32), 4862–4892. doi: 10.2174/1381612823666170704125420 [DOI] [PubMed] [Google Scholar]
- [70].Clark RA, & Klebanoff SJ (1975). Neutrophil-mediated tumor cell cytotoxicity: role of the peroxidase system. J Exp Med, 141(6), 1442–1447. doi: 10.1084/jem.141.6.1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Clark RA, & Klebanoff SJ (1979). Role of the myeloperoxidase-H2O2-halide system in concanavalin A-induced tumor cell killing by human neutrophils. J Immunol, 122(6), 2605–2610. [PubMed] [Google Scholar]
- [72].Clark RA, & Szot S (1981). The myeloperoxidase-hydrogen peroxide-halide system as effector of neutrophil-mediated tumor cell cytotoxicity. J Immunol, 126(4), 1295–1301. [PubMed] [Google Scholar]
- [73].Slivka A, LoBuglio AF, & Weiss SJ (1980). A potential role for hypochlorous acid in granulocyte-mediated tumor cell cytotoxicity. Blood, 55(2), 347–350. [PubMed] [Google Scholar]
- [74].Weiss SJ, & Slivka A (1982). Monocyte and granulocyte-mediated tumor cell destruction. A role for the hydrogen peroxide-myeloperoxidase-chloride system. J Clin Invest, 69(2), 255–262. doi: 10.1172/jci110447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Saito H, Fukumura D, Kurose I, Suematsu M, Tada S, Kagawa T, … Tsuchiya M (1992). Visualization of oxidative processes at the cellular level during neutrophil-mediated cytotoxicity against a human hepatoma cell line, HCC-M. Int J Cancer, 51(1), 124–129. doi: 10.1002/ijc.2910510122 [DOI] [PubMed] [Google Scholar]
- [76].Granot Z, Henke E, Comen EA, King TA, Norton L, & Benezra R (2011). Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell, 20(3), 300–314. doi: 10.1016/j.ccr.2011.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sionov RV, Fainsod-Levi T, Zelter T, Polyansky L, Pham CT, & Granot Z (2019). Neutrophil Cathepsin G and Tumor Cell RAGE Facilitate Neutrophil Anti-Tumor Cytotoxicity. Oncoimmunology, 8(9), e1624129. doi: 10.1080/2162402X.2019.1624129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gershkovitz M, Caspi Y, Fainsod-Levi T, Katz B, Michaeli J, Khawaled S, … Granot Z (2018). TRPM2 Mediates Neutrophil Killing of Disseminated Tumor Cells. Cancer Res, 78(10), 2680–2690. doi: 10.1158/0008-5472.Can-17-3614 [DOI] [PubMed] [Google Scholar]
- [79].Gershkovitz M, Fainsod-Levi T, Khawaled S, Shaul ME, Sionov RV, Cohen-Daniel L, … Granot Z (2018). Microenvironmental Cues Determine Tumor Cell Susceptibility to Neutrophil Cytotoxicity. Cancer Res, 78(17), 5050–5059. doi: 10.1158/0008-5472.Can-18-0540 [DOI] [PubMed] [Google Scholar]
- [80].Gershkovitz M, Fainsod-Levi T, Zelter T, Sionov RV, & Granot Z (2019). TRPM2 modulates neutrophil attraction to murine tumor cells by regulating CXCL2 expression. Cancer Immunol Immunother, 68(1), 33–43. doi: 10.1007/s00262-018-2249-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Koga Y, Matsuzaki A, Suminoe A, Hattori H, & Hara T (2004). Neutrophil-derived TNF-related apoptosis-inducing ligand (TRAIL): a novel mechanism of antitumor effect by neutrophils. Cancer Res, 64(3), 1037–1043. doi: 10.1158/0008-5472.can-03-1808 [DOI] [PubMed] [Google Scholar]
- [82].Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, … Colgan SP (2014). Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity, 40(1), 66–77. doi: 10.1016/j.immuni.2013.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, … Wagner DD (2012). Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A, 109(32), 13076–13081. doi: 10.1073/pnas.1200419109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, … Stockwell BR (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell, 149(5), 1060–1072. doi: 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Karsch-Bluman A, Feiglin A, Arbib E, Stern T, Shoval H, Schwob O, … Benny O (2019). Tissue necrosis and its role in cancer progression. Oncogene, 38(11), 1920–1935. doi: 10.1038/s41388-018-0555-y [DOI] [PubMed] [Google Scholar]
- [86].Scaffidi P, Misteli T, & Bianchi ME (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature, 418(6894), 191–195. doi: 10.1038/nature00858 [DOI] [PubMed] [Google Scholar]
- [87].Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, … Billiar TR (2005). The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med, 201(7), 1135–1143. doi: 10.1084/jem.20042614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, … Schmidt AM (1999). RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell, 97(7), 889–901. doi: 10.1016/s0092-8674(00)80801-6 [DOI] [PubMed] [Google Scholar]
- [89].Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, … Manfredi AA (2004). HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep, 5(8), 825–830. doi: 10.1038/sj.embor.7400205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, … Tracey KJ (1999). HMG-1 as a late mediator of endotoxin lethality in mice. Science, 285(5425), 248–251. doi: 10.1126/science.285.5425.248 [DOI] [PubMed] [Google Scholar]
- [91].Shi Y, Evans JE, & Rock KL (2003). Molecular identification of a danger signal that alerts the immune system to dying cells. Nature, 425(6957), 516–521. doi: 10.1038/nature01991 [DOI] [PubMed] [Google Scholar]
- [92].Gordon TP, Kowanko IC, James M, & Roberts-Thomson PJ (1985). Monosodium urate crystal-induced prostaglandin synthesis in the rat subcutaneous air pouch. Clinical and experimental rheumatology, 3(4). [PubMed] [Google Scholar]
- [93].Martinon F, Petrilli V, Mayor A, Tardivel A, & Tschopp J (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature, 440(7081), 237–241. doi: 10.1038/nature04516 [DOI] [PubMed] [Google Scholar]
- [94].Decker P, Singh-Jasuja H, Haager S, Kotter I, & Rammensee HG (2005). Nucleosome, the main autoantigen in systemic lupus erythematosus, induces direct dendritic cell activation via a MyD88-independent pathway: consequences on inflammation. J Immunol, 174(6), 3326–3334. doi: 10.4049/jimmunol.174.6.3326 [DOI] [PubMed] [Google Scholar]
- [95].Ronnefarth VM, Erbacher AIM, Lamkemeyer T, Madlung J, Nordheim A, Rammensee HG, & Decker P (2006). TLR2/TLR4-Independent Neutrophil Activation and Recruitment upon Endocytosis of Nucleosomes Reveals a New Pathway of Innate Immunity in Systemic Lupus Erythematosus. JOURNAL OF IMMUNOLOGY -BETHESDA-, 177(11), 7740–7749. [DOI] [PubMed] [Google Scholar]
- [96].Decker P, Wolburg H, & Rammensee HG (2003). Nucleosomes induce lymphocyte necrosis. Eur J Immunol, 33(7), 1978–1987. doi: 10.1002/eji.200323703 [DOI] [PubMed] [Google Scholar]
- [97].Basu S, Binder RJ, Suto R, Anderson KM, & Srivastava PK (2000). Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-κB pathway. International Immunology, 12(11), 1539. [DOI] [PubMed] [Google Scholar]
- [98].Panjwani NN, Popova L, & Srivastava PK (2002). Heat shock proteins gp96 and hsp70 activate the release of nitric oxide by APCs. J Immunol, 168(6), 2997–3003. doi: 10.4049/jimmunol.168.6.2997 [DOI] [PubMed] [Google Scholar]
- [99].Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, … Calderwood SK (2000). HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. NATURE MEDICINE, 6, 435–442. [DOI] [PubMed] [Google Scholar]
- [100].Melcher A, Todryk S, Hardwick N, Ford M, Jacobson M, & Vile RG (1998). Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. NATURE MEDICINE, 4(5), 581–587. [DOI] [PubMed] [Google Scholar]
- [101].Binder RJ, Anderson KM, Basu S, & Srivastava PK (2000). Cutting edge: heat shock protein gp96 induces maturation and migration of CD11c+ cells in vivo. Journal of immunology (Baltimore, Md. : 1950), 165(11), 6029–6035. [DOI] [PubMed] [Google Scholar]
- [102].Ishii KJ, Suzuki K, Coban C, Takeshita F, Itoh Y, Matoba H, … Klinman DM (2001). Genomic DNA Released by Dying Cells Induces the Maturation of APCs. J Immunol The Journal of Immunology, 167(5), 2602–2607. [DOI] [PubMed] [Google Scholar]
- [103].Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, & Rifkin IR (2004). Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J Exp Med, 199(12), 1631–1640. doi: 10.1084/jem.20031942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, & Marshak-Rothstein A (2002). Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature, 416(6881), 603–607. doi: 10.1038/416603a [DOI] [PubMed] [Google Scholar]
- [105].Hussain SP, & Harris CC (2007). Inflammation and cancer: an ancient link with novel potentials. Int J Cancer, 121(11), 2373–2380. doi: 10.1002/ijc.23173 [DOI] [PubMed] [Google Scholar]
- [106].Tlsty TD, & Gascard P (2019). Stromal directives can control cancer. Science, 365(6449), 122–123. doi: 10.1126/science.aaw2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Sengupta S, Lohse CM, Leibovich BC, Frank I, Thompson RH, Webster WS, … Kwon ED (2005). Histologic Coagulative Tumor Necrosis as a Prognostic Indicator of Renal Cell Carcinoma Aggressiveness. CANCER -PHILADELPHIA THEN HOBOKEN-, 104(3), 511–520. [DOI] [PubMed] [Google Scholar]
- [108].Gabay C, & Kushner I (1999). Acute-phase proteins and other systemic responses to inflammation. N Engl J Med, 340(6), 448–454. doi: 10.1056/NEJM199902113400607 [DOI] [PubMed] [Google Scholar]
- [109].Beutler B, & Cerami A (1988). The history, properties, and biological effects of cachectin. Biochemistry, 27(20), 7575–7582. doi: 10.1021/bi00420a001 [DOI] [PubMed] [Google Scholar]
- [110].Galluzzi L, Buque A, Kepp O, Zitvogel L, & Kroemer G (2017). Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol, 17(2), 97–111. doi: 10.1038/nri.2016.107 [DOI] [PubMed] [Google Scholar]
- [111].Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, … Krysko DV (2016). Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep, 15(2), 274–287. doi: 10.1016/j.celrep.2016.03.037 [DOI] [PubMed] [Google Scholar]
- [112].Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, … Albert ML (2015). RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science, 350(6258), 328–334. doi: 10.1126/science.aad0395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Friedmann Angeli JP, Krysko DV, & Conrad M (2019). Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. doi: 10.1038/s41568-019-0149-1 [DOI] [PubMed] [Google Scholar]
- [114].Mittal D, Gubin MM, Schreiber RD, & Smyth MJ (2014). New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr Opin Immunol, 27, 16–25. doi: 10.1016/j.coi.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Zhang Y, & Ertl HC (2016). Starved and Asphyxiated: How Can CD8(+) T Cells within a Tumor Microenvironment Prevent Tumor Progression. Front Immunol, 7, 32. doi: 10.3389/fimmu.2016.00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, & Chandel NS (2007). The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol, 177(6), 1029–1036. doi: 10.1083/jcb.200609074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Chen X, Song M, Zhang B, & Zhang Y (2016). Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid Med Cell Longev, 2016, 1580967. doi: 10.1155/2016/1580967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, … Kreutz M (2007). Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood, 109(9), 3812–3819. doi: 10.1182/blood-2006-07-035972 [DOI] [PubMed] [Google Scholar]
- [119].Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di Trapani G, … Rubartelli A (2002). Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci U S A, 99(3), 1491–1496. doi: 10.1073/pnas.022630299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Hangai S, Ao T, Kimura Y, Matsuki K, Kawamura T, Negishi H, … Yanai H (2016). PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc Natl Acad Sci U S A, 113(14), 3844–3849. doi: 10.1073/pnas.1602023113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Cekic C, & Linden J (2016). Purinergic regulation of the immune system. Nat Rev Immunol, 16(3), 177–192. doi: 10.1038/nri.2016.4 [DOI] [PubMed] [Google Scholar]
- [122].Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, … Kroemer G (2015). Combinatorial strategies for the induction of immunogenic cell death. Front Immunol, 6, 187. doi: 10.3389/fimmu.2015.00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Teijeira A, Garasa S, Ochoa MC, Villalba M, Olivera I, Cirella A, … Melero I (2020). IL8, Neutrophils, and NETs in a Collusion against Cancer Immunity and Immunotherapy. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-20-1319 [DOI] [PubMed] [Google Scholar]
- [124].Bhat KP, Salazar KL, Balasubramaniyan V, Wani K, Heathcock L, Hollingsworth F, … Aldape KD (2011). The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev, 25(24), 2594–2609. doi: 10.1101/gad.176800.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Lenting K, Verhaak R, ter Laan M, Wesseling P, & Leenders W (2017). Glioma: experimental models and reality. Acta Neuropathologica, 133(2), 263–282. doi: 10.1007/s00401-017-1671-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Dolma S, Lessnick SL, Hahn WC, & Stockwell BR (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell, 3(3), 285–296. doi: 10.1016/s1535-6108(03)00050-3 [DOI] [PubMed] [Google Scholar]
- [127].Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, … Stockwell BR (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature, 447(7146), 864–868. doi: 10.1038/nature05859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, … Stockwell BR (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell, 156(1–2), 317–331. doi: 10.1016/j.cell.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, & Chi JT (2019). The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep, 28(10), 2501–2508 e2504. doi: 10.1016/j.celrep.2019.07.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Noch E, & Khalili K (2009). Molecular mechanisms of necrosis in glioblastoma: the role of glutamate excitotoxicity. Cancer Biol Ther, 8(19), 1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]